

Abstract
SARS‐CoV‐2 is an emerging coronavirus that causes dysfunctions in multiple human cells and tissues. Studies have looked at the entry of SARS‐CoV‐2 into host cells mediated by the viral spike protein and human receptor ACE2. However, less is known about the cellular immune responses triggered by SARS‐CoV‐2 viral proteins. Here, we show that the nucleocapsid of SARS‐CoV‐2 inhibits host pyroptosis by blocking Gasdermin D (GSDMD) cleavage. SARS‐CoV‐2‐infected monocytes show enhanced cellular interleukin‐1β (IL‐1β) expression, but reduced IL‐1β secretion. While SARS‐CoV‐2 infection promotes activation of the NLRP3 inflammasome and caspase‐1, GSDMD cleavage and pyroptosis are inhibited in infected human monocytes. SARS‐CoV‐2 nucleocapsid protein associates with GSDMD in cells and inhibits GSDMD cleavage in vitro and in vivo. The nucleocapsid binds the GSDMD linker region and hinders GSDMD processing by caspase‐1. These insights into how SARS‐CoV‐2 antagonizes cellular inflammatory responses may open new avenues for treating COVID‐19 in the future.
Keywords: GSDMD, inflammasome, nucleocapsid, pyroptosis, SARS‐CoV‐2
Subject Categories: Autophagy & Cell Death; Microbiology, Virology & Host Pathogen Interaction
Despite caspase‐1 and NLRP3 inflammasome activation, pyroptotic cell death of SARS‐CoV2‐infected human monocytes is prevented by cleavage site masking in the key effector GSDMD by a viral protein.

Introduction
COVID‐19, a disease impelled by the coronavirus SARS‐CoV‐2, has now been becoming a worldwide pandemic (Au et al, 2020). Compared with other coronavirus family members such as SARS‐CoV‐1 and MERS‐CoV, which lead to a high mortality rate, the lethality rate caused by SARS‐CoV‐2 is a little more than 2% (Petrosillo et al, 2020). Although SARS‐CoV‐2 shows 79.5% sequence similarity to SARS‐CoV‐1 (Rossi et al, 2020), its triggered symptoms have pronounced differences from SARS‐CoV‐1‐induced syndromes in respect of time of latent period, spectrum of plasma cytokines and severity of immune cell response (Consiglio et al, 2020; Long et al, 2020; Xu et al, 2020; Zhang et al, 2020; V'Kovski et al, 2021). Cytokine storm is considered to be responsible for the severe symptoms in ICU patients infected with SARS‐CoV‐2 (Zhou et al, 2020). However, a recent study shows that cytokine levels in critically ill COVID‐19 patients are lower than those in sepsis patients (Kox et al, 2020). Whether cytokine storm dominates the pathogenesis of COVID‐19 and what kinds of cells produce these cytokines are still elusive. Many SARS‐CoV‐1‐originated proteins have been shown to interact with the host immune factors to favor the viral survival (Devaraj et al, 2007; Narayanan et al, 2008; Chen et al, 2009; Freundt et al, 2009). Whether SARS‐CoV‐2‐derived proteins exert the same role with their SARS‐CoV‐1 counterparts still needs further investigation.
Inflammasomes are a macromolecular machinery consisting of pro‐caspase‐1, ASC, and inflammasome nucleators such as NOD‐like receptors (NLRs), AIM2, and pyrin. So far, there are five inflammasome‐nucleating NLRs comprising NLRP1, NLRP3, NLRC4, NLRP6, and NLRP9b (Xue et al, 2019). Among these, NLRP3 is of great importance for the myeloid cells to counter infectious pathogens including viruses and bacteria (Kanneganti et al, 2006; Allen et al, 2009; Zhao & Zhao, 2020). Generally, myeloid cells need two signals to fully activate the NLRP3 inflammasome (Latz et al, 2013; Wen et al, 2013; Guo et al, 2015). One signal is LPS that triggers up‐regulations of NLRP3 and pro‐IL‐1β, and the second signals are ATP and nigericin that initiate inflammasome assembly. Inflammasome activation leads to pro‐IL‐1β process and subsequent IL‐1β secretion, which is essential for activating multiple cells including neutrophils, T cells, and B cells (Tominaga et al, 2000). Another result of inflammasome activation is pyroptosis mediated by Gasdermin family members (Fernandes‐Alnemri et al, 2007; Kayagaki et al, 2015; Shi et al, 2015; Wallach et al, 2016). Pyroptosis destroys the living apartment for invaders and serves as a defending mechanism to restrict pathogen spreading (Aglietti & Dueber, 2017). Human monocytes have a high expression level of NLRP3 and own multiple inflammasome‐activating pathways, rendering them reacting quickly to outside stimuli (Wang et al, 2013; Gaidt et al, 2016; Gritsenko et al, 2020). Recently, a single‐cell RNA‐sequencing study identifies monocyte changes in COVID‐19 patient PBMCs (Bost et al, 2020), whether these ready‐to‐go monocytes play a role in COVID‐19 pathogenesis is still unclear, and the exact role of inflammasome in this process also needs to be unraveled.
GSDMD belongs to the Gasdermin family and is cleaved by the caspase‐1 dimers post‐inflammasome activation, leading to cell membrane permeability, cell content leakage and finally cell death termed pyroptosis (Kayagaki et al, 2015; Boucher et al, 2018; Chan & Schroder, 2020). Caspase‐1 has limited cellular targets because of its rigid substrate selection (Ramirez et al, 2018). GSDMD is one of the few caspase‐1 targets inside cells (Kayagaki et al, 2015; Shi et al, 2015). Active caspase‐1 dimers bind to the GSDMD C‐terminus and cleave the tetrapeptide after GSDMD D275 (Wang et al, 2020). Freed GSDMD N‐terminus binds to lipids and oligomerizes spontaneously to form large pores on the membrane (Aglietti et al, 2016; Ding et al, 2016; Liu et al, 2016; Sborgi et al, 2016). Viral proteins could hijack GSDMD to prevent host pyroptosis. EV71 3C cleaves GSDMD between Q193 and G194, leaving a truncated GSDMD N fragment that loses the capacity to form pores on membrane (Lei et al, 2017). Whether SARS‐CoV‐2‐derived proteins restrict GSDMD oligomerization and thereby inhibit pyroptosis still needs further survey.
Here, we show that IL‐1β secretion and pyroptosis are inhibited in SARS‐CoV‐2‐infected human monocytes. SARS‐CoV‐2 nucleocapsid specifically binds GSDMD in monocytes and protects GSDMD from oligomerization. For molecular mechanisms, SARS‐CoV‐2 nucleocapsid binds GSDMD linker region, hindering GSDMD cleavage by the activated caspase‐1 dimers. Mutation of the GSDMD linker region abolishes the cleavage restraint effect of nucleocapsid. Our study uncovers a distinctive feature of nucleocapsid in antagonizing cellular immune responses that might serve as a target for COVID‐19 treatment in the future.
Results
SARS‐CoV‐2‐infected monocytes exhibit abnormal inflammatory status
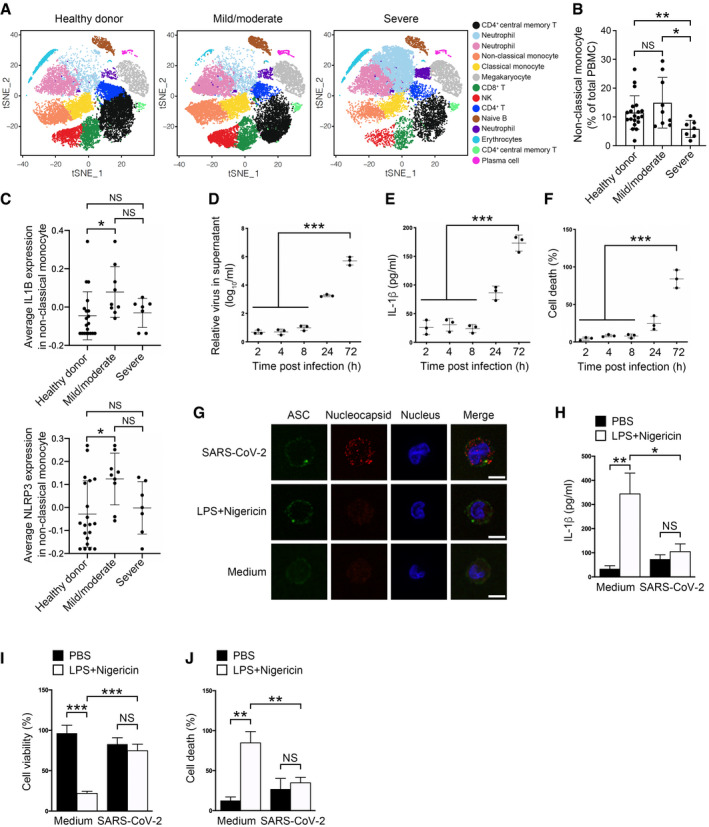
Previous studies showed up‐regulated IL1B expression levels in monocytes of COVID‐19 patients (Wen et al, 2020; Wauters et al, 2021). However, serum IL‐1β levels were not elevated post‐SARS‐CoV‐2 infection, even in severe COVID‐19 patients (Chen et al, 2020; Merad & Martin, 2020). To figure out the cause of this discrepancy, we reanalyzed recently published single‐cell RNA‐sequencing data of COVID‐19 patient PBMCs (GSE163668) (Combes et al, 2021). Monocytes are one of the main cell types secreting IL‐1β, which is processed by activated inflammasomes, especially NLRP3 inflammasome in the case of viral infection (Kanneganti et al, 2006). We noticed that cell number of non‐classical monocytes was not decreased in mild/moderate COVID‐19 patients compared with healthy controls (Fig 1A and B and Appendix Fig S1A). Moreover, IL1B was strongly expressed in non‐classical monocytes of mild/moderate COVID‐19 patients compared with healthy donors (Fig 1C), suggesting an involvement of this cytokine in the SARS‐CoV‐2 infection of mild/moderate patients. Although neutrophils also expressed IL1B, the average IL1B expression levels were not varied significantly among healthy controls and COVID‐19 patients (Appendix Fig S1B). Pro‐IL‐1β is processed by inflammasomes comprising NOD‐like receptors (Guo et al, 2015). Correspondingly, the NOD‐like receptor NLRP3 showed a similar rising trend in mild/moderate non‐classical monocytes as IL1B did (Fig 1C), while other NLRs such as NLRP1, NLRP4, NLRP6, or NLRP9 did not (Appendix Fig S1C). Indeed, inflammasome players such as GSDMD, NEK7, ASC, and caspase‐1 were ubiquitously expressed in human monocytes (Appendix Fig S1D), indicating an intact inflammasome signaling pathway exists in mild/moderate patient monocytes.
Figure 1. IL‐1β secretion is impaired in SARS‐CoV‐2‐infected monocytes.
-
A–CReanalysis of published single‐cell sequencing data (GSE163668) of SARS‐CoV‐2 patient PBMCs. t‐Distributed Stochastic Neighbor Embedding (tSNE)‐based visualization of single cells containing PBMCs from healthy donors, and SARS‐CoV‐2‐infected mild/moderate and severe patients is shown on the left panel, and cluster annotations are shown on the right (A). Percentage of non‐classical monocytes in each sample group were calculated (B). Relative average expression levels of IL‐1B and NLRP3 in non‐classical monocytes of each group were calculated (C). n = 20 for healthy donor, n = 9 for mild/moderate, n = 7 for severe. For (B, C), each dot represents an individual. For (B, C), the Brown–Forsythe test and the Welch ANOVA test were used and data were shown as means ± SD. *P < 0.05 and **P < 0.01; NS, non‐significant.
-
D–FCD14+ monocytes sorted from healthy PBMCs were infected with SARS‐CoV‐2 viruses at an MOI of 1 for 1 h, followed by washing away extracellular viruses and further culture to meet the indicated infection time. Supernatants were collected and subjected to RT–PCR analysis of viral RNAs (D) and ELISA of IL‐1β (E). The percentage of cells undergoing cell death was calculated through checking released LDH in the supernatants (F). For (D–F), Student's t‐test was used and data were shown as means ± SD of three technical replicates. ***P < 0.001; results were repeated three times with similar results.
-
GSorted human monocytes were incubated with control medium or SARS‐CoV‐2 viruses at an MOI of 1 for 1 h, followed by washing away extracellular viruses and further culture for 1 h. Cells were fixed and stained with the indicated antibodies. Otherwise, monocytes were challenged with 1 μg/ml LPS for 3 h and 10 μM nigericin for 30 min, followed by fixing and staining with the indicated antibodies. Treatment with medium from uninfected Vero cells served as a control. Scale bar: 5 μm.
-
H–JSupernatants from control or SARS‐CoV‐2‐infected CD14+ monocytes with or without LPS and nigericin stimulation were collected and subjected to an ELISA for IL‐1β levels (H). Cell viability in cell pellets was examined using a cell viability assay through checking ATP levels inside cells (I). The percentage of cells undergoing pyroptosis was calculated through checking released LDH in the supernatants (J). For (H–J), Student's t‐test was used and data were shown as means ± SD of three technical replicates. *P < 0.05; **P < 0.01; and ***P < 0.001; NS, non‐significant. Results were repeated three times with similar results.
Data information: For (B) and (C), the Brown–Forsythe test and the Welch ANOVA test were used and data were shown as means ± SD. For (D–F) and (H–J), Student's t‐test was used and data were shown as means ± SD of three technical replicates. *P < 0.05; **P < 0.01; and ***P < 0.001; NS, non‐significant. For (D–J), results were repeated three times with similar results.
We therefore sought to determine the inflammatory status in human monocytes infected with SARS‐CoV‐2 viruses. We infected human monocytes with SARS‐CoV‐2 and examined the inflammasome status in these cells. Albeit SARS‐CoV‐2 viruses entered into monocytes and replicated there (Fig 1D), secreted IL‐1β were hardly detected in the supernatant at the early times post‐infection (Fig 1E). Moreover, cell death was not prevalent in SARS‐CoV‐2‐infected cells until 24 h post‐infection (Fig 1F). NLRP3 activation assembles ASC to aggregates that can be used as an inflammasome activation marker (Gaidt et al, 2016). ASC specks appeared in virus‐infected cells as early as 2 h post‐infection (Fig 1G), suggesting an induction of inflammasomes by SARS‐CoV‐2 infection. Nigericin administration following LPS priming serves as a means to activate classical NLRP3 inflammasome and induce pyroptosis in human monocytes (Gaidt et al, 2016). When we isolated human monocytes and checked the protein level of secreted IL‐1β, we found that SARS‐CoV‐2‐infected monocytes released less IL‐1β than controls treated with LPS and nigericin (Fig 1H). Treatment of SARS‐CoV‐2‐infected monocytes with LPS and nigericin did not further increase IL‐1β secretion (Fig 1H). This was in agreement with previous reports that COVID‐19 patients had low IL‐1β levels in the periphery (Chen et al, 2020). IL‐1β is released from cell pores formed by cleaved GSDMD that causes pyroptosis. We then checked the cell viability of these SARS‐CoV‐2‐infected cells. There were more viable cells in SARS‐CoV‐2‐infected monocytes than LPS‐ and nigericin‐stimulated control cells as monitored by calculating ATPs in living cells (Fig 1I). LDH releasing assay also showed less cells underwent pyroptosis in SARS‐CoV‐2‐infected monocytes than control cells with NLRP3 inflammasome activation (Fig 1J). LPS and nigericin administration did not give rise to further cell cytotoxicity in SARS‐CoV‐2‐infected monocytes (Fig 1I and J), suggesting a pyroptosis inhibition in these cells. In all, these results imply that IL‐1β secretion and pyroptosis are blocked in SARS‐CoV‐2‐infected monocytes although the inflammasome apparatus is activated.
SARS‐CoV‐2 nucleocapsid inhibits pyroptosis in human monocytes
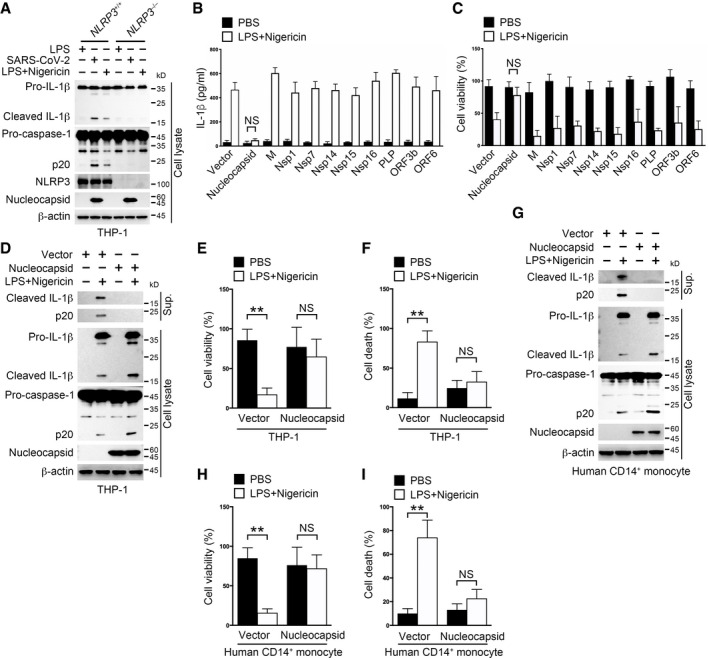
To fully elucidate the mechanism underlying the observed abnormal inflammasome signaling in SARS‐CoV‐2‐infected monocytes, we took use of the canonical inflammasome activator LPS and nigericin to mimic SARS‐CoV‐2‐induced inflammasome activation in a human monocyte cell line THP‐1. Indeed, SARS‐CoV‐2 virus induced an NLRP3‐dependent inflammasome activation just as LPS and nigericin did (Fig 2A). Several SARS‐CoV‐1‐encoded proteins were reported to participate in hosts' innate immune responses (Devaraj et al, 2007; Narayanan et al, 2008; Chen et al, 2009; Freundt et al, 2009). We then selected 10 SARS‐CoV‐1 counterparts in SARS‐CoV‐2 and tested their impacts on inflammasome activation in THP‐1 cells. We generated THP‐1 cells stably expressing these genes (Appendix Fig S2A) and checked their impacts on inflammasome activation post‐LPS and nigericin stimulation. Mature IL‐1β secretion was observed in cells expressing SARS‐CoV‐2 genes except for the nucleocapsid (Fig 2B). Accordingly, THP‐1 cells expressing SARS‐CoV‐2 genes but not nucleocapsid underwent pyroptosis following inflammasome activation (Fig 2C), resembling the phenomenon observed in SARS‐CoV‐2‐infected monocytes (Fig 1H and I). Nucleocapsid is abundantly present in the viral particles and immediately released to host cytosol upon viral infection (Carlson et al, 2020), which might function to restrain cellular processes such as inflammasome activation besides its nucleic acid binding task. To test this hypothesis, we generated THP‐1 cells stably expressing exogenous nucleocapsid and examined the inflammasome signaling events post‐LPS and nigericin stimulation. Although cells expressing nucleocapsid or not had inflammasome activated post‐LPS and nigericin stimulation (Fig 2D), nucleocapsid‐overexpressing cells secreted less IL‐1β and had more viable cells than control cells (Fig 2D–F). Similar phenomena were also observed in another human monocyte cell line U937 cells (Appendix Fig S2B and C). Moreover, SARS‐CoV‐2 nucleocapsid overexpression diminished secreted IL‐1β and increased cell viability both in THP‐1‐derived macrophages and in mouse peritoneal macrophages (Appendix Fig S2D–G), suggesting that SARS‐CoV‐2 nucleocapsid also inhibits pyroptosis in macrophages. Finally, we electroporated plasmids encoding SARS‐CoV‐2 nucleocapsid into human primary monocytes and checked the inflammasome status in these cells. Inflammasomes were activated following administration of NLRP3 inflammasome activators with or without SARS‐CoV‐2 nucleocapsid (Fig 2G). However, pyroptosis was blocked in nucleocapsid‐expressing cells (Fig 2H and I). Altogether, these results indicate that SARS‐CoV‐2 nucleocapsid prohibits pyroptosis in human monocytes.
Figure 2. IL‐1β secretion and pyroptosis are inhibited by SARS‐CoV‐2 nucleocapsid in human monocytes.
-
ANLRP3+/+ and NLRP3 −/− THP‐1 cells were incubated with SARS‐CoV‐2 at an MOI of 1 for 1 h, followed by washing away extracellular viruses and further culture for 3 h. Otherwise, cells were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Cell pellets were collected and immunoblotted with antibodies against the indicated proteins. Treatment with 1 μg/ml LPS for 3 h served as a control.
-
B, CTHP‐1 cells stably expressing the indicated SARS‐CoV‐2 genes were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Supernatants were collected for an ELISA to determine supernatant IL‐1β levels (B), and cell pellets were subjected to a cell viability assay to verify the percentage of viable cells through checking cellular ATP levels (C). For (B, C), Student's t‐test was used and data were shown as means ± SD of three technical replicates. NS, non‐significant.
-
D–FTHP‐1 cells stably expressing the nucleocapsid gene or control vector were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Supernatants and cell pellets were collected and immunoblotted with antibodies against the indicated proteins (D). The percentage of viable cells was determined through checking cellular ATP levels in cell pellets (E). The percentage of cells undergoing pyroptosis was calculated through checking released LDH in the supernatants (F). For (E, F), Student's t‐test was used and data were shown as means ± SD of three technical replicates. **P < 0.01; NS, non‐significant.
-
G–IHuman CD14+ monocytes were electroporated with plasmids encoding the nucleocapsid or control vector for 36 h, followed by stimulation with 10 μM nigericin for 30 min after pretreatment with 1 μg/ml LPS for 3 h. Supernatants and cell pellets were collected and immunoblotted with antibodies against the indicated proteins (G). The percentage of viable cells was determined through checking cellular ATP levels in cell pellets (H). The percentage of cells undergoing pyroptosis was calculated through checking released LDH in the supernatants (I). For (H, I), Student's t‐test was used and data were shown as means ± SD of three technical replicates. **P < 0.01; NS, non‐significant.
Data information: For (B–I), control vector used here was empty vector corresponded to the nucleocapsid‐containing vector. For (B, C, E, F, H, I), Student's t‐test was used and data were shown as means ± SD of three technical replicates. **P < 0.01; NS, non‐significant. Similar results were observed for at least three times.
Source data are available online for this figure.
SARS‐CoV‐2 nucleocapsid binds GSDMD
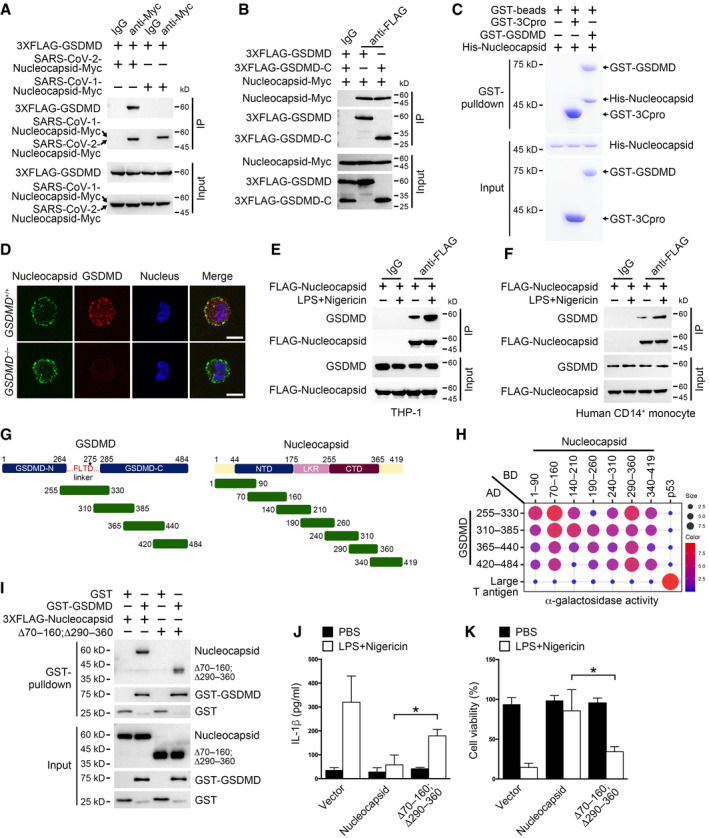
To find out how SARS‐CoV‐2 nucleocapsid participated in monocyte inflammasome regulation, we screened a mouse bone marrow cDNA library using SARS‐CoV‐2 nucleocapsid as a yeast two‐hybrid bait. SARS‐CoV‐2 nucleocapsid did not have autoactivation activity in yeast cells (Appendix Fig S3A). Among the top 24 biggest clones we screened, 14 ones contained cDNA fragments corresponding to GSDMD. Others were four clones for DDX21, three clones for C1orf122, two clones for GBP4, and one clone for G3BP1. In particular, DDX21 and G3BP1 were previously described nucleocapsid interactors (Gordon et al, 2020). The interaction between SARS‐CoV‐2 nucleocapsid and mouse GSDMD was confirmed in HEK293T cells through co‐immunoprecipitation (Appendix Fig S3B). SARS‐CoV‐2 nucleocapsid also interacted with human GSDMD in HEK293T cells, while SARS‐CoV‐1 nucleocapsid did not (Fig 3A). Human GSDMD is processed by caspase‐1 right after D275, freeing GSDMD N‐terminus from its C‐terminus‐mediated autoinhibition (Shi et al, 2015). We then checked which part of GSDMD was responsible for SARS‐CoV‐2 nucleocapsid binding through co‐transfecting nucleocapsid and GSDMD fragments into HEK293T cells. We found that SARS‐CoV‐2 nucleocapsid associated with GSDMD C‐terminus but not the N‐terminus (Appendix Fig S3C). Moreover, we collected GSDMD, its C‐terminus, and nucleocapsid from HEK293T cells individually and incubated these proteins together for an in vitro co‐immunoprecipitation assay. SARS‐CoV‐2 nucleocapsid showed strong affinity for full‐length GSDMD and GSDMD C‐terminus (Fig 3B). To test whether the interaction between SARS‐CoV‐2 nucleocapsid and GSDMD was direct, we purified recombinant SARS‐CoV‐2 nucleocapsid and GSDMD from Escherichia coli and performed a pull‐down assay. We found that SARS‐CoV‐2 nucleocapsid directly interacted with GSDMD as visualized by both Coomassie Blue staining and immunoblotting (Fig 3C and Appendix Fig S3D).
Figure 3. SARS‐CoV‐2 nucleocapsid interacts with GSDMD.
-
APlasmids encoding FLAG‐tagged full‐length GSDMD were co‐transfected with plasmids encoding Myc‐tagged SARS‐CoV‐1 nucleocapsid or SARS‐CoV‐2 nucleocapsid into HEK293T cells for 24 h, followed by immunoprecipitation with antibody against Myc or isotype IgG. Input and precipitates were immunoblotted as indicated.
-
BPlasmids encoding FLAG‐tagged full‐length GSDMD, GSDMD C‐terminus and Myc‐tagged nucleocapsid were transfected into HEK293T cells individually. Exogenous proteins were purified using FLAG or Myc targeting antibodies coupled to magnetic beads 24 h post‐transfection. Purified proteins were then mixed and incubated for 2 h, followed by immunoprecipitation with antibody against FLAG or isotype IgG. Input and precipitates were immunoblotted as indicated.
-
CGST‐GSDMD, His‐tagged nucleocapsid, and control GST‐3C protease (GST‐3Cpro) were purified from Escherichia coli BL21, followed by a GST pull‐down assay. Input and precipitates were visualized by Coomassie Blue staining.
-
DGSDMD+/+ and GSDMD −/− THP‐1 cells were transfected with plasmids encoding nucleocapsid for 24 h, followed by cell fixation, permeabilization, and staining with antibodies against nucleocapsid and GSDMD. The nucleus was counterstained with DAPI. Scale bar: 5 μm.
-
E, FTHP‐1 (E) or human CD14+ monocytes (F) were transfected with plasmids encoding FLAG‐tagged nucleocapsid for 36 h, followed by pretreatment of 1 μg/ml LPS for 3 h and 10 μM nigericin stimulation for 30 min. Cells were lysed and immunoprecipitated with antibody against FLAG or isotype IgG. Input and precipitates were immunoblotted as indicated.
-
GSchemes for GSDMD (left panel) and SARS‐CoV‐2 nucleocapsid (right panel) truncations.
-
HPlasmids encoding DNA binding domain (BD)‐tagged nucleocapsid truncations and activation domain (AD)‐tagged GSDMD C‐terminus truncations were co‐transfected into yeast strain AH109, and double‐positive clones were grown on plates containing X‐α‐gal. The intensity of blue color was calculated and shown as scores for both dot size and color. Plasmids encoding BD‐p53 and AD‐large T antigen were co‐transfected as a positive control.
-
IGST‐GSDMD and control GST were mixed with HEK293T cell lysate containing FLAG‐tagged full‐length nucleocapsid or truncated nucleocapsid lacking amino acids 70–160 and 290–360, followed by a GST pull‐down assay. Input and precipitates were immunoblotted as indicated.
-
J, KTHP‐1 cells stably expressing full‐length nucleocapsid or truncated nucleocapsid lacking amino acids 70–160 and 290–360 were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. An ELISA was performed to determine supernatant IL‐1β levels (J), and a cell viability assay was conducted to verify the percentage of viable cells through checking cellular ATP levels (K). For (J) and (K), control vector used here was empty vector corresponded to the nucleocapsid‐containing vector. For (J) and (K), Student's t‐test was used and data were shown as means ± SD of three technical replicates. *P < 0.05. Results were repeated three times with similar results.
Data information: Control vector used here was empty vector corresponded to the nucleocapsid‐containing vector. Student's t‐test was used, and data were shown as means ± SD of three technical replicates. *P < 0.05. Results were repeated three times with similar results.
Source data are available online for this figure.
To validate the interaction between SARS‐CoV‐2 nucleocapsid and GSDMD under physiological conditions, we transfected plasmids encoding SARS‐CoV‐2 nucleocapsid into THP‐1 cells and checked nucleocapsid's co‐localization with endogenous GSDMD through confocal imaging. In THP‐1 cells, exogenous nucleocapsid co‐localized with GSDMD but not COXIV (Fig 3D and Appendix Fig S3E). Moreover, SARS‐CoV‐2 nucleocapsid interacted with endogenous GSDMD in THP‐1 cells and their association was elevated post‐inflammasome activation (Fig 3E). Moreover, electroporation of plasmids encoding nucleocapsid into human primary monocytes led to the association between nucleocapsid protein and endogenous GSDMD, and inflammasome ignition strengthened this association (Fig 3F). We also found that virus nucleocapsid protein interacted with endogenous GSDMD in human primary monocytes infected with SARS‐CoV‐2 viruses (Appendix Fig S3F).
We then mapped the GSDMD binding regions on SARS‐CoV‐2 nucleocapsid through a yeast two‐hybrid assay. All seven nucleocapsid fragments were shown to interact with the GSDMD C‐terminus (Fig 3G and H), indicating the existence of multiple interfaces between these two proteins. Among the nucleocapsid truncations, fragments containing amino acids 70–160 and 290–360 had the highest binding affinity for GSDMD than others (Fig 3H). We then generated nucleocapsid mutant lacking amino acids 70–160 and 290–360 and tested its binding ability to GSDMD through a pull‐down assay. We found that nucleocapsid lacking amino acids 70–160 and 290–360 had a slightly weaker association with GSDMD than full‐length nucleocapsid (Fig 3I). Furthermore, THP‐1 cells carrying this nucleocapsid mutant had more amount of secreted IL‐1β than full‐length nucleocapsid‐expressing cells post‐inflammasome activation (Appendix Fig S3G and Fig 3J). Consistently, viable cells were decreased in THP‐1 cells expressing nucleocapsid mutant (Fig 3K), suggesting that GSDMD binding by nucleocapsid amino acids 70–160 and 290–360 is essential for nucleocapsid‐mediated pyroptosis inhibition. We generated THP‐1 cells stably expressing nucleocapsid NTD or CTD domain and tested their impacts on the IL‐1β secretion and cell viability. We found that neither NTD nor CTD possessed the ability to suppress IL‐1β secretion or pyroptosis (Appendix Fig S3H–J), suggesting that multiple interfaces of nucleocapsid are involved to inhibit GSDMD. Altogether, our results show that SARS‐CoV‐2 nucleocapsid binds GSDMD.
SARS‐CoV‐2 nucleocapsid inhibits GSDMD cleavage
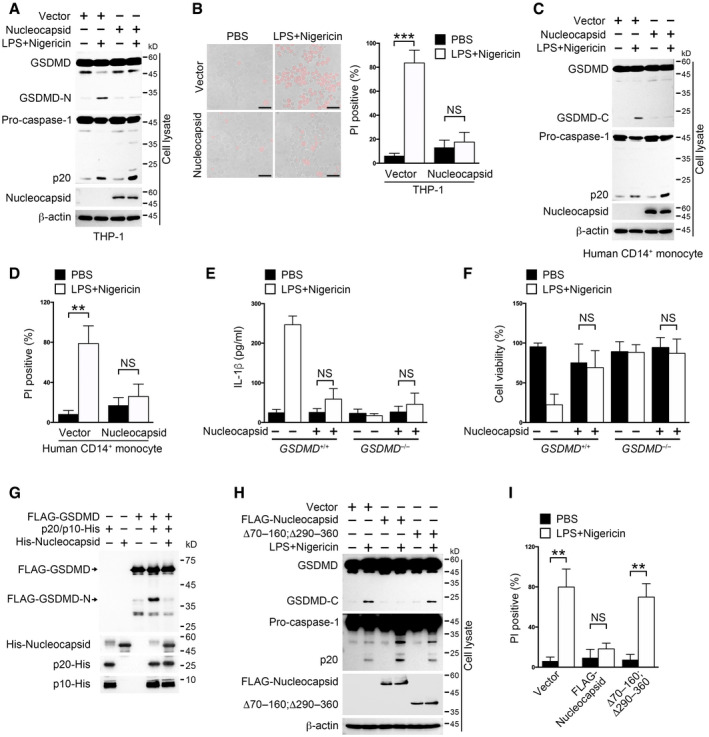
Processed GSDMD N‐terminus‐induced pores on cell membrane are required for mature IL‐1β releasing (Evavold et al, 2018). Considering the association between SARS‐CoV‐2 nucleocapsid and GSDMD, we sought to examine whether nucleocapsid had an effect on GSDMD processing. We generated nucleocapsid‐expressing THP‐1 cells and checked the protein status of GSDMD post‐inflammasome activation. We found that nucleocapsid overexpression inhibited GSDMD cleavage but not caspase‐1 activation (Fig 4A). We generated THP‐1 cells stably expressing SARS‐CoV‐1 nucleocapsid and checked the NLRP3 inflammasome activation status in these cells post‐LPS and nigericin stimulation. The protein levels of caspase‐1 p20 and GSDMD‐cleaved fragment were comparable between SARS‐CoV‐1 nucleocapsid‐expressing cells and control cells, suggesting that SARS‐CoV‐1 nucleocapsid does not inhibit GSDMD maturation (Appendix Fig S4A). Consistently, less cells were undergoing pyroptosis in nucleocapsid‐expressing cells than control ones (Fig 4B). To further validate the inhibitory role of nucleocapsid on GSDMD processing, we transfected nucleocapsid into human primary monocytes and examined GSDMD integrity through immunoblotting. Nucleocapsid overexpression inhibited GSDMD cleavage and pyroptosis in human CD14+ monocytes as the same as in THP‐1 cells (Fig 4C and D). Furthermore, we generated GSDMD knockout THP‐1 cells and introduced plasmids encoding nucleocapsid into these cells (Appendix Fig S4B). Nucleocapsid overexpression had almost the same effects on IL‐1β secretion or cell viability as GSDMD knockout did (Fig 4E and F), suggesting that nucleocapsid effectively targets GSDMD for inhibiting IL‐1β secretion and pyroptosis inside cells.
Figure 4. SARS‐CoV‐2 nucleocapsid protects GSDMD from cleavage by caspase‐1.
-
A, BTHP‐1 cells stably expressing nucleocapsid or control vector were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Cells were either immunoblotted with antibodies against the indicated proteins (A) or stained with propidium iodide (PI) (B, left). Scale bar: 20 μm. The percentage of PI‐positive cells was calculated (B, right). Student's t‐test was used, and data were shown as means ± SD of three technical replicates. ***P < 0.001; NS, non‐significant. Results were repeated three times with similar results.
-
C, DSorted human CD14+ monocytes were electroporated with plasmids encoding nucleocapsid or control vector for 36 h and then stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Cells were immunoblotted with antibodies against the indicated proteins (C). Otherwise, cells were stained with PI and the percentage of PI‐positive cells was calculated (D). Student's t‐test was used, and data were shown as means ± SD of three technical replicates. **P < 0.01; NS, non‐significant. Results were repeated three times with similar results.
-
E, FGSDMD+/+ and GSDMD −/− THP‐1 cells stably expressing nucleocapsid or control vector were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Supernatants were collected for an ELISA to determine IL‐1β levels (E). Cell pellets were subjected to a cell viability assay through checking cellular ATP levels (F). For (E, F), Student's t‐test was used. Data were shown as means ± SD of three technical replicates. NS, non‐significant. Results were repeated three times with similar results.
-
GAn in vitro cleavage assay was performed by incubating GSDMD and caspase‐1 p20/p10 complex with or without the presence of nucleocapsid, followed by immunoblotting with antibodies against the indicated protein tags. FLAG‐GSDMD was immunoprecipitated from HEK293T cells using anti‐FLAG‐coupled magnetic beads. His‐tagged p20, p10, and nucleocapsid were purified from E. coli strain BL21.
-
H, ITHP‐1 cells stably expressing full‐length nucleocapsid or mutant lacking amino acids 70–160 and 290–360 were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Cells were either immunoblotted with antibodies against the indicated proteins (H). Cells were also stained with PI and the percentage of PI‐positive cells was calculated (I). Student's t‐test was used, and data were shown as means ± SD of three technical replicates. **P < 0.01; NS, non‐significant. Results were repeated three times with similar results.
Data information: For (A–F, H, I), control vector used here was empty vector corresponded to the nucleocapsid‐containing vector. For (B, D–F, I), Student's t‐test was used and data were shown as means ± SD of three technical replicates. **P < 0.01 and ***P < 0.001; NS, non‐significant. Similar results were observed for at least three times.
Source data are available online for this figure.
To decipher how nucleocapsid affected GSDMD processing, we purified GSDMD, nucleocapsid, and p20/p10 from HEK293T cells and E. coli, respectively (Shi et al, 2015). An in vitro cleavage assay was performed using these purified proteins (Fig 4G). Nucleocapsid addition kept GSDMD intact from p20/p10‐mediated processing, suggesting that nucleocapsid directly inhibits caspase‐1‐mediated GSDMD cleavage. To test whether the nucleocapsid truncation lacking amino acids 70–160 and 290–360 lost the ability to inhibit GSDMD cleavage, we transfected the nucleocapsid truncation and GSDMD into THP‐1 cells and triggered inflammasome activation using canonical inflammasome activators. We found that the nucleocapsid truncation lost the ability to protect GSDMD from cleavage (Fig 4H). Moreover, pyroptosis was not prohibited by nucleocapsid mutant (Fig 4I), further suggesting that nucleocapsid undermines GSDMD processing in human monocytes. In all, our results indicate that SARS‐CoV‐2 nucleocapsid directly inhibits GSDMD cleavage mediated by caspase‐1.
SARS‐CoV‐2 nucleocapsid protects GSDMD linker from being cut by caspase‐1
The cleavage inhibition of GSDMD by SARS‐CoV‐2 nucleocapsid might be caused by two means. One was that nucleocapsid and the caspase‐1 p20/p10 complex competitively bound GSDMD on the same site and nucleocapsid had a higher binding affinity for GSDMD than p20/p10, avoiding GSDMD being cut by caspase‐1. The other was that SARS‐CoV‐2 nucleocapsid bound around D275 of the GSDMD linker region, prohibiting the p20/p10's recognition of the tetrapeptide. To test these possibilities, we firstly purified recombinant GSDMD, nucleocapsid and p20/p10 from E. coli and performed a GST pull‐down assay. We found that GSDMD‐associated p20/p10 was not diminished upon the increase in nucleocapsid (Fig 5A), suggesting that nucleocapsid does not competitively repel p20/p10 complex from GSDMD. We also examined the p20/p10 binding sites on GSDMD. Based on the previously reported structure of the GSDMD and the p20/p10 complex (Appendix Fig S5A), we speculated those amino acids (aa) 302–310 and 350–368 of GSDMD were required for its interaction with the p20/p10 complex. We performed a GST pull‐down assay with recombinant GSDMD, GSDMD truncations lacking aa 302–310, aa 350–368, or both, and p20/p10. GSDMD truncation without aa 302–310 and aa 350–368 completely lost its capacity to bind the p20/p10 complex (Appendix Fig S5B). Moreover, these GSDMD truncations diminished the cleavage efficacy of the p20/p10 complex in HEK293T cells (Fig 5B). However, GSDMD truncations lacking the p20/p10 interacting regions were still able to interact with nucleocapsid either in an in vitro pull‐down assay or in HEK293T cells (Fig 5C and Appendix Fig S5C), so were the GSDMD C‐terminus truncations (Appendix Fig S5D and E). These results suggest that nucleocapsid‐mediated cleavage inhibition of GSDMD is not caused by the nucleocapsid occupation of p20/p10 binding sites on GSDMD.
Figure 5. SARS‐CoV‐2 nucleocapsid protects GSDMD linker region from caspase‐1 mediated cleavage.
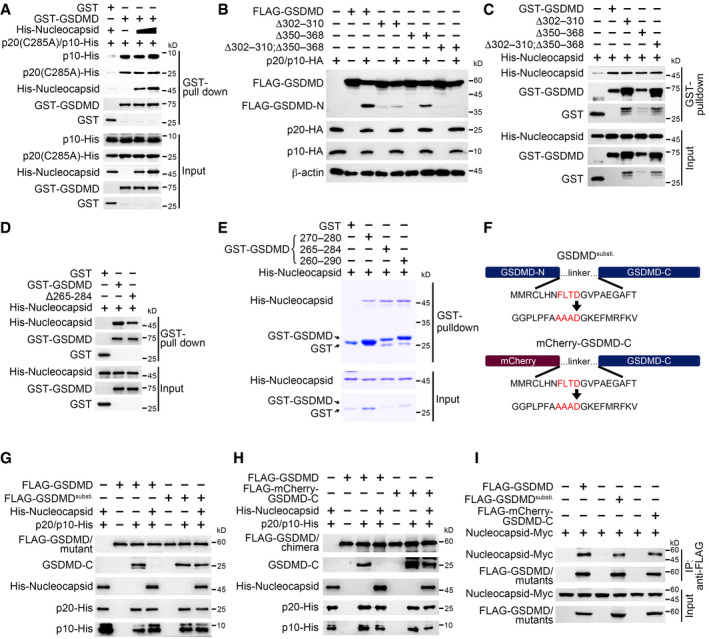
-
AGST or GST‐GSDMD, p20 (C285A)‐His, p10‐His, and increasing amount of His‐nucleocapsid were incubated for a GST pull‐down assay, followed by immunoblotting with antibodies against the indicated protein tags.
-
BA cleavage assay was performed through transfecting GSDMD and its mutants with caspase‐1 p20/p10 complex into HEK293T cells, followed by immunoblotting with antibodies against the indicated protein tags. β‐Actin served as a loading control.
-
CA GST pull‐down assay was performed through incubating GST‐tagged GSDMD and mutants with His‐tagged nucleocapsid, followed by immunoblotting with antibodies against the indicated protein tags. GST‐GSDMD, GST‐GSDMD mutants, and His‐nucleocapsid were purified from Escherichia coli strain BL21.
-
DGST‐GSDMD and its mutant lacking amino acids 265–284 were mixed with His‐tagged nucleocapsid, followed by a GST pull‐down assay. Input and precipitates were immunoblotted with antibodies against the indicated protein tags.
-
EGST‐tagged GSDMD linker containing amino acids 270–280, 265–284, or 260–290 was incubated with His‐tagged nucleocapsid, followed by a GST pull‐down assay visualized with Coomassie blue staining.
-
FSchemes for GSDMD linker substitution and GSDMD chimera. GSDMD linker spanning amino acids 265–284 was replaced by a mCherry sequence except for the amino acid D275 (GSDMDsusti., upper panel). mCherry‐GSDMD‐C chimera was generated by displacing GSDMD N‐terminus with mCherry (lower panel). The linker was also changed as above.
-
G, HWild‐type GSDMD and GSDMDsusti. (G) or mCherry‐GSDMD‐C (H) was purified from HEK293T cells and incubated with recombinant p20/p10 complex and His‐tagged nucleocapsid in a cleavage buffer, followed by immunoblotting with antibodies against the indicated protein tags or antibody against GSDMD C‐terminus.
-
IPlasmids encoding wild‐type GSDMD, GSDMDsusti., or mCherry‐GSDMD‐C were co‐transfected with nucleocapsid into HEK293T cells, followed by immunoprecipitation with antibody against FLAG. Input and precipitates were immunoblotted with antibodies against the indicated protein tags.
Data information: Experiments were repeated three times with similar results.
Source data are available online for this figure.
We then sought to check whether nucleocapsid bound the GSDMD linker region for GSDMD protection. We made a GSDMD truncation lacking the linker (GSDMD Δ265–284) and examined its interaction with nucleocapsid. GSDMD without the linker bound slightly less nucleocapsid than intact GSDMD (Fig 5D and Appendix Fig S5F), suggesting that the GSDMD linker is indeed involved in the association of these two proteins. To test the direct interaction between nucleocapsid and the GSDMD linker, we purified three GST‐fused linkers with different lengths for a GST pull‐down assay. We found that all the three linkers interacted with nucleocapsid regardless of the linker lengths (Fig 5E), implying that GSDMD linker around D275 is specifically recognized by nucleocapsid. To test whether nucleocapsid occupied GSDMD linker to prohibit p20/p10 recognition, we generated GSDMD mutants with the linker substituted with an unrelated sequence while keeping the cleavage site (Fig 5F). We then performed an in vitro cleavage assay using these mutants. We found that full‐length GSDMD with linker substituted was processed normally by the p20/p10 complex even in the presence of nucleocapsid (Fig 5G). Similar results were observed using the GSDMD chimera, which was a fusion of mCherry and GSDMD C‐terminus bridged with a substituted linker (Fig 5F and H). Moreover, the association between GSDMD or chimera and nucleocapsid was slightly diminished when the linker region was replaced (Fig 5I and Appendix Fig S5G). In all, our results show that SARS‐CoV‐2 nucleocapsid directly binds the linker region of GSDMD to protect GSDMD tetrapeptide from being cut by caspase‐1.
SARS‐CoV‐2 inhibits IL‐1β secretion in human monocytes
We then restored WT or linker‐substituted GSDMD into GSDMD knockout THP‐1 cells. We found that the cleavage of WT GSDMD was impaired in nucleocapsid‐expressing cells (Fig 6A). However, the processing of linker‐substituted GSDMD was consistent between vector‐ and nucleocapsid‐expressing cells (Fig 6A), suggesting that nucleocapsid cannot suppress the processing of linker‐substituted GSDMD. Accordingly, IL‐1β was secreted normally in cells expressing nucleocapsid when GSDMD linker was replaced (Fig 6B). Pyroptosis inhibition by nucleocapsid was also abolished in GSDMD linker‐substituted cells (Fig 6C and D). Similarly, SARS‐CoV‐2 infection triggered IL‐1β release in GSDMD linker‐substituted cells but not in GSDMD wild‐type cells (Fig 6E). Moreover, viable cell number of GSDMD linker‐substituted cells post‐SARS‐CoV‐2 infection was less than that of GSDMD wild‐type cells (Fig 6F), further suggesting an involvement of SARS‐CoV‐2 nucleocapsid in antagonizing GSDMD processing in human monocytes.
Figure 6. SARS‐CoV‐2 inhibits IL‐1β secretion in human monocytes.
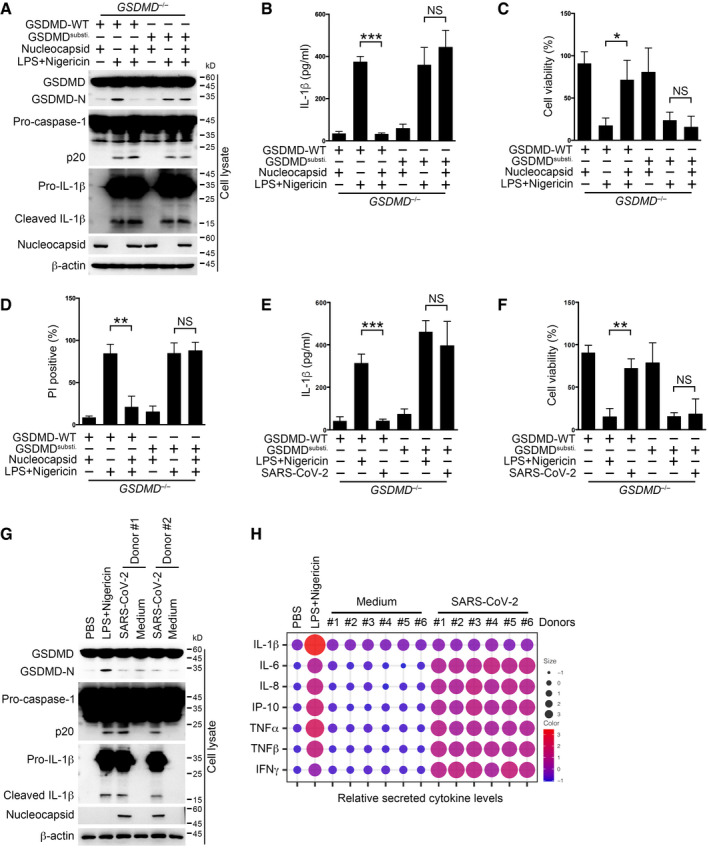
-
A–DGSDMD−/− THP‐1 cells rescued with wild‐type or linker‐substituted GSDMD were transfected with plasmids encoding nucleocapsid or control vector and then stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Cells were immunoblotted with antibodies against the indicated proteins (A), and supernatants were subjected to an ELISA to determine IL‐1β levels (B). Otherwise, cell viability in cell pellets was examined using a cell viability assay through checking ATP levels inside cells (C). Cells were stained with propidium iodide (PI), and the percentage of PI‐positive cells was calculated (D). For (B–D), Student's t‐test was used and data were shown as means ± SD of three technical replicates. *P < 0.05; **P < 0.01; and ***P < 0.001; NS, non‐significant. Results were repeated three times with similar results.
-
E, FGSDMD−/− THP‐1 cells rescued with wild‐type or linker‐substituted GSDMD were infected with SARS‐CoV‐2 at an MOI of 1 for 1 h, followed by washing away extracellular viruses and further culture for 3 h. Otherwise, cells were stimulated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h. Supernatants were subjected to an ELISA to determine IL‐1β levels (E). Otherwise, cell viability in cell pellets was examined using a cell viability assay through checking ATP levels inside cells (F). For (E, F), Student's t‐test was used and data were shown as means ± SD of three technical replicates. **P < 0.01 and ***P < 0.001; NS, non‐significant. Results were repeated three times with similar results.
-
GSorted CD14+ monocytes from donors were infected with SARS‐CoV‐2 at an MOI of 1 for 1 h, followed by washing away extracellular viruses and further culture for 3 h. Cells were immunoblotted with antibodies against the indicated proteins. CD14+ monocytes from healthy donors were treated with 10 μM nigericin for 30 min following pretreatment with 1 μg/ml LPS for 3 h and served as a positive control.
-
HDonor monocytes were infected with SARS‐CoV‐2 at an MOI of 1 for 1 h, followed by washing away extracellular viruses and further culture for 3 h. Supernatants were collected and subjected to a proteome profiling using an antibody array. Intensities for each cytokine were normalized individually and shown together as scores for both dot size and color. LPS‐ and nigericin‐treated healthy donor monocytes served as a positive control. n = 6 for each group.
Data information: For (A–D), control vector used here was empty vector corresponded to the nucleocapsid‐containing vector. For (B–F), Student's t‐test was used and data were shown as means ± SD of three technical replicates. *P < 0.05; **P < 0.01; and ***P < 0.001; NS, non‐significant. For (A–G), experiments were repeated three times with similar results.
Source data are available online for this figure.
Finally, to find out whether nucleocapsid‐mediated GSDMD protection happened in SARS‐CoV‐2 infected human primary monocytes, we collected CD14+ monocytes from healthy donors and examined the cellular signaling events post‐SARS‐CoV‐2 infection through immunoblotting. We found that GSDMD cleavage was inhibited, whereas inflammasomes were activated in SARS‐CoV‐2‐infected human monocytes (Fig 6G). Moreover, we surveyed the secreted cytokine spectrum in SARS‐CoV‐2‐infected monocytes through proteome profiling using an antibody array. We found supernatant IL‐1β level in SARS‐CoV‐2‐treated cells was as low as that in mock‐treated cells, but protein levels of other inflammatory cytokines such as IL‐6 and TNF‐α in SARS‐CoV‐2‐infected cells were higher than those in the control (Fig 6H). Together with the finding that SARS‐CoV‐2‐infected cells were hardly undergoing pyroptosis (Fig 1I and J), our results suggest that SARS‐CoV‐2 nucleocapsid prohibits IL‐1β secretion and pyroptosis in human monocytes.
Discussion
In this study, we have shown that monocytes infected with SARS‐CoV‐2 undergo inflammasome ignition accompanied by caspase‐1 activation. However, IL‐1β secretion and pyroptosis are blocked in these cells. Specifically, SARS‐CoV‐2 nucleocapsid associates with GSDMD and protects GSDMD from being cut by caspase‐1. This interaction between SARS‐CoV‐2‐encoded protein and host factor might explain the long asymptomatic infection of COVID‐19 disease and shed new light on the cure of this global epidemic in the future.
GSDMD recognition by the caspase‐1 p20/p10 complex is recently shown to rely on the GSDMD C‐terminus (Wang et al, 2020). The specific recognition of the GSDMD C‐terminus but not the canonical tetrapeptide perfectly explains the narrow substrate spectrum of inflammatory caspases (Ramirez et al, 2018). In our study, we find that SARS‐CoV‐2 nucleocapsid associates with the GSDMD C‐terminus and the linker region. The interfaces between nucleocapsid and GSDMD are multiple, and the cleavage inhibition of GSDMD requires both the NTD and CTD domains of nucleocapsid, implying a higher tertiary structure exploited by nucleocapsid to tether GSDMD and protect it. This protection is meaningful for the virus in two aspects. Firstly, SARS‐CoV‐2 coronaviruses need a long latent time to reproduce themselves and spread before the immune system finds them (Zhao & Zhao, 2020). Therefore, they need to suppress host immune responses immediately post their entry to host cells. Inflammasomes are activated by multiple exogenous stimuli, and invading viruses are strong activators of the NLRP3 inflammasome in human monocytes (Kanneganti et al, 2006; Allen et al, 2009; Zhao & Zhao, 2020). Inflammasome activation leads to maturation of IL‐1β whose secretion needs GSDMD permeabilization on the membrane (Ding et al, 2016; Gaidt et al, 2016; Chan & Schroder, 2020). GSDMD‐mediated pore forming also results in pyroptosis, releasing various inflammatory cytokines that were unfavorable to virus propagation (Chan & Schroder, 2020). SARS‐CoV‐2 nucleocapsid is released into host cytosol immediately post the entry of viruses. Nucleocapsid binding to GSDMD inhibits pyroptosis and stops the secretion of immune signaling factors to the outside. Secondly, host monocytes could store massive mature IL‐1β in the cytosol due to GSDMD inhibition. Once SARS‐CoV‐2 viruses enter into the lytic phase, temporary releasing of abundant inflammatory cytokines causes severe illness in patients. A recent study shows that a prolonged incubation of SARS‐CoV‐2 induces monocyte cell death (Ferreira et al, 2021), the mechanism of which needs further exploration. Besides, when we looked at the expression levels of inflammasome‐related genes, we found that GSDMD was significantly down‐regulated in non‐classical monocytes of severe patients. GSDMD down‐regulation may lead to other forms of cell death (Zheng et al, 2020), which might contribute to the observed decreases in non‐classical monocytes in severe patients. A better understanding of the intracellular activities of SARS‐CoV‐2 would be beneficial for the control and treatment of this disease.
In all, we find that GSDMD processing is blocked by SARS‐CoV‐2 nucleocapsid protein, which might be a target of future therapy for mild COVID‐19 patients.
Materials and Methods
Antibodies and reagents
Anti‐c‐Myc magnetic beads (88843) were purchased from Pierce. Antibodies against IL‐1β (12703), cleaved IL‐1β (83186), caspase‐1 (3866), Gasdermin D (93709), and ASC (13833) were purchased from Cell Signaling Technology. Antibodies against caspase‐1 (ab207802) and cleaved N‐terminal Gasdermin D (ab215203) were purchased from Abcam. Antibodies against SARS‐CoV‐2 Nucleocapsid (ARC2372) and 6xHis tag (MA121315) were purchased from Invitrogen. Antibodies against FLAG tag (F3165), β‐actin (A1978), and anti‐FLAG M2 magnetic beads (M8823) were purchased from Sigma‐Aldrich. Antibodies against c‐Myc (sc‐40) and GST (sc‐138) were purchased from Santa Cruz Biotechnology. Anti‐HA tag antibody (HX1820) was purchased from Huaxingbio (Beijing). HRP‐conjugated goat anti‐mouse IgG (SA00001‐1) and HRP‐conjugated goat anti‐rabbit IgG (SA00001‐2) were purchased from Proteintech Group. Alexa Fluor Plus 488‐conjugated goat anti‐mouse IgG (A32723) and Alexa Fluor 594‐conjugated goat anti‐rabbit IgG (A32740) were purchased from Invitrogen.
Glutathione Sepharose 4B resin (17075601) was purchased from Cytiva. Protein A/G PLUS‐Agarose beads (sc‐2003) were purchased from Santa Cruz Biotechnology. Propidium iodide staining solution (556463) was purchased from BD. DAPI (2879038) was purchased from PeproTech (BioGems). Nucleic acid transfection enhancer (NATE; lyec‐nate) was purchased from InvivoGen. Human Monocyte Nucleofector Kit was purchased from Lonza. Dropout (DO)/–Leu/‐Trp and DO/–Ade/‐His/‐Leu/‐Trp supplement, SMART MMLV reverse transcriptase (639524), X‐α‐Gal (630462), and carrier DNA (630440) were purchased from Clontech, Takara Bio. Random primer (hexadeoxyribonucleotide mixture, pd(N)6) (3801) was purchased from Takara Bio. Protease inhibitor cocktail (11697498001) was purchased from Roche. Total RNA extraction kit (8034111) was purchased from Dakewe (Beijing). NI‐NTA agarose beads (R90115) were purchased from Invitrogen. LPS (B46894) was purchased from Innochem (Beijing). Nigericin (481990) was purchased from Merck Millipore. Disuccinimidyl suberate (ab141274) was purchased from Abcam. CellTiter‐Glo luminescent cell viability assay kit (G7570) and CytoTox 96 non‐radioactive cytotoxicity assay kit (G1780) were purchased from Promega.
Single‐cell sequencing data reanalysis
Feature matrix data (GSE163668) of healthy donors, SARS‐CoV‐2‐positive mild/moderate and severe, were used (Combes et al, 2021): n = 20 for healthy donors, n = 9 for mild/moderate patients, and n = 7 for severe patients. Reanalysis was performed using Seurat 3.2. Doublets were removed based on UMI numbers and mitochondrial gene percentage in accordance with the following standards: gene expressed in less than three cells; genes inside one cell less than 200 ones; percentage of mitochondrial genes above 8%; and gene numbers per cell more than 8, 500 or less than 200. Variable genes were calculated using the Seurat function “FindVariableGenes” with the following criteria: x cutoff ranging from 0.05 to 8, y cutoff 0.5. 1,000 variable genes among clusters were imported into Seurat “RunPCA” pipeline, followed by non‐linear dimensional reduction of the top 12 PCs using the Seurat function “RunTSNE”. The Seurat function “FindClusters” was used to find clusters with resolution 0.5. For feature plots, normalized data were plotted on the scatter plots of tSNE_1 and tSNE_2 in the form of dot colors.
Monocyte separation
Human bloods were mixed with an equal volume of saline and then added to lymphocyte separation medium, followed by centrifugation and PBMC collection. Human PBMCs were incubated with APC‐conjugated anti‐CD14 antibody for 30 min on ice, followed by incubation with anti‐APC‐conjugated magnetic beads. Monocytes were enriched on a magnetic separator. To test the purity of sorted monocytes, cells were stained with APC‐conjugated anti‐CD14 antibody, followed by FACS examination. Cells with purity above 95% were used for subsequent experiments. Healthy blood samples were provided by the researcher volunteering. Informed consent was obtained from all subjects, and experiments conformed to related principles. The study was licensed by the Ethics Committee of Institute of Microbiology, Chinese Academy of Sciences.
Cell culture and inflammasome activation
Sorted CD14+ monocytes and THP‐1 cells were cultured in RPMI 1640 (Solarbio, Beijing) containing HEPES, l‐glutamine, sodium pyruvate, penicillin–streptomycin, 10% FBS, and 0.05 mM 2‐Mercaptoethanol. HEK293T cells were cultured in DMEM (high glucose, Biological Industries) supplemented with penicillin–streptomycin and 10% FBS. Cells were cultured at 37°C with a 5% CO2 humidified atmosphere. THP‐1 cells were differentiated into macrophages in the presence of 10 ng/ml PMA for 3 days. For inflammasome activation by canonical activators, cells were primed with 1 μg/ml LPS for 3 h, followed by stimulation with 10 μM nigericin for 30 min at 37°C. THP‐1 (TIB‐202) and U937 (CRL‐1593.2) cells were purchased from ATCC.
SARS‐CoV‐2 preparation and infection
The SARS‐CoV‐2 strain hCoV‐19/China/CAS‐B001/2020 (National Microbiology Data Center NMDCN0000102‐3 and GISAID databases EPI_ISL_514256‐7) was isolated and identified by Dr. Yuhai Bi from Institute of Microbiology, Chinese Academy of Sciences. Experiments dealing with SARS‐CoV‐2 were performed in a biosafety level 3 (BSL‐3) laboratory under the approval of Institute of Microbiology, Chinese Academy of Sciences. Viruses were replicated in Vero E6 cells. Virus containing supernatants were collected and filtered through a 100 kD centrifugal filter. Pellets were resuspended with fresh medium and filtered again. Uninfected Vero cell supernatants were collected, filtered and resuspended with fresh medium, serving as control medium. Virus titers were determined by a plaque assay through calculating plaques in semi‐solid viral infected Vero cells. Human monocytes or THP‐1 cells were incubated with SARS‐CoV‐2 at an MOI of 1 for 1 h, followed by washing to remove extracellular viruses. Cells were further cultured for the indicated times before harvest. Otherwise, monocytes were further challenged with 1 μg/ml LPS for 3 h and 10 μM nigericin for 30 min, followed by either cell harvest for immunoblotting or fixing for immunostaining.
Transfection and lentiviral production
Transfection reagent jetPRIME (Polyplus‐transfection) was used to conduct transfection both in HEK293T and THP‐1 cells. HEK293T cells were plated 14 h earlier to achieve a density of about 70% when transfection was performed. THP‐1 cells were transferred into medium containing NATE for at least 30 min at 37°C before performing conventional transfection procedures. The media containing transfection reagent and NATE (for THP‐1) were removed 6 h after transfection. Lentiviral plasmids containing exogenous genes of interest and packaging plasmids pSPAX2 and pMD2.G were transfected into HEK293T cells at a ratio of 4:3:1. 48 h later, and supernatants were collected, followed by centrifugation at 187 g for 5 min and filtration using 0.45‐μm sterile syringe filters (Merck Millipore). The filtrate was then concentrated with a 100 kD Amicon ultra centrifugal filter unit (Merck Millipore). THP‐1 or U937 cells were incubated with lentiviruses for 6 h, followed by culture medium replacement with fresh medium. The medium was replaced with RPMI 1640 containing 2 μg/ml puromycin 48 h post‐infection for 3 days. A Human Monocyte Nucleofector Kit (Lonza) was used to electroporate plasmids into human primary monocytes following the manufacturer's instructions. Cells were cultured for 36 h, followed by LPS and nigericin stimulation.
Knockout cell line generation by CRISPR/Cas9 technology
LentiCRISPRv2 vectors expressing CRISPR‐Cas9 targeting guide RNA (gRNA) designed for target genes were used to generate knockout cell lines. gRNA targeting NLRP3 was 5′‐CTGATTAGTGCTGAGTACCG‐3′. gRNA targeting GSDMD was 5′‐CTTGCTTTAGACGTGCAGCG‐3′. Guide RNAs were designed using the GPP sgRNA Designer on the GPP Web Portal of Broad Institute. The gRNA‐expressing plasmids and packaging plasmids were transfected into HEK293T cells for 48 h, followed by supernatant collection and lentivirus concentration. THP‐1 cells were incubated with lentiviruses at an MOI of 1, followed by culture medium replacement with fresh medium containing 2 μg/ml puromycin 48 h post‐infection. Cells were further cultured in the presence of puromycin for 3 days to form stables.
Cytotoxicity assay
Cells following inflammasome activation were centrifuged at 187 g for 5 min, and supernatants and cell pellets were collected, respectively. The supernatants were subjected to lactate dehydrogenase (LDH) assay to assess cell death using a CytoTox 96 non‐radioactive cytotoxicity assay kit (Promega). Cell pellets were used to measure cell viability by cellular ATP level determination with CellTiter‐Glo luminescent cell viability assay kit (Promega). Both the supernatants and cell pellets were used for immunoblotting analysis.
ELISA
IL‐1β levels in supernatants were analyzed by an ELISA kit (D711068, Sangon Biotech, Shanghai) following the manufacturer's instructions.
RNA extraction and PCR identification
Total RNA extraction kit (Dakewe, Beijing) was used to extract RNAs in THP‐1 cells according to the manufacturer's instructions. For reverse transcription, 1 μg RNA was mixed with 2 μl N6 primers and 7 μl DEPC water, followed by incubation at 72°C for 2 min. The mixture was then chilled on ice for 2 min and then mixed with 4 μl 5× first‐strand buffer (Takara Bio), 2 μl 100 mM DTT, 2 μl 10 mM dNTPs mix (Solarbio, Beijing), and 2 μl SMART MMLV reverse transcriptase (Clontech, Takara Bio). The mixture was incubated under the following conditions: 10 min at 25°C, 1 h at 42°C, and 10 min at 75°C. cDNA samples were diluted for further PCR analysis. Primers for PCR identification were as follows: 5′‐AGGGGAACTTCTCCTGCTAG‐3′ (forward) and 5′‐TAGTGGCAGTACGTTTTTGC‐3′ (reverse) for SARS‐CoV‐2 nucleocapsid identification in patient monocytes; 5′‐TCTGATAATGGACCCCAAAATC‐3′ (forward) and 5′‐GGCCTGAGTTGAGTCAGCACTG‐3′ (reverse) for SARS‐CoV‐2 nucleocapsid identification in THP‐1 cells; 5′‐ATGGCAGATTCCAACGGTACT‐3′ (forward) and 5′‐CTGTACAAGCAAAGCAAT‐3′ (reverse) for SARS‐CoV‐2‐M; 5′‐AACACACGTCCAACTCAGTT‐3′ (forward) and 5′‐CACGCATGAGTTCACGGGTA‐3′ (reverse) for SARS‐CoV‐2‐Nsp1; 5′‐TCTAAAATGTCAGATGTAAA‐3′ (forward) and 5′‐TTGTAAGGTTGCCCTGTT‐3′ (reverse) for SARS‐CoV‐2‐Nsp7; 5′‐ACTGGGTTAC ATCCTACACA‐3′ (forward) and 5′‐CTAAAGCCAGCTGAGATCAT‐3′ (reverse) for SARS‐CoV‐2‐Nsp14; 5′‐AGTTTAGAAAATGTGGCTTT‐3′ (forward) and 5′‐TTGTAATTTTGGGTAAAATGT‐3′ (reverse) for SARS‐CoV‐2‐Nsp15; 5′‐TCTAGTCAAGCGTGGCAAC‐3′ (forward) and 5′‐GTTGTTAACAAGAACATCA‐3′ (reverse) for SARS‐CoV‐2‐Nsp16; 5′‐GAAGTGAGGACTATTAAGGT‐3′ (forward) and 5′‐ATAAGTAACTGGTTTTATGGT‐3′ (reverse) for SARS‐CoV‐2‐PLP; 5′‐ATGATGCCAACTATTTTCT‐3′ (forward) and 5′‐CACTATTGTAAGGTATAC‐3′ (reverse) for SARS‐CoV‐2‐ORF3b; 5′‐ATGTTTCATCTCGTTGACT‐3′ (forward) and 5′‐ATCAATCTCCATTGGTTGCT‐3′ (reverse) for SARS‐CoV‐2‐ORF6; 5′‐TGAAGTTCATCTGCACCACC‐3′ (forward) and 5′‐TCCTCCTTGAAGTCGATGCC‐3′ (reverse) for eGFP; and 5′‐CACCAACTGGGACGACATGGAGAAA‐3′ (forward) and 5′‐TGGCTGGGGTGTTGAAGGTCTCAAA‐3′ (reverse) for ACTB.
Recombinant protein expression and purification
For recombinant expression of p10 and p20 (wild‐type and enzymatically inactive C285A mutant) of caspase‐1, cDNAs encoding proteins aforementioned were cloned into pET‐15b with a C‐terminal 6xHis tag. For recombinant expression of SARS‐CoV‐2‐nucleocapsid, cDNA‐encoding nucleocapsid was cloned into pET‐15b vector with an N‐terminal 6xHis tag. Plasmids were transformed into E. coli strain BL21, and the bacteria grew in LB medium supplemented with 100 μg/ml ampicillin for 8 h at 37°C. Protein expression was induced by 0.1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) through shaking at 120 rpm overnight at 16°C. The whole purification procedure was performed at 4°C. Cells were collected and resuspended in a binding buffer containing 0.5 M NaCl, 20 mM Tris, 5 mM imidazole, and 1% Triton X‐100, pH 7.5, and lysed by sonication. The lysate was centrifuged at 16,200 g for 30 min. Cleared lysate was filtered through sterile syringe filters before passing through a column loaded with Ni‐NTA agarose beads. The column was washed with a wash buffer containing 0.5 M NaCl, 20 mM Tris, and 60 mM imidazole, pH 7.5, and eluted by an elution buffer containing 1 M NaCl, 40 mM Tris, and 2 M imidazole, pH 7.5. For recombinant expression of GSDMD (full‐length and truncated ones), the cDNAs of full‐length human GSDMD, amino acids (aa) 276–484, aa 270–280, aa 265–284, and aa 260–290, truncation lacking aa 265–284, truncation lacking aa 302–310, truncation lacking aa 350–368, and truncation lacking both aa 302–310 and aa 350–368 were cloned into pGEX‐6P‐1 vector. Escherichia coli strain BL21 was transformed and cultured in LB medium containing 100 μg/ml ampicillin for 8 h at 37°C. For recombinant protein expression, the medium was added with IPTG to a final concentration of 0.5 mM and shook at 150 rpm for 3 h at 37°C. Cells were harvested and resuspended with PBS containing 1% Triton X‐100, followed by lysis through sonication. Cell lysates were centrifuged at 16,200 g for 30 min at 4°C and filtered through sterile syringe filters. The filtered lysate was loaded on column containing glutathione Sepharose 4B resin, followed by washing with PBS containing 1% Triton X‐100. The protein was eventually eluted by 1 ml 10 mM l‐glutathione solution. Recombinant PreScission protease was expressed and purified as indicated above.
GST pull‐down assay
Glutathione Sepharose 4B resin was washed twice with PBS and blocked with 1% BSA in PBS for 1 h at 4°C. Purified proteins were mixed with recombinant GST‐tagged proteins and BSA‐blocked glutathione Sepharose 4B resin for 2 h at 4°C. Resins were washed three times with PBS containing 1% Triton X‐100, followed by sample preparation for SDS–PAGE visualized either by Coomassie Blue staining or immunoblotting.
In vitro cleavage assay
In vitro GSDMD cleavage was performed as described (Wang et al, 2020). In brief, 1 μg wild‐type or linker region‐substituted GSDMD together with or without 2 μg recombinant SARS‐CoV‐2 nucleocapsid were incubated with 0.1 mM or 0.5 mM caspase‐1 p20/p10 complex in a 40 μl reaction buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 3 mM EDTA, 0.005% (v/v) CHAPS, and 10 mM DTT for 1 h at 37°C. The mixture was added with SDS loading buffer and boiled for 10 min, followed by standard immunoblotting analysis.
Immunoprecipitation
HEK293T cells were seeded on six‐well culture plates and were transfected with plasmids as indicated 16 h later. 24‐h post‐transfection, cells were washed twice with PBS and lysed in a pre‐chilled buffer containing 150 mM NaCl, 50 mM Tris (pH 7.5), 1 mM EDTA, 1% Triton X‐100, 1% protease inhibitor cocktail, and 10% glycerol for 30 min on ice. Cell lysates were centrifuged at 16,200 g for 10 min at 4°C. Primary antibodies and their isotype control IgG were immobilized to AminoLink Coupling Resin (Thermo Fisher Scientific) following the manufacturer's instructions. Supernatants were incubated with immobilized antibodies for 2 h at 4°C. Resins were washed three times with PBS containing 1% Triton X‐100, followed by immunoprecipitant detachment in 0.1 M glycine–HCl, pH 2.7. Supernatants were neutralized by adding 1/10 the volume of 2 M Tris–HCl, pH 8.0, followed by immunoblotting with the indicated antibodies.
Immunofluorescence
Cells were washed twice with PBS and adhered to covers pre‐coated with poly‐l‐lysine, followed by fixation with 4% paraformaldehyde for 10 min at room temperature. Cells were then washed twice with PBS and permeabilized with PBS containing 1% Triton X‐100 for 10 min, followed by washing with PBS for twice and blockage in 10% normal goat serum for 30 min at 37°C. Cells were incubated with primary antibody for 2 h, followed by washing with PBS for three times and further incubation with fluorescence‐conjugated secondary antibody for 1 h. Nuclei were stained with DAPI. For co‐immunostaining using antibodies with the same origin, Lightning‐Link kits (ab236553 and ab269900, Novus Biologicals) were used to directly link fluorescence to primary antibodies. Cells were visualized through an UltraVIEW VoX imaging system (PerkinElmer).
Propidium iodide staining
Cells were washed twice with PBS and resuspended in 100 μl PBS, followed by addition of 10 μl propidium iodide staining solution. Cells were plated on slides and examined through fluorescence microscopy. At least thirty visual fields were examined for each sample.
Yeast two‐hybrid screening
Yeast strain AH109 was inoculated into 5 ml YPDA medium and shook at 250 rpm for 16 h at 30°C. The overnight culture was transferred to a flask containing 50 ml YPDA and incubated for 3 h at 30°C. Yeast cells were collected by centrifugation at 3,000 g for 10 min and resuspended in a 1XTE/LiAc buffer (pH7.5) containing 0.1 M lithium acetate, 0.1 M Tris–HCl, and 10 mM EDTA. A mixture of plasmid DNA and carrier DNA was added to the yeast cell suspension and vortexed to mix well. A PEG/LiAc buffer containing 40% polyethylene glycol 3350, 0.1 M lithium acetate, 0.1 M Tris–HCl, and 10 mM EDTA was then added to the suspension and vortexed at a high speed. The suspension was shaken at 200 rpm for 30 min at 30°C, followed by addition of DMSO and heat shock for 15 min in a 42°C water bath. Cells were chilled in an ice bath for 2 min and resuspended to plate on appropriate SD agar plates. A mouse bone marrow cDNA library was used for Y2H screening with BD‐tagged nucleocapsid as bait. BD‐tagged nucleocapsid truncations and AD‐tagged GSDMD C‐terminus truncations were co‐transfected into yeast strain AH109, and double‐positive clones were grown on plates containing X‐α‐gal. The intensity of blue color was monitored on a scanner.
Human angiogenesis antibody array
Human Angiogenesis Array Q1000 (RayBiotech) was used to quantify the levels of secreted cytokines in control or SARS‐CoV‐2‐infected monocytes following the manufacturer's instructions. Quantified values for each cytokine were normalized using R function “scale”. Normalized data for all cytokines were then merged together and visualized as dot plot in R 4.0.1.
Statistical analysis
No statistical methods were used to predetermine sample size. Experiments were independently repeated at least three times to achieve statistical significance. No randomization or blinding procedures were used in this study. No samples were excluded from the analysis. Data were shown as means ± SD of three technical replicates. Data with normal distribution determined by Shapiro–Wilk normality test were statistically analyzed by two‐tailed Student's t‐tests if not specified. Bartlett's test was used. The Brown–Forsythe test and the Welch ANOVA test were used for the reanalysis of single‐cell RNA‐sequencing data. Data were analyzed by GraphPad Prism 9.0. P‐values < 0.05 were termed as significant (*P < 0.05; **P < 0.01; and ***P < 0.001); NS means non‐significant where P ≥ 0.05.
Author contributions
JM, FZ, and MZ designed and performed experiments and analyzed data. FS, DY, JM, XZ, WL, YQ, YZ, and DJ performed experiments. SW and PX initiated the study, designed and performed experiments, analyzed data, and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Source Data for Appendix
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Dr. Yuhai Bi (Institute of Microbiology, Chinese Academy of Sciences) for providing SARS‐CoV‐2 strain. We are grateful for the help from staff of BSL‐3 in the Institute of Microbiology, Chinese Academy of Sciences. We thank Qihua He (Peking University Medical and Health Analysis Center) for technical help. This work was supported by the National Key R&D Program of China (2019YFA0111800), Strategic Priority Research Programs of the Chinese Academy of Sciences (XDB29020000), National Natural Science Foundation of China (81922031, 31770939), Key Research Program of Frontier Sciences of Chinese Academy of Sciences (ZDBS‐LY‐SM025), Fok Ying Tung Education Foundation to P.X., and Youth Innovation Promotion Association of CAS to S.W.
The EMBO Journal (2021) 40: e108249.
Contributor Information
Shuo Wang, Email: wangshuo@im.ac.cn.
Pengyan Xia, Email: xiap@pku.edu.cn.
Data availability
This study includes no data deposited in external repositories.
References
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC (2016) GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 113: 7858–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Dueber EC (2017) Recent insights into the molecular mechanisms underlying pyroptosis and Gasdermin family functions. Trends Immunol 38: 261–271 [DOI] [PubMed] [Google Scholar]
- Allen IC, Scull MA, Moore CB, Holl EK, McElvania‐TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30: 556–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au L, Boos LA, Swerdlow A, Byrne F, Shepherd STC, Fendler A, Turajlic S (2020) Cancer, COVID‐19, and antiviral immunity: the CAPTURE study. Cell 183: 4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, Blecher‐Gonen R, Cohen M, Medaglia C, Li Het al (2020) Host‐viral infection maps reveal signatures of severe COVID‐19 patients. Cell 181: 1475–1488.e1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJet al (2018) Caspase‐1 self‐cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 215: 827–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CR, Asfaha JB, Ghent CM, Howard CJ, Hartooni N, Safari M, Frankel AD, Morgan DO (2020) Phosphoregulation of phase separation by the SARS‐CoV‐2 N protein suggests a biophysical basis for its dual functions. Mol Cell 80: 1092–1103.e1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AH, Schroder K (2020) Inflammasome signaling and regulation of interleukin‐1 family cytokines. J Exp Med 217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cai H, Pan J, Xiang N, Tien P, Ahola T, Guo D (2009) Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci USA 106: 3484–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Wu DI, Guo W, Cao Y, Huang DA, Wang H, Wang T, Zhang X, Chen H, Yu Het al (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130: 2620–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes AJ, Courau T, Kuhn NF, Hu KH, Ray A, Chen WS, Chew NW, Cleary SJ, Kushnoor D, Reeder GCet al (2021) Global absence and targeting of protective immune states in severe COVID‐19. Nature 591: 124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consiglio CR, Cotugno N, Sardh F, Pou C, Amodio D, Rodriguez L, Tan Z, Zicari S, Ruggiero A, Pascucci GRet al (2020) The immunology of multisystem inflammatory syndrome in children with COVID‐19. Cell 183: 968–981.e967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, Lin R, Peters CJ, Tseng C‐T, Baker SCet al (2007) Regulation of IRF‐3‐dependent innate immunity by the papain‐like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem 282: 32208–32221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F (2016) Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116 [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC (2018) The pore‐forming protein gasdermin D regulates interleukin‐1 secretion from living macrophages. Immunity 48: 35–44.e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes‐Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase‐1 activation. Cell Death Differ 14: 1590–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AC, Soares VC, de Azevedo‐Quintanilha IG, Dias SSG, Fintelman‐Rodrigues N, Sacramento CQ, Mattos M, de Freitas CS, Temerozo JR, Teixeira Let al (2021) SARS‐CoV‐2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov 7: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt EC, Yu L, Park E, Lenardo MJ, Xu XN (2009) Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J Virol 83: 6631–6640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid‐Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T, Hornung V (2016) Human monocytes engage an alternative inflammasome pathway. Immunity 44: 833–846 [DOI] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DLet al (2020) A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature 583: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsenko A, Yu S, Martin‐Sanchez F, Diaz‐Del‐Olmo I, Nichols EM, Davis DM, Brough D, Lopez‐Castejon G (2020) Priming is dispensable for NLRP3 inflammasome activation in human monocytes in vitro . Front Immunol 11: 565924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti T‐D, Özören N, Body‐Malapel M, Amer A, Park J‐H, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele Pet al (2006) Bacterial RNA and small antiviral compounds activate caspase‐1 through cryopyrin/Nalp3. Nature 440: 233–236 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QTet al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Kox M, Waalders NJB, Kooistra EJ, Gerretsen J, Pickkers P (2020) Cytokine levels in critically Ill patients with COVID‐19 and other conditions. JAMA 324: 1565–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Zhang Z, Xiao X, Qi J, He B, Wang J (2017) Enterovirus 71 inhibits pyroptosis through cleavage of gasdermin D. J Virol 91: e01069‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q‐X, Tang X‐J, Shi Q‐L, Li Q, Deng H‐J, Yuan J, Hu J‐L, Xu W, Zhang Y, Lv F‐Jet al (2020) Clinical and immunological assessment of asymptomatic SARS‐CoV‐2 infections. Nat Med 26: 1200–1204 [DOI] [PubMed] [Google Scholar]
- Merad M, Martin JC (2020) Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol 20: 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, Tseng CT, Makino S (2008) Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J Virol 82: 4471–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosillo N, Viceconte G, Ergonul O, Ippolito G, Petersen E (2020) COVID‐19, SARS and MERS: are they closely related? Clin Microbiol Infect 26: 729–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez MLG, Poreba M, Snipas SJ, Groborz K, Drag M, Salvesen GS (2018) Extensive peptide and natural protein substrate screens reveal that mouse caspase‐11 has much narrower substrate specificity than caspase‐1. J Biol Chem 293: 7058–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi GA, Sacco O, Mancino E, Cristiani L, Midulla F (2020) Differences and similarities between SARS‐CoV and SARS‐CoV‐2: spike receptor‐binding domain recognition and host cell infection with support of cellular serine proteases. Infection 48: 665–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, Hiller S (2016) GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35: 1766–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- Tominaga K, Yoshimoto T, Torigoe K, Kurimoto M, Matsui K, Hada T, Okamura H, Nakanishi K (2000) IL‐12 synergizes with IL‐18 or IL‐1beta for IFN‐gamma production from human T cells. Int Immunol 12: 151–160 [DOI] [PubMed] [Google Scholar]
- V'Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V (2021) Coronavirus biology and replication: implications for SARS‐CoV‐2. Nat Rev Microbiol 19: 155–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Dillon CP, Green DR (2016) Programmed necrosis in inflammation: toward identification of the effector molecules. Science 352: aaf2154 [DOI] [PubMed] [Google Scholar]
- Wang H, Mao L, Meng G (2013) The NLRP3 inflammasome activation in human or mouse cells, sensitivity causes puzzle. Protein Cell 4: 565–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Sun QI, Zhong X, Zeng M, Zeng H, Shi X, Li Z, Wang Y, Zhao Q, Shao Fet al (2020) Structural Mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell 180: 941–955.e920 [DOI] [PubMed] [Google Scholar]
- Wauters E, Van Mol P, Garg AD, Jansen S, Van Herck Y, Vanderbeke L, Bassez A, Boeckx B, Malengier‐Devlies B, Timmerman Aet al (2021) Discriminating mild from critical COVID‐19 by innate and adaptive immune single‐cell profiling of bronchoalveolar lavages. Cell Res 31: 272–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Miao EA, Ting JP (2013) Mechanisms of NOD‐like receptor‐associated inflammasome activation. Immunity 39: 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, Liu X, Xie L, Li J, Ye Jet al (2020) Immune cell profiling of COVID‐19 patients in the recovery stage by single‐cell sequencing. Cell Discov 6: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Han M, Li T, Sun W, Wang D, Fu B, Zhou Y, Zheng X, Yang Y, Li Xet al (2020) Effective treatment of severe COVID‐19 patients with tocilizumab. Proc Natl Acad Sci USA 117: 10970–10975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM (2019) Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol 40: 1035–1052 [DOI] [PubMed] [Google Scholar]
- Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, Jia X, Wu M, Shi B, Xu Set al (2020) Viral and host factors related to the clinical outcome of COVID‐19. Nature 583: 437–440 [DOI] [PubMed] [Google Scholar]
- Zhao C, Zhao W (2020) NLRP3 inflammasome‐A key player in antiviral responses. Front Immunol 11: 211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Williams EP, Malireddi RKS, Karki R, Banoth B, Burton A, Webby R, Channappanavar R, Jonsson CB, Kanneganti TD (2020) Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase‐8/RIPK3 during coronavirus infection. J Biol Chem 295: 14040–14052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, Sun R, Tian Z, Xu X, Wei H (2020) Pathogenic T‐cells and inflammatory monocytes incite inflammatory storms in severe COVID‐19 patients. Natl Sci Rev 7: 998–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Source Data for Appendix
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
This study includes no data deposited in external repositories.