

Abstract
The COVID‐19 (coronavirus disease) global pandemic, caused by the spread of the SARS‐CoV‐2 (severe acute respiratory syndrome coronavirus 2) virus, currently has limited treatment options which include vaccines, anti‐virals, and repurposed therapeutics. With their high specificity, tunability, and biocompatibility, small molecules like peptides are positioned to act as key players in combating SARS‐CoV‐2, and can be readily modified to match viral mutation rate. A recent expansion of the understanding of the viral structure and entry mechanisms has led to the proliferation of therapeutic viral entry inhibitors. In this comprehensive review, inhibitors of SARS and SARS‐CoV‐2 are investigated and discussed based on therapeutic design, inhibitory mechanistic approaches, and common targets. Peptide therapeutics are highlighted, which have demonstrated in vitro or in vivo efficacy, discuss advantages of peptide therapeutics, and common strategies in identifying targets for viral inhibition.
Keywords: coronavirus, peptide therapeutics, SARS‐CoV, SARS‐CoV‐2, SARS‐CoV‐2 mutants
SARS‐CoV‐2 has rapidly spread across the globe, impacting millions. With its rise, the design of peptide inhibitors has expanded swiftly, and in the last 18 months many groups have worked to identify both new viral targets for inhibition and potential therapeutics. Here much of the recent work in the field is covered, and recent computational and experimental data are consolidated.
1. Introduction: From SARS to SARS‐CoV‐2
COVID‐19 (coronavirus disease) was identified as a global pandemic by the World Health Organization on March 11, 2020,[ 1 ] and is caused by viral infection of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2).[ 2 ] The first recorded cases originated in Wuhan, Hubei Province, China in December 2019.[ 3 ] From there, global travel and commerce allowed for rapid global transmission of the virus.
SARS‐CoV‐2 is the most recent of the seven known HCoV (human coronaviruses). Severe Acute Respiratory Syndrome (SARS) is caused by the viral agent SARS‐CoV. Emerging from China in late 2002,[ 4 , 5 ] the SARS pandemic saw 8000 infections and 800 deaths.[ 6 ] Many infections resulted from super‐spreading events in hospitals, and the pandemic was considered resolved in July 2003[ 6 ] through rigorous public health measures, though there were a few isolated breakouts into 2004.[ 7 ] Nine years later, a new widespread coronavirus was identified: Middle Eastern Respiratory Syndrome (MERS).[ 8 ] Unlike SARS‐CoV, MERS circulates to this day, as it does not spread rapidly between humans.[ 9 , 10 ]
The most recent coronavirus, SARS‐CoV‐2, spreads more quickly with higher infectivity than the other coronaviruses, partially because of its higher binding affinity to angiotensin‐converting enzyme‐2 (ACE‐2).[ 11 ] At the point of this publication, we have reached over one year past identification of the index patient. Of all the human coronaviruses, SARS‐CoV‐2 is most genetically similar to SARS‐CoV (79.6%),[ 12 ] though it has a distinct phylogenetic lineage (Table 1 ). The closest known genetic relative is a bat coronavirus (BatCoV RaTG13), with a genome sequence identity of 96.2%.[ 12 ] Despite this knowledge, there have been significant on‐going challenges in fighting newly emerging strains, some of which have notably higher rates of human transmission.[ 13 , 14 ] In addition to viral spread in hospitals and care homes, which are prevalent among all three pandemic HCoV, a high incidence of asymptomatic and pre‐symptomatic[ 15 ] SARS‐CoV‐2 infection drastically increases community spread, as individuals transmit the virus before learning they are symptomatic (if they ever do).[ 16 , 17 , 18 , 19 ]
Table 1.
Comparison of three common and lethal coronaviruses
| SARS‐CoV | MERS | SARS‐CoV‐2 | |
|---|---|---|---|
| Genus | Clade I, lineage B | Clade II, lineage C | Clade I, lineage B |
| Binding receptor | ACE‐2 | Dipeptidyl peptidase 4 | ACE‐2 |
| First identified case date; location | 16 November 2002; Guangdong, China[ 20 ] | 13 June 2012; Jeddah, Saudi Arabia[ 21 ] | 7 December 2019; Wuhan, China[ 22 ] |
| Number of countries affected | 29[ 23 ] | 27[ 24 ] | 216[ 25 ] |
| ICU hospitalization rate | ≈20%[ 26 ] | ≈2.5%[ 27 ] | ≈2%[ 28 ] |
1.1. Physiological Effects of SARS‐CoV‐2
Due to the widespread presence of the ACE‐2 receptor throughout the human body, evidence shows that SARS‐CoV‐2 can impact the respiratory, cardiovascular, gastrointestinal, urinary, oral, pancreatic, and neurological systems.[ 29 ] The presence of the ACE‐2 receptor throughout the human body, along with clinical evidence, suggests SARS‐CoV‐2 has damaging systemic effects.[ 30 ] SARS‐CoV‐2 patients, in addition to presenting with chest pain and dyspnea, can have dysrhythmia and acute left ventricular dysfunction.[ 31 ] For patients with pre‐existing myocarditis and myocardial injury, SARS‐CoV‐2 involves the renin‐angiotensin‐aldosterone system,[ 32 ] thus greatly increasing mortality rate. Even without pre‐existing cardiovascular conditions, patient electrocardiograms (ECG) display abnormalities which resemble acute coronary syndrome.[ 31 ] Additionally, acute heart failure could also be the first symptom that patients present.[ 33 ] The systemic inflammation during infection poses an increased risk for patients with cardiovascular disease, as inflammation of blood vessels can free plaque and create an embolus,[ 34 ] which could then induce acute myocardial infarction, stroke, or clot formation elsewhere in the body.[ 31 , 35 ] The kidneys, while not necessarily directly affected by the disease, are at risk from accumulation injury, hypoxia, clot, and cytokine storm.[ 36 , 37 , 38 , 39 ] SARS‐CoV‐2 is expressed in the gastrointestinal (GI) tract, allowing it to replicate in the human small intestine and shed into human stool,[ 40 , 41 , 42 , 43 ] potentially facilitating fecal‐oral transmission.[ 44 ] In addition, replication of SARS‐CoV‐2 can cause GI damage through ischemic enteritis with irregular necrosis and fibrin thrombus in the arterioles,[ 45 ] as well as cholestasis and small vessel thrombosis. Finally, the liver is at risk due to the hepatotoxicity of the drugs used for treatment, inflammation from pneumonia, cytokine storm, and systemic hypoxic conditions which create reactive oxygen species,[ 46 , 47 , 48 ] all of which can cause moderate to severe liver damage.
1.2. Understanding the Structure‐Virality Bases of SARS‐CoV‐2
Coronaviruses are single stranded, positive sense RNA viruses ranging between 80–220 nm in diameter.[ 49 ] The viral envelope (E) is studded with the Spike glycoproteins (S) that give the members of Coronaviridae family their characteristic appearance.[ 49 ] Hemagglutinin‐esteraste (HE) is also present in the membrane of some betacoronaviridae.[ 50 ] Also embedded within the envelope are the transmembrane non‐glycosylated protein (M) and the envelope protein (E) (Figure 1 ). Within the phospholipid bilayers of the membrane is the nucleocapsid itself, consisting of single positive strand RNA wrapped with nucleocapsid protein (N).[ 49 ] The enclosed RNA acts as both viral genome and messenger RNA once the virus has been taken up by the host cell, allowing for direct translation of viral RNA into protein by the host cell ribosomes.[ 50 ] As with all single positive strand RNA viruses, the RNA encodes for the RNA‐dependent RNA polymerase (RdRP) necessary for replication and protein transcription.[ 50 ] The four structural proteins (N, E, M, and S) are typically highly conserved among all coronaviruses, as is the RdRP encoding region.[ 51 ] Spike protein binds to ACE‐2 (Figure 2 ), mediating receptor binding and membrane fusion through use of the S1 and S2 subunits[ 52 ] on the receptor binding domain (RBD). Following binding, the S2 subunit undergoes a conformational change[ 53 ] and two heptad‐repeat domains (HR1 and HR2) of the S2 subunit (Figure 3 ) mediate key membrane fusion and entry between the virus and the host cell.[ 49 , 50 , 52 , 54 , 67 ] These combined features confer a high proclivity for human infection and successful viral spreading.[ 52 ] Despite some differences between the structure‐virality of SARS‐CoV and SARS‐CoV‐2, they share important genetic and binding similarities[ 55 , 56 ] which may allow for the identification of inhibitors active against both. The structure virality of SARS‐CoV has been well‐described[ 57 , 58 , 59 , 60 , 61 , 62 ] and in‐depth comparisons have been made between it and other coronaviruses such as SARS‐CoV‐2.[ 22 , 25 , 55 , 56 ]
Figure 1.
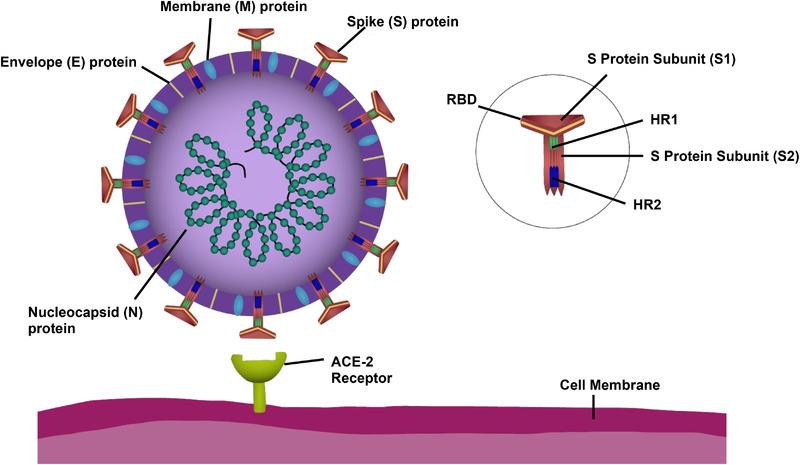
Components of the SARS‐CoV‐2 virus and the host‐cell binding target ACE‐2 receptor. The envelope (E) protein, membrane (M) protein, nucleocapsid (N) protein, and Spike (S) protein are the key structural proteins of SARS‐CoV‐2. The structural proteins (N, E, M, and S) are highly conserved within the family Coronaviridae. The single positive strand nature of SARS‐CoV‐2 and its family members allows for rapid transcription of its RNA and infection of neighboring cells. The receptor binding domain (RBD) of the S protein is made of the S1 and S2 subunits. S2 is further divided into two heptad repeat regions, HR1 and HR2. S2 is essential for viral fusion and entry into the host cell.
Figure 2.
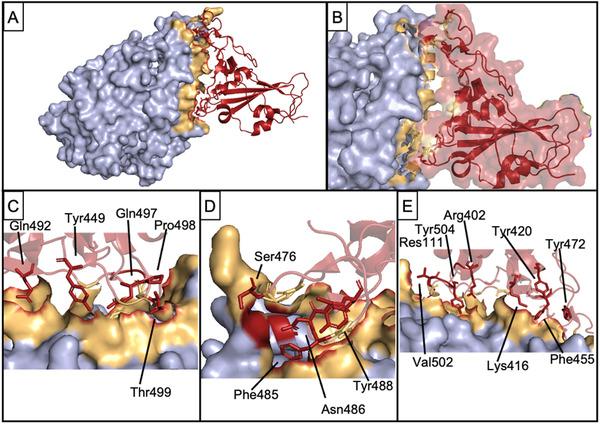
Interaction of Spike RBD and ACE‐2. A) Bound complex between ACE‐2 (light blue) and Spike RBD (red). Tan shows PPI interface on ACE‐2. B) Close‐up view of the interaction interface. C–E) indicate and label crucial residues from Spike RBD which contribute to complex formation. PDB 7DMU.
Figure 3.
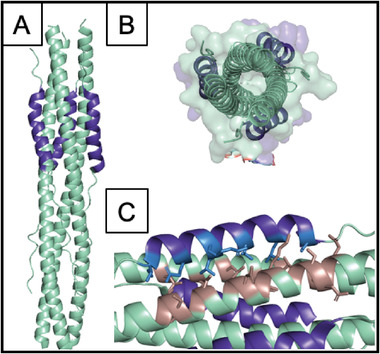
Structure of the HR1‐HR2 trimeric fusion core. A) Cartoon representation showing HR1 in cyan and HR2 in dark blue. B) Top‐down view. C) Key interacting residues between HR2 (side chains shown in light blue) and HR1 (side chains shown in tan). PDB 6LXT.
1.3. Viral Fusion Proteins
Viral fusion proteins are, as a whole, highly conserved from species to species. In the years since the last two HCoV pandemic outbreaks, bats have been widely accepted as the viral reservoir host for pathogenic‐capable coronaviruses.[ 63 ] While this is primarily attributed to the heterogeneity of the bat ACE‐2 receptor and rigorous immune system,[ 64 , 65 ] the homogeneity of human ACE‐2 receptors cannot be ignored, especially as the combination of the Bat‐SCoV‐Spike protein with a human RBD has produced a highly chimeric Spike protein with a high ACE‐2 to RBD affinity.[ 66 ] As for the Spike protein, the target cells and come in close contact to the host cell membrane to allow viral‐cell fusion, integral in the delivery of the viral genome. Such viral fusion proteins are divided into 3 classes based on protein structure.[ 67 ] Our interest lies in the Class I fusion proteins, characterized by α‐helical trimers which consist of a metastable trimer protein (the RBD) and the fusion subunit (which extends into the host cell membrane).[ 68 ] In the case Coronaviridae, the Spike protein is trimeric and S2 is the fusion subunit. After fusion to the cell membrane, endocytosis occurs and the viral membrane fuses to the endosomal membrane.[ 50 ] Viral transcription and replication are mediated much the same as for most single positive strand RNA virus. RdRP is directly translated from the viral genome, and the complementary strand is synthesized.[ 49 ] Aside from RdRP, all other enzymes and proteases are recruited from the host cell itself.
2. An Overview of Current Non‐Peptide Strategies for SARS‐CoV‐2
There are currently 30 vaccines for SARS‐CoV‐2 in various developmental stages.[ 69 ] Out of the 30 developed and developing vaccines, 6 are RNA‐based[ 61 ] and 2 have been Food and Drug Administration‐ (FDA) approved: the mRNA‐1273 (Moderna/US National Institutes of Allergy and Infectious Diesase (NIAID)) and BNT162b1/BNT162b2 (Pfizer Inc./BioNTech) vaccines.[ 70 ] The mRNA‐1273 was developed by Moderna and the NIAID Vaccine Research Center.[ 71 ] For the mRNA‐1273, a lipid nanoparticle is used for the delivery of transcripts coding for the Spike protein of SARS‐CoV‐2.[ 72 ] Compared to placebo, those who received mRNA‐1273 had a 94.1% lower symptomatic COVID‐19 incidence in large Phase 3 clinical trials. After two doses (both for the placebo and vaccine group) were administered to 30 420 participants, 196 cases of symptomatic COVID‐19 were reported: 185 in the placebo‐receiving group and 11 in the vaccine‐receiving group.[ 73 ]
The BNT162b1 and vaccines developed by Pfizer Inc. and BioNTech employ polyethylene glycol (PEG) lipid nanoparticles[ 71 ] containing RNA coding for either SARS‐CoV‐2 RBD antigens (BNT162b1) or SARS‐CoV‐2 Spike Protein antigens (BNT162b2).[ 74 ] Phase 3 clinical trials of these vaccine candidates showed that out of 36 523 participants, 170 participants became infected with COVID‐19 after receiving a second dose. Of these, 162 participants received the placebo and 8 participants received the vaccine, corresponding to a 95% efficacy rate.[ 75 ]
While they have shown good efficacy, RNA vaccines have inherently low stability, relatively short shelf‐life and often require specialized equipment for their storage or transport.[ 76 ] Both FDA‐approved vaccines have low stability and require storage in freezing temperatures, making distribution challenging. As a reference, Pfizer's vaccine requires storage at −70 °C and can be refrigerated for no longer than 5 days, and Moderna's vaccine is stored at −20 °C and can only be refrigerated for 30 days.[ 77 , 78 ] While synthetic peptides also have a low stability,[ 79 ] modifications which confer hierarchical self‐assembly can significantly increase their stability at room temperature, reducing the need for constant refrigeration from production to clinical use.[ 80 , 81 ]
New variants which have arisen present a number of mutations, some in the RBD, which potentially may lower a vaccines’ ability to confer immunity against such newer variants. The SARS‐CoV‐2 virus has many conserved residues with the preceding SARS‐CoV virus,[ 82 , 83 , 84 , 85 ] facilitating a reduced development time for peptide therapeutics specific to SARS‐CoV‐2. In one report, Xia et al. successfully used a peptide inhibitor developed for SARS‐CoV, EK1, to make and test a successful SARS‐CoV‐2 inhibitor merely weeks after the first COVID‐19 outbreak, aided by sequence alignment of the two viral genomes. This homology‐based method can potentially expedite the development of peptide inhibitors for new or variant strains.[ 84 ] Increased recent interest in peptide inhibitors has shortened their development time in comparison to the longer timelines necessary for vaccines; rapid development of stable and effective therapeutics will be crucial to match the rapid mutation rate and emergence of new SARS‐CoV‐2 variants.
While vaccines show great promise, they are not without potential pitfalls. Several vaccines have slowed or entirely halted their production, including the V590 and V591 vaccine candidates developed by Merck.[ 86 , 87 ] These candidates have been discontinued, while on‐going research is now focusing on two therapeutic alternatives (MK‐4482 and MK‐7110), which aim to reduce the inflammatory response to SARS‐CoV‐2.[ 88 ]
2.1. Selected SARS‐CoV‐2 Therapies
Several small molecules have been repurposed, approved, and/or clinically tested in humans. Remdesivir completed clinical trials in June of 2020[ 89 ] and functions by inhibiting RNA polymerase, efficiently prematurely halting COVID‐19 RNA replication.[ 90 ] The drug displays good in vitro promise as a therapeutic candidate for SARS‐CoV and MERS. A double‐blind, randomized, placebo‐controlled trial was conducted to evaluate its efficacy in hospitalized COVID‐19 patients, and showed reduced recovery time and clinical improvements by day 15 as compared to treatment with the placebo.[ 90 ] While a larger scale European study found the differences to less significant, the United States FDA approved Remdesivir for use in late October of 2020. Dexamethasone is an artificial corticosteroid (binding to nuclear steroid receptors) that mimics the anti‐inflammatory properties of a natural hormone produced by the adrenal gland.[ 91 , 92 ] This drug was developed as part of the Randomized Evaluation of COVID‐19 Therapy (RECOVERY) trial in the United Kingdom.[ 93 ] Currently monoclonal antibodies (Table 2 ) are the method of treatment rather than repurposed drugs, and are having significant success in treatment over their repurposed counterparts.
Table 2.
Leading non‐peptide therapeutics undergoing trials for SARS‐CoV‐2
| Therapeutic name | Biological mechanism | Outcome |
|---|---|---|
| Remdesivir | Repurposed RdRP inhibitor for Ebola and Marburg virus |
|
| Ebastine | Repurposed second generation antihistamine H1 agonist | Inhibits T‐cell activation and migration (specifically TNF‐α and GM‐CSF). |
| Danoprevir a) | Repurposed chronic Hepatitis C Protease (NS3/4A) inhibitor | Inhibits chymotrypsin‐like proteases (essential in viral replication) |
| Casirivimab/Imdevimab | Experimental SARS‐CoV‐2 monoclonal antibody |
|
| Bamlanivimab/Etesevimab | Experimental SARS‐CoV‐2 monoclonal antibody |
|
in conjunction with Ritonavir.
3. Advantages of Peptides as Therapeutics
Peptide therapeutics (typically consisting of <5–100 amino acids) display numerous advantages over traditional small molecule drugs and large biologics for treatment against SARS‐CoV‐2. Peptides have unique properties which make them valuable as SARS‐CoV‐2 therapeutics: high specificity and affinity, low toxicity, delivery by multiple routes, low immunogenic activity, short development time, and good long‐term stability under mild storage conditions.[ 94 , 95 , 96 , 97 , 98 , 99 ] These types of therapeutics have gained increasing traction in the clinic, with nearly 63 FDA‐approved peptides (all <100 residues) currently on market,[ 100 ] and more than 400 peptides in global development.[ 95 ] This shift is due in part to the ease, low cost, speed, and modularity of solid‐phase peptide synthesis, which can be performed both automatically and combinatorially.[ 101 , 102 ] Peptide therapeutics are quicker to develop, validate, and approve.[ 103 ] Furthermore, predicting biologically relevant human doses of peptides is generally easier[ 104 ] and more accurate[ 105 , 106 ] than for new small molecule drugs.
Characteristics of peptide therapeutics enable highly specific interactions with residues on target proteins, including a long, flexible backbone that enables optimal positioning of amino acids.[ 95 , 103 ] Protein‐protein interactions (PPIs) tend to occur over large contact surfaces (≈1500–3000 Å2). The contact area between a small molecule therapeutic and its target protein, however, is much smaller (≈300–1000 Å2), a key drawback when using small molecules to target PPIs.[ 107 ] Small molecules typically target deep pockets in folded proteins and are much less effective at binding to the larger, greasier, and flatter surfaces commonly found in PPI interfaces.[ 107 , 108 , 109 , 110 , 111 ] Compounds that bind PPIs, therefore, have historically tended to be more hydrophobic and have higher molecular weights than traditional drugs.[ 107 ] Additionally, single mutations on target sites are more likely to render small molecule drugs non‐functional compared to larger peptides.[ 112 ]
The rapid clearance, low circulation time, and proteolytic susceptibility of peptides do confer an inherently low half‐life for these drugs, though these attributes also prevent their accumulation in tissues, and peptides can be chemically modified to improve stability such as through polyethylene glycol (PEG)‐ylation or adding domains which promote hierarchical self‐assembly.[ 113 , 114 , 115 ] As biologically‐derived therapeutics, peptides are metabolized into benign constituents, avoiding complications of hepatic and renal accumulation typically seen with small molecule drugs, like the production of toxic metabolites and drug–drug interactions.[ 114 , 115 ] These are significant barriers that delay the deployment of novel small molecule drugs. Furthermore, the low toxicity of peptide therapeutics permits safe administration to patients with seriously impaired renal function, which is especially relevant given that renal failure is frequently observed in SARS‐CoV‐2 patients and associated with a greater frequency of complications and in‐hospital mortality.[ 116 , 117 ]
While peptide therapeutics are generally administered through the parenteral route (which has the added benefit of ensuring maximum patient compliance[ 118 ]), alternative technologies allow for the targeted delivery of peptides through oral, transdermal, and intranasal routes.[ 116 ] The intranasal route is especially attractive for delivering therapeutic peptides against SARS‐CoV‐2 due to the massive viral localization within the respiratory tract. Peptides are less likely to prompt a fatal immune response, yet repeatedly demonstrate the ability to induce robust immunity against epitopes associated with disease.[ 111 , 119 , 120 ]
3.1. Rational Design of Peptide Therapeutics
Suitable targets for SARS‐CoV[ 121 , 122 , 123 , 124 , 125 , 126 , 127 ] and SARS‐CoV‐2[ 128 , 129 , 130 , 131 , 132 , 133 , 134 , 135 , 136 , 137 ] peptide therapeutics have been established previously through many disparate methods, many of them through computational or homology studies.
In brief: In silico analysis makes use of molecular simulation programs that employ scoring functions to calculate binding affinity energies of protein–protein interactions, predict and score stable complexes, and determine key interactions which occur at the contact surface of said complex.[ 138 ] Inputs for these programs include experimentally determined receptor and ligand PDB files or files containing an interacting protein–protein complex, though when data is not available for the proteins of interest, protein models can be generated with programs such as I‐TASSER, SWISS‐MODEL, or Rosetta.[ 139 , 140 ] Broadly speaking, there are two classes of protein modelling programs: homology modelling and ab initio modelling, both of which have been used to develop SARS‐CoV‐2 peptide therapeutics. The former predicts protein structures by determining regions of homology for sequences for which the structure is known on the PDB database, giving final structures formed by stitching together distinct regions of homology. This method has typically been used to model Spike peptide binders by themselves since there are several experimentally derived structures of the ACE‐2 receptor now available.[ 137 , 141 ] In one report by Sitthiyotha and Chunsrivirot, Rosetta was used to improve the binding affinity of a short RBD PPI scaffold derived from ACE‐2 (PDB:6M0J).[ 141 ] Molecular dynamics (MD) simulations (AMBER) were used to confirm the predicted improved binding affinity, and in their design the authors sought to avoid mutating residues responsible for stabilizing interactions between the ACE‐2 helix and the SARS‐CoV‐2 RBD (in the end selecting only I21, E23, F28, L39, and L45 as designable residues, as these residues had not been previously implicated in RBD binding).[ 141 ] While this publication was one of the earlier reports for PPIs against SARS‐CoV‐2, many homology‐based designs have now been published to identify peptide therapeutics targeting the RBD.[ 141 ]
Xia et al. used a similar approach in developing a pan‐coronavirus fusion inhibitor which could potentially act as a broad‐spectrum anti‐HCoV drug for newly emerging coronaviruses. In their report, authors noted high sequence homology across HCoV viruses which gives rise to a conserved hydrophobic face on the HR1 region of the S2 subunit, important in the formation of a 6‐HB and mediating initial host‐viral membrane fusion. HR1 homology modelling was achieved through SWISS‐MODEL and PPI:HR1 complexes were further energy minimized to perform MD simulation, ultimately giving rise to peptide inhibitors with activity in the high nm range and low immunogenicity.[ 141 ]
Ab initio modelling relies on the intrinsic properties of the sequence, such as the formation of secondary structure and long‐distance interactions, to identify possible structures. These methods tend to be more effective for robust scaffolds or smaller peptides with a few, simple features and their accuracy can be increased with deep neural network training and application. Approaches like this are particularly effective for small peptides and may have expanded applicability in the development of helical binder libraries for high‐throughput computational and/or in vitro screening, as successfully demonstrated by Cao et. al.[ 173 ] In their report, α helical PPIs were developed, building both from the known ACE‐2 binding site on RBD as well as designed PPIs which identified new binding modes to RBD. This methodology gave rise to ten new inhibitors with excellent binding affinity (100 pm–10 nm), which successfully prevented SARS‐CoV‐2 infection in vitro (IC50 values between 24 pm–35 nm).[ 173 ]
Over the past year several experimentally determined structures of the Spike RBD‐ACE‐2 complex have become available for SARS‐CoV‐2[ 142 , 143 ] and for its homologs in the SARS‐CoV (Figure 4 ) and MERS‐CoV proteomes; this has become a popular therapeutic target (due to its extracellular and opsonizable localization), and has allowed the derivation and testing of inhibitory peptides from fragments of ACE‐2. To date, homology modelling has proven the most feasible and accurate approach given the preponderance of available structures for the Spike RBD‐ACE‐2 interaction and related homologs.
Figure 4.
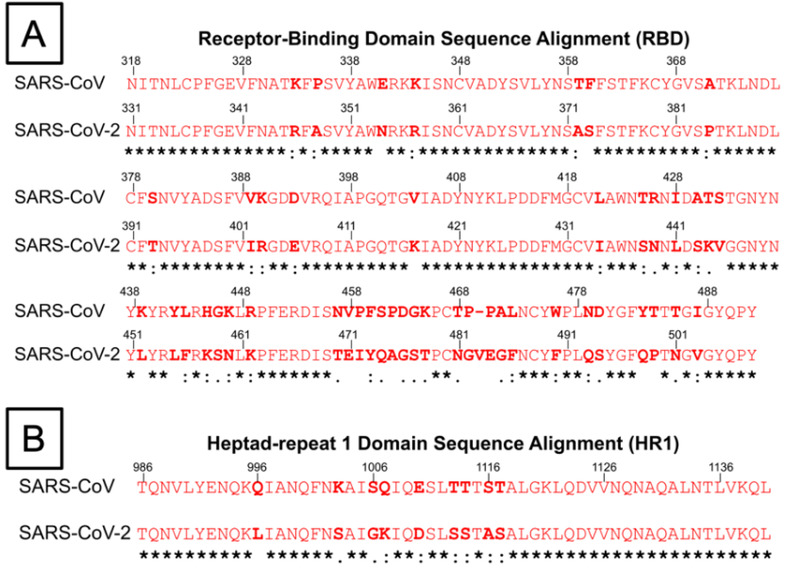
Amino acid sequence alignment of the receptor‐binding domain and heptad repeat 1 (HR1) domain of both SARS‐CoV and SARS‐CoV‐2 virus. Conserved residues between both viruses are marked with asterisks (*), residues with similar properties are marked with a colon (:), while residues with only marginally similar properties are marked with a period (.). A) Amino acid sequence alignment of RBD of SARS‐CoV and SARS‐CoV‐2. The several residue changes in the SARS‐CoV‐2 RBD in comparison to SARS‐CoV allow for higher binding affinity between RBD and ACE‐2 at the RBD–ACE‐2 interface. B) Amino acid sequence alignment of HR1 domains of SARS‐CoV and SARS‐CoV‐2. The residue changes marked within the HR1 domain prompt study into differences in the interactions between HR1 and HR2 domains, which affect 6‐HB formation.
Once a suitable template and model have been obtained they can be evaluated for binding affinity to the target RBD sequence using molecular docking algorithms, which cluster thousands of docked complexes to find the most stable conformation and consider user restraints, steric hindrance, van der Waals energies, electrostatic energies, and de‐solvation energies.[ 144 ] Users can set various restraints, including active and passive residues, contact distances, simulation temperatures, dihedral and hydrogen bond restraints, and residual dipolar couplings to parameterize the conformations which are tested.[ 145 ] Proteins can also be docked with a rigid or flexible backbone depending on application.[ 145 ] Once the molecular docking is complete, protein–protein complex conformations can be evaluated based on the respective scoring function output, as well as the various energies (van der Waals, electrostatic, restraints violation) and error estimates. Molecular docking methods allowed for the identification and development of SARS‐CoV‐2 peptide therapeutics such as HR2 and GST‐removed HR2 peptides as studied by Chu et al. and CP‐1 peptide docked by GRAMM and studied by Liu et al.[ 146 , 147 ]
Molecular dynamics simulations allow for evaluation of the molecular motions of the protein–protein complex to assess stability, solvent and ion effects via equilibration of the protein–protein–solvent–ion system with thermodynamic, steric, and electrostatic force fields. Some molecular simulation programs perform molecular dynamics on macromolecular complexes, which tests the stability of the atoms in a solvated complex with a force field over a period of nanoseconds to microseconds.[ 148 ] A major application of this, especially common with Spike RBD inhibitory peptide discovery, is to iteratively truncate the region of the receptor that makes up the contact surface target protein (in this case Spike) and perform MD simulation with the target protein to test the stability overtime to determine the shortest efficacious sequence. Many studies have taken this approach, and from it the α‐1 helix from ACE‐2 was identified as a stable, peptide inhibitor.[ 149 , 150 ]
Commonly used MD programs such as AMBER and GROMACS[ 151 , 152 ] can calculate values such as RMSD, the stability of individual residues, and the binding energy of a complex, to generate original peptides such as SBP1 and SBP2; follow‐ up studies rationally mutated regions of these peptides derived from ACE‐2 to produce more stable complexes with SARS‐CoV‐2, some establishing novel binding modes as mentioned above.[ 153 ]
Following MD simulations, the importance of any specific residue in peptide conformation, stability, or binding affinity can be evaluated using alanine scanning. Alanine scanning provides a unique advantage of investigating the contribution of each residue toward the binding affinity energy and function of the sequence of interest by using an input protein‐protein complex and mutating each residue to an alanine to quantify changes in binding affinity.[ 128 , 154 ] Alanine scans give a suitable starting point for the discovery of more stable peptides as it can pinpoint the energetic effects of a given residue substitution. Some molecular modeling suites and stand‐alone tools with this algorithm include FoldX, Rosetta, and BUDE Alanine Scan.[ 140 , 155 , 156 ] Favorable mutations can be made on the peptide inhibitor and retested with further alanine scanning and MD simulation until a satisfactory stable inhibitory peptide is found. This type of method was employed in a study which took the α‐1 helix from ACE‐2 and performed alanine scanning to find the most important residues, truncating sections containing the least important residues to make a shorter and more stable peptide for the inhibition of Spike RBD.[ 128 ] Removal of 5 N‐terminal residues generated a new 23mer which was modeled and energy minimized in Chimera, docked in PyDock, HADDOCK, and ZDOCK and then MD simulations performed and analyzed in GROMACS.[ 128 ] This type of analysis can also be used to compare potential PPI interactions or the relative stability of bound complexes. Alanine scanning mutagenesis was used to determine “hotspots” at PPI interfaces, and revealed mutations in SARS‐CoV‐2 relative to SARS‐CoV which confer stronger binding affinity to the ACE‐2 receptor.[ 157 ] An alternative approach utilizes known structural information and residues, which contribute significantly to protein–protein interactions via their side chains—selective mutagenesis in Rosetta at these sites generates a branching tree of targets to be evaluated using MM‐GBSA or other molecular dynamics (MD) methods.[ 137 ] Diverse methods exist for the quantification of a residue's contribution to binding affinity, but other factors such as steric constraints and various energies must also be considered. Iterative molecular dynamics simulations can be applied to resolve the latter issues and other tools such as PRODIGY or PIC can be used to quantify the significance of a particular substitution on the strength of the inhibitor–RBD interaction, as was performed by Sitthiyotha and Chunsrivirot in Rosetta.
Key methods of identifying potential therapeutic targets on SARS‐CoV‐2 commonly include homology studies to previous BCVs, particularly the SARS‐CoV and MERS‐CoV. A particular area of interest in SARS‐CoV‐2 is the Spike protein. To establish and quantify homology, phylogenetic alignment analysis of the various coronavirus clades[ 158 ] is used in combination with published sequence and structural comparisons.[ 141 , 147 , 159 ] Determination of the functional domains of SARS‐CoV‐2 through amino acid alignment with SARS‐CoV has revealed key interactions between HR1 and HR2, providing more specific inhibitory targets.[ 83 , 84 ] Many different in silico techniques, especially sequence alignment and homology‐based protein modeling via SWISS‐MODEL, have helped to identify key methods, models, and potential inhibitory peptide sequences through study of Spike:ACE‐2 interactions.[ 133 , 160 ] In addition, the Wimley and White (WW) interfacial hydrophobicity scale is used to determine lipophilic regions of a potential drug target and was used to identify potential drug targets in the S2 subunit of Spike protein in SARS‐CoV and murine hepatitis virus (MHV).[ 161 ] Crystallography of HR1 and HR2 binding regions with subsequent model construction has yielded targets through structural comparison between SARS‐CoV and MERS‐CoV.[ 162 , 163 , 164 ]
4. SARS‐CoV and SARS‐CoV‐2 Peptide Therapeutics
4.1. Binding Inhibition by Targeting Host Cell ACE‐2 Interaction with Spike Protein RBD
SARS‐CoV and SARS‐CoV‐2 are part of the Betacoronaviridae family.[ 165 ] For both, the Spike protein is classified as a Class I fusion protein with its trimeric α‐helical structure and its use of subunits S1 and S2, which aid in viral attachment and membrane fusion.[ 52 , 67 , 68 ] The first stage of infection with both SARS‐CoV and SARS‐CoV‐2 arises from the PPI between viral Spike protein and the host ACE‐2 receptor, specifically via the RBD present on the S1 subunit (Figure 2A,B).[ 52 , 166 ] After binding, the HR1 and HR2 domains present on the S2 subunit undergo important conformational changes followed by complexation, allowing fusion of the viral and host cell membranes.[ 67 , 68 , 163 ] Circumventing these events by blocking their corresponding PPIs has proven effective in inhibiting SARS‐CoV and SARS‐CoV‐2 entry.
4.2. Peptide Inhibitors Targeting the RBD
Peptides which target the RBD have been identified by a variety of experimental and computational methods. These approaches, however, are not mutually exclusive—computational methods are commonly used to inform mutagenesis or modification of existing peptides for synthesis of novel peptides for use in in vitro or in vivo testing. Numerous peptides derived from the ACE‐2 α‐helix involved in the RBD‐ACE‐2 interaction have undergone modification based on surface‐exposed residues at the contact interface between the two proteins (Figure 2C–E). The RBDs of SARS‐CoV and MERS S‐protein fluctuate between two distinct conformational states, standing and lying (Figure 5 ).[ 167 , 168 ] Lying RBDs are incapable of binding to ACE‐2 and are consequently believed to largely evade host immunity, while RBDs in the standing state bind to ACE‐2.[ 167 ] Similar conformational flipping has also been observed in the Spike RBD of SARS‐CoV‐2.[ 168 , 169 ] Targeting the standing conformation may provide a novel approach for the design of antiviral peptides due to the exposure of the flexible RBD and distal HR1 loop, and has already been proposed as a mechanism of action for anti‐Spike SARS‐CoV and MERS antibodies.[ 167 ] The recent emergence of a SARS‐CoV‐2 variant with the Spike protein amino acid sequence mutation D614G, associated with a more transmittable strain, has placed importance on peptide inhibitors which target the standing conformation.[ 170 ] The viral strain with the G614 residue has been shown to have an increased likelihood to be in the standing, open conformation which favors an interaction with ACE‐2, and could also favor similar interactions for peptide inhibitors targeting the RBD.[ 171 ]
Figure 5.
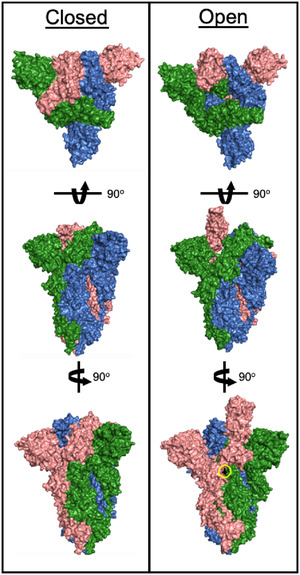
Structure of the SARS‐CoV‐2 Spike RBD. The left hand “closed” conformation does not bind ACE‐2. A conformational switch to the “open” structure can bind ACE‐2 and initiates viral infection. For the D614G strain, a single point mutation (residue in black, circled in yellow) causes Spike RBD to preferentially occupy the “open” conformation. PDB 6ZB4 and 7DK3.
Alanine scanning mutagenesis identified residues of the ACE‐2 receptor that are important for interaction with SARS‐CoV Spike protein, leading to the development of peptide inhibitors derived from ACE‐2 (P1‐P6).[ 154 ] These peptide inhibitors demonstrate potent antiviral activity, especially peptide inhibitor P6, comprised of two discontinuous fragments of ACE‐2 linked with a glycine residue, and effectively inhibits 50% infection at 0.1 µm (Table 3 ).[ 154 ] For SARS‐CoV‐2, peptide inhibitors targeting RBD have also been shown to reduce infection in vitro. In particular, the peptide SBP‐1 developed by the Pentelute Lab of MIT exhibited high binding affinity to the SARS‐CoV‐2 RBD (K D = 1.3 µm).[ 137 , 149 ] Sitthiyotha and Chunsrivirot further optimized this peptide inhibitor using the SBP25‐RBD complex as a template for RosettaDesign‐based mutation and interface analysis. Subsequent MD validation and calculation of ΔΔG based on individual residue mutations enabled identification of several peptides with higher binding affinity—these peptides are characterized by mutations at residues 8 (Phe) and 25 (Leu) and improvements in binding affinity of up to 8.0 kcal mol−1.[ 137 ]
Table 3.
Peptide inhibitors targeting interactions mediated by Spike RBD on SARS‐CoV and SARS‐CoV‐2
| Peptide name | Target | Sequence | Development Stage | Efficacy and/or binding kinetics b) |
|---|---|---|---|---|
| SARS‐CoV | ||||
| P6[ 152 ] | RBD | EEQAKTFLDKFNHEAEDLFYQSSGLGKGDFR | In vitro | IC50 (P) = 0.1 µm |
| HR2‐8[ 81 ] | HR1 | ELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYEQYIK | In vitro | EC50 (V) = 17 µm |
| CP‐1[ 144 ] | HR1 | GINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYE | In vitro | IC50 (V) = 19 µm |
| HR1‐1[ 165 ] | HR2 | NGIGVTQNVLYENQKQIANQFNKAISQIQESLTTTSTA | In vitro | EC50 (V) = 3.68 µm |
| HR2‐18[ 165 ] | HR1 | IQKEIDRLNEVAKNLNESLIDLQELGK | In vitro | EC50 (V) = 5.22 µm |
| HR2‐38[ 187 ] | HR1 | GDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYE | In vitro | IC50 (V) = 5 nm |
| SR9[ 188 ] | HR1 | ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | In vitro | EC50 (V) = 5 nm |
| HR1‐a[ 143 ] | HR2 | YENQKQIANQFNKAISQIQESLTTTSTA | In vitro | EC50 (P) = 1.61 µm |
| GST‐removed‐HR2[ 143 ] | HR1 | DVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYEQYI | In vitro | EC50 (P) = 2.15 µm |
| HR2[ 143 ] | HR1 | ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | In vitro | EC50 (P) = 0.34 µm |
| P6[ 170 ] | HR1 | GINASVVNIQKEIDRLNEVAKNL | In vitro | IC50 (V) = 2.28 µm |
| S471‐503[ 180 ] | ACE‐2 | ALNCYWPLNDYGFYTTTGIGYQPYRVVVLSFEL | In vitro | EC50 (V) = 41.6 µm |
| SP‐10[ 221 ] | ACE‐2 | STSQKSIVAYTM | In vitro | IC50 (B) = 1.88 nm |
| SARS‐CoV‐2 | ||||
| SBP‐1[ 150 ] | RBD | IEEQAKTFLDKFNHEAEDLFYQS | In vitro | K D (L) = 47 nm |
| Inhibitor 1[ 147 ] | RBD | IEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNIT | In silico | N/A |
| 18 AA Peptide[ 151 ] | RBD | FLDKFNHEAEDLFYQSSL | In silico | N/A |
| SPB25F8N [ 137 ] | RBD | IEEQAKTNLDKFNHEAEDLFYQSSL | In silico | N/A |
| SPB25F8N/L25R [ 137 ] | RBD | IEEQAKTNLDKFNHEAEDLFYQSSR | In silico | N/A |
| SPB25L25V [ 137 ] | RBD | IEEQAKTFLDKFNHEAEDLFYQSSV | In silico | N/A |
| AVP0671[ 222 ] | RBD | TWLATRGLLRSPGRYVYFSPSASTWPVGIWTTGELVLGCDAAL | In silico | N/A |
| Peptide 1[ 223 ] | RBD | TVFGLNVWKRYSK‐(βA)‐K(Biotin)‐CONH2 | In vitro | K D (L) = 250 nm |
| AVP1244[ 222 ] | ACE‐2 | GCASRCKAKCAGRRCKGWASAFRGRCYCKCFRC | In silico | N/A |
| P8[ 171 ] | RBD | SALEEQLKTFLDKFMHELEDLLYQLAL | In vitro | IC50 (V) = 46 nm |
| P9[ 171 ] | RBD | SALEEQYKTFLDKFMHELEDLLYQLSL a) | In vitro | IC50 (V) = 53 nm |
| P10[ 171 ] | RBD | SALEEQYKTFLDKFMHELEDLLYQLAL a) | In vitro | IC50 (V) = 42 nm |
| AHB1[ 177 ] | RBD | DEDLEELERLYRKAEEVAKEAKDASRRGDDERAKEQMERAMRLFDQVFELAQELQEKQTDGNRQKATHLDKAVKEAADELYQRVR | In vitro | IC50 (V) = 35 nm |
| AHB2[ 177 ] | RBD | ELEEQVMHVLDQVSELAHELLHKLTGEELERAAYFNWWATEMMLELIKSDDEREIREIEEEARRILEHLEELARK | In vitro | IC50 (V) = 15.5 nm |
| LCB1[ 177 ] | RBD | DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKGDERLLEEAERLLEEVER | In vitro | IC50 (V) = 23.54 pm |
| LCB3[ 177 ] | RBD | NDDELHMLMTDLVYEALHFAKDEEIKKRVFQLFELADKAYKNNDRQKLEKVVEELKELLERLLS | In vitro | IC50 (V) = 48.1 pm |
| [22–44][ 224 ] | RBD | EEQAKTFLDKFNHEAEDLFYQSS | In vitro | IC50 = 1–10 µm |
| [22–57][ 224 ] | RBD | EEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEE | In vitro | IC50 = 1–10 µm |
| 2019‐nCoV HR2P[ 97 ] | HR1 | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | In vitro | IC50 (P) = 0.98 µm |
| EK1[ 96 ] | HR1 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL | In vitro | IC50 (P) = 2375 nm |
| EK1C4[ 96 ] | HR1 | (N)EK1‐GSGSG‐PEG4‐(Chol) | In vitro | IC50 (P) = 15.8 nm |
| IPB02[ 148 ] | HR1 | ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELK‐(Chol) | In vitro | IC50 (P) = 0.08 µm |
| [SARSHRC‐PEG4]2‐chol[ 225 ] | HR1 | [DISGINASWNIQKEIDRLNEVAKNLNESLIDLQEL‐PEG4]2‐(Chol) | In vitro and in vivo | IC50 (V) = 3 nm |
| SARS‐BLOCK peptide 5[ 157 ] | ACE‐2 | Unknown | In vitro | IC95 (P) = 6.66 µm |
| P9[ 171 ] | RBD | SALEEQYKTFLDKFMHELEDLLYQLSL | In vitro | IC50 (V) = 53 nm |
| P10[ 171 ] | RBD | SALEEQYKTFLDKFMHELED LLYQL AL | In vitro | IC50 (V) = 42 nm |
| RBD‐pep2[ 171 ] | RBD | IYQAGSTPCNGVEGFNCYFP | In vitro | N/A |
| DP7[ 172 ] | RBD | VQWRIRVAVIRK | In vitro | IC50 (P) = 73.625 µg mL−1 |
| hACE221‐55A36K‐F40E[ 173 ] | RBD | IEEQAKTFLDKFNHEKEDLEYQSSLASWNYNTNIT | In vitro | IC50 (B) = 3.6 µm |
| SAP1[ 174 ] | RBD | TFLDKFNHEAEDLFYQ | In vitro | IC50 (P) = 2.39 mm |
| SAP2[ 175 ] | RBD | EDLFYQSSL | In vitro | IC50 (P) = 3.72 mm |
| SAP6[ 175 ] | RBD | EDLFYQ | In vitro | IC50 (P) = 1.90 mm |
Note:
is homotyrosine;
V: Virus infection inhibition assay result; P: pseudotype virus with reporter inhibition assay result; B: competitive biotinylated enzyme‐linked immunosorbent assay (ELISA) of Spike protein and ACE‐2 result; L: bio‐layer interferometry (BLI). For BLI/SPR experiments, K D is reported. EC50 and IC50, while representing the same value in this inhibition assay, are shown as initially reported.
Karoyan and coworkers used the similar N‐terminal helix of human ACE‐2 derivation to develop peptide inhibitors (P8‐P10) which prevented SARS‐CoV‐2 infection with 50% inhibition (IC50) in the nanomolar concentration range.[ 172 ]
A similar approach of mutating contact site residues was adopted by Han et al., but was executed using in vitro site‐directed mutagenesis, and identified a peptide with an EC50 of 0.1 µm.[ 154 ] An alternative linker‐based approach relying on alanine scanning and MD was adopted by Han and Kral, leading to the identification of a linker‐based peptide with untested in vitro efficacy but with established computational efficacy.[ 150 ] Other promising peptide candidates which remain to be characterized in vitro include the peptide produced by Baig et al., an 18‐mer designed using alanine scanning and subsequent MD.[ 128 ] Their approach distinguishes itself from others’ work due to the inclusion of toxicity estimates via the ToxinPred server, and additional metric which can be considered when evaluating the properties of new peptides.
Other approaches relying on de novo synthesis of inhibitors chose not to base their starting template on the ACE‐2‐RBD interaction, instead choosing helical binders from a scaffold library and subsequent RIF docking. With this method, it was possible to identify peptides with IC50 values in the nanomolar range.[ 173 ] Peptides LCB1 and LCB3, with an IC50 value in the picomolar concentration range, were identified by this same group, suggesting that de novo synthesis and modelling with downstream binding mode clustering is a viable option going forward.
4.3. Peptide Inhibitors that Target the ACE‐2 Receptor
The ACE‐2 host cell receptor is a membrane bound aminopeptidase that is expressed in heart, lung, kidney, and epithelial cells found in blood vessels and the lower gastrointestinal tract.[ 30 ] As previously described, its interaction with the RBD of SARS‐CoV and SARS‐CoV‐2 is responsible for viral infection as both viruses use ACE‐2 as the entry receptor (Figure 6 ).[ 52 ] Thus, developing peptide inhibitors targeting the ACE‐2 receptor also constitutes much of the research on inhibiting SARS‐CoV‐2 infection (Figure 6).
Figure 6.
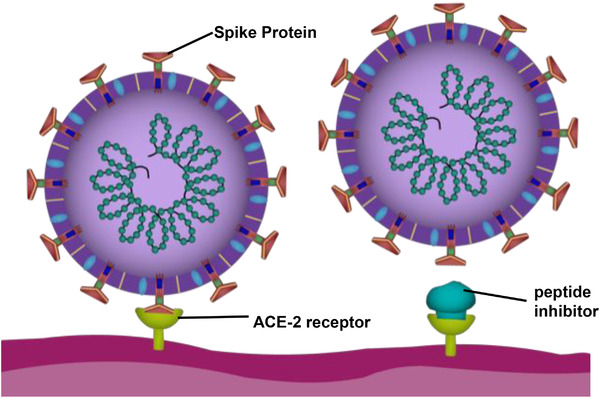
Binding inhibitor mechanism for the SARS‐CoV‐2 virus. To the left, the RBD of the S1 subunit on the Spike protein of SARS‐CoV‐2 binds to the ACE‐2 receptor on the host‐cell, completing the first step of viral infection. To the right, a peptide inhibitor binding to the ACE‐2 receptor, preventing the Spike protein from binding by blocking the ACE‐2 receptor binding.
Wong et al. helped determine the 193‐residue portion of the SARS‐CoV S protein that exhibited binding affinity for ACE‐2, and synthesized a peptide (residues 318–510) which was able to inhibit Spike protein‐pseudotype lentivirus infection with an IC50 of less than 10 nm.[ 174 ] This excellent result prompted others to study this sequence range within ACE‐2 in an effort to develop optimized inhibitors. Struck et al. developed a peptide inhibitor (called RBD‐11b) derived from residues 438–443 of the Spike protein of SARS‐CoV to inhibit its entry; its dissociation constant was K D = 46 µm, signifying high occupation of the ACE‐2 receptor's binding site.[ 175 ] Another peptide inhibitor derived from the SARS‐CoV RBD residues of similar origin was S471‐503, and inhibited plaque formation of SARS‐CoV with an EC50 value of 41.6 µm.[ 176 ]
Watson et al. at LigandAl utilize this same method of attack in developing SARS‐BLOCK, a series of peptide inhibitors that were designed and simulated using RaptorX homology modelling. One of these peptides conferred up to 95% SARS‐CoV‐2 infection inhibition with a concentration as low as 30 nm.[ 177 ] Moreover, since the SARS‐BLOCK peptides prevent association of ACE‐2 to RBD, not only do they inhibit infection, but they also allow neutralizing antibodies to activate an immune response more easily.
4.4. Fusion Inhibition by Targeting Interactions between HR1 and HR2 Domains
Subunit S2 of Spike protein has been shown to help with lipid mixing and subsequent viral‐cell membrane fusion after binding to ACE‐2.[ 53 , 178 ] S2 comprises the highly conserved heptad repeat domains HR1 and HR2, which upon infection, interact to form a stable, antiparallel, α‐helical rod‐like complex.[ 67 , 68 , 161 ] The conformational change that the HR regions in the S2 subunit undergo is important to the membrane fusion process, specifically because it results in the formation of a 6‐HB which facilitates the virus coming in close proximity to the host‐cell to initiate infection (Figure 7 ).[ 84 , 147 , 163 ] While this process is triggered by viral attachment via the S1 subunit to the ACE‐2 host‐cell receptor, nuanced conformational changes contribute to infection, and can be divided into 3 stages involving HR1 and HR2: first, α‐helices form which extend into the host‐cell membrane along with the fusion peptide to expose the hydrophobic HR1 and HR2 regions (into an intermediate pre‐hairpin state) as revealed by detailed circular dichroism (CD) and electron microscopy (EM) investigation.[ 147 , 163 , 179 ] Next, both HR1 and HR2 form trimers, and HR2 binds to HR1 to form the post‐fusion state 6‐HB, also known as the fusion core.[ 147 , 163 , 164 , 179 ] Finally, this fusion core brings the opposing viral and cell membranes close together and helps form fusion pores through which viral genetic material can enter the host cell.[ 147 , 180 ]
Figure 7.
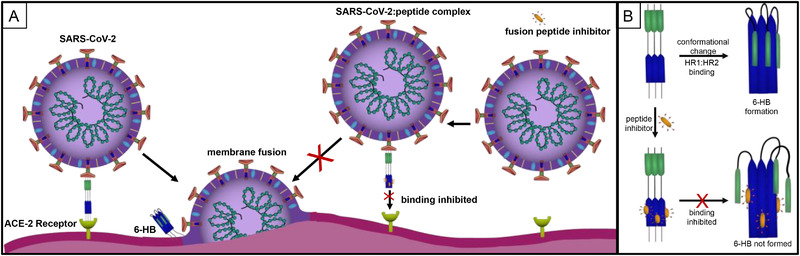
Fusion inhibitor mechanisms for the SARS‐CoV‐2 virus. A) Formation of a 6‐HB initiates viral fusion. B) The HR2 domain, comprised of 3 ɑ‐helices, present within the S2 subunit of SARS‐CoV‐2 interacts with the HR1 domain, ɑ‐helices, to form a six‐helical bundle (6‐HB). This process can be inhibited by the presence of three copies of a fusion peptide inhibitor.
Prior to the formation of the fusion core, while both domains are exposed during their intermediate pre‐hairpin state, HR2 helices bind to the grooves of HR1 helical trimers; in the absence of this binding, the fusion core will not form, and viral infection cannot occur (Figure 7).[ 147 , 163 , 179 ] Peptide‐inhibitors derived from the HR2 region can bind to the exposed intermediate pre‐hairpin HR1 region and prevent viral entry in SARS‐CoV, MHV, HIV‐1, and other viruses such as SARS‐CoV‐2 with class I envelope proteins.[ 127 , 141 , 147 , 163 , 181 , 182 ] CD data suggests that these inhibitors may adopt atypical α‐helix characteristics and are extremely sensitive to sequence additions or deletions—peptide inhibitors based on HR1 or HR2, therefore, will need to largely mirror the native proteins in length, sequence and structure.[ 181 ]
Peptides which target the SARS‐CoV‐2 fusion complex are largely derived from the interaction between HR1 and HR2. Numerous studies have modified regions of these two domains on the Spike protein of coronaviruses to develop inhibitory peptides which can prevent the formation of this fusion complex. These studies have been performed in silico with molecular docking and MD simulations as well as in vitro assays to gauge experimental efficacy. Zhu et al., Xia et al., and Liu et al., are just a few selected examples which successfully developed inhibitory peptides based on HR1 and HR2 to prevent this fusion.[ 84 , 147 , 151 ] These peptides displayed micromolar and nanomolar IC50 and EC50 values (Table 3), and their secondary structures were evaluated by CD. Other teams have introduced mutations to these peptides, including Xia et al. who included specific glutamic acid and lysine residues to increase the solubility and antiviral efficacy of their peptide.[ 141 ] This resulted in a new peptide, EK1, with an IC50 of 2.23 µm, with improved potential for in vivo studies due in part to its increased solubility (Table 3).[ 141 ] Previously, Ujike et al. inserted an X‐EE‐XX‐KK sequence in along different regions of HR2 while studying human immnodeficiency virus (HIV).[ 183 ] This was also done with SARS‐CoV, resulting in lower nanomolar affinity when compared to the original peptide tested (Table 3).
Lipid‐ or cholesterol‐conjugated peptides may also confer certain advantages over their naked peptide analogs, providing additional avenues of study for fusion inhibitory peptides. EK1C4, the cholesterol‐conjugated version of EK1, saw a 67‐fold increase in inhibition (IC50 = 37 nm).[ 83 ] Similar to lipopeptide EK1C4, lipopeptide IPB02 facilitate more significant fusion than its cholesterol‐lacking peptide analogue, IPB01.[ 151 ]
4.5. Comparison of Viral Proteins of SARS‐CoV and SARS‐CoV‐2
Peptide inhibitors have demonstrated success in preventing viral entry into host cells through the targeting of functional domains on the Spike protein, including the RBD in the S1 subunit, and the HR1 and HR2 domains in the S2 subunit. Sequence alignment of the S2 subunits of both SARS‐CoV and SARS‐CoV‐2 shows high conservation, with relative similarity of the HR1 and HR2 domains as 92.6% and 100%, respectively.[ 83 , 84 ] The RBD is less conserved, with around 74% identity.[ 184 ]
The high sequence conservation in Spike protein RBD, HR1, and HR2 between SARS‐CoV and SARS‐CoV‐2 allows for consistent and general targeting between peptide inhibitors of both species. SARS‐CoV‐2 still targets ACE‐2 as its host‐cell receptor, like SARS‐CoV; the variation between the viruses, however, is associated with a higher binding affinity of SARS‐CoV‐2 with the ACE‐2 receptor.[ 151 , 185 ] While there is some sequence variation in the HR1 region, it is still highly conserved in SARS‐CoV‐2, suggesting similar mechanisms of membrane fusion in SARS‐CoV and SARS‐CoV‐2.[ 83 , 133 ] CD analysis revealed that the α‐helical content of peptides derived from the HR1 domain was higher in SARS‐CoV‐2; additionally, the interaction between HR1 of SARS‐CoV‐2 and an HR2 derived peptide had a higher thermostability compared to SARS‐CoV, indicative of a stronger interaction between these domains in SARS‐CoV‐2 versus SARS‐CoV.[ 151 ]
Sequencing and a high level of homology between SARS‐CoV and SARS‐CoV‐2 greatly improved the development time of the peptide inhibitors discussed in this review, and in the future this method will continue to prove useful in identify novel peptide therapeutics for viral entry inhibition.
5. Outlook
The COVID‐19 pandemic has spread rapidly across the globe, and with it the scientific community had faced unprecedented challenges. Over a period of months, millions have died or been infected while scientists have worked continuously to gain a better understanding of the disease and SARS‐CoV‐2 virus. To date, several clinically approved vaccines exist and are being distributed. Previous information gained on SARS‐CoV, such as data on ACE‐2 binding as well as the Spike glycoprotein, has been essential in quickly facilitating SARS‐CoV‐2‐specific research. Treatments like non‐peptide therapeutics largely feature repurposed small molecules, used in previous viral outbreaks, but which lack the specificity and efficacy a newly designed therapeutic could provide, and largely serve as a stop‐gap until more efficacious treatments are identified and approved.
Peptide therapeutics and new promising targets against SARS‐CoV‐2 have been developed through homology studies and computational simulation. Key regions, such as the Spike protein‐ACE‐2 interaction are prime targets for binding inhibitors. Regions like the HR1 and HR2 components of Spike protein have also proven good targets due to their importance in the stabilizing the virus‐cell interaction, though this method requires robust peptide design to counteract the viral fusion mechanism. While holding significant progress, peptide therapeutics still suffer from key limitations associated with: stability (API and formulation[ 99 , 186 , 187 , 188 , 189 ]/in vitro and in vivo[ 190 , 191 , 192 , 193 , 194 ]); susceptibility to proteases[ 190 , 195 , 196 , 197 , 198 , 199 , 200 ] potential immune (IgG) responses[ 120 , 201 , 202 , 203 ] cost at scale[ 204 , 205 , 206 , 207 ] pharmacokinetic bioavailability.[ 208 , 209 , 210 , 211 , 212 ] Notwithstanding, numerous strategies have been explored to overcome these challenges and others, including self‐assembling peptides from our group[ 213 , 214 , 215 , 216 , 217 , 218 , 219 , 220 ] and others.[ 221 , 222 , 223 , 224 , 225 ]
COVID‐19 has tested the limits of current development pathways of traditional and repurposed drugs. Here we have explored the expanding field of therapeutic peptide inhibitors of SARS‐CoV‐2, with a particular focus on Spike protein inhibitors. The speed and gravity of the COVID‐19 pandemic has set a new standard for therapeutic development, with hundreds of therapeutics on the market and under investigation. In recent years peptides have shown promise in this new era of disease, and offer advantages in their versatility, low‐toxicity, specificity, and most importantly, rapid adaptability. The last is clearly seen in the impact SARS‐CoV research has had on aiding development of SARS‐CoV‐2 therapeutics, with many peptides being adapted or loosely based on former investigations. Though the two strains have clear differences, the continuing emergence of coronavirus outbreaks has made obvious the need for specific and adaptable inhibitors that can reliably target conserved regions of emerging strains. While vaccines have been deployed, the potential for peptide therapeutics still remains for treating the millions globally who are or will be infected with SARS‐CoV‐2. Because of their versatility and ease of storage and well‐understood design principles, peptide therapeutics offer a viable strategy to mitigate the severity of the SARS‐CoV‐2 pandemic and future coronavirus outbreaks.
Conflict of Interest
V.A.K. has equity interests in start‐up companies attempting to translate peptides from peptide‐based technological platform.
Acknowledgements
A.A.J. and V.A.K. contributed equally as corresponding authors. J.K. and K.P. contributed equally to this work. This work was supported by grants NIH R15 EY029504 01A1S1 for A.A.J.; NIH R15 EY029504, NSF IIP 1903617, the NJIT Undergraduate Research and Innovation (URI) Program for V.A.K.
Biographies
Disha Panchal is an undergraduate research associate in KumarLab at the New Jersey Institute of Technology in Newark, New Jersey. She is currently pursuing a Bachelor's degree in Biology on a pre‐medical track. Her interests include molecular biology and gene therapy, and her future work aims to develop intracellular delivery mechanisms for nucleic acids.

Jeena Kataria is an undergraduate research associate in KumarLab at the New Jersey Institute of Technology in Newark, New Jersey. Currently, she is working toward a major in Biomedical Engineering and a minor in Applied Mathematics. In the lab, she works on peptide synthesis, complementary therapies for vascular medicine, and prototype development for surgical applications. After graduation, she hopes to attend medical school.

Kamiya Patel is an undergraduate research associate in KumarLab at the New Jersey Institute of Technology in Newark, New Jersey. She is currently pursuing a Bachelor's degree for Biomedical Engineering at the university. Her concentrations within the lab include solid‐phase peptide synthesis and complementary therapies in vascular medicine. Her future endeavors will focus on project management and business in the biopharmaceutical space.

Kaytlyn Crowe is a graduate research associate in KumarLab at the New Jersey Institute of Technology in Newark, New Jersey. She is currently pursuing a Master's degree in Biomedical Engineering at the university. Her concentrations within the lab include slow release of naloxone, dental health, translational medicine, 3D printing, entrepreneurship, as well as literature review research. Her future work will focus on translational medicine in the biopharmaceutical space.

Varun Pai is an undergraduate researcher in the lab of Dr. Neeraj Chauhan at the Public Health Research Institute in Newark, New Jersey. He is currently a pre‐medical student pursuing a Bachelor's degree in Biology at NJIT. His interests include computational modelling, drug discovery, and molecular biology. His future endeavors will focus on microbiology and pathogenesis in infectious disease.

Abdul‐Rahman Azizogli is an undergraduate research assistant in KumarLab at the New Jersey Institute of Technology. He is currently pursuing a Bachelor's of Science in Biology. In the lab, he works on the computational modeling of hydrogel systems and structural characterization via NMR. His future career goals include therapeutic research and clinical medicine.

Neil Kadian is a rising freshman at Yale University and a 2021 graduate of Dr. Ronald E. McNair Academic High School in Jersey City, New Jersey. He plans to earn his bachelor's degree in Molecular, Cellular, and Developmental Biology. His major research interests are in translational medicine, drug delivery, cancer research, and neuroscience. He aspires to pursue a career combining academic, translational, and clinical medicine in the future.

Sreya Sanyal is an undergraduate research associate in KumarLab at the New Jersey Institute of Technology in Newark, New Jersey. She is currently pursuing Bachelor's degrees in Biology and History. In the lab, she works on in vitro and in vivo studies of therapeutic peptide hydrogels addressing high cholesterol and macular degeneration. She is interested in hematology/oncology and her future goals include translational therapeutic research and clinical medicine.

Abhishek Roy is a graduate research assistant in Kumarlab at the New Jersey Institute of Technology in Newark, New Jersey. He is currently a second year Ph.D. student in Biomedical Engineering department at the university. His expertise lies in structural and biophysical characterization of peptides as well as in vivo experiments such as aligned vascularization, pharmocokinetics, and imaging. His future endeavors will focus on transitioning into the pharmaceutical industry.

Joseph Dodd‐o is a graduate research assistant in Kumarlab at the New Jersey Institute of Technology in Newark, New Jersey. He is currently a first year Ph.D. student studying Biomedical Engineering. Joe is currently contributing to research on computational design, synthesis, structural characterization, and in vitro validation of de novo peptide sequences. Joe's future goals include R&D and manufacturing optimization in the oncology pharmaceutical space.

Amanda Acevedo‐Jake is an NIH Individual Diversity fellow working in the KumarLab at NJIT where she studies mimetic peptide hydrogels to promote localized wound healing and tissue regeneration. She obtained her Ph.D. from Rice University in 2017 working in the lab of Prof. Jeff Hartgerink on self‐assembling mimetic collagen peptides. After graduation, she was awarded an MSCA Fellowship to carry out research on stimuli‐responsive rotaxane‐oligonucleotides working with Prof. Ali Tavassoli and Prof. Stephen Goldup.

Vivek Kumar received his B.Sc. Degree in Biomedical Engineering from Northwestern University, and Ph.D. in Bioengineering from the Georgia Institute of Technology. His expertise is in tissue engineering, drug development, and delivery; specific interests lie in inflammation modulation, angiogenesis, and understanding cytokine mimicry. Currently, he is at the New Jersey Institute of Technology as an assistant professor in Biomedical Engineering and Chemical and Materials Engineering, and at Rutgers School of Dental Medicine. Prof. Kumar strives to encourage research involvement at all levels (high school, undergraduate, graduate, and post‐doctoral) and aims to translate technologies in startups.

Contributor Information
Amanda M. Acevedo‐Jake, Email: aa2954@njit.edu.
Vivek A. Kumar, Email: vak@njit.edu.
References
- 1.Listings of WHO's response to COVID‐19, World Health Organization 2020, https://www.who.int/news/item/29-06-2020-covidtimeline.
- 2. Wu F., Zhao S., Yu B., Chen Y. M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., Zhang Y. Z., Nature 2020, 579, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., N. Engl. J. Med. 2020, 382, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drosten C., Günther S., Preiser W., van der Werf S., Brodt H. R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R. A., Berger A., Burguière A. M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J. C., Müller S., Rickerts V., Stürmer M., Vieth S., Klenk H. D., Osterhaus A. D., Schmitz H., Doerr H. W., N. Engl. J. Med. 2003, 348, 1967. [DOI] [PubMed] [Google Scholar]
- 5. Zhong N. S., Zheng B. J., Li Y. M., Poon; Xie Z. H., Chan K. H., Li P. H., Tan S. Y., Chang Q., Xie J. P., Liu X. Q., Xu J., Li D. X., Yuen K. Y., Peiris, Guan Y., Lancet 2003, 362, 1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lam W. K., Zhong N. S., Tan W. C., Respirology 2003, 8, S2.15018125 [Google Scholar]
- 7. Liang W. N., Zhao T., Liu Z. J., Guan B. Y., He X., Liu M., Chen Q., Liu G. F., Wu J., Huang R. G., Xie X. Q., Wu Z. L., Biomed. Environ. Sci. 2006, 19, 445. [PubMed] [Google Scholar]
- 8. Zaki A. M., van Boheemen S., Bestebroer T. M., Osterhaus A. D., Fouchier R. A., N. Engl. J. Med. 2012, 367, 1814. [DOI] [PubMed] [Google Scholar]
- 9. Zumla A., Hui D. S., Perlman S., Lancet 2015, 386, 995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Assiri A., Al‐Tawfiq J. A., Al‐Rabeeah A. A., Al‐Rabiah F. A., Al‐Hajjar S., Al‐Barrak A., Flemban H., Al‐Nassir W. N., Balkhy H. H., Al‐Hakeem R. F., Makhdoom H. Q., Zumla A. I., Memish Z. A., Lancet Infect. Dis. 2013, 13, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brielle E. S., Schneidman‐Duhovny D., Linial M., Viruses 2020, 12, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou P., Yang X.‐L., Wang X.‐G., Hu B., Zhang L., Zhang W., Si H.‐R., Zhu Y., Li B., Huang C.‐L., Chen H.‐D., Chen J., Luo Y., Guo H., Jiang R.‐D., Liu M.‐Q., Chen Y., Shen X.‐R., Wang X., Zheng X.‐S., Zhao K., Chen Q.‐J., Deng F., Liu L.‐L., Yan B., Zhan F.‐X., Wang Y.‐Y., Xiao G.‐F., Shi Z.‐L., Nature 2020, 579, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ou J., Zhou Z., Dai R., Zhang J., Lan W., Zhao S., Wu J., Seto D., Cui L., Zhang G., Zhang Q., bioRxiv 2020, 2020.03.15.991844. [Google Scholar]
- 14. Tang J. W., Bahnfleth W. P., Bluyssen P. M., Buonanno G., Jimenez J. L., Kurnitski J., Li Y., Miller S., Sekhar C., Morawska L., Marr L. C., Melikov A. K., Nazaroff W. W., Nielsen P. V., Tellier R., Wargocki P., Dancer S. J., J. Hosp. Infect. 2021, 110, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee S., Meyler P., Mozel M., Tauh T., Merchant R., Can. J. Anaesth. 2020, 67, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jayaweera M., Perera H., Gunawardana B., Manatunge J., Environ. Res. 2020, 188, 109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sosnowski T. R., Curr. Opin. Colloid Interface Sci. 2021, 54, 101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abbas M., Robalo Nunes T., Martischang R., Zingg W., Iten A., Pittet D., Harbarth S., Antimicrob. Resist. Infect. Control 2021, 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oran D. P., Topol E. J., Ann. Intern. Med. 2020, 173, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. CDC SARS Response Timeline, Centers for Disease Control and Prevention 2013.
- 21. Zaki A. M., van Boheemen S., Bestebroer T. M., Osterhaus A. D. M. E., Fouchier R. A. M., N. Engl. J. Med. 2012, 367, 1814. [DOI] [PubMed] [Google Scholar]
- 22. Zhu Z., Lian X., Su X., Wu W., Marraro G. A., Zeng Y., Respir. Res. 2020, 21, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang G., Chen Q., Xu J., Liu Y., Lim W., Peiris J. S. M., Anderson L. J., Ruan L., Li H., Kan B., Di B., Cheng P., Chan K. H., Erdman D. D., Gu S., Yan X., Liang W., Zhou D., Haynes L., Duan S., Zhang X., Zheng H., Gao Y., Tong S., Li D., Fang L., Qin P., Xu W., Group S. D. W., Emerging Infect. Dis. 2004, 10, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seddiq N., Al‐Qahtani M., Al‐Tawfiq J. A., Bukamal N., Case Rep. Infect. Dis. 2017, 2017, 1262838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y.‐y., Li B.‐r., Ning B.‐t., Front. Immunol. 2020, 11, 2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joynt G. M., Yap H. Y., Curr. Infect. Dis. Rep. 2004, 6, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Al‐Dorzi H. M., Aldawood A. S., Khan R., Baharoon S., Alchin J. D., Matroud A. A., Al Johany S. M., Balkhy H. H., Arabi Y. M., Ann. Intensive Care 2016, 6, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stokes E. K., Zambrano L. D., Anderson K. N., Marder E. P., Raz K. M., Felix S. E. l. B., Tie Y., Fullerton K. E., Morb. Mortal. Wkly. Rep. 2020, 69, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ashraf U. M., Abokor A. A., Edwards J. M., Waigi E. W., Royfman R. S., Hasan S. A., Smedlund K. B., Hardy A. M. G., Chakravarti R., Koch L. G., Physiol. Genomics 2021, 53, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamming I., Timens W., Bulthuis M. L. C., Lely A. T., Navis G. J., van Goor H., J. Pathol. 2004, 203, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Long B., Brady W. J., Koyfman A., Gottlieb M., Am. J. Emerg. Med. 2020, 38, 1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tersalvi G., Vicenzi M., Calabretta D., Biasco L., Pedrazzini G., Winterton D., J. Card. Failure 2020, 26, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng Y.‐Y., Ma Y.‐T., Zhang J.‐Y., Xie X., Nat. Rev. Cardiol. 2020, 17, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atri D., Siddiqi H. K., Lang J. P., Nauffal V., Morrow D. A., Bohula E. A., JACC Basic Transl. Sci. 2020, 5, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Agdamag A. C. C., Edmiston J. B., Charpentier V., Chowdhury M., Fraser M., Maharaj V. R., Francis G. S., Alexy T., Medicina 2020, 56, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martinez‐Rojas M. A., Vega‐Vega O., Bobadilla N. A., Am. J. Physiol. Renal. Physiol. 2020, 318, F1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang M., Xiong H., Chen H., Li Q., Ruan X. Z., Kidney Dis. (Basel) 2021, 7, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Naicker S., Yang C.‐W., Hwang S.‐J., Liu B.‐C., Chen J.‐H., Jha V., Kidney Int. 2020, 97, 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Diao B., Wang C., Wang R., Feng Z., Tan Y., Wang H., Wang C., Liu L., Liu Y., Liu Y., Wang G., Yuan Z., Ren L., Wu Y., Chen Y., medRxiv 2020, 2020.03.04.20031120. [Google Scholar]
- 40. Zhou J., Li C., Liu X., Chiu M. C., Zhao X., Wang D., Wei Y., Lee A., Zhang A. J., Chu H., Cai J.‐P., Yip C. C.‐Y., Chan I. H.‐Y., Wong K. K.‐Y., Tsang O. T.‐Y., Chan K.‐H., Chan J. F.‐W., To K. K.‐W., Chen H., Yuen K. Y., Nat. Med. 2020, 26, 1077. [DOI] [PubMed] [Google Scholar]
- 41. Cheung K. S., Hung I. F. N., Chan P. P. Y., Lung K. C., Tso E., Liu R., Ng Y. Y., Chu M. Y., Chung T. W. H., Tam A. R., Yip C. C. Y., Leung K. H., Fung A. Y., Zhang R. R., Lin Y., Cheng H. M., Zhang A. J. X., To K. K. W., Chan K. H., Yuen K. Y., Leung W. K., Gastroenterology 2020, 159, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holshue M. L., DeBolt C., Lindquist S., Lofy K. H., Wiesman J., Bruce H., Spitters C., Ericson K., Wilkerson S., Tural A., Diaz G., Cohn A., Fox L., Patel A., Gerber S. I., Kim L., Tong S., Lu X., Lindstrom S., Pallansch M. A., Weldon W. C., Biggs H. M., Uyeki T. M., Pillai S. K., N. Engl. J. Med. 2020, 382, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Viana S. D., Nunes S., Reis F., Ageing Res. Rev. 2020, 62, 101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dhama K., Patel S. K., Yatoo M. I., Tiwari R., Sharun K., Dhama J., Natesan S., Malik Y. S., Singh K. P., Harapan H., J. Environ. Manage. 2021, 280, 111825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bhayana R., Som A., Li M. D., Carey D. E., Anderson M. A., Blake M. A., Catalano O., Gee M. S., Hahn P. F., Harisinghani M., Kilcoyne A., Lee S. I., Mojtahed A., Pandharipande P. V., Pierce T. T., Rosman D. A., Saini S., Samir A. E., Simeone J. F., Gervais D. A., Velmahos G., Misdraji J., Kambadakone A., Radiology 2020, 297, E207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feng G., Zheng K. I., Yan Q.‐Q., Rios R. S., Targher G., Byrne C. D., Poucke S. V., Liu W.‐Y., Zheng M.‐H., J. Clin. Transl. Hepatol. 2020, 8, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fan Z., Chen L., Li J., Cheng X., Yang J., Tian C., Zhang Y., Huang S., Liu Z., Cheng J., Clin. Gastroenterol. Hepatol. 2020, 18, 1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu L., Liu J., Lu M., Yang D., Zheng X., Liver Int. 2020, 40, 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park S. E., Clin. Exp. Pediatr. 2020, 63, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Modrow S., Falke D., Truyen U., Schätzl H., in Molecular Virology, Springer‐Verlag, Berlin Heidelberg: 2013, pp. 185–349. [Google Scholar]
- 51. Brian D. A., Baric R. S., Coronavirus Replication Reverse Genet. 2005, 287, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F., Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang Y., Yang C., Xu X. F., Xu W., Liu S. W., Acta Pharmacol. Sin. 2020, 41, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jin Y., Yang H., Ji W., Wu W., Chen S., Zhang W., Duan G., Viruses 2020, 12, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hatmal M. M., Alshaer W., Al‐Hatamleh M. A. I., Hatmal M., Smadi O., Taha M. O., Oweida A. J., Boer J. C., Mohamud R., Plebanski M., Cells 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cueno M. E., Imai K., Front. Med. (Lausanne) 2020, 7, 594439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Groneberg D. A., Hilgenfeld R., Zabel P., Respir. Res. 2005, 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. He Y., Zhou Y., Liu S., Kou Z., Li W., Farzan M., Jiang S., Biochem. Biophys. Res. Commun. 2004, 324, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hofmann H., Pohlmann S., Trends Microbiol. 2004, 12, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tan Y. J., Lim S. G., Hong W., Antiviral Res. 2005, 65, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vijayanand P., Wilkins E., Woodhead M., Clin. Med. 2004, 2, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ziebuhr J., Curr. Opin. Microbiol. 2004, 7, 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bolles M., Donaldson E., Baric R., Curr. Opin. Virol. 2011, 1, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hou Y., Peng C., Yu M., Li Y., Han Z., Li F., Wang L. F., Shi Z., Arch. Virol. 2010, 155, 1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li W., Wong S. K., Li F., Kuhn J. H., Huang I. C., Choe H., Farzan M., J. Virol. 2006, 80, 4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Letko M., Marzi A., Munster V., Nat. Microbiol. 2020, 5, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. White J. M., Delos S. E., Brecher M., Schornberg K., Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bosch B. J., van der Zee R., de Haan C. A., Rottier P. J., J. Virol. 2003, 77, 8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sharma O., Sultan A. A., Ding H., Triggle C. R., Front. Immunol. 2020, 11, 585354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kumar D., Narayanan D. K. L., Kayarohanam S., Fuloria S., Fuloria N., Kumar A., Djearamane S., Wu Y.‐S., Chakravarthi S., Subramaniyan V., Int. J. Pharm. Res. 2021, 13, 4588. [Google Scholar]
- 71. Chilamakuri R., Agarwal S., Cells 2021, 10, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang F., Kream R. M., Stefano G. B., Med. Sci. Monit. 2020, 26, e924700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baden L. R., El Sahly H. M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S. A., Rouphael N., Creech C. B., McGettigan J., Khetan S., Segall N., Solis J., Brosz A., Fierro C., Schwartz H., Neuzil K., Corey L., Gilbert P., Janes H., Follmann D., Marovich M., Mascola J., Polakowski L., Ledgerwood J., Graham B. S., Bennett H., Pajon R., Knightly C., et al., N. Engl. J. Med. 2020, 384, 403.33378609 [Google Scholar]
- 74. Sahin U., Muik A., Derhovanessian E., Vogler I., Kranz L. M., Vormehr M., Baum A., Pascal K., Quandt J., Maurus D., Brachtendorf S., Lörks V., Sikorski J., Hilker R., Becker D., Eller A.‐K., Grützner J., Boesler C., Rosenbaum C., Kühnle M.‐C., Luxemburger U., Kemmer‐Brück A., Langer D., Bexon M., Bolte S., Karikó K., Palanche T., Fischer B., Schultz A., Shi P.‐Y., et al., Nature 2020, 586, 594. [DOI] [PubMed] [Google Scholar]
- 75.Pfizer and BioNTech announce vaccine candidate against covid‐19 achieved success in first interim analysis from phase 3 study, Pfizer, New York: 2020, https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-announce-vaccine-candidate-against. [Google Scholar]
- 76. Crommelin D. J. A., Anchordoquy T. J., Volkin D. B., Jiskoot W., Mastrobattista E., J. Pharm. Sci. 2020, 110, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Holm M. R., Poland G. A., Vaccine 2021, 39, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shahcheraghi S. H., Ayatollahi J., Aljabali A. A., Shastri M. D., Shukla S. D., Chellappan D. K., Jha N. K., Anand K., Katari N. K., Mehta M., Satija S., Dureja H., Mishra V., Almutary A. G., Alnuqaydan A. M., Charbe N., Prasher P., Gupta G., Dua K., Lotfi M., Bakshi H. A., Tambuwala M. M., Ther. Delivery 2021, 3, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jenssen H., Aspmo S. I., Methods Mol. Biol. 2008, 494, 177. [DOI] [PubMed] [Google Scholar]
- 80. Sun T., Han H., Hudalla G. A., Wen Y., Pompano R. R., Collier J. H., Acta Biomater. 2016, 30, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jeong W.‐j., Lee M. S., Lim Y.‐b., Biomacromolecules 2013, 14, 2684. [DOI] [PubMed] [Google Scholar]
- 82. Emerging SARS‐CoV‐2 Variants, Centers for Disease Control and Prevention 2021. [PubMed]
- 83. Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L., Cell Res. 2020, 30, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xia S., Zhu Y., Liu M., Lan Q., Xu W., Wu Y., Ying T., Liu S., Shi Z., Jiang S., Lu L., Cell Mol. Immunol. 2020, 17, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jaimes J. A., André N. M., Chappie J. S., Millet J. K., Whittaker G. R., J. Mol. Biol. 2020, 432, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. A Study to Assess Safety , Tolerability, and Immunogenicity of V591 (COVID‐19 Vaccine) in Healthy Participants (V591‐001), https://ClinicalTrials.gov/show/NCT04498247 (accessed: January 2021).
- 87.Dose Ranging Trial to Assess Safety and Immunogenicity of V590 (COVID‐19 Vaccine) in Healthy Adults (V590‐001), https://ClinicalTrials.gov/show/NCT04569786 (accessed: January 2021).
- 88.Merck Discontinues Development of SARS‐CoV‐2/COVID‐19 Vaccine Candidates; Continues Development of Two Investigational Therapeutic Candidates, Merck, Darmstadt, Germany: 2021. [Google Scholar]
- 89.Study to Evaluate the Safety and Antiviral Activity of Remdesivir (GS‐5734™) in Participants With Moderate Coronavirus Disease (COVID‐19) Compared to Standard of Care Treatment, https://ClinicalTrials.gov/show/NCT04292730 (accessed: February 2021).
- 90. Beigel J. H., Tomashek K. M., Dodd L. E., Mehta A. K., Zingman B. S., Kalil A. C., Hohmann E., Chu H. Y., Luetkemeyer A., Kline S., Lopez de Castilla D., Finberg R. W., Dierberg K., Tapson V., Hsieh L., Patterson T. F., Paredes R., Sweeney D. A., Short W. R., Touloumi G., Lye D. C., Ohmagari N., Oh M.‐d., Ruiz‐Palacios G. M., Benfield T., Fätkenheuer G., Kortepeter M. G., Atmar R. L., Creech C. B., Lundgren J., et al., N. Engl. J. Med. 2020, 383, 1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Abd El‐Aziz T. M., Stockand J. D., Infect. Genet. Evol. 2020, 83, 104327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Compound Summary for CID 5743, Dexamethasone, National Center for Biotechnology Information 2021.
- 93.Randomised Evaluation of COVID‐19 Therapy, https://ClinicalTrials.gov/show/NCT04381936 (accessed: January 2021).
- 94. Jitendra, Sharma P. K., Bansal S., Banik A., Indian J. Pharm. Sci. 2011, 73, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee A. C., Harris J. L., Khanna K. K., Hong J. H., Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar]
- 96. Lau J. L., Dunn M. K., Bioorg. Med. Chem. 2018, 26, 2700. [DOI] [PubMed] [Google Scholar]
- 97. Gorr S. U., Flory C. M., Schumacher R. J., PLoS One 2019, 14, e0216669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lashua L. P., Melvin J. A., Deslouches B., Pilewski J. M., Montelaro R. C., Bomberger J. M., J. Antimicrob. Chemother. 2016, 71, 2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zapadka K. L., Becher F. J., Gomes Dos Santos A. L., Jackson S. E., Interface Focus 2017, 7, 20170030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Usmani S. S., Bedi G., Samuel J. S., Singh S., Kalra S., Kumar P., Ahuja A. A., Sharma M., Gautam A., Raghava G. P. S., PLoS One 2017, 12, e0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Merrifield R. B., J. Am. Chem. Soc. 1963, 85, 2149. [Google Scholar]
- 102. Albericio F., Kruger H. G., Future Med. Chem. 2012, 4, 1527. [DOI] [PubMed] [Google Scholar]
- 103. L. Otvos, Jr. , Wade J. D., Front. Chem. 2014, 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Di L., AAPS J. 2015, 17, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sharma V., McNeill J. H., Br. J. Pharmacol. 2009, 157, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Huh Y., Smith D. E., Feng M. R., Xenobiotica 2011, 41, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wells J. A., McClendon C. L., Nature 2007, 450, 1001. [DOI] [PubMed] [Google Scholar]
- 108. Cunningham A. D., Qvit N., Mochly‐Rosen D., Curr. Opin. Struct. Biol. 2017, 44, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hwang H., Vreven T., Janin J., Weng Z., Proteins 2010, 78, 3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fuller J. C., Burgoyne N. J., Jackson R. M., Drug Discovery Today 2009, 14, 155. [DOI] [PubMed] [Google Scholar]
- 111. Nero T. L., Morton C. J., Holien J. K., Wielens J., Parker M. W., Nat. Rev. Cancer 2014, 14, 248. [DOI] [PubMed] [Google Scholar]
- 112. Sorolla A., Wang E., Golden E., Duffy C., Henriques S. T., Redfern A. D., Blancafort P., Oncogene 2020, 39, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Henninot A., Collins J. C., Nuss J. M., J. Med. Chem. 2018, 61, 1382. [DOI] [PubMed] [Google Scholar]
- 114. Vlieghe P., Lisowski V., Martinez J., Khrestchatisky M., Drug Discovery Today 2010, 15, 40. [DOI] [PubMed] [Google Scholar]
- 115. Marqus S., Pirogova E., Piva T. J., J. Biomed. Sci. 2017, 24, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bajracharya R., Song J. G., Back S. Y., Han H.‐K., Comput. Struct. Biotechnol. J 2019, 17, 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Uribarri A., Núñez‐Gil I. J., Aparisi A., Becerra‐Muñoz V. M., Feltes G., Trabattoni D., Fernández‐Rozas I., Viana‐Llamas M. C., Pepe M., Cerrato E., Capel‐Astrua T., Romero R., Castro‐Mejía A. F., El‐Battrawy I., López‐País J., D'Ascenzo F., Fabregat‐Andres O., Bardají A., Raposeiras‐Roubin S., Marín F., Fernández‐Ortiz A., Macaya C., Estrada V., J. Nephrol. 2020, 33, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mócsai A., Kovács L., Gergely P., BMC Med. 2014, 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gokhale A. S., Satyanarayanajois S., Immunotherapy 2014, 6, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. McGregor D. P., Curr. Opin. Pharmacol. 2008, 8, 616. [DOI] [PubMed] [Google Scholar]
- 121. Akaji K., Konno H., Mitsui H., Teruya K., Shimamoto Y., Hattori Y., Ozaki T., Kusunoki M., Sanjoh A., J. Med. Chem. 2011, 54, 7962. [DOI] [PubMed] [Google Scholar]
- 122. Bosch B. J., Martina B. E., Van Der Zee R., Lepault J., Haijema B. J., Versluis C., Heck A. J., De Groot R., Osterhaus A. D., Rottier P. J., Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chou K.‐C., Wei D.‐Q., Zhong W.‐Z., Biochem. Biophys. Res. Commun. 2003, 308, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Du Q., Wang S., Wei D., Sirois S., Chou K. C., Anal. Biochem. 2005, 337, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ghosh A. K., Xi K., Grum‐Tokars V., Xu X., Ratia K., Fu W., Houser K. V., Baker S. C., Johnson M. E., Mesecar A. D., Bioorg. Med. Chem. Lett. 2007, 17, 5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shuto D., Kasai S., Kimura T., Liu P., Hidaka K., Hamada T., Shibakawa S., Hayashi Y., Hattori C., Szabo B., Ishiura S., Kiso Y., Bioorg. Med. Chem. Lett. 2003, 13, 4273. [DOI] [PubMed] [Google Scholar]
- 127. Zhu J., Xiao G., Xu Y., Yuan F., Zheng C., Liu Y., Yan H., Cole D. K., Bell J. I., Rao Z., Tien P., Gao G. F., Biochem. Biophys. Res. Commun. 2004, 319, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Baig M. S., Alagumuthu M., Rajpoot S., Saqib U., Drugs R&D 2020, 20, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chaturvedi P., Han Y., Kral P., Vukovic L., Adv. Theory Simul. 2020, 3, 2000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Han Y., Kral P., ACS Nano 2020, 14, 5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Karoyan P., Vieillard V., Gomez‐Morales L., Odile E., Guihot A., Luyt C. E., Denis A., Grondin P., Lequin O., Commun. Biol. 2021, 4, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Larue R. C., Xing E., Kenney A. D., Zhang Y., Tuazon J. A., Li J., Yount J. S., Li P. K., Sharma A., Bioconjugate Chem. 2021, 32, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ling R., Dai Y., Huang B., Huang W., Yu J., Lu X., Jiang Y., Peptides 2020, 130, 170328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Maas M. N., Hintzen J. C. J., Loffler P. M. G., Mecinovic J., Chem. Commun. 2021, 57, 3283. [DOI] [PubMed] [Google Scholar]
- 135. Pomplun S., Jbara M., Quartararo A. J., Zhang G., Brown J. S., Lee Y.‐C., Ye X., Hanna S., Pentelute B. L., ACS Cent. Sci. 2021, 7, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Rathod S. B., Prajapati P. B., Punjabi L. B., Prajapati K. N., Chauhan N., Mansuri M. F., In Silico Pharmacol. 2020, 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sitthiyotha T., Chunsrivirot S., J. Phys. Chem. B 2020, 124, 10930. [DOI] [PubMed] [Google Scholar]
- 138. Pagadala N. S., Syed K., Tuszynski J., Biophys. Rev. 2017, 9, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yang J., Zhang Y., Nucleic Acids Res. 2015, 43, W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Alford R. F., Leaver‐Fay A., Jeliazkov J. R., O'Meara M. J., DiMaio F. P., Park H., Shapovalov M. V., Renfrew P. D., Mulligan V. K., Kappel K., Labonte J. W., Pacella M. S., Bonneau R., Bradley P., Dunbrack R. L., Das R., Baker D., Kuhlman B., Kortemme T., Gray J. J., J. Chem. Theory Comput. 2017, 13, 3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Xia S., Yan L., Xu W., Agrawal A. S., Algaissi A., Tseng C.‐T. K., Wang Q., Du L., Tan W., Wilson I. A., Jiang S., Yang B., Lu L., Sci. Adv. 2019, 5, eaav4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q., Science 2020, 367, 1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X., Nature 2020, 581, 215. [DOI] [PubMed] [Google Scholar]
- 144. Huang S.‐Y., Briefings Bioinf. 2018, 19, 982. [DOI] [PubMed] [Google Scholar]
- 145. Salmaso V., Moro S., Front. Pharmacol. 2018, 9, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Chu L.‐H. M., Chan S.‐H., Tsai S.‐N., Wang Y., Cheng C. H.‐K., Wong K.‐B., Waye M. M.‐Y., Ngai S.‐M., J. Cell. Biochem. 2008, 104, 2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C. R., Xiong H., Farmar J., Debnath A. K., Tien P., Jiang S., Lancet 2004, 363, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Durrant J. D., McCammon J. A., BMC Biol. 2011, 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhang G., Pomplun S., Loftis A. R., Tan X., Loas A., Pentelute B. L., bioRxiv 2020, 2020.03.19.999318. [Google Scholar]
- 150. Han Y., Král P., ACS Nano 2020, 14, 5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhu Y., Yu D., Yan H., Chong H., He Y., J. Virol. 2020, 94, e00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Case D. A., Belfon K., Ben‐Shalom I. Y., Brozell S. R., Cerutti D. S., T. E. Cheatham, III , Cruzeiro V. W. D., Darden T. A., Duke R. E., Giambasu G., Gilson M. K., Gohlke H., Goetz A. W., Harris R., Izadi S., Kasava‐ jhala K., Kovalenko A., Krasny R., Kurtzman T., Lee T. S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Man V., Merz K. M., Miao Y., Mikhailovskii O., et al., AMBER 2020, University of California Press, San Francisco, CA: 2020. [Google Scholar]
- 153. Zhang G., Pomplun S., Loftis A. R., Loas A., Pentelute B. L., bioRxiv 2020, 2020.03.19.999318. [Google Scholar]
- 154. Han D. P., Penn‐Nicholson A., Cho M. W., Virology 2006, 350, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L., Nucleic Acids Res. 2005, 33, W382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Wood C. W., Ibarra A. A., Bartlett G. J., Wilson A. J., Woolfson D. N., Sessions R. B., Bioinformatics 2020, 36, 2917. [DOI] [PubMed] [Google Scholar]
- 157. Zou J., Yin J., Fang L., Yang M., Wang T., Wu W., Bellucci M. A., Zhang P., J. Chem. Inf. Model. 2020, 60, 5794. [DOI] [PubMed] [Google Scholar]
- 158. Tiwari M., Mishra D., J. Clin. Virol. 2020, 128, 104441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. B. Sainz, Jr. , Rausch J. M., Gallaher W. R., Garry R. F., Wimley W. C., J. Virol. 2005, 79, 7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Watson A., Rapid simulation and design of synthetic peptide mini‐scaffold vaccines for novel coronavirus (SARS‐CoV‐2) and pandemic preparedness, Ligandel, San Francisco, CA: 2019. [Google Scholar]
- 161. B. Sainz, Jr. , Mossel E. C., Gallaher W. R., Wimley W. C., Peters C. J., Wilson R. B., Garry R. F., Virus Res. 2006, 120, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Huentelman Matthew J., Zubcevic J., Hernández Prada Jose A., Xiao X., Dimitrov Dimiter S., Raizada Mohan K., Ostrov David A., Hypertension 2004, 44, 903. [DOI] [PubMed] [Google Scholar]
- 163. Xu Y., Zhu J., Liu Y., Lou Z., Yuan F., Liu Y., Cole D. K., Ni L., Su N., Qin L., Li X., Bai Z., Bell J. I., Pang H., Tien P., Gao G. F., Rao Z., Biochemistry 2004, 43, 14064. [DOI] [PubMed] [Google Scholar]
- 164. Gao J., Lu G., Qi J., Li Y., Wu Y., Deng Y., Geng H., Li H., Wang Q., Xiao H., Tan W., Yan J., Gao G. F., J. Virol. 2013, 87, 13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ge X.‐Y., Li J.‐L., Yang X.‐L., Chmura A. A., Zhu G., Epstein J. H., Mazet J. K., Hu B., Zhang W., Peng C., Zhang Y.‐J., Luo C.‐M., Tan B., Wang N., Zhu Y., Crameri G., Zhang S.‐Y., Wang L.‐F., Daszak P., Shi Z.‐L., Nature 2013, 503, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hoffmann M., Kleine‐Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.‐H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., Cell 2020, 181, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Yuan Y., Cao D., Zhang Y., Ma J., Qi J., Wang Q., Lu G., Wu Y., Yan J., Shi Y., Zhang X., Gao G. F., Nat. Commun. 2017, 8, 15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Cai Y., Zhang J., Xiao T., Peng H., Sterling S. M., Walsh R. M., Rawson S., Rits‐Volloch S., Chen B., Science 2020, 369, 1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C. L., Abiona O., Graham B. S., McLellan J. S., Science 2020, 367, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Korber B., Fischer W. M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E. E., Bhattacharya T., Foley B., Hastie K. M., Parker M. D., Partridge D. G., Evans C. M., Freeman T. M., de Silva T. I., McDanal C., Perez L. G., Tang H., Moon‐Walker A., Whelan S. P., LaBranche C. C., Saphire E. O., Montefiori D. C., Cell 2020, 182, 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mansbach R. A., Chakraborty S., Nguyen K., Montefiori D. C., Korber B., Gnanakaran S., bioRxiv 2020, 2020.07.26.219741. [Google Scholar]
- 172. Karoyan P., Vieillard V., Odile E., Denis A., Guihot A., Luyt C.‐E., Gómez‐Morales L., Grondin P., Lequin O., bioRxiv 2020, 2020.08.24.264077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Cao L., Goreshnik I., Coventry B., Case J. B., Miller L., Kozodoy L., Chen R. E., Carter L., Walls A. C., Park Y.‐J., Strauch E.‐M., Stewart L., Diamond M. S., Veesler D., Baker D., Science 2020, 370, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Wong S. K., Li W., Moore M. J., Choe H., Farzan M., J. Biol. Chem. 2004, 279, 3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Struck A.‐W., Axmann M., Pfefferle S., Drosten C., Meyer B., Antiviral Res. 2012, 94, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hu H., Li L., Kao R. Y., Kou B., Wang Z., Zhang L., Zhang H., Hao Z., Tsui W. H., Ni A., Cui L., Fan B., Guo F., Rao S., Jiang C., Li Q., Sun M., He W., Liu G., J. Comb. Chem. 2005, 7, 648. [DOI] [PubMed] [Google Scholar]
- 177. Watson A., Ferreira L., Hwang P., Xu J., Stroud R., bioRxiv 2020, 2020.08.06.238915. [Google Scholar]
- 178. Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F., Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Heald‐Sargent T., Gallagher T., Viruses 2012, 4, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Plemper R. K., Curr. Opin. Virol. 2011, 1, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yuan K., Yi L., Chen J., Qu X., Qing T., Rao X., Jiang P., Hu J., Xiong Z., Nie Y., Shi X., Wang W., Ling C., Yin X., Fan K., Lai L., Ding M., Deng H., Biochem. Biophys. Res. Commun. 2004, 319, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Bosch B. J., Martina B. E. E., van der Zee R., Lepault J., Haijema B. J., Versluis C., Heck A. J. R., de Groot R., Osterhaus A. D. M. E., Rottier P. J. M., Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ujike M., Nishikawa H., Otaka A., Yamamoto N., Yamamoto N., Matsuoka M., Kodama E., Fujii N., Taguchi F., J. Virol. 2008, 82, 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Marqus S., Pirogova E., Piva T. J., J. Biomed. Sci. 2017, 24, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S., Zhou Y., Du L., Cell Mol. Immunol. 2020, 17, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Raza F., Zafar H., Zhu Y., Ren Y., Ullah A., Khan A. U., He X., Han H., Aquib M., Boakye‐Yiadom K. O., Ge L., Pharmaceutics 2018, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Cleland J. L., Langer R., Formulation and Delivery of Proteins and Peptides: Design and Development Strategies, vol. 567, ACS Publications, Washington, DC: 1994, pp. 1–19. [Google Scholar]
- 188. Bak A., Leung D., Barrett S. E., Forster S., Minnihan E. C., Leithead A. W., Cunningham J., Toussaint N., Crocker L. S., AAPS J. 2015, 17, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Pearlman R., Wang Y. J., Stability and Characterization of Protein and Peptide Drugs, vol. 567, Springer, New York: 1993, pp. 1–19. [Google Scholar]
- 190. Bottger R., Hoffmann R., Knappe D., PLoS One 2017, 12, e0178943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Fominaya J. B., Bravo J., Rebollo A., Ther. Delivery 2015, 10, 1171. [DOI] [PubMed] [Google Scholar]
- 192. Imanishi S., Katoh T., Yin Y., Yamada M., Kawai M., Suga H., J. Am. Chem. Soc. 2021, 143, 5680. [DOI] [PubMed] [Google Scholar]
- 193. Pernot M., Vanderesse R., Frochot C., Guillemin F., Barberi‐Heyob M., Expert Opin. Drug Metab. Toxicol. 2011, 7, 793. [DOI] [PubMed] [Google Scholar]
- 194. Yang J. Z., Chen W., Borchardt R. T., J. Pharmacol. Exp. Ther. 2002, 303, 840. [DOI] [PubMed] [Google Scholar]
- 195. Stromstedt A. A., Pasupuleti M., Schmidtchen A., Malmsten M., Antimicrob. Agents Chemother. 2009, 53, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Gunasekera S., Muhammad T., Stromstedt A. A., Rosengren K. J., Goransson U., Front. Microbiol. 2020, 11, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Gorris H. H., Bade S., Roeckendorf N., Albers E., Schmidt M. A., Franek M., Frey A., Anal. Chem. Res. 2009, 81, 1580. [DOI] [PubMed] [Google Scholar]
- 198. Li Y., Wu M., Chang Q., Zhao X., RSC Adv. 2018, 8, 22268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Meng H., Kumar K., J. Am. Chem. Soc. 2007, 129, 15615. [DOI] [PubMed] [Google Scholar]
- 200. Zhao Y., Zhang M., Qiu S., Wang J., Peng J., Zhao P., Zhu R., Wang H., Li Y., Wang K., Yan W., Wang R., AMB Express 2016, 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. R. W. Frenck, Jr. , Klein N. P., Kitchin N., Gurtman A., Absalon J., Lockhart S., Perez J. L., Walter E. B., Senders S., Bailey R., Swanson K. A., Ma H., Xu X., Koury K., Kalina W. V., Cooper D., Jennings T., Brandon D. M., Thomas S. J., Tureci O., Tresnan D. B., Mather S., Dormitzer P. R., Sahin U., Jansen K. U., Gruber W. C., Group C. C. T., N. Engl. J. Med. 2021, 385, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Sadoff J., Le Gars M., Shukarev G., Heerwegh D., Truyers C., de Groot A. M., Stoop J., Tete S., Van Damme W., Leroux‐Roels I., Berghmans P. J., Kimmel M., Van Damme P., de Hoon J., Smith W., Stephenson K. E., De Rosa S. C., Cohen K. W., McElrath M. J., Cormier E., Scheper G., Barouch D. H., Hendriks J., Struyf F., Douoguih M., Van Hoof J., Schuitemaker H., N. Engl. J. Med. 2021, 384, 1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Trier N., Hansen P., Houen G., Int. J. Mol. Sci. 2019, 20, 6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Zhao C. X., Dwyer M. D., Yu A. L., Wu Y., Fang S., Middelberg A. P., Biotechnol. Bioeng. 2015, 112, 957. [DOI] [PubMed] [Google Scholar]
- 205. Muttenthaler M., King G. F., Adams D. J., Alewood P. F., Nat. Rev. Drug Discovery 2021, 20, 309. [DOI] [PubMed] [Google Scholar]
- 206. Glaser V., Genet. Eng. Biotechnol. News 2013, 33, 32. [Google Scholar]
- 207. L. Otvos, Jr. , Wade J. D., Front. Chem. 2014, 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Park M., Jardetzky T. S., Barron A. E., Biopolymers 2011, 96, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Han Y., Gao Z., Chen L., Kang L., Huang W., Jin M., Wang Q., Bae Y. H., Acta Pharm. Sin. B 2019, 9, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Ibrahim Y. H. Y., G. Regdon, Jr. , Hamedelniel E. I., Sovany T., DARU J. Pharm. Sci. 2020, 28, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Renukuntla J., Vadlapudi A. D., Patel A., Boddu S. H., Mitra A. K., Int. J. Pharm. 2013, 447, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Zizzari A. T., Pliatsika D., Gall F. M., Fischer T., Riedl R., Drug Discovery Today 2021, 26, 1097. [DOI] [PubMed] [Google Scholar]
- 213. Kumar V. A., Liu Q., Wickremasinghe N. C., Shi S., Cornwright T. T., Deng Y., Azares A., Moore A. N., Acevedo‐Jake A. M., Agudo N. R., Pan S., Woodside D. G., Vanderslice P., Willerson J. T., Dixon R. A., Hartgerink J. D., Biomaterials 2016, 98, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Kumar V. A., Taylor N. L., Shi S., Wang B. K., Jalan A. A., Kang M. K., Wickremasinghe N. C., Hartgerink J. D., ACS Nano 2015, 9, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Kumar V. A., Taylor N. L., Shi S., Wickremasinghe N. C., D'Souza R. N., Hartgerink J. D., Biomaterials 2015, 52, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Kumar V. A., Wang B. K., Kanahara S. M., Exp. Biol. Med. (Maywood) 2016, 241, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Nguyen P. K., Gao W., Patel S. D., Siddiqui Z., Weiner S., Shimizu E., Sarkar B., Kumar V. A., ACS Omega 2018, 3, 5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Sarkar B., Ma X., Agas A., Siddiqui Z., Iglesias‐Montoro P., Nguyen P. K., Kim K. K., Haorah J., Kumar V. A., Chem. Eng. J. 2021, 408, 127295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Siddiqui Z., Sarkar B., Kim K. K., Kadincesme N., Paul R., Kumar A., Kobayashi Y., Roy A., Choudhury M., Yang J., Shimizu E., Kumar V. A., Acta Biomater. 2021, 126, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Wickremasinghe N. C., Kumar V. A., Hartgerink J. D., Biomacromolecules 2014, 15, 3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Pollaro L., Heinis C., MedChemComm 2010, 1, 319. [Google Scholar]
- 222. Ngambenjawong C., Gustafson H. H., Pineda J. M., Kacherovsky N. A., Cieslewicz M., Pun S. H., Theranostics 2016, 6, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Evans B. J., King A. T., Katsifis A., Matesic L., Jamie J. F., Molecules 2020, 25, 2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Gentilucci L., De Marco R., Cerisoli L., Curr. Pharm. Des. 2010, 16, 3185. [DOI] [PubMed] [Google Scholar]
- 225. Di L., AAPS J. 2015, 17, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]