

Abstract
The novel coronavirus Severe Acute Respiratory Syndrome Coronavirus‐2 (SARS‐CoV‐2) or COVID‐19 has caused a worldwide pandemic. The fatal virus has affected the health of human beings as well as the socio‐economic situation all over the world. To date, no concrete medicinal solution has been proposed to combat the viral infection, calling for an urgent, strategic, and cost‐effective drug development approach that may be achievable by applying targeted computational and virtual screening protocols. Immunity is the body's natural defense against disease‐causing pathogens, which can be boosted by consuming plant‐based or natural food products. Active constituents derived from natural sources also scavenge the free radicals and have anti‐inflammatory activities. Herbs and spices have been used for various medicinal purposes. In this study, 2,96 365 natural and synthetic derivatives (ligands) belonging to 102 classes of compounds were obtained from PubChem and assessed on Lipinski's parameters for their potential bioavailability. Out of all the derivatives, 3254 obeyed Lipinski's rule and were virtually screened. The 115 top derivatives were docked against SARS‐CoV‐2, SARS‐CoV, MERS‐CoV, and HCoV‐HKV1 main proteases (Mpros) as receptors using AutoDock Vina, AutoDock, and iGEMDOCK 2.1. The lowest binding energy was exhibited by ligands 2 and 6 against all the four Mpros. The molecular dynamic simulation was also performed with ligand 6 using the GROMACS package. Good bioactivity scores, absorption, distribution, metabolism, excretion, and toxicity profile and drug‐like pharmacokinetic parameters were also obtained. Hydroxychloroquine was used as the control drug.
Keywords: docking, Lipinski, protease, SARS‐CoV‐2, simulation, virtual screening
1. INTRODUCTION
Betacoronaviruses (βCoVs) are the genera of coronaviruses (CoVs), which have received significant importance due to their potent virulence. Human infecting coronaviruses include HCoV‐OC43, HKU and 229E5, Severe Acute Respiratory Syndrome Coronavirus (SARS‐CoV), and the Middle East Respiratory Syndrome Coronavirus (MERS‐CoV). Out of these, the SARS and MERS coronaviruses were highly pathogenic1 and associated with high mortality. In December 2019, the third Severe Acute Respiratory Syndrome Coronavirus‐2 (SARS‐CoV‐2) was identified from China as the newest pathogenic virus of the βCoV lineage.2 By March 2020, the World Health Organization (WHO) declared COVID‐19 a pandemic, which is still unfolding and the scientific fraternity does not have any targeted medicinal solution to combat this disease. The novel coronavirus possesses high mutability, making it susceptible to rapid spread, owing to its ribonucleic genetic material. RNA viruses are usually less stable than their DNA counterparts with high mutation ability3 showing that the virus may mutate and acquire a less treatable form in a short period. Recent epidemiological studies have also confirmed that the mutated SARS‐CoV‐2 may be more contagious.4 The known coronaviruses are associated with acute respiratory distress syndrome leading to reduced lung function, or even death in severe cases. SARS‐CoV‐2 has a lower mortality rate than MERS‐CoV and SARS‐CoV, but has a faster spread rate5 and is difficult to contain. It is crucial to comprehend the host hijacking mechanism for drug development. To date, there are no marketed drugs against SARS‐CoV, SARS‐CoV‐2, and MERS‐CoV, although clinical trials are underway with RNA polymerase (RdRP) inhibitor remdesivir6 and research has suggested that nucleoside analogs may prove beneficial against SARS‐CoV‐2.7 CoVs generally contain 16 non‐structural (nsp‐16) proteins along with spike (S), membrane (M), nucleocapsid (N), and envelop (E) proteins.8 The envelope‐anchored spike protein is a crucial determinant required for the viral interaction and is one of the first components to come in contact with the infected host.9 Previous studies suggest that both SARS‐CoV‐2 and SARS‐CoV cause infection through the association of the Receptor‐binding domain (RBD) to angiotensin‐converting enzyme 2 (ACE2).10 Several molecular targets have been identified in search of new antiviral drugs. The X‐ray structure of SARS‐CoV‐2 has been recently explored for computational analysis. Computational chemistry offers an effective solution in the development of potential lead compounds, in a time and cost‐effective manner. Computational strategies include molecular dynamics (MD) and virtual screening11 and computational tools have been explored to develop probable SARS‐CoV‐2 Mpro inhibitors. SARS‐CoV‐2 Mpro interacts with the active sites and releases the RNA into the host cell and infects the same.12 It replicates itself and mainly affects the respiratory system. Hence, there is an imperative need to identify a SARS‐CoV‐2 Mpro inhibitor. The CoV Mpros are enveloped positive‐stranded RNA viruses with the largest viral RNA genomes ranging between 27 and 31 kb.13 The CoV replicase gene has two polyproteins, termed pp1a and pp1b, which govern the replication and transcription.14 Most maturation cleavage events are governed by the CoV Mpro or 3CL protease or 3CLpro, a three‐domain (domains I to III) cysteine protease.15 The structures of CoV Mpro revealed the formation of an active homodimer,16, 17 the 1‐7 N‐terminal residues of the Mpro have an important role in the proteolytic action18 and the C‐terminal domain is essential for dimerization.19 Computational tools help in understanding the interaction between small molecules, also known as ligands and receptors.20 Molecular docking and MD simulation find application in the assessment of the binding affinity of the probable lead molecule with the receptor. Molecular docking studies predict the binding energy when a ligand binds to the receptor and the interaction affinity.21 MD simulation is used to simulate a protein to know the dynamic behavior of the system. The simulating entities are taken in a box either at a fixed or varying temperature. Replica exchange molecular dynamics (REMD) can be used to study the inhibition of the protease with a variation in temperature.22
The present study is aimed at the development of a probable inhibitor of CoV Mpros, including SARS‐CoV‐2, with the help of in silico and virtual screening of the bioactive constituents (natural and synthetic derivatives) of some commonly occurring herbs and spices against the target protein. Spices and herbs have found a prominent role as flavoring and aroma inducing agents. They are also useful as coloring or preservative agents and used for medicinal purposes.23 They also offer an array of benefits against acute, chronic, and non‐communicable ailments. Their active constituents are rich in sulfur‐containing compounds, alkaloids, tannins, terpenoids, vitamins, flavonoids, etc24 They also contain saponins, enzymes, and minerals, including selenium, copper, manganese, zinc, chromium, iodine, etc. Spices also possess exceptional antioxidant activity as they are rich in phenolic compounds.25 Other than antioxidant potential, the bioactive constituents are also responsible for anti‐inflammatory, anti‐tumorigenic, and anti‐carcinogenic activities along with blood sugar and cholesterol‐lowering activities.26 Studies support that spice consumption reduces the risk of mortality associated with cancer, heart, and respiratory diseases. A recent study in China with 487 375 participants related to the consumption of spicy foods as part of their diet concluded the inclusion of spices in food to improve and maintain health.27 The present study took into account the computational pharmacokinetics and drug‐likeness assessment of the hit molecules, in search of a potential inhibitor of the SARS‐CoV, SARS‐CoV‐2, HCoV‐HKU1, and MERS‐CoV Mpros. The protein‐ligand binding was studied through molecular docking and MD simulation was performed to measure the stability of the best protein‐ligand complex.
2. MATERIAL AND METHODS
The crystal structure of the target protein is the most accurate model for the evaluation of protein‐ligand interaction. Several important criteria need to be considered before the selection of the appropriate crystal structure of the target protein viz. (a) minimum possible resolution or maximum quality of the target protein; (b) identification of the crystal structure based on domain completeness. The full structure of the studied protein must be available, and there should not be any gap between the residues, as partial domain leads to inaccurate analysis, (c) any mutation in the target protein of interest must be avoided, (d) the side chains of the crystal structure must be small so that the ligand does not interact with them, (e) identification of the complete structure of the target protein is necessary, as partial domain would lead to an incorrect evaluation, and the confirmation must be done based on its sequence analysis, (f) presence of crystalline water/co‐factor/ligands also favors the selection of desirable target proteins, as the attachment of a ligand shows that the target protein is mimicking the natural system and the very fact supports the presence of binding sites or active pockets, even though later on they can be easily removed, (g) active sites can be easily identified. Based on these parameters, the SARS‐CoV‐2 (PDB ID: 6LU7), SARS‐CoV (1Q2W), MERS‐CoV (4RSP), and HCoV‐HKV1 (3D23) Mpros were selected from the RSCB protein data bank. The structures of the ligands were obtained from PubChem and assessed on Lipinski's rule of five for their bioavailability. Virtual screening was done through VEGA ZZ 3.2.0., MGL Tools 1.5.6 and AutoDock Vina 1.1.2. AutoDock 4.2.6 and iGEMDOCK 2.1 were used for docking evaluation. The grid parameters used for the molecular docking studies are represented in Table S2 along with their coordinates. The most interactive and suitable grids were, Grid IV for SARS‐CoV, Grid II for HCoV‐HKU1, Grid I for MERS‐CoV, and Grid II for SARS‐CoV‐2 Mpros (Table S1), respectively. The best conformations were identified based on the liberated energy upon interaction with the target Mpros. Top scoring derivatives were analyzed for their drug‐like character on several parameters, including bioactivity score and ADMET profile. Molinspiration 2018 (www.molinspiration.com) was used to predict the bioactivity scores. OSIRIS Property Explorer 5.2.1 was used to assess the toxicity. admetSAR 2.0 (http://lmmd.ecust.edu.cn/admetsar1/predict/) and swissADME (http://www.swissadme.ch/) were used to assess the ADMET properties. BIOVIA Discovery Studio 2017R2 and PyMol 1.8 were used to visualize the structures.
2.1. Identification of ligands
In developing countries like India and China, a plant‐based medicine system has been used for ages for the improvement of health and well‐being. WHO has predicted that the majority of the world population relies upon medicine based on herbal sources.28 Herbs and spices have been used as food additives29 and derived from roots, barks, seeds, fruits, flowers, etc.30 They are used to enhance aroma, as flavoring agents and for various medicinal purposes. Their bioactive constituents have the potential to decrease the risk of diabetes, obesity, cancer, and cardiovascular disease.31 Their bioactive constituents have innumerable medicinal benefits like purgative, laxative, expectorant, carminative, diuretic, etc. Spices have a range of functions and protect the human body against many illnesses and disorders.32 In this study, 2,96,365 plant‐based natural and synthetic derivatives were extracted from PubChem (Table S2), for further exploration of their binding potential with the CoV Mpros. The docking interactions were also studied against the two physiological ligands (MPD and PRD) and hydroxychloroquine (HCQ) as a reference drug.
2.2. Identification of receptor
Spike, envelop, membrane, protease, and nucleocapsid proteins are some of the potential target sites explored for drug designing against coronaviruses. Sixteen nonstructural polyproteins (NSPs) containing PP1a and PP1b are found in the genome of coronavirus encoded by replicase.33 The release of NSPs is facilitated by the protease. The cysteine protease or Mpro or 3‐C like protease (3CLpro) leads to the cleavage at the C‐terminal of the polyprotein. The Cys‐His dyad is responsible for the protease activity.34 The protease cleaves at 11 specific sites in the p1 region of PP1a and PP1ab facilitating replication of the virus and release of the NSPs. Human immunodeficiency virus (HIV) protease inhibitors like lopinavir and ritonavir have shown inhibitory action against Mpros. In this study, the Mpro was selected as the target protein, and through flexible docking, the binding interaction with the selected ligands was unleashed.
2.3. Ligand preparation
Crystalline structures of the selected ligands were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov). Overall, 2,76, 225 derivatives were found in the database. To obtain the potential lead entity, the ligands were filtered on Lipinski's rule of bioavailability, which is essential to be known in a promising drug moiety to know how the dose reaches the bloodstream, upon oral administration.35 Of all the selected ligands, 3254 obeyed Lipinski's rule and were further selected for virtual screening and molecular docking. Further optimization was done through Merck Molecular Force Field and MGL tools 1.5.6 was used for the preparation of ligand parameters. 2D and 3D structures of the ligands were viewed in ChemDraw 3D.
2.4. Receptor preparation
Replicase polyprotein 1a (pp1a) and replicase polyprotein 1b (pp1b) exist in the genome of coronavirus.36 The viral 30 kb RNA genome open reading frame (ORF1a/b) consists of polyproteins (pp1a and pp1ab), accessory, spike, small envelope, matrix, and nucleocapsid proteins. In this study, pp1a was downloaded from the PDB. The open reading frame is important for defining the target genomic sequence.37 Open reading frame 1a (ORF1a) was obtained from the Protein Data Bank. The structural procurement of target proteins was carried out by removing water, hit atom, ligands, and ions. Target proteins were also subjected to structural filtration through Chimera 1.12 and energy minimization was done through the default root mean deviation square (RMSD) and assisted model building with energy refinement (AMBER) force field 14SB. For molecular docking, target structures were modified by adding polar hydrogen atoms and Kollman charges, whereas nonpolar hydrogen atoms were merged. Polar hydrogen atoms are usually involved in hydrogen bonding leading to dipole‐dipole and other weak interactions, whereas non‐polar hydrogens are involved in van der Waals interactions, which do not have very significant importance. The Kollman charges depict the template value for an amino acid, derived from the corresponding electrostatic potential. Although the binding sites of the ligands were identified by the sequence analysis of the PDB file, yet for accurate confirmation and validation, the interior pocket and cavities of the target protein were also obtained from CASTp 3.0 (Computed Atlas of Surface Topography of proteins) (Figure S1).38
2.5. Molecular docking interactions through AutoDock Vina, AutoDock, and iGEMDOCK
Molecular docking helps in establishing the most suitable interaction profile of the ligand with the target protein and is used to estimate the energy released during the interaction. AutoDock is an open‐source and automated docking program developed by Molecular Graphics Lab, Scripps Research Institute, La Jolla, CA 92037, United States. It is applicable for the calculation of the interactions profile of biological macromolecules such as proteins/enzymes and ligands. AutoDock suite offers the minimum binding energy of interaction between the ligand and the receptor protein. The calculation of the binding energy is based on the scoring function, which in turn is based on the AMBER force field as well as linear regression analysis using the Lamarckian genetic algorithm. The default settings of AutoDock are commonly applied to comprehend the interaction profile of the ligand with the target. For each docking application, AutoDock is run several times to have several docked conformations of the ligand with the receptor protein. iGEMDOCK is another software used for molecular docking, virtual screening, and postscreening investigation. The iGEMDOCK success rate is 78% (RMSD <2.0 Å).39 Accurate docking was performed by iGEMDOCK, by setting the population size at 800, 80 generations in 10 number of solutions as the genetic algorithm parameters.
2.6. MD simulation
The ligand 6 (PubChem CID: 20183791) and its complexes with SARS‐CoV‐2, SARS‐CoV‐, MERS‐CoV, and HCoV‐ HKU1 Mpros were subjected to MD simulations to analyze the conformational flexibility and binding stability by using GROMACS (2018.2). The force field CHARMM 36 and CGenFF 4.1 were used to generate the topology, atomic, and charge parameters. The box sizes of complex systems were generated in the cubic unit cell. The box sizes of SARS‐CoV‐2, SARS‐CoV, HCoV‐HKU1, and MERS‐CoV Mpro complex systems were set at 893.32, 831.3, 878.63, and 956.33 nm3, respectively. TIP3P water model was selected for the generation of protein topology files in the CHARMM 36 force field. Further, complexes of the ligand 6 with the target proteases were built and a dodecahedron edge box was generated. An aqueous environment was created around each complex. The system was neutralized using Na+ and Cl− ions. The complex was energetically minimized using the steepest descent algorithm. After the minimization, it was equilibrated at constant temperature (300 K) and pressure (1 bar) to generate a conserve ensemble of Canonical ensemble (NVT): moles (N), volume (V) and temperature (T) and Isothermal‐Isobaric (NPT) ensemble: moles (N), pressure (P) and temperature (T) for 100 ps time. After the equilibration, the complex system was subjected to run for 100 ns time of simulation with a step of 2 femtoseconds (fs) using leapfrog algorithm. After the completion of the runs, the generated trajectories were analyzed for the structural deviation between the ligand and the complex system and graphs were generated using MATLAB.
3. RESULTS AND DISCUSSION
The diagrammatic representation of the study is given as Figure 1.
FIGURE 1.
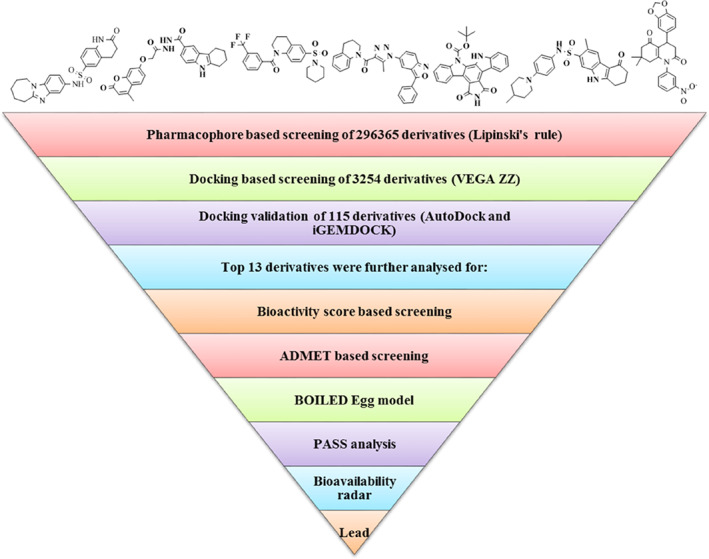
Schematic representation of the study
3.1. Virtual screening
The main aim of virtual screening is to assess a large collection of molecules against a specific target in a time and cost‐effective manner. The virtual screening was performed using VEGA ZZ 3.2.0 and AutoDock Vina with 3254 ligands, out of which 115 best ligands were selected for docking studies.
3.2. Molecular docking
AutoDock Tools were used for the multiple grid docking evaluations and docking validation was done through iGEMDOCK. The results of the best five ligands and the released binding energy upon interaction are represented in Tables 1 and 2. After molecular docking, the top 13 ligands were selected for drug‐likeness analysis (Figure 2). Two physiological ligands ((4 seconds)‐2‐methyl‐2,4‐pentanediol or MPD and n‐[(5‐methylisoxazol‐3‐yl)carbonyl]alanyl‐l‐valyl‐n~1~‐((1r,2z)‐4‐(benzyloxy)‐4‐oxo‐1‐{[(3r)‐2‐oxopyrrolidin‐3 yl]methyl}but‐2‐enyl)‐l leucinamide or PRD) were used as reference and HCQ was used as the control drug. The minimum binding energy was exhibited by ligands 2 and 6 against the CoV Mpros. The chemical structures of the 13 best ligands along with their chemical identification number are given as Figure 2. The docking interactions of the ligands are shown in Figure 3, along with the interacting amino acids (Figure 4). The heteroatoms of the ligands were involved in the hydrogen bonding, with the target proteins. Ligand 2 has two rings and an NH group in its structure. This functional group is well‐known for its tendency to form hydrogen bond and is mainly responsible for the polar interactions between the ligand and the target Mpros. In addition, in the presence of aromatic rings, in its structure, ligand 2 was also involved in non‐polar interactions. In the same way, N and O atoms of triazole and isoxazole rings of the ligand 6 were involved in the formation of hydrogen bonding as well as the dipole‐induce dipole, dispersion force, London dispersion force, or van der Waals forces with the amino acids present in the active site. Owing to the presence of two‐azole rings, ligand 6 may have potential inhibitory action against the viral Mpro. Most interacting amino acid residues are presented in Table S3 and Figure 5, in which hydrogen bond‐forming residues are also included along with the bond distance and interacting atoms. The docking results were further validated through iGEMDOCK, and the results of the best two ligands (2, 6) are summarized in Table S3.
TABLE 1.
The binding energy of hit compound at different grid map of the target protein
| Identified hits | |||||
|---|---|---|---|---|---|
| Derivative | Grid I | Grid II | Grid III | Grid IV | Grid V |
| 1Q2W (SARS‐CoV) | |||||
| 1 | −9.6 | −9.3 | −9.3 | −9.6 | −9.6 |
| 6 | −9.3 | −9.0 | −9.0 | −9.3 | −9.3 |
| 3 | −9.3 | −8.8 | −9.0 | −9.2 | −8.9 |
| 12 | −8.5 | −9.0 | −9.7 | −9.7 | −9.2 |
| 2 | −9.2 | −9.3 | −9.3 | −9.2 | −9.3 |
| 3D23 (HKU1) | |||||
| 11 | −9.5 | −9.5 | −7.2 | −6.9 | −7.1 |
| 1 | −9.4 | −9.8 | −7.4 | −8.4 | −7.7 |
| 6 | −9.2 | −9.4 | −7.8 | −7.4 | −7.8 |
| 5 | −9.4 | −9.4 | −7.0 | −7.4 | −7.4 |
| 12 | −8.7 | −9.8 | −7.9 | −8.4 | −7.4 |
| 4RSP (MERS‐CoV) | |||||
| 9 | −11.0 | −10.2 | −10.9 | −9.8 | −11.0 |
| 10 | −10.8 | −11.0 | −10.9 | −8.3 | −10.8 |
| 8 | −10.6 | −10.5 | −10.6 | −9.9 | −10.6 |
| 11 | −10.4 | −10.4 | −10.3 | −8.0 | −10.3 |
| 7 | −10.2 | −10.1 | −10.1 | −8.9 | −10.1 |
| 6LU7 (SARS‐CoV‐2) | |||||
| 1 | −8.7 | −9.0 | −8.9 | −8.8 | −7.9 |
| 4 | −8.1 | −8.2 | −8.5 | −8.4 | −8.2 |
| 2 | −7.9 | −9.1 | −8.7 | −8.7 | −7.9 |
| 13 | −6.5 | −10.0 | −7.8 | −10.1 | −9.9 |
| 12 | −7.0 | −9.1 | −8.0 | −9.2 | −9.2 |
TABLE 2.
The binding energy of hit compound at most suitable grid map of the target protein (AutoDock)
| Derivative | 1Q2W | 3D23 | 4RSP | 6LU7 | ||||
|---|---|---|---|---|---|---|---|---|
| BE | IC | BE | IC | BE | IC | BE | IC | |
| 1 | −6.4 | 20.42 | −5.41 | 108.14 | −5.7 | 66.59 | −5.53 | 88.97 |
| 2 | −7.1 | 6.2 | −6.93 | 8.32 | −7.2 | 5.3 | −6.12 | 32.9 |
| 3 | −6.5 | 13.45 | −6.70 | 12.32 | −6.50 | 17.17 | −6.06 | 36.38 |
| 4 | −4.18 | 864.86 | −4.55 | 465.33 | −4.41 | 581.85 | −4.80 | 304.76 |
| 5 | −6.45 | 18.76 | −6.30 | 24.03 | −5.44 | 103.15 | −5.88 | 48.83 |
| 6 | −7.06 | 6.68 | −6.83 | 9.84 | −6.85 | 9.53 | −6.17 | 30.07 |
| 7 | −5.81 | 54.83 | −5.59 | 79.75 | −5.38 | 113.65 | −6.65 | 13.32 |
| 8 | −5.92 | 45.47 | −5.04 | 201.18 | −5.88 | 48.81 | −6.21 | 27.83 |
| 9 | −5.95 | 43.46 | −5.93 | 45.06 | −6.67 | 12.88 | −6.16 | 30.65 |
| 10 | −6.61 | 14.28 | −6.58 | 15.09 | −6.55 | 15.86 | −7.76 | 2.05 |
| 11 | −6.81 | 10.24 | −6.88 | 8.99 | −6.31 | 23.9 | −5.35 | 120.58 |
| 12 | −7.29 | 4.57 | −6.21 | 28.1 | −7.07 | 6.58 | −5.96 | 43.1 |
| 13 | −6.08 | 34.84 | −5.58 | 81.69 | −5.69 | 67.24 | −6.93 | 8.32 |
| MPD a | −2.2 | 24.46 | −2.48 | 15.09 | −2.33 | 19.71 | −2.70 | 10.52 |
| PRD a | −3.54 | 2.54 | −4.12 | 1.01 | −3.77 | 1.73 | −3.99 | 1.18 |
| HCQ a | −3.5 | 2.72 | −3.95 | 1.27 | −3.78 | 1.7 | −3.3 | 3.64 |
Note: BE, binding energy; IC, inhibition constant (μM); MPD, ((4 seconds)‐2‐methyl‐2,4‐pentanediol; PRD, n‐[(5‐methylisoxazol‐3‐yl) carbonyl] alanyl‐l‐valyl‐n ~ 1 ~ −((1r,2z)‐4‐(benzyloxy)‐4‐oxo‐1‐{[(3r)‐2‐ oxopyrrolidin‐3 yl] methyl} but‐2‐enyl)‐l‐leucinamide; HCQ, hydroxychloroqui.
IC (mM).
FIGURE 2.
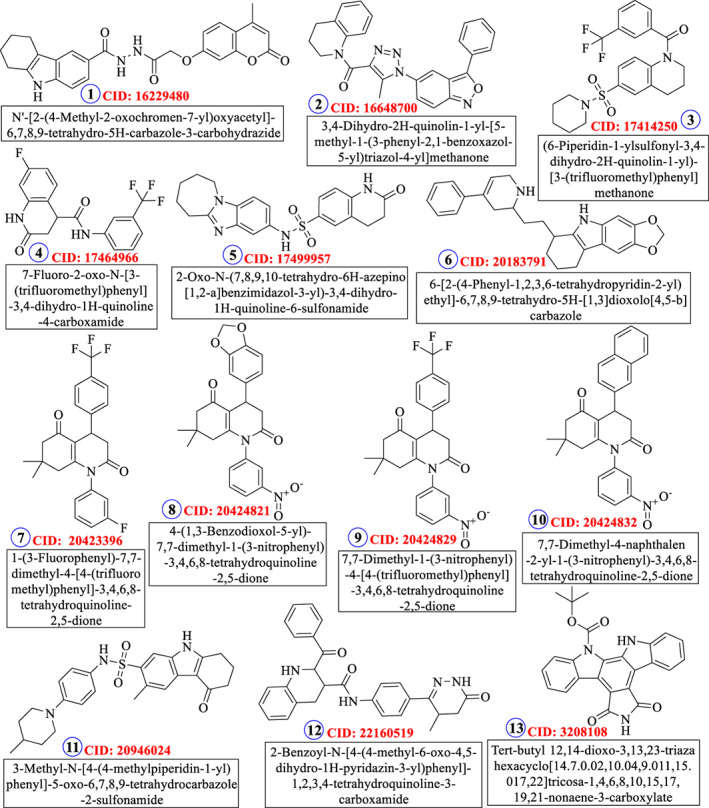
Thirteen best ligands selected based on molecular docking
FIGURE 3.
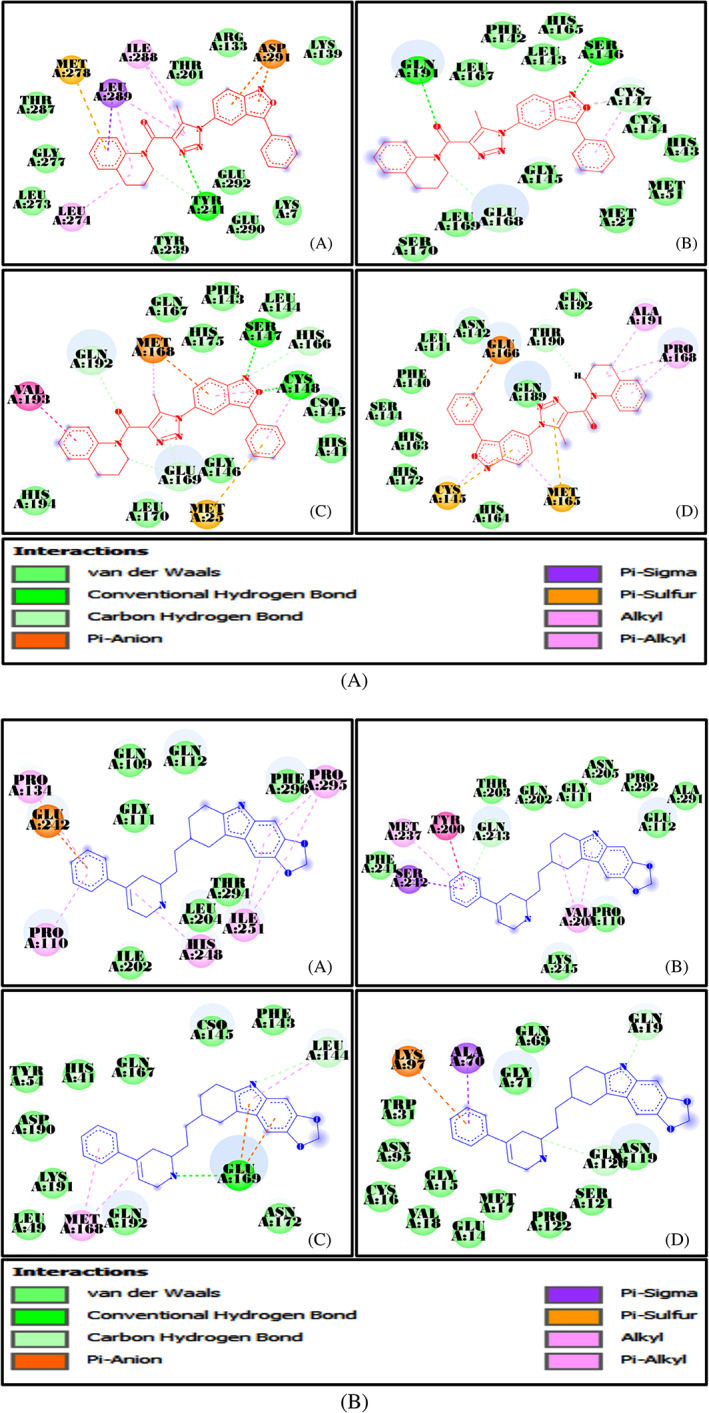
A, Docking interactions of ligand 2 with A: 1Q2W, B: 3D23, C: 4RSP, and D: 6LU7; B, Docking interactions of ligand 6 with A: 1Q2W, B: 3D23, C: 4RSP, and D: 6LU7
FIGURE 4.

Interacting amino acids and released binding energy of ligand 2 and 6
FIGURE 5.
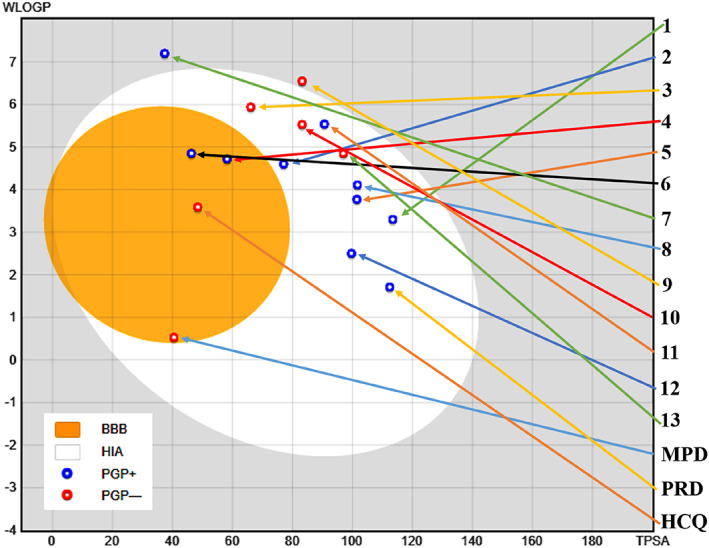
Boiled‐egg's presentation of top 1‐13 derivatives along with physiological ligands (MPD and PRD) and control drug (hydroxychloroquine [HCQ])
The molecular docking results were in tandem with some of the previous computational and virtual screening studies undertaken in search of a probable Mpro inhibitor. However, most of the earlier studies have either used food and drug administration (FDA) approved drugs or naturally occurring phytochemicals as probable Mpro inhibitor (Table 3). The present study mainly explored natural as well as synthetic bioactive constituents of herbs and spices as prospective Mpro inhibitors, not only against SARS‐CoV‐2 but also against the other known coronaviruses. Another differentiating point of the study was the usage of three docking software (AutoDock Vina, AutoDock, and iGEMDOCK) for the binding energy calculation and validation.
TABLE 3.
Some previous virtual studies to identify Mpro inhibitor
| S. No. | Target/PDB ID | Docking tool | Number of ligands | Lead molecule | Binding Energy (kcal/mol) | References |
|---|---|---|---|---|---|---|
| 1 | SARS‐CoV‐2/6LU7 | AutoDock Vina | 100 (Fungal metabolites) | Pyranonigrin A | −7.3 | 40 |
| 2 |
SARS‐CoV‐2 3CLpro/6Y2F; SARS‐CoV 3CLpro/3TNT; MERS‐CoV 3CL/5WKK |
AutoDock Vina | 263 (Phytochemicals) and 75 (FDA approved antiviral drugs) | Glecaprevir (DB13879) |
−9.3 (6Y2F); −9.7 (3TNT); |
41 |
| Daclatasvir (DB09102) |
−9.2 (6Y2F); −9.3 (3TNT); |
|||||
| Paritaprevir (DB09297) |
−9.3 (6Y2F); −8.7(5WKK) |
|||||
| Atazanavir (DB01072) | −9.2 (3TNT); −9.1(5WKK) | |||||
| Vincapusine (11646359) |
−7.5 (6Y2F); −8.3 (3TNT); −9.7(5WKK) |
|||||
| Alloyohimbine (120716) |
−8.0 (6Y2F); −9.0 (3TNT); −8.6(5WKK) |
|||||
| Gummadiol (10308017) |
−7.8 (6Y2F); −8.4 (3TNT); −8.4(5WKK) |
|||||
| 18‐Hydroxy‐3‐epi‐alpha‐yohimbine (102004710) |
−8.1 (6Y2F); −8.0 (3TNT) |
|||||
| 3 | SARS‐CoV‐2 Mpro/6Y2F | AutoDock Vina | 77 (FDA approved antiviral drugs) | Lopinavir‐Ritonavir | −10.6 | 42 |
| Tipranavir | −8.7 | |||||
| Raltegravir | −8.3 | |||||
| 4 | SARS‐CoV‐2 Mpro/5R7Y | AutoDock Vina | 61 (SARS 3CLpro inhibitors) | Molecule ID 19 | −8.6 | 43 |
| Molecule ID 43 | −8.6 | |||||
| Molecule ID 44 | −8.5 | |||||
| Molecule ID45 | −8.6 | |||||
| Molecule ID 49 | −8.5 | |||||
| Molecule ID 58 | −8.8 | |||||
| 5 | SARS‐CoV‐2/6LU7 | Glide | 3992 (FDA approved drugs) and 175 815 (Asinex BioDesign Library) | Ritonavir | −8.01 | 44 |
| Disubstituted pyrazole‐based (1) | −9.02 | |||||
| Disubstituted pyrazole‐based (2) | −8.725 | |||||
| cyclic amide based (12) | −8.273 | |||||
| 6 | SARS‐CoV‐2 Mpro/7BQY | AutoDock Vina | >100 | Camptothecin gimatecan | −11.07 | 45 |
| Lamellarin D | −11.32 | |||||
| Leopolic acid A | −12.22 | |||||
| Stachys ehrenbergii | −13.07 | |||||
| 7 | SARS‐CoV‐2 Mpro/6LU7 | AutoDock Vina | 1051 (FDA approved drugs) | R428 | −10.5 | 46 |
| Teniposide | −9.8 | |||||
| VS5584 | −9.4 | |||||
| Setileuton | −8.5 |
3.3. Pharmacokinetic study of the hit molecules
3.3.1. Bioactivity score
Molinspiration was used for the analysis of bioactivity score against known drug targets like G protein‐coupled receptors (GPCR) ligand, ion channel modulator (ICM), nuclear receptor ligand (NRL), kinase inhibitor (KI), protease inhibitor (PI), and enzyme inhibitor (EI). The software has a broad range of cheminformatics tools associated with the molecular operation and requires simple handling and uses .smiles/.sdf file (http://www.molinspiration.com). Molinspiration does molecular calculation with the help of the existing information in the molecular database and controls fragment‐based virtual screening.47 A drug target can be a receptor protein, which is usually essential for the natural system. Some most acceptable drug targets are ‐
GPCR
Enzymes (mainly protein kinases, proteases, esterases, and phosphatases)
Ligand‐gated ion channel
Ion channels Modulators
Almost all the top 13 ligands displayed moderate to good bioactivity score, showing their potential drug‐like nature. Ligand 6 and 13 had positive bioactivity score against all the selected drug targets (Table 4).
TABLE 4.
Bioactivity score
| Derivative | GPCR | ICM | KI | NRL | PI | EI |
|---|---|---|---|---|---|---|
| 1 | −0.33 | −0.76 | −0.41 | −0.45 | −0.38 | −0.24 |
| 2 | −0.14 | −0.34 | −0.25 | −0.42 | −0.41 | −0.38 |
| 3 | 0.10 | −0.22 | −0.21 | −0.01 | 0.14 | −0.09 |
| 4 | −0.09 | −0.22 | −0.26 | −0.16 | −0.10 | −0.16 |
| 5 | 0.04 | −0.26 | −0.37 | −0.33 | −0.07 | −0.09 |
| 6 | 0.53 | 0.29 | 0.13 | 0.06 | 0.23 | 0.21 |
| 7 | −0.46 | −0.50 | −0.85 | −0.43 | −0.47 | −0.44 |
| 8 | −0.62 | −0.67 | −1.07 | −0.70 | −0.62 | −0.53 |
| 9 | −0.54 | −0.51 | −0.94 | −0.51 | −0.53 | −0.48 |
| 10 | −0.56 | −0.57 | −0.96 | −0.59 | −0.52 | −0.46 |
| 11 | 0.10 | −0.20 | −0.27 | −0.18 | −0.10 | −0.01 |
| 12 | −0.14 | −0.47 | −0.59 | −0.47 | −0.04 | −0.10 |
| 13 | 0.14 | 0.07 | 0.51 | 0.12 | 0.01 | 0.46 |
Abbreviations: EI, enzyme inhibitor; GPCR, GProtein‐coupled receptor; ICM, ion channel modulator; KI, kinase inhibitor; NRL, nuclear receptor ligand; PI, protease inhibitor.
3.4. Pharmacokinetic and drug‐likeness assessment
Evaluation of ADMET properties is a prerequisite for the pre‐clinical analysis.48 The drug development procedure involves substantial time and money. Computational and in silico prediction for important pharmacokinetic properties is an accurate and time‐effective process. Different computational models have been used to predict ADMET (absorption, distribution, mechanism, excretion, and toxicity) parameters of lead compounds during the early stage of drug discovery, which may facilitate the development of new drugs with better ADMET properties. admetSAR is a free online database, that is, regularly updated and provides access to 96 000 unique compounds covering more than 50 categories with ADMET‐allied properties49 such as blood‐brain barrier (BBB) penetration, human intestinal absorption (HIA). The BBB restricts the passage of substances in the central nervous system and also regulates the movement of substances that are crucial for the healthy maintenance of the CNS and its functioning, thereby acting as a mediator between the CNS and blood. For effective drug development, it is almost essential to understand the physiology of BBB and the ability of the drug to cross over it. HIA is an important ADME parameter because of its relevance associated with the transportation of the drug to its target. The intestinal absorption of a molecule also influences the bioavailability of the drug. SwissADME is another web‐based free tool, established by the molecular modeling group of the Swiss Institute of Bioinformatics (SIB). Cheminformatics has established many molecular descriptors based on chemical structures, including oral bioavailability and carcinogenicity.50, 51 Ames toxicity assessment, which is used to assess the potential mutagenicity associated with a test chemical by treating the mutated Salmonella typhimurium strains with it. The top 13 ligands had positive HIA and BBB crossing potential. No carcinogenicity was found to be associated with any of the 13 ligands (Table 5).
TABLE 5.
Absorption, distribution, mechanism, excretion, and toxicity (ADMET) analysis
| Derivative | HIA | HOB | Caco‐2 | PPB | BBB | AMES | Carcino | Biodeg |
|---|---|---|---|---|---|---|---|---|
| 1 | +/0.9850 | −/0.6286 | −/0.8385 | 0.936 | +/0.9401 | −/0.5700 | −/0.9714 | −/0.8750 |
| 2 | +/0.9683 | +/0.5571 | −/0.7407 | 1.090 | +/0.9890 | −/0.5900 | −/0.8857 | −/0.9500 |
| 3 | +/0.9708 | +/0.5143 | −/0.6371 | 1.116 | +/0.9793 | −/0.6800 | −/0.8031 | −/0.7500 |
| 4 | +/0.9514 | +/0.6714 | −/0.5257 | 1.199 | +/0.9855 | −/0.5900 | −/0.9286 | −/0.9500 |
| 5 | +/0.9791 | +/0.6571 | −/0.8786 | 1.115 | +/0.9749 | −/0.6800 | −/0.7745 | −/0.9250 |
| 6 | +/0.9866 | −/0.6286 | −/0.5850 | 0.874 | +/0.9857 | −/1.0000 | −/0.5900 | −/0.8500 |
| 7 | +/0.9785 | +/0.5714 | +/0.5949 | 1.321 | +/0.9779 | −/0.6300 | −/0.8000 | −/0.9000 |
| 8 | +/0.9409 | −/0.5286 | −/0.6278 | 1.272 | +/0.9730 | +/0.6600 | −/0.8714 | −/0.9250 |
| 9 | +/0.9490 | +/0.6571 | −/0.6630 | 1.330 | +/0.9740 | −/0.6900 | −/0.8000 | −/0.9250 |
| 10 | +/0.9618 | +/0.6286 | −/0.5962 | 1.250 | +/0.9730 | +/0.5400 | −/0.8000 | −/0.9750 |
| 11 | +/0.9878 | +/0.7857 | −/0.7812 | 1.347 | +/0.9781 | −/0.6700 | −/0.7429 | −/0.9500 |
| 12 | +/0.9899 | +/0.6714 | −/0.8151 | 0.947 | +/0.9763 | −/0.5200 | −/0.8286 | −/0.8750 |
| 13 | +/0.9660 | −/0.5000 | +/0.5349 | 0.953 | +/0.9698 | +/0.6600 | −/0.8714 | −/0.9000 |
Abbreviations: AMES, Ames mutagenesis; BBB, blood–brain barrier; Biodeg, biodegradability; Caco‐2, Caco‐2 permeability; Carcino, carcinogenesis; HIA, human intestinal absorption; HOB, human oral bioavailability; PPB, plasma protein binding (Unit = 100%).
To assess the HIA and BBB crossing ability, the Egan's egg was used to create the BOILED egg model,52 which accurately predicts the physicochemical descriptors. The BOILED‐Egg's yolk represents the physicochemical space for a high probability of BBB permeability, whereas the Egg‐white represents the physicochemical space for high HIA probability. The blue points on the plot are predicted to be actively effluxed by P‐gp (PGP+) and red dots show the nonsubstrate of P‐gp (PGP‐). P‐gp protects the central nervous system from xenobiotics.53 Points' located within the BOILED‐Egg's yolk represent passive permeation of the molecules through the BBB as well as passive absorption by the gastrointestinal tract. The ligand 6, lied inside the white ellipse and the BOILED‐Egg's yolk, as shown by the findings (Figure 5).54
Several parameters like molecular weight (MW), polar surface area, etc. influence the drug‐likeness character. Large molecules have difficulty crossing over the biological membranes55 and it has been reported that 80% of the known drugs have their MW below 450 Da. The Polar surface area and the number of hydrogen bonds increase with the MW.56 Hydrogen bonding also has a vital significance in biological systems, as extensive hydrogen bonding causes a delay in the movement of drugs from gut to blood. Drugs having partial rigid character generally possess good to excellent oral bioavailability.57 LogP represents the solubility of the proposed compounds affecting the capacity of a molecule to pass through the cell membrane.58 The MW of all the 13 ligands was within the limit of 500 and TPSA was below 140 Å2 (Table 6).
TABLE 6.
Pharmacokinetic parameters
| Derivative | MW | nRB | nOH | nOHNH | miLogP | TPSA | Abs% | nAtoms | MR | Lipinski violation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 445.48 | 5 | 8 | 3 | 3.70 | 113.43 | 69.87 | 33 | 113.56 | 0 |
| 2 | 435.49 | 4 | 5 | 0 | 5.13 | 77.06 | 82.41 | 33 | 129.04 | 0 |
| 3 | 452.50 | 4 | 5 | 0 | 4.65 | 57.69 | 89.10 | 31 | 118.17 | 0 |
| 4 | 352.29 | 3 | 4 | 2 | 2.98 | 58.20 | 88.92 | 25 | 85.34 | 0 |
| 5 | 410.50 | 3 | 7 | 2 | 3.10 | 93.09 | 76.88 | 29 | 115.48 | 0 |
| 6 | 400.52 | 4 | 4 | 2 | 5.36 | 46.29 | 93.03 | 30 | 124.85 | 0 |
| 7 | 431.43 | 3 | 3 | 0 | 5.22 | 37.38 | 96.10 | 31 | 111.73 | 1 |
| 8 | 434.45 | 3 | 8 | 0 | 4.01 | 101.67 | 73.92 | 32 | 121.66 | 0 |
| 9 | 458.44 | 4 | 6 | 0 | 5.02 | 83.20 | 80.30 | 33 | 120.60 | 0 |
| 10 | 440.50 | 3 | 6 | 0 | 5.30 | 83.20 | 80.20 | 33 | 133.10 | 0 |
| 11 | 451.59 | 4 | 6 | 2 | 4.80 | 82.27 | 80.62 | 32 | 132.49 | 0 |
| 12 | 466.54 | 5 | 7 | 3 | 4.63 | 99.66 | 74.62 | 35 | 145.90 | 0 |
| 13 | 425.44 | 2 | 7 | 2 | 4.85 | 96.96 | 75.55 | 32 | 126.59 | 0 |
Abbreviations: Abs %, percentage of absorption; miLog P, logarithm of compound partition coefficient between n‐octanol and water; MR, molecular refractivity; MW, molecular weight; nAtoms, number of atoms; nON, number hydrogen bond acceptor; nOHNH, number hydrogen bond donor; nRB, number of rotatable bonds; TPSA, topological polar surface area.
The bioavailability radar is a pictorial representation and assessment of drug‐likeness of a molecule. It is plotted for six physicochemical properties viz. LIPO (Lipophility): −0.7 < XLOGP3 < +5.0; SIZE: 150 g/mol < MW < 500 g/mol; POLAR (Polarity): 20Å2 < TPSA <130 Å2; INSOLU (Insolubility): 0 < Log S (ESOL) < 6; INSATU (Insaturation): 0.25 < Fraction Csp3 < 1; FLEX (Flexibility): 0 < Num, rotatable bonds <9. The pink area represents the optimal range for each parameter. The colored zone in the radar shows the best suited physiochemical space for oral bioavailability. The bioavailability radar predicted promising results for ligand 6 (Figure 6).
FIGURE 6.
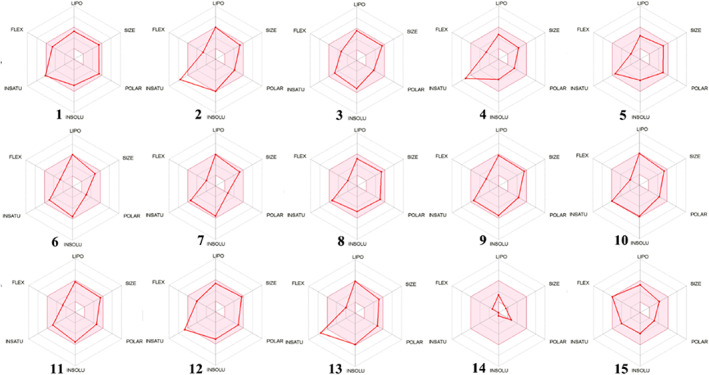
Representation of bioavailability radar for the selected lead derivatives
3.5. MD simulation analysis
MD simulation is performed to know the movement of atoms and molecules over a certain time. The simulation predicts the dynamic behavior of the interaction between the target and receptor. The results may be used to analyze the ligand's confirmation and energy change upon interaction. The docked Mpro complexes with ligand 6 were simulated using the GROMACS package. The stability of the protein‐ligand conformation can be estimated by the deviations observed during the simulation time. The smaller the deviations, stable would be the protein structure.59, 60 The RMSD analysis is used for the assessment of the deviation in the backbone structure of the protein during the simulation time. The smaller the deviation between the starting and final conformations, the more stable would be the structure of the protein. RMSD was calculated for the C‐alpha (Cα) to check the stability. Simulated complex systems showed no significant change in the Cα atoms of the ligated SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 Mpros from the initial structure, whereas the ligated HCoV‐ HKU1 Mpro showed a significant change. The complexes of SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2 Mpros with the ligand 6 showed the maximum deviation around 0.20 nm from the initial structure, whereas the complex of HCoV‐HKU1 Mpro with ligand 6 showed a deviation greater than 0.45 nm, depicting the instability of the HCoV‐HKU1 complex in comparison to the other proteins. The overall complex trajectories were stable during the time of simulation (Figure 7A). The RMSD plot of the ligand suggested its stable interaction with the target Mpros during the time of simulation except for MERS‐CoV Mpro, which showed a deviation of 1.2 nm (Figure 7B). The high deviation in the ligand RMSD plot of MERS‐CoV Mpro might be due to the unstable interaction with the protein, as the trajectory visualization showed that the ligand moved to positions other than the binding pocket.
FIGURE 7.
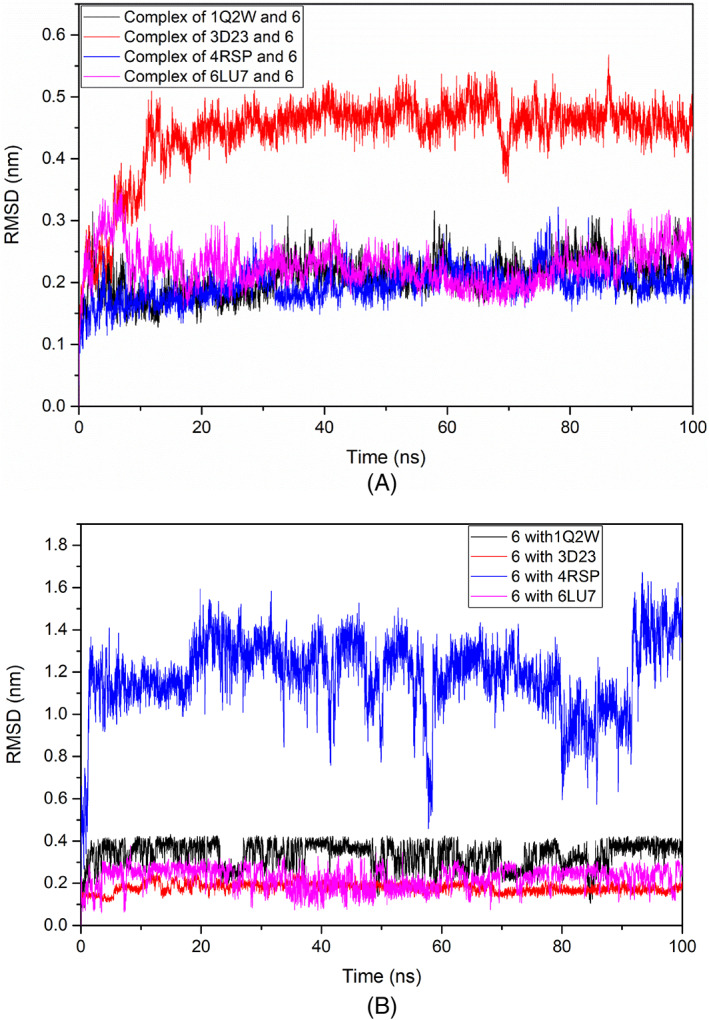
Root mean deviation square (RMSD) plots of: A, target‐ligand complexes; B, ligand in the complexes
The radius of gyration (Rg) depicts the atoms mass‐weighted root mean square distance of atoms from the center of mass and shows the stability of the complex. The fitness of folding, compactness, and target's structure, for the simulation time of 100 ns are presented in the Rg plot. During the simulation time, the ligated MERS‐CoV and SARS‐ CoV‐2 Mpros showed constant Rg values of 2.22 nm and the ligated SARS‐CoV Mpro showed less fluctuation in the Rg value viz. 2.25 nm, as compared to the initial time to 35 ns. The ligated HCoV‐HKU1 Mpro showed a higher Rg value viz., 2.30 nm after the initial time (5 ns) (Figure 8). The higher the Rg value, the greater would be the change in the structural compactness and folding of the protein.
FIGURE 8.
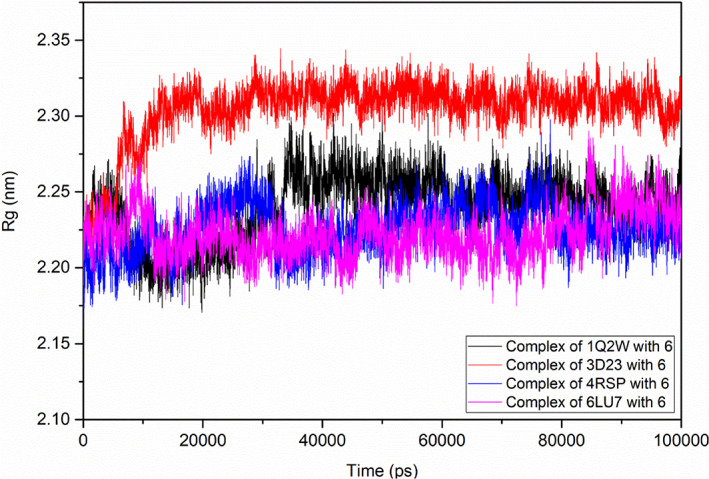
Rg plot of the selected target‐ligand complex
Solvent‐accessible surface area (SASA) of ligated Mpros was studied to assess the changes in the hydrophobic and hydrophilic residues. The target‐ligand complexes showed almost similar SASA values between 168 and 170 nm2, except ligated SARS‐CoV Mpro, showing a lesser value (163 nm2) during the simulation time (Figure 9).
FIGURE 9.
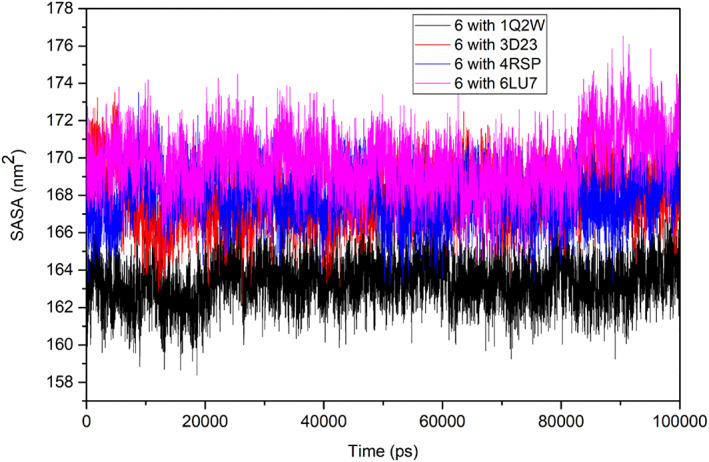
Solvent‐accessible surface area (SASA) plot of the ligand 6
To further analyze the complex's structural stability and the effect of the ligand on the flexibility of the structure of the target protein, the root mean square fluctuation (RMSF), per residue of all the target proteins' backbone, was studied. To understand the changes related to the ligand, the RMSF plots of the ligated Mpros were generated for ligand 6 and evaluated for atom wise fluctuations in the molecule. The RMSF plot suggested that the target‐ligand complex had higher fluctuations and low stability (Figures 10 and 11).
FIGURE 10.
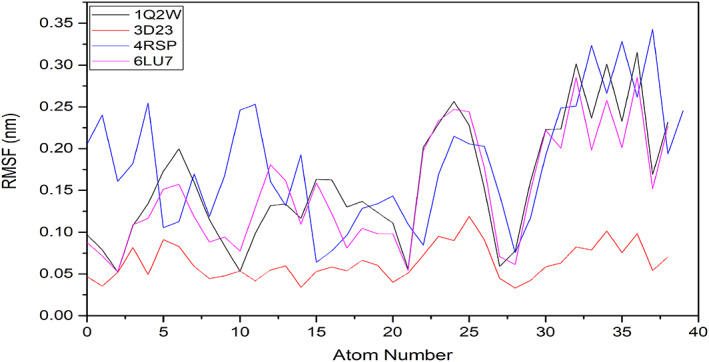
Root mean square fluctuation (RMSF) plot generated for A, Mpros; B, ligated Mpros
FIGURE 11.
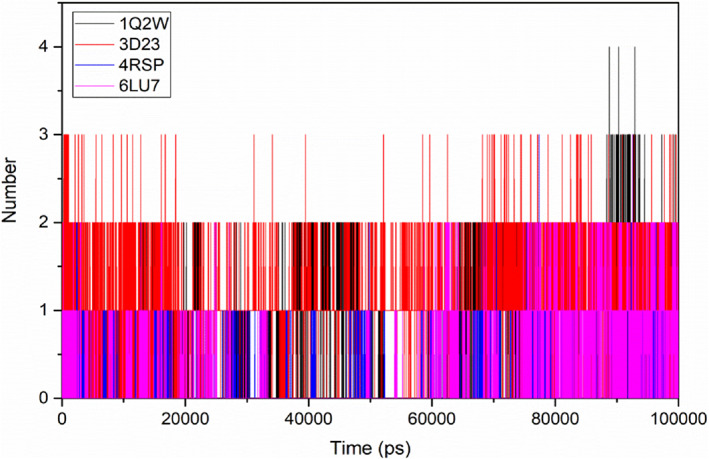
Number of hydrogen bonds between the target‐ligand complexes during the simulation time
The interaction strength between the ligand and the target was quantified by the analysis of hydrogen bonding and non‐bonding energies, throughout the simulation time. Ligand 6 interacted and formed hydrogen bonds with the target Mpros for the whole simulation time (Figure 11).
Interaction energy and stability of the complexes were studied by the analysis of Coulomb and Lennard‐Jones interaction energies. The ligand 6 exhibited better Coulombic‐Short Range (Coulumb‐SR) binding energy (−175 kJ/mol) with SARS‐CoV and HCoV‐ HKU1 Mpros. The Coulumb‐SR interactions with SARS‐CoV‐2 Mpro showed binding energy of 100 kJ/mol from the starting to 10 ns and between 60 and 100 ns (Figure 12A). Between 10 and 60 ns, the ligand did not show any Coulumb‐SR binding with MERS‐CoV Mpro. Ligand 6 showed better Lennard‐Jones interaction energy (Figure 12B) with all the target proteases during the time of the simulation. Overall, the binding energies between ligand 6 and the target Mpros suggested stable binding and interaction.
FIGURE 12.
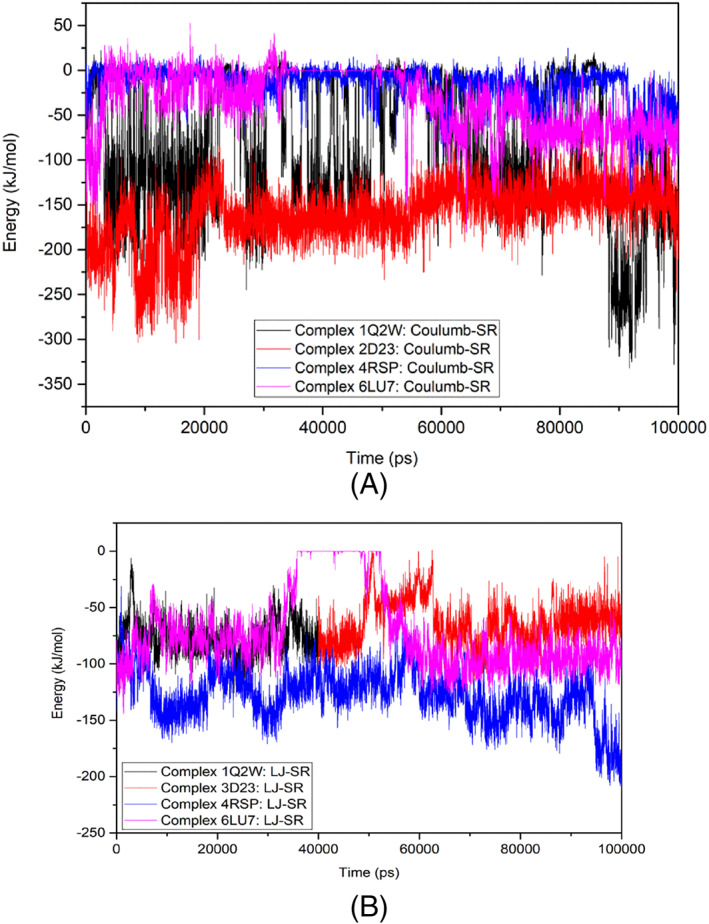
Interaction energy and binding stability of the target‐ligand complexes: A, Coulomb‐SR energy; B, Lennard‐Jones‐SR energy
4. CONCLUSION
The main protease enzyme is chymotrypsin‐like cysteine, which plays an important role in governing the replication and transcription of coronavirus. In this study, virtual interaction of main proteases of SARS‐CoV‐2, SARS‐CoV, MERS‐CoV, and HCoV‐HKV1 with active constituents of some common herbs and spices, as the plant‐based medicinal system is as old as mankind itself. The derivatives were explored and filtered through Lipinski's parameters. HCQ was used as a reference drug. The interactions showed positive results with some of the derivatives and the best interaction was observed in the case of dihydroquinoline derivative, 3,4‐Dihydro‐2H‐quinolin‐1‐yl‐[5‐methyl‐1‐(3‐phenyl‐2,1‐benzoxazol‐5‐yl)triazole‐4 yl]methanone and obtained from Flinder rose (caper), belonging to Capparidacea family and used as a seasoning agent and the fruit is used as a pickle and 6‐[2‐(4‐Phenyl‐1,2,3,6‐tetrahydropyridin‐2‐yl)ethyl]‐6,7,8,9‐tetrahydro‐5H‐[1,3]dioxolo[4,5‐b]carbazole, a potential derivative of carbazole, which is obtained from sweet neem belonging to Rutaceae family is also a potent antioxidant agent. The findings pave way for further exploration of the active constituents of natural plant‐based resources for an effective treatment process to combat SARS‐CoV‐2 and other microbial infections with minimal side effects.
CONFLICT OF INTEREST
The authors declare they have no competing interests.
Supporting information
Data S1. Supporting Information
ACKNOWLEDGEMENTS
The authors are thankful to the Research and Development Committee, Integral University for the support. Dr A. M. Alanazi acknowledges the Research Supporting Project Number (RSP‐2020/261), King Saud University, Riyadh, Saudi Arabia. Dr Nidhi Mishra is thankful to the Central Computing facility, IIIT‐Allahabad.
Azad I, Khan T, Maurya AK, Irfan Azad M, Mishra N, Alanazi AM. Identification of Severe Acute Respiratory Syndrome Coronavirus‐2 inhibitors through in silico structure‐based virtual screening and molecular interaction studies. J Mol Recognit. 2021;34(10):e2918. 10.1002/jmr.2918
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article
REFERENCES
- 1.Yin Y, Wunderink RG. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology. 2018;23(2):130‐137. 10.1111/resp.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Z, Xiao X, Wei X, et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS‐CoV‐2. J Med Virol. 2020;92(6):595‐601. 10.1002/jmv.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uskokovic V. Why have nanotechnologies been underutilized in the global uprising against the coronavirus pandemic? Nanomedicine (Lond). 2020;15(17):1719‐1734. 10.2217/nnm-2020-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long SW, Olsen RJ, Christensen PA, et al. Molecular architecture of early dissemination and massive second wave of the SARS‐CoV‐2 virus in a major metropolitan. mBio. 2020;11(6):e02707‐e02720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gates B. Responding to Covid‐19 a once‐in‐a‐century pandemic? N Engl J Med. 2020;382:1677‐1679. [DOI] [PubMed] [Google Scholar]
- 6.Sheahan TP, Sims AC, Leist SR, et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS‐CoV. Nat Commun. 2020;11:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheahan TP, Sims AC, Zhou S, et al. An orally bioavailable broad‐spectrum antiviral inhibits SARS‐CoV‐2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med. 2020;12:eabb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang S, Hillyer C, Du L. Neutralizing antibodies against SARSCoV‐2 and other human coronaviruses. Trends Immunol. 2020;41(5):355‐359. 10.1016/j.it.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang, Y., Yang, C., Xu, Xf. et al. Structural and functional properties of SARS‐CoV‐2 spike protein: potential antivirus drug development for COVID‐19. Acta Pharmacol Sin. 2020;41:1141‐1149. 10.1038/s41401-020-0485-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215‐220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 11.Aris S‐N‐AM, Rahman MZA, Rahman RNZRA, et al. Identification of potential riboflavin synthase inhibitors by virtual screening and molecular dynamics simulation studies. J King Saud Univ Sci. 2021;33(2):101270. 10.1016/j.jksus.2020.101270. [DOI] [Google Scholar]
- 12.Kumar N, Awasthi A, Kumari A, et al. Antitussive noscapine and antiviral drug conjugates as arsenal against COVID‐19: a comprehensive chemoinformatics analysis. J Biomol Struct Dyn. 2020;20:1‐16. 10.1080/07391102.2020.1808072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maier HJ, Bickerton E, Britton P. Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses. 2015;1282:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebuhr J. The coronavirus replicase. Curr Top Microbiol Immunol. 2005;287:57‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus‐encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81:853‐879. [DOI] [PubMed] [Google Scholar]
- 16.Yang H, Yang M, Ding Y, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci U S A. 2003;100:13190‐13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science. 2003;300:1763‐1767. [DOI] [PubMed] [Google Scholar]
- 18.Xue X, Yang H, Shen W, et al. Production of authentic SARS‐CoV M(pro) with enhanced activity: application as a novel tag‐cleavage endopeptidase for protein overproduction. J Mol Biol. 2007;366:965‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J, Wei Z, Song J. Dissection study on the severe acute respiratory syndrome 3C‐like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J Biol Chem. 2004;279:24765‐24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajendran V, Gopalakrishnan C, Sethumadhavan R. Pathological role of a point mutation (T315I) in BCR‐ABL1 protein—a computational insight. J Cell Biochem. 2018;119(1):918‐925. 10.1002/jcb.26257. [DOI] [PubMed] [Google Scholar]
- 21.Purohit R. Role of ELA region in auto‐activation of mutant KIT receptor: a molecular dynamics simulation insight. J Biomol Struct Dyn. 2014;32(7):1033‐1046. 10.1080/07391102.2013.803264. [DOI] [PubMed] [Google Scholar]
- 22.Sarma P, Shekhar N, Prajapat M, Avti P, Kaur H, Kumar S. In‐silico homology assisted identification of inhibitor of RNA binding against 2019‐nCoV N‐protein (N terminal domain). J Biomol Struct Dynam. 2020;1‐9. 10.1080/07391102.2020.1753580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang TA. Health benefits of culinary herbs and spices. J AOAC Int. 2019;102(2):395‐411. 10.5740/jaoacint.18-0418. [DOI] [PubMed] [Google Scholar]
- 24.Yashin A, Yashin Y, Xia X, Nemzer B. Antioxidant activity of spices and their impact on human health: a review. Antioxidants (Basel). 2017;6(3):70. 10.3390/antiox6030070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neveu V, Perez‐Jiménez J, Vos F, et al. Phenol‐Explorer: an online comprehensive database on polyphenol contents in foods. Database (Oxford). 2010;2010:bap024. 10.1093/database/bap024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Craig WJ. Health‐promoting properties of common herbs. Am J Clin Nutr. 1999;70(3):491S‐499S. 10.1093/ajcn/70.3.491s. [DOI] [PubMed] [Google Scholar]
- 27.Lv J, Qi L, Yu C, et al. Consumption of spicy foods and total and cause specific mortality: population‐based cohort study. BMJ. 2015;351:h3942. 10.1136/bmj.h3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan K. Some aspects of toxic contaminants in herbal medicines. Chemosphere. 2003;52:1361‐1371. 10.1016/S0045-6535(03)00471-5. [DOI] [PubMed] [Google Scholar]
- 29.Lai PK, Roy J. Antimicrobial and chemopreventive properties of herbs and spices. Curr Med Chem. 2004;11:1451‐1460. 10.2174/0929867043365107. [DOI] [PubMed] [Google Scholar]
- 30.Herman L. Herb & Spice Companion: The Complete Guide to over 100 Herbs & Spices. New York, NY: Wellfleet Press; 2015. [Google Scholar]
- 31.Anderson JJ, Anthony MS, Cline JM, et al. Health potential of soy isoflavones for menopausal women. Publ Health Nutr. 1999;2:489‐504. 10.1017/s1368980099000671. [DOI] [PubMed] [Google Scholar]
- 32.Sachan A, Kumar S, Kumari K, et al. nMedicinal uses of spices used in our traditional culture: worldwide. J Med Plants Stud. 2018;6:116‐122. [Google Scholar]
- 33.Lindner HA, Fotouhi‐Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R. The papain‐like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol. 2005;79:15199‐15208. 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimamoto Y, Hattori Y, Kobayashi K, et al. Fused‐ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors. Bioorg Med Chem. 2015;23:876‐890. 10.1016/j.bmc.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3‐25. 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 36.Mielech AM, Kilianski A, Baez‐Santos YM, Mesecar AD, Baker SC. MERS‐CoV papain‐like protease has deISGylating and deubiquitinating activities. Virology. 2014;450–451:64‐70. 10.1016/j.virol.2013.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nomburg J, Meyerson M, DeCaprio JA. Noncanonical junctions in subgenomic RNAs of SARS‐CoV‐2 lead to variant open reading frames. bioRxiv. 2020. 10.1101/2020.04.28.066951. [DOI] [Google Scholar]
- 38.Khan T, Lawrence AJ, Azad I, Raza S, Joshi S, Khan AR. Computational drug designing and prediction of important parameters using in silico methods—a review. Curr Comp‐Aided Drug Des. 2019;15:384‐397. 10.2174/1573399815666190326120006. [DOI] [PubMed] [Google Scholar]
- 39.Azad I, Akhter Y, Khan T, et al. Synthesis, quantum chemical study, AIM simulation, in silico ADMET profile analysis, Molecular Docking and antioxidant activity assessment of aminofuran derivatives. J Mol Str. 2019;1203:127285. 10.1016/j.molstruc.2019.127285. [DOI] [Google Scholar]
- 40.Rao P, Shukla A, Parmar P, et al. Reckoning a fungal metabolite, Pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS‐CoV‐2 virus identified using docking and molecular dynamics simulation. Biophys Chem. 2020;264:106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bahadur GA, Ajmal AM, Lee J, Abul FM, Mashay A‐AK. Structure‐based virtual screening of phytochemicals and repurposing of FDA approved antiviral drugs unravels lead molecules as potential inhibitors of coronavirus 3C‐like protease enzyme. J King Saud Univ Sci. 2020;32(6):2845‐2853. 10.1016/j.jksus.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yogesh K, Harvijay S, Chirag NP. In silico prediction of potential inhibitors for the Main protease of SARS‐CoV‐2 using molecular docking and dynamics simulation‐based drug‐repurposing. J Infect Public Health. 2020;13:1210‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Motiwale M, Yadav NS, Kumar S, et al. Finding potent inhibitors for COVID‐19 main protease (Mpro): an in silico approach using SARS‐CoV‐3CL protease inhibitors for combating CORONA. J Biomol Struct Dyn. 2020;8:1‐12. 10.1080/07391102.2020.1829501. [DOI] [PubMed] [Google Scholar]
- 44.Kanhed AM, Patel DV, Teli DM, Patel NR, Chhabria MT, Yadav MR. Identification of potential Mpro inhibitors for the treatment of COVID‐19 by using systematic virtual screening approach. Mol Divers. 2020;31:1‐19. 10.1007/s11030-020-10130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mazzini S, Musso L, Dallavalle S, Artali R. Putative SARS‐CoV‐2 Mpro inhibitors from an in‐house library of natural and nature‐inspired products: a virtual screening and molecular docking study. Molecules. 2020;25(16):3745. 10.3390/molecules25163745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bharadwaj S, Azhar EI, Kamal MA, et al. SARS‐CoV‐2 Mpro inhibitors: identification of anti‐SARS‐CoV‐2 Mpro compounds from FDA approved drugs. J Biomol Struct Dyn. 2020;5:1‐16. 10.1080/07391102.2020.1842807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan T, Ahmad R, Azad I, Raza S, Joshi S, Khan AR. Computer‐aided drug design and virtual screening of targeted combinatorial libraries of mixed‐ligand transition metal complexes of 2‐butanone thiosemicarbazone. Comput Biol Chem. 2018;75:178‐195. 10.1016/j.compbiolchem.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Nisha CM, Kumar A, Vimal A, Bai BM, Pal D, Kumar A. Docking and ADMET prediction of few GSK‐3 inhibitors divulges 6‐bromoindirubin‐3‐oxime as a potential inhibitor. J Mol Graph Model. 2016;65:100‐107. 10.1016/j.jmgm.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 49.Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Info Model. 2012;52:3099‐3105. 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 50.Yang H, Lou C, Sun L, et al. admetSAR 2.0: web‐service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2018;35:1067‐1069. [DOI] [PubMed] [Google Scholar]
- 51.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug‐likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daina A, Zoete V. A boiled‐egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem Med Chem. 2016;11:1117‐1121. 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szakacs G, Varadi A, Ozvegy‐Laczka C, et al. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME‐Tox). Drug Dis Today. 2008;13:379‐393. 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 54.Isyaku Y, Uzairu A, Uba S. Computational studies of a series of 2‐substituted phenyl‐2‐oxo‐, 2‐hydroxyl‐ and 2‐acylloxyethylsulfonamides as potent anti‐fungal agents. Heliyon. 2020;6:e03724. 10.1016/j.heliyon.2020.e03724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garcia‐Sosa AT, Maran U, Hetenyi C. Molecular property filters describing pharmacokinetics and drug binding. Curr Med Chem. 2012;19:1646‐1662. [DOI] [PubMed] [Google Scholar]
- 56.Almi I, Belaidi S, Melkemi N, Bouzidi D. Chemical reactivity, drug‐likeness and structure activity/property relationship studies of 2,1,3‐benzoxadiazole derivatives as anti‐cancer activity. J Bionano. 2018;12:49‐57. [Google Scholar]
- 57.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615‐2623. [DOI] [PubMed] [Google Scholar]
- 58.Azad I, Nasibullah M, Khan T, Hassan F, Akhter Y. Exploring the novel heterocyclic derivatives as lead molecules for design and development of potent anticancer agents. J Mol Graph Model. 2018;81:211‐228. 10.1016/j.jmgm.2018.02.013. [DOI] [PubMed] [Google Scholar]
- 59.Aier I, Varadwaj P, Raj U. Structural insights into conformational stability of both wild‐type and mutant EZH2 receptor. Sci Rep. 2016;6:34984. 10.1038/srep34984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maurya AK, Mulpuru V, Mishra N. Discovery of novel coumarin analogs against the α‐glucosidase protein target of diabetes mellitus: pharmacophore‐based QSAR, docking, and molecular dynamics simulation studies. ACS Omega. 2020;5(50):32234‐32249. 10.1021/acsomega.0c03871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article