

Graphical abstract

Keyword: COVID-19, SARS-COV-2, SARS-COV, ACE2, Spike protein, Molecular dynamics
Abstract
The SARS-CoV-2 coronavirus (COVID-19) that is causing the massive global pandemic exhibits similar human cell invasion mechanism as the coronavirus SARS-CoV, which had significantly lower fatalities. The cell membrane protein Angiotensin-converting enzyme 2 (ACE2) is the initiation point for both the coronavirus infections in humans. Here, we model the molecular interactions and mechanical properties of ACE2 with both SARS-CoV and COVID-19 spike protein receptor-binding domains (RBD). We report that the COVID-19 spike RBD interacts with ACE2 more strongly and at only two protein residues, as compared to multi-residue interaction of the SARS-CoV. Although both coronaviruses stiffen the ACE2, the impact of COVID-19 is six times larger, which points towards differences in the severity of the reported respiratory distress. The recognition of specific residues of ACE2 attachments to coronaviruses is important as the residues suggest potential sites of intervention to inhibit attachment and subsequent entry of the COVID-19 into human host cells
1. Introduction
Coronaviruses have been posing mild to serious health concerns for the public since their discovery in 1965 [1]. These large positive-stranded RNA viruses were named due to their crown-like appearance observed using electron microscopy [2]. About 200 different coronaviruses have been discovered to date, that infects different creatures, including bats, birds, cattle, dogs, pigs, rodents, monkeys, humans, etc. [3], [4], [5], [6]. The seven coronaviruses found among humans are HCoV-229E, HCoV-NL63, HCoV-OC43, HKU1, SARS-CoV, MERS-CoV, and SARS-CoV-2 [7], [8]. Among the mentioned coronaviruses, the first four are commonly found and generally cause symptoms of common cold, while the uncommonly found latter three can be much more deadly, causing severe pneumonia. In 2002–03, the SARS-CoV (Severe Acute Respiratory Syndrome) infected around 8000 people around the globe and caused 774 deaths[9]. Since its first emergence in 2012, the MERS-CoV (Middle East Respiratory Syndrome) has infected 2494 people with a fatality of 858 in 27 countries [10]. The SARS-CoV-2, also known as COVID-19, the recently emerged pandemic, was first reported in Wuhan, China, in late December 2019 [11], [12]. According to the World Health Organization (WHO), the number of globally confirmed cases of COVID-19 is 209,201,939 with 4,390,467 fatalities in 216 countries (as of August 20, 2021) [13].
The SARS-CoV and SARS-CoV-2 are closely related coronaviruses that are classified as beta-coronaviruses, and both have originated in bats [8], [14]. The SARS-CoV-2 genome exhibits a 80% identity match with the SARS-CoV genome [15]. Another remarkable similarity between them is their host cell entry mechanism. Both coronaviruses utilize spike glycoproteins (S) to enter host cells by binding with cell surface Angiotensin-Converting Enzyme 2 (ACE2) receptors though their spike (S) genes share only 75% sequence similarity [8], [15], [16], [17], [18]. Spike glycoprotein (S), one of the four structural proteins of coronaviruses, is a class I virus membrane fusion protein [19]. The large ectodomain of spike protein comprises of receptor-binding domain S1 and membrane fusion domain S2 [20], [21], [22]. Both the N-terminal domain (NTD) and the C-terminal domain (CTD) of the S1 subunit are attributed to viral host receptor attachment of different coronaviruses [23], [24], [25]. The S2 subunit, the most conserved region of the spike protein, carries the fusion peptide (FP) along with two heptad repeats (HR1 and HR2) for performing viral and host membrane fusion [19], [26].
The ACE2, an essential carboxypeptidase of the renin-angiotensin system (RAS), plays a crucial role in maintaining cardiovascular homeostasis [27]. This Type I membrane protein is primarily expressed in the heart, kidneys, intestine, and lungs [28], [29]. As a homolog of ACE, it negatively regulates the RAS system by cleaving AngI into Ang1-9 and AngII into Ang1-7 [28], [30]. Inside a healthy human lung, alveolar epithelial Type II cells are characterized by abundant expression of ACE2 [31]. Downregulation of ACE2 in these cells causes severe lung injury that may be associated with acute respiratory distress syndrome (ARDS) occurring from alveolar collapse due to increased surface tension [32], [33], [34]. Both SARS-CoV and SARS-CoV-2 infection have been shown to cause ARDS in severely ill patients [35], [36]. The introduction of host cell infection by these coronaviruses is marked by the molecular interaction of the spike glycoprotein (S) receptor-binding domain (RBD) with ACE2 cell receptor [37]. This interaction ultimately leads to the invasion of the host cell by the virus replicating machinery.
The in vivo folding behavior of proteins contributes to their effective functioning [38], [39], and variation in temperature and pH impacts the folded conformation [40], [41]. Cellular motion-induced mechanical stretching in the extracellular matrix, muscle, and cell receptors also result in protein unfolding [42], [43], [44]. As downregulation of ACE2 cell receptors with cyclic stretching of human lung epithelial cells may be associated with ARDS in case of coronavirus infections (both SARS-CoV and SARS-CoV-2), the molecular interactions and unfolding pathway of ACE2 with and without the presence of spike receptor-binding domain can highlight the deviation of ACE2 behavior due to viral infections [32], [45], [46]. This change in behavior can be modeled through pairwise non-bonded interactions and mechanical response to external forces. Molecular dynamics (MD) simulation is a computational technique that predicts the time-dependent behavior of a molecular system in terms of energy (bonded and non-bonded) and conformation. MD simulations have been employed to investigate different material systems i.e., oil shale [47], [48], [49], swelling clays [50], [51], and proteins [52]. The interactions within coronaviral RNA dependent RNA polymerase (RdRp) have also been analyzed using MD simulations [53]. Steered molecular dynamics (SMD) is an in silico mechanobiological methodology for investigating the mechanical response of proteins during unfolding as well as the unbinding procedure of ligands from them [54], [55], [56]. In the current study, we report molecular dynamics simulations and steered molecular dynamics simulations of human ACE2 in the proximity of both SARS-CoV and SARS-CoV-2 spike (S) protein receptor-binding domain (RBD) to determine their pairwise non-bonded interactions and effect of these interactions on the mechanical response of ACE2 respectively. We also utilize SMD to explore the binding forces of coronavirus spike RBDs to ACE2. Since ACE2 is the primary cellular receptor for the SARS-CoV and the SARS-CoV-2, any changes in the mechanisms of attachment of ACE2 with SARS-CoV and SARS-CoV-2 spike (S) protein receptor-binding domains (RBD) is relevant to the understanding of the host cell invasion and for developing interventions to prevent attachment.
2. Methodology
2.1. The SARS-CoV ACE2 and SARS-CoV-2 ACE2 interaction model construction
The initial three-dimensional structures of SARS-CoV spike RBD with ACE2 and SARS-CoV-2 spike RBD with ACE2 have been obtained from RCSB Protein Data Bank. The SARS-CoV model has been developed using X-ray diffraction data [57] while the model for SARS-CoV-2 was constructed using cryo-Electron Microscopy data [58]. Both of these models were experimentally validated before submitting to Protein Data Bank. The corresponding PDB ID of SARS-CoV spike RBD-ACE2 complex and SARS-CoV-2 spike RBD-ACE2 complex are 2AJF and 6M17, respectively [57], [58]. The models were chosen due to their availability and similarity i.e., both models utilized ACE2 homodimer. The SARS-CoV complex model (2AJF) contains two spike-RBD chains (E and F) bound with two ACE2 protein chains (A and B) (Fig. 1 ).
Fig. 1.

Equilibrated structure of human ACE2 with SARS-CoV spike RBD where two chains of SARS-CoV spike RBD (chain E and F) attach to the peptidase domains (PD) of ACE2 homodimer (chain A and B), i.e., chain E attaches on the PD of chain A, and chain F attaches on the PD of chain B. Chain A, B, E and F are represented by red, blue, orange and green colored segments respectively. The initial model has been obtained from RCSB Protein Data Bank (PDB ID: 2AJF) and solvated in a water box. Water molecules are removed from display for the visual clarity of molecular structures. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The SARS-CoV-2 model comprises of two spike-RBD chains (E and F) with ACE2 dimer (chain B and D) along with the neutral amino acid transporter B0AT1 (chain A and C). As the primary objective of this study is to investigate the interactions between coronavirus spike RBD and ACE2, we have removed the amino acid transporter B0AT1 from model 6 M17 (Fig. 2 ).
Fig. 2.
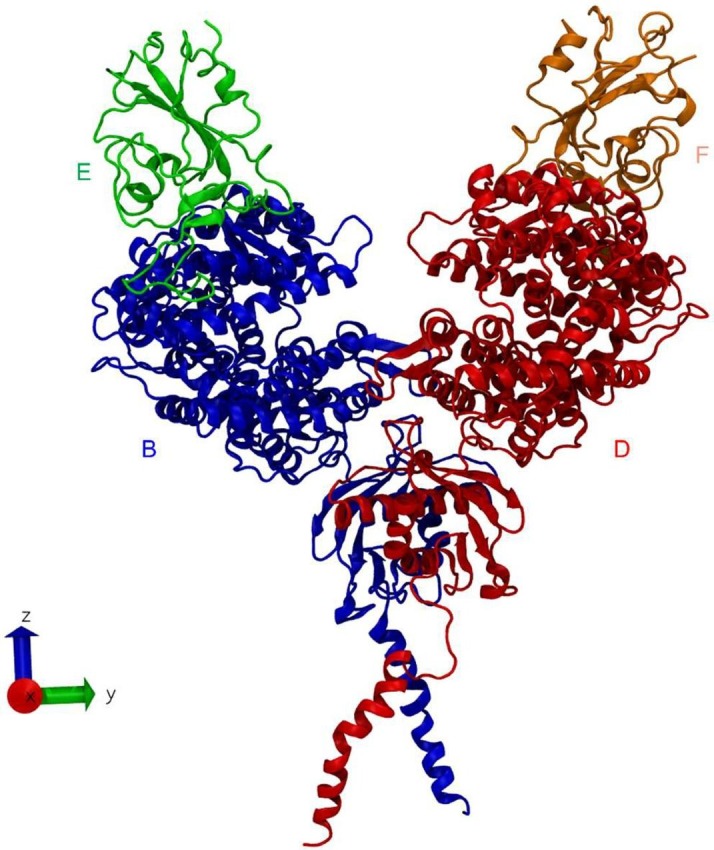
Equilibrated structure of full-length human ACE2 (both peptidase and collectrin domains) with SARS-CoV-2 spike RBD where two chains of SARS-CoV-2 spike RBD (chain E and F) attach to the N-terminal peptidase domains (PD) of ACE2 homodimer (chain B and D), i.e., chain E attaches on the PD of chain B, and chain F attaches on the PD of chain D. Chain B, D, E and F are represented by blue, red, green and orange colored segments respectively. The initial model has been obtained from RCSB Protein Data Bank (PDB ID: 6 M17) with neutral amino acid transporter removed. The modified model was solvated in a water box and then minimized and equilibrated. Water molecules are removed from display for the visual clarity of molecular structures. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Further, the AutoPSF plugin of Visual Molecular Dynamics (VMD 1.9.3) has been used for adding H-atoms and assigning partial charges to both models. These protein models were then solvated in a water box of 5 Å thickness. The final dimensions of solvated SARS-CoV spike RBD-ACE2 complex are 118.75 Å × 118.78 Å × 145.36 Å comprising a total of 192,502 atoms. The solvated model of full-length ACE2 with SARS-CoV-2 spike RBD has the dimensions of 106.06 Å × 146.66 Å × 200.60 Å with 295,174 atoms.
2.2. Molecular dynamics and steered molecular dynamics simulations of SARS-CoV and SARS-CoV-2 RBD interaction with ACE2
Molecular dynamics (MD) and steered molecular dynamics (SMD) simulations were performed using NAMD 2.12, a parallel molecular dynamics code [59]. NAMD was developed by the Theoretical and Computational Biophysics Group at the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign. All the parameters were obtained from CHARMm (Chemistry at HARvard Macromolecular Mechanics) force field [60]. It consists of functions and constants to define energy expression. CHARMm uses both bonded and non-bonded interaction terms. In this study, we utilize non-bonded interactions. At first, both models are minimized at 0 K temperature and 0 bar pressure using conjugate-gradient method [61]. Further, both models are brought to 310 K temperature and 1.01325 bar pressure to mimic the human physiological condition. The models are run for five ns using a timestep of 0.5 fs until they reach the equilibrium condition. Thermodynamic and conformational equilibration of structures are characterized by total energy, and root mean squared deviation (RMSD), respectively. These equilibrated models are further utilized for non-bonded energy calculations and steered molecular dynamics (SMD) simulations. Constant velocity SMD of ACE2 is done to assess its mechanical behavior by pulling its one terminal while keeping the other terminal fixed. In case of SARS-CoV model, the N-terminal was pulled and the C-terminal was kept fixed with and without the presence of spike RBD (Fig. 3 a). In the SARS-CoV-2 model, the boundary atom of peptidase domain (residue id 615) remained fixed while pulling its N-terminal (Fig. 4 a). A constant velocity pulling of the spike RBD is utilized to probe its pull-off force from ACE2. The C-terminal of spike RBD was pulled with a velocity of 0.01 Å/fs by keeping the distant terminal (C-terminal for SARS-CoV and peptidase domain terminal for SARS-CoV-2) of ACE2 fixed (Fig. 3b and Fig. 4b). All the SMD simulations employ a spring constant of 7 kcal/mol/Å2 and a pulling velocity of 0.01 Å/fs. All the simulations were performed at the Center for Computationally Assisted Science and Technology (CCAST), a parallel computing facility at North Dakota State University. Each simulation utilized one node, dual-socket Intel Xeon 2670v2 “Ivy Bridge” 2.5 GHz with 64 GB DDR3 RAM at 1866 MHz and 50 processors.
Fig. 3.

(a) The constant-velocity pulling of ACE2 in the presence of SARS-CoV spike RBD. The structures of ACE2 homodimer (chain A and B) with SARS-CoV spike RBD (chain E and F) are used. Chain A, B, E, and F are represented by red, blue, orange, and green colored segments, respectively. The N-terminal of ACE2 chain B is pulled along the X-axis with a constant velocity by keeping its C-terminal fixed both in the presence and absence of spike RBD chain F. (b) The constant-velocity pulling of spike RBD chain F for unbinding from ACE2 chain B performed by pulling the C-terminal of spike RBD chain F while keeping the C-terminal of ACE2 chain B fixed. Water molecules are removed from display for the visual clarification of molecular structures. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.

(a) The constant-velocity pulling of ACE2 in the presence of SARS-CoV-2 spike RBD. The equilibrated structures of ACE2 homodimer (chain B and D) with SARS-CoV-2 spike RBD (chain E and F) have been employed to explore this behavior. Chain B, D, E, and F are represented by blue, red, green, and orange-colored segments, respectively. The N-terminal of ACE2 chain B was pulled along the Z-axis with a constant velocity by keeping it's peptidase domain terminal fixed both in the presence and absence of spike RBD chain E. (b) The constant-velocity pulling of spike RBD chain E towards its unbinding from ACE2 chain B. It has been performed by pulling the C-terminal of SARS-CoV-2 spike RBD chain E while keeping the peptidase domain terminal of ACE2 chain B fixed. Water molecules are removed from display for the visual clarification of molecular structures. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Results
3.1. Interaction energies of the SARS-CoV spike RBD with human ACE2 complex
The full-length ectodomain structure of human ACE2 is characterized by the claw-like N-terminal peptidase domain and C-terminal collectrin domain [62]. In this case, the N-terminal peptidase domain of ACE2 serves as the cellular receptor of concave surfaced SARS-CoV spike receptor-binding domain (RBD) [57]. The SARS-CoV spike RBD is 174 residues long, with the terminal residues being cysteine (CYS) and glutamic acid (GLU) [57]. Each unit of ACE2 homodimer consists of 597 residues with serine (SER) and aspartic acid (ASP) as the terminal residues. The spike RBD attaches to each ACE2 protomer resulting in the complex formation with two spike protein chains (E and F) with ACE2 homodimer (A and B), as shown in Fig. 1. Further, chain E attaches to peptidase domain (PD) of chain A, while chain F attaches similarly to the chain B. We define the total non-bonded interaction energies between two molecular entities as the sum of electrostatic and Van der Waals (VDW) interaction energies between the entities.
The negative values of non-bonded interaction energies constitute attractive interactions, and the positive values are repulsive interactions. In order to determine the interactions between human ACE2 and SARS-CoV spike RBD, non-bonded interaction energies have been computed between chain B of ACE2 and chain F of spike RBD (Fig. 5 a and Supplementary Table 1).
Fig. 5.
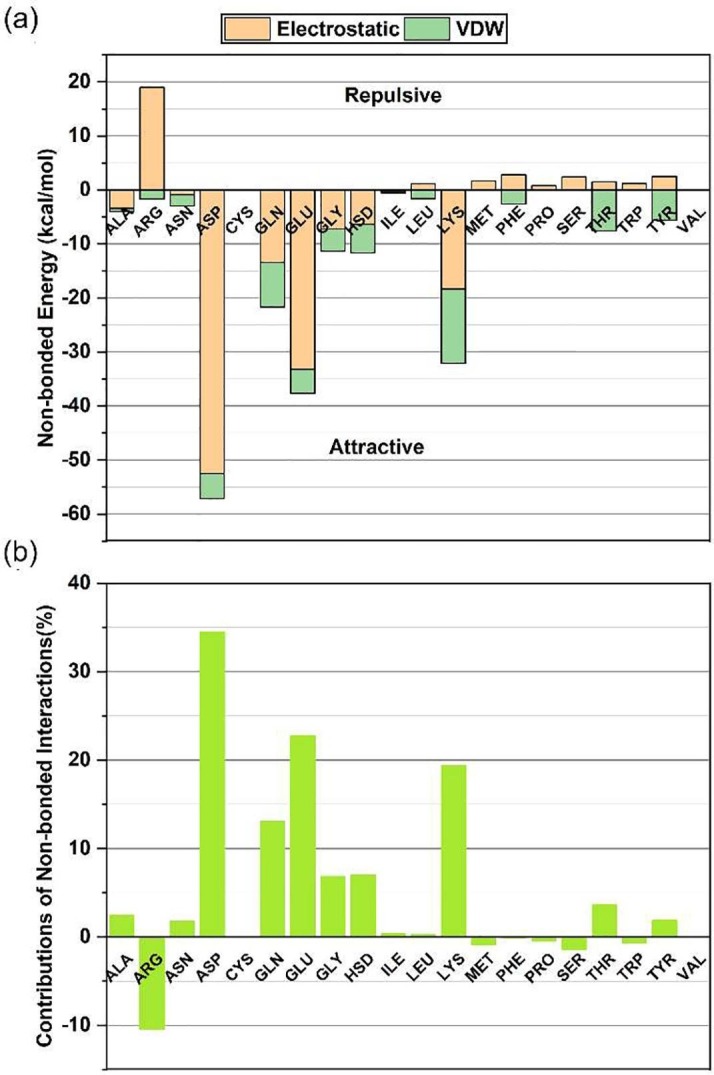
(a) The non-bonded interaction energies of ACE2 residues with SARS-CoV spike RBD. Negative and positive magnitudes of interaction energies represent the attractive and repulsive interactions, respectively. Here, non-bonded interactions are measured between chain B of ACE2 and chain F of SARS-CoV spike RBD. (b) The relative percentage contributions of non-bonded interactions by ACE2 residues with SARS-CoV spike RBD. Four residues of ACE2 (ASP > GLU > LYS > GLN) have significant interactions (attractive) with SARS-CoV spike RBD as they contribute about 89% of the total interactions altogether. ARG is the only residue of ACE2 that interacts significantly but repulsively with SARS-CoV spike RBD.
The total non-bonded interactions between them are −166 kcal/mol, with electrostatic and VDW interactions of −103 kcal/mol and −63 kcal/mol, respectively. Among the 20 different types of residues (amino acids) of ACE2, four different residues interact significantly with spike RBD. Aspartic acid (−57 kcal/mol), glutamic acid (−38 kcal/mol), lysine (−32 kcal/mol) and glutamine (−22 kcal/mol) of ACE2 contribute approximately 89% of the attractive non-bonded interactions with SARS-CoV spike RBD (Fig. 5(b)). Other significantly interacting residues of human ACE2 are histidine (−12 kcal/mol), glycine (−11 kcal/mol), and arginine (+17 kcal/mol). The non-bonded interactions of specific residues of ACE2 with SARS-CoV spike RBD are provided in Supplementary Table 5. These specific residues have been chosen based on the suggestion of structure resolving study [57] and they been shown to contribute 29.2% (−48.5 kcal/mol) of the total interaction energy.
In terms of the secondary structure, turn and coil components of ACE2 contribute to the majority (62%) of the non-bonded interactions (−103 kcal/mol). The rest of the interactions originate primarily from the alpha-helices of ACE2 (Supplementary Table 2). In terms of the tertiary structure, more than 99% of the interactions arise from the polar residues of ACE2. Also, the non-bonded interactions between the two chains of ACE2 (A and B) are calculated as −285 kcal/mol in the absence of SARS-CoV spike RBD. This interaction energy is reduced to −107 kcal/mol in the ACE2-spike RBD complex. In both cases, the interactions are predominantly electrostatic.
3.2. The binding force of the SARS-CoV spike RBD with human ACE2 complex
The constant velocity steered molecular dynamics (SMD) [59], [63] method has been used to investigate the mechanical response of ACE2 through its modeling of unfolding to external loading. As shown in Fig. 6 a, Chain B of ACE2 is stretched at constant velocity (0.01 Å/fs) both in the absence and presence of SARS-CoV spike RBD chain F. The molecular stretching is performed by pulling the N-terminal of chain B while keeping the C-terminal fixed. The force–displacement plot (Fig. 6b) represents the stretching pathway of chain B, where the peaks represent the unfolding of alpha helices, small turns, and coils (breaking of H-bonds). In the absence of SARS-CoV spike RBD, the unfolding of the α1 helix of ACE2 occurs at a force of 6624 pN with a corresponding displacement of 101 Å. The highest peak force, 8408 pN at a displacement of 172 Å, represents the unfolding of α2 helix. The linear stretching of the protein chain characterizes the post-peak downhill region of this peak where no unfolding of coil or helix occurs. In the presence of spike RBD chain F, the breaking of α1 and α2 helices occur at 7425 pN and 10,021 pN at displacements of 104 Å and 184 Å, respectively. The pulling of ACE2 causes the translation of spike RBD in the same direction.
Fig. 6.

(a) Mechanical response of ACE2 chain B with the attachment of SARS-CoV spike RBD chain F due to constant-velocity pulling. (b) The force–displacement plot of ACE2 with and without bound spike RBD. The plot for bound spike has been corrected for the rigid motion of spike RBD by subtracting the amount of force accountable for this motion. The peaks in force–displacement plots of ACE2 characterize the unwinding of helices (H-bond breaking). The presence of spike RBD increases the required force to unwind the helices/coils, thus increasing the stiffness of ACE2. (c) Pulling off SARS-CoV spike RBD chain F from ACE2 peptidase domain of chain B. (d) Force-displacement plot of pulling of spike RBD chain F for unbinding from ACE2 chain B. The first peak specifies the partial separation of spike RBD (chain F) while its complete separation from ACE2 (chain B) is marked by the second peak. Beyond this point, all the minor peaks and other features in the plot represent the linear stretching of spike RBD.
The binding force of SARS-CoV spike RBD and the human ACE2 has also been explored utilizing the constant velocity SMD simulations. The C-terminal of SARS-CoV spike RBD chain F is pulled away from ACE2 chain B to determine the amount of force required to separate the spike RBD from the spike RBD-ACE2 complex (Fig. 6(c)). The detachment of the spike RBD from ACE2 is seen as two important unlatching events at about 20 Å and 121 Å displacement, which are indicated by two sharp peaks in the force–displacement curve. The first peak is observed at 8030 pN force at 20 Å (Fig. 6(d)). The downward slope is characterized by the linear stretching of RBD chain F with no unfolding of coils or helices. Beyond the minimum at 61 Å displacement, the RBD starts to move away from ACE2 due to pulling. The second peak in the plot at a displacement of 121 Å occurs at a force of 10,454 pN and results in complete detachment of the spike RBD from the ACE2 complex. Beyond the second peak, all the smaller peaks and valleys in the plot are related only to the response of spike RBD chain F to pulling.
3.3. Interaction energies of the SARS-CoV-2 (Covid-19) spike RBD with human ACE2
The spike (S) protein of SARS-CoV-2 attaches to the peptidase domain (PD) of human ACE2 [58]. Due to the homodimerization of ACE2, two spike (S) protein receptor-binding domains (RBDs) attach to ACE2 dimer, where each PD binds with one RBD. Chain B and D of ACE2 are attached to spike RBD chain E and F, respectively (Fig. 2). The SARS-CoV-2 spike RBD consists of 183 residues with cysteine (CYS) at the N-terminal and leucine (LEU) at the C-terminal. The ACE2 protomer is a full-length model having 748 residues, which N-terminal is isoleucine (ILE), and C-terminal is arginine (ARG) [58]. The peptidase domain consists of about 80% of the total residues of ACE2 (residues 21 to 615). The non-bonded interactions between every residue of chain B of ACE2 and chain E of SARS-CoV-2 spike RBD have been computed to probe the interactions within the SARS-CoV-2 spike RBD-ACE2 complex and are shown in Fig. 7 (a) and Supplementary Table 3. The total non-bonded interactions between them (chain B of ACE2 and SARS-CoV-2 spike RBD chain E) are observed to be −356 kcal/mol with electrostatic and VDW interactions of −289 kcal/mol and −67 kcal/mol respectively. Among the 20 different residues of ACE2, two residues contribute to almost 86% of the total non-bonded attractive interactions between ACE2 and SARS-CoV-2 RBD (Fig. 7(b)). The non-bonded interactions of specific residues of ACE2 with SARS-CoV-2 spike RBD are provided in Supplementary Table 5. These specific residues have been chosen based on the suggestion of structure resolving study [58] and they been shown to contribute 43.2% (−153.8 kcal/mol) of the total interactions.
Fig. 7.
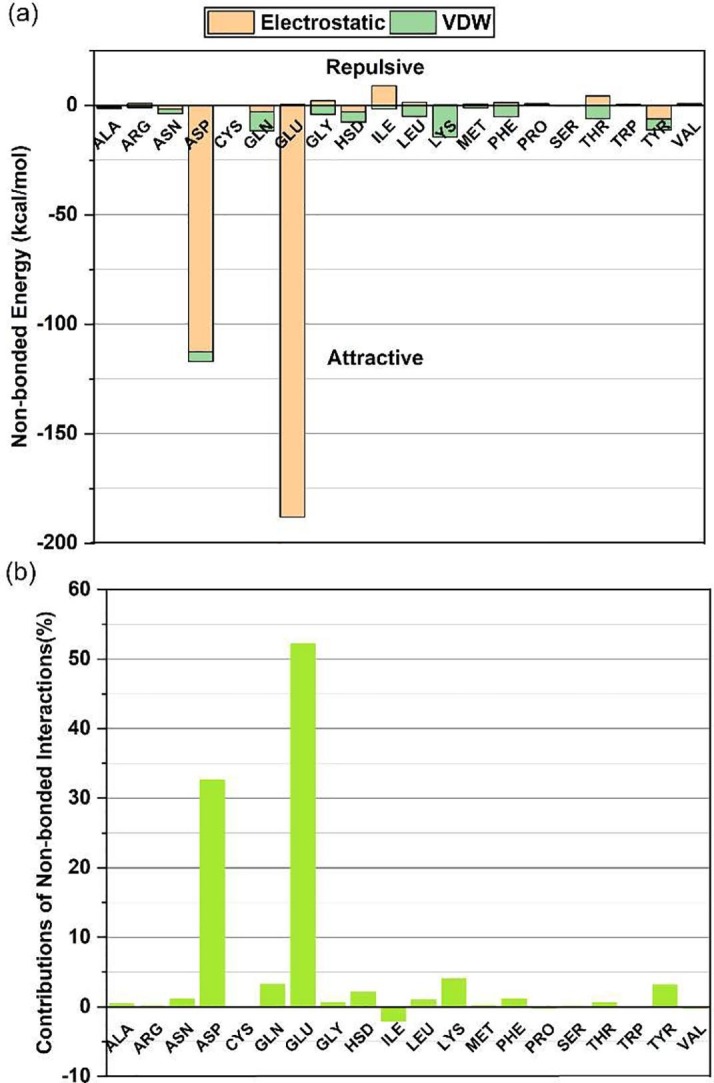
(a) The non-bonded interactions of ACE2 residues with SARS-CoV-2 spike RBD. Negative and positive magnitudes of interactions represent attractive and repulsive interactions, respectively. Here, non-bonded interactions are measured between chain B of ACE2 and chain E of SARS-CoV-2 spike RBD. (b) The relative percentage contributions of non-bonded interactions of ACE2 residues with SARS-CoV-2 spike RBD. Electrostatic interactions yield more than 80% of the non-bonded interactions. Two residues of ACE2 (GLU > ASP) have significant attractive interaction with SARS-CoV-2 spike RBD as they contribute about 86% of the total interaction energy.
The glutamic acid (GLU) alone contributes to more than 52% of the interaction energy (−188 kcal/mol), and Aspartic acid (ASP) contributes to about 33% of the interaction energy (−117 kcal/mol) with SARS-CoV-2 spike RBD. Lysine (LYS), glutamine (GLN), and tyrosine (TYR) are other significantly interacting residues of ACE2. Based on protein secondary structure, the helices of ACE2 contribute to 94% of the attractive interaction energy with SARS-CoV-2 spike RBD while beta-strands and turns produce the remainder (Supplementary Table 4). Polar residues of ACE2 participate in more than 99% of these interactions. Also, the total non-bonded interactions within the ACE2 dimer (between chain B and D) are observed to be −648 kcal/mol and −808 kcal/mol in the absence and presence of SARS-CoV-2 spike RBD respectively. In both cases, the electrostatic interactions produce more than 80% of the total interactions.
3.4. Binding force of the SARS-CoV-2 (COVID-19) spike RBD with human ACE2 complex
The mechanical response of ACE2 chain B is determined utilizing constant-velocity SMD simulations both in the absence and the presence of SARS- CoV-2 spike RBD chain E as shown in Fig. 8 a. This is performed by pulling the N-terminal of human ACE2 chain B while keeping its peptidase domain terminal (residue 615) fixed. The SARS-CoV spike RBD-ACE2 model contains only the peptidase domain of ACE2. In order to be consistent with the SARS-CoV model, we have fixed the peptidase domain terminal of full length ACE2 of SARS-CoV-2 spike RBD-ACE2 model. The first peak in the force–displacement plot of ACE2 without spike RBD indicates the beginning of α1 helix unfolding (breaking of H-bonds) (Fig. 8(b)). The peak of 3803 pN at a displacement of 112 Å marks the complete unfolding of α1 helix. The unfolding of α2 helix is accomplished at 4286 pN force at a displacement of 251 Å. The subsequent peak of 5456 pN is the result of the unfolding of α3 helix at the displacement of 539 Å, while downward slopes represent the linear stretching of protein without the breaking of coils or helices. The presence of spike RBD in the proximity of ACE2 causes an increase in the peak helix-unfolding forces by a significant amount. In the SARS-CoV-2 spike RBD-ACE2 complex, the unfolding forces of the α1, α2, and α3 helices are computed as 5346 pN, 4370 pN, and 6492 pN at displacements of 102 Å, 254 Å, and 530 Å respectively. The spike RBD chain E also moves in the pulling direction along with the peptidase domain of ACE2.
Fig. 8.

(a) Mechanical response of ACE2 chain B with the attachment of SARS-CoV-2 spike RBD chain E. (b) The force–displacement plot of ACE2 with and without bound spike RBD. The plot for bound spike has been corrected for the rigid motion of RBD by subtracting the amount of force accountable for this motion. The peaks in force–displacement plots of ACE2 characterize the unwinding of helices (H-bond breaking). The presence of spike RBD increases the required force to unwind the helices/coils, thus increasing the stiffness of ACE2. (c) Pulling off SARS-CoV-2 spike RBD chain E from ACE2 peptidase domain of chain B. (d) Force-displacement plot of pulling of spike RBD chain E towards its unbinding from ACE2 chain B. The first peak specifies the partial separation of spike RBD (chain E) while its complete separation from ACE2 (chain B) is marked by the second peak. Beyond this point, all the minor peaks and other features in the plot represent the linear stretching of spike RBD.
The C-terminal of SARS-CoV-2 spike RBD chain E is pulled away from ACE2 chain B to evaluate the binding force of spike RBD within the SARS-CoV-2 spike RBD-ACE2 complex (Fig. 8c). The detachment of the spike RBD from ACE2 is characterized by two unlatching events at about 46.2 Å and 129 Å, which are indicated by two sharp peaks in the force–displacement curve. The first peak has a maximum force of 6713.18 pN at 46.2 Å displacement (Fig. 8d). The highest peak of 7759.95 pN at a displacement of 129 Å signifies the complete separation of spike RBD chain E from the SARS-CoV-2 spike RBD-ACE2 complex. The remainder of the plot is the mechanical response of spike RBD chain E alone.
4. Discussion
The simulations indicate that electrostatic interactions dominate the non-bonded interactions between coronavirus spike RBD and ACE2 for both the SARS-CoV-2 and SARS-CoV. One of the major differences between the two coronaviruses spike-RBD interaction with ACE2 is that in the case of SARS-CoV-2, the majority of attractive interaction energies are primarily mediated by just two residues of ACE2; GLU and ASP, whereas, for SARS-CoV, the attractive interaction energies are spread out over four residues GLU, ASP, LYS and GLN (Fig. 5a & b and Fig. 7a & b). The two residue versus multi-residue interaction of the SARS-CoV-2 and SARS-CoV spike RBDs with the human ACE2 is likely to cause significant differences in the attachment of the two coronaviruses with human ACE2. These residues are generally classified as charged or polar residues attributed to salt-bridge and H-bond formations. Further, more than 99% of the spike-ACE2 interactions arise from polar residues of ACE2. The spike-ACE2 interactions for SARS-CoV-2 (−356 kcal/mol) are more than twice the interactions of SARS-CoV spike-ACE2 (−166 kcal/mol). Structural observations in a recent report also suggest higher interactions of SAR-CoV-2 RBD with ACE2 as compared to SARS-CoV RBD [58]. The higher interactions of SARS-CoV-2 are also reflected by the secondary structure of ACE2. In the SARS-CoV-2 spike RBD-ACE2 complex, the majority of the interaction energy arises from the ACE2 helices. On the other hand, in the SARS-CoV spike RBD-ACE2 complex, more than half of the interaction energy is caused by ACE2 turns. Another potential source of differences in interactions arises from the fact that the interacting ACE2 surface areas are 6059 Å2 and 5595 Å2 with SARS-CoV-2 spike RBD and SARS-CoV spike RBD respectively. A higher interacting surface area of ACE2 results in higher interactions with SARS-CoV-2 spike RBD. A lower dissociation constant (K D) is suggested from recently reported surface plasmon resonance experiments for the SARS-CoV-2 spike RBD binding of ACE2 (15 nM) [64] as compared to that also reported from surface plasmon experiments for SARS-CoV (185.1 nM) [65]. A lower dissociation constant represents higher binding affinity, i.e., SARS-CoV-2 spike has 10–15 fold higher binding affinity with ACE2 than SARS-CoV as per the above mentioned published studies[64], [65], [66]. The simulations presented here indicate a higher attractive non-bonded interaction energy for the SARS CoV-2 spike RBD with ACE (−356 kcal/mole) as compared to that for SARS-CoV RBD (−166 kcal/mole) which is in agreement with the surface plasmon experiments. The surface plasmon experiments are conducted by binding immobilized RBD on a sensor chip followed by injecting varying concentrations of ACE2 [64], [65], and studying the association and dissociation of ACE2 to the RBD. Measuring binding energy of the molecular entities that form RBD and ACE2 complex provides a different but complementary view in the attachment of the ACE2 to the coronavirus RBD. The investigation of nine specific residues of ACE2 in terms of their interactions with coronaviral spike RBDs reveal some interesting phenomena. These nine residues cumulatively exhibited almost 3 times higher non-bonded interactions with SARS-CoV-2 spike RBD (−153.3 kcal/mol) relative to SARS-CoV spike RBD (−48.5 kcal/mol). Among these residues, D30 has been found to be most intercating (−97.5 kcal/mol) with SARS-CoV-2 while K353 interacted (−35.3 kcal/mol) most with SARS-CoV. Certain residues (K31, M82, R357) were found to be oppositely interacting (attraction/repulsion) between SARS-CoV and SARS-CoV-2 spike RBD.
The mechanical response of ACE2 is obtained from the force–displacement plot of the complex using constant velocity pulling (Figs. 6b and 8b). The sharp peaks in the plot result from the unfolding of helices or coils in ACE2. The ACE2 force–displacement curve shifts upward, i.e., increased force is needed to cause the same displacement when the coronaviruses are attached to the ACE2 for both the coronaviruses. This force increment is the result of spike RBD binding interactions with the ACE2 peptidase domain (PD) that results in changes to the ACE2 unfolding behavior making the response stiffer. The force needed to pull the RBD by itself is found to be around 1000 pN, and this magnitude of the force is subtracted from the net force displacement of the RBD-ACE2 complex for both the coronaviruses. The plots shown in Figs. 6b and 8b are corrected for the force needed to pull just the RBD. From these observations, it is inferred that the presence of spike RBD makes ACE2 stiffer for both coronaviruses resulting in higher helix/coil unfolding forces. It is to be noted that the ACE2 model utilized with SARS-CoV spike RBD contains only the peptidase domain, while the ACE2 model utilized with SARS-CoV-2 spike RBD contains both peptidase and the collectrin domain [57], [58]. In both cases, the majority of the ACE2 models (spike-RBD ACE2 interacting regions) used are identical, with differences in the back end of the model. The collectrin domain at the back end of ACE2 in the SARS-CoV-2 model is far from spike RBD and does not affect the interactions with the spike RBD. However, in order to compare the force–displacement response of the ACE2 for the two coronavirus attachments, the peptidase domain terminal of full length ACE2 was fixed in the SMD simulations of SARS-CoV-2 spike RBD-ACE2 model. This particular action nullifies the impact of backend portion (collectrin domain) of ACE2 on the external loading response. The comparison of the mechanical response of ACE2 is done by computing the relative changes in ACE2 stiffness due to spike RBD attachment for both coronaviruses. The force displacement response of the ACE2 peptidase domain differs between the two models due to the differences in pulling direction and peptidase domain conformation with spike RBD. Therefore, the direct comparison of ACE2 stiffness is not done between SARS-CoV and SARS-CoV-2 spike attachement. Instead, relative change in stiffness of ACE2 as a result of RBD attachment are calculated. The stiffness was calculated from the force–displacement curves by finding the slope of the line connecting first peak of the force–displacement curve to the origin from each of the plots in Figs. 6b and 8b. The relative change in stiffness was calculated as the ratio of change in stiffness due to spike attachment of ACE2 stiffnesses with spike RBD and the initial stiffness of ACE2 stiffness without spike RBD. The ACE2 exhibits a 54% increase in stiffness (from 33.95 pN/ Å to 52.41 pN/ Å) with the attachment of SARS-CoV-2 RBD, while only a 9% increase in stiffness (from 65.58 pN/ Å to 71.39 pN/ Å) of ACE2 is observed with the attachment of SARS-CoV RBD. The downregulation of ACE2 cell receptors is known to be responsible for ARDS [32], [33]. Dramatic differences are seen in the outcome of the ARDS severity in the SARS-CoV and SARS-CoV-2 infected patients. Here we observe a vivid difference in the change in stiffness of ACE2 (54% versus 9%) between SARS-CoV-2 and SARS-CoV attachment to ACE2. Change in stiffness suggest changes to the unfolding mechanisms of proteins. These altered unfolding characteristics or changes in structure are highly related to biological functionality. Thus, the observed significant differences in stiffness of ACE2 on the coronavirus attachments could point towards differences in biological response of ACE2 and therefore the vast differences in severity of ARDS in the two coronaviruses; resulting in 774 deaths due to SARS-CoV versus over 4,390,467deaths (as of August 20, 2021) worldwide due to the SARS-CoV-2.
The binding force of spike RBD towards the ACE2 cell receptor is evaluated from the force–displacement behavior obtained by pulling the spike RBD at a constant velocity. We observe two peaks in the force–displacement plots for both coronaviruses resulting from two specific unlatching events that lead to the detachment of the spike RBD from ACE2. Both the spike RBDs from the two coronaviruses exhibit a two-step unlatching, leading to detachment, as shown in Supplementary Figs. 1 and 2. The SARS-CoV-2 spike RBD shows unlatching events at 6713.18 pN and 7759.95 pN as compared to 8030 pN and 10,454 pN, for the SARS-CoV spike RBD. Thus unlatching of the SARS-CoV-2 from ACE2 occurs at a lower force than the SARS-CoV. The mechanical pull-off of the coronavirus RBD from ACE2 enabled using SMD is a different phenomenon than measurement of dissociation constant using surface plasmon resonance [64], [65]. On pulling the spike RBD from the attachment to ACE2, conformational changes begin in the RBD, which progressively reduce interaction energies at the ACE2-spike RBD interface. These changes are influenced by factors like ACE2-RBD interface area, two-residue interactions of SARS-CoV-2 spike RBD as opposed to multi-residue interaction of SARS-CoV spike RBD with ACE2, RBD structure, unfolding rate of RBD, ACE2 conformation etc. As seen in Figs. 6d and 8d, the pulled terminal of SARS-CoV-2 spike RBD needed to be displaced by a larger distance (580 Å) than SARS-CoV (500 Å) for the force magnitude during pulling to become zero, indicating the influence of factors described above on the deformation. It appears that these factors likely cause faster reduction of non-bonded interactions in spike-ACE2 complex during the pulling of SARS-CoV-2 RBD compared to SARS-CoV RBD, resulting in lower unlatching force of SARS-CoV-2 spike RBD.
Since SMD simulations presented here mimic single protein pulling experiments with AFM, it is suggested that future AFM experiments that evaluate binding mechanisms of these complexes consider the effect of the unfolding of spike RBD. SMD simulations are thus a useful methodology to observe an accurate sequence of events in the pulling away of the spike RBD from ACE2. Understanding the important biological consequence of the formation of the ACE2-coronavirus spike RBD complex is aided by this additional viewpoint of stiffening of ACE2 on attachment to coronavirus, and the pulling force of the spike RBD from the ACE2-coronavirus spike RBD complex.
5. Conclusions
This study utilizes computational techniques to explain the initial host cell response due to coronavirus infections. It has been performed by capturing the molecular interactions and changes in the mechanical response of coronavirus cellular receptor angiotensin-converting enzyme 2 (ACE2) in the presence of SARS-CoV and SARS-CoV-2 spike (S) protein receptor-binding domain (RBD). Molecular dynamics (MD) simulation has been employed to determine the non-bonded interactions, while steered molecular dynamics (SMD) was used to describe the mechanical response of ACE2. The binding force of coronavirus spike (S) RBDs from the ACE2 has also been investigated by using SMD. The major findings of our study are summarized below:
-
•
Of the attractive non-bonded interactions of SARS-CoV-2 RBD with ACE2, 86% result from just two ACE2 residues; GLU and ASP. On the other hand, 89% of the SARS-CoV spike RBD interaction energy is spread over four ACE2 residues, including LYS and GLN, besides GLU and ASP. These observations suggest potential sites of intervention to inhibit attachment of spike RBD to ACE2.
-
•
The non-bonded interaction energies between SARS-CoV-2 spike RBD and ACE2 are more than twice the interaction energies between SARS-CoV spike RBD and ACE2.
-
•
The pull-off force of the spike RBD from the ACE2 is higher in magnitude for the SARS-CoV. On pulling the spike RBD from the attachment of ACE2, continuous conformational changes begin in the RBD, which progressively reduce interaction energies at the ACE2-spike RBD interface and hence influence total pull-off force.
-
•
The attachment of spike RBD with ACE2 results in the stiffening of ACE2 for both SARS-CoV-2 and SARS-CoV. The relative change in stiffness due to the attachment of spike RBD is higher for SARS-CoV-2 (54%) compared to the SARS-CoV (9%). The significantly larger relative stiffness of ACE2 on the SARS-CoV-2 attachment as compared to the SARS-CoV attachment points towards differences in the biological response of ACE2.
Since the host entry modes of the two coronaviruses compared here are similar, it is interesting to note the differences in the mechanisms of interactions, two ACE2 residues for SARS-CoV-2 versus multiple attachment residues for SARS-CoV. The stronger non-bonded interaction energies between SARS-CoV-2 and ACE2 result in a much stiffer ACE2 on attachment to the coronavirus spike RBD than for the SARS-CoV. Overall, the evaluation of these mechanisms of attachment and the resulting binding forces are critical to the development of therapies beyond vaccines that prevent the attachment and subsequent entry into host cells. Further mechanobiological studies that relate mechanical changes to the severity of the ARDS would provide a definitive answer to this important health concern.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors gratefully acknowledge the use of computational resources at the North Dakota State University Center for Computationally Assisted Science and Technology (NSF MRI grants #1229316, #2019077and NSF OIA NDACES-1946202). Discussions on the physiological consequences and biological ramifications with Preeya Katti, St. George’s University School of Medicine are acknowledged.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.chemphys.2021.111353.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Tyrrell D.A.J., Bynoe M.L. Cultivation of viruses from a high proportion of patients with colds. Lancet. 1966;1(7428):76. doi: 10.1016/s0140-6736(66)92364-6. [DOI] [PubMed] [Google Scholar]
- 2.Holmes K.V., Lai M.M.C. Third ed. Fundamental Virology; 1996. Coronaviridae: The Viruses and their Replication; pp. 541–559. [Google Scholar]
- 3.Tong S., Conrardy C., Ruone S., Kuzmin I.V., Guo X., Tao Y., Niezgoda M., Haynes L., Agwanda B., Breiman R.F., Anderson L.J., Rupprecht C.E. Detection of Novel SARS-like and Other Coronaviruses in Bats from Kenya. Emerging Infectious Dis. 2009;15(3):482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woo P.C.Y., Lau S.K.P., Lam C.S.F., Lau C.C.Y., Tsang A.K.L., Lau J.H.N., Bai R., Teng J.L.L., Tsang C.C.C., Wang M., Zheng B.-J., Chan K.-H., Yuen K.-Y. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012;86(7):3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee A., Kulcsar K., Misra V., Frieman M., Mossman K. Bats and coronaviruses. Viruses. 2019;11(1):41. doi: 10.3390/v11010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L., Liu B., Yang J., Jin Q. DBatVir: the database of bat-associated viruses. Database. 2014;2014 doi: 10.1093/database/bau021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A.R. Fehr, S. Perlman, Coronaviruses: An Overview of Their Replication and Pathogenesis, Coronaviruses: Methods and Protocols 1282 (2015) 1-23. [DOI] [PMC free article] [PubMed]
- 8.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020 doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.M. Chan-Yeung, R.H. Xu, SARS: Epidemiology, Respirology 8 (2003) S9-S14. [DOI] [PMC free article] [PubMed]
- 10.Lee J., Chowell G., Jung E. A dynamic compartmental model for the Middle East respiratory syndrome outbreak in the Republic of Korea: a retrospective analysis on control interventions and superspreading events. J. Theor. Biol. 2016;408:118–126. doi: 10.1016/j.jtbi.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan W., Zhao X., Ma X., Wang W., Niu P., Xu W., Gao G.F., Wu G.Z. A novel coronavirus genome identified in a cluster of pneumonia cases—Wuhan, China 2019–2020. China CDC Weekly. 2020;2(4):61–62. [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu D., Zhang W., Wang X., Li B., Yang J., Song X., Zhao B., Huang W., Shi R., Lu P., Niu F., Zhan X., Ma D., Wang W., Xu G., Wu G.F., Gao W., Tan C. China Novel, A Novel Coronavirus from Patients with Pneumonia in China, 2019. New Engl. J. Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.W.H. Organization, Coronavirus disease 2019 (COVID-19): situation report, (2021).
- 14.Hu B., Zeng L.-P., Yang X.-L., Ge X.-Y., Zhang W., Li B., Xie J.-Z., Shen X.-R., Zhang Y.-Z., Wang N., Luo D.-S., Zheng X.-S., Wang M.-N., Daszak P., Wang L.-F., Cui J., Shi Z.-L., Drosten C. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017;13(11) doi: 10.1371/journal.ppat.1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu R.J., Zhao X., Li J., Niu P.H., Yang B., Wu H.L., Wang W.L., Song H., Huang B.Y., Zhu N., Bi Y.H., Ma X.J., Zhan F.X., Wang L., Hu T., Zhou H., Hu Z.H., Zhou W.M., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J.Y., Xie Z.H., Ma J.M., Liu W.J., Wang D.Y., Xu W.B., Holmes E.C., Gao G.F., Wu G.Z., Chen W.J., Shi W.F., Tan W.J. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., Müller M.A., Drosten C., Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020:1–6. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 19.Bosch B.J., van der Zee R., de Haan C.A.M., Rottier P.J.M. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77(16):8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirchdoerfer R.N., Cottrell C.A., Wang N., Pallesen J., Yassine H.M., Turner H.L., Corbett K.S., Graham B.S., McLellan J.S., Ward A.B. Pre-fusion structure of a human coronavirus spike protein. Nature. 2016;531(7592):118–121. doi: 10.1038/nature17200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walls A.C., Tortorici M.A., Bosch B.-J., Frenz B., Rottier P.J.M., DiMaio F., Rey Félix.A., Veesler D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature. 2016;531(7592):114–117. doi: 10.1038/nature16988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beniac D.R., Andonov A., Grudeski E., Booth T.F. Architecture of the SARS coronavirus prefusion spike. Nat. Struct. Mol. Biol. 2006;13(8):751–752. doi: 10.1038/nsmb1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C., Tang J., Ma Y., Liang X., Yang Y., Peng G., Qi Q., Jiang S., Li J., Du L., Li F., Perlman S. Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. J. Virol. 2015;89(11):6121–6125. doi: 10.1128/JVI.00430-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B.-X., Ma G.-P., Ge J.-W., Li Y.-J. Porcine Aminopeptidase N is a functional receptor for the PEDV Coronavirus. Chinese J. Virol. 2009;25(3):220–225. [PubMed] [Google Scholar]
- 25.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279(5):3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallagher T.M., Buchmeier M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279(2):371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W., McKinnie S.M.K., Farhan M., Paul M., McDonald T., McLean B., Llorens-Cortes C., Hazra S., Murray A.G., Vederas J.C., Oudit G.Y. Angiotensin-converting enzyme 2 metabolizes and partially inactivates Pyr-Apelin-13 and apelin-17 physiological effects in the cardiovascular system. Hypertension. 2016;68(2):365–377. doi: 10.1161/HYPERTENSIONAHA.115.06892. [DOI] [PubMed] [Google Scholar]
- 28.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R.E., Acton S. A novel angiotensin converting enzyme related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87(5):E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 29.Hamming I., Timens W., Bulthuis M.L.C., Lely A.T., Navis G.J., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203(2):631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme - cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275(43):33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y., Zhao Z., Wang Y., Zhou Y., Ma Y., Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. BioRxiv. 2020 doi: 10.1164/rccm.202001-0179LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong-Poi H., Crackower M.A., Fukamizu A., Hui C.-C., Hein L., Uhlig S., Slutsky A.S., Jiang C., Penninger J.M. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobbs L.G. Pulmonary surfactant. Annu. Rev. Med. 1989;40(1):431–446. doi: 10.1146/annurev.me.40.020189.002243. [DOI] [PubMed] [Google Scholar]
- 34.Ni W., Yang X., Yang D., Bao J., Li R., Xiao Y., Hou C., Wang H., Liu J., Yang D., Xu Y., Cao Z., Gao Z. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care. 2020;24(1):1–10. doi: 10.1186/s13054-020-03120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S.X., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., Grp S.W. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 36.Yang X., Yu Y., Xu J., Shu H., Xia J.A., Liu H., Wu Y., Zhang L., Yu Z., Fang M., Yu T., Wang Y., Pan S., Zou X., Yuan S., Shang Y. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir. Med. 2020;8(5):475–481. doi: 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofmann H., Pöhlmann S. Cellular entry of the SARS coronavirus. Trends Microbiol. 2004;12(10):466–472. doi: 10.1016/j.tim.2004.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onuchic José.N., Luthey-Schulten Z., Wolynes P.G. Theory of protein folding: the energy landscape perspective. Annu. Rev. Phys. Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 39.Pain R.H. IRL Press at Oxford University Press; Oxford; New York: 1994. Mechanisms of Protein Folding. [Google Scholar]
- 40.Brooks Iii C.L. Simulations of protein folding and unfolding. Curr. Opin. Struct. Biol. 1998;8(2):222–226. doi: 10.1016/s0959-440x(98)80043-2. [DOI] [PubMed] [Google Scholar]
- 41.Duan Y., Kollman P.A. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282(5389):740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 42.Kellermayer M.S.Z., Smith S.B., Granzier H.L., Bustamante C. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 1997;276(5315):1112–1116. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- 43.Rief M., Gautel M., Oesterhelt F., Fernandez J.M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276(5315):1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 44.Tskhovrebova L., Trinick J., Sleep J.A., Simmons R.M. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 1997;387(6630):308–312. doi: 10.1038/387308a0. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H., Baker A. Recombinant human ACE2: acing out angiotensin II in ARDS therapy. Crit. Care. 2017;21 doi: 10.1186/s13054-017-1882-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang R., Pan Y., Fanelli V., Wu S., Luo A.A., Islam D., Han B., Mao P., Ghazarian M., Zeng W., Spieth P.M., Wang D., Khang J., Mo H., Liu X., Uhlig S., Liu M., Laffey J., Slutsky A.S., Li Y., Zhang H. Mechanical stress and the induction of lung fibrosis via the midkine signaling pathway. Am. J. Respir. Crit. Care Med. 2015;192(3):315–323. doi: 10.1164/rccm.201412-2326OC. [DOI] [PubMed] [Google Scholar]
- 47.Faisal H.M.N., Katti K.S., Katti D.R. An insight into quartz mineral interactions with kerogen in Green River oil shale. Int. J. Coal Geol. 2021;238:103729. [Google Scholar]
- 48.Faisal H.M.N., Katti K.S., Katti D.R. Modeling the behavior of organic kerogen in the proximity of calcite mineral by molecular dynamics simulations. Energy Fuels. 2020;34(3):2849–2860. [Google Scholar]
- 49.D.R. Katti, K.S. Katti, K. Thapa, N. Faisal, Modeling the Nanoscale Kerogen Inclusions in Green River Oil Shale, Poromechanics VI, pp. 1968-1975.
- 50.Faisal H.M.N., Katti K.S., Katti D.R. Molecular mechanics of the swelling clay tactoid under compression, tension and shear. Appl. Clay Sci. 2021;200 [Google Scholar]
- 51.Katti D.R., Thapa K.B., Faisal H.M.N., Katti K. International Conference of the International Association for Computer Methods and Advances in Geomechanics. Springer; 2021. Molecular origin of compressibility and shear strength of swelling clays; pp. 641–647. [Google Scholar]
- 52.Jaswandkar S.V., Faisal H.M.N., Katti K.S., Katti D.R. Dissociation mechanisms of G-actin subunits govern deformation response of actin filament. Biomacromolecules. 2021;22(2):907–917. doi: 10.1021/acs.biomac.0c01602. [DOI] [PubMed] [Google Scholar]
- 53.Faisal H.M.N., Katti K.S., Katti D.R. Differences in interactions within viral replication complexes of SARS-CoV-2 (COVID-19) and SARS-CoV coronaviruses control RNA replication ability. Jom. 2021;73(6):1684–1695. doi: 10.1007/s11837-021-04662-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Isralewitz B., Baudry J., Gullingsrud J., Kosztin D., Schulten K. Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model. 2001;19(1):13–25. doi: 10.1016/s1093-3263(00)00133-9. [DOI] [PubMed] [Google Scholar]
- 55.Binnig G., Quate C.F., Gerber C. Atomic force microscope. Phys. Rev. Lett. 1986;56(9):930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 56.Svoboda K., Block S.M. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 1994;23(1):247–285. doi: 10.1146/annurev.bb.23.060194.001335. [DOI] [PubMed] [Google Scholar]
- 57.Li F., Li W.H., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 58.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., MacKerell A.D. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Payne M.C., Teter M.P., Allan D.C., Arias T.A., Joannopoulos J.D. Iterative minimization techniques for abinitio total-energy calculations - molecular-dynamics and conjugate gradients. Rev. Mod. Phys. 1992;64(4):1045–1097. [Google Scholar]
- 62.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., Dales N.A., Patane M.A., Pantoliano M.W. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279(17):17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Izrailev S., Stepaniants S., Isralewitz B., Kosztin D., Lu H., Molnar F., Wriggers W., Schulten K. In: Computational Molecular Dynamics: Challenges. Deuflhard P., Hermans J., Leimkuhler B., Mark A.E., Reich S., Skeel R.D., editors. Methods; Ideas, Springer, Berlin Heidelberg, Berlin, Heidelberg: 1999. Steered Molecular Dynamics; pp. 39–65. [Google Scholar]
- 64.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kirchdoerfer R.N., Wang N., Pallesen J., Wrapp D., Turner H.L., Cottrell C.A., Corbett K.S., Graham B.S., McLellan J.S., Ward A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-34171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geng Y.-J., Wei Z.-Y., Qian H.-Y., Huang J., Lodato R., Castriotta R.J. Pathophysiological characteristics and therapeutic approaches for pulmonary injury and cardiovascular complications of coronavirus disease 2019. Cardiovasc. Pathol. 2020;47 doi: 10.1016/j.carpath.2020.107228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.