

Abstract
The antiretroviral drug favipiravir (FPV) inhibits RNA‐dependent RNA polymerase. It has been developed for the treatment of the novel coronavirus (severe acute respiratory syndrome coronavirus 2) infection disease, coronavirus disease 2019 (COVID‐19). However, its pharmacokinetics in patients with COVID‐19 is poorly understood. In this study, we measured FPV serum concentration by liquid chromatography–tandem mass spectrometry and conducted population pharmacokinetic analysis. A total of 39 patients were enrolled in the study: 33 were administered FPV 1600 mg twice daily (b.i.d.) on the first day followed by 600 mg b.i.d., and 6 were administered FPV 1800 mg b.i.d. on the first day followed by 800 mg or 600 mg b.i.d. The median age was 68 years (range, 27–89 years), 31 (79.5%) patients were men, median body surface area (BSA) was 1.72 m2 (range, 1.11–2.2 m2), and 10 (25.6%) patients required invasive mechanical ventilation (IMV) at the start of FPV. A total of 204 serum concentrations were available for pharmacokinetic analysis. A one‐compartment model with first‐order elimination was used to describe the pharmacokinetics. The estimated mean clearance/bioavailability (CL/F) and distribution volume/bioavailability (V/F) were 5.11 L/h and 41.6 L, respectively. Covariate analysis revealed that CL/F was significantly related to dosage, IMV use, and BSA. A simulation study showed that the 1600 mg/600 mg b.i.d. regimen was insufficient for the treatment of COVID‐19 targeting the 50% effective concentration (9.7 µg/mL), especially in patients with larger BSA and/or IMV. A higher FPV dosage is required for COVID‐19, but dose‐dependent nonlinear pharmacokinetics may cause an unexpected significant pharmacokinetic change and drug toxicity. Further studies are warranted to explore the optimal FPV regimen.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
We previously reported that favipiravir (FPV) concentrations in patients who are critically ill with coronavirus disease 2019 (COVID‐19) was much lower than that in healthy volunteers. However, the factors affecting FPV pharmacokinetics in this patient population are poorly understood.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study explored the factors affecting the pharmacokinetics (PK) of FPV in patients with coronavirus disease 2019 (COVID‐19) by conducting a population PK analysis.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The clearance/bioavailability of FPV was significantly related to FPV dosage, invasive mechanical ventilation (IMV) use, and body surface area (BSA). The 1600 mg/600 mg b.i.d. regimen was considered insufficient for the treatment of COVID‐19 targeting the 50% effective concentration (9.7 µg/mL), especially in patients with larger BSA and/or IMV.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The results should be used for the selection of the dosing regimen and when planning future clinical trials of FPV in patients with COVID‐19.
INTRODUCTION
Favipiravir (FPV) is an RNA‐dependent RNA polymerase inhibitor that acts on a broad spectrum of viral RNA polymerases. 1 It was originally developed for resistant influenza virus infections but is now being developed as a treatment for severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection disease, coronavirus disease 2019 (COVID‐19). 2 , 3 Because of the unexpected rapid spread of the virus worldwide, which caused the COVID‐19 pandemic, FPV has been used for the treatment of COVID‐19; however, the pharmacokinetics (PK) of FPV in these populations are still largely unknown. 4 Therefore, PK studies of FPV in patients with COVID‐19 are needed to ensure its safety and efficiency in this patient population.
We previously reported a case series of FPV PK in patients with severe COVID‐19. The concentration of FPV in seven patients who required invasive mechanistic ventilation (IMV) was much lower than that in healthy volunteers previously reported. The concentrations were also lower than the 50% effective concentration (EC50; 9.7 µg/mL) of FPV for SARS‐CoV‐2, and we reported that this was a great concern in the treatment. 5
Despite this, the factors affecting FPV PK in patients with COVID‐19 remain unclear. Thus, in the present study, we performed a population PK (PPK) analysis of FPV in patients with COVID‐19 to characterize the PK of FPV in this patient population.
METHODS
Study patients
Patients with COVID‐19 administered FPV tablets (AVIGAN® tablet 200 mg; Toyama Chemical Co., Ltd., Tokyo, Japan) between March 19, 2020 and May 23, 2020, in Kobe City Medical Center General Hospital were enrolled in this retrospective observation study. Patient characteristics including age, sex, body weight (BW), height (HT), body surface area (BSA), body mass index (BMI), serum aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), serum creatinine (SCr), comedications, and clinical status after starting FPV were investigated. Clinical status was evaluated using a seven‐category ordinal scale as follows: (1) nonhospitalization, no limitation of activities; (2) nonhospitalization, limitation of activities; (3) hospitalization, not requiring oxygen; (4) hospitalization, requiring oxygen by mask or nasal prongs; (5) hospitalization, requiring noninvasive ventilation and/or high‐flow oxygen; (6) hospitalization, requiring oxygen (invasive) and/or extracorporeal membrane oxygenation; and (7) death.
This study was conducted in accordance with the Declaration of Helsinki and approved by the institutional ethics board (approval number, Zn200418; approval date, March 31, 2020). Informed consent was obtained from all study participants or their families.
FPV administration
In this retrospective study, patients were administered 1600 mg FPV twice daily (b.i.d.) on Day 1, followed by 600 mg b.i.d. from Day 2 to Day 5 (or more if needed, up to 14 days). Some patients who started FPV in another hospital and were then transferred to our hospital were administered 1800 mg b.i.d. on Day 1, followed by 800 mg b.i.d., and changed to 600 mg b.i.d. in our hospital. Patients on mechanical ventilation were administered suspensions of FPV tablets through a nasogastric tube.
PK measurement
This was a retrospective PK study using stored residual serum samples of routine clinical practice. All samples were obtained for clinical laboratory tests for general care, and residual serum samples stored at −20°C within 12 h after laboratory tests were used for PK measurement (stability was confirmed 5 , 6 ). All FPV dosages, dosing times, and blood sampling times were recorded. FPV serum concentration was determined using a validated liquid chromatography–tandem mass spectrometry assay. The samples were measured using two calibration ranges (5–1000 and 500–100,000 ng/mL) to avoid carryover. The limit of quantification (LOQ) was 5 ng/ml. The assay precision (relative standard deviation percentage; n = 5) and accuracy (relative error percentage; n = 5) were 3.1%–7.4% and 97.1%–102.1%, respectively.
PK analysis
The nonlinear mixed effects modeling program NONMEM version 7.41 (ICON Development Solutions) was used to analyze the FPV PK data. The structure model was assumed to be a one‐compartment model with first‐order elimination because the peak‐time PK data were limited. PK parameters, clearance/bioavailability (CL/F), and distribution volume/F (V/F) were estimated by the first‐order conditional estimation method with interaction. The interindividual variability was described by proportional error model and included in CL/F as the following equation:
where θi is the PK parameter estimate of individual i, θ tv is the typical value of the PK parameter, and ηi is the interindividual random effect of individual i with a mean of 0 and variance of ω 2. The proportional error model, additional error model, and combined error model were tested to describe residual errors using the following equation:
where Yobs,ij and Ypred,ij are the jth observation and prediction of the FPV concentration, respectively, for the ith individual, and εadd,ij and εprop,ij are the additive and proportional residual error with a mean of 0 and variance of σ 2, respectively. The base model was selected based on the likelihood ratio test (LRT) of change in the object function value (OFV).
Covariate analysis
Covariate analysis was conducted using a stepwise method based on the LRT to evaluate the factors influencing FPV PK. Potential covariates (age, sex, BW, HT, BSA, BMI, AST, ALT, SCr, comedications, clinical status, and FPV dosage) were added to the base model (forward inclusion criteria; p < 0.05). After a full covariate model was developed, each factor was removed in the backward elimination step (backward elimination criteria; p < 0.01). The following equation was used for categorical (0 or 1) and continuous covariate values, respectively:
where θi is the PK parameter estimate of the individual (i) and θtv is the typical PK parameter in the study population. θcov is the covariate coefficient, COVi is the individual (i) value of covariates, and COVmedian is the median value of continuous covariates in the study population. Clinical status and FPV dosage (as the last administered dosage at the time) changed over time, and these factors were incorporated as time‐varying covariates. The final model was determined according to the LRT and clinical relevance.
Final model evaluation
To identify potential bias due to the model structure, goodness‐of‐fit plots were generated. Population prediction (PRED) versus observation plots, individual prediction (IPRED) versus observation plots, conditional weighted residuals (CWRES) versus PRED plots, and CWRES versus time after first dose plots were visually assessed. A nonparametric bootstrap resampling method was used to evaluate the precision and robustness of the estimated PK parameters. A total of 1000 resamplings were run, and the medians and 95% confidence intervals (CI) were estimated. To evaluate the performance of the final PPK model, a prediction‐corrected visual predictive check (pcVPC) was performed. The 1000 data sets were replicated and simulated using the final model, and the distribution of the simulated concentrations was then graphically compared with that of the observations. Negative concentrations were truncated by LOQ.
Simulation study
PK simulation was performed 1000 times using the final model with σ fixed at 0. The PK of the following three types of regimens were simulated: 1600 mg b.i.d. followed by 600 mg b.i.d., 3200 mg b.i.d. followed by 1200 mg b.i.d., and 1600 mg b.i.d. Furthermore, a simulation using the published data of previous clinical studies 7 , 8 , 9 reporting FPV concentrations was performed.
RESULTS
Patient baseline characteristics
A total of 39 patients were included in this observational study. All patients were diagnosed with COVID‐19 using real‐time polymerase chain reaction. Their baseline characteristics are summarized in Table 1. The median age was 68 years (range, 27–89 years), number of male patients was 31 (79.5%), and median BSA was 1.72 m2 (range, 1.14–2.2 m2). A total of 10 patients required IMV at the initiation of FPV (25.6%), and 7 patients (17.9%) had worn IMV after starting FPV. Two patients were weaned off IMV during FPV treatment. A total of 33 patients were administered 1600 mg b.i.d./600 mg b.i.d., and the others were administered 1800 mg b.i.d./800 mg or 600 mg b.i.d.
TABLE 1.
Patient baseline characteristics at the beginning of favipiravir treatment
| Median or n | Range or % | |
|---|---|---|
| Patients | 39 | |
| Age, years | 68 | 27–89 |
| Sex, male/female | 31/8 | 79.5/20.5 |
| Body weight, kg | 64 | 29–100 |
| Height, cm | 168 | 144–182 |
| Body surface area, m2 | 1.72 | 1.14–2.20 |
| Body mass index, kg/m2 | 23.5 | 12.9–30.9 |
| SCr, mg/dL | 0.81 | 0.81–10.71 |
| AST, IU/L | 45 | 13–155 |
| ALT, IU/L | 30 | 8–109 |
| WHO ordinal score | ||
| 7 hospitalized, invasive oxygen | 10 | 25.6 |
| 6 hospitalized, high‐flow oxygen | 0 | 0 |
| 5 hospitalized, requiring oxygen | 24 | 61.5 |
| 4 hospitalized, not requiring oxygen | 5 | 12.8 |
| Favipiravir regimen | ||
| 1600/600 mg b.i.d. | 33 | 84.6 |
| 1800/800 or 600 mg b.i.d. | 6 | 15.4 |
Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; b.i.d., twice daily; SCr, serum creatinine; WHO, World Health Organization.
PPK modeling
Base model
Among the 219 samples assessed, 204 FPV serum concentrations (median 5 points [range, 1–13] per patient) were used for the PPK analysis. Samples were obtained at 0‒6 h (11 points), 6.1‒12 h (125 points), 12.1‒24 h (30 points), and >24 h (38 points) from the last administration. A total of 15 samples (6.8%) below LOQ at 0‒6 h (2 points), 12.1‒24 h (2 points), and >24 h (11 points) were excluded as missing values. The PK data were fitted to a one‐compartment model with a first‐order elimination model. Among the tested residual error models, the combined residual error model had the smallest OFV and was thus selected as the base model (Table 2). As incorporation of interindividual error with V/F did not significantly reduce OFV (ΔOFV = −2.331, p = 0.127), an interindividual error was only considered for CL/F.
TABLE 2.
Covariate analysis (base model, forward inclusion steps, and full models)
| Step | OFV | dOFV | Model | p value | |
|---|---|---|---|---|---|
| Base model | 0 | 4221.276 | Base model with additive error | ||
| 0 | 3514.076 | Base model with additive error | |||
| 0 | 3413.232 | Base model with combined error | |||
| Forward inclusion | 1 | 3413.54 | 0.308 | AGE on CL/F | 0.579 |
| 2 | 3411.053 | −2.179 | SEX on CL/F | 0.140 | |
| 3 | 3408.898 | −4.334 | WT on CL/F | 0.037 | |
| 4 | 3405.29 | −7.942 | HT on CL/F | 0.005 | |
| 5 | 3407.302 | −5.93 | BSA on CL/F | 0.015 | |
| 6 | 3410.911 | −2.321 | BMI on CL/F | 0.128 | |
| 7 | 3413.22 | −0.012 | SCR on CL/F | 0.913 | |
| 8 | 3410.32 | −2.912 | AST on CL/F | 0.088 | |
| 9 | 3405.562 | −7.67 | ALT on CL/F | 0.006 | |
| 10 | 3392.264 | −20.968 | IMV on CL/F | <0.001 | |
| 11 | 3411.028 | −2.204 | Baseline IMV on CL/F | 0.138 | |
| 12 | 3411.554 | −1.678 | Amlodipine on CL/F | 0.195 | |
| 13 | 3412.65 | −0.582 | Metoclopramide on CL/F | 0.446 | |
| 14 | 3411.039 | −2.193 | Fentanyl on CL/F | 0.139 | |
| 15 | 3412.984 | −0.248 | Rocuronium on CL/F | 0.619 | |
| 16 | 3411.039 | −2.193 | Propofol on CL/F | 0.139 | |
| 17 | 3411.776 | −1.456 | Dexmedetomidine on CL/F | 0.228 | |
| 18 | 3377.174 | −36.058 | Dose on CL/F | <0.001 | |
| Full model | 13 | 3349.983 | −60.785 | ALT, WT, IMV, and dose on CL/F (Full Model 1) | <0.001 |
| 13 | 3346.866 | −64.032 | ALT, HT, IMV, and dose on CL/F (Full Model 2) | <0.001 | |
| 13 | 3348.498 | −62.875 | ALT, BSA, IMV, and dose on CL/F (Full Model 3) | <0.001 |
Bold values indicate statistical significance p < 0.05.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; BSA, body surface area; CL/F, clearance/bioavailability; dOFV, differences in objective function values; HT, height; IMV, invasive mechanistic ventilation; OFV, object function value; SCR, serum creatinine; WT, body weight.
Covariate analysis
In the forward inclusion steps (p value < 0.05), physical constitution (BW, HT, and BSA), ALT, IMV (clinical status = 7, or not), comedications, and FPV dosage (dose) were included as covariates of CL/F (Table 2). Aldehyde oxidases inhibitor 4 (amlodipine [n = 8] and metoclopramide [n = 5]) and IMV‐related drugs (fentanyl [n = 19], rocuronium [n = 14], propofol [n = 19], and dexmedetomidine [n = 8]) were included as comedications suspected to have drug–drug interactions. The physical constitution was correlated with each other, and three full models (Full Models 1–3) were generated (Table 2). In backward elimination (p < 0.01), elimination of ALT and BW did not significantly reduce OFV. The elimination of HT, BSA, IMV, and dose significantly reduced OFV (Table 3). Between HT and BSA, we concluded that BSA was a better covariate for representing physical constitution than HT only; thus, BSA, IMV, and dose were retained in the final model. The PK parameters estimated for the final model are shown in Table 4. CL/F is represented by the following equation:
TABLE 3.
Covariate analysis (backward elimination and the final model)
| Step | OFV | dOFV | Elimination | Model | p value | |
|---|---|---|---|---|---|---|
| Base model | 0 | 4221.276 | Base model with additive error | |||
| Full model | 13 | 3349.983 | −60.785 | ALT, WT, IMV, and dose on CL/F | ||
| 13 | 3346.866 | −64.032 | ALT, HT, IMV, and dose on CL/F | |||
| 13 | 3348.498 | −62.875 | ALT, BSA, IMV, and dose on CL/F | |||
| Backward elimination | 13 | 3352.447 | 2.464 | −ALT | WT, IMV, and dose on CL/F | 0.116 |
| 13 | 3349.2 | 2.334 | −ALT | HT, IMV, and dose on CL/F | 0.127 | |
| 13 | 3350.357 | 1.859 | −ALT | BSA, IMV, and dose on CL/F (final model) | 0.172 | |
| 14 | 3358.452 | 6.005 | −ALT and WT | IMV and dose on CL/F | 0.014 | |
| 14 | 3358.452 | 9.252 | −ALT and HT | IMV and dose on CL/F | 0.002 | |
| 14 | 3358.452 | 8.095 | −ALT and BSA | IMV and dose on CL/F | 0.004 | |
| 15 | 3377.174 | 18.722 | −ALT, WT, and IMV | Dose on CL/F | < 0.001 |
Bold values indicate statistical significance p < 0.05.
Abbreviations: ALT, alanine aminotransferase; BSA, body surface area; CL/F, clearance/bioavailability; dOFV, differences in objective function values; HT, height; IMV, invasive mechanistic ventilation; OFV, object function value; WT, body weight.
TABLE 4.
Pharmacokinetics parameters on the final model and results of nonparametric bootstrap
| Structure model parameters | Mean | RSE (%) | Shrinkage (%) | Bootstrap (n = 983/1000) | ||||
|---|---|---|---|---|---|---|---|---|
| Median | 2.5th | – | 97.5th | |||||
| CL/F, L/h | 5.11 | 24.10 | 5.29 | 2.91 | – | 9.75 | ||
| V/F, L/h | 41.6 | 29.60 | 42.2 | 15.1 | – | 84.3 | ||
| Dose on CL/F | –0.61 | 42.80 | −0.60 | −0.20 | – | −3.34 | ||
| IMV on CL/F | 1.71 | 16.80 | 1.63 | 0.98 | – | 2.11 | ||
| BSA on CL/F | 2.22 | 34.60 | 2.22 | 0.75 | 3.94 | |||
| Interindividual variability model parameters | ||||||||
| ω2 CL/F | 0.355 | 26.10 | 2.40 | 0.331 | 0.161 | – | 0.584 | |
| Residual error model parameters | ||||||||
| σ2 proportional error | 0.749 | 13.10 | 3.20 | 0.718 | 0.549 | – | 0.906 | |
| σ2 additive error | 764 | 51.80 | 744 | 125 | – | 1675 | ||
Abbreviations: BSA, body surface area; CL/F, clearance/bioavailability; IMV, invasive mechanistic ventilation; V/F, distribution volume/bioavailability; ω2, interindividual variability; σ2, intraindividual variability.
FPV dosage was negatively correlated with CL/F, indicating dose‐dependent nonlinear PK. Patients who required IMV (clinical status = 7, IMV = 1) had a greater CL than those who did not (IMV = 0). BSA was positively correlated with CL/F.
Model evaluation
Goodness‐of‐fit plots with the final model are shown in Figure 1. Observations versus PRED or IPRED plots were symmetrically distributed around the diagonal line, indicating good model predictivity. No systematic bias was observed in the CWRES versus PRED or time after the first dose plots. Thus, no specifications on the structure model or residual model were found. The means and 95% CIs of the final PK parameters by the nonparametric bootstrap analysis are shown in Table 2. The success rate was 98.3%, and all median values were close to the model estimates and showed the robustness of the model PK parameters. The pcVPC showed that the observation value was in good agreement with the simulated concentration of the model (Figure 2).
FIGURE 1.
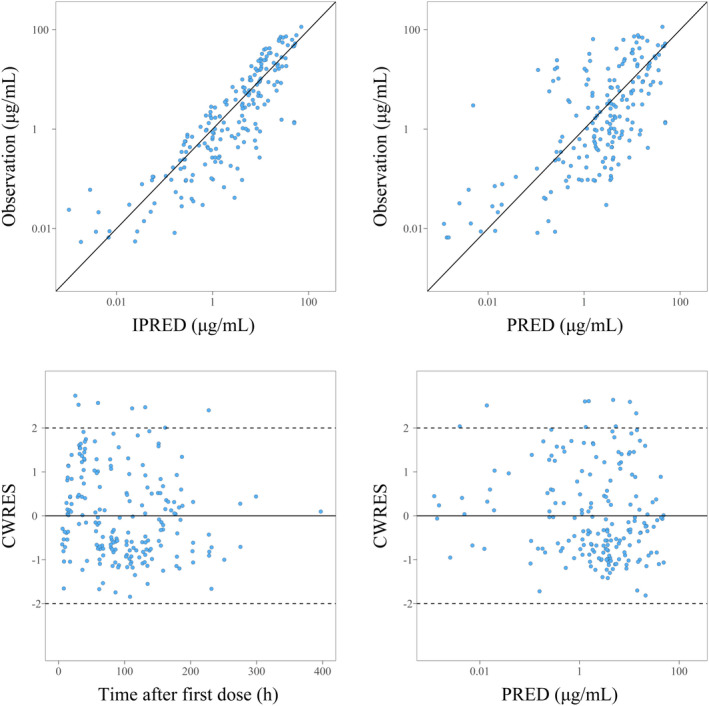
Goodness‐of‐fit plots of the final model. Population prediction (PRED) versus observation plots (upper right), individual prediction (IPRED) versus observation plots (upper left), conditional weighted residuals (CWRES) versus PRED plots (lower right), and CWRES versus time after first dose plots (lower left) are presented.
FIGURE 2.
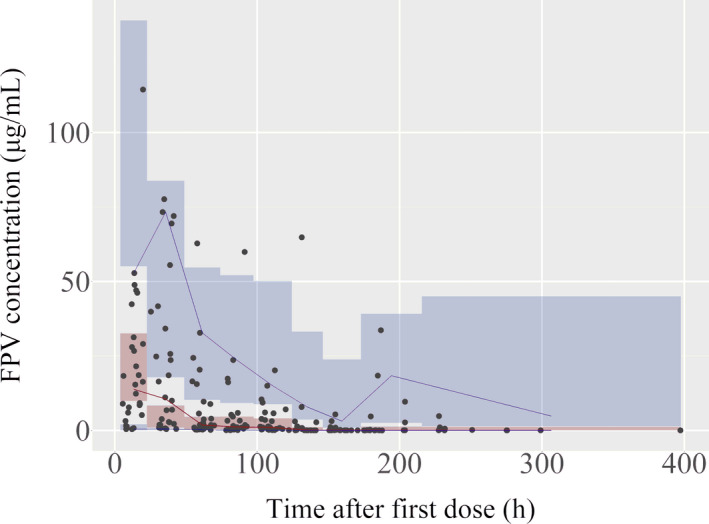
Prediction‐corrected visual predictive checks of the final model. Red and blue lines are the median and the 5th to 95th percentiles of the prediction‐corrected observed favipiravir (FPV) concentration in each bin, respectively. Red and blue areas are the 90% confidence interval for the median value of the model prediction and the 5th and 95th percentiles of each bin. Dots indicate the prediction‐corrected observations. Negative concentrations were truncated by limit of quantification.
Simulation study
Results of simulations using the final PPK model are shown in Figure 3. The mean and 5th–95th percentile values of the three types of regimens on IMV or non‐IMV and BSA = 1.72 or 2.20 (this was the median or upper value in the study population) are presented. The mean trough concentration of the 1600/600 mg b.i.d. regimen was lower than the EC50 reported in SARS‐CoV‐2 (9.7 µg/mL) 10 with any simulation; therefore, the regimen may be insufficient for the treatment of COVID‐19. In patients who required IMV, the mean trough concentration of the doublet regimen (3200/1200 mg b.i.d.) was also lower than the EC50, and that of the 1600 b.i.d. regimen only exceeded the EC50 in patients with median BSA = 1.72 and not in those with BSA = 2.20. In patients who did not require IMV, the mean trough concentration of the 1600 b.i.d. regimens only exceeded the EC50 in patients with BSA = 2.20.
FIGURE 3.
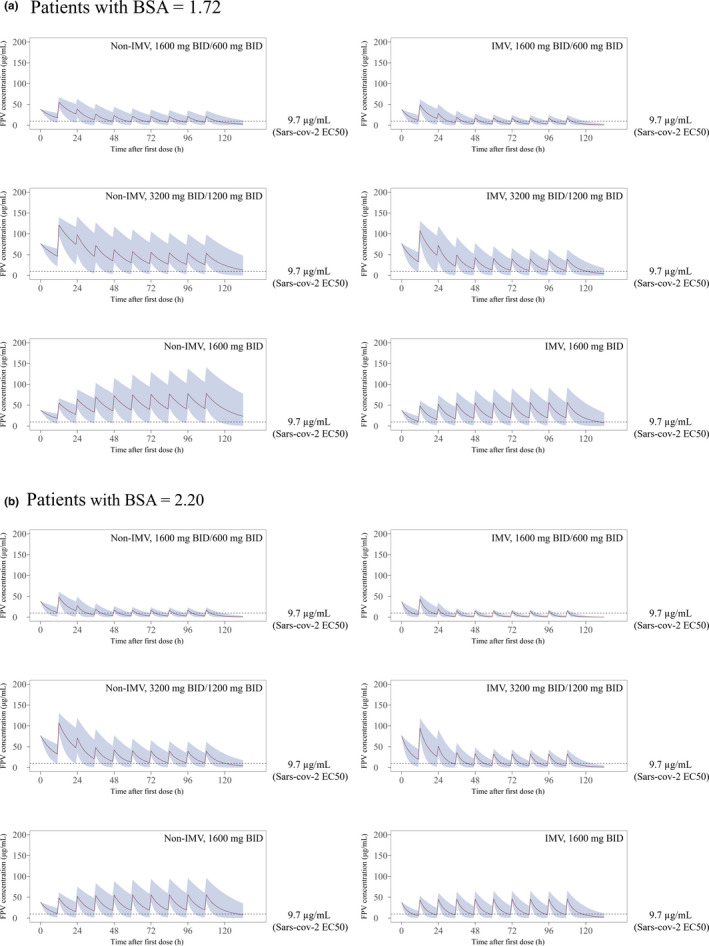
Effect of three types of regimens on patients with or without invasive mechanistic ventilation (IMV) as simulated using the final model. (a) Patients with body surface area (BSA) = 1.72 and (b) patients with BSA = 2.20 are presented. Red solid line indicates the mean value, blue area indicates the 5th and 95th percentiles of the prediction, and the dotted line indicates the 50% effective concentration (EC50) of favipiravir (FPV) for severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; 9.7 µg/mL). BID, twice daily.
In addition, simulation results using the published data of previous clinical studies 7 , 8 , 9 are shown in Figures S1–S3. Although the simulation was based on some assumptions, the final model could adaptably describe the observations of previous studies.
DISCUSSION
In this study, we characterized the PK of FPV in patients with COVID‐19 using a PPK analysis. This is the first report of a PPK analysis of FPV in patients with COVID‐19. The FPV PK was adeptly described by the one‐compartment first‐order elimination model, and the PK parameters were comparable with those of the one‐compartment first‐order elimination model reported previously. 9 CL/F was significantly related to IMV use, FPV dosage, and BSA in the covariate analysis. Model validation results revealed that the final model had good precision and robustness. The simulation with the final model indicated that the 1600/600 mg b.i.d. regimen may be insufficient for the treatment of COVID‐19 targeting the EC50 (9.7 µg/mL), suggesting that a higher dosage is needed for treatment.
The result that patients who required IMV had higher CL/F and exhibited low FPV concentrations was consistent with that of previous studies. PK case studies in patients with severe influenza virus infection 11 and COVID‐19 8 requiring IMV have been reported, and their FPV concentrations were lower than those reported in other previous studies. We have also reported seven patients who were critically ill with COVID‐19 treated with FPV. 5 Although the cause could not be specified, IMV was only significantly incorporated as a time‐varying covariate of CL/F and not a baseline status in the covariate analysis. This suggested that IMV‐related factors (such as biological changes or combination drugs) affected FPV metabolism or absorption. Patients with severe COVID‐19 require deep sedation to prevent lung injury, which might decrease drug absorption.
Interestingly, the FPV dosage was negatively related to CL/F. The loading regimen was used for FPV treatment, and the CL/F after the administration of 1600 mg FPV was 0.55 (=1600/600−0.61)‐fold lower than that after the administration of 600 mg FPV. This resulted in a relatively high FPV concentration on the first day, which decreased thereafter. This was also reported in other studies in patients with Ebola virus disease 7 and severe influenza infection. 9 In both studies, the loading regimen was used, and patients showed unexpected drops of FPV concentrations. In a later study on severe influenza infection, a PPK analysis of FPV was conducted with time included as a covariate to explain the drop. 9 In the present study, we used FPV dosage as a covariate, as it may be able to explain the dose‐dependent autoinhibition of metabolic enzymes (aldehyde oxidases) 12 as reported in nonhuman primates. 12 Although this is not confirmative because we have not separately examined the administrated dosage, it may be useful as one of the possibilities of dosing design for the future development of FPV treatment. Dose‐dependent nonlinear PK is observed in many drugs, and it causes unexcepted overexposure and toxicity. 13 , 14 , 15 , 16
We conducted a simulation study of three potential regimens of COVID‐19, and among them, the 1600/600 mg b.i.d. regimen was likely to be insufficient for COVID‐19 treatment, and the 1600 mg b.i.d. regimen only exceeded the EC50 in patients with BSA = 2.20 and patients without IMV. This is an extrapolation simulation using the overdosage range of available PK data, and the PK of any high‐dose FPV regimen cannot actually be predicted from our data. However, treatment for COVID‐19 is still limited, and high‐dose FPV is worth considering as also indicated by other reports. 9 , 17 In a recently reported animal model of COVID‐19, FPV was suggested to have an antiviral effect at only relatively high plasma and lung concentrations in the model. 18 Intracellular metabolites (favipiravir‐ribofuranosyl‐5'‐triphosphate) of FPV were reported to be dose (concentrations)‐dependent. 19 Thus, low blood concentrations less than the EC50 of SARS‐CoV‐2 were not so promising in terms of the antiviral effect. However, they should be carefully tested by monitoring the PK of FPV to avoid overexposure.
Finally, there were several limitations to this study. First, this was a retrospective observational study using sparse PK data; thus, interindividual variability of the absorption rate and V/F could not be estimated. In addition, a PPK model describing FPV metabolism (e.g., Michaelis‐Menten elimination) could not be constructed. A planned and detailed PK study is required for the accurate dosing design of FPV. Second, we did not discuss the efficiency of FPV against COVID‐19 because patients were receiving various drugs and had various symptoms; therefore, the antiviral effect of FPV could not be evaluated. Further studies are needed to examine the relationship between the PK variability and the pharmacodynamics of FPV.
In conclusion, PPK analysis of FPV revealed that CL/F was significantly related to FPV dosage, IMV use, and BSA. The 1600/600 mg b.i.d. regimen seemed insufficient for the treatment of COVID‐19 targeting the EC50 (9.7 µg/mL), especially in patients with larger BSA and/or IMV. A higher FPV dosage is required for COVID‐19, but dose‐dependent nonlinear PK may cause an unexpected significant PK change and drug toxicity. Further studies are warranted to explore the optimal FPV regimen.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
K.I. wrote the manuscript. K.I., A.N., M.E., H.I., N.M., S.F., K.T., and T.H. designed the research. K.I., A.N., H.F., and R.T. performed the research. K.I. analyzed the data.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
We thank all of the patients who participated in this study and their families, the individuals who expedited rapid approval from the Ethics Committee and collection of blood samples, and the frontline medical staff treating patients with COVID‐19 in Kobe City Medical Center General Hospital.
Irie K, Nakagawa A, Fujita H, et al. Population pharmacokinetics of favipiravir in patients with COVID‐19. CPT Pharmacometrics Syst Pharmacol. 2021;10:1161–1170. 10.1002/psp4.12685
Funding information
No funding was received for this work.
REFERENCES
- 1. Furuta Y, Komeno T, Nakamura T. Favipiravir (T‐705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:449‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cai Q, Yang M, Liu D, et al. Experimental treatment with favipiravir for COVID‐19: An open‐label control study. Engineering (Beijing). 2020;6:1192‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doi Y, Hibino M, Hase R, et al. A prospective, randomized, open‐label trial of early versus late favipiravir therapy in hospitalized patients with COVID‐19. Antimicrob Agents Chemother. 2020;64(12):e01897‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Du YX, Chen XP. Favipiravir: Pharmacokinetics and concerns about clinical trials for 2019‐nCoV infection. Clin Pharmacol Ther. 2020;108:242‐247. [DOI] [PubMed] [Google Scholar]
- 5. Irie K, Nakagawa A, Fujita H, et al. Pharmacokinetics of favipiravir in critically ill patients with COVID‐19. Clin Transl Sci. 2020;13:880‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Habler K, Brügel M, Teupser D, et al. Simultaneous quantification of seven repurposed COVID‐19 drugs remdesivir (plus metabolite GS‐441524), chloroquine, hydroxychloroquine, lopinavir, ritonavir, favipiravir and azithromycin by a two‐dimensional isotope dilution LC‐MS/MS method in human serum. J Pharm Biomed Anal. 2021;196:113935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nguyen THT, Guedj J, Anglaret X, et al. Favipiravir pharmacokinetics in Ebola‐Infected patients of the JIKI trial reveals concentrations lower than targeted. PLoS Negl Trop Dis. 2017;11:e0005389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hirai D, Yamashita D, Seta K. Favipiravir for COVID‐19 in a patient on hemodialysis. Am J Kidney Dis. 2021;77:153‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y, Zhong WU, Salam A, et al. Phase 2a, open‐label, dose‐escalating, multi‐center pharmacokinetic study of favipiravir (T‐705) in combination with oseltamivir in patients with severe influenza. EBioMedicine. 2020;62:103125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30:269‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Favié LM, Murk JL, Meijer A, Nijstad AL, van Maarseveen EM, Sikma MA. Pharmacokinetics of favipiravir during continuous venovenous haemofiltration in a critically ill patient with influenza. Antivir Ther. 2018;2018(23):457‐461. [DOI] [PubMed] [Google Scholar]
- 12. Madelain V, Guedj J, Mentré F, et al. Favipiravir pharmacokinetics in nonhuman primates and insights for future efficacy studies of hemorrhagic fever viruses. Antimicrob Agents Chemother. 2017;61:e01305‐e01316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ludden TM. Nonlinear pharmacokinetics: clinical Implications. Clin Pharmacokinet. 1991;20:429‐446. [DOI] [PubMed] [Google Scholar]
- 14. Lin JH. Dose‐dependent pharmacokinetics: experimental observations and theoretical considerations. Biopharm Drug Dispos. 1994;15:1‐31. [DOI] [PubMed] [Google Scholar]
- 15. Powis G, Ames MM, Kovach JS. Dose‐dependent pharmacokinetics and cancer chemotherapy. Cancer Chemother Pharmacol. 1981;6:1‐9. [DOI] [PubMed] [Google Scholar]
- 16. Watanabe PG, Young JD, Gehring PJ. The importance of non‐linear (dose‐dependent) pharmacokinetics in hazard assessment. J Environ Pathol Toxicol 1:147‐159. [PubMed] [Google Scholar]
- 17. Eloy P, Solas C, Touret F, et al. Dose rationale for favipiravir use in patients infected with SARS‐CoV‐2. Clin Pharmacol Ther. 2020;108:188. [DOI] [PubMed] [Google Scholar]
- 18. Smee DF, Hurst BL, Egawa H, Takahashi K, Kadota T, Furuta Y. Intracellular metabolism of favipiravir (T‐705) in uninfected and influenza A (H5N1) virus‐infected cells. J Antimicrob Chemother 2009;64(4):741‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Driouich J‐S, Cochin M, Lingas G, et al. Favipiravir antiviral efficacy against SARS‐CoV‐2 in a hamster model. Nat Commun. 2021;12:1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material