

Abstract
A critical step to evaluate the potential in vivo antiviral activity of a drug is to connect the in vivo exposure to its in vitro antiviral activity. The Anti–SARS‐CoV‐2 Repurposing Drug Database is a database that includes both in vitro anti–SARS‐CoV‐2 activity and in vivo pharmacokinetic data to facilitate the extrapolation from in vitro antiviral activity to potential in vivo antiviral activity for a large set of drugs/compounds. In addition to serving as a data source for in vitro anti–SARS‐CoV‐2 activity and in vivo pharmacokinetic information, the database is also a calculation tool that can be used to compare the in vitro antiviral activity with in vivo drug exposure to identify potential anti–SARS‐CoV‐2 drugs. Continuous development and expansion are feasible with the public availability of this database.
INTRODUCTION
Since the outbreak of coronavirus disease of 2019 (COVID‐19) in December 2019, the disease has infected more than 99.3 million people and caused more than 2.13 million deaths worldwide as of January 25, 2021. A tremendous amount of effort has been devoted to finding treatments for COVID‐19. It generally takes from a few years to decades to bring a new drug to the market with acceptable safety and efficacy profiles. To shorten the development timeline for COVID‐19 treatments, the drug development community is searching for potential candidates among approved drugs or drugs under investigation for other indications to take advantage of their known safety profiles. One essential property of the candidate compound is to inhibit the virus causing COVID‐19, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2).
Developing a drug for anti–SARS‐CoV‐2 involves multiple steps of efforts, such as understanding the coronavirus life cycle at a molecular level, identifying potential drug target(s), screening for drugs using in vitro assays, and testing the lead drugs in preclinical species and finally in humans for efficacy and safety. Among the process of developing a repurposed anti–SARS‐CoV‐2 drug, translating the in vitro findings to in vivo performance is a key step to improving development efficiency.
We have previously discussed the considerations when linking in vitro to in vivo antiviral activity where understanding the mechanism/site of action, and the drug's absorption, distribution, metabolism, and excretion (ADME) is key.1 In this article, we present our effort on constructing a database for anti–SARS‐CoV‐2 repurposing. The database consists of information on in vitro anti–SARS‐CoV‐2 activity and relevant in vivo pharmacokinetic data as well as analyses comparing in vitro antiviral activity with in vivo exposure. The outcome from this early estimation of the candidate is just the first step toward a rational drug development including appropriate additional preclinical studies and early clinical studies defining dosing based on the impact on viral loads.
CONSTRUCTION AND CONTENT
The Anti–SARS‐CoV‐2 Repurposing Drug Database was created using data from published articles (April to November 2020), publicly available new drug application (NDA) reviews, and drug labels from Drugs@FDA for approved drugs in the United States.
In vitro anti–SARS‐CoV‐2 activity data collection
We searched literature published from April to November 2020 for information regarding in vitro anti–SARS‐CoV‐2 activity of approved drugs or molecules that are currently under development for various indications. Any drug/molecule with the drug concentration that inhibits 50% of the virus (EC50) found in the report was included in the database. In the database, we included reported EC50; cytotoxic concentration, the drug concentration that reduces the cell viability by 50% (CC50); the cell line information reported in the paper; and the references. The the drug concentration that inhibits 90% of the virus (EC90) is not commonly reported. For drugs/compounds where EC90 values were not reported, we estimated the EC90 assuming the underlying antiviral effect follows a maximal effect (Emax) model (Equation (1)). This approach generally overestimates the true EC90 value and underestimates the inhibition potential.
The antiviral activity assay conditions that were used to evaluate drugs’ anti–SARS‐CoV‐2 activity varied from laboratory to laboratory. In general, to determine the EC50, cells are seeded in experimental plates prior to the infection for approximately 1 day. Drugs may be introduced to cells either prior to or after infection. Cells are infected with SARS‐CoV‐2 at various virus concentrations to reach different multiplicity of infection (MOI). Infected cells are incubated with drugs at a series of concentrations for 24, 48 hours, or longer. At the end of drug incubation, supernatants are removed, and cells are fixed for virus quantification. Various methods have been used for virus quantification, including quantitative real‐time reverse transcription–polymerase chain reaction (qRT‐PCR), immunofluorescence‐based imaging, cytopathic effect, or other methods.
In the antiviral and cell viability assays, the final concentration that exerts antiviral effect may differ from the initially added concentration (nominal concentration) due to cellular sequestration and nonspecific binding. The reported EC50 values could be affected by these factors because the EC50 values are generally estimated based on the initial nominal concentrations and not corrected for nonspecific binding.2 For highly protein bound drugs or drugs that are accumulated inside the cells, the true EC50 values could be different from the reported EC50 values.2
The readers are encouraged to read the original articles cited in the database for in vitro antiviral studies as the experimental conditions may affect the EC50 values being reported, such as the type of cell lines (included in the database), the cell density, the MOI, the time that drugs are introduced to the infected cells (prior to or after infection), the incubation times, and the methods being used to quantify virus.
Drug data collection
The drugs/compounds with reported EC50 values toward SARS‐CoV‐2 were split into two categories (and two spreadsheets): pharmacokinetic (PK) data available and PK data not available. For drugs/compounds with PK data, we collected the NDA numbers (if approved in the United States), molecular weight (MW), PK parameters and the corresponding dose and dosage form, unbound fraction in the plasma (fup), approved indications or drug class, and COVID‐19–related clinical trials up to March 1, 2021, from the clinicaltrials.gov website. There are a few drugs in this sheet that are not approved in the United States but in other countries. The indications for those drugs are collected based on literature search and should be viewed with caution. For PK parameters, the maximal concentration (Cmax) values obtained at the highest dose level are generally collected. Although this is an unusual approach for antiviral drugs, the highest exposure (i.e., Cmax at the high dose level) was collected with the intention to include as many potential candidates as possible. For drugs where the fup values are not found, a value of one is assumed and used as an input for in vivo exposure and in vitro antiviral activity comparison. This assumption serves as a conservative scenario to provide the highest possible free drug concentration for potential in vivo antiviral activity.
For drugs/compounds without reported PK information, we used the same data set format but left the missing information blank. The users can input PK information when it becomes available and compare in vivo drug exposure with in vitro EC50 using the same equations incorporated in the data set for drugs/compounds with PK information.
In vivo drug exposure and in vitro anti–SARS‐CoV‐2 activity comparison
To achieve antiviral activity, it is expected that the in vivo drug exposure should be comparable to or higher than the in vitro concentration demonstrating sufficient antiviral activity, and the desirable exposure should be maintained for a certain period of time. We have previously discussed in detail the general considerations connecting in vitro antiviral activity to in vivo drug exposure for prediction of antiviral effect using hydroxychloroquine as an example.1 In this study, we applied the same method to a large database of drugs/compounds to assess their potential in vivo anti–SARS‐CoV‐2 effect. Based on the collected in vitro anti–SARS‐CoV‐2 EC50 and PK data, fup*PK/EC50 can be calculated for each drug where fup is the unbound plasma fraction, PK is the PK parameter which usually is the Cmax value, and EC50 is the in vitro drug concentration that inhibits 50% of the viral replication.
For antiviral drugs, such as antiretroviral agents, it is generally expected that the plasma antiviral drug concentrations need to remain above the protein‐adjusted EC90 to increase the chances of clinical benefit.2 Therefore, the trough concentration (Ctrough or Cmin) is a more relevant and commonly used PK metric to be compared with the in vitro EC90. In this database, we collected Cmax because Cmax is a more commonly reported PK parameter compared with Cmin, which is easier to obtain clinically. In addition, Cmin can be derived based on a PK model if Cmax suggests potential in vivo antiviral activity to justify further investigation of Cmin. In addition, for majority of the drugs/compounds, Cmax is enough to show that in vivo exposure cannot reach the in vitro EC50.
For each drug, more than one fup*PK/EC50 value could be calculated if there were more than one EC50 value reported. PK value is another source of variability. However, we only included one PK value that was generally obtained at the highest dose level for each drug as the purpose of the database is for a fast screening to identify potential anti–SARS‐CoV‐2 compounds by comparing the in vivo drug exposure with its in vitro antiviral activity.
The readers are encouraged to conduct additional assessment once they identify a drug that has high in vivo drug exposure compared with its in vitro EC50. For each drug, it is critical to understand the mechanism of action, the site of action, the active moiety, and the drug's ADME properties. For example, some of the drugs included in the database are prodrugs (e.g., remdesivir, nitazoxanide). Not all of these considerations are included in the database, but some of them are discussed next in case examples.
UTILITY
The database construction and utility are illustrated in Figure 1. Briefly, the database can be used for the following purposes:
A source for a quick search for anti–SARS‐CoV‐2 EC50 values
A source for a quick identification of drugs with in vivo exposure higher than in vitro anti–SARS‐CoV‐2 EC50
A calculation tool to compare the in vitro antiviral activity with in vivo drug exposure
FIGURE 1.
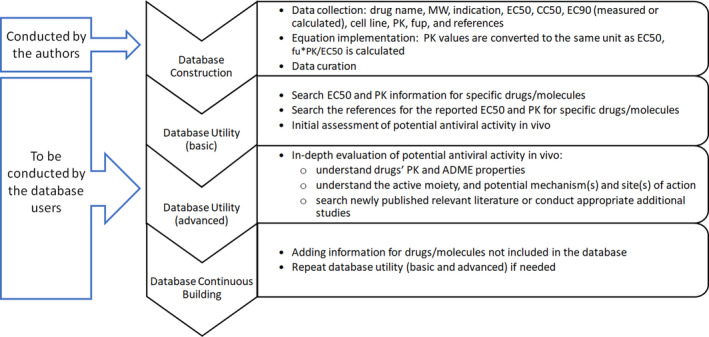
Illustration of database construction and utility. ADME, absorption, distribution, metabolism, and excretion; CC50, the drug concentration that reduces the total cell number by 50% values; EC50, the drug concentration that inhibits 50% of the virus; EC90, the drug concentration that inhibits 90% of the virus; fup, unbound fraction in plasma; MW, molecular weight; PK, pharmacokinetic
It should be noted that the authors are not making a statement about whether these drugs are expected to work for COVID‐19 and are only compiling available information for drugs that have been assessed in vitro for SARS‐CoV‐2.
Database overview
There are two spreadsheets in the database, namely, “with PK” (the drugs/compounds with PK information) and “without PK” (the compounds without PK information). In the “with PK” spreadsheet, there are 113 drugs/compounds with both PK and EC50 data available. In the “without PK” spreadsheet, there are 83 compounds with EC50 data but not PK data. We are interested in the drugs/compounds with low EC50 values (potent in vitro antiviral activity) and high fup*PK/EC50 values (high in vivo exposure relative to in vitro antiviral potency).
It should be noticed that there are a few drugs where the reported EC50 values were very low (such as lisinopril, metformin, ouabain, and valproic acid). When we examined the exposure–response (ER) curves for those drugs, it appeared that the curves were flat, suggesting that the Emax values were very close to the minimal inhibition effect when there is no drug available, suggesting that those drugs are not good candidates even if their in vivo exposures are well above the reported in vitro EC50 values. Relying on fup*PK/EC50 alone to judge antiviral activity requires an implicit assumption that high enough fup*PK/EC50 should lead to almost complete inhibition of virus replication (i.e., Emax ~=100%). These few examples highlight the importance of examining the raw in vitro exposure–response curves to ensure the validity of the implicit assumption about Emax. There are also a few drugs where the CC50 (50% cytotoxic concentration) value was close to the EC50 value, suggesting that the drug is too toxic at the efficacious exposure level and there is no safe and effective therapeutic window. Further exploration is not suggested for those drugs. We have indicated “flat ER curve” and “toxic,” respectively, for those drugs/compounds.
Excluding the aforementioned drugs, in the “with PK” spreadsheet, there are 21 drugs with EC50 values less than 1 µM (Table 1) and 8 drugs/compounds with fup*PK/EC50 values larger than one (Table 1). We selected EC50 of 1 µM and fup*PK/EC50 greater than one as the cutoff values with the intent to include as many compounds as possible for further evaluation, not the best goal but a permissive approach.
TABLE 1.
Summary of drugs with EC50 < 1 µM, fup*PK/EC50 > 1, and fup < 0.01
| Drugs in PK with EC50 < 1 µM | Drugs in PK with fup*PK/EC50 > 1 | Drugs in PK with fup < 0.01 |
|---|---|---|
| Amodiaquine | Atazanavir | Azilsartan |
| Astemizole | Chloroquine | Ciclesonide |
| Atazanavir | Favipiravir | Clofazimine |
| Bromhexine | Nafamostat | Dolutegravir |
| Camostat | Naquotinib | Droloxifene |
| Chloroquine | Nitazoxanide | Eltrombopag |
| Clofazimine | Remdesivir | Indomethacin |
| Cyclosporine | Tetrandrine | Ivacaftor |
| Dacomitinib | Lopinavir | |
| Digitoxin | Lusutrombopag | |
| Digoxin | Midostaurin | |
| Hanfangchin A (Tetrandrine) | Nitazoxanide | |
| Hexachlorophene | Osimertinib mesylate | |
| Hydroxychloroquine | Pazopanib | |
| Nafamostat | Pimozide | |
| Naquotinib | Pioglitazone | |
| Niclosamide | Piperaquine | |
| Pioglitazone | Tetrandrine | |
| Pyronaridine | Thioridazine | |
| Remdesivir | Tipranavir | |
| Tetrandrine | Toremifene |
Abbreviations: EC50, the drug concentration that inhibits 50% of the virus; fup, unbound fraction in plasma; PK, pharmacokinetic.
In the “with PK” spreadsheet, 21 of 113 drugs are highly protein bound (with fup values < = 1%). Considering that nonspecific binding to the cells are not routinely measured in the in vitro antiviral studies, the “true” in vitro EC50 values could be lower for those drugs. The effect can be minimized if the cells were preincubated with the treatment drugs. Nevertheless, the values of fup*PK/EC50 for highly protein bound compounds should be interpreted with caution and the readers are encouraged to read the original articles on how the EC50 values were measured and understand the drug's ADME properties.
In the next section, we selected a few compounds with fup*PK/EC50 values larger than one as case examples for further discussion. The users are encouraged to explore the database and identify potential compounds for further assessment. The users may download the database and continuously add relevant information and new entries as they become available.
Case study: atazanavir, atazanavir/ritonavir
Atazanavir (MW = 705g/mol) is a protease inhibitor approved for the treatment of human immunodeficiency virus type‐1 (HIV‐1) in combination with other antiretroviral agents.3 The recommended dose for atazanavir depends on the treatment history of the patient and the use of other coadministered drugs. Ritonavir is required with several atazanavir dosage regimens. The recommended atazanavir/ritonavir dosage in treatment‐naïve adult patients is 300 mg/100 mg once daily.
Atazanavir's anti–SARS‐CoV‐2 activity was measured in vitro by various groups.4, 5, 6 The reported EC50 ranged from 0.2 to >50 µM (Table 2) using the monkey kidney epithelial Vero cells,4, 5 VeroE6 expressing transmembrane serine protease 2 (TMPRSS2),6 or a human pulmonary epithelial cell line5 in the presence5 or absence4, 5 of ritonavir. In the article published by Jeon et al.,4 atazanavir did not show anti–SARS‐CoV‐2 activity with an EC50 > 50 µM in Vero cells infected with a MOI of 0.0125. Yamamoto et al.6 tested anti–SARS‐CoV‐2 activity for several HIV‐1 protease inhibitors including atazanavir in VeroE6 cells expressing TMPRSS2 (VeroE6/TMPRSS2) and found the EC50 for atazanavir to be 9.36 µM. Fintelman‐Rodrigues et al.5 reported EC50 values of 2.0 µM and 0.5 µM for atazanavir and atazanavir in combination with ritonavir (atazanavir/ritonavir), respectively, in Vero cells. The authors further tested the anti–SARS‐CoV‐2 activity in a human epithelial pulmonary cell line (A549). Atazanavir showed about 10‐fold lower EC50 (0.22 µM) in A549 compared with the EC50 (2.0 µM) obtained in Vero cells. Atazanavir/ritonavir showed similar EC50 (0.6 µM) in A549 compared with the EC50 (0.5 µM) obtained in Vero cells. Fintelman‐Rodrigues et al.5 hypothesized that atazanavir binds to the SARS‐CoV‐2 major protease (Mpro) and inhibits the SARS‐CoV‐2 Mpro enzymatic activity.
TABLE 2.
Summary of in vitro studies for atazanavir and atazanavir/ritonavir
| Drug | EC50 (µM) | CC50 (µM) | Cell line | Postinfection treatment time (h) | MOI | Virus quantification method | fu*PK/EC50 | Reference |
|---|---|---|---|---|---|---|---|---|
| Atazanavir | >50 | >50 | Vero | 72 | 0.05 | Viral cytopathic effect | <0.02 | 4 |
| Atazanavir | 0.22 | 312 | Human epithelial pulmonary (A549) | 48 | 0.01 | qRT‐PCR | 4.69 | 5 |
| Atazanavir | 2.0 | 312 | Vero | 48 | 0.01 | qRT‐PCR | 0.52 | 5 |
| Atazanavir | 9.36 | >81 | VeroE6/TMPRSS2 | 24 | 0.01 | qRT‐PCR | 0.11 | 6 |
| Atazanavir/ritonavir | 0.5 | 280 | Vero | 48 | 0.01 | qRT‐PCR | 2.07 | 5 |
| Atazanavir/ritonavir | 0.6 | 280 | Human epithelial pulmonary (A549) | 48 | 0.01 | qRT‐PCR | 1.72 | 5 |
Abbreviations: CC50, the drug concentration that reduces the total cell number by 50%; EC50, the drug concentration that inhibits 50% of the virus; MOI, multiplicity of infection; PK, pharmacokinetic; qRT‐PCR, quantitative real‐time reverse transcription–polymerase chain reaction; TMPRSS2, transmembrane serine protease 2.
Following multiple‐dose oral administration of 400 mg atazanavir once daily under the fed state in healthy volunteers, the geometric mean Cmax and Cmin were 5199 and 159 ng/mL (i.e., 7.38 and 0.23 µM), respectively. Atazanavir is 86% bound to human serum proteins, and protein binding is independent of concentration. Atazanavir is metabolized by cytochrome P450 (CYP) 3A (CYP3A). When it was coadministered with ritonavir, a strong CYP3A inhibitor, the Cmax and area under the concentration‐time curve (AUC) of atazanavir was increased by 86% and 238%, respectively, compared with atazanavir being administered alone. The Cmin of atazanavir was increased by approximately 12‐fold when atazanavir was administered with ritonavir compared with it being administered alone.
Comparing the unbound plasma concentration using Cmax (Cmax,u) to the in vitro anti–SARS‐CoV‐2 EC50, the ratio can be above one when the EC50 is in the range of nanomolar (nM). However, if we consider the unbound plasma concentration using Cmin (Cmin,u) to the in vitro anti–SARS‐CoV‐2 EC50 values, the Cmin,u (0.23*14%*12 = 0.39 µM) obtained when atazanavir was administered with ritonavir is barely higher than the lowest reported EC50 (0.22 µM). When atazanavir is coadministered with ritonavir, the Cmin was increased by approximately 12‐fold. Comparing Cmin,u of atazanavir coadministered with ritonavir to the lowest in vitro anti–SARS‐CoV‐2 EC50 value (0.22 µM), the ratio is barely above one.
In this case example, we compared atazanavir in vivo exposure with its in vitro EC50. Depending on the in vitro EC50 value, in vivo atazanavir can reach in vitro EC50 at certain time points, such as the time span around the time where the highest plasma concentration is observed following administration. It is common that a large range of in vitro EC50 values can be reported for one compound as we observed in the database. It is essential to understand the reasons for the differences if possible and consider all the relevant EC50 values. To justify a potentially efficacious dosing regimen, a more common strategy is to compare Cmin,u with in vitro EC90. Fup*PK/EC50 should only serve as a screening metric. A search in ClinicalTrials.gov identified an ongoing randomized, open‐label phase II trial to investigate the efficacy and safety of atazanavir/ritonavir plus nitazoxanide for the treatment of COVID‐19 (ClinicalTrials.gov identifier: NCT04459286). The purpose of this example is to illustrate how to potentially use this database as a starting point in repurposing anti–SARS‐CoV‐2 drugs.
Case study: nafamostat mesylate
Nafamostat mesylate is a serine protease inhibitor approved in Japan and Korea, but not in the United States, for the treatment of acute pancreatitis. It is a weak anticoagulant. Early in vitro studies conducted using Vero cells did not identify nafamostat as a potential anti–SARS‐CoV‐2 candidate due to the observed high EC50 values (Table 3). Later studies exploring the effect cell types on the anti–SARS‐CoV‐2 inhibition potential identified that nafamostat showed several 100‐fold higher potencies in inhibiting SARS‐CoV‐2 in Calu‐3 cells. As shown in Table 3, the EC50 values reported in Vero cells are generally above 10 µM, whereas the EC50 values reported in Calu‐3 cells are generally <12 nM, but one reported about 3 µM.
TABLE 3.
Summary of in vitro studies for nafamostat
| Drug | EC50 (µM) | CC50 (µM) | Cell line | Postinfection treatment time (h) | MOI | Virus quantification method | fu*PK/EC50 | Reference |
|---|---|---|---|---|---|---|---|---|
| Nafamostat | 22.5 | >100 | VeroE6 | 48 | 0.05 | qRT‐PCR | 0.0115 | 17 |
| Nafamostat | 39.54 | 3639.15 | VeroE6 | 48 | 0.025 | Immunofluorescencea | 0.0066 | 18 |
| Nafamostat | 31.6 | N.R. | VeroE6/TMPRSS2 | 72 (pretreatment) | 0.01 | Cytopathic effect | 0.0082 | 9 |
| Nafamostat | >100 | N.R. | VeroE6/TMPRSS2 | 72 (no pretreatment) | 0.01 | Cytopathic effect | <0.0026 | 9 |
| Nafamostat | 3.16 | N.R. | Calu‐3 | 120 (no pretreatment) | 0.01/0.1 | Cytopathic effect | 0.0821 | 9 |
| Nafamostat | 0.0068 | N.R. | Calu‐3 | 120 (pretreatment) | 0.01 | Cytopathic effect | 38.14 | 9 |
| Nafamostat | 0.0115 | N.R. | Calu‐3 | 120 (pretreatment) | 0.1 | Cytopathic effect | 22.55 | 9 |
| Nafamostat | 13.88 | N.R. | Vero | N.R. | N.R. | N.R. | 0.0187 | 19 |
| Nafamostat | 0.0022 | >25 | Calu‐3 | 24 | 0.1 | Immunofluorescencea | 117.89 | 19 |
Abbreviations: CC50, the drug concentration that reduces the total cell number by 50%; EC50, the drug concentration that inhibits 50% of the virus; MOI, multiplicity of infection; N.R., not reported; PK, pharmacokinetic; qRT‐PCR, quantitative real‐time reverse transcription–polymerase chain reaction; TMPRSS2, transmembrane serine protease 2.
Labeling the viral N protein; VeroE6/TMPRSS2: VeroE6 cells expressing TMPRSS2.
It has been reported that SARS‐CoV‐2 use the angiotensin‐converting enzyme 2 (ACE2) for binding and the serine protease TMPRSS2 or lysosomal proteases for the spike (S) protein priming.7 Lung epithelium‐derived Calu‐3 cells, but not Vero cells, endogenously express ACE2. Nafamostat inhibits the TMPRSS2, one of the pathways for SARS‐CoV‐2 to activate spike for entry into the host cells.8 Yamamoto et al.9 also showed that to obtain low EC50 values, Calu‐3 cells need to be incubated with nafamostat prior to infection (the pretreatment group; Table 3).
Following the administration of 40 mg nafamostat mesylate by an intravenous infusion for 90 min, the plasma levels were 79–90 ng/mL (i.e., 0.23–0.26 µM). Nafamostat mesylate has a short half‐life of 8 min in humans.10 The nafamostat plasma levels are relatively higher than the EC50 values measured in Calu‐3 cells. However, there are a few caveats in this comparison. First, the unbound fraction in plasma is assumed to be one as we did not find the fup value for nafamostat. Second, the half‐life of nafamostat is too short to maintain the plasma concentration levels to be effective unless continuous intravenous dosing was used. Nevertheless, there are a few clinical trials ongoing to evaluate the clinical efficacy of nafamostat in treating COVID‐19 based on a search via clinicaltrials.gov. There is also ongoing effort in developing nafamostat inhalation product.10
Case study: remdesivir
Remdesivir is a nucleotide analog RNA polymerase inhibitor approved for the treatment of COVID‐19 requiring hospitalization.11 Remdesivir is a prodrug and is metabolized to a nucleoside monophosphate intermediate in cells by carboxylesterase 1 (CES1) and/or cathepsin A. The nucleoside monophosphate is subsequently phosphorylated by cellular kinases to form the nucleoside triphosphate metabolite (GS‐443902), which is the pharmacologically active moiety.
Prior to the approval of remdesivir, there were multiple studies reporting the anti–SARS‐CoV‐2 EC50 values based on remdesivir concentrations. The reported EC50 values range from 0.002 to 23.15 µM measured in Vero, human lung epithelial Calu‐3, human embryonic kidney 293T (HEK293T) cells expressing ACE2 (HEK293T/ACE2), and the human hepatocyte Huh cell lines or Huh expressing ACE2 (Huh7/ACE2) using various viral quantification methods (Table 4). The two lowest EC50 values were measured in Huh cells. EC50 values less than 1 µM were reported in all the tested cell lines including the Vero, human lung epithelial Calu‐3, HEK293T/ACE2, Huh7.5, and Huh7/ACE2 cell lines using immunofluorescence imaging or qRT‐PCR viral quantification methods.
TABLE 4.
Summary of in vitro studies for remdesivir
| Drug | EC50 (µM) | CC50 (µM) | Cell line | Postinfection treatment time (h) | MOI | Virus quantification method | fu*PK/EC50 | Reference |
|---|---|---|---|---|---|---|---|---|
| Remdesivir | 0.77 | >100 | VeroE6 | 48 | 0.05 | qRT‐PCR | 0.58 | 17 |
| Remdesivir | 1.65 | N.D. | VeroE6 | 72 | 0.002 | qRT‐PCR | 0.27 | 20 |
| Remdesivir | 8.24 | >50 | Vero | 72 | 0.05 | Viral cytopathic effect | 0.05 | 4 |
| Remdesivir | 11.41 | >25 | Vero | 24 | 0.0125 | Immunofluorescencea | 0.04 | 4 |
| Remdesivir | 23.15 | >100 | VeroE6 | 48 | 0.02 | TCID | 0.02 | 21 |
| Remdesivir | 26.90 | >100 | VeroE6 | 48 | 0.02 | qRT‐PCR | 0.02 | 21 |
| Remdesivir | 0.002 | >40 | Huh7.5 | 30 | 1 | Imagingb | 221.94 | 22 |
| Remdesivir | 0.457 | >40 | Vero | 30 | 1 | Imagingb | 0.97 | 22 |
| Remdesivir | 0.005 | >40 | Calu‐3 | 48 | 0.5 | Imagingb | 88.78 | 22 |
| Remdesivir | 1.3 | >50 | Calu‐3 | 24 | 0.1 | Immunofluorescencea | 0.34 | 19 |
| Remdesivir | 0.62 | >2.5 | VeroE6 | 24 | 0.1 | Immunofluorescencea | 0.72 | 23 |
| Remdesivir | 0.0072 | 0.72 | HEK293T/ACE2 | 24 | 0.3 | Immunofluorescencea | 61.65 | 23 |
| Remdesivir | 0.0026 | 0.98 | Huh7/ACE2 | 24 | 0.2 | Immunofluorescencea | 170.72 | 23 |
Abbreviations: ACE2, angiotensin‐converting enzyme 2; CC50, the drug concentration that reduces the total cell number by 50%; EC50, the drug concentration that inhibits 50% of the virus; HEK293T, human embryonic kidney 293T; MOI, multiplicity of infection; N.D., not determined; PK, pharmacokinetic; qRT‐PCR, quantitative real‐time reverse transcription–polymerase chain reaction; N.D., not determined; TCID, tissue culture infectious dose.
Labeling the viral N protein.
Labeling viral dsRNA and spike protein.
In vivo, following a 30‐min intravenous infusion of 100 mg remdesivir once daily, the plasma Cmax and Cmax,u of remdesivir was 2229 ng/mL (i.e., 3.7 µM) and 267.5 ng/mL (i.e., 0.44 µM), respectively. The Cmin of remdesivir was not detectable.11 The two inactive metabolites, GS‐441524 and GS‐704277, were detectable, but the active metabolite, GS‐443902, was not detectable in plasma. The proposed intracellular metabolic scheme and anti–SARS‐CoV‐2 mechanism can be found in the remdesivir clinical pharmacology review.12 Briefly, remdesivir enters cells via passive diffusion, which is then metabolized to GS‐704277 by CES1. GS‐704277 is converted to GS‐441524‐monphosphate, which is phosphorylated to the active triphosphate GS‐443902 or dephosphorylated to GS‐441524.12 The active triphosphate metabolite (GS‐443902) is highly ionized and difficult to diffuse across the cell membrane and therefore accumulates in the cells. Studies in human macrophages in vitro suggest that intracellular levels of the triphosphate may exceed 100 µM.13
Comparing the Cmax,u following intravenous infusion of 100 mg remdesivir with the in vitro EC50 values, the ratios of Cmax,u to EC50 ranged from 0.02 to 222 depending on the reported EC50 value measured based on the remdesivir concentrations. This is a rough comparison that does not account for the mechanism and site of action of remdesivir. Remdesivir is a prodrug. The active moiety, GS‐443902, is formed and accumulates inside the cells and cannot be detected in plasma following the recommended dosing regimen. It inhibits the SARS‐CoV‐2 RNA‐dependent RNA polymerase, which is essential for viral replication. The in vitro study by Riva et al.23 suggested that the antiviral potency of remdesivir depends on the cell type where relatively higher potency was observed in HEK293T/ACE2 and Huh7/ACE2 cells compared with the Vero cells. The antiviral potency in human lung epithelial Calu‐3 varied by 260‐fold (0.005 µM vs. 1.3 µM) as reported by two research groups (Table 4). In the labeling of remdesivir, it is reported that the EC50 values of remdesivir were 0.0099 µM and 0.28 µM in primary human airway epithelial cells after 48 hours of treatment and Calu‐3 cells after 72 hours of treatment, respectively.
CES1, the primary enzyme for the metabolism of remdesivir, is expressed in numerous human tissues with high expression in the liver, gallbladder, and lung. Therefore, it is suspected that the rate and extent of the formation and accumulation of the active metabolite depends on the cell types in different tissues/organs in vivo. Directly comparing the unbound plasma remdesivir to its in vitro EC50 value may not be the best way to estimate the in vivo antiviral activity. Nevertheless, the efficacy of remdesivir in treating COVID‐19 was demonstrated in the pivotal phase III trial, ACTT‐1, and supported by the other two phase III trials, GS‐US‐540–5773 and GS‐US‐540–5774.14
DISCUSSION
We present the Anti–SARS‐CoV‐2 Repurposing Drug Database, which includes both in vitro anti–SARS‐CoV‐2 activity and in vivo PK data for potential in vivo antiviral efficacy assessment. This database includes 113 drugs with PK data information and 83 compounds without PK data. This database is available in the Microsoft Excel (.xlsx) format via the Clinical Pharmacology and Therapeutics (CPT): Pharmacometrics & Systems Pharmacology (PSP) journal website.
The users are encouraged to read the original articles where the data came from because we were not able to include all of the details. For example, many in vitro study experimental conditions are not included in the database, such as the preincubation time, postinfection incubation time, MOI, and virus quantification methods, all of which may affect the EC50 measurement. It is also critical to examine the exposure–response curve that was used to estimate EC50 because a flat dose–response curve may provide a misleading EC50 estimate. The cell line information was included in the database, which is another important factor affecting the EC50 values. The EC50 values measured with human lung epithelial cells may be more relevant to the in vivo conditions compared with those measured with Vero cells. Of note, the EC50 data collected in the current database are tested against the original SARS‐CoV‐2 strain. With the emerging of new variants, updated EC50 values tested against the new variants can be added to the database when they become available.
For the PK information, we only included one exposure measure and fup. For a majority of the drugs, the exposure measure included was Cmax measured in plasma with the intent to not dismiss a potential drug too quickly. The approved dose may not be the maximum tolerated dose for some drugs, and therefore additional dose levels may be studied against SARS‐CoV‐2 depending on the safety margins. It is also critical for the investigator to conduct additional evaluation, such as assessing Cmin and EC90, which may eventually dismiss the drug. There are a few drugs with a subscript ‘b,’ indicating the drug concentration was measured in blood. The blood concentration should be converted to plasma concentration as the free plasma concentration is the most relevant exposure metric as we have previously discussed extensively.1 The plasma protein binding (fup) is not always reported but is critical derivation of drug concentration at the site of action. For drugs/compounds without fup information, a value of fup = 1 was assumed with the intent to identify as many potentially effective drugs as possible at the first place. The users should be cautious when interpreting the calculation using the fup of 1 and may consider other method, such as an in silico approach, to estimate the fup.
Although SARS‐CoV‐2 affects many organs, the respiratory tract is a major site of infection. Understanding the intracellular distribution and penetration in the epithelial lining fluid (ELF) can be imperative to evaluate the in vivo antiviral activity. Previous studies for anti‐infective agents suggested that the ELF to plasma concentration ratios can be >1 or ≤1.15, 16 For agents that showed ELF to plasma concentration ratios >1, potential explanations are the transporter involvement and technical issues associated with ELF concentration measurement.15
Drug–drug interaction (DDI) is another clinical pharmacology aspect that could be important but has not been extensively discussed in the article. In the case of atazanavir, the in vitro anti–SARS‐CoV‐2 EC50 values differed by 3–4‐fold in the absence and presence of ritonavir with mixed trend (Table 2). Atazanavir showed a lower EC50 value (0.22 µM) compared with atazanavir/ritonavir (0.6 µM) in the human epithelial pulmonary cells, but higher EC50 value (2.0 vs. 0.5 µM) in the Vero cells (Table 2). The reasons for the differences are unknown. In vivo, ritonavir increases the Cmax and AUC of atazanavir by 86% and 238%, respectively. Although DDIs are generally well studied in the original programs, evaluating the potential DDIs in anti–SARS‐CoV‐2 activities is also warranted.
The Anti–SARS‐CoV‐2 Repurposing Drug Database is a comprehensive database that compiles both in vitro and in vivo data as a first step for evaluation of drug in vivo anti–SARS‐CoV‐2 potential. Additional considerations are illustrated by three case examples when investigators use this database for further evaluation. The public availability of this database may facilitate the drug searching and development for potential COVID‐19 treatment.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
DISCLAIMER
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the U.S. Food and Drug Administration.
Supporting information
Supplementary Material
Zhang X, Yang Y, Grimstein M, et al. Anti–SARS‐CoV‐2 Repurposing Drug Database: Clinical Pharmacology Considerations. CPT Pharmacometrics Syst Pharmacol. 2021;10:973–982. 10.1002/psp4.12681
Funding information
No funding was received for this work.
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the U.S. Food and Drug Administration.
REFERENCES
- 1.Fan J, Zhang X, Liu J, et al. Connecting hydroxychloroquine in vitro antiviral activity to in vivo concentration for prediction of antiviral effect: a critical step in treating patients with coronavirus disease 2019. Clin Infect Dis. 2020;71(12):3232–3236. 10.1093/cid/ciaa623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boffito M, Back DJ, Flexner C, et al. Toward consensus on correct interpretation of protein binding in plasma and other biological matrices for COVID‐19 therapeutic development. Clin Pharmacol Ther. 2021;110(1):64–68. 10.1002/cpt.2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.NDA 021567 atazanavir USPI. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021567s044,206352s008lbl.pdf. Published 2020. Accessed December 9, 2020.
- 4.Jeon S, Ko M, Lee J, et al. Identification of antiviral drug candidates against SARS‐CoV‐2 from FDA‐approved drugs. Antimicrob Agents Chemother. 2020;64(7):e00819‐20. 10.1128/AAC.00819-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fintelman‐Rodrigues N, Sacramento CQ, Ribeiro Lima C, et al. Atazanavir, alone or in combination with ritonavir, inhibits SARS‐CoV‐2 replication and proinflammatory cytokine production. Antimicrob Agents Chemother. 2020;64. 10.1128/AAC.00825-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto N, Matsuyama S, Hoshino T, Yamamoto N. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv. 2020. 10.1101/2020.04.06.026476 [DOI] [Google Scholar]
- 7.Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann M, Schroeder S, Kleine‐Weber H, Müller MA, Drosten C, Pöhlmann S. Nafamostat mesylate blocks activation of SARS‐CoV‐2: new treatment option for COVID‐19. Antimicrob Agents Chemother. 2020;64(6):e00754‐20. 10.1128/AAC.00754-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto M, Kiso M, Sakai‐Tagawa Y, et al. The anticoagulant nafamostat potently inhibits SARS‐CoV‐2 S protein‐mediated fusion in a cell fusion assay system and viral infection in vitro in a cell‐type‐dependent manner. Viruses. 2020;12(6):629. 10.3390/v12060629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okajima K, Uchiba M, Murakami K. Nafamostat Mesilate. Cardiovasc Drug Rev. 1995;13(1):51‐65. [Google Scholar]
- 11.NDA 214787 remdesivir USPI. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214787Orig1s000lbl.pdf. Published 2021. Accessed March 2, 2021
- 12.NDA 214787 remdesivir clinical pharmacology review(s). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000ClinpharmR.pdf. Published 2021. Accessed March 2, 2021.
- 13.Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS‐5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381‐385. 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.NDA 214787 summary review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000Sumr.pdf. Published 2021. Accessed March 2, 2021.
- 15.Rodvold KA, George JM, Yoo L. Penetration of anti‐infective agents into pulmonary epithelial lining fluid: focus on antibacterial agents. Clin Pharmacokinet. 2011;50:637‐664. 10.2165/11594090-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 16.Rodvold KA, Yoo L, George JM. Penetration of anti‐infective agents into pulmonary epithelial lining fluid: focus on antifungal, antitubercular and miscellaneous anti‐infective agents. Clin Pharmacokinet. 2011;50:689‐704. 10.2165/11592900-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30:269‐271. 10.1038/s41422-020-0282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459‐468. 10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko M, Jeon S, Ryu WS, Kim S. Comparative analysis of antiviral efficacy of FDA‐approved drugs against SARS‐CoV‐2 in human lung cells. J Med Virol. 2021;93(3):1403‐1408. 10.1002/jmv.26397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Touret F, Giilles M, Barral K, et al. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS‐CoV‐2 replication. Sci Rep. 2020;10(1):13093. 10.1038/s41598-020-70143-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choy KT, Wong AY, Kaewpreedee P, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS‐CoV‐2 replication in vitro. Antiviral Res. 2020;178:104786. 10.1016/j.antiviral.2020.104786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dittmar M, Lee JS, Whig K, et al. Drug repurposing screens reveal cell‐type‐specific entry pathways and FDA‐approved drugs active against SARS‐Cov‐2. Cell Rep. 2021;35(1):108959. 10.1101/2020.06.19.161042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riva L, Yuan S, Yin X, et al. Discovery of SARS‐CoV‐2 antiviral drugs through large‐scale compound repurposing. Nature. 2020;586(7827):113‐119. 10.1038/s41586-020-2577-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material