

Abstract
Dendritic cell (DC) vaccines are used for cancer and infectious diseases, albeit with limited efficacy. Modulating the formation of DC–T‐cell synapses may greatly increase their efficacy. The effects of graphene oxide (GO) nanosheets on DCs and DC–T‐cell synapse formation are evaluated. In particular, size‐dependent interactions are observed between GO nanosheets and DCs. GOs with diameters of >1 µm (L‐GOs) demonstrate strong adherence to the DC surface, inducing cytoskeletal reorganization via the RhoA‐ROCK‐MLC pathway, while relatively small GOs (≈500 nm) are predominantly internalized by DCs. Furthermore, L‐GO treatment enhances DC–T‐cell synapse formation via cytoskeleton‐dependent membrane positioning of integrin ICAM‐1. L‐GO acts as a “nanozipper,” facilitating the aggregation of DC–T‐cell clusters to produce a stable microenvironment for T cell activation. Importantly, L‐GO‐adjuvanted DCs promote robust cytotoxic T cell immune responses against SARS‐CoV‐2 spike 1, leading to >99.7% viral RNA clearance in mice infected with a clinically isolated SARS‐CoV‐2 strain. These findings highlight the potential value of nanomaterials as DC vaccine adjuvants for modulating DC–T‐cell synapse formation and provide a basis for the development of effective COVID‐19 vaccines.
Keywords: DC–T‐cell synapses, dendritic cells, ICAM‐1, SARS‐CoV‐2
A novel material‐based method of regulating dendritic cell (DC)–T‐cell synaptic contact for adoptive DC vaccine engineering is reported. Graphene oxide (GO) nanosheets serve as a “nanozipper” or “double‐sided tape” facilitating the formation and aggregation of DC–T‐cell immune synapses to elicit robust cytotoxic T cell immune responses against SARS‐CoV‐2 infection. Cytoskeleton‐dependent membrane‐positioning of intercellular cell adhesion molecule‐1 (ICAM‐1) might be the underlying mechanism.
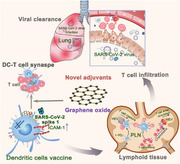
1. Introduction
Dendritic cells (DCs), one of the most potent antigen‐presenting cells to initiate naïve T cells, have long been harnessed for immunotherapies to elicit antigen‐specific T cell responses to eradicate infections or tumors.[ 1 ] They can also be employed to enhance and reestablish immune functions in immunocompromised individuals. Ex‐vivo‐prepared DCs must be stimulated by adjuvants to become functionally competent for T cell activation.[ 1 , 2 ] Adjuvant screening and optimization are typically guided by the “two‐signal” theory, with an emphasis on improving antigen phagocytosis and presentation by DCs (the antigen signal),[ 3 ] promoting the expression of the costimulatory molecules (the costimulatory signal) or cytokine secretion.[ 4 ] Currently, the most well‐established GMP‐compliant adjuvants (i.e., cytokine cocktails) are designed to augment the co‐stimulatory signal of DC–T‐cell interactions by up‐regulating costimulatory molecules on DCs and promoting the secretion of pro‐inflammatory cytokines.[ 5 ] Nevertheless, in patients with cancer receiving standard DC immunotherapy, the overall response rate of ≤15%[ 6 ] is far below expectations, indicating that other key factors may determine DC–T‐cell interactions.
Accumulating evidence shows the formation of stable of DC–T‐cell membrane conjunction is another indispensable “pre‐signal” for T cell priming, which is termed as immune synapse (IS).[ 7 ] IS formation is a tightly regulated biological process involving the redistribution of multiple adhesion molecules (e.g., ICAM‐1, neuropilin, plexins, and spinophilin) to the cell periphery.[ 8 ] The adhesion molecules in an orderly structure construct a peripheral adhesion ring junction around the TCR‐peptide‐loaded MHC cluster to stabilize the DC–T‐cell interaction.[ 7 , 9 ] Sustained TCR engagement initiates a phosphorylation cascade, triggering multiple downstream branching signaling pathways for T cell proliferation and activation.[ 10 ] Thus, modulating IS formation between DC and T cells is likely to be an effective novel strategy for optimizing DC vaccines.
Graphene oxide (GO) possesses beneficial physiochemical properties for diverse biomedical applications.[ 11 ] Increasing evidence indicates that GOs have varying size‐dependent interactions with the cell plasma membrane.[ 12 ] We have also found that larger‐sized GO sheets are inclined to be located at the edges of macrophages, instead of being internalized, likely corresponding to the adsorption or adherence of GOs on the plasma membrane.[ 13 ] Moreover, membrane‐coated GOs may directly act on adhesion molecules, transducing outside‐in signaling involved in multiple biological processes.[ 12 , 14 ] These findings collectively suggest that GOs impact DC–T‐cell IS formation and hence affect the efficiency of T cell priming by DCs.
In this study, we comprehensively investigated the effects of GO on the biological and functional properties of DCs, with an emphasis on DC–T‐cell synapse formation. In particular, we systematically tested the protective efficacy of a GO‐adjuvanted DC vaccine against the newly emerged SARS‐CoV‐2. This coronavirus is the cause of an ongoing global health crisis, necessitating the rapid development of effective vaccines to terminate the pandemic.[ 15 ] Recently, an adaptive Phase IB‐II trial was started to evaluate the safety and efficacy of an autologous DC‐based vaccine in preventing COVID‐19.[ 16 ] The ex vivo generated and functionally competent DC vaccines are expected to offer a unique opportunity to improve the protection of elderly and immune‐compromised individuals.[ 17 ] This work is expected to provide first‐hand information for the evaluation of SARS‐CoV‐2‐specific DC vaccines and to offer insights into DC vaccine engineering based on the strategy of promoting DC–T‐cell communication.
2. Results and Discussion
We characterized the morphological features of two differently sized GOs by atomic force microscopy (AFM; Figure 1a). Single‐ or two‐layer (bilayer with a height of 0.8–1.6 nm) nanosheets were obtained with an average dimension of ≈500 nm for the small GOs (denoted S‐GOs) and ≈1.00 µm for the larger counterparts (L‐GOs). Raman spectra showed similar basal structure profiles with characteristic D‐peaks (1331 cm−1) and G‐peaks (1596 cm−1) for both GO samples (Figure S1, Supporting Information). A similar ratio of intensity of the D band (ID)/intensity of the G band (IG) was observed for the S‐ and L‐GOs, indicating comparable structural defects and extents of surface oxidation in the two GO samples. X‐ray photoelectron spectroscopy (XPS) analysis (Figure 1b and Table S1, Supporting Information) revealed the presence of functional groups (C—C/C=C, C—OH, C=O, and O=C—OH) and the C/O ratios were both calculated to be 2.19, with a material chemical purity of 99.8%. Together with similar zeta potentials (Table S1, Supporting Information), these results indicated that S‐GO and L‐GO had comparable physicochemical and structural properties, except for lateral dimensions.
Figure 1.
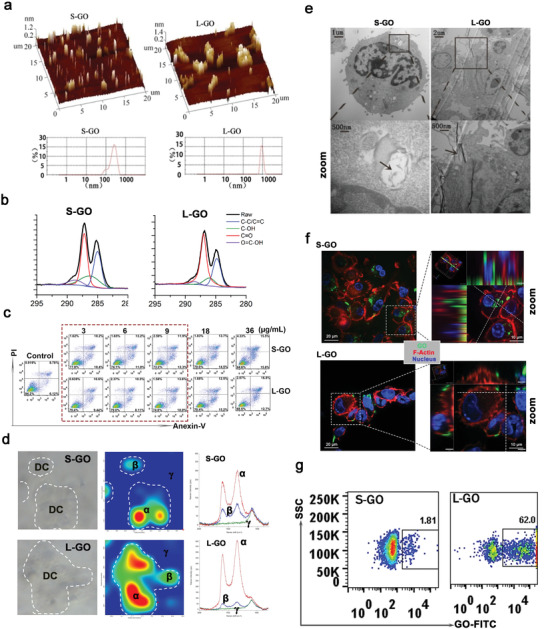
Characterization of GOs and their interactions with DCs. a) AFM images of GO nanosheets. b) XRD pattern of GOs. c) Apoptosis analysis of DCs after treatment with GOs at a dose of 3–36 µg mL−1. d) Confocal Raman mapping image of GO‐treated DCs at a dose of 9 µg mL−1. The Raman spectra for denoted positions (α, β, γ) in the middle panel are presented in the right panel. e) TEM image of GO‐treated DCs. The black arrows in the zoomed image point to the internalized or membrane‐adhered GOs. f) Images obtained by confocal microscopy. Green: FITC‐labeled GOs; Red: F‐actin stained by rhodamine‐conjugated phalloidin. g) The detection of GO adhered to DC membrane by FACS. Representative results from two or three replicates are shown.
Next, various doses of GO were coincubated with DCs to test their cytotoxicity. Cells were double labeled with propidium iodide and Annexin V (Figure 1c) and evaluated by a 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) cytotoxicity assay (Figure S2, Supporting Information). DCs could tolerate GO treatment at 9 µg mL− 1, with a viability of >90%. Confocal Raman mapping (Figure 1d) indicated that both S‐ and L‐GOs exhibit high levels of accumulation in DC‐located regions. Transmission electron microscopy (TEM; Figure 1e) confirmed that S‐GOs were predominately internalized by DCs and localized in the phagosomes or lysosomes, whereas L‐GOs showed strong adherence to the surface plasma membrane with less phagocytosis. Utilizing FITC‐covalently modified GOs, confocal microscopy (Figure 1f) corroborated the TEM findings, confirming the different distributions of the two differently sized GOs in DCs. Furthermore, a fluorescence‐activated cell sorting (FACS) analysis demonstrated that a higher percentage of DCs (>60%) were detected with L‐GOs (Figure 1g), indicating a high affinity between L‐GOs and DCs.
To gain insight into whether GOs affect DC–T‐cell interactions, CD8α+ T cells from OT‐I TCR transgenic mice were cocultured with GO‐pretreated and OVA257–264‐bearing DCs. The dynamic interaction between T cells and DCs was recorded by confocal imaging for a period of more than 100 min (Figure 2a and Videos S1–S3, Supporting Information). The contact area between DCs and T cells and period of interaction were calculated. The direct DC–T‐cell contact area in the L‐GO‐treated group was approximately fourfold higher than that in untreated DCs and approximately twofold higher than that in S‐GO‐treated DCs (Figure 2b). Moreover, >80% of DC–T‐cell clusters persisted for over 100 min in the L‐GO‐treated group, compared to 46% in the untreated group and 17% in the S‐GO‐treated group (Figure 2c). These data demonstrated that the pretreatment of DCs with L‐GOs dramatically increased contact with T cells and synapse formation.
Figure 2.
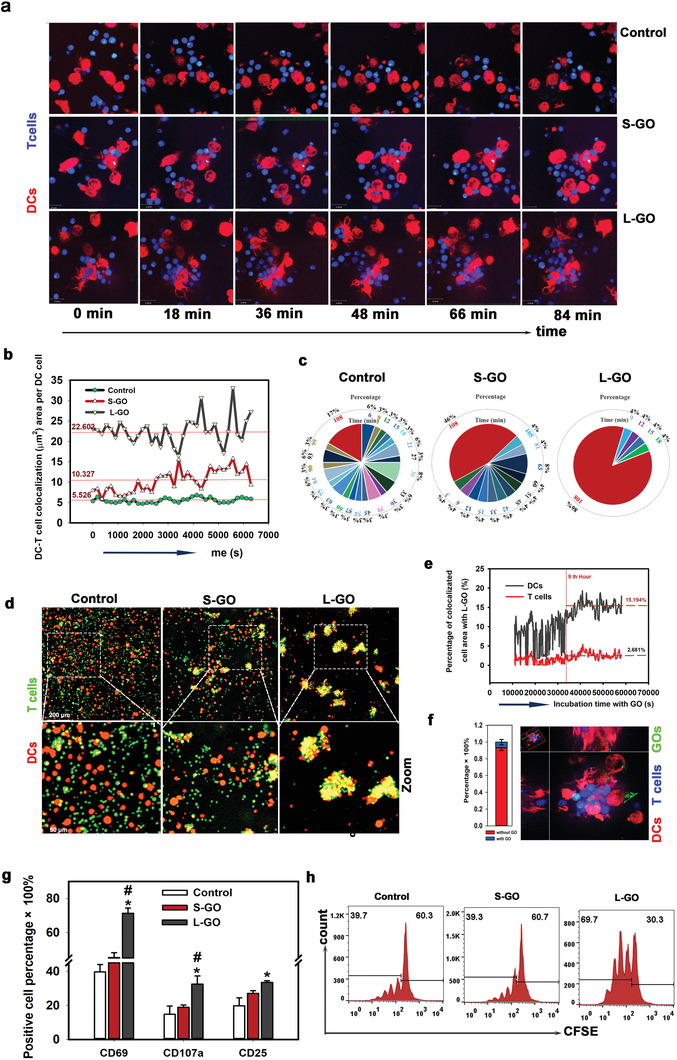
L‐GOs enhanced DC–T‐cell synaptic contact and T cell priming. a) Dynamic interaction between DCs and T cells by confocal imaging over 100 min. Red: DCs from tdTomato transgenic mice; Blue: CD8+ T cells from OT‐I transgenic mice. b) The direct contact area of DC–T‐cells determined by calculating the co‐localized areas visualized using light channels 561 nm and 405 nm in dynamic microscopy images. c) Proportional distribution of the duration of DC–T‐cell adhesion. d) Images of DC–T‐cell clusters. Red: DCs; Green: T cells. e) L‐GO absorption on respective DCs and T cells determined by calculating the co‐localized areas visualized using light channel 488 nm (FITC‐labeled L‐GOs) with 561 nm (tdTomato + DCs) and 405 nm (CellTrack‐Blue‐labeled T cells). f) Proportion of L‐GO‐containing DC–T‐cell synapses. Left: statistical analysis; right: representative image of co‐localization of L‐GOs, DCs, and T cells. g) DCs induced T cell activation, as indicated by FACS analyses of CD69, CD107a, and CD25. T cells were gated by antibody of FITC‐anti‐CD8α+. Data are presented as means ± s.d; n = 3–5; The Shapiro–Wilk test was used to evaluate normality and the Holm–Sidak method was used for multiple comparisons. *p < 0.05 compared with the control group. #p < 0.05 compared with the S‐GO group. h) T cell proliferation by detecting CFSE dye dilution. Representative results from two or three replicates are shown.
Many extraordinarily large and long‐lasting DC–T‐cell clusters were also observed in L‐GO‐mediated synapse formation (Figure 2d). To determine how L‐GOs interact with DCs and T cells, FITC‐labeled L‐GOs were added to the DC–T‐cell coincubation system and were observed for 20 h (Video S4, Supporting Information) for quantitative analyses of the adherence of L‐GOs to DC and T cells. Within the first 9 h after the addition of L‐GO, the binding of L‐GO to the DC membrane was temporary and unstable in an active cycle of adsorption–dissociation–adsorption (Figure 2e). Notably, adherence stabilized 9 h later, and an average of 15.2% of DC membranes was coated with L‐GOs. In contrast, only 2.3% of the T cell membranes showed adsorbed L‐GOs, suggesting that the binding affinity of L‐GO to T cells was substantially lower than that to DCs (Figure 2e and Figure S3, Supporting Information). This biased L‐GO binding to the DC membrane decreased the possibility that L‐GOs physically bridge DC and T cell membranes. As expected, less than 5% of the area of DC and T cell co‐localization contained L‐GOs, revealing that most L‐GOs did not remain between cells (Figure 2f). This finding also indicated that the adherence of L‐GOs would not interrupt molecule pairing between DCs and T cells. Nevertheless, the high binding affinity of DCs indeed contributed to DC–T‐cell clustering. With increasing time, more L‐GOs were attached to DCs and, simultaneously, the membrane‐adhered L‐GOs continued to combine with adjacent DCs or DC–T‐cell clusters (Figure S4 and Video S4, Supporting Information), finally forming a large DC–T‐cell cluster. In this process, L‐GOs functioned as “nanozippers” or “double‐sided tape” to physically construct a stable microenvironment for DC–T‐cell interactions, further evidenced by Video S5, Supporting Information. Collectively, these data provide the first evidence that L‐GOs show dramatic selectivity for adhering to different cell membranes. Their high binding affinity with the DC membrane contributed to DC–T‐cell clustering and their low binding affinity with T cells prevented interference with DC–T‐cell interactions.
Upon activation, T cells sequentially upregulate a series of inducible T cell activation antigens, including early CD69 and late CD25, to facilitate CD8+ T cell proliferation.[ 18 ] CD107a (LAMP‐1) is a marker of the cytotoxic activity of T cells.[ 19 ] Our data indicated that CD69, CD107a, and CD25 on CD8α+ T cells were all highly upregulated after co‐culture with L‐GO‐treated DCs for 48 h (Figure 2g). However, S‐GO‐treated DCs did not demonstrate significant increases in the levels of these markers. In addition, T cells in the L‐GO‐treated group showed a twofold higher efficiency in CFSE dye dilution in daughter cells than that of their counterparts (Figure 2h), indicating a highly elevated proliferation ability. Altogether, these findings suggest that L‐GO treatment greatly prolonged DC–T‐cell synaptic interactions and increased T cell activation and proliferation. Notably, free GOs of both sizes had little effect on T cell activation (Figure S5, Supporting Information). Moreover, L‐GO treatment did not directly promote Th1 cytokine secretion, allostimulatory molecule expression (Figure S6, Supporting Information) and OVA257–264 presentation (Figure S7, Supporting Information) of DCs. So, L‐GO‐promoted DC–T‐cell IS formation may dramatically contribute to the enhanced T cell responses, at least in the initial stage of DC–T‐cell interaction. Besides, we detected an obviously upregulated CD40 and MHC I expression on L‐GO treated DCs after being incubated with T cells for 48 h (Figure S8, Supporting Information), indicating there may be a positive feedback mechanism for L‐GO adjuvanted DC–T‐cell interaction, and all those mechanisms worked together to maximize T cell activation.
As mentioned above, L‐GOs have a bifunctional role in mediating DC–T‐cell interactions by: i) increasing direct contact between DCs and T cells, and ii) promoting their clustering. The latter is associated with the high binding affinity of L‐GOs with the DC membrane; however, it is still unknown how L‐GOs enhance the direct synaptic contact between DCs and T cells. The finding that L‐GOs did not physically bridge the DC–T‐cell membrane suggested that they likely triggered a molecular mechanism that increased DC–T‐cell adhesion. Adhesion molecules that maintain interactions between cells are essential for forming the supramolecular activation cluster of the synapse.[ 20 ] Transcriptional changes in adhesion molecules associated with DC–T‐cell adhesion were measured by RNA sequencing (Figure S9, Supporting Information). Of 21 adhesion molecules with known roles in adhesion, 3 were upregulated at the RNA level in GO‐treated DCs: ICAM‐1, neurophilin‐1 (NP1), and plexin‐A2. Further analysis by FACS, with LPS stimulation as a positive control, confirmed that ICAM‐1 levels are significantly elevated on the DC surface after treatment with L‐GOs (Figure 3a), whereas NP1 and plexin‐A2 remained at very low levels and were not affected by GO treatment. Furthermore, the cellular localization of ICAM‐1 was visualized by immunostaining (Figure 3b). Untreated‐ or S‐GO‐treated DCs showed a striking phenomenon wherein the strongly stained areas were mainly intracellular and scattered in or on the surface of vesicle‐like structures. In sharp contrast, ICAM‐1 was mainly located on the plasma membrane of L‐GO‐treated DCs, suggesting that L‐GO treatment promoted ICAM‐1 translocation from the cytosol to the DC surface.
Figure 3.
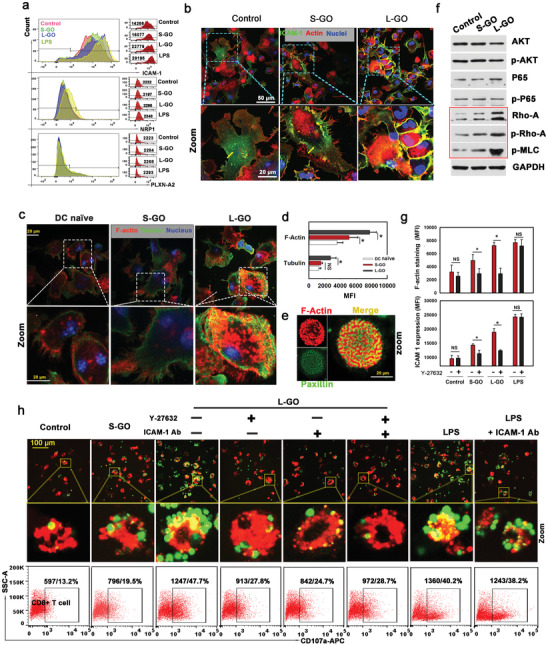
L‐GO‐induced translocation of ICAM‐1 promotes DC–T‐cell adhesion. a) Levels of ICAM‐1, neurophilin‐1 (NP1), and plexin‐A2 expression on the DC surface were detected by FACS. LPS treatment (100 ng mL−1) was set as a positive control. b) Cellular distribution of ICAM‐1 in DCs visualized by immunofluorescence staining. Green: ICAM‐1; red: F‐actin; blue: nuclei. c) Staining of F‐actin and β‐tubulin in GO‐treated DCs. red: F‐actin; green: tubulin; blue: nuclei. d) Statistical analysis of the average fluorescence intensity of F‐actin and microtubules; data are presented as means ± s.d.; n = 5; *p < 0.05 compared with the control group; #p < 0.05 compared with the small graphene oxide (S‐GO) group, evaluated by the Holm–Sidak method. e) Focal adhesion plaques detected by combined F‐actin (red) and paxillin (green) staining. f) Activation of the Rho‐ROCK‐MLC signaling pathway detected by Western blotting. g) Effect of blocking the Rho‐ROCK signaling pathway by Y27632 on DC cytoskeletal organization (top) and ICAM‐1 translocation (bottom); data are presented as means ± s.d.; n = 7–12; *p < 0.05 by one‐tailed t‐test; NS: not significant. h) Effect of Y27632 pretreatment or ICAM‐1 blocking on DC–T‐cell synapse formation and CD107a expression on CD8+ T cells. Red: OVA‐bearing DCs stained by DiI dye; green: OT‐1‐mice‐derived CD8+ T cells labeled by CSFE. Representative results from two or three replicates are shown.
A previous study has indicated that ICAM‐1 in DCs is highly mobile, showing continuous internalization and recycling back to the surface of the cell membrane, and this process is highly dependent on the organization of the cytoskeleton.[ 21 ] We visualized the organization of microfilaments and microtubules in DCs. Substantially stronger F‐actin and β‐tubulin staining was observed in L‐GO‐treated DCs than in untreated DCs (Figure 3c,d). In addition, a large number of specialized actin microstructures formed within the cell‐plate interface by combined F‐actin and paxillin staining (Figure 3e), confirming the dramatically enhanced cytoskeletal organization in L‐GO‐adjuvanted DCs. PI3K‐Akt, NF‐κB, and RhoA‐ROCK signaling pathways, which are closely related to cytoskeletal organization, were examined (Figure 3f). AKT, p65, and their activated forms (p‐AKT and p‐p65) did not differ in DCs before and after GO treatment. In contrast, the levels of ROCK and downstream p‐MLC increased substantially after L‐GO treatment. In addition, Y27632, a specific inhibitor of ROCK, significantly suppressed the L‐GO‐induced polymerization of both F‐actin and β‐tubulin (Figure 3g, upper panel) and decreased the membrane positioning of ICAM‐1 (Figure 3g, lower panel). Finally, Y27632 pretreatment, ICAM‐1 blocking, or their combination dramatically impaired L‐GO‐induced DC–T‐cell synapse formation (Figure 3h, up panel) and T cell activation by detecting CD107a expression (Figure 3h below panel). However, blocking ICAM‐1 exerted a very limited effect on LPS‐induced DC–T‐cell conjugates, implying that ICAM‐1 is specifically indispensable for L‐GO‐induced DC–T‐cell IS formation. Collectively, the mechanistic insights into L‐GO‐induced IS formation can be summarized as follows. L‐GO promotes the polymerization of F‐actin and β‐tubulin, especially via the RhoA‐ROCK signaling pathway, and the well‐assembled cytoskeleton accelerates ICAM‐1 transportation to the cell surface. Finally, ICAM‐1, as a critical component of peripheral SMAC, contributes to DC–T‐cell membrane conjunction by pairing with LFA‐1 on T cells.[ 8c ] However, the mechanism by which L‐GOs promote DC cytoskeletal organization remains to be elucidated. L‐GOs may directly act on integrins, as reported previously,[ 12 , 14 ] thereby transducing outside‐in signaling to produce an intriguing interaction between cytoskeletal reorganization and adhesion control at the IS.[ 22 ] The effect of L‐GOs may alternatively be related to their ability to generate reactive oxygen species (ROS). L‐GOs had a higher carbon radical content than that of their small counterparts (Figure S10, Supporting Information), which might contribute to their strong biological oxidative potential, validated by ROS production in vitro.[ 23 ] The elevation of ROS can directly promote the phosphorylation of ROCK to enhance the cytoskeletal organization in DCs.[ 24 ]
The above findings indicate that L‐GOs are promising adjuvants for DC vaccines based on their ability to effectively promote DC–T‐cell conjunction and thereby to promote T cell activation. To further verify this, OVA257–264 (OVAp)‐specific DC vaccines with L‐GO as an adjuvant were prepared. A cytokine‐cocktail (denoted “C‐C”), the most commonly used DC vaccine adjuvant, was used as a control. Mice were vaccinated according to the schematic diagram shown in Figure 4a. Popliteal lymph nodes (PLNs) were taken on day 12 to evaluate OVAp‐specific CD8+ T‐cell priming. Strikingly, the numbers of OVAp‐specific CD8+ T cells were 20‐ and 15‐fold higher for L‐GO‐adjuvanted DC vaccines than for the cytokine cocktail and S‐GO‐adjuvanted DCs, respectively (Figure 4b‐i,c). Furthermore, two to threefold more CD8+ T cells expressed the activation markers CD69 or CD44 in the L‐GO group (Figure 4b‐ii,iii,d) than in the cytokine‐cocktail group. Antigen‐recall experiments based on the detection of intracellular IFN‐γ and TNF‐α (Figure 4b‐iv,v,e) in CD8+ T cells after ex vivo re‐stimulation with OVAp corroborated the above results and further confirmed the antigen‐specific CD8+ T cells in immunized mice. Taken together, these data suggest that the in vivo administration of L‐GO‐adjuvanted DC vaccines primed a robust antigen‐specific CD8+ T cell response, which far exceeded the response to the conventional adjuvanted DC vaccine.
Figure 4.
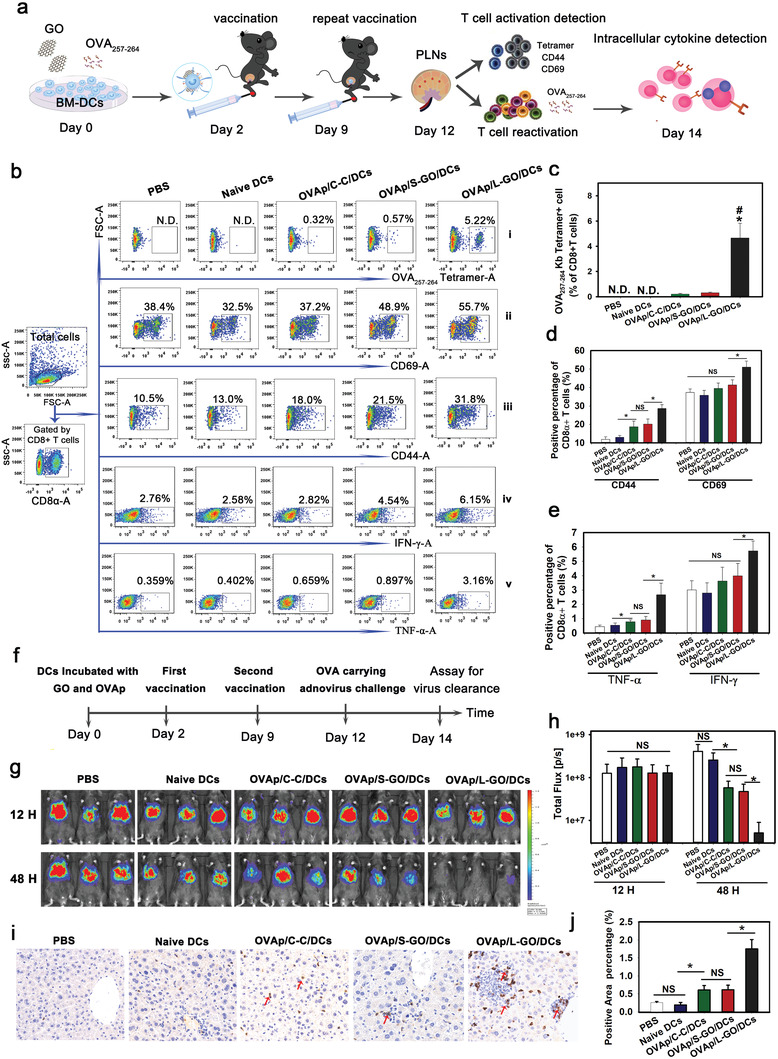
In vivo T cell priming and antiviral effect of DC vaccines. a) Schematic diagram of DC vaccination process. b) CD8+ T cells primed by DC vaccines. The OVA257–264‐specific CD8+ T cells in PLNs were detected by OVA tetramer (i), activated CD8+ T cells were detected by staining CD69 and CD44 (ii,iii), and the intracellular cytokines IFN‐γ and TNF‐α (iv,v) were detected by FACS after the antigen‐recall experiment. Statistical analyses are summarized in (c)–(e). f) Schematic diagram of DC vaccination and viral challenge. Data are presented as means ± s.d.; n = 5; Tukey tests were used for multiple comparisons; N.D.: not detectable; *p < 0.05 compared to the OVAp/C‐C/DCs group, #p < 0.05 compared to OVAp/S‐GO/DCs. g,h) Bioluminescence imaging and statistical analysis of AdFLO infection in the liver. Red arrow: CD8+ T cells. i) Infiltration of CTLs into the liver detected by immunohistochemical staining. j) Statistical analysis of T cell infiltration; data are presented as means ± s.d.; n = 6; *p < 0.05 by the Holm–Sidak method; NS: not significant. Representative results from two or three replicates are shown.
To test the protective efficacy of GO‐adjuvanted DC vaccines against viral infection, we constructed a recombinant adenovirus of serotype 5 (AdFLO) bearing OVA (target gene) and firefly luciferase (marker gene). The intravenous delivery of AdFLO predominantly leads to liver infection, which can be quantified by bioluminescence imaging.[ 25 ] The immunization and viral challenge followed the schedule shown in Figure 4f. We observed a comparable light intensity in all immunized mice 12 h after viral challenge, indicating that the viral inoculation was uniform and endogenous CTLs had not yet exerted an obvious killing effect at this time point (Figure 4g,h). At 48 h, we observed a dramatically decreased viral load in the L‐GO‐adjuvanted group, with an approximately sixfold higher clearance efficiency than that of the cytokine‐cocktail‐adjuvanted group. Consistently, significantly higher infiltration of CTLs was found in the inflammatory lesions of liver sections in mice injected with L‐GO‐adjuvanted DCs (Figure 4i,j), confirming the robust antigen‐specific cytolytic immune response. Furthermore, the protective efficacy of L‐GO adjuvanted DC vaccines was significantly discounted by Y27632 pretreatment, indicating the Rho‐ROCK‐MLC‐ICAM‐1 positing is also critical to L‐GO mediated in vivo DC–T‐cell interaction (Figure S11, Supporting Information). In addition, as the Y‐27632 treatment had little effect on LPS adjuvanted DC function, indicating the forementioned signal pathway is specifically indispensable for L‐GO adjuvanted DCs.
The above results demonstrated that L‐GO‐adjuvanted DCs elicited robust antigen‐specific CD8+ T cell responses and could be potential adoptive cell vaccines to protect against viral infection. Recently, an adaptive Phase IB‐II trial to evaluate the safety and efficacy of an autologous DC‐based vaccine in preventing COVID‐19 in adults was started.[ 16 ] This trial emphasized the recruitment of elderly patients and others at a higher risk of poor outcomes after COVID‐19 infection. The rationale lies in the fact that aged individuals exhibit a reduction in certain DC populations[ 17 , 26 ] and obvious dysfunctions of DCs, including impaired phagocytic capacity,[ 27 ] altered cytokine production,[ 28 ] and a failure to activate naïve CD8+ T cells into cytotoxic T cells.[ 29 ] The dysfunction of DCs might compromise the protective efficacy of vaccines currently under development, as reported for other vaccines.[ 17a,b ] In this regard, ex vivo‐generated and functionally competent DC vaccines might offer a unique opportunity to improve the protection of elderly and immune‐compromised individuals against the ongoing COVID‐19 pandemic. Encouraged by the remarkable T cell‐priming capacity of L‐GO‐adjuvanted DCs, we further evaluated whether this approach could be employed to defend against SARS‐CoV‐2 infection. DCs adjuvanted with L‐GOs or cytokine‐cocktail were ex vivo pulsed with spike 1 proteins from SARS‐CoV‐2 and adoptively injected into recipient mice following the vaccination procedure shown in Figure S12, Supporting Information. After two immunizations, recombinant vectors carrying Spike1‐IRES‐Fluc were directionally delivered to the lungs of recipient mice. The reduction in Fluc light intensity was calculated to quantify anti‐spike 1 CTL functions. All immunized mice showed equivalent luminous efficacies at 12 h posttransfection (Figure 5a,b). At 48 h, the light intensity in mice immunized with Spike 1‐pulsed DCs was dramatically lower than that of mock‐treated animals, indicating the elicitation of spike 1‐specific CTLs. In addition, mice immunized with L‐GO‐adjuvanted DCs showed maximum light reduction; light intensity was more than twofold lower than that observed for cytokine‐cocktail‐adjuvanted DCs. Apoptosis dependent on Fas ligand (FasL)/Fas or TNF is one of the essential pathways by which CTLs kill antigen‐expressing target cells. TdT‐mediated dUTP nick‐end labeling (TUNEL) (Figure S13, Supporting Information) (Figure 5c, left panel) demonstrated significantly higher levels of apoptosis of lung epithelial cells in mice immunized with L‐GO‐adjuvanted DCs. In addition, immunohistochemical staining of infiltrated CD8α+ T cells (Figure 5c, right panel) corroborated the apoptosis results, showing that CTL infiltration was highest in the L‐GO‐adjuvanted group.
Figure 5.
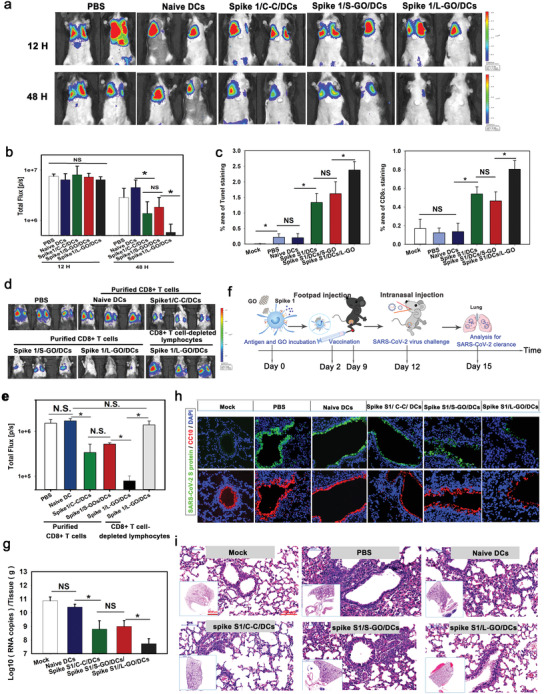
The priming of CTLs by DC vaccines against SARS‐CoV‐2. a) Bioluminescence imaging and b) statistical analysis of the clearance of spike 1 in the lungs. Data are presented as means ± s.d.; n = 5; the Holm–Sidak method was used for multiple comparisons; N.S.: not significant. c) Statistical data for TUNEL (Left panel) and CD8+ T cell staining of lung sections. Data are presented as means ± s.d.; n = 5; *p < 0.05 by the Holm–Sidak method; NS: not significant. d) Bioluminescence imaging of the clearance of spike 1 and e) a statistical analysis for adoptively transfused purified CD8+T cells isolated from spleens and PLNs of immunized mice. Data are presented as means ± s.d. n = 4; the Holm–Sidak method was used for multiple comparisons; *p < 0.05; NS: not significant. f) Schematic diagram of vaccination and viral challenge. g) Quantification of SARS‐CoV‐2 viral RNA in lungs of infected mice. Data are presented as means ± s.d.; n = 4–6; the Holm–Sidak method was used for multiple comparisons and t‐tests were used for pairwise comparisons; *p < 0.05; NS: not significant. h) Visualization of spike 1 and CC10 expression in lung sections. Green: SARS‐CoV‐2 Spike 1; red: CC10+ cub cell; blue: nuclei. i) Hematoxylin & eosin staining of lung sections. n = 4–6.
An adoptive cell transfer experiment was employed to confirm the therapeutic effects of the elicited anti‐spike 1 CTLs to eradicate target cells, following the scheme summarized in Figure S14 (Supporting Information). Significant enlargements of the spleen, PLNs, and inguinal lymph nodes (ILNs) were found in mice immunized with spike 1‐pulsed DCs (Figure S15, Supporting Information), especially with L‐GO‐adjuvanted DCs. Next, 5 × 106 purified CD8+ T cells pooled from the spleen and LNs were adoptively injected into recipient mice. At 24 h, light signal reduction was greatest in mice that received CD8+ T cells from the L‐GO‐adjuvanted group (Figure 5d,e). Importantly, mice that had received the “CD8+ T cell depleting lymphocytes” from mice immunized by L‐GO‐adjuvanted DC vaccines showed no significant light decrease, confirming that the CD8+ T cells were the key mediator for the observed protectivity. These data collectively indicate that L‐GO‐adjuvanted DC vaccines, excelling over the conventional “C–C” group, induced robust cytotoxic T cell immune responses against SARS‐CoV‐2 and protected recipients by promptly eradicating antigen‐bearing target cells.
A mouse‐adapted strain of a clinical SARS‐CoV‐2 isolate was used to validate the protective efficacy of L‐GO‐adjuvanted DC vaccines. This adapted SARS‐CoV‐2 strain contains an N501Y mutation at the receptor‐binding domain of the spike protein and can replicate and cause interstitial pneumonia in BALB/c mice.[ 30 ] Mice were injected with two doses of DC vaccines at a 1‐week interval (Figure 5f), and 3 d after the last vaccination, all immunized mice were intranasally challenged with 1.6 × 104 pfu of the adapted SARS‐CoV‐2 at passage 6 (called MASCp6). All mice were sacrificed 3 d after viral inoculation for virological and histopathological analyses. High copy numbers of viral RNAs (>1010/tissue [g]) were detected in the lungs of mock‐treated mice (unimmunized or vaccinated with naïve DCs). The cytokine‐cocktail or S‐GO‐adjuvanted DC vaccination reduced the RNA load by approximately 100‐fold. In contrast, L‐GO‐adjuvanted DC vaccines achieved >1500‐fold RNA load reduction, that is, more than 99.7% of viral copies were cleared from lung tissues (Figure 5g). Moreover, levels of SARS‐CoV‐2 spike protein were high in the lungs of mock‐treated mice, predominantly co‐localized with CC10+ club cells and distributed in bronchi and bronchioles (Figure 5h). In contrast, a dramatically weakened fluorescence signal was detected in immunized mice, with the weakest signal obtained from the L‐GO‐adjuvanted group. Consistently, MASCp6‐induced pulmonary damage was attenuated in mice vaccinated with L‐GO‐adjuvanted DCs (Figure 5i). It is worth noting that the enhanced protective efficiency of L‐GO adjuvanted DC vaccines in the SARS‐CoV‐2 challenged mice model may benefit from both the humoral and cellular immune responses. SARS‐CoV‐2 spike1 specific IgG could be detected in the serum of all immunized mice by spike 1‐bearing DCs (Figure S16, Supporting Information). In addition, L‐GO adjuvanted DCs induced a much higher level of IgG than S‐GO or cytokine‐cocktail adjuvanted DCs, suggesting the L‐GO increased DC–T‐cell interaction may not be confined to CD8+ T cells.
Finally, the protective efficacy of DC vaccines adjuvanted by L‐GOs, GM‐CSF, cytokine‐cocktail and LPS, respectively, was also compared in parallel based on the mice model in Figure 5a. L‐GO‐adjuvanted DCs achieve a comparable vaccine efficacy with LPS‐adjuvanted ones and have more advantages than the currently used cytokine‐cocktail and GM‐CSF adjuvanted ones in priming in vivo spike1 specific CTLs (Figure S17, Supporting Information).
Indeed, similar to all graphene‐based materials, GOs also face the challenge of going through preclinical tests to accelerate conversion. Nevertheless, as an adjuvant of ex vivo prepared DC vaccines, >90% GO was removed by centrifugal washing before injection in vivo. This means that <1.0 µg of GO was subcutaneously injected per mouse, which was three orders of magnitude lower than the well‐tolerated dose in vivo according to a previous report.[ 31 ] In addition, the route of subcutaneous delivery can avoid the direct entry of GOs into the systemic circulation, greatly reducing potential hazards. A complete blood cell count (Table S2, Supporting Information) one week after the last DC vaccination revealed no statistically significant differences from values obtained for untreated mice. Moreover, no obvious histological changes were observed in the liver, lung, kidney, spleen, brain, pancreas, heart, intestine, footpad (the injected position), or PLN of L‐GO‐adjuvanted mice (Figure S18, Supporting Information). Thus, L‐GO could be considered an exploitable nanoadjuvant of the adoptive DCs vaccine that can be potentially used for future biomedical applications, although much more research is needed.
The following three pathways were involved in L‐GO‐enhanced ability of DCs to prime endogenous T cells. i) L‐GO‐induced DC cytoskeletal reorganization allows increased DC draining to LNs (Figure S19, Supporting Information), thereby increasing the probability that DCs encounter matched naïve T cells. ii) Highly expressed ICAM‐1 on DCs resulting from membrane positioning can aid in arresting more continuously passing naïve T cells in LNs and in stabilizing the contact of DC–T‐cells via ICAM‐1/LFA‐1 pairing (Figure S20, Supporting Information). iii) Due to the high binding affinity, a part of DC‐absorbed L‐GOs also drained to LNs along with migratory DCs (Figure S21, Supporting Information), which can also act as a cellular zipper to promote the formation of mega DC–T‐cell clusters in LNs (Figure S20, Supporting Information), thus providing sustained stimuli for in vivo T cell activation.
3. Conclusion
The overall process and mechanism of action of GOs in the current study are presented in Figure 6 . The lateral size of the GO nanosheets largely determined the interaction dynamics between GOs and DCs. GOs with diameters of >1 µm demonstrated strong adherence to the surface of the DC membrane and thus promoted cytoskeleton reorganization specifically via the RhoA‐ROCK‐MLC signaling pathway. The formation of ultra‐large DC–T‐cell synaptic clusters was observed in vitro, where CD8+T cell activation and proliferation were dramatically augmented. L‐GO‐induced and cytoskeleton‐dependent ICAM‐1 translocation from the cytosol to the DC surface contributed to enhanced DC–T‐cell adhesion. In this respect, L‐GOs are expected to be universal and promising candidates as adjuvants for DC vaccines. As‐prepared DCs induced more than a 20‐fold higher antigen‐specific CD8+ T cell response than cytokine‐cocktail adjuvanted DCs and achieved a substantially higher protective effect against SARS‐CoV‐2 infection, resulting in >99.7% viral RNA clearance from lung tissues. These robust anti‐SARS‐CoV‐2 immune responses induced by DC vaccines could provide a favorable reference for developing personalized antiviral therapy to fight against the COVID‐19 global pandemic. More importantly, our systematical investigation of the interaction between GOs and DCs revealed, for the first time, the crucial roles of nanosheet‐shaped 2D materials in promoting DC–T‐cell IS assembly, providing new insights for DC vaccine engineering based on the strategy of promoting DC–T‐cell communication.
Figure 6.
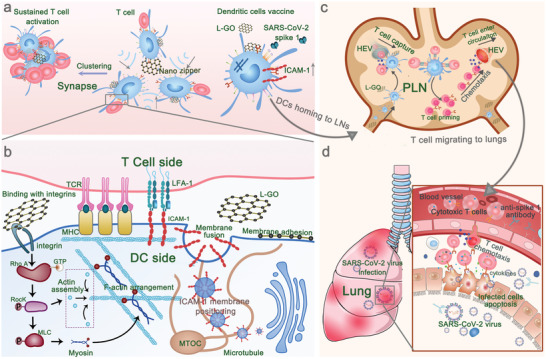
Schematic illustration of large‐sized graphene oxide enhancing DC–T‐cell synaptic contact and magnifying DC vaccine efficiency against SARS‐CoV‐2. a) The high binding affinity of L‐GOs with DC membrane promoted DC–T‐cell clustering. b) L‐GOs enhance DC–T‐cell synapse formation via cytoskeleton‐dependent membrane‐positioning of integrin ICAM‐1. c) The footpad‐injected DC vaccines prime T cells in PLN. d) SARS‐CoV‐2 spike‐1‐specific cytotoxic T cells and anti‐SARS‐CoV‐2 spike 1 antibodies corporately clear the infections.
4. Experimental Section
All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Committee on Animal Care and Use of the Academy of Military Medical Sciences (Approval No.: IACUC‐DWZX‐2020‐002).
Data were analyzed using Sigmaplot Software (Version 12, San Diego, CA, USA). A Dunnett's t‐test or one‐way analysis of variance (ANOVA) was used to analyze the normally distributed data. Nonparametric testing was used to analyze the non‐normally distributed data. P < 0.05 was considered to indicate a significant difference.
Details of the used reagents, antibodies, and methods can be found in the Supporting Information.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supplemental Video 1
Supplemental Video 2
Supplemental Video 3
Supplemental Video 4
Supplemental Video 5
Acknowledgements
Q.Z. and H.G. contributed equally to this work. This work was funded by the National Natural Science Foundation of China (81770196, 81970166, 81903624, and 81701583) and Mega‐Project of Science Research (2017ZX10304402‐003‐004).
Zhou Q., Gu H., Sun S., Zhang Y., Hou Y., Li C., Zhao Y., Ma P., Lv L., Aji S., Sun S., Wang X., Zhan L., Large‐Sized Graphene Oxide Nanosheets Increase DC–T‐Cell Synaptic Contact and the Efficacy of DC Vaccines against SARS‐CoV‐2. Adv. Mater. 2021, 33, 2102528. 10.1002/adma.202102528
Contributor Information
Shihui Sun, Email: sunsh01@163.com.
Xiaohui Wang, Email: wangxh@bmi.ac.cn.
Linsheng Zhan, Email: zhanls@bmi.ac.cn.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.a) Wculek S. K., Cueto F. J., Mujal A. M., Melero I., Krummel M. F., Sancho D., Nat. Rev. Immunol. 2020, 20, 7; [DOI] [PubMed] [Google Scholar]; b) Steinman R. M., Annu. Rev. Immunol. 2012, 30, 1; [DOI] [PubMed] [Google Scholar]; c) da Silva L. T., Santillo B. T., de Almeida A., Duarte A., Oshiro T. M., Front. Immunol. 2018, 9, 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Ho N. I., Huis In 't Veld L. G. M., Raaijmakers T. K., Adema G. J., Front. Immunol. 2018, 9, 2874; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhong H., Shurin M. R., Han B., Expert Rev. Vaccines 2007, 6, 333. [DOI] [PubMed] [Google Scholar]
- 3.a) Qian C., Yang L. J., Cui H., Front. Pharmacol. 2020, 11, 960; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou Q., Zhang Y., Du J., Li Y., Zhou Y., Fu Q., Zhang J., Wang X., Zhan L., ACS Nano 2016, 10, 2678. [DOI] [PubMed] [Google Scholar]
- 4.a) Wilgenhof S., Corthals J., Heirman C., van Baren N., Lucas S., Kvistborg P., Thielemans K., Neyns B., J. Clin. Oncol. 2016, 34, 1330; [DOI] [PubMed] [Google Scholar]; b) Pearson F. E., Chang K., Minoda Y., Rojas I. M. L., Haigh O. L., Daraj G., Tullett K. M., Radford K. J., Immunol. Cell Biol. 2018, 96, 390. [DOI] [PubMed] [Google Scholar]
- 5.a) Jonuleit H., Kuhn U., Muller G., Steinbrink K., Paragnik L., Schmitt E., Knop J., Enk A. H., Eur. J. Immunol. 1997, 27, 3135; [DOI] [PubMed] [Google Scholar]; b) Sutherland S. I. M., Ju X., Horvath L. G., Clark G. J., Front. Immunol. 2021, 12, 641307; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Alcaide E. G., Krishnarajah S., Junker F., Vaccines 2021, 9, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Harari A., Graciotti M., Bassani‐Sternberg M., Kandalaft L. E., Nat. Rev. Drug Discovery 2020, 19, 635; [DOI] [PubMed] [Google Scholar]; b) Amon L., Hatscher L., Heger L., Dudziak D., Lehmann C. H. K., Pharmaceutics 2020, 12, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Benvenuti F., Front. Immunol. 2016, 7, 70; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Alcaraz‐Serna A., Bustos‐Moran E., Fernandez‐Delgado I., Calzada‐Fraile D., Torralba D., Marina‐Zarate E., Lorenzo‐Vivas E., Vazquez E., de Alburquerque J. B., Ruef N., Gomez M. J., Sanchez‐Cabo F., Dopazo A., Stein J. V., Ramiro A., Sanchez‐Madrid F., Sci. Adv. 2021, 7, eabb9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Calvo V., Izquierdo M., Front. Cell Dev. Biol. 2021, 9, 629097; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Leithner A., Altenburger L. M., Hauschild R., Assen F. P., Rottner K., Stradal T. E. B., Diz‐Munoz A., Stein J. V., Sixt M., J. Cell Biol. 2021, 220, e202006081; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Comrie W. A., Li S., Boyle S., Burkhardt J. K., J. Cell Biol. 2015, 208, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L., Science 1999, 285, 221; [PubMed] [Google Scholar]; b) Yu Y., Smoligovets A. A., Groves J. T., J. Cell Sci. 2013, 126, 1049; [DOI] [PubMed] [Google Scholar]; c) Kumari S., Mak M., Poh Y. C., Tohme M., Watson N., Melo M., Janssen E., Dustin M., Geha R., Irvine D. J., EMBO J. 2020, 39, e102783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Razvag Y., Neve‐Oz Y., Sajman J., Yakovian O., Reches M., Sherman E., Cell Rep. 2019, 29, 3506. [DOI] [PubMed] [Google Scholar]
- 11.a) Aguilar‐Bolados H., Contreras‐Cid A., Yazdani‐Pedram M., Acosta‐Villavicencio G., Flores M., Fuentealba P., Neira‐Carrillo A., Verdejo R., Lopez‐Manchado M. A., J. Colloid Interface Sci. 2018, 524, 219; [DOI] [PubMed] [Google Scholar]; b) Romero‐Aburto R., Narayanan T. N., Nagaoka Y., Hasumura T., Mitcham T. M., Fukuda T., Cox P. J., Bouchard R. R., Maekawa T., Kumar D. S., Torti S. V., Mani S. A., Ajayan P. M., Adv. Mater. 2013, 25, 5632; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Han J., Kim Y. S., Lim M. Y., Kim H. Y., Kong S., Kang M., Choo Y. W., Jun J. H., Ryu S., Jeong H. Y., Park J., Jeong G. J., Lee J. C., Eom G. H., Ahn Y., Kim B. S., ACS Nano 2018, 12, 1959; [DOI] [PubMed] [Google Scholar]; d) Daniyal M., Liu B., Wang W., Curr. Med. Chem. 2020, 27, 3665. [DOI] [PubMed] [Google Scholar]
- 12.a) Sun C., Wakefield D. L., Han Y., Muller D. A., Holowka D. A., Baird B. A., Dichtel W. R., Chem 2016, 1, 273; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhu J., Xu M., Gao M., Zhang Z., Xu Y., Xia T., Liu S., ACS Nano 2017, 11, 2637; [DOI] [PubMed] [Google Scholar]; c) Vranic S., Rodrigues A. F., Buggio M., Newman L., White M. R. H., Spiller D. G., Bussy C., Kostarelos K., ACS Nano 2018, 12, 1373. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y., Ma C., Wang Z., Zhou Q., Sun S., Ma P., Lv L., Jiang X., Wang X., Zhan L., Nanoscale 2020, 12, 8147. [DOI] [PubMed] [Google Scholar]
- 14.a) Gonzalez P. A., Carreno L. J., Figueroa C. A., Kalergis A. M., Cytokine Growth Factor Rev. 2007, 18, 19; [DOI] [PubMed] [Google Scholar]; b) Gonzalez P. A., Carreno L. J., Cespedes P. F., Bueno S. M., Riedel C. A., Kalergis A. M., Clin. Dev. Immunol. 2013, 2013, 450291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Umakanthan S., Sahu P., Ranade A. V., Bukelo M. M., Rao J. S., Abrahao‐Machado L. F., Dahal S., Kumar H., Kv D., Postgrad. Med. J. 2020, 96, 753; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Soleimanpour S., Yaghoubi A., Expert Rev. Vaccines 2021, 20, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aivita Biomedical, Inc, Clin Trials.gov Identifier: NCT04386252 2020. Phase Ib‐II trial of dendritic cell vaccine to prevent COVID‐19 in adults. https://clinicaltrials.gov/ct2/show/NCT04386252 (accessed: June 2021).
- 17.a) de Gomensoro E., Del Giudice G., Doherty T. M., Ann. Med. 2018, 50, 181; [DOI] [PubMed] [Google Scholar]; b) Agrawal A., Gupta S., Ageing Res. Rev. 2011, 10, 336; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sibanda M., Meyer J. C., Mahlaba K. J., Burnett R. J., Front. Public Health 2021, 9, 635266; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Anastasiou O. E., Heger D., Vaccines 2021, 9, 169.33671437 [Google Scholar]
- 18.a) Maimela N. R., Liu S., Zhang Y., Comput. Struct. Biotechnol. J 2019, 17, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fumarola C., Bonelli M. A., Petronini P. G., Alfieri R. R., Biochem. Pharmacol. 2014, 90, 197. [DOI] [PubMed] [Google Scholar]
- 19.a) Wattrang E., Dalgaard T. S., Norup L. R., Kjaerup R. B., Lunden A., Juul‐Madsen H. R., J. Immunol. Methods 2015, 419, 35; [DOI] [PubMed] [Google Scholar]; b) Wiechmann A., Wilde B., Tyczynski B., Amann K., Abdulahad W. H., Kribben A., Lang K. S., Witzke O., Dolff S., Front. Med. 2021, 8, 556776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Suzuki K., Okuno T., Yamamoto M., Pasterkamp R. J., Takegahara N., Takamatsu H., Kitao T., Takagi J., Rennert P. D., Kolodkin A. L., Kumanogoh A., Kikutani H., Nature 2007, 446, 680; [DOI] [PubMed] [Google Scholar]; b) Cemerski S., Shaw A., Curr. Opin. Immunol. 2006, 18, 298; [DOI] [PubMed] [Google Scholar]; c) Katakai T., Habiro K., Kinashi T., J. Immunol. 2013, 191, 1188. [DOI] [PubMed] [Google Scholar]
- 21.a) Jo J. H., Kwon M. S., Choi H. O., Oh H. M., Kim H. J., Jun C. D., J. Cell. Biochem. 2010, 111, 1125; [DOI] [PubMed] [Google Scholar]; b) Malinova D., Fritzsche M., Nowosad C. R., Armer H., Munro P. M., Blundell M. P., Charras G., Tolar P., Bouma G., Thrasher A. J., J. Leukocyte Biol. 2016, 99, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Makrogianneli K., Carlin L. M., Keppler M. D., Matthews D. R., Ofo E., Coolen A., Ameer‐Beg S. M., Barber P. R., Vojnovic B., Ng T., Mol. Cell. Biol. 2009, 29, 2997; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bros M., Haas K., Moll L., Grabbe S., Cells 2019, 8, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pelin M., Fusco L., Martin C., Sosa S., Frontinan‐Rubio J., Gonzalez‐Dominguez J. M., Duran‐Prado M., Vazquez E., Prato M., Tubaro A., Nanoscale 2018, 10, 11820. [DOI] [PubMed] [Google Scholar]
- 24. Zhou Z., Zhao J., Hu K., Hou X., Sun X., Pan X., Wang X., Li N., Yang Z., Zhang F., Zhou Q., Zhan L., Int. J. Radiat. Oncol., Biol., Phys. 2021, 109, 95. [DOI] [PubMed] [Google Scholar]
- 25. Stabenow D., Frings M., Truck C., Gartner K., Forster I., Kurts C., Tuting T., Odenthal M., Dienes H. P., Cederbrant K., Protzer U., Knolle P. A., Hepatology 2010, 51, 1430. [DOI] [PubMed] [Google Scholar]
- 26. Wong C. P., Magnusson K. R., Ho E., Exp. Gerontol. 2010, 45, 163. [DOI] [PubMed] [Google Scholar]
- 27.a) Agrawal A., Agrawal S., Cao J. N., Su H., Osann K., Gupta S., J. Immunol. 2007, 178, 6912; [DOI] [PubMed] [Google Scholar]; b) Channappanavar R., Perlman S., J. Clin. Invest. 2020, 130, 6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Agrawal A., Agrawal S., Gupta S., Front. Immunol. 2017, 8, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Zacca E. R., Crespo M. I., Acland R. P., Roselli E., Nunez N. G., Maccioni M., Maletto B. A., Pistoresi‐Palencia M. C., Moron G., PLoS One 2015, 10, e0140672; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rodriguez‐Garcia M., Fortier J. M., Barr F. D., Wira C. R., Aging Cell 2018, 17, e12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gu H., Chen Q., Yang G., He L., Fan H., Deng Y. Q., Wang Y., Teng Y., Zhao Z., Cui Y., Li Y., Li X. F., Li J., Zhang N. N., Yang X., Chen S., Guo Y., Zhao G., Wang X., Luo D. Y., Wang H., Yang X., Li Y., Han G., He Y., Zhou X., Geng S., Sheng X., Jiang S., Sun S., Qin C. F., Zhou Y., Science 2020, 369, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.a) Kanakia S., Toussaint J. D., Mullick Chowdhury S., Tembulkar T., Lee S., Jiang Y. P., Lin R. Z., Shroyer K. R., Moore W., Sitharaman B., Biomaterials 2014, 35, 7022; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Newman L., Jasim D. A., Prestat E., Lozano N., de Lazaro I., Nam Y., Assas B. M., Pennock J., Haigh S. J., Bussy C., Kostarelos K., ACS Nano 2020, 14, 10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Video 1
Supplemental Video 2
Supplemental Video 3
Supplemental Video 4
Supplemental Video 5
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.