

Abstract
The contributions that the R-Ras subfamily [R-Ras, R-Ras2/teratocarcinoma 21 (TC21), and M-Ras] of small GTP-binding proteins make to normal and aberrant cellular functions have historically been poorly understood. However, this has begun to change with the realization that all three R-Ras subfamily members are occasionally mutated in Noonan syndrome (NS), a RASopathy characterized by the development of hematopoietic neoplasms and abnormalities affecting the immune, cardiovascular, and nervous systems. Consistent with the abnormalities seen in NS, a host of new studies have implicated R-Ras proteins in physiological and pathologic changes in cellular morphology, adhesion, and migration in the cardiovascular, immune, and nervous systems. These changes include regulating the migration and homing of mature and immature immune cells, vascular stabilization, clotting, and axonal and dendritic outgrowth during nervous system development. Dysregulated R-Ras signaling has also been linked to the pathogenesis of cardiovascular disease, intellectual disabilities, and human cancers. This review discusses the structure and regulation of R-Ras proteins and our current understanding of the signaling pathways that they regulate. It explores the phenotype of NS patients and their implications for the R-Ras subfamily functions. Next, it covers recent discoveries regarding physiological and pathologic R-Ras functions in key organ systems. Finally, it discusses how R-Ras signaling is dysregulated in cancers and mechanisms by which this may promote neoplasia.
The Ras superfamily of small GTP-binding proteins includes >100 proteins that regulate diverse cellular functions, such as proliferation, survival, cytoskeletal dynamics, and molecular and vesicular transport.1 To date, the classic Ras proteins [Harvey Ras viral oncogene homolog (H-Ras), Kirsten Ras viral oncogene homolog (K-Ras), and neuroblastoma Ras (N-Ras] have been most extensively studied. Like other family members, classic Ras proteins play essential roles in intracellular signaling, promoting proliferation, survival, migration, and/or differentiation, and, when inappropriately activated, pathophysiology. Classic Ras proteins function as molecular switches that are activated when guanine nucleotide exchange factors (GEFs), which are themselves activated by stimuli such as growth factor signaling,2, 3, 4 interact with Ras and induce conformational changes that promote GDP release and subsequent GTP binding. The duration of Ras activation is, in turn, limited by GTPase-activating proteins (GAPs), which stimulate an intrinsic GTPase activity in Ras, triggering GTP hydrolysis and inactivation.2 Unfortunately, these finely balanced regulatory systems are often disrupted in human cancers,5 by either the overexpression of classic Ras isoforms or activating mutations in genes encoding these proteins, resulting in prolonged Ras-mediated signaling. Classic Ras proteins can also be inappropriately activated by loss-of-function mutations in Ras GAP genes such as NF1, as seen in the hereditary tumor susceptibility syndrome neurofibromatosis type 1 and in several tumor types that occur sporadically in the general population.
The closest relatives of the classic Ras proteins are members of the R-Ras subfamily (the R in R-Ras denotes related). In contrast to classic Ras proteins, the physiological and pathologic functions of the R-Ras subfamily are poorly understood. However, in recent years, these proteins have drawn increasing attention, and it is becoming clear that they also regulate key functions, including changes in cell morphology, adhesion, and migration. This review focuses on the physiological and pathologic consequences of the activation of R-Ras subfamily proteins. It first briefly considers the structure and regulation of R-Ras subfamily proteins and our current understanding of the signaling cascades they regulate; for additional details, please refer to some excellent reviews that provide more in-depth information.6,7 Next, the consequences of mutations in R-Ras genes in specific variants of Noonan syndrome (NS) and the early insights they provide regarding the function(s) of R-Ras subfamily members are discussed. Finally, the review discusses our current understanding of the role that R-Ras subfamily proteins play in cardiovascular disease, immune system function, nervous system development, and the pathogenesis of human cancers, all of which are states that are highly dependent on changes in cellular morphology, adhesion, and migration.
Structure and Regulation of the R-Ras Subfamily and Signaling Cascades Controlled by R-Ras Proteins
The R-Ras subfamily of small GTP-binding proteins has three members: R-Ras, R-Ras2/TC21, and M-Ras. These three proteins have approximately 55% sequence similarity with the classic Ras subfamily8,9 (Figure 1, A and B). R-Ras was discovered in 1987, when a human genomic library was screened under low stringency hybridization conditions with an H-Ras probe. In 1990, R-Ras2 was cloned first from a human teratocarcinoma cell line using degenerate oligonucleotides recognizing a region conserved in Ras proteins and subsequently from an ovarian carcinoma cDNA library.10 Seven years later, M-Ras (muscle Ras viral oncogene homolog) was identified and cloned from the skeletal muscle cell line C2 and human embryonic lung fibroblasts using probes derived from R-Ras and R-Ras2 sequences.8 Cluster analyses comparing the sequences of the R-Ras subfamily proteins with the classic Ras subfamily demonstrate that the R-Ras proteins are distinct from the classic Ras proteins and cluster with one another (Figure 1C). This largely reflects differences in the N- and C-terminal regions of R-Ras and classic Ras proteins. Each R-Ras subfamily member has unique N-terminal regions absent in classic Ras proteins, and their C-terminal hypervariable regions also differ from those found in classic Ras proteins (Figure 1, A, D, and E).11 The latter regions affect the distinct localization of different R-Ras proteins, which, in turn, affects their cellular functions.11,12 The C-terminal region also plays a key role in the activation of downstream signaling pathways. As evidence of this, proteins in which the C-terminus of H-Ras and R-Ras was replaced with an R-Ras or H-Ras tail, respectively, functioned most like their tail donor11; R-Ras also has a proline PXXP motif in its C-terminus that is a putative Src homology 3 binding motif. In contrast to the N- and C-terminal variable regions, the effector domains of classic and R-Ras proteins are highly similar.
Figure 1.

A–C: Shared domains of Ras proteins (A) and sequence alignment showing R-Ras and classic Ras protein sequences colored by amino acid class (B) with annotations for conserved signaling domains and known downstream effectors [Raf, phosphatidylinositol 3-kinase (PI3K), Rac, and Rho; C]. A and B: Purple bars under the alignment indicate the sites of the G1 box (amino acids 36 to 43, 21 to 28, and 20 to 27 in R-Ras1, R-Ras2, and M-Ras, respectively), the G2 box (a single amino acid: 61, 46, and 45 in R-Ras1, R-Ras2, and M-Ras, respectively), the G3 box (amino acids 83 to 86, 68 to 71, and 67 to 70 in R-Ras1, R-Ras2, and M-Ras, respectively), the G4 box (amino acids 142 to 145, 127 to 130, and 126 to 129 in R-Ras1, R-Ras2, and M-Ras, respectively), and the G5 box (amino acids 172 to 174, 157 to 159, and 156 to 158 in R-Ras1, R-Ras2, and M-Ras, respectively). Yellow bars beneath the alignment indicate the sites of switch region I (amino acids 59 to 66, 44 to 51, and 43 to 50 in R-Ras1, R-Ras2, and M-Ras, respectively) and II (amino acids 85 to 103, 70 to 88, and 69 to 87 in R-Ras1, R-Ras2, and M-Ras, respectively). The green bar indicates the distinct amino termini of R-Ras subfamily proteins, whereas the orange bar indicates their distinct carboxy termini. Asterisks indicate regions that are required for the activation of downstream signaling proteins (C). D: Cluster analysis showing relationships between R-Ras and classic Ras members, with distance signifying divergence. E: R-Ras subfamily protein sequence alignment, showing levels of sequence identity between members, summarized using black bar height below the alignment, with higher bars signifying more conservation. Alignments and phylogenetic tree were generated using MegaAlign Pro 15 (DNAStar, Madison, WI). FliI, Flightless 1; FLNa, Filamin A.
The canonical R-Ras, R-Ras2, and M-Ras proproteins have similar sizes (218, 204, and 208 amino acids, respectively). Following translation, three amino acid residues at the carboxy terminus of these proteins are cleaved from the proprotein, and the protein is modified by the addition of lipid molecules to the new carboxy terminal amino acid. The particular lipid that is added differs among these proteins. R-Ras is geranylgeranylated and palmitoylated, whereas M-Ras is geranylgeranylated. R-Ras2, like the classic Ras proteins, is palmitoylated and farnesylated, which potentially explains why R-Ras2 localizes to the same lipid domains as classic Ras proteins and, unlike R-Ras, strongly activates extracellular signal-regulated kinase signaling. Addition of these lipid moieties allows the mature R-Ras protein to become membrane-associated, which is essential for their function. In contrast, the differences in the lipidation of R-Ras and H-Ras result in distinct localization of these proteins, which may explain why they have opposite effects in cells (see below).
To date, most studies have focused on the function of the mature lipidated forms of R-Ras, R-Ras2, and M-Ras. However, although R-Ras is represented by a single isoform, this is not true of R-Ras2 and M-Ras. Because of the production of alternatively spliced transcripts, three R-Ras2 isoforms exist, including the canonical form noted above and two other isoforms with shortened N-termini. Likewise, the gene encoding M-Ras produces six variant mRNA transcripts that encode either the canonical protein or a variant protein with a shortened N-terminus. The functions of the R-Ras2 and M-Ras protein variants with shortened N-termini are at present poorly understood. With the exception of their N- and C-terminal regions, the structure of the canonical isoforms of R-Ras, R-Ras2, and M-Ras are similar. All three R-Ras proteins contain conserved regions that form a GTP-binding pocket. Although in proximity in the mature folded protein, the amino acid residues that form this pocket are dispersed along the linear sequence of the protein, being divided into five segments known as the G1, G2, G3, G4, and G5 boxes (Figure 1, A, B, and D). Two other highly conserved regions that partially overlap with the G2 and G3 boxes are also present in all three R-Ras subfamily members. These segments, which are known as switch domains I and II, are essential for the transition between the activated and inactivated conformations of R-Ras proteins. According to the loaded spring hypothesis, the γ-phosphate of GTP forms hydrogen bonds with invariant threonine and glycine residues in switch regions I and II, respectively. This hydrogen bonding restrains the movement of switch regions I and II, thereby locking the Ras protein into the active conformation. Cleavage of the GTP γ-phosphate and the resulting loss of these hydrogen bonds allow the protein to relax into the inactive GDP-bound conformation. Of note, switch region I is also essential for the activation of Raf and phosphatidylinositol 3-kinase (PI3K). The more variable amino and carboxy terminal regions also play an important role in signal transduction. The amino terminus of R-Ras is required for the activation of Rac,13 which is essential for cell migration, whereas the carboxy terminus of R-Ras targets the protein to focal adhesions14 and facilitates RhoA activation that induces changes in migration and morphology.15
A host of activating GEFs and inactivating GAPs regulate the activity of R-Ras subfamily proteins. Given the sequence similarities between classic Ras and R-Ras subfamily proteins, it is not surprising that some of these GEFs and GAPs regulate the activity of proteins in both Ras subfamilies, whereas others specifically control R-Ras proteins. For instance, 10 known GEFs can activate H-Ras and/or R-Ras (Figure 2A). These GEFs include two that solely activate H-Ras [Son of Sevenless (SOS) 1 and Ras guanyl nucleotide-releasing protein 1 (RasGRP1)], three that only activate R-Ras [breast cancer antiestrogen resistance 3 (BCAR3), Crk SH3-binding guanine nucleotide-releasing protein (C3G), and RasGRP2], and five that are capable of activating both H-Ras and R-Ras [SOS2, Ras guanine nucleotide releasing factor (RasGRF1), RasGRF2, RasGRP3, and RasGRP4].2 However, there are some differences between the GEFs that activate R-Ras and the GEFs that activate other proteins of the R-Ras subfamily. For instance, R-Ras is the only member of the R-Ras subfamily that does not interact with SOS1, an important regulator of classic Ras proteins.2 On the other hand, the regulation of M-Ras is more aligned with classic Ras proteins, with Sos1 and Sos2 both being capable of activating M-Ras.2 As with the GEFs, R-Ras proteins are regulated by a unique profile of GAPs; some of these GAPs are also capable of inactivating H-Ras [eg, neurofibromin 1 (NF1)], whereas others specifically target R-Ras (eg, R-RasGAP) (Figure 2B).2,16 As with the GEFs, though, there are some differences as to which GAPs inactivate different R-Ras subfamily members. For example, R-RasGAP inactivates R-Ras and R-Ras2 but not M-Ras.2 Given the large number of proteins involved, it is likely that our current understanding of the regulation of R-Ras subfamily proteins by GEFs and GAPs is still incomplete.
Figure 2.

Venn diagrams of guanine nucleotide exchange factors (GEFs; A) and GTPase-activating proteins (GAPs; B) that regulate the R-Ras subfamily member R-Ras1 and the classic Ras protein H-Ras. Common regulators are shown in the overlapping center (purple), and differential regulators are shown in separate red (R-Ras) and blue (H-Ras) regions of each Venn diagram.2 SOS, Son of Sevenless.
R-Ras proteins share identical effector domains, yet they are able to activate redundant and distinct cytoplasmic signaling proteins, diversifying their physiological effects (Figure 1B). For instance, M-Ras is the only R-Ras subfamily member that activates suppressor, homolog, of clear 2 (SHOC2), which, through protein phosphatase 1c (PP1c), can activate cellular Raf proto-oncogene (CRAF), albeit weakly. It has also been suggested that M-Ras cooperates with proteins in the classic Ras subfamily to activate the mitogen-activated protein kinase (MAPK) signaling pathway, thereby promoting proliferation and transformation of cells.2,8,17 R-Ras2, like classic Ras proteins, can strongly activate extracellular signal-regulated kinase and the MAPK pathway. Conversely, R-Ras has a predilection to function through the PI3K pathway, with a notably lower affinity for Raf relative to the classic Ras proteins.16, 17, 18, 19
One of the unique functions attributed to the R-Ras subfamily is R-Ras's inside-out activation of integrins, in which intracellular signaling generates a balance between adhesion and migration. However, the mechanism(s) by which R-Ras activates integrins is unclear, and a direct interaction between R-Ras and integrins has not yet been demonstrated. We do know that family with sequence similarity 38, member A (FAM38A) activates R-Ras, which leads to β1 integrin activation and increased adhesiveness.20 Plexin-B1 regulates R-Ras via its GAP domain, suppressing extracellular matrix–dependent R-Ras activation and β1 integrin activation in vitro, leading to decreased cell migration.21 Direct phosphorylation of Tyr66 within R-Ras’ effector domain by EphB2 or Src also blocks R-Ras activation of integrins, decreasing adhesion.22,23 This aligns with previous work showing that activated R-Ras promotes adhesion via integrin activity, whereas dominant negative R-Ras [R-Ras(43N)] suppresses adhesion.24,25 This is in contrast with H-Ras, which suppresses integrin activation.12,26
The C-terminus of R-Ras also plays an important role in integrin activation. Unlike other Ras proteins, R-Ras has a proline-rich region containing an Src homology 3 binding domain at its C-terminus. A study using H-Ras/R-Ras chimeras showed that this hypervariable C-terminus is critical for integrin activation and R-Ras membrane targeting.11,14,27 The C-terminal has also proven vital to actin and integrin regulation, via binding to Flightless 1 or Filamin A.28, 29, 30 R-Ras must be active for this binding, and its C-terminal proline-rich region Src homology 3 domain must be intact for cytoskeletal rearrangement, including formation of cellular extensions involved in migration.29,30 Interestingly, the unique N-terminus of R-Ras is not necessary for interactions with integrins or focal adhesions but is necessary for interaction with Rac. Removal of the N-terminus results in less Rac activation and cell proliferation, but more integrin activation and cell migration, suggesting that the N-terminus may have a negative effect on migration.14 The variable roles of the R-Ras N- and C-terminal regions suggest that R-Ras regulation of integrin signaling is complex and likely reflects a balance between the pro-adhesive and promigratory effects of different R-Ras domains.
Given the significance of classic Ras proteins in human diseases, it is reasonable to predict that perturbation of R-Ras subfamily signaling will interfere with normal cellular function. However, because R-Ras subfamily proteins activate signaling cascades that only partially overlap with those activated by classic Ras proteins, alterations in R-Ras activation is expected to produce abnormalities distinct from classic Ras. This is borne out by the abnormalities observed in subtypes of a genetic disease known as Noonan syndrome. Intriguingly, these observations align with recent findings about the role of R-Ras proteins in the pathogenesis of other human diseases commonly encountered in the general population.
R-Ras Subfamily Functions in Human Physiology and Disease
Noonan Syndrome
The consequences of R-Ras subfamily mutations are illustrated by specific subtypes of NS, a multisystem disease that is classified as a RASopathy. Mutations in R-Ras,31 R-Ras2 [Noonan syndrome 12; Online Mendelian Inheritance in Man (OMIM, https://www.omim.org) number 618614], and M-Ras (Noonan syndrome 11; OMIM number 618499), although extremely rare, are found in patients with NS comorbidities, such as hypertrophic cardiomyopathy, congenital heart defects, juvenile myelomonocytic leukemia, and acute myelogenous leukemia. Genetically, the mutations causing NS are classified as gain of function and are inherited in an autosomal-dominant manner or occur de novo. The cytoplasmic signaling cascades that are activated downstream of mutated R-Ras proteins and drive the abnormalities encountered in NS are incompletely understood. However, the pathogenesis of these abnormalities is consistently dependent on AKT activation and, in a more stimulus-dependent manner, MAPK signaling.32, 33, 34, 35, 36, 37 Mutation of R-Ras subfamily members are responsible for relatively few NS cases, with the PTPN11 (Noonan syndrome 1; OMIM number 163950) and SOS1 (Noonan syndrome 4; OMIM number 610733) genes being more commonly mutated. However, M-Ras is activated by mutated Sos1, which raises the question of whether M-Ras is indirectly involved in the pathogenesis of Noonan syndrome 4.33,34,37
Other phenotypic findings in NS are extremely variable, but may include visual, auditory, and cognitive defects, facial dimorphisms, skeletal malformations, defective growth, short stature, and increased cancer susceptibility.33 Specific M-Ras and R-Ras2 mutants, recently identified and validated using zebrafish models, are linked with a stronger craniofacial phenotype, including macrocephaly, suggesting central nervous system (CNS) involvement.32,35 Clinical findings in patients with mutations in M-Ras also include cardiovascular and hematologic defects, such as bleeding disorders, congenital heart disease, pulmonary valve stenosis, and hypertrophic cardiomyopathy.37 Taken together, clinical manifestations of NS associated with R-Ras subfamily mutations suggest these proteins play important roles in the cardiovascular, immune, and nervous systems.
Cardiovascular System
R-Ras subfamily signaling also contributes to pathology in the cardiovascular system. Multiple genome-wide association studies have identified single-nucleotide polymorphisms (SNPs) in the MRAS locus that are associated with either increased risk for the development of coronary artery disease or protective effects against coronary artery disease.38, 39, 40 Interestingly, these observations may be ancestry-specific, as studies focused solely on Han Chinese found that the effect of these MRAS SNPs on the development of coronary artery disease was statistically insignificant.41,42 However, caution is merited, as Lu et al42 found a much smaller frequency of the previously identified SNPs in the Han Chinese population studied and cautioned that the resulting potentially lower power might fail to capture a significant association. Nonetheless, a similar study in a Czech population found that MRAS SNP variants were not associated with acute coronary syndrome or cardiovascular disease mortality, supporting the idea that the associations observed in initial studies are ancestry-dependent.43 Further study of these associations in additional populations is merited.
Genome-wide association study analyses have also linked MRAS SNPs with dyslipidemia, an important risk factor for coronary artery disease. Alshahid et al40 examined a wider array of associations, linking MRAS variants to obesity, hypercholesterolemia, and hypertriglyceridemia, which was associated with increased levels of both low-density lipoprotein and high-density lipoprotein. In contrast, studies by Freyer et al44 showed no difference in atherosclerotic plaque formation in genetically engineered mice with an MRAS knockout,40 raising the question of whether the human MRAS SNP variants noted above have gain-of-function effects. Genome-wide association studies examining the impact of M-Ras on cerebral ischemic infarcts found no linkage to increased risk of infarction in Han Chinese patients. However, these same studies found some MRAS SNP variants were associated with risk for increased cerebral infarct area.39 Together, these associations suggest MRAS has a complicated relationship to the pathology of atherosclerotic disease and ischemic infarcts that may prove challenging to decipher.
In contrast, R-Ras is clearly functionally linked to vascular stability. R-Ras is a negative regulator of vascular proliferation and is down-regulated in regenerating vessels.45 R-Ras is instead most highly expressed in mature, quiescent vasculature, specifically in fully differentiated vascular smooth muscle cells in arterioles and arteries and endothelial cells in capillaries.46 Down-regulation of R-Ras activity by p120RasGAP, independent of classic Ras signaling, is essential for sprouting, angiogenesis, vascular regeneration, and wound healing.45 R-Ras plays a role in vivo during endothelial tubulogenesis, inducing lumen formation via Akt activation as opposed to H-Ras's known effects on limiting endothelial proliferation during tubulogenesis.47 This suggests a unique role for the R-Ras–Akt axis in ensuring vessel patency. R-Ras, together with vascular endothelial growth factor, stabilizes microtubules during lumenogenesis, allowing proper blood flow and perfusion through regenerating capillaries.47 Activated R-Ras also attenuates vascular endothelial growth factor signaling by blocking internalization of stimulated vascular endothelial growth factor receptors. This receptor blockade prevents new vessel sprouting, but interestingly, does not trigger the destruction of recently formed vessels, such as is typically observed with total vascular endothelial growth factor blockade, suggesting that a divergent regulatory signaling cascade is in play.48,49
In addition to its effects on tubulogenesis, R-Ras plays a positive role in maintaining the functionality of normal mature vasculature. Blood vessels that lack R-Ras have impaired endothelial barrier function, leading to edema from leaked proteins and reduced perfusion efficacy.50,51 This vessel leakiness is partially caused by semaphorin-induced disassembly of adhesive structures and decreased vascular endothelial cadherin (VE-cadherin) accumulation at endothelial adherens junctions. This reflects the fact that R-Ras–mediated phosphorylation of VE-cadherin at Ser665 regulates the internalization of VE-cadherin. R-Ras also enhances pericyte-endothelial cell interactions, so R-Ras knockout results in a weakening of these interactions and a loss of small vessel integrity.48 Conversely, gain-of-function R-Ras mutants enhance endothelial barrier function and increased pericyte coverage, which blocks plasma leakage and provides improved perfusion.52 Recent work shows that regulation of R-Ras in vasculature involves dynamic depalmitoylation by acyl-protein thioesterase 1 (APT-1), which halts membrane trafficking and disrupts adherens junctions; dysregulation of R-Ras palmitoylation results in unstable vasculature seen in hyperglycemia in type II diabetes mellitus models in vivo.53 These effects have also been observed in the context of angiogenesis in malignant tumors. R-Ras critically regulates tumor vascularization and perfusion, as Rras-null animals show higher tumor vascularity than controls.46 Overall, R-Ras provides a delicate balance, inhibiting pathologic angiogenesis while maintaining mature vessel function.
R-Ras2 also regulates the integrity of the vascular system in humans and mice via its action in platelets. R-Ras2 regulates platelet activation and aggregation as well as thrombus formation and stability in response to stimulation by glycoprotein VI-FcRgamma (GPVI-FcRγ), which potentially functions through integrins.54 Rras2-deficient platelets form unstable thrombi in response to endothelial injury and are over three times more likely to embolize than controls, leading to increased severity of stroke and atherosclerosis.54
Immune System Function and Dysfunction
A growing body of evidence indicates R-Ras subfamily members play multiple important roles in immune system function. Lymphocytes (T and B cells) and monocytes produced in the bone marrow are strategically transported in the blood to sites of maturation for specific defenses or directly to sites of infection to mount an innate immune response. Knockout of Rras2 leads to lymphopenia in mice, and knockout of any of the three R-Ras subfamily members results in a decrease in effector T and/or B cells. Innate immune responses involving macrophages and their precursor monocytes are also altered, which may affect pathologic processes, such as the formation of atherosclerotic plaques.44,55 In one study, Apoe knockout mice, which form atherosclerotic plaques when fed a high-fat diet, were compared with Apoe/MRAS dual-knockout mice to see how plaque formation was affected.40 Interestingly, although there was not a difference in plaque size, there were significantly fewer macrophages infiltrating the plaque, suggesting that MRAS knockout reduces the migration, adhesion, invasion, or absolute number of macrophages.40 Studies assessing how macrophage functionality is affected by M-Ras activation showed that M-Ras is concentrated in the early-forming phagosome membrane in vitro and that macrophages expressing active or inactive M-Ras mutants show enhanced or inhibited phagosome formation and uptake, respectively.44,56 Phagosome formation, macrophages' key method of neutralizing foreign invaders, is spatiotemporally regulated via the actin cytoskeleton, consistent with the proposed role of the R-Ras subfamily in regulating cytoskeletal remodeling.56 This likely also explains why MRAS loss alters the number of macrophages in atherosclerotic plaques as M-Ras is essential for monocyte binding to and interaction with endothelial cells.44,57 Monocyte infiltration of the vessel wall is a triggering event for plaque formation and is followed by the transformation of monocytes into lipid-rich foam cells that both sequester oxidized lipoproteins and recruit other inflammatory cells. Purified hematopoietic stem cells transduced with a constitutively active M-Ras G22V mutant grow in the absence of growth factors and form immortalized cells that quickly differentiate first into adherent cells, resembling macrophages, and then almost exclusively into mast cells.58 This suggests that M-Ras can promote hematopoietic transformation. Hematopoietic stem cells transduced with the G22V M-Ras mutant transplanted into micelead to the development of mastocytosis and mast-cell leukemia with splenomegaly and hepatomegaly.58
Dendritic cells, another element of the mononuclear phagocytic system that bridges the innate and adaptive immune systems, are also significantly affected by R-Ras loss.59 The dendritic cells of Rras-null mice have significantly impaired function, with decreased ability to prime allogenic and antigen-specific T cells by forming stable immunologic synapses.59 Splenic dendritic cells lacking R-Ras have 73% less effective priming of T cells in response to antigen exposure, leading to significantly reduced T-cell proliferation (62% to 89% lower in Rras knockout mice than controls).59 This may reflect the diminished ability of Rras-null dendritic cells to spread and interact with fibronectin, associated changes in morphology, defects in vesicular trafficking, or lower expression of major histocompatibility complex class II, CD86, and toll-like receptor 4 (which are necessary for maturation and activation in response to antigen stimulation).59 Mice in which T-cell proliferation or maturation is impaired by Rras loss have inefficient immune system responses, leaving them immunocompromised and less able to fight off infection.
A lack of R-Ras and R-Ras2 cripples adaptive immune responses. Rras-null mice have a 30% to 40% reduction in the total number of T cells in their spleen and lymph nodes. As noted above, Rras2-null mice are lymphopenic, indicating that proliferation and/or survival of lymphocytes is impaired.55,60,61 Interestingly, the attenuation of lymphocyte proliferation can be reversed with cytokine stimulation in Rras-null mice but not in Rras2-null mice, and those that survive show limited functionality and decreased surface expression of L-selectin/CD62L. Some of these effects are related to constitutive interactions between R-Ras2 and T-cell receptors, which support the survival and maintenance of mature T cells by consistently transmitting low-grade signaling that is impaired with the loss of R-Ras2.55,60 T-cell receptor activation and internalization is critical for the phagocytosis of larger particles, including the uptake of major histocompatibility complex–associated antigen from antigen-presenting cells.61 These processes rely on a delicate balance of R-Ras2 activation. Introduction of constitutively active G23V or inactive G28N mutants, despite their antipodal actions, both cause decreased T-cell receptor internalization, although only the active G23V mutant causes widespread lamellipodia formation and membrane protrusions, suggesting an effect on cytoskeletal remodeling.61 This remodeling is also essential for migration and homing of T cells, as demonstrated by the fact that Rras-null mice have a large reduction in the number of T cells trafficked to lymph nodes, resulting in less prominent T-cell regions in the paracortex.60 A recent study by Sawada et al62 indicated that T-cell priming may be affected by R-Ras trafficking in endothelial cells, which provides an intriguing link between the action of this protein in the vascular and immune systems. Using an endothelial cell–specific R-Ras knockout mouse model, they demonstrated a loss of inflammation-induced T-cell diapedesis and homing to lymph nodes, decreasing their priming by dendritic cells and hindering anti-tumor activity against implanted melanomas.62 Interestingly, hematopoietic progenitors have a comparable defect in homing after entering the peripheral blood circulation yet have increased mobilization away from the bone marrow following Rras loss, indicating that cell type–specific downstream signaling leads to unique biological effects on cell movement and migration.63
The proliferation and development of B cells, the antibody-producing cells of the adaptive immune system, are greatly affected by the loss of Rras2 and, to a lesser degree, by Rras loss. Although Rras-null mice develop nearly normal numbers of B cells, Rras2-null mice have markedly decreased B-cell populations and lower PI3K signaling downstream of B-cell receptors.55,64 This leads to diminished generation and expansion of germinal centers within the lymph nodes of Rras2-null mice and decreased adaptive immunity.60,64 Interestingly, Rras2-null mice also have dysfunctional mitochondrial transcription and mitochondrial DNA replication of genes needed for aerobic glucose metabolism in B cells, which likely impairs cellular metabolism.64 Like other immune cells, loss of Rras2 or MRAS causes defects in B-cell adhesion and migration, leading to decreased homing of B cells to lymph nodes.40,63 Rras2-null B cells are also functionally limited in their ability to produce antibodies in response to T-dependent antigens, but not T-independent antigens, suggesting that dysfunctional lymphocyte interactions lead to specific immunodeficiencies.60,64
The complex role of R-Ras subfamily proteins in immune function is also demonstrable in models of autoimmune disease. Autoimmune encephalomyelitis is greatly attenuated in Rras-null mice, although, curiously, this knockout is correlated with a significant increase, rather than a decrease, in the proliferation of natural regulatory T cells.65 In this case, the expression and activation of R-Ras inhibits autoimmunity, due to positively regulating regulatory T-cell development.65 Overall, the actions of the R-Ras subfamily are complex and widespread in the immune system, and both losses and gains of function can lead to pathology.
Nervous System Development and Function
In vitro studies suggest that neurons in the CNS are reliant on R-Ras subfamily signaling for the regulation of axonal and dendritic outgrowth and pruning. The brain, especially during development, is a hotspot for M-Ras expression, although all R-Ras subfamily members are expressed in the CNS. Loss of R-Ras or M-Ras leads to abnormal differentiation and impedes neurite formation in nerve growth factor–stimulated PC12 cells, a commonly used model of neuronal differentation.66,67 M-Ras and R-Ras2 activate both PI3K and MAPK signaling in PC12 cells; despite sharing identical effector sites, R-Ras binds and activates PI3K in these cells but does not activate MAPK, like R-Ras2 and M-Ras.24,67,68
Plexin B1, the receptor for semaphorin 4D, regulates both R-Ras and M-Ras to exert tight control of directional movement and outgrowth of neurites from CNS neurons in vitro.69, 70, 71, 72 These studies show that activation and inhibition of R-Ras yields a dynamic balance necessary for axonal development, remodeling, growth, and pruning in cultured neurons.72 Binding of semaphorin 4D to plexin B1 results in R-Ras inhibition that allows neurite retraction and growth cone collapse in vitro. Although R-Ras appears to influence axonal formation and growth via the regulation of a dynamic balance of activation and suppression, M-Ras expression is up-regulated at a later stage of neural development and regulates dendrite formation.69 Suppression of M-Ras activity by semaphorin 4D or direct knockdown of M-Ras results in abnormal dendrite morphology, prevention of dendrite outgrowth, and blockade of the normal reduction of F-actin in distal dendrites.69,71 Conversely, constitutively active mutant M-Ras enhances dendrite outgrowth and branching.69,70,73 In contrast to these in vitro findings, however, Worzfeld et al74 found that neither R-Ras nor M-Ras was a key player in plexin B's neurodevelopmental effects in vivo. These observations indicate that the in vitro results described above must be further assessed in vivo.
The R-Ras subfamily also exerts effects on CNS glia in vitro and in vivo. R-Ras and R-Ras2 are necessary for oligodendrocyte differentiation in vivo and in vitro as well as myelination, which is essential for saltatory conduction.75 Knockdown of Rras or Rras2 increases the percentage of immature oligodendrocytes and decreases the total number of oligodendrocytes in white matter tracts. Simultaneous loss of Rras and Rras2 results in an even greater reduction in oligodendrocytes and an even higher proportion of immature glia. In comparison to Rras−/− and Rras2−/− mice, R-Ras−/−;R-Ras2−/− animals show severe hypomyelination, indicating that R-Ras and R-Ras2 cooperatively promote oligodendrocyte maturation and myelination.75
R-Ras subfamily proteins also regulate the migration of Schwann cells, the myelinating glia of the peripheral nervous system. This was shown using Schwann cells from genetically engineered mice with ablation of the Nf1 locus. Schwann cells express both R-Ras and R-Ras2.76 Relative to wild-type Schwann cells derived from mouse embryos, Nf1+/− and Nf1−/− Schwann cells show twofold and fourfold higher migration, respectively. These promigratory effects appear to be mediated by R-Ras2, as an activated R-Ras2 mutant, but not an activated R-Ras mutant, increases migration, whereas dominant negative R-Ras, which inhibits R-Ras and R-Ras2, inhibits migration. Inhibitors of either the PI3K or the MAPK pathways also inhibited migration, indicating that these R-Ras2–regulated pathways are essential for migration. In contrast, Keely et al68 used a similar approach to implicate both R-Ras and R-Ras2 in the migration of breast epithelial cells and argued that the migration of these cells is mediated via integrin regulation. These observations suggest that the role of R-Ras and R-Ras2 in cellular migration differs, depending on the cellular context. The observation that Nf1 loss enhances the effect of R-Ras2 activation also raises the question of whether the activation of R-Ras subfamily members in NF1-null neoplasms contributes to the pathogenesis of these tumors.
Cancer
The transforming ability of R-Ras proteins has been studied since their discovery. In terms of proliferation, R-Ras2 has the most potent effects, enhancing proliferation to a degree comparable to that observed with classic Ras proteins.67,77 In cancer, R-Ras subfamily members have been implicated in anchorage-independent growth, increased invasiveness, stimulation of angiogenesis, and amplified tumorigenicity in vivo.46,52,67,78 The effects are context-specific, with R-Ras subfamily hyperactivation promoting migration and invasion in some tumor types, while inhibiting it via adhesion in others.28,68,79 In contrast to classic Ras proteins, mutations of genes encoding R-Ras subfamily members are rarely documented in human cancers. Instead, overexpression of R-Ras, R-Ras2, and/or M-Ras is more common in various cancer types55,64,80 (Table 180, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96). Many cancers that overexpress members of the R-Ras family are highly aggressive and frequently metastasize; given the effects of R-Ras proteins on proliferation, migration, and invasion, it is reasonable to postulate that overexpression of R-Ras proteins contributes to metastasis and/or invasion. Regarding human cancers, neurofibromin, the protein encoded by the NF1 locus, acts as a GAP for both classic Ras and R-Ras proteins. Consequently, NF1 loss potentially results in hyperactivation of R-Ras subfamily proteins, which may contribute to tumorigenesis. However, this possibility has not yet been carefully examined in NF1-null neoplasms.
Table 1.
R-Ras in Cancer
| Cancer type | R-Ras1 | R-Ras2 | M-Ras | Reference |
|---|---|---|---|---|
| Breast | −/+ | − | − | 19,68,80, 81, 82, 83, 84 |
| CNS | − | − | 85,86 | |
| Skin | − | 28,87 | ||
| Ovarian | −/+ | 88 | ||
| Endometrial | − | 89 | ||
| Cervical | − | 90 | ||
| Prostate | − | 79 | ||
| Oral | − | 91 | ||
| Esophagus | − | 92 | ||
| Stomach | − | − | − | 8 |
| Liver | − | 93 | ||
| Renal | − | 94 | ||
| Heme/lymph | − | − | 58,95 | |
| Osteosarcoma/sarcoma | − | 78 | ||
| Head and neck | − | 96 |
Empty cells indicate that an effect has not yet been identified.
+, Tumor suppressive; −, tumor promoting.
CNS, central nervous system.
Breast cancer provides a good example of the potential role of different R-Ras proteins in neoplasia, although it must be cautioned that most of these studies have relied on cell culture models. All three R-Ras family members have been studied in the context of breast cancers.68,80,83,84 Compared with estrogen receptor–positive breast cancers, estrogen receptor–negative breast carcinomas show up-regulation of M-Ras expression. Introduction of a constitutively active M-Ras mutant into MCF7 cells results in proliferation that is no longer estrogen-dependent, indicating that increased M-Ras signaling renders these cells hormone-independent.80,81 In addition, mouse mammary epithelial cells expressing an activated M-Ras mutant grow in an anchorage-independent manner in the absence of serum and are capable of epithelial-mesenchymal transition in vitro.97 Constitutively active R-Ras and R-Ras2 also enhance cell protrusions and migration of cultured breast cancer cells in vitro; knockdown with siRNAs confirmed that endogenous R-Ras is required for these effects.19,27,68
Although all R-Ras family members influence cancer cell movement, they may do so by different mechanisms. Luo et al93 showed that R-Ras2 promotes migration and metastasis in hepatocellular carcinoma by decreasing and increasing the expression of E- and N-cadherin, respectively. The introduction of activated M-Ras mutants (G22V or Q71L) into mammary epithelial cells similarly triggers down-regulation of E-cadherin, which promotes cellular movement.97 R-Ras appears to uniquely regulate cell adhesion and migration via direct plexin B interactions, filamin-A binding, and integrin transport and regulation; this contrasts with R-Ras2 and M-Ras, which instead function through cadherins and other signaling pathways.24,28,98 It must be pointed out that R-Ras actions are dependent on cellular context. R-Ras2 functions much like classic Ras in some cases, acting as a driver of tumor progression; in breast cancer, it plays a role in both tumorigenesis and metastasis.82 Conversely, some investigators have reported that R-Ras has tumor suppressor-like activities (reduced proliferation and migration) in MCF7 cells and is linked to progression-free survival in patients.99,100
R-Ras subfamily proteins can both directly promote the transformation of hematopoietic elements, and in nonhematologic neoplasms, indirectly promote tumorigenesis by altering the microenvironment. R-Ras proteins have been implicated in the pathogenesis of mast cell leukemia58 and chronic myelogenous leukemia95 (Table 1). R-Ras hyperactivation or overexpression can also hinder the normal function of lymphocytes targeting tumors, thereby indirectly promoting tumor growth. As evidence of this, loss of R-Ras or R-Ras2 results in the production of dendritic cells unable to present tumor antigens to activate an immune response and anti-tumor T cells that cannot proliferate or home to the tumor site; both of these events result in tumor growth that is unchecked by the immune system.55,59,60
The vascular system is also intrinsically linked to cancer progression and metastasis. As noted above, Rras-deficient mice have leaky vessels with impaired adherens junctions, allowing the escape of proteins and fluid from the blood supply and easier passage of tumor cells into the bloodstream. This results in a microenvironment that is conducive to the high energy demands of a rapidly growing tumor and increased tumor cell exit from a primary tumor that facilitates metastasis.52,101 Consistent with this, melanoma cells subcutaneously xenografted into Rras-null B6;129Sv mice grow faster and achieve greater tumor volumes than are seen when the same tumor cells are xenografted into wild-type B6;129Sv mice.52 The R-Ras subfamily potentially exerts numerous direct and indirect effects that can impact intratumoral angiogenesis and metastasis. Given the importance that adhesion and migration have for the sprouting, growth, and homing of new vascular elements, these R-Ras subfamily functions are likely important for the expansion of vascular trees within growing neoplasms.
Summary
A dynamic balance between adhesion and movement is essential for cellular functions, such as extension of the axonal and dendritic trees of neurons, the migration of other types of neoplastic and nonneoplastic cells, and metastasis. It is increasingly apparent that proteins in the R-Ras subfamily are key mediators of these processes. We are also beginning to understand the partners that they utilize to modulate these physiological functions (Figure 3). These partner proteins include several that have long been implicated in cellular migration and morphologic changes, such as the MAPK and AKT signaling cascades. Other small GTP-binding proteins, such as RhoA and Rac, are also key modulators of these processes. However, more recently, the function of other molecules, such as E- and N-cadherin, key integrins, and the semaphorin/plexin signaling complex has been linked to R-Ras proteins and cellular migration. Much remains to be discovered about other signaling pathways that are regulated by R-Ras proteins, the nuances by which R-Ras subfamily members regulate their associated signaling partners, and how these nuances translate into physiological and pathologic changes. Delving further into the similarities and differences between members of the R-Ras subfamily will be essential for discerning the mechanisms by which these proteins contribute to pathology and developing effective new therapies targeting R-Ras–regulated signaling pathways.
Figure 3.
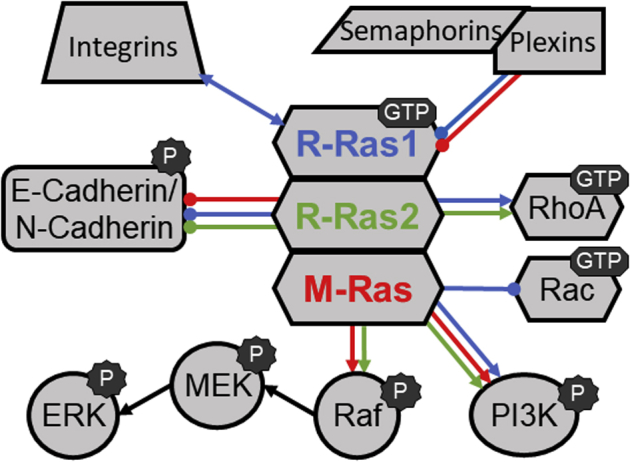
Summary of the signaling pathways that have regulatory interactions with R-Ras subfamily members. The colors of the lines connecting R-Ras proteins with other signaling molecules indicate interactions medicated by R-Ras1 (blue), R-Ras2 (green), or M-Ras (red). Arrowheads on the lines indicate that the interactions are activating, whereas circles on the ends of the lines indicate that the interactions are inhibitory. ERK, extracellular signal-regulated kinase; MEK, MAPK/ERK kinase; PI3K, phosphatidylinositol-3 kinase.
Footnotes
Supported by the National Institute of Neurological Diseases and Stroke grants R01 NS048353 and R01 NS109655 (S.L.C.); the National Cancer Institute grants R01 CA122804 (S.L.C.) and F30 CA247139 (S.M.W.); the Department of Defense grants X81XWH-09-1-0086, W81XWH-11-1-0498, W81XWH-12-1-0164, W81XWH-14-1-0073, and W81XWH-15-1-0193 (S.L.C.); and The Children’s Tumor Foundation grants 2014-04-001 and 2015-05-007 (S.L.C.).
Disclosures: None declared.
References
- 1.Goitre L., Trapani E., Trabalzini L., Retta S.F. The Ras superfamily of small GTPases: the unlocked secrets. Methods Mol Biol. 2014;1120:1–18. doi: 10.1007/978-1-62703-791-4_1. [DOI] [PubMed] [Google Scholar]
- 2.Ohba Y., Mochizuki N., Yamashita S., Chan A.M., Schrader J.W., Hattori S., Nagashima K., Matsuda M. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J Biol Chem. 2000;275:20020–20026. doi: 10.1074/jbc.M000981200. [DOI] [PubMed] [Google Scholar]
- 3.Emmanuel C., Chiew Y.E., George J., Etemadmoghadam D., Anglesio M.S., Sharma R., Russell P., Kennedy C., Fereday S., Hung J., Galletta L., Hogg R., Wain G.V., Brand A., Balleine R., MacConaill L., Palescandolo E., Hunter S.M., Campbell I., Dobrovic A., Wong S.Q., Do H., Clarke C.L., Harnett P.R., Bowtell D.D., deFazio A., Australian Ovarian Cancer Study (AOCS) Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clin Cancer Res. 2014;20:6618–6630. doi: 10.1158/1078-0432.CCR-14-1292. [DOI] [PubMed] [Google Scholar]
- 4.Orita S., Kaibuchi K., Kuroda S., Shimizu K., Nakanishi H., Takai Y. Comparison of kinetic properties between two mammalian ras p21 GDP/GTP exchange proteins, ras guanine nucleotide-releasing factor and smg GDP dissociation stimulation. J Biol Chem. 1993;268:25542–25546. [PubMed] [Google Scholar]
- 5.Hobbs G.A., Der C.J., Rossman K.L. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–1292. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu W.N., Yan M., Chan A.M. A thirty-year quest for a role of R-Ras in cancer: from an oncogene to a multitasking GTPase. Cancer Lett. 2017;403:59–65. doi: 10.1016/j.canlet.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Alcover-Sanchez B., Garcia-Martin G., Wandosell F., Cubelos B. R-Ras GTPases signaling role in myelin neurodegenerative diseases. Int J Mol Sci. 2020;21:5911. doi: 10.3390/ijms21165911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimmelman A., Tolkacheva T., Lorenzi M.V., Osada M., Chan A.M. Identification and characterization of R-ras3: a novel member of the RAS gene family with a non-ubiquitous pattern of tissue distribution. Oncogene. 1997;15:2675–2685. doi: 10.1038/sj.onc.1201674. [DOI] [PubMed] [Google Scholar]
- 9.Lowe D.G., Goeddel D.V. Heterologous expression and characterization of the human R-ras gene product. Mol Cell Biol. 1987;7:2845–2856. doi: 10.1128/mcb.7.8.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan A.M., Miki T., Meyers K.A., Aaronson S.A. A human oncogene of the RAS superfamily unmasked by expression cDNA cloning. Proc Natl Acad Sci U S A. 1994;91:7558–7562. doi: 10.1073/pnas.91.16.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansen M., Prior I.A., Hughes P.E., Oertli B., Chou F.L., Willumsen B.M., Hancock J.F., Ginsberg M.H. C-terminal sequences in R-Ras are involved in integrin regulation and in plasma membrane microdomain distribution. Biochem Biophys Res Commun. 2003;311:829–838. doi: 10.1016/j.bbrc.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 12.Sethi T., Ginsberg M.H., Downward J., Hughes P.E. The small GTP-binding protein R-Ras can influence integrin activation by antagonizing a Ras/Raf-initiated integrin suppression pathway. Mol Biol Cell. 1999;10:1799–1809. doi: 10.1091/mbc.10.6.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holly S.P., Larson M.K., Parise L.V. The unique N-terminus of R-ras is required for Rac activation and precise regulation of cell migration. Mol Biol Cell. 2005;16:2458–2469. doi: 10.1091/mbc.E03-12-0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furuhjelm J., Peranen J. The C-terminal end of R-Ras contains a focal adhesion targeting signal. J Cell Sci. 2003;116:3729–3738. doi: 10.1242/jcs.00689. [DOI] [PubMed] [Google Scholar]
- 15.Jeong H.W., Nam J.O., Kim I.S. The COOH-terminal end of R-Ras alters the motility and morphology of breast epithelial cells through Rho/Rho-kinase. Cancer Res. 2005;65:507–515. [PubMed] [Google Scholar]
- 16.Huff S.Y., Quilliam L.A., Cox A.D., Der C.J. R-Ras is regulated by activators and effectors distinct from those that control Ras function. Oncogene. 1997;14:133–143. doi: 10.1038/sj.onc.1200815. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Viciana P., Sabatier C., McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–4954. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosario M., Paterson H.F., Marshall C.J. Activation of the Ral and phosphatidylinositol 3' kinase signaling pathways by the ras-related protein TC21. Mol Cell Biol. 2001;21:3750–3762. doi: 10.1128/MCB.21.11.3750-3762.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wozniak M.A., Kwong L., Chodniewicz D., Klemke R.L., Keely P.J. R-Ras controls membrane protrusion and cell migration through the spatial regulation of Rac and Rho. Mol Biol Cell. 2005;16:84–96. doi: 10.1091/mbc.E04-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McHugh B.J., Murdoch A., Haslett C., Sethi T. Loss of the integrin-activating transmembrane protein Fam38A (Piezo1) promotes a switch to a reduced integrin-dependent mode of cell migration. PLoS One. 2012;7:e40346. doi: 10.1371/journal.pone.0040346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oinuma I., Ito Y., Katoh H., Negishi M. Semaphorin 4D/plexin-B1 stimulates PTEN activity through R-Ras GTPase-activating protein activity, inducing growth cone collapse in hippocampal neurons. J Biol Chem. 2010;285:28200–28209. doi: 10.1074/jbc.M110.147546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou J.X., Liu Y., Pasquale E.B., Ruoslahti E. Activated SRC oncogene phosphorylates R-ras and suppresses integrin activity. J Biol Chem. 2002;277:1824–1827. doi: 10.1074/jbc.M103133200. [DOI] [PubMed] [Google Scholar]
- 23.Zou J.X., Wang B., Kalo M.S., Zisch A.H., Pasquale E.B., Ruoslahti E. An Eph receptor regulates integrin activity through R-Ras. Proc Natl Acad Sci U S A. 1999;96:13813–13818. doi: 10.1073/pnas.96.24.13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z., Vuori K., Wang H., Reed J.C., Ruoslahti E. Integrin activation by R-ras. Cell. 1996;85:61–69. doi: 10.1016/s0092-8674(00)81082-x. [DOI] [PubMed] [Google Scholar]
- 25.Wang B., Zou J.X., Ek-Rylander B., Ruoslahti E. R-Ras contains a proline-rich site that binds to SH3 domains and is required for integrin activation by R-Ras. J Biol Chem. 2000;275:5222–5227. doi: 10.1074/jbc.275.7.5222. [DOI] [PubMed] [Google Scholar]
- 26.Hughes P.E., Renshaw M.W., Pfaff M., Forsyth J., Keivens V.M., Schwartz M.A., Ginsberg M.H. Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell. 1997;88:521–530. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- 27.Conklin M.W., Ada-Nguema A., Parsons M., Riching K.M., Keely P.J. R-Ras regulates beta1-integrin trafficking via effects on membrane ruffling and endocytosis. BMC Cell Biol. 2010;11:14. doi: 10.1186/1471-2121-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gawecka J.E., Griffiths G.S., Ek-Rylander B., Ramos J.W., Matter M.L. R-Ras regulates migration through an interaction with filamin A in melanoma cells. PLoS One. 2010;5:e11269. doi: 10.1371/journal.pone.0011269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arora P.D., He T., Ng K., McCulloch C.A. The leucine-rich region of Flightless I interacts with R-ras to regulate cell extension formation. Mol Biol Cell. 2018;29:2481–2493. doi: 10.1091/mbc.E18-03-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arora P.D., Nakajima K., Nanda A., Plaha A., Wilde A., Sacks D.B., McCulloch C.A. Flightless anchors IQGAP1 and R-ras to mediate cell extension formation and matrix remodeling. Mol Biol Cell. 2020;31:1595–1610. doi: 10.1091/mbc.E19-10-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flex E., Jaiswal M., Pantaleoni F., Martinelli S., Strullu M., Fansa E.K. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet. 2014;23:4315–4327. doi: 10.1093/hmg/ddu148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki H., Takenouchi T., Uehara T., Takasago S., Ihara S., Yoshihashi H., Kosaki K. Severe Noonan syndrome phenotype associated with a germline Q71R MRAS variant: a recurrent substitution in RAS homologs in various cancers. Am J Med Genet A. 2019;179:1628–1630. doi: 10.1002/ajmg.a.61261. [DOI] [PubMed] [Google Scholar]
- 33.Higgins E.M., Bos J.M., Mason-Suares H., Tester D.J., Ackerman J.P., MacRae C.A., Sol-Church K., Gripp K.W., Urrutia R., Ackerman M.J. Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight. 2017;2:e91225. doi: 10.1172/jci.insight.91225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young L.C., Hartig N., Boned Del Rio I., Sari S., Ringham-Terry B., Wainwright J.R., Jones G.G., McCormick F., Rodriguez-Viciana P. SHOC2-MRAS-PP1 complex positively regulates RAF activity and contributes to Noonan syndrome pathogenesis. Proc Natl Acad Sci U S A. 2018;115:E10576–E10585. doi: 10.1073/pnas.1720352115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niihori T., Nagai K., Fujita A., Ohashi H., Okamoto N., Okada S., Harada A., Kihara H., Arbogast T., Funayama R., Shirota M., Nakayama K., Abe T., Inoue S.I., Tsai I.C., Matsumoto N., Davis E.E., Katsanis N., Aoki Y. Germline-activating RRAS2 mutations cause Noonan syndrome. Am J Hum Genet. 2019;104:1233–1240. doi: 10.1016/j.ajhg.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capri Y., Flex E., Krumbach O.H.F., Carpentieri G., Cecchetti S., Lissewski C., Rezaei Adariani S., Schanze D., Brinkmann J., Piard J., Pantaleoni F., Lepri F.R., Goh E.S., Chong K., Stieglitz E., Meyer J., Kuechler A., Bramswig N.C., Sacharow S., Strullu M., Vial Y., Vignal C., Kensah G., Cuturilo G., Kazemein Jasemi N.S., Dvorsky R., Monaghan K.G., Vincent L.M., Cave H., Verloes A., Ahmadian M.R., Tartaglia M., Zenker M. Activating mutations of RRAS2 are a rare cause of Noonan syndrome. Am J Hum Genet. 2019;104:1223–1232. doi: 10.1016/j.ajhg.2019.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motta M., Fidan M., Bellacchio E., Pantaleoni F., Schneider-Heieck K., Coppola S., Borck G., Salviati L., Zenker M., Cirstea I.C., Tartaglia M. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet. 2019;28:1007–1022. doi: 10.1093/hmg/ddy412. [DOI] [PubMed] [Google Scholar]
- 38.Erdmann J., Grosshennig A., Braund P.S., Konig I.R., Hengstenberg C., Hall A.S. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song Y., Ma R., Zhang H. The influence of MRAS gene variants on ischemic stroke and serum lipid levels in Chinese Han population. Medicine (Baltimore) 2019;98:e18065. doi: 10.1097/MD.0000000000018065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alshahid M., Wakil S.M., Al-Najai M., Muiya N.P., Elhawari S., Gueco D., Andres E., Hagos S., Mazhar N., Meyer B.F., Dzimiri N. New susceptibility locus for obesity and dyslipidaemia on chromosome 3q22.3. Hum Genom. 2013;7:15. doi: 10.1186/1479-7364-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehta N.N. A genome-wide association study in Europeans and South Asians identifies 5 new loci for coronary artery disease. Circ Cardiovasc Genet. 2011;4:465–466. doi: 10.1161/CIRCGENETICS.111.960989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu X., Wang L., Chen S., He L., Yang X., Shi Y. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. 2012;44:890–894. doi: 10.1038/ng.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hubacek J.A., Stanek V., Gebauerova M., Ceska R., Adamkova V., Lanska V., Pitha J. MRAS gene marker rs9818870 is not associated with acute coronary syndrome in the Czech population and does not predict mortality in males after acute coronary syndrome. Adv Clin Exp Med. 2017;26:1213–1217. doi: 10.17219/acem/67460. [DOI] [PubMed] [Google Scholar]
- 44.Freyer J., Behrensen M., Aherrahrou Z., Berbee J., Schunkert H., Erdmann J. MRas-knock-out mouse: B-cell phenotype and reduced macrophage infiltration in atherosclerotic plaques at the aortic root. Eur Heart J. 2013;34 442–442. [Google Scholar]
- 45.Xu L., Komatsu M. Promoter cloning and characterization of the anti-vascular proliferation gene, R-ras: role of Ets- and Sp-binding motifs. J Biol Chem. 2009;284:2706–2718. doi: 10.1074/jbc.M808184200. [DOI] [PubMed] [Google Scholar]
- 46.Komatsu M., Ruoslahti E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat Med. 2005;11:1346–1350. doi: 10.1038/nm1324. [DOI] [PubMed] [Google Scholar]
- 47.Li F., Sawada J., Komatsu M. R-Ras-Akt axis induces endothelial lumenogenesis and regulates the patency of regenerating vasculature. Nat Commun. 2017;8:1720. doi: 10.1038/s41467-017-01865-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawada J., Li F., Komatsu M. R-Ras protein inhibits autophosphorylation of vascular endothelial growth factor receptor 2 in endothelial cells and suppresses receptor activation in tumor vasculature. J Biol Chem. 2015;290:8133–8145. doi: 10.1074/jbc.M114.591511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sawada J., Li F., Komatsu M. R-Ras inhibits VEGF-induced p38MAPK activation and HSP27 phosphorylation in endothelial cells. J Vasc Res. 2015;52:347–359. doi: 10.1159/000444526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwashita S., Kobayashi M., Kubo Y., Hinohara Y., Sezaki M., Nakamura K., Suzuki-Migishima R., Yokoyama M., Sato S., Fukuda M., Ohba M., Kato C., Adachi E., Song S.Y. Versatile roles of R-Ras GAP in neurite formation of PC12 cells and embryonic vascular development. J Biol Chem. 2007;282:3413–3417. doi: 10.1074/jbc.C600293200. [DOI] [PubMed] [Google Scholar]
- 51.Vahatupa M., Prince S., Vataja S., Mertimo T., Kataja M., Kinnunen K., Marjomaki V., Uusitalo H., Komatsu M., Jarvinen T.A., Uusitalo-Jarvinen H. Lack of R-Ras leads to increased vascular permeability in ischemic retinopathy. Invest Ophthalmol Vis Sci. 2016;57:4898–4909. doi: 10.1167/iovs.16-19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sawada J., Komatsu M. Normalization of tumor vasculature by R-Ras. Cell Cycle. 2012;11:4285–4286. doi: 10.4161/cc.22465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei X., Adak S., Zayed M., Yin L., Feng C., Speck S.L., Kathayat R.S., Zhang Q., Dickinson B.C., Semenkovich C.F. Endothelial palmitoylation cycling coordinates vessel remodeling in peripheral artery disease. Circ Res. 2020;127:249–265. doi: 10.1161/CIRCRESAHA.120.316752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janapati S., Wurtzel J., Dangelmaier C., Manne B.K., Bhavanasi D., Kostyak J.C., Kim S., Holinstat M., Kunapuli S.P., Goldfinger L.E. TC21/RRas2 regulates glycoprotein VI-FcRgamma-mediated platelet activation and thrombus stability. J Thromb Haemost. 2018:1632–1645. doi: 10.1111/jth.14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delgado P., Cubelos B., Calleja E., Martinez-Martin N., Cipres A., Merida I., Bellas C., Bustelo X.R., Alarcon B. Essential function for the GTPase TC21 in homeostatic antigen receptor signaling. Nat Immunol. 2009;10:880–888. doi: 10.1038/ni.1749. [DOI] [PubMed] [Google Scholar]
- 56.Egami Y., Araki N. Transient recruitment of M-Ras GTPase to phagocytic cups in RAW264 macrophages during FcgammaR-mediated phagocytosis. Microscopy (Oxf) 2018;67:68–74. doi: 10.1093/jmicro/dfx131. [DOI] [PubMed] [Google Scholar]
- 57.Cole A.L., Subbanagounder G., Mukhopadhyay S., Berliner J.A., Vora D.K. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arterioscler Thromb Vasc Biol. 2003;23:1384–1390. doi: 10.1161/01.ATV.0000081215.45714.71. [DOI] [PubMed] [Google Scholar]
- 58.Guo X., Stratton L., Schrader J.W. Expression of activated M-Ras in hemopoietic stem cells initiates leukemogenic transformation, immortalization and preferential generation of mast cells. Oncogene. 2006;25:4241–4244. doi: 10.1038/sj.onc.1209452. [DOI] [PubMed] [Google Scholar]
- 59.Singh G., Hashimoto D., Yan X., Helft J., Park P.J., Ma G., Qiao R.F., Kennedy C.R., Chen S.H., Merad M., Chan A.M. R-Ras is required for murine dendritic cell maturation and CD4+ T-cell priming. Blood. 2012;119:1693–1701. doi: 10.1182/blood-2011-05-357319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan X., Yan M., Guo Y., Singh G., Chen Y., Yu M., Wang D., Hillery C.A., Chan A.M. R-Ras regulates murine T cell migration and intercellular adhesion molecule-1 binding. PLoS One. 2015;10:e0145218. doi: 10.1371/journal.pone.0145218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinez-Martin N., Fernandez-Arenas E., Cemerski S., Delgado P., Turner M., Heuser J., Irvine D.J., Huang B., Bustelo X.R., Shaw A., Alarcon B. T cell receptor internalization from the immunological synapse is mediated by TC21 and RhoG GTPase-dependent phagocytosis. Immunity. 2011;35:208–222. doi: 10.1016/j.immuni.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sawada J., Perrot C.Y., Chen L., Fournier-Goss A.E., Oyer J., Copik A., Komatsu M. High endothelial venules accelerate naive t-cell recruitment by tumor necrosis factor-alpha-mediated R-Ras up-regulation. Am J Pathol. 2021;191:396–414. doi: 10.1016/j.ajpath.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shang X., Cancelas J.A., Li L., Guo F., Liu W., Johnson J.F., Ficker A., Daria D., Geiger H., Ratner N., Zheng Y. R-Ras and Rac GTPase cross-talk regulates hematopoietic progenitor cell migration, homing, and mobilization. J Biol Chem. 2011;286:24068–24078. doi: 10.1074/jbc.M111.226951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mendoza P., Martinez-Martin N., Bovolenta E.R., Reyes-Garau D., Hernansanz-Agustin P., Delgado P., Diaz-Munoz M.D., Oeste C.L., Fernandez-Pisonero I., Castellano E., Martinez-Ruiz A., Alonso-Lopez D., Santos E., Bustelo X.R., Kurosaki T., Alarcon B. R-Ras2 is required for germinal center formation to aid B cells during energetically demanding processes. Sci Signal. 2018;11:eaal1506. doi: 10.1126/scisignal.aal1506. [DOI] [PubMed] [Google Scholar]
- 65.Ray A., Basu S., Miller N.M., Chan A.M., Dittel B.N. An increase in tolerogenic dendritic cell and natural regulatory T cell numbers during experimental autoimmune encephalomyelitis in Rras-/- mice results in attenuated disease. J Immunol. 2014;192:5109–5117. doi: 10.4049/jimmunol.1302254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun P., Watanabe H., Takano K., Yokoyama T., Fujisawa J., Endo T. Sustained activation of M-Ras induced by nerve growth factor is essential for neuronal differentiation of PC12 cells. Genes Cells. 2006;11:1097–1113. doi: 10.1111/j.1365-2443.2006.01002.x. [DOI] [PubMed] [Google Scholar]
- 67.Kimmelman A.C., Nunez Rodriguez N., Chan A.M. R-Ras3/M-Ras induces neuronal differentiation of PC12 cells through cell-type-specific activation of the mitogen-activated protein kinase cascade. Mol Cell Biol. 2002;22:5946–5961. doi: 10.1128/MCB.22.16.5946-5961.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keely P.J., Rusyn E.V., Cox A.D., Parise L.V. R-Ras signals through specific integrin alpha cytoplasmic domains to promote migration and invasion of breast epithelial cells. J Cell Biol. 1999;145:1077–1088. doi: 10.1083/jcb.145.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saito Y., Oinuma I., Fujimoto S., Negishi M. Plexin-B1 is a GTPase activating protein for M-Ras, remodelling dendrite morphology. EMBO Rep. 2009;10:614–621. doi: 10.1038/embor.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ito Y., Oinuma I., Katoh H., Kaibuchi K., Negishi M. Sema4D/plexin-B1 activates GSK-3beta through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep. 2006;7:704–709. doi: 10.1038/sj.embor.7400737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tasaka G., Negishi M., Oinuma I. Semaphorin 4D/plexin-B1-mediated M-Ras GAP activity regulates actin-based dendrite remodeling through lamellipodin. J Neurosci. 2012;32:8293–8305. doi: 10.1523/JNEUROSCI.0799-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oinuma I., Katoh H., Negishi M. Molecular dissection of the semaphorin 4D receptor plexin-B1-stimulated R-Ras GTPase-activating protein activity and neurite remodeling in hippocampal neurons. J Neurosci. 2004;24:11473–11480. doi: 10.1523/JNEUROSCI.3257-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yukawa K., Tanaka T., Bai T., Ueyama T., Owada-Makabe K., Tsubota Y., Maeda M., Suzuki K., Kikutani H., Kumanogoh A. Semaphorin 4A induces growth cone collapse of hippocampal neurons in a Rho/Rho-kinase-dependent manner. Int J Mol Med. 2005;16:115–118. [PubMed] [Google Scholar]
- 74.Worzfeld T., Swiercz J.M., Senturk A., Genz B., Korostylev A., Deng S., Xia J., Hoshino M., Epstein J.A., Chan A.M., Vollmar B., Acker-Palmer A., Kuner R., Offermanns S. Genetic dissection of plexin signaling in vivo. Proc Natl Acad Sci U S A. 2014;111:2194–2199. doi: 10.1073/pnas.1308418111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanz-Rodriguez M., Gruart A., Escudero-Ramirez J., de Castro F., Delgado-Garcia J.M., Wandosell F., Cubelos B. R-Ras1 and R-Ras2 are essential for oligodendrocyte differentiation and survival for correct myelination in the central nervous system. J Neurosci. 2018;38:5096–5110. doi: 10.1523/JNEUROSCI.3364-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang Y., Rangwala F., Fulkerson P.C., Ling B., Reed E., Cox A.D., Kamholz J., Ratner N. Role of TC21/R-Ras2 in enhanced migration of neurofibromin-deficient Schwann cells. Oncogene. 2004;23:368–378. doi: 10.1038/sj.onc.1207075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Graham S.M., Oldham S.M., Martin C.B., Drugan J.K., Zohn I.E., Campbell S., Der C.J. TC21 and Ras share indistinguishable transforming and differentiating activities. Oncogene. 1999;18:2107–2116. doi: 10.1038/sj.onc.1202517. [DOI] [PubMed] [Google Scholar]
- 78.Young L.C., Hartig N., Munoz-Alegre M., Oses-Prieto J.A., Durdu S., Bender S., Vijayakumar V., Vietri Rudan M., Gewinner C., Henderson S., Jathoul A.P., Ghatrora R., Lythgoe M.F., Burlingame A.L., Rodriguez-Viciana P. An MRAS, SHOC2, and SCRIB complex coordinates ERK pathway activation with polarity and tumorigenic growth. Mol Cell. 2013;52:679–692. doi: 10.1016/j.molcel.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 79.Wong O.G., Nitkunan T., Oinuma I., Zhou C., Blanc V., Brown R.S., Bott S.R., Nariculam J., Box G., Munson P., Constantinou J., Feneley M.R., Klocker H., Eccles S.A., Negishi M., Freeman A., Masters J.R., Williamson M. Plexin-B1 mutations in prostate cancer. Proc Natl Acad Sci U S A. 2007;104:19040–19045. doi: 10.1073/pnas.0702544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castro A.F., Campos T., Babcock J.T., Armijo M.E., Martinez-Conde A., Pincheira R., Quilliam L.A. M-Ras induces Ral and JNK activation to regulate MEK/ERK-independent gene expression in MCF-7 breast cancer cells. J Cell Biochem. 2012;113:1253–1264. doi: 10.1002/jcb.23458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu Y., Feig L.A. Involvement of R-Ras and Ral GTPases in estrogen-independent proliferation of breast cancer cells. Oncogene. 2002;21:7557–7568. doi: 10.1038/sj.onc.1205961. [DOI] [PubMed] [Google Scholar]
- 82.Larive R.M., Moriggi G., Menacho-Marquez M., Canamero M., de Alava E., Alarcon B., Dosil M., Bustelo X.R. Contribution of the R-Ras2 GTP-binding protein to primary breast tumorigenesis and late-stage metastatic disease. Nat Commun. 2014;5:3881. doi: 10.1038/ncomms4881. [DOI] [PubMed] [Google Scholar]
- 83.Clark G.J., Kinch M.S., Gilmer T.M., Burridge K., Der C.J. Overexpression of the Ras-related TC21/R-Ras2 protein may contribute to the development of human breast cancers. Oncogene. 1996;12:169–176. [PubMed] [Google Scholar]
- 84.Barker K.T., Crompton M.R. Ras-related TC21 is activated by mutation in a breast cancer cell line, but infrequently in breast carcinomas in vivo. Br J Cancer. 1998;78:296–300. doi: 10.1038/bjc.1998.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gutierrez-Erlandsson S., Herrero-Vidal P., Fernandez-Alfara M., Hernandez-Garcia S., Gonzalo-Flores S., Mudarra-Rubio A., Fresno M., Cubelos B. R-RAS2 overexpression in tumors of the human central nervous system. Mol Cancer. 2013;12:127. doi: 10.1186/1476-4598-12-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakada M., Niska J.A., Tran N.L., McDonough W.S., Berens M.E. EphB2/R-Ras signaling regulates glioma cell adhesion, growth, and invasion. Am J Pathol. 2005;167:565–576. doi: 10.1016/S0002-9440(10)62998-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Y., Soong J., Mohanty S., Xu L., Scott G. The neural guidance receptor plexin C1 delays melanoma progression. Oncogene. 2013;32:4941–4949. doi: 10.1038/onc.2012.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luo R.Z., Fang X., Marquez R., Liu S.Y., Mills G.B., Liao W.S., Yu Y., Bast R.C. ARHI is a Ras-related small G-protein with a novel N-terminal extension that inhibits growth of ovarian and breast cancers. Oncogene. 2003;22:2897–2909. doi: 10.1038/sj.onc.1206380. [DOI] [PubMed] [Google Scholar]
- 89.Huang Y., Saez R., Chao L., Santos E., Aaronson S.A., Chan A.M. A novel insertional mutation in the TC21 gene activates its transforming activity in a human leiomyosarcoma cell line. Oncogene. 1995;11:1255–1260. [PubMed] [Google Scholar]
- 90.Rincon-Arano H., Rosales R., Mora N., Rodriguez-Castaneda A., Rosales C. R-Ras promotes tumor growth of cervical epithelial cells. Cancer. 2003;97:575–585. doi: 10.1002/cncr.11093. [DOI] [PubMed] [Google Scholar]
- 91.Macha M.A., Matta A., Sriram U., Thakkar A., Shukla N.K., Datta Gupta S., Ralhan R. Clinical significance of TC21 overexpression in oral cancer. J Oral Pathol Med. 2010;39:477–485. doi: 10.1111/j.1600-0714.2009.00854.x. [DOI] [PubMed] [Google Scholar]
- 92.Sharma R., Sud N., Chattopadhyay T.K., Ralhan R. TC21/R-Ras2 upregulation in esophageal tumorigenesis: potential diagnostic implications. Oncology. 2005;69:10–18. doi: 10.1159/000087283. [DOI] [PubMed] [Google Scholar]
- 93.Luo H., Hao X., Ge C., Zhao F., Zhu M., Chen T., Yao M., He X., Li J. TC21 promotes cell motility and metastasis by regulating the expression of E-cadherin and N-cadherin in hepatocellular carcinoma. Int J Oncol. 2010;37:853–859. doi: 10.3892/ijo_00000736. [DOI] [PubMed] [Google Scholar]
- 94.Román J.J.G., Garay G.O., Saenz P., Escuredo K., Ibayondo C.S., Gutkind S., Junquera C., Simón L., Martínez A., Luna J.L.F. Plexin B1 is downregulated in renal cell carcinomas and modulates cell growth. Transl Res. 2008;151:134–140. doi: 10.1016/j.trsl.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 95.Radich J.P., Dai H., Mao M., Oehler V., Schelter J., Druker B., Sawyers C., Shah N., Stock W., Willman C.L., Friend S., Linsley P.S. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103:2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xiao R., Shi L., Yang T., Zhang M., Wang H.-Y., Mai S.-J. Identification of RRAS gene related to nasopharyngeal carcinoma based on pathway and network-based analyses. Transl Cancer Res. 2019;8:664–675. doi: 10.21037/tcr.2019.04.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ward K.R., Zhang K.X., Somasiri A.M., Roskelley C.D., Schrader J.W. Expression of activated M-Ras in a murine mammary epithelial cell line induces epithelial-mesenchymal transition and tumorigenesis. Oncogene. 2004;23:1187–1196. doi: 10.1038/sj.onc.1207226. [DOI] [PubMed] [Google Scholar]
- 98.Sandri C., Caccavari F., Valdembri D., Camillo C., Veltel S., Santambrogio M., Lanzetti L., Bussolino F., Ivaska J., Serini G. The R-Ras/RIN2/Rab5 complex controls endothelial cell adhesion and morphogenesis via active integrin endocytosis and Rac signaling. Cell Res. 2012;22:1479–1501. doi: 10.1038/cr.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song J., Zheng B., Bu X., Fei Y., Shi S. Negative association of R-Ras activation and breast cancer development. Oncol Rep. 2014;31:2776–2784. doi: 10.3892/or.2014.3121. [DOI] [PubMed] [Google Scholar]
- 100.Xu L., Gao Y., Chen Y., Xiao Y., He Q., Qiu H., Ge W. Quantitative proteomics reveals that distant recurrence-associated protein R-Ras and transgelin predict post-surgical survival in patients with stage III colorectal cancer. Oncotarget. 2016;7:43868–43893. doi: 10.18632/oncotarget.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sawada J., Urakami T., Li F., Urakami A., Zhu W., Fukuda M., Li D.Y., Ruoslahti E., Komatsu M. Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell. 2012;22:235–249. doi: 10.1016/j.ccr.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]