

Abstract
Nowadays, massive genomics and transcriptomics data can be generated at the single-cell level. However, proteomics in this setting is still a big challenge. Despite the great improvements in sensitivity and performance of mass spectrometry instruments and the better knowledge on sample preparation processing, it is widely acknowledged that multistep proteomics workflows may lead to substantial sample loss, especially when working with paucicellular samples. Still, in clinical fields, frequently limited sample amounts are available for downstream analysis, thereby hampering comprehensive characterization at protein level. To aim at better protein and peptide recoveries, we compare existing and novel approaches in the multistep sample preparation protocols for mass spectrometry studies, from sample collection, cell lysis, protein quantification, and electrophoresis/staining to protein digestion, peptide recovery, and LC-MS/MS instruments. From this critical evaluation, we conclude that the recent innovations and technologies, together with high quality management of samples, make proteomics on paucicellular samples possible, which will have immediate impact for the proteomics community.
Keywords: single-cell proteomics, sample preparation, mass spectrometry, paucicellular sample
Introduction
Comprehensive characterization of cells is key to understanding biological processes, also in disease and during response to treatment. Substantial advances have been made in the genomics and transcriptomics fields, facilitating highly detailed description of genes and transcripts at the single-cell level.1−3 However, proteome analysis in this setting, and even from very low cell numbers, is still a challenge.4 Even though remarkable progress has been made in the proteomics field, more specifically in mass spectrometry (MS) by improving sensitivity and performance of instruments and sample preparation strategies,5−8 there are still several limitations.
From cell sample collection to peptide analysis in MS, multiple processing steps can result in sample loss, reduced protein recovery, and limited protein identification.9 These limitations become more prominent when working with paucicellular samples.
In this review, we provide a thorough description of the key points to consider when performing a multistep sample preparation protocol for liquid chromatography (LC)-MS/MS analysis with small sample inputs. More specifically, sample collection, cell lysis, protein quantification, electrophoresis/staining, protein digestion, peptide recovery, and LC-MS/MS instrument topics are discussed. Furthermore, novel approaches around single-cell proteomics are described. Altogether, we present a detailed guide not only for new users of proteomics technologies but also for more experienced researchers interested in the latest advancements in the field.
Proteomics Workflow for Paucicellular Samples
Sample preparation for in-depth high-throughput LC-MS/MS analysis requires multiple steps (Figure 1) that must be performed as accurately as possible to ensure reliability and reproducibility. Indeed, sample loss, which can happen along the entire process, can be very dramatic when handling few hundreds of cells. In the next sections, we present a detailed description of the key points to consider when working with paucicellular samples.
Figure 1.

Proteomics workflow. The main steps for sample preparation are depicted. For each phase, a summary of the best conditions for working with quantity-limited samples is shown. FCS, fetal calf serum; MS, mass spectrometry. Created with BioRender.com.
Cell Sample Collection
From the moment of paucicellular sample collection, several questions arise on how to proceed to minimize sample loss, especially when cell sorting is performed and tube-to-tube transfer is required. Starting the sample preparation for proteomics analysis may differ from, e.g., tissue pieces (requiring a dissociation step)10,11 to purified cell populations (for instance via fluorescence activated cell sorting, FACS).12−14 In any case, several parameters must be considered to mitigate the apoptosis phenomenon that can be triggered during these procedures. This is particularly relevant in proteomics studies, as apoptosis results not only in cell death (implying sample loss) but also in altered proteome profiles that disturb the actual cell state picture.15 Although optimal conditions should be defined for each sample type, here we present general guidelines.
Cell Viability Medium
This medium must ensure the highest cell viability. Depending on the cell type, different commercial media (e.g., RPMI, DMEM) combined with supplements (e.g., bovine serum albumin, BSA, or fetal calf serum, FCS) offer an optimal environment for cell survival during sample preparation, e.g., during cell sorting procedures which can take hours. Additionally, small chemical molecules as protease/phosphatase inhibitors can be added to avoid protein degradation. Nevertheless, the nature of such inhibitors must be considered since they could hamper downstream analysis, e.g., usage of serine protease inhibitors will impair subsequent trypsinization required for shotgun MS, if proper sample washing is not performed before protein digestion. Furthermore, the cell viability medium can include proteins as DNase to avoid cell clumping caused by released DNA.16 However, it is well-known in the proteomics field that adding extra proteins to the samples is not ideal, as they will interfere with the downstream analysis by hindering the detection of the sample proteins of interest.17 For this reason, finding the right balance between further protein identification and cell survival is crucial, since dying/apoptotic cells will show a different proteome. In these situations, a thorough definition of the cell medium must be performed and/or testing diverse cell media might be considered, making it sometimes necessary to choose for an option resulting in less proteins identified but at the highest quality. For instance, our experience for sorted cells has indicated that using a low FCS concentration (10%) for paucicellular samples (∼300 cells after sorting) involving long sorting times (>5 h) does not have a great impact in the proteomics analysis, especially if postsorting cell washing is performed appropriately, but results in high-quality and cell-specific identifications (see Cell Washing).
Temperature
Regardless the sample type to be processed (in-bulk or purified), temperature must be regulated along all steps. This is especially critical before cell lysis, as cells must be kept intact, not only from a structural point of view but also from a molecular perspective. A low temperature (4 °C) will help to slow down metabolic activities that can lead to biochemical changes altering gene expression, transcription, and consequent protein profiles.18 Also, freeze/thaw cycles must be avoided at this stage, as cells can be very sensitive to temperature variations not surviving the storage,19 which can be very dramatic when the starting sample material is very limited.
Cell Washing
Cell washing steps are also critical in paucicellular settings since cell pellets are not visible and the chance of accidentally discarding them after centrifugation is increased. Nonetheless, cells must always be washed in a buffer solution to remove cell debris, contaminants and/or BSA/FCS-containing medium.20 Moreover, centrifugation speed must be adapted to the cell type. Overall, the widely accepted recommendation for mammalian cells is to centrifuge at 300–500g for 5 min to avoid cell disruption. However, it is important to mention that this very low speed might result in loose cell pellets and subsequent sample loss, so extreme caution must be taken while washing.
-
▶
Tip – Conclusion: To avoid cell death and sample loss: (a) process fresh cell samples as soon as possible; (b) keep the cells in a medium with the right balance between extra molecules (e.g., protease/phosphatase inhibitors), additional protein content, and cell viable environment (e.g., RPMI + 10% FCS); (c) in between steps, keep cells at 4 °C and avoid freeze/thaw cycles; (d) wash cells at 300g for 5 min in cold PBS Na+/K+ at pH 7.4 (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) for a maximum of 3 rounds; and (e) proceed immediately with the next step or store cell pellets at −80 °C using snap freezing techniques.
Cell Lysis
In recent years, a plethora of protein extraction methods have been defined, mainly focusing on macroscale samples (i.e., hundreds of thousands of cells) and leaving a gap toward research with limited cell numbers.21 Also, depending on the cell type, the protein(s) of interest, their location, and further downstream applications, specific methods might be required.
The optimal cell lysis condition primarily depends on the structure and organization of the cell. For instance, bacterial cell contents are separated from the outside by much thicker layers (cell membrane and cell wall)22 compared to mammalian cells (only cell membrane),23 requiring harsh cell lysis conditions to release their cell contents. On the other hand, the composition of the mammalian cell membranes also affects the lysis efficiency. These membranes consist of phospholipid bilayers that are originally made of highly hydrophobic fatty acid and hydrophilic glycerol moieties, and contain cholesterol molecules in between, strengthening the bilayers and reducing the cell permeability. The cholesterol distribution depends on the cell type, implying that cells containing more cholesterol require stronger cell lysis protocols.24,25 Thus, the broad cell diversity requires the development of different methods to be applied according to the cell features. Classically, cell lysis methods have been classified as mechanical and nonmechanical.21
Mechanical Lysis Methods
These methods are forceful and especially suited for tissues and cells with a thick cell membrane and/or a cell wall, as a high shear force is produced. Commonly used mechanical lysis strategies employ the high-pressure homogenizer, ultrasonication, and bead mill.21,26 These methods can be quite attractive for high quantities of cells as they offer high lysis efficiency.21 However, mechanical equipment has to be carefully used as complications during the process can occur; e.g., heating produced while lysing might denature the proteins in the sample.26 Cross-contamination is also one of their main drawbacks, and thorough cleaning of equipment is required after each usage. For these reasons, mechanical lysis methods are not ideal for processing paucicellular samples.27
Nonmechanical Lysis Methods
These gentler approaches are classified as (i) physical, (ii) biological, and (iii) chemical. (i) Well-known physical approaches use heat, pressure, and sonication. For instance, the freeze–thaw method employs heat force to disturb cells by recurrent freezing and thawing cycles. Despite the simplicity of this strategy, freeze–thaw cycles can be time-consuming, and not useful to extract temperature-sensitive proteins. Regarding the methods relying on pressure, the osmosis produced by the usage of salt-containing lysis buffers can burst cells as a consequence of the difference in salt concentrations in and outside the cell.21,28 Thus, a common hypotonic lysis buffer containing 10 mEq/L of both Cl– and K+ ions, 15 mEq/L of Na+ and 1 mEq/L of Mg2+ ions will produce cell swelling and subsequent cell burst (assuming the intracellular ionic strength is ∼200 mEq/L).29,30 (ii) Biological methods use different enzymes (e.g., lysozyme for Gram-positive bacteria) and proteases to specifically break down the cell membrane and release its content.21 (iii) In the chemical methods, different buffers are used to lyse the cells and release the protein contents. Detergents, the main active ingredient of these buffers in terms of lysis,28, permeabilize the cell membranes by disrupting the hydrophilic–hydrophobic interactions of the phospholipid bilayers. Commonly used ionic and nonionic detergents in protein lysis buffers are, e.g., sodium dodecyl sulfate (SDS) and CHAPS, Triton X and Tween, respectively.21 SDS is well-known for its high efficiency in cellular lysis; however, it can negatively influence protein digestion and LC-MS analysis.31−33 Since removal of detergents prior to LC-MS/MS analysis is crucial to prevent contamination of mass spectrometers, different methods have been developed to eliminate them (e.g., filter-aided sample preparation, FASP).34,35 Nonetheless, including such cleanup approaches is an extra step that may increase the chance of protein loss, which is not ideal in a microproteomics setting. On top of detergents, additives as salts (e.g., NaCl), metal chelators (e.g., EDTA, to prevent oxidation), and protease/phosphatase inhibitors (to prevent protein degradation and loss of posttranslational modifications, PTMs) can be added to stabilize the proteins. Also, reducing agents as dithiothreitol (DTT) and β-mercaptoethanol are important additives to protect proteins from oxidation caused by metal ions, especially cysteine (Cys) residues.36,37 Likewise, chaotropic agents (e.g., urea, guanidine), which have the ability to damage the hydrogen bonds of water molecules and thereby weakening the hydrophobic interactions between proteins,21 are added to disrupt cell integrity and release proteins.
Also, not only the lysis method but the complexity of the procedure are key parameters when working with paucicellular samples. Then, sample transfer from tube-to-tube during processing should be avoided. Thus, multiple protocols (e.g., in-StageTip digestion (iST) and single-pot solid-phase-enhanced sample preparation (SP3); see New Approaches: Advances and Their Applications for more information) that minimize cell and protein/peptide losses while processing have been described.9,38
Finally, combining different nonmechanical methods can be effective in extracting proteins in a microproteomics setup. For example, Bensaddek and colleagues successfully extracted proteins combining chemical (chaotropic agents) and physical methods (freeze–thaw and sonication).39
-
▶
Tip – Conclusion: To get efficient lysis from low cell numbers (less than a few thousands of cells): (a) start by thawing your frozen cell pellet (if necessary) in lukewarm water; (b) avoid mechanical-based methods; (c) better to use sonication, enzymes (in a balanced concentration), lysis buffers, or a combination of them; and (d) perform all lysis steps in a single pot to avoid tube-to-tube transfer and related sample losses.
Protein Quantification
In a proteomics workflow, total protein quantification provides information about the lysis efficiency and protein content in each sample, which will determine the performance of downstream applications (e.g., Western Blot or LC-MS/MS analysis). Protein quantification techniques can be grouped in colorimetric and fluorescent methods (Table 1).
Table 1. Protein Quantification Methods and Their Main Features.
| assay name | residue/complex measured (wavelength) | protein detection range (μg/mL) | advantages | incompatibilities | references |
|---|---|---|---|---|---|
| Colorimetric | |||||
| Ultraviolet absorbance | Aromatic residues: Tyr, Trp, Phe (280 nm) | 20–3000 | Easy and quick | Protein mixtures; Some detergents and nucleic acids | (28,40,42) |
| Bradford | Dye-protein complex: Coomassie Brilliant Blue G-250 and Arg, His, Trp, Tyr and Phe (595 nm) | 0.2–1500 | Compatible with reducing and chaotropic reagents | Limited sample material; Some detergents | (28,40,42,47,48) |
| Biuret | Reduced copper-protein complex (540 nm) | 1000–10 000 | Does not rely on the amino acid composition of the protein | Ammonium salts, sodium phosphate, and glucose | (28,40−42) |
| Lowry | Folin-Ciocalteu reagent + Biuret complex (650–750 nm) | 2–1000 | Easy and quick | Reducing agents, strong acids, and EDTA | (28,42−44) |
| BCA | BCA + Biuret complex (562 nm) | 0.5–20 (microBCA); 20–2000 (standard BCA) | Compatible with high detergent concentrations and chaotropic agents | Reducing and chelating agents | (28,40−42,45) |
| Fluorescent | |||||
| Fluorescent assay (e.g., BisANS) | Protein-fluorescent dye complex | 0.02–100 | Optimal for limited sample material; Compatible with chelators, reducing reagents, salts, free nucleotides, solvents, DNA, and protein inhibitors | Large amount of detergents | (28,49) |
Colorimetric Quantification Assays
Here, the change in the color of the dye upon protein-dye interaction is measured to quantify the proteins. Currently, there are multiple approaches, of which BCA, Bradford assays, and UV absorbance at 280 nm are the most commonly used.
Biuret
This assay, based on the reduction of copper after interaction with peptide bonds in an alkaline environment resulting in a purplish-violet complex that absorbs light at 540 nm, is the oldest and least sensitive (1–10 mg/mL protein) colorimetric quantification method.28,40−42 The Biuret assay is not compatible with ammonium salts, sodium phosphate, and glucose.28 Aiming at optimizing its sensitivity, the Biuret assay was further developed into the Lowry and bicinchoninic acid (BCA) approaches.40,42
Lowry
It was introduced by O.H. Lowry by including the Folin–Ciocalteu (Folin phenol) reagent in the Biuret assay.43 After the cuprous-protein complex is formed, there is an extra interaction with the Folin–Ciocalteu reagent producing a blue-green color that can be detected at 650–750 nm.44 This modification results in a higher sensitivity (∼2–1000 μg/mL protein detected, 100× more sensitive than the Biuret assay). Among others, reducing agents, strong acids, and EDTA are incompatible with the assay.28,42
BCA
This assay, developed by P.K. Smith in 1985, combines the principle of the Biuret reaction and the interaction of BCA with cuprous ions, resulting in an intense purple color that can be measured at 562 nm.45 This modification shows a greater quantitative sensitivity (i.e., 0.5–20 μg/mL for microBCA and 20–2000 μg/mL for standard BCA)28,40,45 and more compatibility with interfering compounds, as high detergent concentrations (up to 5%) and chaotropic agents.28,42,45 However, reducing and chelating agents as DTT and EDTA may interfere with the assay.41,42
Ultraviolet Absorbance
A well-known way to quantify proteins is by using ultraviolet (UV) absorbance measurement around 280 nm. At this wavelength, light is absorbed from the aromatic tyrosine (Tyr), tryptophan (Trp), and to lesser extent phenylalanine (Phe) residues,42 detecting protein amounts from 20 to 3000 μg/mL.28,40 Modern spectrophotometers (e.g., NanoDrop) calculate protein concentration based on the assumption that 1 optical density (OD) unit (at 280 nm) correspond to 1 mg/mL of protein, which is not accurate for most proteins.46 Conversely, protein determination based on the Beer–Lambert law, in which both the absorbance and the molar extinction coefficient (specific for each protein as it depends on the amino acid composition) are considered,40 will more accurately calculate the protein concentration based on the empirical A280 value of the sample. Although it is an easy and quick method, several components from the lysis buffer might affect the measurement as they present a high UV absorbance (e.g., Triton X and nucleic acids).40,42
Bradford
This popular protein quantification assay was established by M.M. Bradford in 1976.47 It is based on the usage of the Coomassie Brilliant Blue G-250 dye that binds, under acidic conditions, to arginine (Arg), histidine (His), Trp, Tyr, and Phe residues in proteins. After the formation of the dye-protein complex, the reddish-brown Coomassie dye becomes blue showing maximum absorbance at 595 nm47,48 and detecting proteins in the 0.2–1500 μg/mL range.28,40,42 This assay is compatible with reducing, chelating, and chaotropic agents; however, detergents can disturb the measurements and quantification relies on the presence of aromatic residues in the protein sequence.28,40,47
Fluorescent Quantification Assays
Here, the increase in fluorescence signal of the dye upon protein-dye binding is measured. Fluorescence-based protein quantification assays offer superior sensitivity (0.02–100 μg/mL protein) compared to colorimetric assays.28 In general, fluorescent assays include a dye molecule that can bind specific protein regions resulting in a fluorescence signal that can be measured by a fluorometer. Recently, Dakti et al.49 developed a novel assay based on a disulfonic acid dipotassium salt (BisANS) fluorescent dye showing high sensitivity (0.28–100 μg/mL) in a broad spectrum. In addition, this method does not present incompatibilities with chelators, detergents, and protein inhibitors, which favors its application in diverse samples. However, the pricing of this methodology and the requirement of specific measuring instruments can be considered as limiting factors.
During the past years, many companies have optimized the original assays offering quantification methods with higher sensitivities, reduced sample volumes, and better compatibility with a broad range of detergents, chaotropes, reducing and chelating agents.
In conclusion, there is not an ideal protein quantification assay for all types of samples, as each assay has its pros and cons. For a successful quantification, assay interfering components in the protein sample (e.g., detergents) have to be considered. Furthermore, total assay time, reproducibility, sensitivity, and required sample volume are critical for the selection of an appropriate protein quantification assay.
-
▶
Tip – Conclusion: For protein quantification from low cell numbers, fluorescent-based protein quantification assays offer the greatest sensitivity.
In-Gel Protein Visualization: Gel Staining Methods
In-gel protein separation (electrophoresis) and visualization are essential parts of the proteomics workflow. A single electrophoresis staining provides information about similarities and differences in protein content between samples and the protein quantity can be estimated by the intensity of the staining. As yet, diverse dyes are available which can be selected for specific research questions based on reproducibility, detection limit, correlation between dye intensity and protein quantity, compatibility with downstream applications, e.g., MS, easy usage, and affordability criteria.50 In-gel protein staining methods can be grouped in colorimetric and fluorescent detection approaches.
Colorimetric Protein Visualization Methods
They are based on dyes visible to the naked eye.
Coomassie Brilliant Blue (CBB)
This disulfonated triphenylmethane dye binds to proteins under acidic conditions through electrostatic interaction with basic amino acids (primarily Arg, lysine (Lys), and His), and with aromatic amino acids (Trp, Tyr, and Phe) by hydrophobic interactions.51 In general, proteins in the range of approximately 8–100 ng can be detected.52,53 The CBB dye exists in two forms: R-250 and G-250, the reddish and dimethylated greenish hue variants, respectively. The Coomassie G-250 form is usually employed for the quantification of proteins, e.g., Bradford, whereas the R-250 is used for the staining of SDS polyacrylamide gels.54 However, since free CBB dye molecules also stain the protein-free areas of the gel via diffusion, a destaining step must be performed to visualize the protein bands. An improved CBB protocol, known as Colloidal Coomassie staining, reduces the free dye by adding high concentration of ammonium sulfate salt which forms colloidal particles (that cannot diffuse into the gel) with the CBB in the alcoholic-acidic solution.51,55,56 Overall, the CBB staining is easy to use, cheap, robust, and compatible with MS.50−52,54
Silver Staining
From all colorimetric dyes, silver staining is the most sensitive protein visualization method (detection limit ∼1 ng protein).51,57,58 This technique involves multiple steps: (a) fixation, to immobilize proteins and remove interfering compounds, (b) sensitization, to increase the contrast of the staining, (c) silver impregnation, by using a silver nitrate or a silver-ammonia complex solution, (d) development, where protein-bound silver ions are reduced in the presence of carbonate to metallic silver resulting in brown-black colored protein bands, and (e) stopping development, to stop the staining before a high background is developed by washing out the excess dye.52,59 Even though the sensitivity of this method is ideal for protein visualization in gels, there are several disadvantages. These include MS incompatibility and low staining reproducibility resulting in band intensity variations due to no end point staining, also depending on the formulation and thickness of the gel.50,51,54,55,58
Fluorescent Protein Visualization Methods
In recent years, different fluorescent stains that can be used during or after electrophoresis have become commercially available (e.g., silver-aggregation-induced emission (AIE) stain,60 SYPRO Ruby stain,54,60 and Deep Purple dye54,61). In general, these methods present similar or slightly better sensitivity than the silver staining approach,51 and they do not involve any chemical modification of the proteins, making them more suitable for downstream applications as MS and Western Blot. Furthermore, fluorescent stainings are quick and easy to perform and do not require the use of toxic reagents; however, an imager is always needed to visualize the proteins, which together with the fluorescent reagents result in a more expensive method compared to colorimetric approaches.62
-
▶
Tip – Conclusion: To stain limited protein amounts in gels, select optimized silver or fluorescent staining methods as they provide higher sensitivity levels.
Protein Digestion
Protein digestion is an important step in the MS-based identification of proteins. Depending on the format, it can be grouped into in-solution and in-gel protein digestion. In both cases, denaturation, reduction, and alkylation of proteins can be performed before the actual protein digestion. In the denaturation step, the secondary and tertiary protein structures are disrupted by strong chaotropic agents such as urea or guanidine. Then, the disulfide bonds between the sulfhydryl groups of cysteine side chains are reduced by reducing agents, e.g., DTT. Alkylating agents as iodoacetamide (IAA) prevent the free sulfhydryl groups from reforming disulfide bonds. Regarding protein digestion, it can be either nonenzymatic or enzymatic. The former implies the disruption of amino acid bonds without using enzymes;63 for instance, via a weak acid solution that hydrolyzes the aspartic acid (Asp) residues,64 cyanogen bromide cleavage at the N-terminus of methionine (Met),65 or hydroxylamine cleaving at asparagine (Asn) and glycine (Gly) bonds.66 These approaches are fast and temperature-dependent as enzymatic methods.67 The enzymatic digestion involves the usage of enzymes, such as trypsin, Lys-C or chymotrypsin, among others, that present different specificities.68 Thus, trypsin cleaves at the C-terminal end of Lys and Arg residues, as long as there is no proline (Pro) residue on the carboxyl side of the cleavage site,69 whereas Lys-C cleaves proteins on the carboxyl-end of Lys residues, regardless the presence of Pro70 and chymotrypsin cleaves peptide amide bonds close to large hydrophobic amino acids (Tyr, Trp, Phe). Despite being temperature-dependent, enzyme-based digestion methods are robust and efficient.68
Regarding the format, in-solution protein digestion can be optimal for low protein amounts since its separation in a gel cannot be fully optimal for further processing.27 However, detergents, salts and other contaminants present in the sample can interfere with the digestion and the downstream applications (e.g., MS analysis). To overcome this, methods such as FASP34 and SP36,38 have been recently developed to allow the removal of these components and facilitate the protein digestion process (see New Approaches: Advances and Their Applications for more information). On the other hand, in-gel protein digestion is ideal for complex samples (e.g., protein lysates) as the electrophoretic separation of the proteins improves the resolution and identification and sample components interfering with the MS analysis (i.e., detergents, salts) are removed during the in-gel processing. Moreover, gel bands corresponding to proteins presenting specific molecular weights and isoelectric points can be individually processed reducing the complexity of the analysis. However, protein recovery from the gel can be suboptimal, especially depending on the protein staining used.27
-
▶
Tip – Conclusion: For protein digestion from paucicellular samples, perform in-solution enzymatic digestion methods in single-pots. Also make sure to adjust the enzyme:protein ratio to allow efficient protein digestion.
Peptide Recovery
The quality, sensitivity, and robustness of the MS analysis are highly influenced by the sample cleanup. Particularly, detergents, chaotropes, salt, and enzymes (after protein digestion) have to be removed before the peptides are measured.71 For such removal, column- or bead-based approaches can be applied.
Columns
After loading the peptide sample in the column, the peptides bind to it allowing for removal of contaminants such as detergents or salts. Then, purified peptides are eluted, ready to be analyzed. There are many different columns depending on their chemistries, which allow for different types of bindings. For quantity-limited peptide samples, microcolumns are an excellent choice, since they prevent loss of peptides.9,35,72
Beads
There is an extensive variety of beads according to their sizes and chemistries. In general, beads are incubated with the peptide sample to prompt the binding of these molecules to the bead surface. Then, washing and removal of contaminants can be performed using the appropriate buffer conditions to later elute the purified peptides.6,73 For proteomics assays where limited amounts of peptides are involved, beads with magnetic properties would be a better choice as the processing system is cleaner in sense of higher purification degree and less sample loss.6,38,73,74
Once peptides are purified, they can be quantified for further analysis. This step is quite challenging in a limited-sample setting and several attempts have been made to develop sensitive quantification methods in the very low range. Similar to protein quantification methods, there are colorimetric and fluorescent peptide quantification assays. For example, in the colorimetric group, a modified BCA assay is available to quantify peptide mixtures. The principle of this test is identical with the BCA protein assay (see Protein Quantification for more information); however, the formula of reagents in the peptide assay is modified and optimized to quantify peptides with more sensitivity.75 As with protein quantification, fluorescent peptide assays offer a greater sensitivity than colorimetric assays. In general, they include specific peptide labeling via, e.g., amine-reactive fluorescent reagents.
-
▶
Tip – Conclusion: For peptide recovery from paucicellular samples, opt for bead-based strategies and single-pot processing for a better output and minimized sample losses.
LC-MS/MS
Several parameters related to the LC-MS/MS measurement are highly critical to get optimal results. The type of chromatographic column, the gradient time, and the MS instrumentation, among others, have a huge impact on the results.76,77
LC Separation
In general, columns are composed of a chemical moiety which determines the polarity (e.g., alkyl chains of either 8 or 18 carbons, C8 or C18 respectively), attached to a matrix support, usually composed of silica beads.78 For selecting the most appropriate column, the complexity, abundancy, solubility, size, and pH of the target peptide and the entire mixture have to be considered. Columns are available in a variety of materials, chemistries, column particle sizes and lengths suitable for all types of protein separation.79,80 Regarding the column length, the optimal dimensions would be those allowing for a quick separation of peptides together with the greatest peak capacity (i.e., number of peaks that can be put into the gradient time frame with a resolution of ∼1). In general, the longer the column, the better the separation.79,81 However, it is important to note that there is a limit in peak capacity, no matter how long the column is (e.g., a 150 mm column only reports 3% higher peak capacity than a 100 mm column).81 Likewise, the longer the gradient time, the greater the peak capacity.79 Also, it has been shown that smaller particle sizes (1.7 vs 3 μm) improve peptide analysis regardless the gradient time and the column length.82 Finally, flow rate also plays a role in the chromatographic separation,81 with the nanoflow-based systems (nanoLC mode) being preferred for increased sensitivity and separation.79 This system is especially relevant for limited amounts of samples, where a capillary column is used to reduce the sample quantity demand by separating the peptides at nL/min flow rates.82
MS Instrumentation
After chromatographic separation of peptides, these are ionized and measured in an initial scan, followed by fragmentation and remeasurement in subsequent scan(s). To achieve the best ionization, fragmentation, and measurement, different configurations can be defined.78 The production of ions may be maximized by using the appropriate nebulization process (e.g., ESI, MALDI, APCI).76 For the fragmentation, approaches as collision-induced dissociation (CID) or electron transfer dissociation (ETD) are widely used.83 Then, the measurement is performed by analyzers as quadrupole ion traps (Q) or Orbitraps. The latter, consisting of a coaxial inner electrode surrounded by a barrel-shaped outer electrode, provides high mass resolution, high dynamic range, and high mass accuracy detection. Usually, MS instruments are composed of a combination of two or more analyzers (e.g., QQQ, Q-Orbitrap).76,78,83
-
▶
Tip – Conclusion: For an improved chromatographic performance: (a) combine long columns (>100 mm) with sufficiently long gradient times (>120 min) and nanoflow rates. (b) For highly reliable and accurate protein identification, MS configurations integrating efficient ionization methods and analyzers with high resolutions and mass accuracies must be employed.
Others
The material of the consumables used during sample processing for proteomics, especially in a microproteomics setting, is a key aspect often neglected. Commonly used tubes contain hydrophobic surfaces that favor the unspecific binding of proteins and peptides, contributing to significant sample losses. Comprehensive studies reported that, in general, polypropylene plastic and borosilicate glass show lower binding and higher peptide recoveries, being highly recommended for proteomics analyses.84−86 In this regard, different tubes presenting less hydrophobicity (and then minimizing the surface absorption by proteins/peptides), known as low-absorption tubes, have been commercialized in recent years (e.g., protein LoBind tubes from Eppendorf). Moreover, choosing tubes at low surface area-to-solution volume ratios might be indicated, especially when working with low cell/protein/peptide amounts, as relatively less of them will bind to the tube surfaces minimizing the sample loss.86
-
▶
Tip – Conclusion: For paucicellular samples use (a) low protein absorption materials made of polypropylene or borosilicate and (b) tubes at low surface area-to-solution volume ratios.
New Approaches: Advances and Their Applications
As stated in this review, the study of proteomes from paucicellular samples, or even at the single-cell level, is still a big challenge. Multiple attempts have been carried out aiming at the development of effective LC-MS/MS-based approaches for high-throughput proteomics analysis from low cell numbers and at the single-cell level. However, the small cell sizes, their complexity, and the broad concentration range of proteins (10–15–10–9 M) are still limiting factors. Nevertheless, some of these strategies have shown promising results (Table 2) and are described below.
Table 2. Examples of Studies Applying Advanced Approaches for Mass Spectrometry-Based Analysis in Single and/or Low Cell Numbersa.
| technology | cell type description | sample amount (no. of cells, protein amount, sample volume) | no. of identified proteins | mass spectrometer | reference |
|---|---|---|---|---|---|
| SP3 | Human oocytes | 100 cells | 2154 | Orbitrap Velosf | (91) |
| Human oocytes | 1 cell (∼100 ng) | ∼445 | Orbitrap Velosf | (91) | |
| Mouse BMDM | 25 000 cell (∼1 μg) | 3152 | Synapt G2-S HDg | (92) | |
| HeLa cells | 1 μg | 3300 | |||
| Somaless retinal axons from 100 eye explants from Xenopus laevis embryos | ∼2 μg | >1000 | Orbitrap Velosf, Q-Exactivef, Orbitrap Fusionf | (93) | |
| Human distal lung resections | 10 mg wet weight | 2412 | Q-Exactive Plusf | (94) | |
| Tubules or glomeruli from human kidney | ∼200 cells | >2000 | Q-Exactive Plusf | (95) | |
| Microorganism mixture (55% B. subtilis + 35% E. coli + 10% S. cerevisiae) | 1 × 107 CFU (0.3 μg) | 1932 | Q-Exactive HFf | (96) | |
| SCoPE-MS | Mouse embryonic stem cells | Single cells | ∼1000 | Orbitrap Elitef | (97) |
| Human monocytes/macrophages | 1018 cells | 2700 | Q-Exactivef | (98) | |
| AML cell model | Single cells | 1000 | Orbitrap Explorisf | (99) | |
| In-StageTip (iST) | S. cerevisiae | 20 μg | 4570 | Q-Exactivef | (9) |
| HeLa cells | 20 μg | 9667 | Q-Exactivef | (9) | |
| Human peripheral blood | 1 μL (×15) | 313 | Q Exactive HFf | (100) | |
| Mouse BMDM | 25 000 cell (∼1 μg) | 2343 | Synapt G2-S HDg | (92) | |
| HeLa cells | 1 μg | 3020 | Synapt G2-S HDg | (92) | |
| Pediatric urine samples | 0.5 mL (∼130 μg) | 1199 | Q Exactive HFf | (101) | |
| Microfluidics | E. colib | 50 ng | 799 | Orbitrap Velosf | (102) |
| THP-1 | 1000 cells | 346–911 | Orbitrap Elitef | (103) | |
| THP-1 | 100 cells | 275–549 | Orbitrap Elitef | (103) | |
| HeLac | 140 cells | ∼3000 | Orbitrap Fusionf | (104) | |
| HeLac | Single cells | 670 | Orbitrap Fusionf | (105) | |
| HeLac | Single cells | 1056 | Orbitrap Eclipsef | (106) | |
| Jurkatd | ∼500 cells | 2500 | Q-Exactive Plusf | (107) | |
| Single mouse oocytee | 1 cell | 355 | Orbitrap Elitef | (108) | |
| Xenopus laevis embryo | 16-cell embryo | 112 | Orbitrap Fusionf | (109) | |
| B and T cellsc | ∼130 cells | 1095 | Orbitrap Explorisf | (110) | |
| HeLac | 70/770 cells | 170/620 | Orbitrap Lumosf | (111) | |
| MCF10Ac | Single cells | 256 | Orbitrap Fusionf | (112) | |
| AMLc | 152 cells | 2558 | Orbitrap Fusionf | (112) | |
| Murine cellsc | 72 single cells | 2300 | Orbitrap Fusionf | (113) | |
| Laser capture microdissection-coupled | Tomato roots (cortical, epidermal) | 5000–7000 cells | 744–1313 | Orbitrap Elitef, Orbitrap Fusionf | (114) |
| Rat brain cortex | 10–18 cells | ∼1000 | Orbitrap Fusionf | (115) | |
| Human motor neurons/interneurons | Single cells | ∼1000 | Orbitrap Eclipsef | (106) | |
| Prostate tumoral cells (spiked) | 1–5 cells | 164–607 | Orbitrap Fusionf | (116) |
AML, acute myeloid leukemia; BMDM, bone marrow-derived macrophages; CFU, colony-forming unit; SP3, single-pot solid-phase-enhanced sample preparation; SCoPE-MS, single cell proteomics by mass spectrometry.
Capillary zone electrophoresis-tandem mass spectrometry, CZE-MS/MS.
nanoPOTS/autoPOTS.
Digital microfluidics combined with SP3, DMF-SP3.
Nanoliter-scale oil-air-droplet (OAD) chip.
By Thermo Scientific (Waltham, MA, US).
By Waters Corporation (Mildford, MA, US).
SP3 Technology
The SP3 protocol6,38 provides a quick, efficient, scalable, high-throughput, flexible, and unbiased sample preparation method in a single tube for ultrasensitive proteomic analysis, especially from quantity-limited samples. The method is based on the usage of paramagnetic beads (Figure 2A), building on previous developments performed in nanodiamond87 and solid-phase reversible technologies.88 Specifically, carboxylate-coated paramagnetic beads, presenting a hydrophilic surface, combined with the addition of an organic solvent, promote the immobilization of proteins and peptides on the bead surface (principle similar to both hydrophilic interaction chromatography HILIC89 and ERLIC90). This immobilization allows for further processing (e.g., contaminant removal, protein digestion, peptide cleanup, desalting, fractionation, concentration, and chemical labeling) minimizing sample losses and maximizing throughput.38 One of the major advantages of this workflow is the no-need for protocol modifications (e.g., for cell lysis or peptide recovery) since potential contaminants (e.g., detergents, chaotropes, salts, buffers, solvents, and acids) are removed during SP3 incubation. Also, this protocol can be scaled down to single-cell analysis and the technical noise can be reduced improving the reproducibility of the technique. However, there are several limitations related to the usage of beads, as intact protein recovery from them can sometimes be challenging and bead aggregation can hamper some steps of the process. Also, special attention must be taken when working with high chromatin-concentrated samples since this molecule can negatively affect the performance.6
Figure 2.
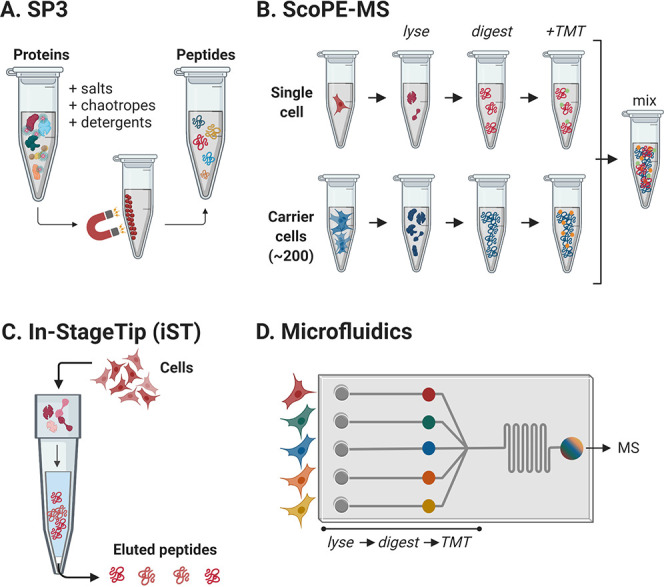
Advanced approaches for mass spectrometry-based analysis of single and/or low cell numbers. (A) The SP3 (single-pot solid-phase-enhanced sample preparation) strategy employs magnetic beads throughout the multistep workflow. (B) The ScoPE-MS (single cell proteomics by mass spectrometry) combines carrier cells with single-cell analysis to improve peptide identification. (C) In the iST method, the full proteomics workflow is performed within a tip. (D) Microfluidics allows for sample processing of single cells in microchip devices. Panel B is adapted from Budnik et al., 2018 [ref (97), Copyright 2018 by Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)]. Created with BioRender.com.
The SP3 method has been extensively applied during the past years, becoming the outstanding technology in diverse fields (Table 2). For instance, Sielaff et al.92 performed an extensive comparison between this method and the FASP and iST approaches processing 1–20 μg of HeLa lysate protein. Although the three protocols performed well in the high range, when handling samples ∼1 μg, the SP3 method showed the best performance and reproducibility. They also applied this bead-based approach to FACS-sorted tumor-associated macrophages quantifying >2900 proteins. Also, Hayoun and colleagues96 tested the performance of denaturing electrophoresis/in-gel proteolysis, suspension-trapping filter-based (S-Trap) approach and SP3 on different microorganisms observing a clear better output for the latter method.
Finally, additional developments have been accomplished by others aiming at extending the applicability of the SP3 technology. Thus, Dagley et al.117 built the Universal Solid-Phase Protein Preparation (USP3) method to facilitate high-throughput sample preparation for bottom-up and top-down MS analysis. With this adapted approach, they profiled 1800 proteoforms from 50 μg of HeLa protein lysate.
SCoPE-MS
The single cell proteomics by MS (SCoPE-MS) approach97 was developed aiming at quantifying proteome heterogeneities from single mammalian cells (Figure 2B). This new strategy focuses on small but crucial deviations, according to the authors, from a typical standard protocol for bulk LC-MS/MS. Thus, the SCoPE-MS technology includes the following: (i) replacement of chemical lysis with mechanical lysis (focused acoustic sonication) to obviate chemicals undermining peptide separation and ionization, (ii) inclusion of carrier cells to provide enough sample for measurement, (iii) elimination of cleanup steps to avoid extra sample losses, (iv) mixing of peptides of interest with TMT-based labeled carrier peptides to minimize peptide loss due to adhesion to the nanoLC column surface, (v) preselection of peptide ions (from both multiple single cells and carrier cells) having the same m/z ratios in the MS1 scan to be analyzed by the MS2 scan (thus, more peptide ions can be fragmented increasing the efficiency), and (vi) decreasing the MS scan times (<milliseconds) and increasing the ion accumulation times to improve identification and quantification.
The SCoPE-MS method has been tested in mouse embryonic stem cells reporting the quantification of over 1000 proteins with ≥105 copies/cell; however, the authors claim that further improvements in the sensitivity and speed of MS could enhance the technology favoring the identification of proteins at 1000 copies/cell.97 Despite the promising features of this approach, including a “carrier proteome” in the workflow might be an issue depending on how similar the carrier cell and the cell of interest are.118 In fact, it has been defined that carrier levels should be limited to 20× the number of cells of interest to get reliable results.119 Recently, additional developments resulted in SCOPE2, a more affordable method where cell lysis is performed with a freeze-heat cycle in pure water instead of sonication. This improvement allowed for a better recovery (2700 proteins quantified from 1018 monocytes/macrophages);98 nevertheless, how proteins without ion content can be solubilized in pure water might still be a challenge for this approach.118 Aiming at increasing the high-throughput characterization, maximizing quantitative accuracy and integrating FACS-based single-cell sorting, Schoof et al. modified the SCoPE-MS approach by switching the pure water by a TFE-based lysis buffer and introducing reduction and alkylation steps. These changes allowed for the identification of ∼1000 proteins per single cell.99
In-StageTip (iST) Platform
The iST approach9 is a simple in-solution processing method that requires minimal handling and can be virtually applied to any range of sample quantities. It is based on in-pipet-tip-built affinity chromatography columns (Figure 2C) that are used for full processing of samples (i.e., lysis, denaturation, reduction, alkylation, protein digestion, peptide elution, and fractionation). However, not all types of reagents (e.g., detergents as SDS) are compatible as they can hamper proteolysis and, therefore, downstream processing.6 Despite this, the iST method can be automated and scaled, offering highly reliable and reproducible results.9 Similarly, the on-microsolid-phase extraction tip (OmSET)-based sample preparation120 has recently been developed for processing down to 200 cells with promising results. In this case, a minimized sample processing volume (1–3 μL) is required and no additional sample cleanup steps are needed.
Microfluidics- and Microchip-Based MS Approaches
Microfluidic chips for cell analysis are a promising approach as they require reduced sample amounts and reagents while offering high throughput analysis, high reproducibility, large parallelization, easy operability, and low-cost121−123 (Figure 2D). In the past years, these microchips have been implemented in different fields (e.g., cell culture, microscopy and single-cell analysis). However, their combination with MS (a.k.a. chip-mass spectrometry) has resulted in a major analytical platform for cell analysis due to its broad applicability, sensitivity, and specificity.7 Particularly, big efforts have been performed to develop chip-MALDI MS and chip-ESI MS platforms. In this context, the single-cell mass spectrometric method (SCMS)124 is able to print sample droplets (nL) containing single cells onto a MALDI plate to further determine the metabolic patterns of different populations.7,125 In the same line, Yang et al.126 combined a removable microfluidic chip (for in situ protein digestion and tag labeling) to a MALDI-Time Of Flight (TOF)-MS equipment. Regarding chip-ESI-MS, diverse approaches have been developed, including capillary electrophoresis (CE)-ESI-MS, which allows the proteome analysis of single cells previously separated via micro- and nanofluidic channels. Thus, over 800 protein groups were identified from 50 ng of E. coli digest102 and metabolic patterns for different embryo anatomical parts were defined from only 8 cells.127 A high-resolution version of this system (CE-ESI-HRMS) allowed for the label-free quantification of ∼112 protein groups from 16-cell Xenopus laevis embryo.109 Other chip-ESI-MS technologies involve the generation of sample droplets to be further processed on the microchip (droplet-ESI-MS) or the combination of paper chromatography and ambient ionization (paper spray-ESI-MS), with a potential application in medical research (e.g., for the analysis of imatinib effect in blood samples128,129 or detection of nicotine alkaloids in biological fluids).130 Highly promising is the CE-ESI-MS-based microfluidic device for continuous lysis of single cells (12 cells/min) combined with MS which permits automated real-time analysis of individual cells.131 For quantitative studies on cell metabolism, Chen et al. developed a stable isotope labeling assisted microfluidic chip-ESI-MS (SIL-chip-ESI-MS) by integrating a microfluidic system for injection of culture medium and drugs, wells for cell culture, on-chip columns for sample preparation and an ESI-MS.132
Also in this group of microfluidics-based MS, the nanoPOTS104,105 platform is included, where microfluidic sample processing, cell sorting and ultrasensitive nanoLC-MS are combined to achieve the proteome analysis from a few tens of cells (up to 3000 proteins from 140 cells). Coupling this nanoPOTS approach to TMT labeling, Dou and colleagues113 identified 2300 proteins from 72 single murine cells in <2 days, depicting the versatility of the microfluidics platform. It is also remarkable the combination of digital microfluidics with the SP3 platform (DMF-SP3),107 in which the SP3 technology is performed in a microfluidic device allowing the identification of 2500 proteins from ∼500 Jurkat T cells. Likewise, coupling nanoPOTS to high-field asymmetric ion mobility spectrometry (FAIMS) has resulted in a 2.3 fold increase in identification from single HeLa cells (1056 vs 459 proteins for with and without FAIMS coupling, respectively).106 Also, nanoPOTS has been combined with a top-down approach to study PTMs from small inputs,111 resulting in the identification of 170 proteoforms from 70 HeLa cells (∼10 ng of protein). An upscaled version of the nanoPOTS, termed autoPOTS (automated preparation in one pot for trace samples),110 was recently developed to analyze samples in the low-microliter range. This adapted approach resulted in only a 25% identification reduction at the single-cell level, while allowing for the usage of commercially available liquid handlers, favoring its implementation in many more laboratories compared to nanoPOTS. Similarly, the developers of nanoPOTS have defined a workflow where the platform is directly linked by means of an autosampler to the LC-MS/MS system.112 Thus, throughput and robustness can be significantly amplified, as their results show (256 proteins identified per single MCF10A cells, 24 single cells analyzed per day). Additionally, Li and colleagues developed the so-called nanoliter-scale oil-air-droplet (OAD)108 chip for performing sample pretreatment and injection of single cells with minimum sample loss and high efficiency (355 proteins identified from a single mouse oocyte). Similar results (i.e., 328 proteins identified in one HeLa cell) were found by using the iPAD-1,133 a sample processing method performed within a 2 nL capillary in only 1 h of time.
Laser Capture Microdissection-Coupled Approaches
Alternative ways of processing small cell inputs include the isolation of single cells by laser capture microdissection (LCM), where tissue cells are visualized under the microscope and isolated by directly harvesting or indirect enrichment by cutting away the unwanted cells.134 Zhu and colleagues coupled the LCM to LC-MS/MS analysis for the study of 5000–7000 cortical and epidermal tomato root-derived cells resulting in the identification of 744 and 1313 protein groups, respectively.114 Although the recovery might not seem very efficient, the main value of this combined platform is the reduced cross-contamination from tissue adjacent cells. Similarly, Zhu et al.115 linked the LCM to nanoPOTS to quantitatively study the rat brain cortex tissue corroborating the promising applicability of such workflow (∼1000 proteins from 10 to 18 cells). The same combination was used to analyze 1–5 spiked prostate tumoral cells resulting in the identification of 164 to 607 protein groups.116 Also, the combination of LCM with the above-mentioned FAIMS reported outstanding results, as up to 1012 and 1085 proteins were identified from single human motor neurons and interneurons, respectively.106
Conclusion
Despite approaches as single-cell RNA sequencing that have been implemented in the lab routine, the analysis of the full protein maps at a single-cell level is still very challenging. Here, we have discussed and summarized several relevant attempts that provide promising results when working with paucicellular samples (less than a few thousands of cells). However, the ultimate success will result from combining highly sensitive MS instrumentation and optimal sample preparation approaches. Thus, not only advanced methodologies are needed, but also good laboratory practices during the multistep and complex proteomics processing, without neglecting small but crucial parameters, such as the selection of the material of consumables and the need of working in a keratin-free environment to reduce potential contaminants.
Although it seems that only a genome-wide amplification method for proteins could resolve the above-mentioned challenge, it is evident that this will not be easily feasible, mainly because protein amplifications strategies will not likely be proportional for the different proteins. For this reason, the proteomics field must continue progressing and integrating knowledge from other areas as transcriptomics, bioinformatics, physics, and artificial intelligence aiming at single-cell proteomics analysis, as this knowledge will significantly benefit the understanding of the life that surrounds us.
Acknowledgments
The presented work was funded by the European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme with an ERC Advanced Grant (ERC-2015-AdG 695655, TiMaScan).
Glossary
Abbreviations
- Arg
arginine
- Asn
asparagine
- Asp
aspartic acid
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- CBB
Coomassie Brilliant Blue
- CE
capillary electrophoresis
- CID
collision-induced dissociation
- Cys
cysteine
- DTT
dithiothreitol
- ETD
electron transfer dissociation
- FACS
fluorescence-activated cell sorting
- FASP
filter-aided sample preparation
- FCS
fetal calf serum
- Gly
glycine
- His
histidine
- IAA
iodoacetamide
- iST
in-StageTip digestion
- LCM
laser capture microdissection
- LC
liquid chromatography
- Lys
lysine
- Met
methionine
- MS
mass spectrometry
- Phe
phenylalanine
- Pro
proline
- Q
quadrupole
- SCMS
single-cell mass spectrometric method
- SCoPE-MS
single cell proteomics by mass spectrometry
- SDS
sodium dodecyl sulfate
- SP3
single-pot solid-phase-enhanced sample preparation
- Trp
tryptophan
- Tyr
tyrosine
- USP3
Universal Solid-Phase Protein Preparation
- UV
ultraviolet.
The authors declare no competing financial interest.
References
- Hwang B.; Lee J. H.; Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50 (8), 96. 10.1038/s12276-018-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema E.; Bernstein B. E.; Buenrostro J. D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2019, 51 (1), 19–25. 10.1038/s41588-018-0290-x. [DOI] [PubMed] [Google Scholar]
- Stuart T.; Satija R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20 (5), 257–272. 10.1038/s41576-019-0093-7. [DOI] [PubMed] [Google Scholar]
- Labib M.; Kelley S. O. Single-cell analysis targeting the proteome. Nature Reviews Chemistry 2020, 4 (3), 143–158. 10.1038/s41570-020-0162-7. [DOI] [PubMed] [Google Scholar]
- Couvillion S. P.; Zhu Y.; Nagy G.; Adkins J. N.; Ansong C.; Renslow R. S.; Piehowski P. D.; Ibrahim Y. M.; Kelly R. T.; Metz T. O. New mass spectrometry technologies contributing towards comprehensive and high throughput omics analyses of single cells. Analyst 2019, 144 (3), 794–807. 10.1039/C8AN01574K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C. S.; Moggridge S.; Müller T.; Sorensen P. H.; Morin G. B.; Krijgsveld J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14 (1), 68–85. 10.1038/s41596-018-0082-x. [DOI] [PubMed] [Google Scholar]
- Mao S.; Li W.; Zhang Q.; Zhang W.; Huang Q.; Lin J.-M. Cell analysis on chip-mass spectrometry. TrAC, Trends Anal. Chem. 2018, 107, 43–59. 10.1016/j.trac.2018.06.019. [DOI] [Google Scholar]
- Yang L.; George J.; Wang J. Deep Profiling of Cellular Heterogeneity by Emerging Single-Cell Proteomic Technologies. Proteomics 2020, 20, e1900226 10.1002/pmic.201900226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak N. A.; Pichler G.; Paron I.; Nagaraj N.; Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11 (3), 319–24. 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Scheuermann S.; Schäfer A.; Langejürgen J.; Reis C. A step towards enzyme-free tissue dissociation. Current Directions in Biomedical Engineering 2019, 5 (1), 545. 10.1515/cdbme-2019-0137. [DOI] [Google Scholar]
- Vieira Braga F. A.; Miragaia R. J. Tissue Handling and Dissociation for Single-Cell RNA-Seq. Methods Mol. Biol. 2019, 1979, 9–21. 10.1007/978-1-4939-9240-9_2. [DOI] [PubMed] [Google Scholar]
- Herzenberg L. A.; Parks D.; Sahaf B.; Perez O.; Roederer M.; Herzenberg L. A. The History and Future of the Fluorescence Activated Cell Sorter and Flow Cytometry: A View from Stanford. Clin. Chem. 2002, 48 (10), 1819–1827. 10.1093/clinchem/48.10.1819. [DOI] [PubMed] [Google Scholar]
- Tung J. W.; Heydari K.; Tirouvanziam R.; Sahaf B.; Parks D. R.; Herzenberg L. A.; Herzenberg L. A. Modern flow cytometry: a practical approach. Clin Lab Med. 2007, 27 (3), 453–68. 10.1016/j.cll.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A.; Schoendube J.; Zimmermann S.; Steeb M.; Zengerle R.; Koltay P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015, 16 (8), 16897–16919. 10.3390/ijms160816897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arntzen M. Ø.; Thiede B. ApoptoProteomics, an integrated database for analysis of proteomics data obtained from apoptotic cells. Molecular & cellular proteomics: MCP 2012, 11 (2), M111.010447. 10.1074/mcp.M111.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawula M.; Hawthorne G.; Smith G. T.; Hill H. M. Best Practice in Biological Sample Collection, Processing, and Storage for LC-MS in Bioanalysis of Drugs. In Handbook of LC-MS Bioanalysis 2013, 139–164. 10.1002/9781118671276.ch13. [DOI] [Google Scholar]
- Hodge K.; Have S. T.; Hutton L.; Lamond A. I. Cleaning up the masses: exclusion lists to reduce contamination with HPLC-MS/MS. J. Proteomics 2013, 88, 92–103. 10.1016/j.jprot.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamlage B.; Maldonado S. G.; Bethan B.; Peter E.; Schmitz O.; Liebenberg V.; Schatz P. Quality Markers Addressing Preanalytical Variations of Blood and Plasma Processing Identified by Broad and Targeted Metabolite Profiling. Clin. Chem. 2014, 60 (2), 399–412. 10.1373/clinchem.2013.211979. [DOI] [PubMed] [Google Scholar]
- Mitchell B. L.; Yasui Y.; Li C. I.; Fitzpatrick A. L.; Lampe P. D. Impact of freeze-thaw cycles and storage time on plasma samples used in mass spectrometry based biomarker discovery projects. Cancer Inf. 2005, 1 (1), 98–104. 10.1177/117693510500100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter D.; Dehghani A.; Steen H.. Optimization of Cell Lysis and Protein Digestion Protocols for Protein Analysis by LC-MS/MS. In Proteomic Profiling: Methods and Protocols; Posch A., Ed.; Springer: New York, NY, 2015; pp 259–273. [DOI] [PubMed] [Google Scholar]
- Shehadul Islam M.; Aryasomayajula A.; Selvaganapathy P. R. A Review on Macroscale and Microscale Cell Lysis Methods. Micromachines 2017, 8 (3), 83. 10.3390/mi8030083. [DOI] [Google Scholar]
- Kuhn A.The Bacterial Cell Wall and Membrane—A Treasure Chest for Antibiotic Targets. In Bacterial Cell Walls and Membranes; Kuhn A., Ed.; Springer International Publishing: Cham, 2019; pp 1–5. [DOI] [PubMed] [Google Scholar]
- Rose G. G.A current interpretation of the anatomy of the mammalian cell. In Mammalian Cell Membranes; Jamieson G. A., Robinson D. M., Eds.; Butterworth-Heinemann, 1976; pp 1–30. [Google Scholar]
- Harayama T.; Riezman H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19 (5), 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Raffy S.; Teissié J. Control of Lipid Membrane Stability by Cholesterol Content. Biophys. J. 1999, 76 (4), 2072–2080. 10.1016/S0006-3495(99)77363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg S. Mechanical/physical methods of cell disruption and tissue homogenization. Methods Mol. Biol. 2008, 424, 3–22. 10.1007/978-1-60327-064-9_1. [DOI] [PubMed] [Google Scholar]
- Feist P.; Hummon A. B. Proteomic challenges: sample preparation techniques for microgram-quantity protein analysis from biological samples. Int. J. Mol. Sci. 2015, 16 (2), 3537–63. 10.3390/ijms16023537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cañas B.; Piñeiro C.; Calvo E.; López-Ferrer D.; Gallardo J. M. Trends in sample preparation for classical and second generation proteomics. J. Chromatogr A 2007, 1153 (1–2), 235–58. 10.1016/j.chroma.2007.01.045. [DOI] [PubMed] [Google Scholar]
- DeCaprio J.; Kohl T. O. Using Dounce Homogenization to Lyse Cells for Immunoprecipitation. Cold Spring Harb Protoc 2019, 2019 (7), pdb.prot098574. 10.1101/pdb.prot098574. [DOI] [PubMed] [Google Scholar]
- Senichkin V. V.; Prokhorova E. A.; Zhivotovsky B.; Kopeina G. S. Simple and Efficient Protocol for Subcellular Fractionation of Normal and Apoptotic Cells. Cells 2021, 10 (4), 852. 10.3390/cells10040852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa T.; Hung L.; McGinley M. G.; Rohde M. F.; Narhi L. O. Induced resistance of trypsin to sodium dodecylsulfate upon complex formation with trypsin inhibitor. J. Protein Chem. 1992, 11 (2), 171–6. 10.1007/BF01025222. [DOI] [PubMed] [Google Scholar]
- Botelho D.; Wall M. J.; Vieira D. B.; Fitzsimmons S.; Liu F.; Doucette A. Top-down and bottom-up proteomics of SDS-containing solutions following mass-based separation. J. Proteome Res. 2010, 9 (6), 2863–70. 10.1021/pr900949p. [DOI] [PubMed] [Google Scholar]
- Yu Y. Q.; Gilar M.; Lee P. J.; Bouvier E. S.; Gebler J. C. Enzyme-friendly, mass spectrometry-compatible surfactant for in-solution enzymatic digestion of proteins. Anal. Chem. 2003, 75 (21), 6023–8. 10.1021/ac0346196. [DOI] [PubMed] [Google Scholar]
- Wiśniewski J. R. Filter-Aided Sample Preparation for Proteome Analysis. Methods Mol. Biol. 2018, 1841, 3–10. 10.1007/978-1-4939-8695-8_1. [DOI] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6 (5), 359–62. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Cleland W. W. Dithiothreitol, a new protective reagent for SH groups. Biochemistry 1964, 3, 480–2. 10.1021/bi00892a002. [DOI] [PubMed] [Google Scholar]
- Deutscher M. P. Maintaining protein stability. Methods Enzymol. 2009, 463, 121–7. 10.1016/S0076-6879(09)63010-X. [DOI] [PubMed] [Google Scholar]
- Hughes C. S.; Foehr S.; Garfield D. A.; Furlong E. E.; Steinmetz L. M.; Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10 (10), 757. 10.15252/msb.20145625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaddek D.; Narayan V.; Nicolas A.; Murillo A. B.; Gartner A.; Kenyon C. J.; Lamond A. I. Micro-proteomics with iterative data analysis: Proteome analysis in C. elegans at the single worm level. Proteomics 2016, 16 (3), 381–392. 10.1002/pmic.201500264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble J. E.; Bailey M. J. Quantitation of protein. Methods Enzymol. 2009, 463, 73–95. 10.1016/S0076-6879(09)63008-1. [DOI] [PubMed] [Google Scholar]
- Sapan C. V.; Lundblad R. L.; Price N. C. Colorimetric protein assay techniques. Biotechnol. Appl. Biochem. 1999, 29 (2), 99–108. [PubMed] [Google Scholar]
- Goldring J. P. Protein quantification methods to determine protein concentration prior to electrophoresis. Methods Mol. Biol. 2012, 869, 29–35. 10.1007/978-1-61779-821-4_3. [DOI] [PubMed] [Google Scholar]
- Lowry O. H.; Rosebrough N. J.; Farr A. L.; Randall R. J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193 (1), 265–75. 10.1016/S0021-9258(19)52451-6. [DOI] [PubMed] [Google Scholar]
- Stoscheck C. M. Quantitation of protein. Methods Enzymol. 1990, 182, 50–68. 10.1016/0076-6879(90)82008-P. [DOI] [PubMed] [Google Scholar]
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150 (1), 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Desjardins P.; Hansen J. B.; Allen M. Microvolume spectrophotometric and fluorometric determination of protein concentration. Curr. Protoc Protein Sci. 2009, 55, 10. 10.1002/0471140864.ps0310s55. [DOI] [PubMed] [Google Scholar]; Chapter 3, Unit 3.
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–54. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Compton S. J.; Jones C. G. Mechanism of dye response and interference in the Bradford protein assay. Anal. Biochem. 1985, 151 (2), 369–74. 10.1016/0003-2697(85)90190-3. [DOI] [PubMed] [Google Scholar]
- Datki Z.; Olah Z.; Macsai L.; Pakaski M.; Galik B.; Mihaly G.; Kalman J. Application of BisANS fluorescent dye for developing a novel protein assay. PLoS One 2019, 14 (4), e0215863 10.1371/journal.pone.0215863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauci V. J.; Wright E. P.; Coorssen J. R. Quantitative proteomics: assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 2011, 4 (1), 3–29. 10.1007/s12154-010-0043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram R. K.; Balasubramaniyan N.; Sundaram P. Protein stains and applications. Methods Mol. Biol. 2012, 869, 451–64. 10.1007/978-1-61779-821-4_39. [DOI] [PubMed] [Google Scholar]
- Miller I.; Crawford J.; Gianazza E. Protein stains for proteomic applications: which, when, why?. Proteomics 2006, 6 (20), 5385–408. 10.1002/pmic.200600323. [DOI] [PubMed] [Google Scholar]
- Patton W. F. Detection technologies in proteome analysis. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2002, 771 (1–2), 3–31. 10.1016/S1570-0232(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Chevalier F. Standard Dyes for Total Protein Staining in Gel-Based Proteomic Analysis. Materials 2010, 3 (10), 4784–4792. 10.3390/ma3104784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyballa N.; Metzger S. Fast and sensitive coomassie staining in quantitative proteomics. Methods Mol. Biol. 2012, 893, 47–59. 10.1007/978-1-61779-885-6_4. [DOI] [PubMed] [Google Scholar]
- Neuhoff V.; Stamm R.; Eibl H. Clear background and highly sensitive protein staining with Coomassie Blue dyes in polyacrylamide gels: A systematic analysis. Electrophoresis 1985, 6 (9), 427–448. 10.1002/elps.1150060905. [DOI] [Google Scholar]
- Chevalier F.; Rofidal V.; Rossignol M. Visible and fluorescent staining of two-dimensional gels. Methods Mol. Biol. 2006, 355, 145–56. 10.1385/1-59745-227-0:145. [DOI] [PubMed] [Google Scholar]
- Harris L. R.; Churchward M. A.; Butt R. H.; Coorssen J. R. Assessing detection methods for gel-based proteomic analyses. J. Proteome Res. 2007, 6 (4), 1418–25. 10.1021/pr0700246. [DOI] [PubMed] [Google Scholar]
- Chevallet M.; Luche S.; Rabilloud T. Silver staining of proteins in polyacrylamide gels. Nat. Protoc. 2006, 1 (4), 1852–8. 10.1038/nprot.2006.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S.; Wong A. Y. H.; Kwok R. T. K.; Li Y.; Su H.; Lam J. W. Y.; Chen S.; Tang B. Z. Fluorogenic Ag(+) -Tetrazolate Aggregation Enables Efficient Fluorescent Biological Silver Staining. Angew. Chem., Int. Ed. 2018, 57 (20), 5750–5753. 10.1002/anie.201801653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdeldin S.; Enany S.; Yoshida Y.; Xu B.; Zhang Y.; Zureena Z.; Lokamani I.; Yaoita E.; Yamamoto T. Basics and recent advances of two dimensional- polyacrylamide gel electrophoresis. Clin. Proteomics 2014, 11 (1), 16. 10.1186/1559-0275-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum E.Fluorescent Staining of Gels. In Protein Gel Detection and Imaging: Methods and Protocols; Kurien B. T., Scofield R. H., Eds.; Springer: New York, NY, 2018; pp 87–94. [Google Scholar]
- Basile F.; Hauser N. Rapid online nonenzymatic protein digestion combining microwave heating acid hydrolysis and electrochemical oxidation. Anal. Chem. 2011, 83 (1), 359–67. 10.1021/ac1024705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis A. S. Cleavage at aspartic acid. Methods Enzymol. 1983, 91, 324–32. 10.1016/S0076-6879(83)91030-3. [DOI] [PubMed] [Google Scholar]
- Rodríguez J. C.; Wong L.; Jennings P. A. The solvent in CNBr cleavage reactions determines the fragmentation efficiency of ketosteroid isomerase fusion proteins used in the production of recombinant peptides. Protein Expression Purif. 2003, 28 (2), 224–231. 10.1016/S1046-5928(02)00700-3. [DOI] [PubMed] [Google Scholar]
- Bornstein P.; Balian G. Cleavage at Asn-Gly bonds with hydroxylamine. Methods Enzymol. 1977, 47, 132–45. 10.1016/0076-6879(77)47016-2. [DOI] [PubMed] [Google Scholar]
- Chen L. C.; Kinoshita M.; Noda M.; Ninomiya S.; Hiraoka K. Rapid Online Non-Enzymatic Protein Digestion Analysis with High Pressure Superheated ESI-MS. J. Am. Soc. Mass Spectrom. 2015, 26 (7), 1085–91. 10.1007/s13361-015-1111-4. [DOI] [PubMed] [Google Scholar]
- Giansanti P.; Tsiatsiani L.; Low T. Y.; Heck A. J. R. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11 (5), 993–1006. 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- Hustoft H. K.; Malerød H.; Wilson S.; Reubsaet L.; Lundanes E.; Greibrokk T.. A Critical Review of Trypsin Digestion for LC-MS Based Proteomics. In Integrative Proteomics; Leung H.-C., Ed.; InTech, 2012; pp 73–92. [Google Scholar]
- Raijmakers R.; Neerincx P.; Mohammed S.; Heck A. J. R. Cleavage specificities of the brother and sister proteases Lys-C and Lys-N. Chem. Commun. 2009, 46 (46), 8827–8829. 10.1039/c0cc02523b. [DOI] [PubMed] [Google Scholar]
- Gundry R. L.; White M. Y.; Murray C. I.; Kane L. A.; Fu Q.; Stanley B. A.; Van Eyk J. E. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Current protocols in molecular biology 2009, 25. 10.1002/0471142727.mb1025s88. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chapter 10, Unit 10.
- Rappsilber J.; Ishihama Y.; Mann M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75 (3), 663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Safarik I.; Safarikova M. Magnetic techniques for the isolation and purification of proteins and peptides. BioMagnetic Research and Technology 2004, 2 (1), 7. 10.1186/1477-044X-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas M.; Pereckas M.; Jones Lipinski R. A.; Ashwood C.; Gundry R. L. SP2: Rapid and Automatable Contaminant Removal from Peptide Samples for Proteomic Analyses. J. Proteome Res. 2019, 18 (4), 1644–1656. 10.1021/acs.jproteome.8b00916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor K. N.; Barry D. T.; Rees R. C.; Dodi I. A.; McArdle S. E.; Creaser C. S.; Bonner P. L. Estimation of peptide concentration by a modified bicinchoninic acid assay. Anal. Biochem. 2009, 393 (1), 138–140. 10.1016/j.ab.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Sargent M.; Sage A.; Wolff C.; Mussell C.; Neville D.; Lord G.; Saeed M.; Lad R.; Godfrey R.; Hird S.; Barwick V.. Instrumentation. In Guide to Achieving Reliable Quantitative LC-MS Measurements; Sargent M., Ed.; RSC Analytical Methods Committee, 2013; pp 5–11. [Google Scholar]
- Shishkova E.; Hebert A. S.; Coon J. J. Now, More Than Ever, Proteomics Needs Better Chromatography. Cell systems 2016, 3 (4), 321–324. 10.1016/j.cels.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley R.Instrumentation for LC-MS/MS in Proteomics. In LC-MS/MS in Proteomics: Methods and Applications; Cutillas P. R., Timms J. F., Eds.; Humana Press: Totowa, NJ, 2010; pp 47–60. [Google Scholar]
- Hsieh E. J.; Bereman M. S.; Durand S.; Valaskovic G. A.; MacCoss M. J. Effects of column and gradient lengths on peak capacity and peptide identification in nanoflow LC-MS/MS of complex proteomic samples. J. Am. Soc. Mass Spectrom. 2013, 24 (1), 148–153. 10.1007/s13361-012-0508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder L. R.; Kirkland J. J.; Glajch J. L.. The Column. In Practical HPLC Method Development, 2nd ed.; John Wiley & Sons, 1997; pp 174–232. [Google Scholar]
- Petersson P.; Frank A.; Heaton J.; Euerby M. R. Maximizing peak capacity and separation speed in liquid chromatography. J. Sep. Sci. 2008, 31 (13), 2346–57. 10.1002/jssc.200800064. [DOI] [PubMed] [Google Scholar]
- Liu H.; Finch J. W.; Lavallee M. J.; Collamati R. A.; Benevides C. C.; Gebler J. C. Effects of column length, particle size, gradient length and flow rate on peak capacity of nano-scale liquid chromatography for peptide separations. J. Chromatogr A 2007, 1147 (1), 30–6. 10.1016/j.chroma.2007.02.016. [DOI] [PubMed] [Google Scholar]
- de Hoffmann E.Mass Spectrometry. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley Online Library, 2005; pp 1–20. [Google Scholar]
- Goebel-Stengel M.; Stengel A.; Taché Y.; Reeve J. R. Jr. The importance of using the optimal plasticware and glassware in studies involving peptides. Anal. Biochem. 2011, 414 (1), 38–46. 10.1016/j.ab.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraut A.; Marcellin M.; Adrait A.; Kuhn L.; Louwagie M.; Kieffer-Jaquinod S.; Lebert D.; Masselon C. D.; Dupuis A.; Bruley C.; Jaquinod M.; Garin J.; Gallagher-Gambarelli M. Peptide storage: are you getting the best return on your investment? Defining optimal storage conditions for proteomics samples. J. Proteome Res. 2009, 8 (7), 3778–85. 10.1021/pr900095u. [DOI] [PubMed] [Google Scholar]
- Kristensen K.; Henriksen J. R.; Andresen T. L. Adsorption of cationic peptides to solid surfaces of glass and plastic. PLoS One 2015, 10 (5), e0122419 10.1371/journal.pone.0122419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-H.; Lee S.-C.; Sabu S.; Fang H.-C.; Chung S.-C.; Han C.-C.; Chang H.-C. Solid-Phase Extraction and Elution on Diamond (SPEED): A Fast and General Platform for Proteome Analysis with Mass Spectrometry. Anal. Chem. 2006, 78 (12), 4228–4234. 10.1021/ac052085y. [DOI] [PubMed] [Google Scholar]
- DeAngelis M. M.; Wang D. G.; Hawkins T. L. Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 1995, 23 (22), 4742–4743. 10.1093/nar/23.22.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert A. J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. Journal of Chromatography A 1990, 499, 177–196. 10.1016/S0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- Alpert A. J. Electrostatic Repulsion Hydrophilic Interaction Chromatography for Isocratic Separation of Charged Solutes and Selective Isolation of Phosphopeptides. Anal. Chem. 2008, 80 (1), 62–76. 10.1021/ac070997p. [DOI] [PubMed] [Google Scholar]
- Virant-Klun I.; Leicht S.; Hughes C.; Krijgsveld J. Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell Proteomics 2016, 15 (8), 2616–27. 10.1074/mcp.M115.056887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sielaff M.; Kuharev J.; Bohn T.; Hahlbrock J.; Bopp T.; Tenzer S.; Distler U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16 (11), 4060–4072. 10.1021/acs.jproteome.7b00433. [DOI] [PubMed] [Google Scholar]
- Cagnetta R.; Frese C. K.; Shigeoka T.; Krijgsveld J.; Holt C. E. Rapid Cue-Specific Remodeling of the Nascent Axonal Proteome. Neuron 2018, 99 (1), 29–46. 10.1016/j.neuron.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åhrman E.; Hallgren O.; Malmström L.; Hedström U.; Malmström A.; Bjermer L.; Zhou X.-H.; Westergren-Thorsson G.; Malmström J. Quantitative proteomic characterization of the lung extracellular matrix in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. J. Proteomics 2018, 189, 23–33. 10.1016/j.jprot.2018.02.027. [DOI] [PubMed] [Google Scholar]
- Höhne M.; Frese C. K.; Grahammer F.; Dafinger C.; Ciarimboli G.; Butt L.; Binz J.; Hackl M. J.; Rahmatollahi M.; Kann M.; Schneider S.; Altintas M. M.; Schermer B.; Reinheckel T.; Göbel H.; Reiser J.; Huber T. B.; Kramann R.; Seeger-Nukpezah T.; Liebau M. C.; Beck B. B.; Benzing T.; Beyer A.; Rinschen M. M. Single-nephron proteomes connect morphology and function in proteinuric kidney disease. Kidney Int. 2018, 93 (6), 1308–1319. 10.1016/j.kint.2017.12.012. [DOI] [PubMed] [Google Scholar]
- Hayoun K.; Gouveia D.; Grenga L.; Pible O.; Armengaud J.; Alpha-Bazin B. Evaluation of Sample Preparation Methods for Fast Proteotyping of Microorganisms by Tandem Mass Spectrometry. Front. Microbiol. 2019, 10, 1985. 10.3389/fmicb.2019.01985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19 (1), 161. 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht H.; Emmott E.; Petelski A. A.; Huffman R. G.; Perlman D. H.; Serra M.; Kharchenko P.; Koller A.; Slavov N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22 (1), 50. 10.1186/s13059-021-02267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoof E. M.; Furtwängler B.; Üresin N.; Rapin N.; Savickas S.; Gentil C.; Lechman E.; Auf dem Keller U.; Dick J. E.; Porse B. T. Quantitative Single-Cell Proteomics as a Tool to Characterize Cellular Hierarchies. Nat. Commun. 2021, 12, 3341. 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer P. E.; Kulak N. A.; Pichler G.; Holdt L. M.; Teupser D.; Mann M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst 2016, 2 (3), 185–95. 10.1016/j.cels.2016.02.015. [DOI] [PubMed] [Google Scholar]
- Ding H.; Fazelinia H.; Spruce L. A.; Weiss D. A.; Zderic S. A.; Seeholzer S. H. Urine Proteomics: Evaluation of Different Sample Preparation Workflows for Quantitative, Reproducible, and Improved Depth of Analysis. J. Proteome Res. 2020, 19 (4), 1857–1862. 10.1021/acs.jproteome.9b00772. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Yan X.; Sun L.; Zhu G.; Dovichi N. J. Detachable strong cation exchange monolith, integrated with capillary zone electrophoresis and coupled with pH gradient elution, produces improved sensitivity and numbers of peptide identifications during bottom-up analysis of complex proteomes. Anal. Chem. 2015, 87 (8), 4572–7. 10.1021/acs.analchem.5b00789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga K.; Katoh Y.; Nagase K.; Igarashi K. Microproteomics with microfluidic-based cell sorting: Application to 1000 and 100 immune cells. Proteomics 2017, 17 (13–14), 1600420. 10.1002/pmic.201600420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Piehowski P. D.; Zhao R.; Chen J.; Shen Y.; Moore R. J.; Shukla A. K.; Petyuk V. A.; Campbell-Thompson M.; Mathews C. E.; Smith R. D.; Qian W.-J.; Kelly R. T. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018, 9 (1), 882. 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Clair G.; Chrisler W. B.; Shen Y.; Zhao R.; Shukla A. K.; Moore R. J.; Misra R. S.; Pryhuber G. S.; Smith R. D.; Ansong C.; Kelly R. T. Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angew. Chem., Int. Ed. 2018, 57 (38), 12370–12374. 10.1002/anie.201802843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y.; Motamedchaboki K.; Misal S. A.; Liang Y.; Guise A. J.; Truong T.; Huguet R.; Plowey E. D.; Zhu Y.; Lopez-Ferrer D.; Kelly R. T. Ultrasensitive single-cell proteomics workflow identifies > 1000 protein groups per mammalian cell. Chemical Science 2021, 12 (3), 1001–1006. 10.1039/D0SC03636F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipert J.; Tholey A. Miniaturized sample preparation on a digital microfluidics device for sensitive bottom-up microproteomics of mammalian cells using magnetic beads and mass spectrometry-compatible surfactants. Lab Chip 2019, 19 (20), 3490–3498. 10.1039/C9LC00715F. [DOI] [PubMed] [Google Scholar]
- Li Z. Y.; Huang M.; Wang X. K.; Zhu Y.; Li J. S.; Wong C. C. L.; Fang Q. Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem. 2018, 90 (8), 5430–5438. 10.1021/acs.analchem.8b00661. [DOI] [PubMed] [Google Scholar]
- Lombard-Banek C.; Reddy S.; Moody S. A.; Nemes P. Label-free Quantification of Proteins in Single Embryonic Cells with Neural Fate in the Cleavage-Stage Frog (Xenopus laevis) Embryo using Capillary Electrophoresis Electrospray Ionization High-Resolution Mass Spectrometry (CE-ESI-HRMS). Mol. Cell Proteomics 2016, 15 (8), 2756–68. 10.1074/mcp.M115.057760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Acor H.; McCown M. A.; Nwosu A. J.; Boekweg H.; Axtell N. B.; Truong T.; Cong Y.; Payne S. H.; Kelly R. T. Fully Automated Sample Processing and Analysis Workflow for Low-Input Proteome Profiling. Anal. Chem. 2021, 93 (3), 1658–1666. 10.1021/acs.analchem.0c04240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Uwugiaren N.; Williams S. M.; Moore R. J.; Zhao R.; Goodlett D.; Dapic I.; Pasa-Tolic L.; Zhu Y. Sensitive Top-Down Proteomics Analysis of a Low Number of Mammalian Cells Using a Nanodroplet Sample Processing Platform. Anal. Chem. 2020, 92 (10), 7087–7095. 10.1021/acs.analchem.0c00467. [DOI] [PubMed] [Google Scholar]
- Williams S. M.; Liyu A. V.; Tsai C.-F.; Moore R. J.; Orton D. J.; Chrisler W. B.; Gaffrey M. J.; Liu T.; Smith R. D.; Kelly R. T.; Pasa-Tolic L.; Zhu Y. Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography-Mass Spectrometry for High-Throughput Single-Cell Proteomics. Anal. Chem. 2020, 92 (15), 10588–10596. 10.1021/acs.analchem.0c01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou M.; Clair G.; Tsai C.-F.; Xu K.; Chrisler W. B.; Sontag R. L.; Zhao R.; Moore R. J.; Liu T.; Pasa-Tolic L.; Smith R. D.; Shi T.; Adkins J. N.; Qian W.-J.; Kelly R. T.; Ansong C.; Zhu Y. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem. 2019, 91 (20), 13119–13127. 10.1021/acs.analchem.9b03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Li H.; Bhatti S.; Zhou S.; Yang Y.; Fish T.; Thannhauser T. W. Development of a laser capture microscope-based single-cell-type proteomics tool for studying proteomes of individual cell layers of plant roots. Hortic. Res. 2016, 3, 16026. 10.1038/hortres.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Dou M.; Piehowski P. D.; Liang Y.; Wang F.; Chu R. K.; Chrisler W. B.; Smith J. N.; Schwarz K. C.; Shen Y.; Shukla A. K.; Moore R. J.; Smith R. D.; Qian W. J.; Kelly R. T. Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol. Cell Proteomics 2018, 17 (9), 1864–1874. 10.1074/mcp.TIR118.000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Podolak J.; Zhao R.; Shukla A. K.; Moore R. J.; Thomas G. V.; Kelly R. T. Proteome Profiling of 1 to 5 Spiked Circulating Tumor Cells Isolated from Whole Blood Using Immunodensity Enrichment, Laser Capture Microdissection, Nanodroplet Sample Processing, and Ultrasensitive nanoLC-MS. Anal. Chem. 2018, 90 (20), 11756–11759. 10.1021/acs.analchem.8b03268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagley L. F.; Infusini G.; Larsen R. H.; Sandow J. J.; Webb A. I. Universal Solid-Phase Protein Preparation (USP(3)) for Bottom-up and Top-down Proteomics. J. Proteome Res. 2019, 18 (7), 2915–2924. 10.1021/acs.jproteome.9b00217. [DOI] [PubMed] [Google Scholar]
- Marx V. A dream of single-cell proteomics. Nat. Methods 2019, 16 (9), 809–812. 10.1038/s41592-019-0540-6. [DOI] [PubMed] [Google Scholar]
- Cheung T. K.; Lee C.-Y.; Bayer F. P.; McCoy A.; Kuster B.; Rose C. M. Defining the carrier proteome limit for single-cell proteomics. Nat. Methods 2021, 18 (1), 76–83. 10.1038/s41592-020-01002-5. [DOI] [PubMed] [Google Scholar]
- Kostas J. C.; Gregus M.; Schejbal J.; Ray S.; Ivanov A. R. Simple and Efficient Microsolid-Phase Extraction Tip-Based Sample Preparation Workflow to Enable Sensitive Proteomic Profiling of Limited Samples (200 to 10,000 Cells). J. Proteome Res. 2021, 20 (3), 1676–1688. 10.1021/acs.jproteome.0c00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.; Chen D.; Li S.; Ou X.; Liu B.-F. Microfluidics towards single cell resolution protein analysis. TrAC, Trends Anal. Chem. 2019, 117, 2–12. 10.1016/j.trac.2019.06.022. [DOI] [Google Scholar]
- He X.; Chen Q.; Zhang Y.; Lin J.-M. Recent advances in microchip-mass spectrometry for biological analysis. TrAC, Trends Anal. Chem. 2014, 53, 84–97. 10.1016/j.trac.2013.09.013. [DOI] [Google Scholar]
- Lu Y.; Yang L.; Wei W.; Shi Q. Microchip-based single-cell functional proteomics for biomedical applications. Lab Chip 2017, 17 (7), 1250–1263. 10.1039/C7LC00037E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantonico A.; Urban P. L.; Fagerer S. R.; Balabin R. M.; Zenobi R. Single-cell MALDI-MS as an analytical tool for studying intrapopulation metabolic heterogeneity of unicellular organisms. Anal. Chem. 2010, 82 (17), 7394–400. 10.1021/ac1015326. [DOI] [PubMed] [Google Scholar]
- Korenaga A.; Chen F.; Li H.; Uchiyama K.; Lin J.-M. Inkjet automated single cells and matrices printing system for matrix-assisted laser desorption/ionization mass spectrometry. Talanta 2017, 162, 474–478. 10.1016/j.talanta.2016.10.055. [DOI] [PubMed] [Google Scholar]
- Yang M.; Chao T. C.; Nelson R.; Ros A. Direct detection of peptides and proteins on a microfluidic platform with MALDI mass spectrometry. Anal. Bioanal. Chem. 2012, 404 (6–7), 1681–9. 10.1007/s00216-012-6257-3. [DOI] [PubMed] [Google Scholar]
- Onjiko R. M.; Plotnick D. O.; Moody S. A.; Nemes P. Metabolic Comparison of Dorsal versus Ventral Cells Directly in the Live 8-cell Frog Embryo by Microprobe Single-cell CE-ESI-MS. Anal. Methods 2017, 9 (34), 4964–4970. 10.1039/C7AY00834A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Wang H.; Manicke N. E.; Lin J. M.; Cooks R. G.; Ouyang Z. Development, characterization, and application of paper spray ionization. Anal. Chem. 2010, 82 (6), 2463–71. 10.1021/ac902854g. [DOI] [PubMed] [Google Scholar]
- Wang H.; Liu J.; Cooks R. G.; Ouyang Z. Paper spray for direct analysis of complex mixtures using mass spectrometry. Angew. Chem., Int. Ed. 2010, 49 (5), 877–80. 10.1002/anie.200906314. [DOI] [PubMed] [Google Scholar]
- Wang H.; Ren Y.; McLuckey M. N.; Manicke N. E.; Park J.; Zheng L.; Shi R.; Cooks R. G.; Ouyang Z. Direct quantitative analysis of nicotine alkaloids from biofluid samples using paper spray mass spectrometry. Anal. Chem. 2013, 85 (23), 11540–11544. 10.1021/ac402798m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellors J. S.; Jorabchi K.; Smith L. M.; Ramsey J. M. Integrated microfluidic device for automated single cell analysis using electrophoretic separation and electrospray ionization mass spectrometry. Anal. Chem. 2010, 82 (3), 967–73. 10.1021/ac902218y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Wu J.; Zhang Y.; Lin J. M. Qualitative and quantitative analysis of tumor cell metabolism via stable isotope labeling assisted microfluidic chip electrospray ionization mass spectrometry. Anal. Chem. 2012, 84 (3), 1695–701. 10.1021/ac300003k. [DOI] [PubMed] [Google Scholar]
- Shao X.; Wang X.; Guan S.; Lin H.; Yan G.; Gao M.; Deng C.; Zhang X. Integrated Proteome Analysis Device for Fast Single-Cell Protein Profiling. Anal. Chem. 2018, 90 (23), 14003–14010. 10.1021/acs.analchem.8b03692. [DOI] [PubMed] [Google Scholar]
- Espina V.; Milia J.; Wu G.; Cowherd S.; Liotta L. A. Laser capture microdissection. Methods Mol. Biol. 2006, 319, 213–29. 10.1007/978-1-59259-993-6_10. [DOI] [PubMed] [Google Scholar]