

Abstract
Abstract
The rapid advances in sequencing technology have led to an explosion of sequence data. Sequence alignment is the central and fundamental problem in many sequence analysis procedure, while local alignment is often the kernel of these algorithms. Usually, Smith–Waterman algorithm is used to find the best subsequence match between given sequences. However, the high time complexity makes the algorithm time-consuming. A lot of approaches have been developed to accelerate and parallelize it, such as vector-level parallelization, thread-level parallelization, process-level parallelization, and heterogeneous acceleration, but the current researches seem unsystematic, which hinders the further research of parallelizing the algorithm. In this paper, we summarize the current research status of parallel local alignments and describe the data layout in these work. Based on the research status, we emphasize large-scale genomic comparisons. By surveying some typical alignment tools’ performance, we discuss some possible directions in the future. We hope our work will provide the developers of the alignment tool with technical principle support, and help researchers choose proper alignment tools.
Graphic abstract

Keywords: Smith–Waterman algorithm, Vector-level parallelization, Intra-sequence alignment, Inter-sequence alignment
Introduction
Sequence alignment is one of the most significant techniques in bioinformatics. The result of sequence alignment is the basis of many other steps. It can be used to find the differences and similarities between aligned sequences [1], which is the premise of biological sequence recognition, structure prediction, and function analysis [2]. Taking the coronavirus (COVID-19) as an example, scientists identified its common features by aligning it against other viruses [3]. However, sequence alignment is a highly time-consuming task. In recent years, as the parallel alignment algorithm has continuously matured and optimized, the computation time has reduced significantly.
Depending on the alignment method used, sequence alignment algorithms can generally be classified into two main types: global alignment and local alignment [4]. In 1970, Saul B. Needlman and Christian D. Wunsch proposed the Needleman–Wunsch (NW) algorithm to find the best match in the entire sequences [5]. Then, in 1981, based on the NW algorithm, Temple F. Smith and Michael S. Waterman developed a local alignment algorithm, which was afterward called the Smith–Waterman (SW) algorithm, to find the best subsequence match between sequence pairs [6]. Both the NW and SW algorithms apply dynamic programming [7] (DP) to compute the sequence alignment, which makes the two algorithms have quadratic time complexities. Accordingly, large-scale sequence alignment is computationally demanding [4, 8]. Significant efforts have been made to accelerate the process. Most of these tasks are accelerated by parallel-computing, including vector-level, thread-level, process-level, and heterogeneous parallelization. In this review, we surveyed these parallel approaches of the SW algorithms and the scopes of their applications. Then, we discussed the development trend of the alignment tools.
The rest of this paper is organized as follows. Section 2 reviews the principle of the SW algorithm and existing common parallel approaches. Section 3 analyzes the parallelization of the SW algorithm based on different parallel approaches. Section 4 presents the discussion.
Preliminaries
Smith–Waterman Procedure
For a given sequence S, define S[i] as the i-th character, while S[i, j] denotes the substring from position i to position j. and denote the target sequence and query sequence, respectively. We assume that the target sequence is placed horizontally, while the query sequence is placed vertically in the SW alignment matrix. is the horizontal coordinate and is the vertical coordinate.
The SW algorithm is used to find the best subsequence match between the sequence and . This algorithm consists of two phases: matrix filling and backtracking. The first phase calculates the alignment matrix between sequences and , while the second phase searches for the best subsequence match.
The original SW algorithm uses linear gap costs to calculate the alignment matrix. Gotoh [9] modified the SW algorithm with affined gap costs. The algorithm can be defined as follows.
| 1 |
where indicates the alignment score of and . and denotes the influence of the previous column and row on the current score, respectively. and are the two symbols of the gap open and extension penalty, respectively. is the scoring matrix, it is usually used to calculate the matching or mismatching scores between the symbols and . Additionally, define , and to be equal to 0 when the index i or j is less than 1.
Once the computation of the alignment matrix is complete, we assume that the cell (, ) stores the optimal alignment score. The backtracking phase would start from (, ) until it reaches a cell with a value equal to zero.
Compared with the other phase in this algorithm, the alignment matrix calculation is far more time-consuming. With the length of the target and query sequence and of m and n (assuming ), the computational and space complexity of the algorithm is equal to O(mn) and O(m), respectively, [10].
Parallel Technologies
Based on the vector-level, thread-level, process-level, and heterogeneous parallelization, existing parallel technologies can be classified into four categories, as outlined below.
Vector-Level
Single Instruction Multiple Data (SIMD), also known as vector-level parallelization, uses a controller to control multiple processors and simultaneously perform the same operation on each group of data to achieve spatial parallelism [11]. In short, it executes one instruction to process multiple data at the same time. Figure 1 illustrates the difference between the scalar and vector operations. The scalar operation can only operate one pair of data points at a time; by contrast, the vector addition instruction can operate eight pairs of data points simultaneously.
Fig. 1.
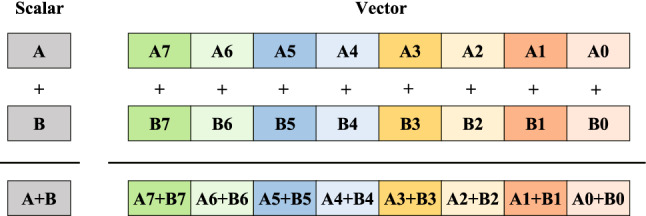
Comparison between scalar operation and vector operation
Intel was the first to introduce Multi Media Extensions (MMX) in 1996. MMX pioneered the SIMD instruction sets and can process 64 bits of data at a time. In 1999, Intel upgraded the MMX and introduced streaming SIMD Extensions (SSE). SEE expanded the vector processing capacity from 64 bits to 128 bits [12]. Subsequently, Intel developed the SSE2 (2000), SSE3 (2004), and SSE4 (2007) instruction sets. In 2008, Intel developed Advanced Vector Extensions (AVX), which expanded the register size to 256 bits and increased the floating-point performance up to twice that of the SSE.
Thread-Level
Multiple Instructions, Multiple Data (MIMD) computers can be divided into the shared memory system and the distributed memory system. In the shared memory system, all computing units share a memory area. Figure 2A presents a simple model of the shared memory system.
Fig. 2.

Distributed and shared memory system model
Thread-level parallelization approaches such as POSIX Threads (Pthreads) are based on a shared memory architecture [13]. It contains a library that can be linked to a C program. Pthreads creates and controls threads through a set of custom APIs [13, 14]. Compared with other thread-level parallelization tools, such as OpenMP [13, 15, 16], Pthreads is a low-level API. This makes it more challenging to program, but more efficient during execution.
Process-Level
Unlike the shared memory system, each core in this system can only access the memory is associated with in the distributed memory system. Figure 2B presents a simple model of the distributed memory system.
Message-Passing Interface (MPI) is a cross-language communication protocol. This is a process-level parallelization approach that is commonly used to achieve parallels in distributed memory systems. Message-passing refers to cases in which each process has an independent stack and code segment when executed in parallel. As independent programs, the information interaction between processes can be completed by explicitly calling communication functions [13]. Notably, MPI is a programming interface standard, not a specific programming language.
Heterogeneous Parallel
Nowadays, many computer clusters contain multiple high-performance processing units. Different processing units can perform different computational tasks, which provides the possibility for heterogeneous parallels. Compared with homogeneous parallels, heterogeneous parallels are focused more on specificity. This approach typically comprises a standard processing unit and a battery of specialized processing units.
Popular heterogeneous accelerators include FPGA, GPU, Xeon Phi, etc. FPGA is a type of circuit that allows for programming by users after manufacturing. It utilizes hardware description language (HDL) for this programming, thus accomplishing a specific task [17]. GPU consists of a large number of streaming multiprocessors (SM). An SM comprises multiple streaming processors (SP), along with other resources, such as a warp scheduler, register, and shared memory. More specific instructions and tasks are processed on the SP; as its function is similar to that of threads in the CPU [18], the amount of SP determines the GPU’s parallel processing capability [19]. Xeon Phi coprocessor [20, 21] is often used as a high-performance computing (HPC) accelerator card. It uses a set of shared-memory coprocessors to accomplish parallelism, which provides a solid foundation to parallel the Smith–Waterman algorithm [22, 23]. However, the heterogeneous parallel needs to be accommodated among different devices, which makes it more cumbersome when programming.
The above parallel technologies implement parallel computing of data by different means. The main advantage is that they can process a large number of data simultaneously, thus significantly reduces the running time of the program. Noteworthily, parallel programs have some extra overheads at runtimes, such as context switching, memory, and communication overhead. These overheads will have some impact on program performance. Generally speaking, when processing the mass of data, the running time of these overheads accounts for a small proportion and has little impact on the results of the program. But for the situation with small amounts of data, the time wasted on these overheads can be close to or even more than the program runs. This will severely degrade program performance.
Parallelization on Smith–Waterman Algorithm
Vector-level Parallelization
As described in Eq. 1, the calculation of depends on the value of , and . Meanwhile, the calculation of depends on the value of and , the calculation of depends on that of and . By observing the position of the above cell, we can determine that each cell’s value depends on the upper-left, left and upper adjacent cells’ values [24]. Figure 3A presents the data dependencies of each cell in the Smith-Waterman algorithm.
Fig. 3.

Data dependencies in the alignment matrix
Existing solutions aimed at eliminating data dependencies can be classified into the intra-sequence alignment and the inter-sequence alignment [25]. In the remainder of this subsection, we illustrate these two solutions in detail.
Intra-sequence Parallelization
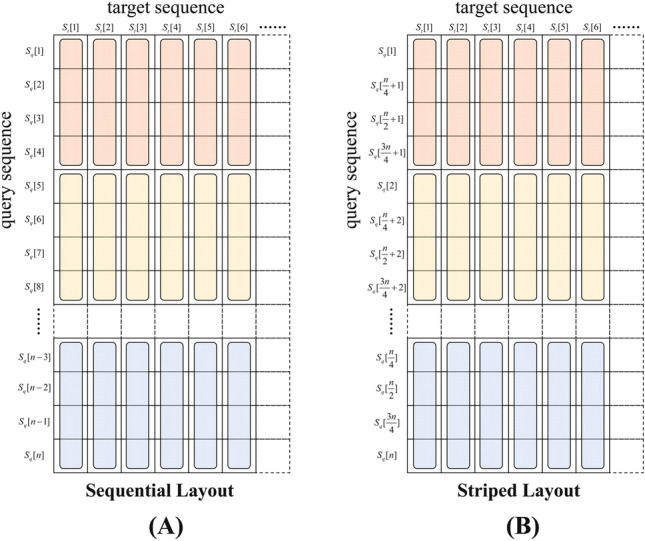
The intra-sequence alignment focuses on accelerating alignment of one single pair of sequences. Figure 4 presents the three main layouts of intra-sequence alignment: anti-diagonal layout [26] (A), sequential layout [24] (B), and striped layout [27] (C).
Fig. 4.

Three intra-sequence alignment approaches
Anti-diagonal Layout was first introduced by Wozniak in 1997 [26]. Figure 3A shows the calculation of and . By examining the data dependencies of the two cells, it is not difficult to discover that the calculation of these two cells is independent of each other which means that they can be executed in parallel. From the above, moreover, we can conclude that the cells that lie in the anti-diagonal direction are independent of these cells. This provides a theoretical basis for parallel computing of the SM algorithm.
For the given alignment matrix, its size is equal to . We define as the diagonal index. For the diagonal d, the starting row index and the ending row index can be calculated as follows:
| 2 |
Based on Eq. 2, the starting column index and the ending column can be calculated as and , respectively, while the number of cells on diagonal d is . We need to run the for-loop d times in the total computation. In each for-loop, moreover, the pieces of data need to be calculated. Due to that, may sometimes not be divisible by the number of elements processed in the SIMD register, we add some dummy symbols to the target and query sequence, which will not influence the final result of the alignment matrix.
When computing any cell (i, j) on the diagonal d, the value of on the diagonal and the values of , , and on the diagonal are needed. Therefore, the program allocates four intermediate buffers to store the values of H on the diagonal , H, E, and F on the diagonal , respectively, [22]. The advantage of this anti-diagonal layout is that it eliminates the data dependencies in the alignment matrix. The drawback here is obvious: namely, it does a large amount of useless computations, which results in a massive waste of computing resources.
Anti-diagonal layout implements the parallel computation of values in the alignment matrix. It does not change the computational and space complexity of SW algorithm. The result is proved correct by Wozniak [26].
Sequential Layout was first proposed by Rognes and Seeberg in 2000 [24]. Figure 4B presents the sketch map of the sequential layout.
| 3 |
In each for-loop of the sequential layout, one residue of the target sequence is aligned to a whole piece of the query sequence. To accelerate the process of alignment, the query sequence is divided into segments of equal length Seg, where the length of Seg is equal to the number that the SIMD register can process at a time. We assume that the SIMD register can process pieces of elements simultaneously. Thus, the length of each segment l is 4, and the segment’s number can be calculated as . Suppose that the query sequence is not divisible by l, in that case, some dummy symbols are added as padding, which will also not influence the final result. The general expression of the query sequence segments is as follows: , where k is the segment index. Figure 5A and Eq. 3 present the query sequence segment layout when , on the condition that n is divisible by l. Each segment in the equation is processed by a SIMD register.
Fig. 5.
Sequential layout and striped layout
As Fig. 3B illustrates, the red arrows show the data dependencies on the diagonal direction, the blue ones show the dependencies in the horizontal direction, and the green ones show it in the vertical direction. Suppose we want to calculate the values in for column j, which contains , , and . We require one vector to store the values of H for cell , cell , cell and cell , two vectors to store the values of H and E for cell , cell , cell and cell , and two vectors to store the values of H and F for cell , cell , cell and cell . By observing the computational dependencies, the values of the four cells in the vector of diagonal H, horizontal H and horizontal E can be calculated and stored in the intermediate buffer in advance, enabling the four cells’ values in the vector of current H to be calculated in parallel. However, in the vertical H and F, the value of each cell in the vector relies on the value of the upper cell.
One feasible solution is the Lazy-F evaluation [27] (sometimes referred to as SWAT-like optimization [25]). The core concept behind this approach is as follows. Equation 1 shows that the calculation of relays on the value of and . By checking the values of and in the alignment matrix, it can be determined that the values of most cells are below . If each value of the four cells in the vector is less than , then can be ignored when computing , which would greatly simplify the computations [24, 28]. For cases in which these values are above the threshold, a Lazy-F loop is added to go through and correct the .
Algorithm 1 outlines the pseudocode of the sequential layout. The algorithm’s procedure can be divided into two key phases: the outer loop and the inner loop. The outer loop is responsible for the target sequence. All values in the first F vector are set to 0 on every column in the inner loop. Subsequently, we follow the pseudocode to calculate the H vector. After the calculation is complete, each element in the H vectors is checked and the errors are corrected during the Lazy-F loop.
Sequential Layout also needs to calculate the whole alignment matrix, so its computational complexity remains the same as O(mn), and the space complexity also equals O(m). Rognes and Seeberg verified this algorithm’s correctness and it has better performance than previous algorithms [25].
Striped Layout is a modified version of the sequential layout. In 2007, Farrar refined the sequential layout and developed the striped layout [27], which follows the main idea of the sequential layout but changes the arrangement of the query sequence. Figure 4C presents the basic form of the striped layout. For ease of comprehension, the order of the query sequence is rearranged in the vertical coordinate, as shown in Fig. 5B.
| 4 |
Due to the different arrangement of the query sequence, the notations are redefined. In the striped layout, the division of the query sequence follows the idea of the sequential layout. But each segment’s length l changes which equals to , where r denotes the number of elements capable of being processed at one time by the SIMD register. For the query sequences that are not divisible by r, some dummy symbols are padded into them. The query segments are defined as = , , , , , where represents the index of segments. The vector takes in charge of the elements which have the same index i in segments on column j. Equation 4 outlines the segment layout when ; here, elements marked in red are processed by the vector.
| 5 |
The rearrangement of the query sequence also leads to changes in the positions of the elements in the scoring matrix M. To make the general formula of vector easier to understand, we use the symbol M(i, j) to represent the former symbol . Accordingly, the vector stores the values of , , , , M(kl, j). Equation 5 shows the vectors of the scoring matrix M in column j when .
Here, the calculation of is the sum of and on column j. To simplify the calculation process, the program preallocates two buffers to store the intermediate H vectors [27]. We use the notations and to represent the two buffers. They are used to store the previous and current column’s H vectors, respectively. Once on a loop, stores the previous column’s H vectors, and stores the current ones. Then, on the next loop, they swapped the values between each other. Now stores the current column’s H vectors, and is ready to store the next column’s H vectors. The remaining loops can be constructed in the same manner.
Figure 6A presents the data dependencies of the H vectors, except for the first one, on the adjacent columns. Each element in the vector depends on the value of the same position in the vector. Figure 6B presents the elements’ data dependencies in the last H vector of each column. To align the values between the two vectors, the values are needed to shift to the left in the last H vector of the previous column. Figure 7 illustrates the left shifting operation of the last H vector on column . The left figure presents the dependencies of each value before shifting, while the right figure presents the situation after shifting.
Fig. 6.

Data dependencies of matrix H and E in striped layout
Fig. 7.
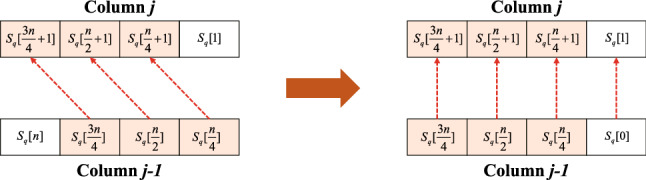
Data dependencies of the first and last H vectors between the adjacent columns
Figure 6C illustrates the dependencies of each vector in matrix E. Values of the elements in each vector in the current column rely on that of the same position in the previous column. Therefore, we simply need to load the result of E vectors in the previous column when calculating the E vectors in the current column.
Figure 8A presents the data dependencies between the adjacent F vectors on the same column, while Fig. 8B shows the same dependencies between the first and the last. To align the last F vector to the first, its values are shifted to the left (the same operation as that in the last H vector). The operation of shifting values in the last F vector is illustrated in Fig. 8C. By observing the cells in the matrix, it can be found that most values of F remain at zero, while H is less than the threshold [27, 28]. Therefore, Lazy-F evaluation is implemented to eliminate the data dependencies between the F vectors.
Fig. 8.

Data dependencies of the F vectors on each column
Based on the discussion above, the striped layout is a modification of the sequential layout. Algorithm 2 presents the pseudocode of the striped layout. It can be divided into three phases: processing the target sequence, processing the query sequence, and correcting the values in H, respectively.
By comparing the striped layout with the sequential layout, it can be determined that the procedure of the sequential layout consists of two key phases: the outer loop and the inner loop. Moreover, the Lazy-F loop is nested in the inner loop. The inner loop first initializes the F vector to zero in the sequential layout and computes the H vector. Subsequently, in each iteration of the inner loop, the Lazy-F loop corrects the values after computing the F vector. The striped layout modifies the procedure; specifically, it takes the Lazy-F loop as a separate loop [27, 29]. Therefore, the Lazy-F loop corrects the errors after the inner loop is finished. The separation of the Lazy-F loop relieves the computation pressure of the iterations in the inner loop. And it also improves the efficiency of the Lazy-F loop.
Due to that striped layout implements a similar computing process as the sequential layout, its computational and space complexity remains the same as O(mn) and O(m). The experiment done by Farrar showed that striped layout also does not change the SW algorithm’s correctness [27].
Farrar compared the performance of anti-diagonal layout, sequential layout, and striped layout in 2006, by testing these three layouts on the same computer configuration with scoring matrices BLOSUM62 and BLOSUM50. When using the same and , the anti-diagonal, sequential, and striped layout complete the search with an average of 352 MCUPS (million cell updates per second), 816 MCUPS, and 2553 MCUPS separately on the scoring matrices BLOSUM62. And on the scoring matrices BLOSUM50, the speed of their average search is 351 MCUPS, 374 MCUPS, and 1817 MCUPS separately [27]. The results show that despite the influence of the scoring matrix, the striped layout performs best among the three intra-sequence alignment layouts. The sequential layout’s performance is far behind the striped layout but still better than the anti-diagonal layout.
Inter-sequence Parallelization
The intra-sequence alignment is typically used to accelerate the alignment between one pair of the target and query sequences. In the actual scenario, multiple target sequences are sometimes aligned to one or many query sequences. The inter-sequence alignment is proposed to handle these two cases. To facilitate better comprehension of the inter-sequence alignment, we first define two notations and , which represent the number of target and query sequences, respectively.
Many-to-one Layout was first proposed by Alpern et al. in 1995 [30]. It is applied to the case in which multiple target sequences are aligned to one target sequence [30]; in other words, the target and query sequence satisfy the condition where and .
Figure 9A illustrates the case of the many-to-one layout where and . In this figure, there are four alignment matrices, each of which have the same query sequence but different target sequences. We assume that each vector stores four values. The values of the same position in the alignment matrices are stored in a vector [25]; thus, the red, orange, blue and green cells in the figure are processed simultaneously.
Fig. 9.

Inter-sequence alignment
Due to that, the target sequence file may have multiple sequences. The target sequence is usually read in the order it is presented in the original file. Figure 10 illustrates how the many-to-one layout processes the target sequence in parallel. Each vector processes the residues of the same position in the four target sequences. The first four vectors processed are depicted in red, orange, blue, and green in this figure. Each target sequence is partitioned into blocks of equal length. Four blocks in the vertical direction are processed as a group. For the target sequences that are not an integer multiple of four, padding is added in the form of dummy symbols, which are revealed as dashes on a grey background. The black triangle indicates the start of new target sequences [25].
Fig. 10.

Blocks of target sequence computed simultaneously
Many-to-many Layout is a modification of the many-to-one layout [31]. It is applied to the case in which multiple target sequences are aligned to multiple query sequences. The target and query sequences satisfy the condition in which and .
Figure 9B illustrates the differences between the two inter-sequence layouts. In the many-to-many layout, each target sequence is aligned to one query sequence. In this figure, four target sequences are aligned to four query sequences, respectively. Each pair of target and query sequences has an alignment matrix [31]. The values of the same position in the alignment matrix are stored in a SIMD vector. Therefore, this approach enables four pairs of target and query sequences to be aligned in parallel.
Compared with the many-to-one layout, the many-to-many layout is more significant in practical scenarios. There are two major reasons for this. First, many sequence alignments are among the multiple target and query sequences. Second, some sequence alignment tools align multiple target sequences to certain candidate positions of the query sequences.
The two inter-sequence alignment layouts both facilitate the alignment of multiple pairs of sequences in parallel. They have different application scenarios depending on their underlying principle. The main advantage of the inter-sequence alignment is that it eliminates all data dependencies between the sequences to be aligned. These two layouts realized the parallel processing of data, so the computational and space complexity of them does not change as well. Rognes and Rahn verified the correctness of these two layouts, respectively, with the alignment tool SWIPE and SeqAn [31, 32].
SWIPE implements the many-to-one inter-sequence layout. The experiments done by Rognes show that SWIPE is more than twice as fast as the striped layout on the same computer configuration. Furthermore, its performance is less affected by the scoring matrix and query length [32]. SeqAn is a frequently used many-to-many inter-sequence layout. Its performance is very close to SWIPE when choosing the same instruction set [31]. Although the inter-sequence alignment may have a faster speed compared with the intra-sequence alignment, they can not accelerate the alignment between one pair of the query and target sequence. Therefore, the choice of specific layouts depends on the actual scenario.
Thread-Level Parallelization
In an attempt to further improve the parallel degree of the sequence alignment tools, many of them have implemented thread-level parallelization (e.g. KSW, KSW2 [33, 34], libssa [25], SeqAn [31], SWIPE [25], SWPS3 [35], etc). Modern processors typically have multiple computing cores. Moreover, with hyper-threading (HT) technology, a processor may have more logical cores than its physical cores. A sequence alignment tool usually obtains optimal performance when the number of threads is equal to the number of logical cores.
Thread-level parallelization consists of two main schemes. The first of these follows the concept of the inter-sequence alignment. In this scheme, multiple pairs of the target and query sequences are aligned simultaneously. The set of these pairs are divided into many subsets; here, the number of subsets usually equals the number of threads. Each thread is responsible for the sequences it allocated. Therefore, the distribution of the sequences should be considered to ensure load balancing.
The second scheme focuses on speeding up the alignment between one pair of target and query sequences, in a way that more closely resembles the intra-sequence alignment. This scheme partitions the alignment matrix into a lot of tiles, and each thread processes a tile. The tiled scheme processes these tiles in the anti-diagonal direction [4] to eliminate the data dependencies. Each tile has the size , while the number of the tile is equal to . In each tile, vector-level parallelization is deployed to speed up the computation. However, this scheme has to add some dummy symbol padding into the target and query sequence to ensure the alignment matrix is divisible by the tile, which will lead to a waste of computing resources.
Process-Level Parallelization
Process-level parallelization can solve the single-node performance bottleneck problem. The implementation of MPI enables the task to be distributed to multiple nodes, which significantly reduces the workload of a single node. The first mode works to distribute the sequences into different nodes. Each node is responsible for the computation of alignment matrices it distributed. The second mode follows the idea of the tiled scheme in the thread-level parallelization.
Generally speaking, there are two possible approaches to sequence distribution [36]. The first of these approaches is by number. The sequence file consists of many pieces of sequences. We assume that the number of the sequences equals seqNum. Each node processes pieces of sequences in this approach (where p is the number of nodes). However, each sequence’s length in the source file is typically unequal, which leads to load imbalance. One feasible solution is to distribute the sequences by their pointers. In this approach, the of residues are aligned in each node, where fileSize is the sequence file’s data size. The drawback of this approach is that it requires redirecting the pointer of each node to the head of a sequence after distribution. Compared with the first approach, this approach runs faster and more efficiently.
The second mode is also developed with the goal of accelerating the computation in one alignment matrix. Based on the tiled scheme, it assembles multiple tiles into a block with a size equal to . An alignment matrix is then divided into blocks, after which each block is further partitioned into tiles. Each node is responsible for the computation of a block. In each block, the computation follows the concept of the tiled scheme in thread-level parallelization. For cases in which the alignment matrix is not divisible by a block, some dummy symbol padding are added, which will lead to a waste of computing resources. Moreover, the communication among the nodes will also slow down the performance.
Heterogeneous Parallelization
The sequence alignment tool comprises multiple phases. In other phases, due to the complexity of the operations involved, CPU is usually utilized for processing. While the local alignment phase is typically the most time-consuming but relatively simple. We could also use other devices, such as GPU, FPGA, and Xeon Phi, etc.
CUDASW++ [37], CUDAlign 3.0 [38], manymap [39], ADEPT [40], etc. combine the CPU and GPU to accelerate the alignment. Due to the fact that GPU has multiple SPs, and with the implementation on GPU clusters [41], it can achieve more parallel capability than CPU [42]. GPU is an efficient platform for long-read alignment tasks, but it does not perform very well when processing short-read alignment tasks. Moreover, the memory access between the CPU and GPU is a key link in this heterogeneous architecture. An unreasonable level of memory access will lead to performance degradation [43].
SWIFOLD [32] and OSWALD [44] implement FPGA with OpenCL to reduce the computational times of the SW algorithm. Due to FPGA’s strong computational capability [45], both of the alignment tools can be used for short-read alignment tasks. Compared with the CPU and GPU platform, the FPGA platform typically achieves better performance per watt [46]. With the help of OpenCL [47], they have better portability and lower programming cost than the traditional FPGA alignment tools. However, good versatility and large memory requirements [48] also come at the expense of performance to a certain extent.
SWAPHI-LS [22] first uses the Xeon Phis to accelerate the alignment for long read tasks. With MPI, it can accelerate the computation among multiple nodes. In each node, SWAPHI-LS can achieve thread-level and vector-level parallelization. Other tools, such as XSW [23], SWIMM [49], and MICA [50], also implement the Xeon Phis for acceleration, and all achieve good throughput. However, the waste of computing resources caused by the communication among multiple cores and the memory required of the program are also significant issues to be considered.
In conclusion, a general framework for parallelizing the local alignment can be described as follows: first, distributing the sequences by their pointers to each node with MPI; subsequently, in each node, the sequences are distributed into multiple computing devices. For devices, such as FPAG, GPU, etc., OpenCL is used to coordinate the tasks among them. For devices like CPU or Xeon Phis, vector-level parallelization is implemented to accelerate the alignment in each thread.
Typical Alignment Tools
Many alignment tools have been used in real life, Table 1 lists some typical tools based on different methods.
Table 1.
Some typical alignment tools
| Tool name | Time | Architecture | Methods | Hardware | Speed (GCUPS) |
|---|---|---|---|---|---|
| STRIPED | 2006 | CPU | Striped | Dual Intel Xeon X5650 CPU @ 2.67 GHz | 14.7 |
| SWIPE | 2011 | CPU | Many-to-one | Dual Intel Xeon X5650 CPU @ 2.67 GHz | 106.2 |
| Intel Xeon E5-2695 v3 @2.3GHz | 220.0 | ||||
| SeqAn | 2018 | CPU | Many-to-many | Dual Intel Xeon Gold 6148 CPU @2.4GHz | 194.1 |
| SWAPHI-LS | 2014 | Xeon Phi | Anti-diagonal | Xeon Phi 5110P @1.05GHz | 29.2 |
| XSW | 2014 | Xeon Phi | Many-to-one | Xeon Phi 3120P @1.1GHz | 50.0 |
| CUDASW++ 3.0 | 2013 | CPU + GPU | Many-to-one | Xeon E5-2670 @2.6GHz + Tesla K20c | 298.8 |
| Xeon E5-2695 v3 @2.3GHz + Tesla K20c | 206.2 | ||||
| OSWALD | 2015 | CPU + FPGA | Many-to-one | Xeon E5-2670 @2.6GHz + Altera Stratix V | 178.9 |
| Xeon E5-2695 v3 @2.3GHz + Altera Stratix V | 401.1 | ||||
| SWIMM | 2015 | CPU + Xeon Phi | Many-to-one | Xeon E5-2670 @2.6GHz | 127.5 |
| Xeon E5-2695 v3 @2.3GHz | 354.8 | ||||
| Xeon E5-2670 @2.6GHz + Xeon Phi 3120P @1.1GHz | 165.5 | ||||
| Xeon E5-2695 v3 @2.3GHz + Xeon Phi 3120P @1.1GHz | 450.5 |
Rognes compared STRIPED and SWIPE with the database produced by the formatdb tool in 2011. The results showed that SWIPE with the many-to-one inter-sequence layout performs better than STRIPED with the striped intra-sequence layout [24]. Rahn test SeqAn using the PacBio-Real dataset. The speed of SeqAn with AVX512 reached 192.14 GCUPS. This indicates that the many-to-many layout has a similar performance towards the many-to-one layout [31], the main difference between them is that they apply to different scenarios.
SWAPHI-LS and XSW are two typical alignment tools based on Xeon Phi. The former implements the anti-diagonal layout, while the latter uses the many-to-one layout. Liu et al. and Wang et al. test the SWAPHI-LS with the NCBI Nucleotide database and XSW with the Swiss-Prot database, respectively. The result is obvious, XSW performs much better than SWAPHI-LS [23]. The reason is that the anti-diagonal layout needs to add some dummy symbols to the alignment matrix, which results in a waste of computing resources.
CUDASW++ 3.0, OSWALD, and SWIMM are hybrid CPU-GPU, CPU-FPGA, and CPU-Xeon Phi alignment tools, respectively. Notably, CUDASW++ 3.0 implements the method wavefront on GPU whose algorithm works the same way as anti-diagonal. Rucci et al. test the performance of the three above tools with the Swiss-Prot database in 2015. He used two different host CPUs in the test. They also test these tools on small, medium, and large datasets with different input sequence size [32, 44]. The results show that parallel computing using pure CPUs has reached the bottleneck of performance, and the integration of different computing devices can bring further performance improvement. The performance of hybrid tools varies towards different scenarios, which can be concluded as follows:
The hybrid CPU-GPU tool CUDASW++ 3.0 provides good performance rates for large sequence size. It is an efficient tool for large datasets and similar sequence pairs. This is because GPU has many processing units, which makes it more suitable for processing simple and large amounts of data [32]. Meanwhile, GPU’s price is relatively friendly.
The hybrid CPU-FPGA tool OSWALD’s performance is independent of sequence size and similarity. And it performs good when processing small and medium datasets [32, 44].
The hybrid CPU-Xeon Phi tool SWIMM has a similar performance towards OSWALD when using a high-performance host CPU. The main advantage of SWIMM is the good portability wihch make it more programmer-friendly [51]. But it has poor energy efficiency due to the communication overhead. It can be a good choice when power is not a priority [44].
Conclusion
Parallel computing is a feasible solution to the processing of ever-growing sequence data. In this review, we revised the existing methods of parallelizing the Smith–Waterman algorithm. We specifically analyze the approaches of vector-level parallelization and introduce some typical alignment tools. This work can provide the developers of the alignment tool with basic technical principle support, and help researchers in this area choose proper alignment tools for different scenarios.
Many existing sequence alignment tools have realized the combination of more than one parallelization method. Future work may focus on the integration of multiple kinds of parallelization. In view of each kind of parallelization specialty, the sequence alignment tools need to provide a general API for users to choose from. Another development trend is to develop a customized SW algorithm hardware accelerator. The hardware/algorithm co-designed accelerator can fully utilize the computing performance of components and saves more memory resources than existing alignment tools, which leads to better performance, especially when processing computational demanding tasks.
Acknowledgements
This work was funded by the National Key R&D Program of China (Grant Nos. 2020YFA0709803, 2018YFB0204301) and NSFC Grants (Grant Nos. 62102427, 61972408 and 61772543).
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Yingbo Cui, Email: cuiyingbomail@163.com.
Ang Zhang, Email: zhangang@me.com.
References
- 1.Khan MI, Kamal MS, Chowdhury L. Msupda: a memory efficient algorithm for sequence alignment. Interdiscip Sci Comput Life Sci. 2016;8(1):84–94. doi: 10.1007/s12539-015-0275-8. [DOI] [PubMed] [Google Scholar]
- 2.Kirkness EF, Bafna V, Halpern AL, Levy S, Remington K, Rusch DB, Delcher AL, Pop M, Wang W, Fraser CM, et al. The dog genome: survey sequencing and comparative analysis. Science. 2003;301(5641):1898–1903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- 3.Issa M, Elaziz MA. Analyzing COVID-19 virus based on enhanced fragmented biological local aligner using improved ions motion optimization algorithm. Appl Soft Comput. 2020;96:106683. doi: 10.1016/j.asoc.2020.106683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Schmidt B. Gswabe: faster gpu-accelerated sequence alignment with optimal alignment retrieval for short dna sequences. Concurr Comput Pract Exp. 2015;27(4):958–972. doi: 10.1002/cpe.3371. [DOI] [Google Scholar]
- 5.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48(3):443–453. doi: 10.1016/b978-0-12-131200-8.50031-9. [DOI] [PubMed] [Google Scholar]
- 6.Smith TF, Waterman MS, et al. Identification of common molecular subsequences. J Mol Biol. 1981;147(1):195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 7.Eddy SR. What is dynamic programming? Nat Biotechnol. 2004;22(7):909–910. doi: 10.1038/nbt0704-909. [DOI] [PubMed] [Google Scholar]
- 8.Daily J. Parasail: Simd c library for global, semi-global, and local pairwise sequence alignments. BMC Bioinform. 2016;17(1):1–11. doi: 10.1186/s12859-016-0930-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gotoh O. An improved algorithm for matching biological sequences. J Mol Biol. 1982;162(3):705–708. doi: 10.1016/0022-2836(82)90398-9. [DOI] [PubMed] [Google Scholar]
- 10.Kucherov G. Evolution of biosequence search algorithms: a brief survey. Bioinformatics. 2019;35(19):3547–3552. doi: 10.1093/bioinformatics/btz272. [DOI] [PubMed] [Google Scholar]
- 11.R Intel. C++ compiler 18.0 developer guide and reference, 2019
- 12.Intel Intel. and ia-32 architectures software developer’s manual. Volume 3A: System Programming Guide, Part 1(64):64
- 13.Hennessy JL, Patterson DA. Computer architecture: a quantitative approach. Amsterdam: Elsevier; 2011. [Google Scholar]
- 14.Butenhof DR. Programming with POSIX threads. Boston: Addison-Wesley Professional; 1997. [Google Scholar]
- 15.Dagum L, Menon R. Openmp: an industry standard api for shared-memory programming. IEEE Comput Sci Eng. 1998;5(1):46–55. doi: 10.1109/99.660313. [DOI] [Google Scholar]
- 16.Pacheco P. An introduction to parallel programming. Amsterdam: Elsevier; 2011. [Google Scholar]
- 17.Oliver T, Schmidt B, Nathan D, Clemens R, Maskell D. Using reconfigurable hardware to accelerate multiple sequence alignment with clustalw. Bioinformatics. 2005;21(16):3431–3432. doi: 10.1093/bioinformatics/bti508. [DOI] [PubMed] [Google Scholar]
- 18.Khajeh-Saeed A, Poole S, Perot JB. Acceleration of the smith-waterman algorithm using single and multiple graphics processors. J Comput Phys. 2010;11:4247–4258. doi: 10.1016/j.jcp.2010.02.009. [DOI] [Google Scholar]
- 19.Manavski SA, Valle G. Cuda compatible gpu cards as efficient hardware accelerators for smith-waterman sequence alignment. BMC Bioinform. 2008;9(2):1–9. doi: 10.1186/1471-2105-9-s2-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao M, Lee W-P, Garrison EP, Marth GT. Ssw library: an simd smith-waterman c/c++ library for use in genomic applications. PLoS One. 2013;8(12):e82138. doi: 10.1371/journal.pone.0082138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui Y, Liao X, Zhu X, Wang B, Peng S (2014) mbwa: A massively parallel sequence reads aligner. In: 8th International Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB 2014). Springer, pp 113–120. 10.1007/978-3-319-07581-5_14
- 22.Y Liu, T-T Tran, F Lauenroth, B Schmidt (2014) Swaphi-ls: Smith-waterman algorithm on xeon phi coprocessors for long dna sequences. In: 2014 IEEE International Conference on Cluster Computing (CLUSTER). IEEE, pp 257–265. 10.1109/cluster.2014.6968772
- 23.L Wang, Y Chan, X Duan, H Lan, X Meng, W Liu (2014) Xsw: Accelerating biological database search on xeon phi. In: 2014 IEEE International Parallel & Distributed Processing Symposium Workshops. IEEE, pp 950–957. 10.1109/ipdpsw.2014.108
- 24.Rognes T, Seeberg E. Six-fold speed-up of smith-waterman sequence database searches using parallel processing on common microprocessors. Bioinformatics. 2000;16(8):699–706. doi: 10.1093/bioinformatics/16.8.699. [DOI] [PubMed] [Google Scholar]
- 25.Rognes T. Faster smith-waterman database searches with inter-sequence simd parallelisation. BMC Bioinform. 2011;12(1):1–11. doi: 10.1186/1471-2105-12-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wozniak A. Using video-oriented instructions to speed up sequence comparison. Bioinformatics. 1997;13(2):145–150. doi: 10.1093/bioinformatics/13.2.145. [DOI] [PubMed] [Google Scholar]
- 27.Farrar M. Striped smith-waterman speeds database searches six times over other simd implementations. Bioinformatics. 2007;23(2):156–161. doi: 10.1093/bioinformatics/btl582. [DOI] [PubMed] [Google Scholar]
- 28.Snytsar R (2019) De (con) struction of the lazy-f loop: improving performance of smith waterman alignment. In: 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE). IEEE, pp 7–10. 10.1109/bibe.2019.00011
- 29.Glenn H, Dave S, Mike U, Darrell B et al (2001) The microarchitecture of the pentium® 4 processor. In: Intel technology journal, Citeseer
- 30.Alpern B, Carter L, Gatlin KS (1995) Microparallelism and high-performance protein matching. In: Supercomputing’95: Proceedings of the 1995 ACM/IEEE Conference on Supercomputing. IEEE, p 24. 10.1145/224170.224222
- 31.Rahn R, Budach S, Costanza P, Ehrhardt M, Hancox J, Reinert K. Generic accelerated sequence alignment in seqan using vectorization and multi-threading. Bioinformatics. 2018;34(20):3437–3445. doi: 10.1093/bioinformatics/bty380. [DOI] [PubMed] [Google Scholar]
- 32.Rucci E, Garcia C, Botella G, De Giusti A, Naiouf M, Prieto-Matias M. Swifold: Smith-waterman implementation on fpga with opencl for long dna sequences. BMC Syst Biol. 2018;12(5):43–53. doi: 10.1186/s12918-018-0614-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki H, Kasahara M. Introducing difference recurrence relations for faster semi-global alignment of long sequences. BMC Bioinform. 2018;19(1):33–47. doi: 10.1186/s12859-018-2014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szalkowski A, Ledergerber C, Krähenbühl P, Dessimoz C. Swps3-fast multi-threaded vectorized smith-waterman for ibm cell/be and× 86/sse2. BMC Res Notes. 2008;1(1):1–4. doi: 10.1186/1756-0500-1-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters D, Luo X, Qiu K, Liang P. Speeding up large-scale next generation sequencing data analysis with pbwa. J Appl Bioinform Comput Biol. 2012;1(1):1–6. doi: 10.4172/2329-9533.1000101. [DOI] [Google Scholar]
- 37.Liu Y, Wirawan A, Schmidt B. Cudasw++ 3.0: accelerating smith-waterman protein database search by coupling cpu and gpu simd instructions. BMC Bioinform. 2013;14(1):1–10. doi: 10.1186/1471-2105-14-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Edans FO, Miranda G, de Melo ACMA, Martorell X, Ayguadé E (2014) Cudalign 3.0: Parallel biological sequence comparison in large gpu clusters. In: 2014 14th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing. IEEE, pp 160–169. doi: 10.1109/ccgrid.2014.18
- 39.Feng Z, Qiu S, Wang L, Luo Q (2019) Accelerating long read alignment on three processors. In: Proceedings of the 48th International Conference on Parallel Processing, pp 1–10. 10.1145/3337821.3337918
- 40.Awan MG, Deslippe J, Buluc A, Selvitopi O, Hofmeyr S, Oliker L, Yelick K. Adept: a domain independent sequence alignment strategy for gpu architectures. BMC Bioinform. 2020;21(1):1–29. doi: 10.1186/s12859-020-03720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okada D, Ino F, Hagihara K. Accelerating the smith-waterman algorithm with interpair pruning and band optimization for the all-pairs comparison of base sequences. BMC Bioinform. 2015;16(1):1–15. doi: 10.1186/s12859-015-0744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Payne JL, Sinnott-Armstrong NA, Moore JH. Exploiting graphics processing units for computational biology and bioinformatics. Interdiscip Sci Comput Life Sci. 2010;2(3):213–220. doi: 10.1007/s12539-010-0002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pirkelbauer P, Lin P-H, Vanderbruggen T, Liao C (2020) Xplacer: Automatic analysis of data access patterns on heterogeneous cpu/gpu systems. In: 2020 IEEE International Parallel and Distributed Processing Symposium (IPDPS). IEEE, pp 997–1007. 10.1109/ipdps47924.2020.00106
- 44.Rucci E, Garcia C, Botella G, De Giusti AE, Naiouf M, Prieto-Matias M. Oswald: O pencl smith-waterman on a ltera’s fpga for large protein databases. Int J High Perform Comput Appl. 2018;32(3):337–350. doi: 10.1177/1094342016654215. [DOI] [Google Scholar]
- 45.Chen B, Xu Y, Yang J, Jiang H (2010) A new parallel method of smith-waterman algorithm on a heterogeneous platform. In: International Conference on Algorithms and Architectures for Parallel Processing. Springer, pp 79–90. doi: 10.1007/978-3-642-13119-6_7
- 46.Fei X, Dan Z, Lina L, Xin M, Chunlei Z. Fpgasw: accelerating large-scale smith-waterman sequence alignment application with backtracking on fpga linear systolic array. Interdiscip Sci Comput Life Sci. 2018;10(1):176–188. doi: 10.1007/s12539-017-0225-8. [DOI] [PubMed] [Google Scholar]
- 47.Stone JE, Gohara D, Shi G. Opencl: a parallel programming standard for heterogeneous computing systems. Comput Sci Eng. 2010;12(3):66. doi: 10.1109/mcse.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y-L, Chang B-Y, Yang C-H, Chiueh T-D. A high-throughput fpga accelerator for short-read mapping of the whole human genome. IEEE Trans Parallel Distrib Syst. 2021;32(6):1465–1478. doi: 10.1109/tpds.2021.3051011. [DOI] [Google Scholar]
- 49.Rucci E, García C, Botella G, De Giusti A, Naiouf M, Prieto-Matías M. An energy-aware performance analysis of swimm: smith-waterman implementation on intel’s multicore and manycore architectures. Concurr Comput Pract Exp. 2015;27(18):5517–5537. doi: 10.1002/cpe.3598. [DOI] [Google Scholar]
- 50.Luo R, Cheung J, Edward Wu, Wang H, Chan S-H, Law W-C, He G, Chang Y, Liu C-M, Zhou D, et al. Mica: a fast short-read aligner that takes full advantage of many integrated core architecture (mic) BMC Bioinform. 2015;16(7):1–8. doi: 10.1186/1471-2105-16-s7-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou Y, Zhu Y, Li Y, Fang-Xiang W, Wang J. Parallel computing for genome sequence processing. Brief Bioinform. 2021 doi: 10.1093/bib/bbab070. [DOI] [PubMed] [Google Scholar]