

Abstract
Presentation of antigens by human leukocyte antigen (HLA) complexes at the cell surface is a key process in the immune response. The α-chain, containing the peptide-binding groove, is one of the most polymorphic proteins in the proteome. All HLA class I α-chains carry a conserved N-glycosylation site, but little is known about its nature and function. Here, we report an in-depth characterization of N-glycosylation features of HLA class I molecules. We observe that different HLA-A α-chains carry similar glycosylation, distinctly different from the HLA-B, HLA-C, and HLA-F α-chains. Although HLA-A displays the broadest variety of glycan characteristics, HLA-B α-chains carry mostly mature glycans, and HLA-C and HLA-F α-chains carry predominantly high-mannose glycans. We expected these glycosylation features to be directly linked to cellular localization of the HLA complexes. Indeed, analyzing HLA class I complexes from crude plasma and inner membrane-enriched fractions confirmed that most HLA-B complexes can be found at the plasma membrane, while most HLA-C and HLA-F molecules reside in the endoplasmic reticulum and Golgi membrane, and HLA-A molecules are more equally distributed over these cellular compartments. This allotype-specific cellular distribution of HLA molecules should be taken into account when analyzing peptide antigen presentation by immunopeptidomics.
Keywords: glycobiology, glycoproteins, human leukocyte antigen (HLA), major histocompatibility complex (MHC), mass spectrometry (MS), cellular localization
Introduction
Major histocompatibility complex class I molecules, in humans termed human leukocyte antigen (HLA) complexes, play a key role in our immune system, as they provide a means to present foreign peptide antigens at the cellular surface, providing a signal to our T-cells to eliminate them. Following their initial discovery as the main factor in defining successful organ transplants, HLA complexes and peptide presentation processes have been studied extensively over the last few decades.1−3 As a result of this large body of research, we have quite a coherent picture of how HLA class I molecules function, from which we can largely understand their critical role in transplantation,4,5 autoimmunity,6−9 bacterial and viral infections,10−13 and more recently tumor immunotherapy.14,15
The cellular processes underlying HLA class I antigen presentation are well understood.16 Briefly, the endogenously synthesized class I peptide ligands of antigens are generated primarily by cytosolic and nuclear proteasomes. These peptides are translocated to the endoplasmic reticulum (ER), where they are loaded into the peptide-binding groove of HLA class I molecules by means of the peptide-loading complex (PLC). After an antigen has been loaded, the stable and functional HLA class I complex comprises a heavy α-chain, a β2-microglobulin (β2m) chain, and the loaded peptide. These complexes traverse out of the ER, into the Golgi, en route to the cell surface. Upon incorporation into the plasma membrane (PM), the peptide can be presented to CD8+ T cells, thereby allowing the T cells to identify and eliminate pathogen-infected cells or cancer cells.17,18
Beyond this generic description, HLA class I complexes are actually extremely diverse. This diversity starts at the DNA level, as in the human genome, six genes encode for six different HLA class I heavy chains; the more abundantly expressed classical genes A, B, and C and the non-classical genes E, F, and G. The classical HLA-A, B, and C genes are highly polymorphic, where several thousands of alleles have been identified across the population.19 The most frequent polymorphisms are found in the sequence regions that encode the peptide-binding groove, highlighted by green boxes20,21 (see Figure 1A). These polymorphisms allow for a wide range of specificities for peptide binding, which consequently allows a diverse set of peptides to be presented at the cell surface. Figure 1A summarizes sequence polymorphisms that are observed in some of the most common HLA A, B, and C allotypes.22 The sequence homology of these proteins is also visualized through the phylogenetic tree (depicted in Figure 1B), from which it is clear that allotypes belonging to separate classical genes do cluster more together. A much more extensive phylogenetic tree analysis of HLA allotypes is provided in Figure S1.
Figure 1.
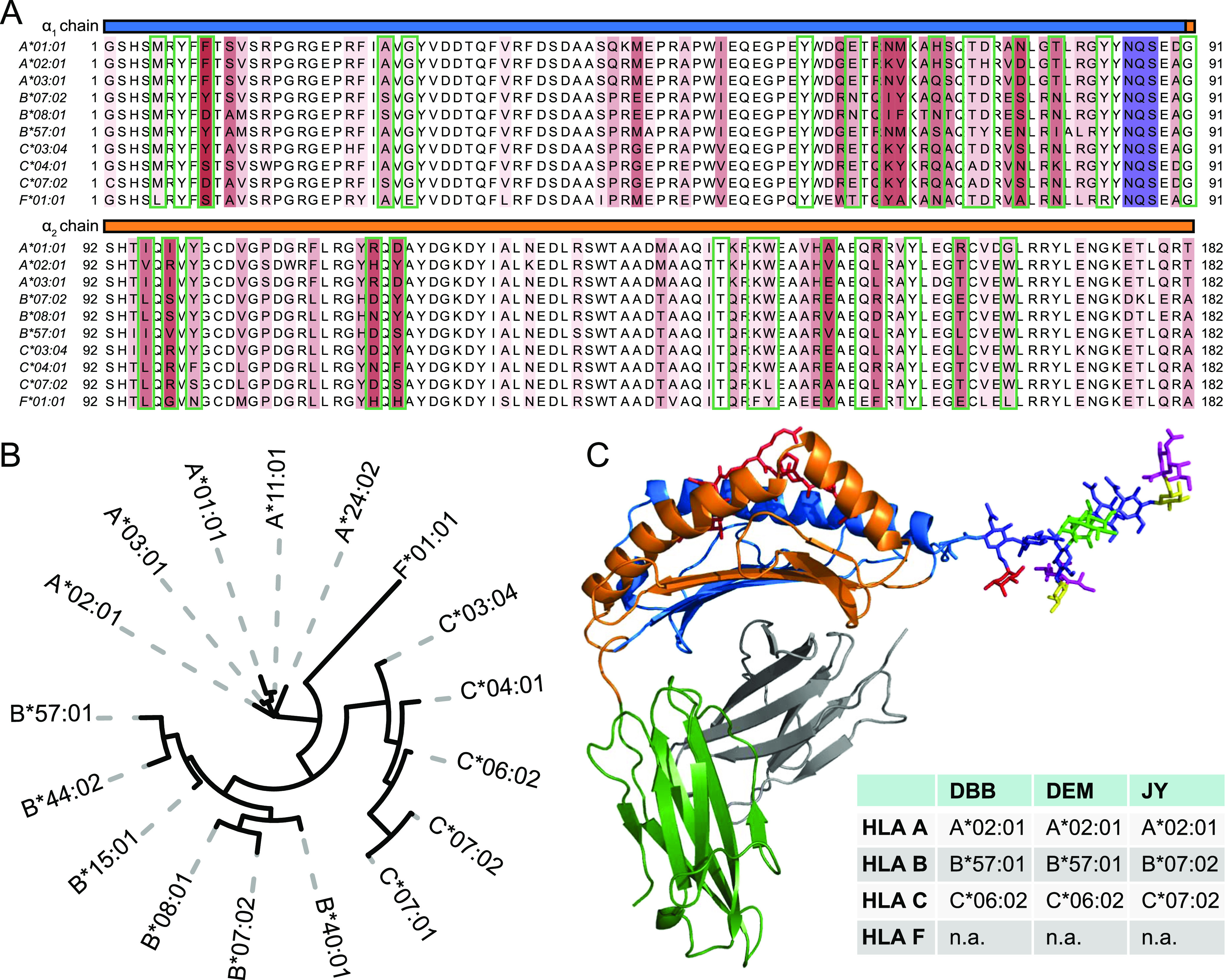
Sequence and structural characteristics of HLA class I molecules. (A) Multiple sequence alignment of the mature form of several common classical HLA A, B, and C genes and HLA F*01:01. The α1 and α2 domains of the sequences are shown starting at position 1 of the mature sequence and ending at position 182. Above the amino acid sequence, the α1 and α2 domains are highlighted in blue and orange. Positions that display amino acid variation between allotypes are highlighted in red with darker tints indicating a higher degree of sequence variation. The conserved N-linked glycosylation site motif is highlighted in purple. Amino acids involved in forming the peptide-binding groove are boxed in green, (B) phylogenetic tree visualization of the multiple sequence alignment depicted in 1A. Although all HLA class I genes display high homology, the allotypes still cluster per distinct classical gene type. See Figure S1 for a much more extensive phylogenetic analysis of HLA class I heavy-chain allotypes. (C) Structural model of the HLA-A*02:01 molecule (pdb: 1I4F). Chains within the HLA molecule are colored: α1-blue, α2-orange, α3-green, β2m-gray, and binding peptide-red. Attached to Asn86 is modeled on scale a reported detected bisected glycan structure.
Notwithstanding these widespread and frequently occurring polymorphisms, a particular region in the sequence, conserved over all reported alleles,19 is highlighted in purple in Figure 1A. This highlighted NxS/T motif is a well-known motif for N-linked glycosylation. It has been established that the asparagine 86 site is indeed glycosylated;23 however, the nature of the glycans attached and their functional role have not been extensively studied. In Figure 1C, a structural model of the HLA class I complex is depicted, with attached to it, modeled to scale, a commonly observed mature glycan structure. It is apparent from this structural model that the N-glycan is localized in the proximity of the peptide-binding groove, and likely when the molecule is at the PM, the glycan can be exposed to the outside of the cell, and interact with external factors, including factors of T cell origin.
Cellular protein N-glycosylation is a complex multistep process in which many enzymes in the ER and Golgi are involved. Therefore, HLA complexes will undergo their glycosylation while traversing through the ER and Golgi compartments. The rate of this traversal has been studied by pulse-chase assays, which use endoglycosidase H (Endo H), an enzyme that digests specifically high-mannose and hybrid/asymmetric glycans (on HLA) to assess the rate through the ER and early Golgi.24−27 To describe the N-glycosylation pathway in short, during this process a Glc3Man9GlcNAc2 glycan is first transferred to the target protein, during or shortly after protein synthesis and translocation into the ER.28 Subsequently, this structure is trimmed, losing two Glc residues to form Glc1Man9GlcNAc2. Next, if the protein is folded correctly, the last Glc residue is removed, and the protein can continue to the Golgi. In the ER or early cis-Golgi, additional Man residues can be removed. This leaves only the Man3GlcNAc2 core that is found in nearly all N-linked glycans. Subsequently in the medial- and trans-Golgi, processing occurs to form complex type glycans with antennae that can include several GlcNAc, Fuc, Gal, and SA residues.29−31 It is expected that a protein is transported to the cell surface only after full glycan maturation has occurred.
Here, we monitored in detail the HLA gene-specific glycosylation patterns of HLA class I molecules of three different cell lines, all homozygous for the classical HLA genes A, B, and C. The cell lines and the allotypes they harbor are shown in Figure 1C. Using a glycopeptide-centric proteomics approach for each distinct HLA heavy-chain, unique glycopeptides could be identified harboring distinct glycoforms. Quantifying these different glycoforms revealed that each of the HLA allotypes was modified with a distinct glycoform profile, which we expected to be directly linked to their differential cellular distribution. When further dissected with subcellular fractionation, we could indeed observe distinctive glycosylations that correlate with differential PM presentation. In the studied JY cells for instance, our data indicate that HLA-B complexes reside nearly exclusively at the PM, whereas a large part of the HLA-A complexes are also observed in inner membrane (IM) compartments. This correlates with our findings that HLA-B has exclusively mature bisected and extended glycans, while HLA-A has a considerable fraction of immature high-mannose glycans. As far as we know, this is the first time that HLA class I glycoform profiling is taken as an indicator to assess HLA cellular localization. Although the correlation between glycosylation and localization was expected, our approach allows us to directly sample subcellular localization of each HLA allotype considering glycosylation as a localization marker, and shows that each HLA allotype can have a unique subcellular distribution despite their very high molecular and structural similarities.
Experimental Procedures
Cell Culture
The B-lymphoblastoid cell line JY (containing HLA-A*02:01, HLA-B*07:02, HLA-C*07:02) was cultured in RPMI 1640 medium (+glutamine, Gibco) supplemented with 10% fetal bovine serum, 50 U/mL penicillin (Gibco), and 50 μg/mL streptomycin (Gibco) in a humidified atmosphere at 37 °C with 5% CO2. The B-lymphoblastoid cell lines DEM (containing HLA-A*02:01, HLA-B*57:01, and HLA-C*06:02) and DBB (containing HLA-A*02:01, HLA-B*57:01, HLA-C*06:02) were cultured in RPMI 1640 medium (+glutamine) supplemented with 15% fetal bovine serum, 1 mM sodium pyruvate (Gibco), 50 U/mL penicillin, and 50 μg/mL streptomycin in a humidified atmosphere at 37 °C with 5% CO2.
HLA Class I Immuno-Affinity Purification and Protein Digestion
Per cell line, 5 × 108 cells were harvested by centrifugation and washed three times with phosphate-buffered saline (PBS). The cells were lysed in 10 mL Pierce IP lysis buffer (Thermo Scientific) supplemented with 1× complete protease inhibitor cocktail (Roche Diagnostics), 50 μg/mL DNAse I (Sigma-Aldrich) and 50 μg/mL RNAse A (Sigma-Aldrich) per gram cell pellet for 1.5 h at 4 °C. The lysate was cleared by centrifugation for 1 h at 18,000g at 4 °C. The protein concentration was determined using the BCA assay (Pierce). HLA class I immuno-affinity purification was performed as described by Demmers et al.32 In short, HLA class I complexes were immunopurified using 0.5 mg W6/32 antibody33 coupled to 125 μL protein A/G beads (Santa Cruz) from 25 mg whole cell (WC) lysate. For the PM and IM fractions, 0.16 mg W6/32 antibody coupled to 40 μL protein A/G beads was used for ∼160 and ∼60 μg input, respectively. To prevent coelution, the antibodies were cross-linked to protein A/G beads. Incubation took place at 4 °C for approximately 16 h. After immuno-affinity purification, the beads were washed with 40 mL of cold PBS. HLA class I complexes and peptide ligands were eluted with 10% acetic acid. The peptide ligands were separated from the HLA molecules using 10 kDa molecular weight cutoff filters (Millipore). The HLA molecules were denatured in 8 M urea in 500 mM ammonium bicarbonate with 1× complete ethylenediaminetetraacetic acid -free protease inhibitor cocktail (Roche Diagnostics). The HLA proteins were reduced, alkylated, and digested with 50 ng trypsin. The digested peptides were loaded onto C18 SEPPAK columns (Supelco) in 0.1% formic acid and eluted after cleanup with 80% acetonitrile (ACN) in 0.1% formic acid. The samples were dried by vacuum centrifugation and reconstituted in 2% formic acid prior to LC–MS/MS analysis.
Cellular Fractionation
PM fractionation was performed by gentle homogenization in an isotonic environment, in the absence of detergents. Homogenized JY cells were applied to a dextran gradient for phase separation (by differential partitioning) into the PM fraction (low-density top fraction) or IM fraction (high-density bottom layer). Intermediate fractions where liquid–liquid mixing could occur were carefully and generously discarded. A similar approach has been published previously.34
Mass Spectrometry Analysis
Glycoproteomics data were acquired using an Agilent 1290 UHPLC (Agilent) coupled to an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific). Proteomics data were acquired using an equivalent UHPLC setup on a Q-exactive HF (Thermo Scientific). Peptides were first trapped on a 2 cm × 100 μm Reprosil C18 trap column (3 μm particle size), followed by separation on a 50 cm × 75 μm Poroshell EC-C18 analytical column (2.7 μm). Trapping was performed for 5 min 0.1% formic acid (solvent A) and eluted with a gradient using 80% ACN with 0.1% formic acid (solvent B). Gradient: 13% B to 40% B over 48 min and 40% B to 100% over 1 min holding at 100% B for 4 min.
For proteomics, the mass spectrometer was operated in data-dependent mode using a top 15 method. Full MS scans were captured using 60,000 resolution at 200 m/z, mass range 310–1600 m/z, with an AGC target of 3 × 106, and a max injection time of 20 ms. MS/MS scans were triggered on charge states 2–5 and excluded for 12 s after selection. A selection window of 1.4 m/z was used, followed by higher-energy collisional dissociation (HCD) fragmentation at 27 normalized collisional energy. Fragmentation scans were captured at 30,000 resolution, with a fixed first mass of 120 m/z, with an AGC target of 1 × 105 and a 50 ms max injection time.
For glycoproteomics, the mass spectrometer was operated in a product ion-triggered data-dependent mode using a 3 s cycle type. Full MS scans were captured on the Orbitrap at 60,000 resolution at 200 m/z, mass range 350–2000 m/z, with an AGC target of 4 × 105, and a max injection time of 50 ms. MS/MS scans were triggered on charge states 2–8 and excluded for 30 s after selection. A selection window of 1.6 m/z was used, followed by fragmentation using HCD at 30% collisional energy. Fragmentation scans were captured on the Orbitrap at 30,000 resolution, mass range 120–4000 m/z. For stepping HCD and EThcD-triggering methods, a targeted mass trigger was configured with a mass list of common glycan oxonium ion fragments as reported by Reiding, et al.35 When at least three ions were detected, another MS/MS scan was triggered on the same precursor. These scans used the same isolation and resolution parameters. For EThcD, calibrated charge-dependent ETD parameters were used and 25% supplemental activation, stepping HCD used collisional energies of 10, 25, and 40%. EThcD and stepping HCD scans used an AGC target of 400% and a 250 ms max injection time.
Data Analysis
Data analysis of proteomics data was performed using MaxQuant (v.1.6.17.0) and the Andromeda search engine. Data were searched against a SwissProt human database (20,431 entries, downloaded on Sept 18, 2019) appended with the specific HLA allotypes found in each cell line. Enzyme specificity was set to trypsin and up to two missed cleavages were allowed. Fixed modification of carbamidomethyl at C and variable modification oxidation at M and N-terminal acetylation were used. An FDR rate of 0.01 was set for both protein and peptide identification. Label-free quantification was performed using IBAQ.36,37
Data analysis of the glycoproteomics data was performed using PMi-Byonic (Protein Metrics) (v3.6). Byonic settings were as follows, cleavage site: fully specific, C-terminal of RK, with up to two missed cleavages. 10 ppm precursor mass tolerance for both HCD and EThcD was applied with a fragment mass tolerance of 20 ppm. Selected modifications: oxidation, variable at M and carbamidomethyl, fixed at C. The Byonic N-glycan 132 human database was used for identification of glycosylation. Precursor isotope off by x was set to “too high or low (narrow)” with a maximum precursor mass of 10,000 Da. Precursor assignment was computed from MS with a maximum of 2 precursors per MS/MS. A 1% FDR (or 20 reverse count) cutoff was used with decoys added in the database. For the protein database, a fasta file was created containing all HLA class I and class II allotypes for the specific cell lines. Subsequent analysis was performed with Python 3.8 using Pandas 1.1.3,38 Numpy 1.19.2,39 and Matplotlib 3.3.2,40 and Seaborn 0.11. Glycopeptides were selected with a minimum Byonic score of 150 and absolute log probability higher than 1. In addition, a glycan needed to be detected at least six times per HLA protein per cell line. Glycans were assigned categories based on compositional requirements. Paucimannose: HexNAc ≤ 2, Hex ≤ 3. High mannose: HexNAc = 2, Hex > 3, hybrid/asymmetric: HexNAc = 3, diantennary: HexNAc = 4, bisected: HexNAc ≥ 5, Hex ≤ 5, and extended: HexNAc ≥ 5, Hex > 5. Distributions of glycan categories were calculated based on glycan peptide spectral match (PSM) counts.
Results
Abundances of HLA Class I Proteins and Their Glycosylation Profiles in WC Lysates
To investigate the glycosylation of HLA class I heavy-chain molecules in detail, we selected a panel of three human cell lines namely DBB, DEM, and JY. These cell lines are all Epstein–Barr virus-transformed immortalized B lymphoblastoid cells of the ECACC HLA-typed collection, and were selected as they all express HLA class I at a reasonably high level. All three cell lines are homozygous for HLA class I, meaning that each only expresses a single A, B, and C allotype. DBB and DEM both express HLA-A*02:01, B*57:01, and C*06:02. JY and HLA-A*02:01, B*07:02, and C*07:02 (Figure 1C). These allotypes are among some of the most common allotypes found in the Caucasian population and these cell lines are routinely used as model systems for studying HLA molecules and their peptide ligandomes.41−43
The analysis was started by enriching all HLA class I molecules from WC lysates by immuno-affinity purification using the pan-HLA class I W6/32 antibody.33 The purified HLA complexes were digested by trypsin, and all resulting peptides were subjected to LC–MS/MS. In a first set of shotgun proteomics analyses, the relative abundance of the different HLA molecules was assessed in the three studied cell lines, using only the unique non-glycosylated HLA peptides. To achieve relative quantification, the IBAQ intensities of the HLA proteins in a sample were summed and the intensity of each HLA protein in that sample was normalized to this summed intensity. Clearly, these data revealed that in all cell lines, the order of abundance in the WC lysates was HLA-A > HLA-B > HLA-C > HLA-F (see Figure 2A and the Tables S1–S3, unique peptides used for quantification are listed in the Tables S6–S8). In contrast to the typical data-dependent shotgun proteomics approaches, a glycopeptide-targeted product-ion-triggered fragmentation strategy was next employed. Precursor ions were selected and fragmented using HCD as is also the case in standard shotgun methods. However, if the resulting MS/MS scan contained glycan-specific oxonium ions, an additional EThcD or stepping-energy HCD scan was triggered on the same, likely glycopeptide, precursor ion.35 In this targeted approach and enabled by the high sequence coverage, we could confidently assign a variety of glycopeptides for each HLA allotype, based on the small but unique protein sequence features. Because the HLA class I molecules were among the most abundant proteins in the sample, as a result of the immuno-affinity enrichment, no further enrichment of glycopeptides was needed to analyze the glycopeptides.
Figure 2.

HLA abundances and glycosylation in the cell lines DBB, DEM, and JY. (A) Normalized protein abundances of different HLA class I molecules immunopurified from WC lysates determined by proteomics, using solely unique non-glycopeptides for quantification. Within the bars, the non-normalized values are depicted. (B) Categorization and color-coding of glycan classes. The six glycan categories are paucimannose (dark blue), high-mannose (green), hybrid/asymmetric (purple), diantennary (light blue), bisected (orange), and extended (red). (C) Stacked bar plots depicting the distribution of glycans for each HLA class I gene in each cell line. Within the bars, the horizontal white depicts the standard deviation averaged over nine injection replicates. On the right of each bar, values indicate the cumulative number of glycopeptides detected across all injection replicates.
Based on the mass shift induced by the glycans to the peptide backbone, combined with the known biosynthetic knowledge about human protein glycosylation, we next annotated glycan compositions to all identified HLA glycopeptides, which were assigned to a smaller number of categories as depicted and color-coded, schematically in Figure 2B. Among these six categories, high-mannose glycans (green) are characterized by having only Man residues extending the Man3GlcNAc2 core shared by all N-glycans. Complex type glycans instead have branches extending the core that are initiated by GlcNAc, which can still be further elongated. Additionally, complex type glycans often contain one or more Fuc and SA residues. Hybrid/asymmetric glycans (purple) display one Man-extended branch and one complex type branch. Diantennary glycans (light blue) instead have two complex type branches. Extended glycans (red) have more than two complex type branches. Bisected glycans (orange) are complex type glycans carrying a bisecting GlcNAc residue attached to the β-Man of the core. Finally, paucimannose and truncated glycans (dark blue) are small mannose glycans or truncated glycans that do not have the complete N glycan core.
We next assessed the relative abundance of these different categories per individual HLA glycopeptide based on the number of spectral counts (PSMs) we observed in our LC MS/MS analysis,27 the detected glycopeptides and glycans are listed in Tables S11–S13 and S16–S18, respectively. These results are depicted in Figure 2C using the color codes for the different glycan categories, with errors bars obtained from replicate measurements. A more detailed overview of the observed glycan compositions is provided in Figure S2. At first glance, although there is some similarity, this analysis revealed that glycoprofiles of allotypes can be quite distinct, even when they are expressed in the same cell. It should be noted that the site occupancy of the glycan is 100%, a fact we could also reproduce by measuring the intact mass of HLA-A by LC–MS. Consequently, non-glycosylated versions of the glycopeptides were not detected in any experiments.
HLA-A*02:01 is abundantly expressed by all three cell lines we investigated. Our data reveal that the glycosylation profile on HLA-A*02:01 is nearly identical in all three cell lines and dominated by bisected (orange) and extended (red) glycans. However, a considerable fraction (25–30% based on the spectral counts) of the HLA-A molecules carry smaller and/or simpler glycans, mostly high-mannose (green) but also some paucimannose (dark-blue) glycans. The DBB and DEM cell lines express B*57:01, the glycosylation of which in these two cell lines is nearly identical and resembles closely that of HLA-A*02:01. In sharp contrast, HLA-B*07:02 in JY cells did harbor a distinct glycosylation pattern, which features almost exclusively bisected (orange) and extended (red) glycans. Distinctively, many more HLA-C molecules, that is, HLA-C*07:02 in JY cells, and HLA-C*06:02 in both DBB and DEM cells (estimated to be around 50% based on spectral counts), are detected with high-mannose glycans (green). The HLA-C glycosylation patterns are quite similar in all three studied cell lines, but different from the other HLA genes. Finally, we also detected, while substantially less-abundant, unique glycopeptides originating from HLA-F in all three cell lines. However, we were not able to annotate the exact HLA-F allele. HLA-F, being one of the non-classical HLA genes, is much less polymorphic (only six different forms have been reported), and all these variants share the same sequence for the part covering the tryptic glycopeptides studied here. In addition, according to our proteomics data, HLA-F expression is quite low, with the DBB cell line forming a noticeable exception (Figure 1A). HLA-F independent from the source cell of origin, was found to be nearly exclusively harboring high-mannose type glycans (green). We did not consider the two other non-classical HLA genes; HLA-E and HLA-G. Sporadically a few unique peptides of HLA-E were detected in our analyses, although their abundances were substantially lower compared to the other HLAs. No unique glycopeptides were detected for HLA-E. No unique peptides for HLA-G were detected in any of our analyses.
Annotation of Glycan Structures on HLA Glycopeptides
LC–MS/MS on glycopeptides is well suited to determine the most likely glycan composition. How these carbohydrate moieties are linked together in the glycan structure, however, is much harder to reveal by mass spectrometric means. Nonetheless, in exceptional cases, LC–MS/MS can also shed light on how a glycan is structured, as nicely described for bisecting glycans by Dang et al.,44 using diagnostic fragment ions. Following a similar strategy, we depict in Figure 3A, an EThcD fragmentation spectrum for a glycopeptide derived from the heavy chain of HLA B*57:01, observing diagnostic fragment ions indicating glycan bisection. In detail, in Figure 3A at m/z 1168.7 (z = 5+), the intact precursor is shown, annotated with “M”. From these data, we determined the intact glycan to have the SA2Gal2Man3GlcNAc5Fuc1 composition. In the EThcD spectrum, a fragment at m/z 1051.5 (z = 4+) is detected, assigned to the peptide plus the glycan lacking 4 hexoses, 2 HexNAcs, and 2 NeuAcs. The mass loss observed for this fragment ion indicates the loss of both antennae on the glycan. After cleaving off both antennae, only a peptide + GlcNAc3Man1Fuc1 fragment remains. This fragment is unique to bisecting glycans, suggesting the bisecting structure of the glycan we observed on HLAs.
Figure 3.
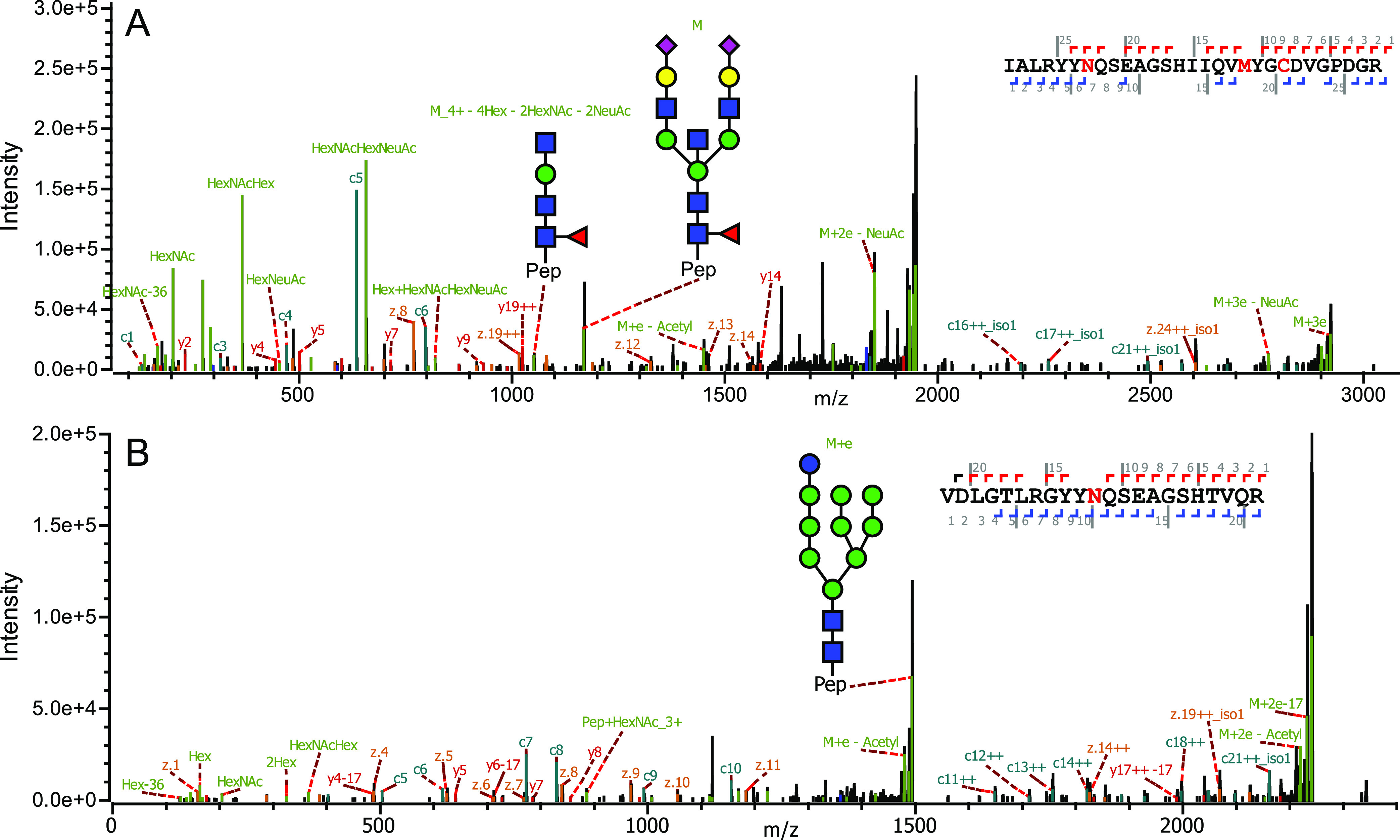
Examples of annotated HLA heavy-chain glycopeptides. (A) EThcD MS/MS spectrum of a HLA-B*57:01-derived bisected glycopeptide (DBB cell line). (A) Precursor glycopeptide (M), m/z 1168.7 (z = 5+), together with its annotated glycan structure are indicated. The fragment ion at m/z 1051.5 (z = 4+), with its annotated glycan structure, verifies the proposed bisected structure. Several additional peptide backbone and glycan fragment ions used for the identification of the glycopeptide are annotated. (B) EThcD MS2 spectrum of a HLA-A*02:01-derived high-mannose glycopeptide (DBB cell line). The annotated glycan structure is indicated. Several additional peptide backbone and glycan fragment ions used for the identification of the glycopeptide are annotated. See Figure S3 for additional MS/MS spectra of different HLA glycopeptides.
In Figure 3B, the EThcD spectrum of a HLA-A*02:01-derived glycopeptide harboring the high-mannose glycan Glc1Man9GlcNAc2 is shown. High-mannose glycans can have up to 9 Man residues, thus the 10th hexose is accredited to a Glc. The high-mannose glycan Glc1Man9GlcNAc2 is thought to be important for binding of the HLA heavy chain to the folding chaperone calreticulin in the ER. Calreticulin interacts optimally with monoglucosylated HLA class I heavy chains, whatever their state of assembly with light chains and peptide, and inhibits their aggregation.45,46 It is expected that proteins harboring these glycans are localized in the ER. As our experiments were carried out on WC lysates, these data hint at that we analyze both internal as well as cell surface-expressed HLA molecules. In Figure S3, several more MS/MS spectra are provided highlighting different glycopeptides detected in our measurements.
Relative Abundances of HLA Class I Proteins and Their Glycosylation Profiles in the Plasma Membrane versus the Inner Membrane
Following their initial synthesis by the ribosome, HLA molecules reside in and traverse through the various inner-membrane compartments (ER and Golgi) of the cell, where they are properly folded and loaded with peptide on their way to the PM. During this process, maturation of the HLA heavy-chain glycan occurs by the various glyco-enzymes present in these compartments.29−31 The data presented above were all resulting from HLA affinity enrichments performed on WC lysates. However, our data did indicate that HLA molecules within a single cell may be differentially glycosylated, which we expect to be linked to the compartmental localization of the HLA molecules in and on the cell. In essence, we utilize the glycosylation characteristic as a proxy for the subcellular distribution of the HLA molecules. Therefore, we extended our analysis performing the HLA affinity enrichment separately on (crude) PM and IM (mostly ER and Golgi) fractions of the cells. This prefractionation comes at the expense of sensitivity, as the pull-downs generally require quite some starting material. Consequently, in the fractionated samples, we were able to detect only the highest abundant glycopeptides, yielding a less-accurate representation of the glycan repertoire. Although some sensitivity is lost, it should still show whether the glycosylation accurately represents the distributions of HLA molecules. We performed this analysis on the JY cells, as in the WC lysates of these cells we observed the most striking differences in glycosylation patterns between HLA-A, B, C, and F (Figure 2). A proteomics-based evaluation of the subcellular fractionation, as described previously,47 assessing the enrichment of protein UniProt keywords in the inner and PM fractions (Figure S4) revealed successful fractionation.
Again, as described above, we assessed by standard shotgun proteomics, quantifying by solely using unique peptides, the relative abundance of the different HLA molecules in the plasma and inner-membrane fraction and compared that to that observed in the WC lysates (see Figure 4A, and Tables S3–S5. The unique peptides used for quantification are listed in Tables S8–S10). A list of other abundantly detected proteins is provided as Supporting Information table. These data clearly revealed that while HLA-A is quite abundant in both the plasma and IM fractions, HLA-B is nearly uniquely enriched in the PM. In contrast, HLA-C was found to be enriched in the IM fraction. Moreover, while HLA-F could not be detected in the WC and PM, it was reasonably abundant in the IM fraction.
Figure 4.
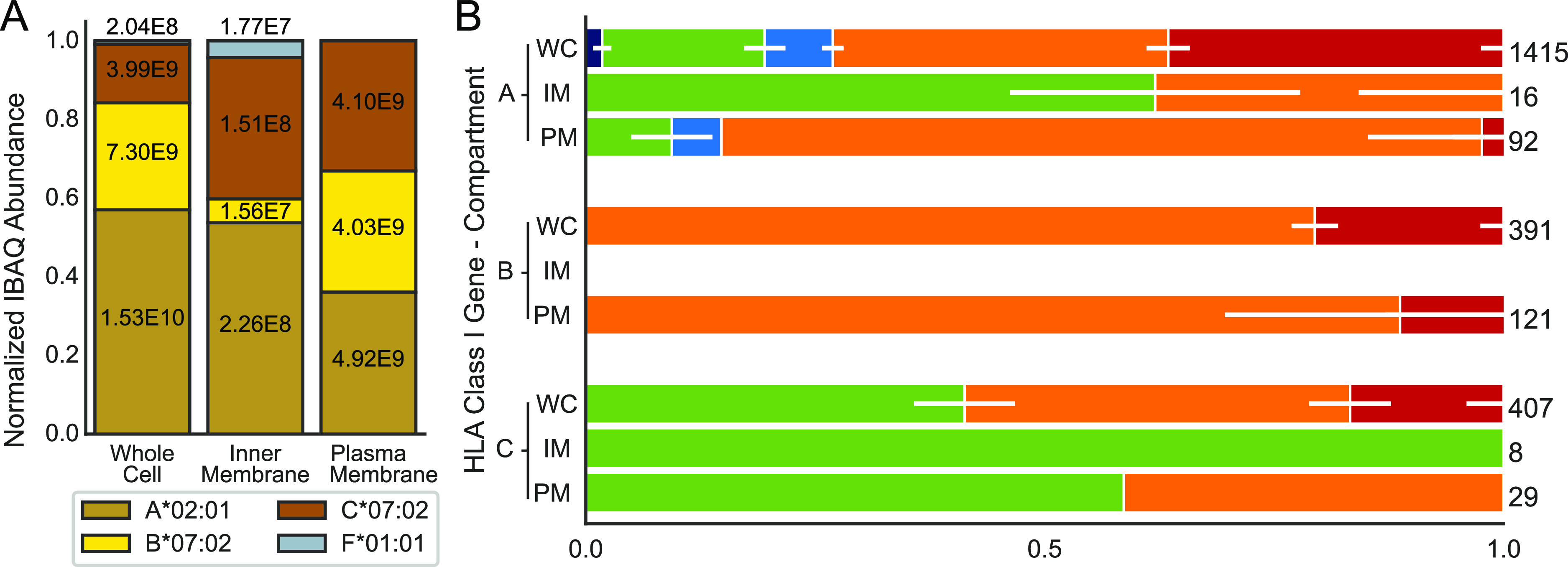
HLA abundances and glycosylation in different compartments of the JY cells. (A) Normalized protein abundances of different HLA class I genes in WC lysates, and the IM (ER and Golgi) and PM fractions determined by proteomics, using solely unique non-glycopeptides for quantification. Within the bars, the non-normalized values are depicted. (B) Stacked bar plots depicting the distribution of glycans for each HLA class I gene in each of the three analyzed fractions. Abbreviations WC, IM, and PM are used to specify the compartments. Within the bars, the horizontal white lines depict the standard deviation averaged over nine injection replicates for the WC and three injection replicates for IM and PM preparations. On the right of each bar, values indicate the cumulative number of glycopeptides detected across all injection replicates. Due to the low abundance of HLA-B in the IM fraction, and for HLA-F, no glycopeptides could be detected following the cellular fractionation. The WC data presented in this figure are identical to the JY cell data presented in Figure 2.
Next, we focused on potential compartment-specific HLA heavy-chain glycosylation (Figure 4B), and for the glycopeptide analysis, we again performed immuno-affinity purification on the HLA molecules. We were only able to make that comparison for the classical HLA class I genes A, B, and C, as the abundance of HLA-F was too low for detection of sufficient glycopeptides. Additionally, although we detected a few HLA-B unique peptides in the shotgun experiments for the inner-membrane fraction, we did not detect glycopeptides of these molecules in the IM fraction, likely due to the low abundance of these molecules in that fraction (Figure 4A). In Figure 4B, the same glycan categorization and color-coding is used as in Figure 2B, and additionally the data for the WC lysate shown in Figure 2B are replicated in Figure 4B for comparison with the data obtained for the IM and PM fractions. The data presented in Figure 4B clearly show that complex glycans represented by the bisected (orange) and extended (red) categories are enriched in the PM fraction, especially for HLA-A and HLA-B. Conversely, high-mannose glycans are found to be enriched in the IM fraction, especially for HLA-A and HLA-C. The fact that we do not detect paucimannose and diantennary glycosylated glycopeptides in the IM and PM fractions may likely be attributed to our lower sensitivity in these experiments due to the lower amount of starting material prior to the affinity purification. The detected glycopeptides and glycans are given in Tables S13–S15 and S18–S20. Notwithstanding these sensitivity issues, the data presented in Figure 4 clearly reveal that the pool of HLA molecules is indeed distributed over the different compartments, and these HLA molecules carry compartment “specific” glycans.
Discussion
Here, we report an in-depth characterization of the N-glycosylation features of HLA class I molecules, affinity purified from WC lysates and enriched PM and IM fractions. We make the striking observation that different allotypes also display different glycosylation patterns, which is noteworthy considering that HLA molecules of different allotypes are nearly identical in sequence and structure, except for a few key residues in the peptide-binding groove. Cumulatively, the data presented here hint at a substantial diversity in the distribution of HLA class I complexes over the different membrane compartments of the cell. By using glycosylation as a proxy for subcellular localization, we are able to show that different allotypes are distinctly distributed over the compartments and that a considerable amount of HLA molecules may reside intracellularly.
Based on the observed glycosylation patterns, it appears that HLA-B complexes, especially in JY cells, are mostly rather mature in their glycosylation and reside largely on the PM of the cell. In sharp contrast, the population of HLA-C molecules are largely enriched in less-mature high-mannose glycans, and mostly residing in the inner-membrane ER/cis-Golgi compartments. Even more so, HLA-F is exclusively modified with high-mannose glycans and seems largely retained in the IM compartments of the cell, as proposed previously.48 The population of HLA-A complexes in the cell display a glycan distribution ranging from paucimannose, high-mannose, diantennary, to fully matured bisected and extended glycans, and seems also to be more widely distributed over the PM and the IM compartments. The distinctive glycosylation and cellular localization of HLA class I proteins as observed in this study is schematically summarized in Figure 5. These observations are made in our model JY cells, but are also conserved to some extent in the DBB and DEM cells that we analyzed in parallel.
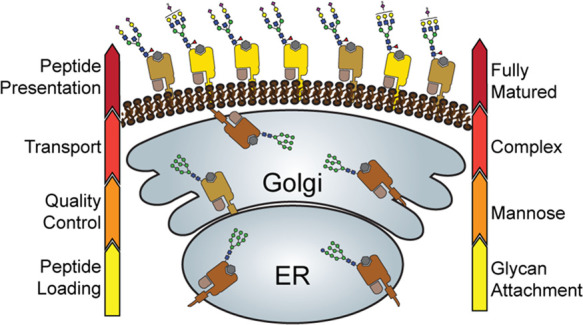
Figure 5.
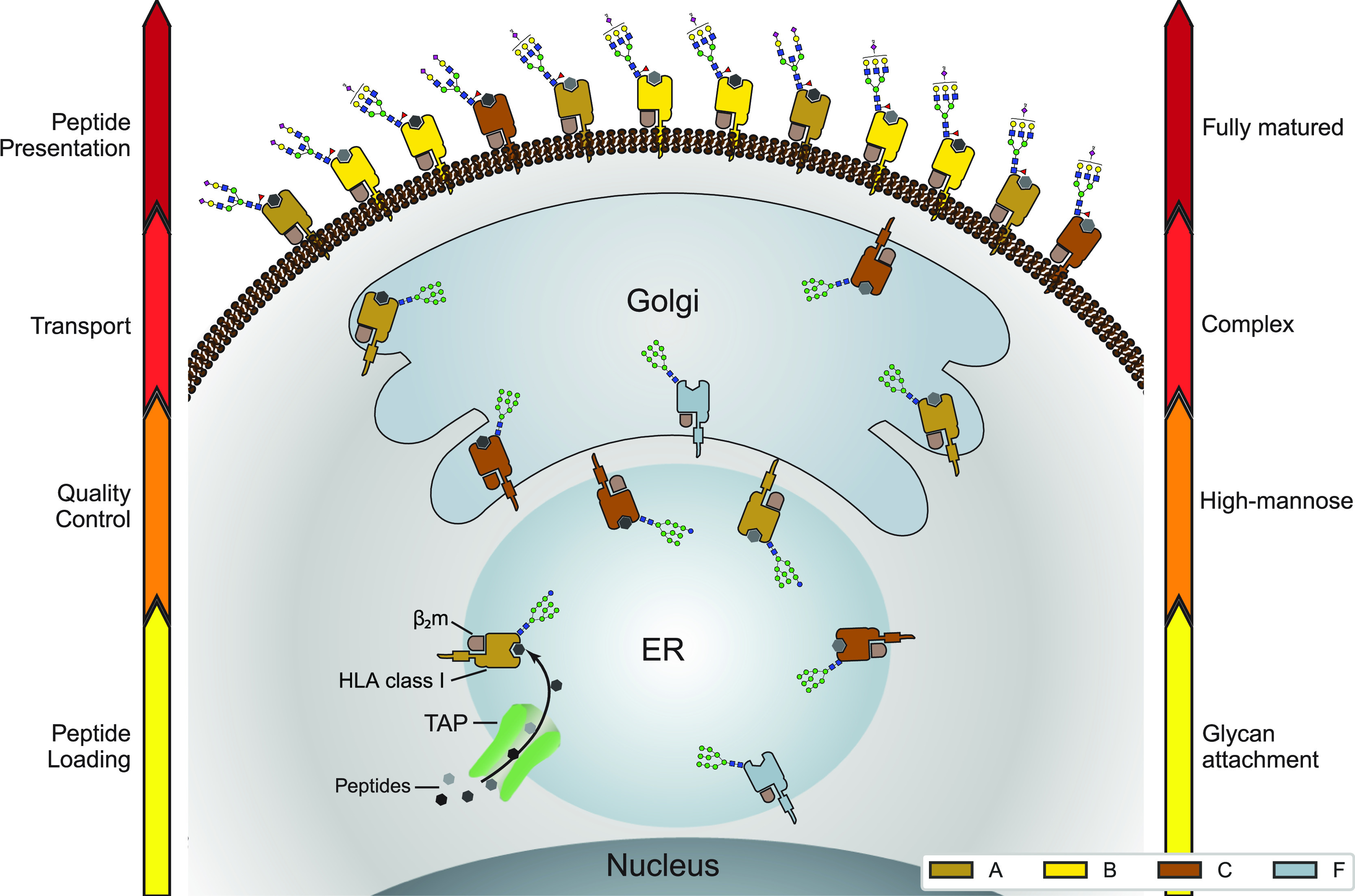
Distinctive glycosylation and cellular localization of HLA class I proteins. Starting as nascent chains synthesized by cellular ribosomes, HLA heavy chains traverse through specific IM compartments to get properly folded, trimmed, associated with the β2m chain, loaded with the peptide antigens, and glycosylated. These processes occur largely sequential and require chaperones, the PLC and a variety of glycoenzymes, residing in the different subcompartments of the ER and Golgi. Fully assembled, peptide-loaded, and maturely glycosylated HLA complexes make it to the cell surface becoming embedded in the PM, where they present the peptides to the T-cell receptors of CD8+ T-cells. The arrows on either side of the schematic indicate the stages of HLA antigen expression (left) and glycosylation maturation (right). The HLA complexes of different class I genes are color coded corresponding to the colors used in Figures 2A and 4A. The number of copies presented illustrates roughly their relative distribution over the IM compartments and PM in JY cells. Moreover, prototypical HLA glycans observed uniquely in the IM fraction and PM fractions are depicted as well.
Previous reports have also looked at the functional role for the N-linked glycosylation on HLA heavy chains. It has been reported that the glycan Glc1Man9GlcNAc2 is the specific structure that is recognized by the folding chaperones calnexin and calreticulin,49 which form a part of the PLC.50 Consequently, the absence of the HLA glycan, accomplished by mutating the asparagine 86, has been shown to completely eliminate PLC activity in vitro.51 A specific functional role of the glycan when the HLA molecule is at the cell surface has not been reported. Except for the discovery that sialylation modulates cell surface stability.52 This observation is not surprising considering the generalized role of sialic acid for extracellular proteins.53,54
While it is well known that HLA complexes can reside either at the PM or at the IMs of the ER and Golgi compartments, the exact distribution of different class I allotypes over these compartments has not been investigated in detail. Evidently, this distribution is determined by a variety of factors. Formation of suboptimally loaded HLA molecules can be retained intracellularly or exhibit a shorter half-life. For example, when HLA is loaded with a low affinity peptide.55 Furthermore, certain sequences exhibit a preference for TAP, which consequently promotes PLC formation, ultimately leading to increased cell surface expression.56 There is also the TAP-independent route for peptide loading utilizing the protein TAPBPR that has different affinities for certain HLA allotypes.57 An extreme case has been identified, where a single natural polymorphism between two allotypes B*44:02 and B*44:05 has strong implications for peptide loading, where the presence of tapasin even lowers the affinity of peptides that can be loaded on B*44:05.25,26 However, whether such factors could contribute to the substantial observed differences in subcellular localization between allotypes as shown here is unclear.
In support of our observations, Ryan and Cobb summarized that there are noticeable differences in glycosylation between HLA-A/HLA-B and HLA-C.58 They argue that the distinct N-glycans found on HLA-C may correlate with a functional role separate from those of HLA-A and HLA-B. They argued that, in line with our data, HLA-C is less abundant at the cell surface, but also binds a more restricted repertoire of peptide antigens, suggesting a reduced role in antigen presentation to CD8+ T cells. Indeed, HLA-C may play a more prominent role in interaction with NK-cell receptors.59 However, they did not report a difference between HLA-A and HLA-B glycosylation. It has been mostly assumed that HLA-A and HLA-B are functionally very alike. Therefore, we assume that the observed differences are caused by a variance in efficiency at which these allotypes traverse through the secretory pathway.
A well-defined functional role for HLA-F has yet to be established. The few reports available hint at its predominant cellular presence in the IM compartments and being dominantly decorated with high-mannose/hybrid N-glycans.48 However, it has also been reported that HLA-F is a high-affinity ligand for NK-cell receptor KIR3DS1, which suggests that HLA-F could play a role similar to HLA-C,60 although we did not detect it at the cell surface.
Although we describe primarily the differences in glycosylation of HLA molecules originating from different class I genes, there may also be differences within allotypes of a specific class I gene. For instance, our data show a substantial difference in glycosylation between the allotypes of HLA-B (Figure 2). Although B*07:02 in the JY cell harbors exclusively mature bisected (orange) and extended (red) glycans, the HLA-B*57:01 heavy-chain molecules in both the DBB and DEM cells show a broader distribution. Next to harboring these bisected and extended mature glycans, a substantial proportion of (up to 25% by spectral count) diantennary (light blue) and high-mannose (green) glycans. Evidently, this could additionally be a result of the different cell lines used in this study, with their different compartmental organization and glycoenzymes they may harbor. In that sense, it may be interesting to extend our glycoprofiling analysis to a well-defined cell line expressing just a single HLA allele as reported by Abelin et al.61 Nevertheless, it is also tempting to speculate about the observed differences with respect to HLA-B*57:01 and B*07:02, which differ in the Bw motifs they contain. The Bw motif is a public epitope (an epitope shared by multiple allotypes), for HLA molecules that play a major role in transplant rejection.62 Two motifs are recognized, Bw4 and Bw6,63 which are found on all HLA-B and some HLA-A heavy chains, and are determined by the amino acid sequence surrounding the N-linked glycosylation site. HLA-B*57:01 of the DBB and DEM cells displays a Bw4 motif, while B*07:02 of JY cells displays a Bw6 motif. HLA molecules harboring the Bw4 motif have been shown to elicit a stronger NK-cell response64 and the glycan attached to the HLA heavy chain is required for the NK-cell interaction.65 There may thus be a link between this Bw motif, the HLA glycosylation pattern, and the interaction with NK-cell KIR cells.
Our findings should impact the important field of HLA peptide ligandome analyses. In these analyses, the focus lies on pathogen-derived or cancer-related neo-antigens, presented by HLA molecules. These antigens are of great importance in the development of pathogen- and antitumor-targeting vaccines.14 The two main experimental approaches to retrieve peptide-antigens from HLA molecules are immunoprecipitation from WC extracts or mild acid elution on intact cells.66 In the latter approach, only the ligandome present at the cell surface should be sampled, whereas with the former approach, the intercellular HLA complexes are probed as well. Based on our observations, the two approaches could chart distinctive ligandomes particularly for HLA allotypes, which can have a considerable part of their molecules residing intracellularly. Each approach is clearly associated with different strengths and potential bias;67 also a definitive consensus is still elusive, and may be context-dependent.
In conclusion, through an in-depth analysis of the glycosylation profiles on HLA class I molecules, originating from three different cell lines, covering four different class I genes and six different allotypes, we observed that they all exhibit different glycosylation profiles. It would be interesting to expand such analysis to more cell lines covering a broader range of allotypes, and/or even primary cells, to probe whether such observations are general or very much cell type specific. We show that the HLA glycan-signature reflects the cellular organization, and is distinct for the same HLA molecules residing at the PM or the IMs (ER and Golgi). Although this is fully in line with what would be expected based on our knowledge of protein cellular glycosylation, analyzing glycoform profiles may thus provide a direct quantitative assessment of the cellular distribution of specific HLA class I molecules. As the peptide repertoire presented at the cell surface is sampled by T-cells, our observations are significant for the analysis and interpretation of HLA peptide ligandomes.
Data Availability
The mass spectrometry proteomics and glycoproteomics data and accompanying processed search files have been deposited to the ProteomeXchange Consortium via the PRIDE68 partner repository with the data set identifier PXD023684.
Acknowledgments
We acknowledge support for this research through the NWO-funded Large-scale Infrastructures program X-Omics (Project 184.034.019) embedded in the Netherlands Proteomics Center and the NWO Gravitation program Institute for Chemical Immunology (ICI00003). We thank Dr. Stefan Stevanović (University of Tubingen, Germany) for providing the pan-HLA antibody W6/32. We thank Dr. Bas van Breukelen (Utrecht University) for help in constructing Figure 5.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00466.
Phylogenetic tree visualization of the most observed HLA class I heavy chains; abundance distribution of glycan for each HLA class I gene in each cell line; additional examples of annotated MS/MS spectra of HLA glycopeptides; and proteomics-based validation of the subcellular fractionation (PDF)
DBB top 100 identified proteins with respective IBAQ values; DEM top 100 identified proteins with respective IBAQ values; top 100 identified proteins with respective IBAQ values;PM proteome with respective IBAQ values; and IM proteome with respective IBAQ values (XLSX)
DBB—HLA unique peptides used for protein quantification; DEM—HLA unique peptides used for protein quantification; JY—HLA unique peptides used for protein quantification; JY PM—HLA unique peptides used for protein quantification; JY IM—HLA unique peptides used for protein quantification; DBB—HLA-detected glycopeptides; DEM—HLA-detected glycopeptides; JY—HLA-detected glycopeptides; JY PM—HLA-detected glycopeptides; JY IM—HLA-detected glycopeptides; DBB—HLA glycans reported by Byonic; DEM—HLA glycans reported by Byonic; JY—HLA glycans reported by Byonic; JY PM—HLA glycans reported by Byonic; and JY IM—HLA glycans reported by Byonic (XLSX)
Author Contributions
§ M.H. and L.C.D. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Vyas J. M.; Van der Veen A. G.; Ploegh H. L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008, 8, 607–618. 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefjes J.; et al. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- Klein J.; Sato A. The HLA system. N. Engl. J. Med. 2000, 343, 702–709. 10.1056/nejm200009073431006. [DOI] [PubMed] [Google Scholar]
- Bradley B. A. The role of HLA matching in transplantation. Immunol. Lett. 1991, 29, 55–59. 10.1016/0165-2478(91)90199-k. [DOI] [PubMed] [Google Scholar]
- Montgomery R. A.; et al. HLA in transplantation. Nat. Rev. Nephrol. 2018, 14, 558–570. 10.1038/s41581-018-0039-x. [DOI] [PubMed] [Google Scholar]
- Gough S. C.; Simmonds M. J. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genomics 2007, 8, 453–465. 10.2174/138920207783591690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanelli E.; Breedveld F. C.; de Vries R. R. P. HLA association with autoimmune disease: a failure to protect?. Rheumatology (Oxford) 2000, 39, 1060–1066. 10.1093/rheumatology/39.10.1060. [DOI] [PubMed] [Google Scholar]
- Bodis G.; Toth V.; Schwarting A. Role of Human Leukocyte Antigens (HLA) in Autoimmune Diseases. Rheumatol. Ther. 2018, 5, 5–20. 10.1007/s40744-018-0100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñiz-Castrillo S.; Vogrig A.; Honnorat J. Associations between HLA and autoimmune neurological diseases with autoantibodies. Autoimmun. Highlights 2020, 11, 2. 10.1186/s13317-019-0124-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra N. K.; Kaur G. MHC-based vaccination approaches: progress and perspectives. Expet Rev. Mol. Med. 2003, 5, 1–17. 10.1017/s1462399403005957. [DOI] [PubMed] [Google Scholar]
- Blackwell J. M.; Jamieson S. E.; Burgner D. HLA and infectious diseases. Clin. Microbiol. Rev. 2009, 22, 370–385. 10.1128/cmr.00048-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa E. E.; et al. HLA-associated protection of lymphocytes during influenza virus infection. Virol. J. 2020, 17, 128. 10.1186/s12985-020-01406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulder P. J. R.; Walker B. D. HIV and HLA class I: an evolving relationship. Immunity 2012, 37, 426–440. 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher T. N.; Schreiber R. D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Bassani-Sternberg M.; Coukos G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17. 10.1016/j.coi.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Neefjes J. J.; Schumacher T. N. M.; Ploegh H. L. Assembly and intracellular transport of major histocompatibility complex molecules. Curr. Opin. Cell Biol. 1991, 3, 601–609. 10.1016/0955-0674(91)90029-x. [DOI] [PubMed] [Google Scholar]
- Demmers L. C.; et al. Single-cell derived tumor organoids display diversity in HLA class I peptide presentation. Nat. Commun. 2020, 11, 5338. 10.1038/s41467-020-19142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone P.; et al. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. 10.1093/jnci/djt184. [DOI] [PubMed] [Google Scholar]
- Robinson J.; et al. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015, 43, D423–D431. 10.1093/nar/gku1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham P.; et al. Nature of polymorphism in HLA-A, -B, and -C molecules. Proc Natl Acad Sci U S A 1988, 85, 4005–4009. 10.1073/pnas.85.11.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deutekom H. W. M.; Keşmir C. Zooming into the binding groove of HLA molecules: which positions and which substitutions change peptide binding most?. Immunogenetics 2015, 67, 425–436. 10.1007/s00251-015-0849-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Galarza F. F.; et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020, 48, D783–D788. 10.1093/nar/gkz1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber L. D.; et al. Unusual uniformity of the N-linked oligosaccharides of HLA-A, -B, and -C glycoproteins. J. Immunol. 1996, 156, 3275–3284. [PubMed] [Google Scholar]
- Scull K. E.; et al. Secreted HLA recapitulates the immunopeptidome and allows in-depth coverage of HLA A*02:01 ligands. Mol. Immunol. 2012, 51, 136–142. 10.1016/j.molimm.2012.02.117. [DOI] [PubMed] [Google Scholar]
- Williams A. P.; et al. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity 2002, 16, 509–520. 10.1016/s1074-7613(02)00304-7. [DOI] [PubMed] [Google Scholar]
- Bailey A.; et al. Selector function of MHC I molecules is determined by protein plasticity. Sci. Rep. 2015, 5, 14928. 10.1038/srep14928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren D. H.; et al. Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics 2010, 7, 39–53. 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- Aebi M.; et al. N-glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 2010, 35, 74–82. 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Varki A.; et al. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor (NY), 2015–2017. Bookshelf ID: NBK310274. [PubMed] [Google Scholar]
- Moremen K. W.; Tiemeyer M.; Nairn A. V. Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P. Golgi glycosylation. Cold Spring Harbor Perspect. Biol. 2011, 3, a005199. 10.1101/cshperspect.a005199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmers L. C.; Heck A. J. R.; Wu W. Pre-fractionation Extends but also Creates a Bias in the Detectable HLA Class Ι Ligandome. J. Proteome Res. 2019, 18, 1634–1643. 10.1021/acs.jproteome.8b00821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnstable C.; et al. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 1978, 14, 9–20. 10.1016/0092-8674(78)90296-9. [DOI] [PubMed] [Google Scholar]
- Mezzadra R.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. 10.1038/nature23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiding K. R.; et al. The benefits of hybrid fragmentation methods for glycoproteomics. Trac. Trends Anal. Chem. 2018, 108, 260–268. 10.1016/j.trac.2018.09.007. [DOI] [Google Scholar]
- Schwanhäusser B.; et al. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Cox J.; et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. 10.1074/mcp.m113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney W.Data structures for statistical computing in python. Proceedings of the 9th Python in Science Conference, Austin, TX, 2010.
- van der Walt S.; Colbert S. C.; Varoquaux G. The NumPy array: a structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. 10.1109/mcse.2011.37. [DOI] [Google Scholar]
- Hunter J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. 10.1109/mcse.2007.55. [DOI] [Google Scholar]
- Chong C.; et al. High-throughput and Sensitive Immunopeptidomics Platform Reveals Profound Interferonγ-Mediated Remodeling of the Human Leukocyte Antigen (HLA) Ligandome. Mol. Cell. Proteomics 2018, 17, 533–548. 10.1074/mcp.tir117.000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M.; et al. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics 2015, 14, 658–673. 10.1074/mcp.m114.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen J.; et al. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc Natl Acad Sci U.S.A. 2009, 106, 2776–2781. 10.1073/pnas.0812901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L.; et al. Recognition of Bisecting N-Glycans on Intact Glycopeptides by Two Characteristic Ions in Tandem Mass Spectra. Anal. Chem. 2019, 91, 5478–5482. 10.1021/acs.analchem.8b05639. [DOI] [PubMed] [Google Scholar]
- Wearsch P. A.; et al. Major histocompatibility complex class I molecules expressed with monoglucosylated N-linked glycans bind calreticulin independently of their assembly status. J. Biol. Chem. 2004, 279, 25112–25121. 10.1074/jbc.m401721200. [DOI] [PubMed] [Google Scholar]
- Culina S.; et al. Calreticulin promotes folding of functional human leukocyte antigen class I molecules in vitro. J. Biol. Chem. 2004, 279, 54210–54215. 10.1074/jbc.m410841200. [DOI] [PubMed] [Google Scholar]
- Armony G.; Heck A. J. R.; Wu W. Extracellular crosslinking mass spectrometry reveals HLA class I–HLA class II interactions on the cell surface. Mol. Immunol. 2021, 136, 16–25. 10.1016/j.molimm.2021.05.010. [DOI] [PubMed] [Google Scholar]
- Lee N.; Ishitani A.; Geraghty D. E. HLA-F is a surface marker on activated lymphocytes. Eur. J. Immunol. 2010, 40, 2308–2318. 10.1002/eji.201040348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov G.; et al. Structural basis of carbohydrate recognition by calreticulin. J. Biol. Chem. 2010, 285, 38612–38620. 10.1074/jbc.m110.168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C.; Tampé R. Proofreading of Peptide-MHC Complexes through Dynamic Multivalent Interactions. Front. Immunol. 2017, 8, 65. 10.3389/fimmu.2017.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearsch P. A.; Peaper D. R.; Cresswell P. Essential glycan-dependent interactions optimize MHC class I peptide loading. Proc Natl Acad Sci U.S.A. 2011, 108, 4950–4955. 10.1073/pnas.1102524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Z.; et al. MHC Class I Stability is Modulated by Cell Surface Sialylation in Human Dendritic Cells. Pharmaceutics 2020, 12, 249. 10.3390/pharmaceutics12030249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell A. G.; et al. The role of sialic acid in determining the survival of glycoproteins in the circulation. J. Biol. Chem. 1971, 246, 1461–1467. 10.1016/s0021-9258(19)76994-4. [DOI] [PubMed] [Google Scholar]
- Bork K.; Horstkorte R.; Weidemann W. Increasing the sialylation of therapeutic glycoproteins: the potential of the sialic acid biosynthetic pathway. J. Pharm. Sci. 2009, 98, 3499–3508. 10.1002/jps.21684. [DOI] [PubMed] [Google Scholar]
- Springer S. Transport and quality control of MHC class I molecules in the early secretory pathway. Curr. Opin. Immunol. 2015, 34, 83–90. 10.1016/j.coi.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Yarzabek B.; et al. Variations in HLA-B cell surface expression, half-life and extracellular antigen receptivity. eLife 2018, 7, e34961 10.7554/elife.34961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov G. I.; et al. Interaction of TAPBPR, a tapasin homolog, with MHC-I molecules promotes peptide editing. Proc Natl Acad Sci U S A 2016, 113, E1006–E1015. 10.1073/pnas.1519894113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan S. O.; Cobb B. A. Roles for major histocompatibility complex glycosylation in immune function. Semin. Immunopathol. 2012, 34, 425–441. 10.1007/s00281-012-0309-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais M. E.; Dong T.; Rowland-Jones S. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player?. Immunology 2011, 133, 1–7. 10.1111/j.1365-2567.2011.03422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W. F.; et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat. Immunol. 2016, 17, 1067–1074. 10.1038/ni.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abelin J. G.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz C. T. Human leukocyte antigen Bw4 and Bw6 epitopes recognized by antibodies and natural killer cells. Curr. Opin. Organ Transplant. 2014, 19, 436–441. 10.1097/mot.0000000000000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rood J. J.; Van Leeuwen A. Leukocyte Grouping. a Method and Its Application. J. Clin. Invest. 1963, 42, 1382–1390. 10.1172/jci104822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr W. H.; Pando M. J.; Parham P. KIR3DL1 polymorphisms that affect NK cell inhibition by HLA-Bw4 ligand. J. Immunol. 2005, 175, 5222–5229. 10.4049/jimmunol.175.8.5222. [DOI] [PubMed] [Google Scholar]
- Salzberger W.; et al. Influence of Glycosylation Inhibition on the Binding of KIR3DL1 to HLA-B*57:01. PLoS One 2015, 10, e0145324 10.1371/journal.pone.0145324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenmann L. K.; Marcu A.; Stevanović S. Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology 2018, 154, 331–345. 10.1111/imm.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm T.; et al. Mild Acid Elution and MHC Immunoaffinity Chromatography Reveal Similar Albeit Not Identical Profiles of the HLA Class I Immunopeptidome. J. Proteome Res. 2021, 20, 289–304. 10.1021/acs.jproteome.0c00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics and glycoproteomics data and accompanying processed search files have been deposited to the ProteomeXchange Consortium via the PRIDE68 partner repository with the data set identifier PXD023684.