

Abstract
Increasing numbers of people are living with osteoarthritis (OA) due to aging and obesity, creating an urgent need for effective treatment and preventions. Two top risk factors for OA, age and obesity, are associated with endoplasmic reticulum (ER) stress. The I-ERS mouse, an ER stress–driven model of primary OA, was developed to study the role of ER stress in primary OA susceptibility. The I-ERS mouse has the unique ability to induce ER stress in healthy adult articular chondrocytes and cartilage, driving joint degeneration that mimics early primary OA. In this study, ER stress–induced damage occurred gradually and stimulated joint degeneration with OA characteristics including increased matrix metalloproteinase activity, inflammation, senescence, chondrocyte death, decreased proteoglycans, autophagy block, and gait dysfunction. Consistent with human OA, intense exercise hastened and increased the level of ER stress–induced joint damage. Notably, loss of a critical ER stress response protein (CHOP) largely ameliorated ER stress–stimulated OA outcomes including preserving proteoglycan content, reducing inflammation, and relieving autophagy block. Resveratrol diminished ER stress–induced joint degeneration by decreasing CHOP, TNFα, IL-1β, MMP-13, pS6, number of TUNEL-positive chondrocytes, and senescence marker p16 INK4a. The finding, that a dietary supplement can prevent ER stressed–induced joint degeneration in mice, provides a preclinical foundation to potentially develop a prevention strategy for those at high risk to develop OA.
Osteoarthritis (OA) is a common condition affecting 240 million people worldwide, significantly impacting quality of life due to pain and functional limitations.1 Moreover, the number of people living with OA is rising at an alarming rate, primarily due to population aging and obesity.2, 3, 4 Although the understanding of mechanisms of secondary and/or post-traumatic OA (PTOA) has increased due to research from animal model studies, less progress has been made in understanding primary and/or idiopathic OA.5 The causes and risk factors of primary OA are heterogeneous and complex, limiting the efforts to develop disease-modifying osteoarthritis drugs. One promising therapeutic target and important area of research is the association between ER stress in articular cartilage chondrocytes and initiation of primary and/or idiopathic OA.6 Age-related primary OA results from the accumulation of advanced glycation end-products, which stimulate ER stress and/or chondrocyte senescence.7 Injection of advanced glycation end-products into mouse joints caused ER stress and chondrocyte apoptosis, and led to degradation of articular cartilage, whereas an inhibitor of advanced glycation end-product formation was protective.8 ER stress in obesity is triggered by the excessive demand for insulin, which drives protein synthesis beyond the ER capacity, leading to an increase in unfolded and misfolded protein.9 A recent study showed that diet-induced obesity in mice leads to ER stress and joint damage that was ameliorated by phenylbutyric acid, an ER stress reduction drug.10 Taken together, these findings show that the two most common primary OA risk factors, age and obesity, both involve ER stress in the initiation of joint destruction.
Disturbances of ER homoeostasis leads to activation of the unfolded protein response, a protective mechanism involving chaperone proteins to clear the accumulation of proteins in the ERs and preserve cell viability.11, 12, 13 However, as chondrocytes age, the level of chaperones declines as does the ability of chondrocytes to cope with ER stress.14 When folding and degradation mechanisms fail to restore ER homoeostasis, chronic ER stress develops and stimulates C/EBP homologous protein (CHOP), a proapoptotic transcription factor through the protein kinase R-like endoplasmic reticulum kinase branch of the unfolded protein response.15,16 Studies have shown that the number of chondrocytes in OA cartilage expressing CHOP positively correlates with the level of cartilage degeneration and number of apoptotic chondrocytes.17 Importantly, the loss of CHOP protects against advanced glycation end-product ER stress–induced OA and surgically induced OA,8,18 indicating that CHOP plays an important and/or central role in ER stress–stimulated joint degeneration. Suppressing ER stress in chondrocytes with salubrinal decreased inflammation-driven up-regulation of a critical degradative enzyme activity [matrix metalloproteinase 13 (MMP-13)] in an animal model of OA.19 MMP-13 is at the top of an OA destruction hierarchy in which articular cartilage is degraded leading to ulceration and erosion of cartilage, and ultimately, synovitis.20 These important findings suggest that ER stress reduction should be targeted as an OA treatment and/or prevention strategy. Although ER stress has been linked to primary OA initiation, this information has not yet been utilized to develop prevention and/or treatment strategies or to define the point of reversible pathology in ER stress–stimulated joint degeneration. This information is critical for moving from the bench to the bedside with an ER stress–targeted disease-modifying osteoarthritis drug.
To address this translational knowledge gap, the inducible-ER stress (I-ERS) mouse model was developed as an ER stress-inducible OA model, using tetracycline-inducible expression of a misfolded protein, mutant cartilage oligomeric matrix protein (mutant-COMP).21 OA markers of pathology (proteoglycan content, inflammation, degradative enzyme MMP-13, blockage of autophagy, chondrocyte death, chondrocyte senescence, and gait disturbances) were examined in I-ERS + doxycycline (DOX) articular cartilage. A major advantage of this system is the capacity to turn on and off the expression of misfolded protein in adult I-ERS mice. Using this model, ER stress was shown to be a key underlying mechanism of OA joint damage, and resveratrol ameliorated cartilage degeneration.
Materials and Methods
Generation of Bigenic Mice
The mice used in this study are designated I-ERS (inducible ER stress) because they were used to study the induction of ER stress in adult mice and its effect on articular cartilage. As previously described, this bigenic inducible mouse line was generated using two plasmids: cartilage oligomeric matrix protein (pTRE-COMP) [human COMP+FLAG tag coding sequence driven by the tetracycline responsive element promoter (TRE)] and reverse tetracycline-controlled transactivator (rtTA) coding sequence driven by a type II collagen promoter (pTET-On-Col II).21 These two constructs together express mutant-COMP in cartilage mice in the presence of DOX.21, 22, 23 Mice were PCR genotyped to verify the presence of both transgenes.21 DOX was administered in drinking water (500 ng/mL) from 16 to 20 weeks of age because it is less stressful than daily injection or gavage feeding for a long period of time. Resveratrol (0.25 g/L) was administered through drinking water from 16 to 20 weeks of age (Reserveages Organics, Gainesville, FL).
DOX administered from birth to 4 weeks causes a dwarfing phenotype, and with longer expression of mutant-COMP, joint degeneration occurs (data not shown). By contrast, I-ERS mice were not administered DOX during skeletal growth (birth to 10 weeks), and the size of I-ERS mice was comparable to controls. DOX was administered at 16 weeks after skeletal growth was complete to evaluate the impact of ER stress on joint health in adult mice in the absence of growth deficiency.
Housing and Husbandry
These studies were approved by the institutional animal care and use committee of The University of Texas Health Science Center at Houston. Mus musculus control C57BL∖6 and bigenic I-ERS mice were housed with a maximum of five animals per cage. Animals were housed in a 12:12 light/dark cycle: lights on from 7 AM to 7 PM) at 21°C to 25°C, and animals were euthanized with carbon dioxide.
Exercise Protocol
Mice were acclimated to the treadmill for 1 week before exercise regimen began. Acclimation was performed as follows: day 1: run 17 cm per second for 5 minutes in three sessions with 5-minute breaks in between; day 2: run for 10 minutes in three sessions with 5-minute breaks in between—first session run at 17 cm per second, second session run at 17 cm per second increasing speed to 25 cm per second, and third session run at 25 cm per second; day 3: run for 20 minutes in two sessions with a 5-minute break in between—first session run at 17 cm per second for 10 minutes increasing speed to 25 cm per second for 10 minutes, and second session run at 25 cm per second; day 4: run for 30 minutes in two sessions with a 5-minute break in between—first session run at 17 cm per second for 10 minutes, increasing speed to 25 cm per second, and second session run at 35 cm per second; day 5: run for 1 hour—first 10 minutes run at 17 cm per second then increasing speed to 35 cm per second for the remainder of the time. Mice ran 5 days a week for total of 1 hour with speed increasing over a 30-minute period at a five degree incline. This intense exercise was designed to stimulate mechanical stress on the joints. Running began with 10 minutes at 17 cm per second, then speed was increased from 17 cm per second to 42 cm per second over a 20-minute period, and a final 30 minutes of running at 42 cm per second. The resting pad was equipped with a shock grid that was disabled after 5 seconds, and the Harvard Apparatus treadmill (model: LE-8710MTS; Harvard Apparatus, Holliston, MA) recorded the time each mouse spent running. Mice were exercised in the laboratory. At least nine control and I-ERS mice underwent the exercise protocol ± DOX. All mice were moved to the lab at 8:00 AM, and running occurred between 9:00 AM and 12:30 PM.
OA Scoring
OA scoring was performed on 10 different sections divided into quadrants (medial femoral condyle, medial tibial plateau, lateral femoral condyle, lateral tibial plateau). Scores ranged from 0 for normal cartilage to 6 for lesions reaching the calcified cartilage for >75% of the quadrant width and may be presented as a total score or each score listed individually.24 The traditional OARSI (Osteoarthritis Research Society International) scoring covers a wide range of OA pathology, and in this study, OA scoring was modified to optimize evaluation of early OA pathology. Here, four areas, synovium, bone and/or cartilage, and tibial and femoral articular cartilage, were scored from 0 to 3 on Safranin O–stained sections. In this modified OA scoring system, a score of 0 indicated normal or no damage; 1, mild damage; 2, moderate damage; and 3, denotes severe damage. Synovitis, bone and/or cartilage damage, and proteoglycan of tibia and femur were scored individually, and all scores summed with a maximal score of 12. Synovitis was defined as: mild, increase in thickness of synovial lining and increase in stromal area; moderate, increase in stromal density; or severe, synovial lining is thickened with further increase of stromal cellular density. Bone and/or cartilage damage was defined as: normal, surface was smooth; mild, minor erosion of the surface; moderate, presence of remodeling with minor erosion; or severe, major erosion. Proteoglycans of the articular cartilage of the tibia and femur was classified as normal if staining was even through to the subchondral bone, mild when staining was thinned, moderate with thinning of proteoglycan stained layer and absence of staining in some areas, or severe if there was widespread loss of proteoglycan staining. Ten mice per experimental group was used for each time point to provide 80% to 90% power to detect a minimal difference of 2 or 3 units. All scoring was performed blindly (F.H.G.). Section depth, thickness, fixation, and decalcification conditions were all identical for all limbs analyzed. Kruskal-Wallis test was used to evaluate distribution of OA scores across at six experiment groups with post hoc Dunn's test comparing I-ERS DOX run with all other groups.
Immunohistochemistry
Hind limbs from male I-ERS and C57BL∖6 control mice were collected, and articular cartilage was prepared and analyzed as previously described.21 Briefly, the limbs were identically fixed in 95% ethanol followed by decalcification in Immunocal (StatLab, McKinney, TX) for 1 week, and pepsin (1 mg/mL in 0.1 N HCl) was used for antigen retrieval for immunostaining with human COMP (MA1-20221, 1:100; Thermo Fisher Scientific, Waltham, MA), IL-1 (ab7632, 1:200; Abcam, Cambridge, UK), tumor necrosis factor α (TNFα) (ab6671, 1:200; Abcam), PDI (SC-20132, 1:100; Santa Cruz Biotechnology, Dallas, TX), p16 INK4a (ab189034, 1:200; Abcam), pS6 (2215S rabbit polyclonal, 1:200; Cell Signaling Technology, Danvers, MA), MMP-13 (ab39012, 1:50; Abcam), or 10% w/v formalin for terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL). Sections of the same thickness were incubated with antibodies overnight at 4°C for immunostaining, and species-specific secondary antibodies were incubated with sections for 50 minutes. Coverslips were mounted with ProLong Gold anti-fade reagent (Molecular Probes, Eugene, OR). For proteoglycan stains, samples were deparaffinized and hydrated in distilled water, and then stained with Safranin O (477-73-6; Spectrum Chemical, New Brunswick, NJ) or Alcian blue 8GX (A-3157; Sigma Aldrich, St. Louis, MO) according to the manufacturer's protocol. Immunostaining was performed on 10 mice in each group.
MMP Imaging
Mice were treated with depilatory cream to remove hair prior to imaging. MMP sense 680 reagent was injected in the tail vein, and animals were imaged 24 hours later per manufacturer's instructions with an uninjected control and imaged on an IVIS Spectrum In Vivo Imaging System (PerkinElmer, Waltham, MA) according to the manufacturer’s protocol. MMPsense signal was assessed blindly (J.W.) by positioning a circle of a standard size around the knee (in all samples), and radiance efficiency was generated from IVIS Living Image software version 4.2 (Caliper Live Sciences, Waltham, MA). Five mice were included per group, which had the power to detect a difference of 30% or greater. U-test was used to evaluate MMP activity in control and I-ERS mice. Mice were imaged between 4 and 6 PM.
TUNEL Quantification
The number of TUNEL-positive chondrocytes was compared with the total number of growth plate chondrocytes (DAPI, nuclei stain) to calculate the percentage of TUNEL-positive chondrocytes using ImageJ software version 1.5aj8 (NIH, Bethesda, MD; https://imagej.nih.gov/ij). Three articular cartilage sections from each mouse were counted from 10 mice. Differences due to treadmill exercise and age were assessed by analysis of variance, and post hoc two-way comparisons were analyzed by Tukey tests.
Gait Analysis
Gait was analyzed using a DigiGait treadmill system (Mouse Specifics Inc., Boston, MA) following the protocol described for collagen-induced OA.25 Previously, it has been shown that running during gait analysis was necessary to uncover subtle changes in ambulatory function that are associated with OA.26 Briefly, video camera images of mice running through a transparent belt were captured and analyzed. At least 3 seconds of uphill 15-degree running continuous strides were used to calculate and measure gait parameters with DigiGait software version 12. Mice were run for 25 cm per second, and seven parameters for gait analysis including ataxia coefficient, brake stride percentage, brake stance percentage, hind limb shared stance time, propel stance percentage, swing duration covariance, and stride length variance were measured. Gait analysis was performed in the animal behavioral testing room, and assessments were performed by software, so blinding was not used.
DigiGait recommended three mice per group to measure ethanol-induced ataxia or running speed (Mouse Specifics Inc., https://mousespecifics.com/digitgait-protocols, last accessed February 22, 2021). Based on this information, five mice per group were analyzed because OA gait dysfunction causes more subtle changes compared with ethanol-induced ataxia. Analysis of variance was performed followed by pairwise comparisons with Kruskal-Wallis and post hoc Dunn's analysis. All mice were moved to the behavior room at 8:00 AM, and DigiGait analysis occurred between 9:00 AM and 12:30 PM.
Results
Mutant-COMP Is Retained in the ER and Stimulates ER Stress in Adult Articular Chondrocytes
Previous studies demonstrated that misfolded-COMP accumulated in the ER of growth plate chondrocytes, stimulating increased CHOP expression, indicating that misfolded-COMP generates ER stress.21, 22, 23,27 To determine whether ER stress in adult articular chondrocytes can generate OA-like joint degeneration in healthy articular cartilage, misfolded-COMP was induced in 16-week–old mice, and joints were evaluated at 20 weeks. Misfolded-COMP, expressed in response to DOX was retained in the ER (Figure 1) of adult articular I-ERS chondrocytes, whereas none was detected in control C57BL/6 articular chondrocytes (Figure 1). ER retention of misfolded-COMP in I-ERS articular chondrocytes stimulated an increase in CHOP (Figure 1L), phosphorylated-eIF2α (Supplemental Figure S1, C and D), and activating transcription factor 4 (data not shown). Moreover, glucose-regulated protein 78/binding immunoglobulin protein was slightly elevated in I-ERS mice with DOX from 16 to 20 weeks, whereas glucose-regulated protein of 94 kDa was unchanged (data not shown). Collectively, this indicated that ER stress is induced in adult articular chondrocytes similar to that previously reported in juvenile growth plate chondrocytes as a result of misfolded-protein.22
Figure 1.

Human mutant-COMP is retained in endoplasmic reticulum (ER) of I-ERS murine articular chondrocytes and stimulates CHOP expression. Control (A–C, G) and I-ERS (D–F, H) tibial articular cartilage was immunostained using human COMP-specific antibodies (16 and 20 weeks) or CHOP (I–N, 16 and 20 weeks). Mutant-COMP was not expressed in control articular chondrocytes (A–C, G), minimal signal was present in I-ERS mice in the absence of doxycycline (DOX) (D and E), but was expressed and retained in ER of articular chondrocytes of I-ERS mice administered DOX (F and H). Low magnification image of C is shown in G, and F is shown in H. CHOP signal was not observed at 16 or 20 weeks in controls or at 16 or 20 weeks in I-ERS mice in the absence of DOX (I–M). CHOP was present in articular chondrocytes of I-ERS in the presence of DOX, which correlates with the presence of ER accumulation of mutant-COMP (N). DOX treatment was from 16 to 20 weeks. Scale bars = 100 μm (A–N).
Importantly, there was no detectable misfolded-COMP in the extracellular matrix of articular cartilage of I-ERS mice (Figure 1), and the abundance of type II collagen was not perturbed (data not shown), indicating that the type II collagen extracellular matrix (articular cartilage) was not compromised by the presence of misfolded protein. Type IX collagen and matrilin 3 were reduced in the I-ERS articular cartilage with 4 weeks of mutant-COMP expression, and this may impact degeneration (data not shown).
Markers of Primary OA Are Present in ER Stress–Induced Adult I-ERS Articular Cartilage
Next, joint health of I-ERS mice after 4 weeks of ER stress induction was evaluated to establish whether ER stress produced joint destruction similar to OA damage. The loss of proteoglycans in the articular cartilage of I-ERS mice was first evaluated because this early-stage pathological change precedes thinning of articular cartilage.28, 29, 30, 31, 32 Proteoglycans in the articular cartilage of I-ERS mice at 16 and 20 weeks was assessed using Safranin O (Figure 2) and Alcian blue (Supplemental Figure S2) staining. In the absence of DOX, the I-ERS articular cartilage was comparable to the control (Figure 2, A, B, E, and F, and Supplemental Figure S2, A and C). ER stress from 16 to 20 weeks in adult I-ERS mice resulted in a loss of proteoglycans at the articular surface (Figure 2, C and G, and Supplemental Figure S2, B and D). Four weeks of mechanical stress (intense running) in addition to ER stress caused a dramatic decrease in proteoglycans in the I-ERS femoral and tibial articular cartilages (Figure 2, D, G, and H). Moreover, dramatic loss of proteoglycans in the transitional and deep zone of articular cartilage with ER stress and strenuous treadmill running in I-ERS mice was observed (Figure 2H).
Figure 2.

Proteoglycans are less abundant in I-ERS mice treated with doxycycline (DOX) from 16 to 20 weeks and exercised. Safranin O staining of control and I-ERS tibias collected at 20 weeks in the presence and absence of DOX and exercise regimen from 16 to 20 weeks. An abundant and rich proteoglycan layer was observed in all control articular cartilage (E–H). The proteoglycan layer in I-ERS mice without DOX was normal (A and B), but diminished, in the I-ERS mice with DOX (C) and exercise (D). Treadmill exercise (RUN) was from 16 to 20 weeks. Scale bars = 500 μm (A–H).
MMP activity, a marker for OA degenerative processes, was evaluated using an MMP substrate that fluoresces when cleaved by either MMP-2, -3, -6, or -13. MMP activity was significantly higher in I-ERS mice with DOX from 16 to 20 weeks of age (P < 0.00005) (median, 1.3 × 108, range, 4.6 × 107 to 9.5 × 108) compared with controls (median, 4.2 × 107, range, 6.8 × 106 to 5.7 × 107) (Figure 3B). The intensity of the signal indicated higher MMP activity with representative images of both control and I-ERS + DOX (Figure 3A). Consistent with elevated MMP activity, MMP-13 immunostaining was higher in I-ERS mice with DOX ± exercise compared with control or I-ERS without DOX (Figure 3, C–J). In addition to higher levels of MMP, ER stress increased TNFα and IL-1, markers of inflammation, in I-ERS mice with DOX ± exercise compared with controls and I-ERS mice without DOX (Figure 3, K–Z). These findings demonstrate that both OA-associated cytokines and MMP activity were elevated in I-ERS articular chondrocytes with the induction of ER stress.
Figure 3.

Cartilage degeneration markers, inflammatory proteins, and mTORC1 signaling are elevated in I-ERS mice treated with doxycycline (DOX) from 16 to 20 weeks. Control and I-ERS mice were administered DOX from 16 to 20 weeks and subjected to MMPsense 680 imaging of the hind limbs at 20 weeks (A and B) to assess MMP-2, -3, -9, and -13 activity in vivo or MMP-13 immunostaining (C–J). Total radiance efficiency is plotted in B, with examples of control and I-ERS + DOX hind limbs shown in A. A significant difference between control and I-ERS MMP activity was detected. Control radiant efficiency median was 42.07 × 106 (range, 6.79 × 106 to 57.05 × 106) compared with I-ERS 131.45 × 106 (range, 45.62 × 106 to 953.00 × 106). Control and I-ERS tibial articular cartilage, immunostained with MMP-13 at 16 weeks prior to treatment and at 20 weeks with DOX ± exercised (C–J). I-ERS mice with DOX or DOX and exercise increases the level of MMP-13 observed in articular chondrocytes (E and F), whereas I-ERS without DOX and controls show low levels of MMP-13 signal (C, D, G–J). Control and I-ERS tibial articular cartilage was immunostained with TNFα (K–R), IL-1β (S–Z), and pS6 (AA–HH) at 16 weeks prior to DOX treatment and at 20 weeks with DOX ± exercise. The levels of proinflammatory cytokines TNFα and IL-1β and pS6 were increased in DOX-treated I-ERS mice (M, U, CC) and in those treated with DOX and subjected to exercise (N, V, DD). TNFα, IL-1β, and pS6 immunostaining in control murine articular cartilage was minimal or absent (O–R, W–Z, EE–HH). Treadmill exercise (RUN) was from 16 to 20 weeks. Dotted line marks the margin of articular cartilage, and asterisks mark areas of immunosignal. ∗∗∗∗P < 0.00005 (U-test). Scale bars: 100 μm (C–HH).
Autophagy recycling of cellular components is essential to maintain chondrocyte metabolism and cellular health and importantly, autophagy is repressed in OA.33 mTORC1 regulates synthesis of cellular components and autophagy.34 To evaluate whether autophagy was blocked in the I-ERS articular chondrocytes, mTORC1 activity was evaluated through phosphorylated S6 ribosomal protein (pS6), a readout for mTORC1 activity.35 Autophagy was suppressed (high levels of mTORC1 signaling, pS6) in I-ERS mice + DOX (Figure 3, X–HH) compared with controls. This finding is consistent with autophagy repression previously demonstrated in human OA.33
OA is associated with high levels of articular chondrocyte death and increased chondrocyte senescence that impairs cellular function.36 At 16 weeks, the percentage of TUNEL-positive articular chondrocytes was equivalent between control and I-ERS mice without DOX (median, I-ERS 6.7%, control 5.6%) (data not shown). At 20 weeks, in the absence of DOX, TUNEL-positive chondrocytes were slightly elevated in I-ERS mice compared with controls (median, I-ERS 11.2%, control 5.2%; P ≤ 0.05) (Figure 4). A very small amount of mutant-COMP may be synthesized in the absence of DOX and may account for the slightly higher percentage of TUNEL-positive articular chondrocytes in 20-week–old mice compared with controls (Supplemental Table S1). However, Safranin O staining of I-ERS mice at 9 months (no DOX) showed that articular cartilage proteoglycan content was not appreciably different from controls, suggesting that I-ERS mice were not substantially more prone to joint degeneration in the absence of ER stress than control mice (data not shown).
Figure 4.

Inducing endoplasmic reticulum (ER) stress in the I-ERS mice results in an increase in TUNEL-positive chondrocytes and p16 INK4a senescence marker in articular cartilage. A: Sections from the hind limb articular cartilage of at least 10 animals per group were stained with TUNEL and DAPI. The number of TUNEL-positive articular chondrocytes was divided by the total number of chondrocytes to calculate the average percentage of TUNEL-positive chondrocytes for each group. Only the I-ERS mice show significantly increased TUNEL-positive chondrocytes in the articular cartilage from the knee joint. The differences in percentage of TUNEL-positive chondrocytes were statistically significant between I-ERS and control mice while accounting for doxycycline (DOX), mechanical stress from running, and age of the mice (analysis of variance P < 0.00005). Significant effects of the mutation and an interaction between I-ERS and DOX was identified by four-way analysis of variance model (P = 0.0021). Age of the mice or running did not significantly influence the percentage of TUNEL-positive chondrocytes. A significant difference in the distribution of the percentage TUNEL-positive chondrocytes was identified (P = 0.0001). Significant differences in percentage of TUNEL-positive chondrocytes are shown here. Additional information is available in Supplemental Table S1. B–I: Control and I-ERS tibial articular cartilage were immunostained with p16 INK4a at 16 weeks prior to DOX treatment and at 20 weeks with −DOX and +DOX ± exercise. The level of p16 INK4a was increased in I-ERS mice with DOX and intense exercise (E). DOX treatment was from 16 to 20 weeks, and treadmill exercise (RUN) was from 16 to 20 weeks. Dashed lines mark the margin of articular cartilage, and asterisks mark areas of specific immunosignal. Data are expressed as means ± SD. ∗P < 0.05, ∗∗∗P < 0.0005 (post hoc Dunn test). Scale bars = 100 μm (B–I).
Importantly, the increase in the percentage of TUNEL-positive chondrocytes in I-ERS mice with DOX was dramatically different than controls (8% control + DOX; 31% I-ERS + DOX). If the contribution of I-ERS genotype (6%) is removed, the percentage of TUNEL-positive chondrocytes in I-ERS mice with DOX was still threefold higher than controls (31% to 6% = 25% divided by 8% control = 3.125-fold) indicating that I-ERS mice had substantially increased articular chondrocyte death (Figure 4) (P ≤ 0.0005). OA initiation and progression is linked to articular chondrocyte death that is driven by cellular stresses29,37, 38, 39, 40 including oxidative, ER stress, and TNFα and IL-1β inflammatory processes.41 In the I-ERS mice (+DOX), higher levels of articular chondrocyte death occurred along with TNFα/IL-1β inflammation and ER stress, suggesting these processes are associated with articular chondrocyte death in the I-ERS mice. Interestingly, senescence marker levels (p16 INK4a) were elevated only in I-ERS mice on DOX and exercise (Figure 4), suggesting that mechanical stress along with the presence of ER stress increases joint damage, resulting in faster progression of OA.
Strenuous Exercise Exacerbates OA, Causing Abnormal Gait and Pain in I-ERS Mice
The OARSI semiquantitative scoring system was modified to evaluate the level of knee joint damage in I-ERS mice (Materials and Methods). Synovitis, bone and/or cartilage damage, and proteoglycan content of the tibia and femur was scored.24 Intense mechanical stress from treadmill running increased the OA score in the I-ERS + DOX above all other experimental groups (median score, 5.5; P ≤ 0.0058) (Figure 5). Supplemental Table S2 shows that the increased OA score in the I-ERS (+DOX and strenuous exercise) was from significantly less proteoglycan in the articular cartilage of the femur, more bone and/or cartilage damage, and greater synovitis. This increase in OA score with strenuous exercise occurred even though the I-ERS + DOX ran approximately half that of the control time. C57BL∖6 control mice without DOX ran the longest with an average of 45 minutes, and the I-ERS with DOX ran the least, with an average of 25 minutes, despite shock applied from the resting pad (data not shown). Intense exercise clearly exacerbated the ER stress joint damage in I-ERS mice, while having very little impact on control mice (Figures 2, 3, 4, and 5), indicating that I-ERS mice were more susceptible to intense mechanical stress, which is an OA risk factor. ER stress alone for 4 weeks in I-ERS mice did not generate extensive structural damage as shown by the OA score of 2.5 (Figure 5). However, ER stress reduced proteoglycan content and increased inflammation, both signs of early stage OA (Figures 2 and 3).
Figure 5.

Articular cartilage damage is higher and gait irregular with doxycycline (DOX) and exercise. Tibial and femoral articular cartilage, synovium, and bone and/or cartilage were each scored from 0 to 3 using a maximal total score of 12. Individual osteoarthritis (OA) scores were plotted, and median and first and fourth quartiles are indicated by bars. OA score of I-ERS mice in the presence of DOX and exercise (RUN) was higher than all other groups. Details of the score in each group is shown in Supplemental Table S2. A significant difference in the distribution of the OA scores across the six experimental group was identified (Kruskal-Wallis test P = 0.0137). The mutant DOX run group is the only one that was different from all the other groups (post hoc Dunn test P < 0.0058). Gait was evaluated on 3 seconds of continuous strides running at 25 cm per second at five degrees uphill in control and I-ERS mice at 20 weeks ± DOX ± exercise. Ataxia coefficient, brake stride percentage, brake stance percentage, hind limb shared stance time, propel stance percentage, swing duration covariance, and stride length variance were evaluated. Endoplasmic reticulum (ER) stress in I-ERS mice increased ataxia coefficient, and ER stress and exercise increased hind limb shared stance time. DOX treatment was from 16 to 20 weeks, and treadmill exercise (RUN) was from 16 to 20 weeks. ∗∗∗P < 0.005 I-ERS + DOX + RUN versus all other groups. OARSI, Osteoarthritis Research Society International.
Joint pain is a hallmark of OA. Although structural changes in the joints lead to gait dysfunction and joint pain,42 the degree of joint damage is not necessarily associated with the level of pain. This was demonstrated in a study that showed knee pain from OA or experimentally imposed pain in absence of joint damage alters gait, indicating that pain itself modifies gait.37,43 To assess joint pain in I-ERS mice during ambulation, changes in gait were used as a proxy for joint pain. Gait was analyzed using the DigiGait system (Mouse Specifics) to uncover subtle changes in ambulatory function typically found in OA. Sixteen gait parameters were analyzed, with ataxia coefficient and shared hind limb stance time significantly changed. Ataxia coefficient was increased in I-ERS mice + DOX ± treadmill exercise, indicating an irregular gait was associated with ER stress–induced joint damage (data not shown; I-ERS + DOX − run = 0.46, control + DOX − run = 0.31; P ≤ 0.05; and I-ERS + DOX + run = 0.55 and control + DOX + run = 0.3; P ≤ 0.05). Strenuous exercise in combination with ER stress increased hind limb shared stance time in the I-ERS mice (data not shown; hind limb shared stance time I-ERS + DOX + run = 0.0825 seconds; control + DOX + run = 0.065 seconds; P ≤ 0.05). Hind limb shared stance time is the amount of time that both limbs are in contact with the surface. Shared stance time increases with joint pain because distribution of body weight over both limbs reduces pain.26 These measures of gait disturbance suggest that ER stress–induced joint damage in the presence of intense exercise exacerbates joint pain.
Absence of CHOP Reduces OA in I-ERS Mice
Loss of CHOP is protective in the DMM (destabilization of the medial meniscus) surgically induced OA mouse model.18 Because CHOP is an essential molecule for ER stress signaling, I-ERS/CHOP–null (I-ERS/CHOP−/−) mice were analyzed for the presence of OA markers. ER stress induction in I-ERS/CHOP−/− (+DOX) mice resulted in more proteoglycan in the articular cartilage (Safranin O) (Figure 6) than in the I-ERS mice (+DOX). Intracellular mutant-COMP accumulated in the I-ERS articular chondrocytes, but not in those of I-ERS/CHOP−/− mice (Figure 6, A and B). In the absence of CHOP, misfolded protein does not accumulate because autophagy clears the protein as it is synthesized (Figure 6, B and J), thereby alleviating ER stress. This leads to decreased TNFα and IL-1β (Figure 6, F and H) preserving chondrocyte function and increasing proteoglycans (Figure 6D) in the articular cartilage of I-ERS/CHOP−/− mice. These findings establish that articular cartilage OA outcomes in the I-ERS mice result from ER stress mediated through CHOP, as a result of the accumulation of misfolded protein.
Figure 6.

Loss of CHOP dampens markers of osteoarthritis (OA) pathology. I-ERS and I-ERS/CHOP−/− tibial articular cartilage were immunostained with mutant human COMP, Safranin O, TNFα, IL-1β, or phosphoS6 (pS6) at 20 weeks with doxycycline (DOX). Proteoglycan levels were higher in articular cartilage of I-ERS/CHOP−/− compared with I-ERS (D compared with C). The level of intracellular mutant-COMP, TNFα, IL-1β, and pS6 were increased in DOX-treated I-ERS mice (A, E, G, I) compared with I-ERS/CHOP−/− (B, F, H, J). DOX treatment was from 16 to 20 weeks. Dashed lines mark the margin of articular cartilage. Scale bars: 100 μm (A–J).
Resveratrol Prevents OA Pathology in I-ERS Mice
Resveratrol was first described as an antioxidant but more recently has been recognized for its anti-inflammatory properties reducing multiple proinflammatory cytokines and chemokines including TNFα, TNFβ, IL-1β, IL-6, IL-8, IL-17, and COX.44 Resveratrol treatment reduces ER stress in a variety of conditions including pseudoachondroplasia, polycystic ovary disease, brain injury, diabetic nephropathy, Parkinson disease, and chronic obstructive pulmonary disease.45, 46, 47, 48, 49, 50 Based on these studies, resveratrol treatment was tested to determine whether it could prevent and/or reduce joint degeneration in I-ERS mice. Resveratrol treatment in I-ERS + DOX mice dramatically reduced markers of degeneration and ER stress (CHOP) (Figure 7, D–F) associated with mutant-COMP retention (Figure. 7, A–C). Resveratrol prevented inflammation (TNFα and IL-1β) (Figure 7, G–L), which stimulates MMP-13 expression in chondrocytes51 and thus decreased MMP-13 in I-ERS articular cartilage (Figure 7, M–O). Resveratrol directly promoted autophagy by inhibiting mTORC1 activity,52 and consistent with this function, resveratrol reduced the autophagy block (pS6) (Figure 7, P–R) in the I-ERS articular cartilage chondrocytes. The reduction of chondrocyte stress decreased chondrocyte death (TUNEL) (Figure 7, S–U) and senescence (p16 INK4a) (Figure 7, V–X) in I-ERS mice. ER stress, inflammation, degradative enzyme MMP-13, chondrocyte death, and senescence in articular cartilage were normalized with resveratrol treatment.
Figure 7.

Resveratrol treatment prevents articular cartilage damage in I-ERS mice. Control I-ERS and resveratrol-treated I-ERS tibial articular cartilage was immunostained with mutant human COMP, CHOP, TNFα, IL-1β, MMP-13, phosphoS6 (pS6), or p16 INK4a at 20 weeks with doxycycline (DOX), and TUNEL staining (green signal; blue DAPI nuclei marker) was performed. The level of intracellular mutant-COMP, CHOP, TNFα, IL-1β, MMP-13, pS6, TUNEL, and p16 INK4a immunostaining were increased in DOX-treated I-ERS mice (B, E, H, K, N, Q, T, W) compared with I-ERS treated with resveratrol (C, F, I, L, O, R, U, X) or controls (A, D, G, J, M, P, S, V). DOX treatment was from 16 to 20 weeks, and resveratrol treatment (+Res) was from 16 to 20 weeks. Dotted lines mark the margin of articular cartilage. Scale bars: 100 μm (A–X).
Discussion
The results of this study demonstrate that ER stress in adult articular chondrocytes leads to OA pathologic susceptibility and establishes the I-ERS mouse as a model of ER stress-induced primary OA. Although ER stress (no intense exercise) for 4 weeks induced joint degeneration consistent with early primary OA, one limitation is the lack of information about the effect of longer periods of ER stress, although we anticipate that it will stimulate cartilage and/or bone damage. The induction of ER stress, targeted to articular chondrocytes of adult mice for 4 weeks, was sufficient to stimulate markers of joint degeneration consistent with the development of early-stage primary OA. The decrease in murine I-ERS articular cartilage proteoglycan levels, increased presence of MMPs, OA-associated inflammatory markers, and articular chondrocyte death, are direct evidence of OA in the I-ERS mouse. The findings that ER stress induced an OA-like joint destruction are consistent with recent work demonstrating that high-fat diet–induced OA in mice was driven directly by ER stress, and not solely by mechanical forces from the excess weight.10 Importantly, ER stress–induced joint degeneration was prevented by oral resveratrol treatment that reduced ER stress, inflammation, MMP13, senescence, and reactivation of autophagy (Figure 6). This significant outcome extends the findings of prior studies showing that resveratrol prevents OA caused by a high-fat diet.53, 54, 55 Resveratrol is a promising preventative approach; however, because there are no current diagnostic methods to precisely identify individuals who will develop primary OA prior to the onset of obvious symptoms, a treatment that can reverse joint damage is needed.
Resveratrol, in two clinical trials, decreased pain with age-related osteoarthritis and improved menopause-related quality of life in women when administered for either 14 weeks or 24 months.56,57 Two other trials showed that resveratrol, added to meloxicam therapy for knee OA, decreased pain more than meloxicam alone.58,59 These encouraging clinical trials need to be expanded to evaluate joint health before and after resveratrol administration to assess whether resveratrol can halt and/or heal joint damage. Moreover, studies in OA model systems collectively show that resveratrol prevents and/or delays onset and/or progression and pain by reducing inflammation (TNF-α, IL-1β, IL-6, and IL-18), oxidative stress, degradative enzymes (including, but not limited to, MMP-13), chondrocyte apoptosis, and by promoting autophagy in chondrocytes.53, 54, 55,60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71 Despite these promising outcomes, progress toward clinical translation has been slow. Supplement-related research suffers from the lack of product standardization that is required for drugs and has been a major roadblock. Nevertheless, this promising research has the potential to provide a safe, effective, and economic OA therapeutic that could provide relief or millions of individuals suffering from OA.
The results of these studies in the I-ERS/CHOP−/− mice demonstrate that CHOP is an essential player in ER stress signaling and the downstream pathology including inflammation, oxidative stress, and autophagy block (Figure 6, F, H, and J, and Figure 1, C, D, I, and L).72 Importantly, inflammation, oxidative stress, and autophagy block have all been identified in OA cartilage.73,74 MMP-13, TNFα, IL-1β, and senescence marker (p16 INK4a) were primarily restricted to the superficial zone of the articular cartilage of I-ERS with ER stress, whereas pS6, which indicates block of autophagy, was in the intermediate zone (Figures 3 and 4). The implications for joint degeneration associated with these zonal differences are not clear. The chondrocytes in the superficial zone may have higher levels of inflammatory markers due to exposure to cytokines in the adjacent synovial fluid. The diminished levels of proteoglycans in the intermediate and deep layers may correlate with compromised matrix synthesis with the onset of ER stress. Mutant-COMP was not detectable in the I-ERS murine articular cartilage extracellular matrix, and type II collagen was not altered (data not shown). However, unlike established OA murine models, matrilin 3 and type IX collagen were less abundant in the articular cartilage of I-ERS mice and may enhance the pathologic process (data not shown). Loss of CHOP in the I-ERS/CHOP−/− mice largely ameliorated the joint destruction despite the continued expression of mutant COMP, demonstrating that the pathology was driven by ER stress signaling through CHOP and not the presence of misfolded protein (Figure 6). This work establishes that ER stress from the accumulation of a mutant protein drives joint degeneration in adult I-ERS mice.
Repetitive mechanical stress is a risk factor for developing OA in humans.75 These studies show that strenuous exercise exacerbated ER stress–induced joint degeneration (Figures 2, 3, 4, and 5), similar to that reported in another murine study.75 The combination of strenuous exercise and ER stress generated structural damage, with an OA score of 5.5 that was significantly higher than the ER stress alone OA score of 2.5 (P ≤ 0.0058). Proteoglycans were more abundant in the superficial zone compared with the transitional and deep zones of the articular cartilage in I-ERS + DOX and mechanical stress.
Running in this study was used as one proxy for pain. The I-ERS mice ran only 55% as much as controls, despite experiencing shock on the rest pad, which should stimulate the mouse to run. Interestingly, although I-ERS mice ran less than controls, mechanical stress substantially increased joint destruction, indicating that ER-stressed articular cartilage was more vulnerable to strenuous mechanical forces. Importantly, gait disturbances, also a proxy for pain, were observed with joint degeneration induced in the ER stress alone and ER stress with strenuous exercise groups. Irregular gait (increased ataxia coefficient) was found in the I-ERS mice (with ER stress and ± intense exercise) and is consistent with a shuffling gait reported in mice with OA.76 Moreover, ER and mechanical stress in I-ERS mice increased hind limb shared stance time (the amount of time that both hind limbs are on the treadmill belt simultaneously) suggesting the presence of hind limb pain associated with ambulation (Results). This finding is consistent with a study in rodents showing bilateral joint injuries correlated with increased limb shared stance time.26 Moreover, I-ERS gait results are similar to those reported for hip and knee OA in humans77 and with OA-like gait changes found with experimentally imposed pain in healthy human volunteers.43
The I-ERS mouse provides a noninvasive model system of primary OA to study the mechanisms and/or factors involved in the establishment, propagation, and late stages of OA diseases and as such has several advantages over other OA models. Three genetically predisposed mouse models, STR/ort, C57BL/6, and Del1, are used most frequently to study primary OA, and each has strengths and weaknesses. STR/ort mice are more susceptible to premature aging and PTOA due to increases in β-catenin and decreases in WNT signaling.78 Although a low level of WNT signaling has been shown to cause OA in STR/ort mice, elevated WNT signaling in both experimental and human OA exacerbates joint damage by inducing MMPs and aggrecanases.79 The STR/ort OA model shows that appropriate WNT signaling is necessary to maintain healthy cartilage. C57BL/6 mice are predisposed to age-related OA caused by a decrease in matrix gene expression and an increase in immune and defense responses similar to humans.80 The Del1 mice harbor a short deletion in type II collagen that compromises the integrity of the cartilage.81 In contrast to the Del1 mouse, the I-ERS mouse starts with healthy articular cartilage that is subsequently damaged by ER stress, providing a more physiological model. Although the STR/ort, C57BL/6, and Del1 OA models have been very useful in understanding some risk factors involved in primary OA, the I-ERS mouse provides a complementary approach to study ER stress–induced primary OA. An important advantage of the I-ERS mouse is the ability to turn on and off induction of ER stress (a physiological stress involved in OA), and this feature allows critical questions to be addressed: i) determining whether ER stress can be resolved after cessation of induction, ii) identifying whether there is a critical therapeutic intervention window in which ER stress can be halted and development of intervention strategies based on suppression of ER stress, and iii) identifying cellular mechanisms critical to recovery and defining factors involved in healing.
Joint degeneration in I-ERS mice was gradual, based on the presence of OA histological markers, but only limited structural damage was identified by lack of increased OA score after 4 weeks of ER stress. Longer periods of ER stress induction in I-ERS mice would likely induce structural degeneration. Both primary and PTOA develop over many years, and therefore, slower progression of joint degeneration in the I-ERS mouse is consistent with human OA. Because the I-ERS mouse requires weeks of ER stress induction to develop joint disease, it is most suitable for primary OA investigations rather than PTOA studies.
Development of OA therapies and/or effective prevention strategies is crucial because the prevalence of knee OA has doubled in the last 70 years, due to populations living longer and obesity rates rising.82 ER stress increases with aging because protein folding is compromised by declining levels of folding enzymes and chaperones,83,84 and by the presence of free fatty acids and reactive oxygen species in obesity.85 The connection between ER stress and aging and/or obesity, both OA risk factors, illustrates the necessity for a comprehensive understanding of the role of ER stress in primary OA in order to drive development of therapies that halt and/or reverse progression of joint degeneration.8,14,17,85,86 The I-ERS mouse model provides an important model system in which to obtain a more complete understanding of primary OA. The finding that resveratrol prevents ER stress–induced joint degeneration in the I-ERS mouse along with the observation that resveratrol prevents OA induced in animal models by type II diabetes,70 high-fat diet,53,54 monosodium iodoacetate,67 and surgery55 indicates that resveratrol may be able to prevent and/or ameliorate primary and PTOA.
Acknowledgment
We thank Dr. Joseph L. Alcorn for manuscript editing assistance and helpful discussions.
Author Contributions
J.T.H. oversaw the project and prepared the manuscript; A.C.V., J.W., F.Co., M.G.H., F.Ch., and F.H.G. did the experiments; K.L.P. managed the project, designed research, analyzed data, and prepared the manuscript; all authors read and approved the final manuscript. K.L.P. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIAMS) Award 5R01AR057117-10 (J.T.H. and K.L.P.), the Leah Lewis Family Foundation (J.T.H.), and The Rolanette and Berdon Lawrence Bone Disease Program of Texas (K.L.P.).
K.L.P. is first and senior author.
Disclosures: None declared.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2021.05.016.
Supplemental Data
Supplemental Figure S1.
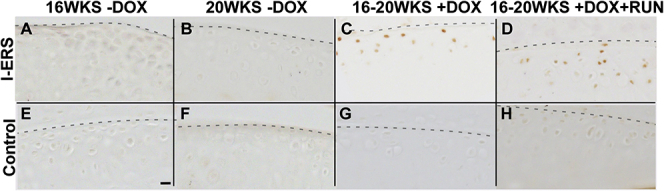
The endoplasmic reticulum (ER) stress marker, phosphorylated-eIF2α, is present in I-ERS mice administered doxycycline (DOX). Control (A–D) and I-ERS (E–H) tibial articular cartilage was immunostained using phosphor-eIF2α antibodies (16 and 20 weeks). Phosphor-eIF2α is not detected in control mice (E–H) or I-ERS mice in the absence of DOX (A–B), but is observed in articular chondrocytes of I-ERS mice administered DOX (C–D). Dashed line marks the margin of articular cartilage. Scale bar = 100 µm.
Supplemental Figure S2.
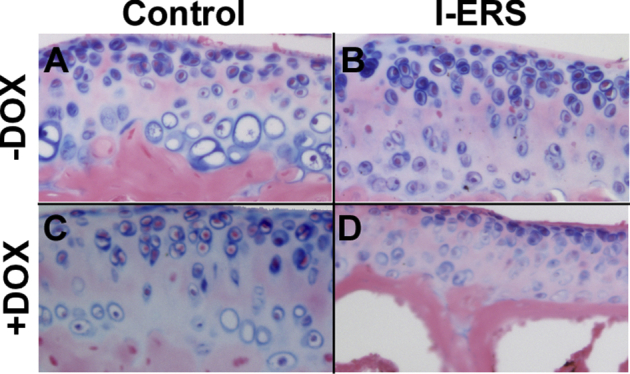
Proteoglycans were reduced in I-ERS mice administered doxycycline (DOX) from 16 to 20 weeks. Control (A and C) and I-ERS (B and D) tibias at 20 weeks ± DOX from 16 to 20 weeks were collected, fixed, deparaffinized, and stained with Alcian blue. Endoplasmic reticulum (ER) stress in I-ERS mice decreases Alcian blue staining in the articular cartilage. Original magnification, ×40.
References
- 1.Hawker G.A. Osteoarthritis is a serious disease. Clin Exp Rheumatol. 2019;37(Suppl 120):3–6. [PubMed] [Google Scholar]
- 2.Johnson V.L., Hunter D.J. The epidemiology of osteoarthritis. Best Pract Res Clin Rheumatol. 2014;28:5–15. doi: 10.1016/j.berh.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Neogi T., Zhang Y. Epidemiology of osteoarthritis. Rheum Dis Clin North Am. 2013;39:1–19. doi: 10.1016/j.rdc.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y., Jordan J.M. Epidemiology of osteoarthritis. Clin Geriatr Med. 2010;26:355–369. doi: 10.1016/j.cger.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishnan Y., Grodzinsky A.J. Cartilage diseases. Matrix Biol. 2018;71-72:51–69. doi: 10.1016/j.matbio.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes A., Oxford A.E., Tawara K., Jorcyk C.L., Oxford J.T. Endoplasmic reticulum stress and unfolded protein response in cartilage pathophysiology; contributing factors to apoptosis and osteoarthritis. Int J Mol Sci. 2017;18:665. doi: 10.3390/ijms18030665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bijlsma J.W., Berenbaum F., Lafeber F.P. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377:2115–2126. doi: 10.1016/S0140-6736(11)60243-2. [DOI] [PubMed] [Google Scholar]
- 8.Yamabe S., Hirose J., Uehara Y., Okada T., Okamoto N., Oka K., Taniwaki T., Mizuta H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 2013;280:1617–1629. doi: 10.1111/febs.12170. [DOI] [PubMed] [Google Scholar]
- 9.Ramos-Lopez O., Riezu-Boj J.I., Milagro F.I., Moreno-Aliaga M.J., Martinez J.A., project Mena Endoplasmic reticulum stress epigenetics is related to adiposity, dyslipidemia, and insulin resistance. Adipocyte. 2018;7:137–142. doi: 10.1080/21623945.2018.1447731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan L., Harper L., McNulty M.A., Carlson C.S., Yammani R.R. High-fat diet induces endoplasmic reticulum stress to promote chondrocyte apoptosis in mouse knee joints. FASEB J. 2020;34:5818–5826. doi: 10.1096/fj.201902746R. [DOI] [PubMed] [Google Scholar]
- 11.Fribley A., Zhang K., Kaufman R.J. Regulation of apoptosis by the unfolded protein response. Methods Mol Biol. 2009;559:191–204. doi: 10.1007/978-1-60327-017-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman R.J., Back S.H., Song B., Han J., Hassler J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in beta-cells. Diabetes Obes Metab. 2010;12(Suppl 2):99–107. doi: 10.1111/j.1463-1326.2010.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malhotra J.D., Kaufman R.J. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown M.K., Naidoo N. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. 2012;3:263. doi: 10.3389/fphys.2012.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim R., Emi M., Tanabe K., Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 16.Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R.T., Remotti H., Stevens J.L., Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takada K., Hirose J., Senba K., Yamabe S., Oike Y., Gotoh T., Mizuta H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int J Exp Pathol. 2011;92:232–242. doi: 10.1111/j.1365-2613.2010.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uehara Y., Hirose J., Yamabe S., Okamoto N., Okada T., Oyadomari S., Mizuta H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthritis Cartilage. 2014;22:1007–1017. doi: 10.1016/j.joca.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 19.Hamamura K., Lin C.C., Yokota H. Salubrinal reduces expression and activity of MMP13 in chondrocytes. Osteoarthritis Cartilage. 2013;21:764–772. doi: 10.1016/j.joca.2013.02.657. [DOI] [PubMed] [Google Scholar]
- 20.Mehana E.E., Khafaga A.F., El-Blehi S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: an updated review. Life Sci. 2019;234:116786. doi: 10.1016/j.lfs.2019.116786. [DOI] [PubMed] [Google Scholar]
- 21.Posey K.L., Veerisetty A.C., Liu P., Wang H.R., Poindexter B.J., Bick R., Alcorn J.L., Hecht J.T. An inducible cartilage oligomeric matrix protein mouse model recapitulates human pseudoachondroplasia phenotype. Am J Pathol. 2009;175:1555–1563. doi: 10.2353/ajpath.2009.090184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Posey K.L., Coustry F., Veerisetty A.C., Liu P., Alcorn J.L., Hecht J.T. Chop (Ddit3) is essential for D469del-COMP retention and cell death in chondrocytes in an inducible transgenic mouse model of pseudoachondroplasia. Am J Pathol. 2012;180:727–737. doi: 10.1016/j.ajpath.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posey K.L., Coustry F., Veerisetty A.C., Liu P., Alcorn J.L., Hecht J.T. Chondrocyte-specific pathology during skeletal growth and therapeutics in a murine model of pseudoachondroplasia. J Bone Miner Res. 2014;29:1258–1268. doi: 10.1002/jbmr.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glasson S.S., Chambers M.G., Van Den Berg W.B., Little C.B. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18(Suppl 3):S17–S23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 25.Vincelette J., Xu Y., Zhang L.N., Schaefer C.J., Vergona R., Sullivan M.E., Hampton T.G., Wang Y.X. Gait analysis in a murine model of collagen-induced arthritis. Arthritis Res Ther. 2007;9:R123. doi: 10.1186/ar2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs B.Y., Kloefkorn H.E., Allen K.D. Gait analysis methods for rodent models of osteoarthritis. Curr Pain Headache Rep. 2014;18:456. doi: 10.1007/s11916-014-0456-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coustry F., Posey K.L., Liu P., Alcorn J.L., Hecht J.T. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am J Pathol. 2012;180:738–748. doi: 10.1016/j.ajpath.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calvo E., Palacios I., Delgado E., Sanchez-Pernaute O., Largo R., Egido J., Herrero-Beaumont G. Histopathological correlation of cartilage swelling detected by magnetic resonance imaging in early experimental osteoarthritis. Osteoarthritis Cartilage. 2004;12:878–886. doi: 10.1016/j.joca.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Madry H., Luyten F.P., Facchini A. Biological aspects of early osteoarthritis. Knee Surg Sports Traumatol Arthrosc. 2012;20:407–422. doi: 10.1007/s00167-011-1705-8. [DOI] [PubMed] [Google Scholar]
- 30.Ruan M.Z., Erez A., Guse K., Dawson B., Bertin T., Chen Y., Jiang M.M., Yustein J., Gannon F., Lee B.H. Proteoglycan 4 expression protects against the development of osteoarthritis. Sci Transl Med. 2013;5:176ra34. doi: 10.1126/scitranslmed.3005409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei L., Fleming B.C., Sun X., Teeple E., Wu W., Jay G.D., Elsaid K.A., Luo J., Machan J.T., Chen Q. Comparison of differential biomarkers of osteoarthritis with and without posttraumatic injury in the Hartley guinea pig model. J Orthop Res. 2010;28:900–906. doi: 10.1002/jor.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin W., Park J.I., Loeser R.F. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J Biol Chem. 2009;284:31972–31981. doi: 10.1074/jbc.M109.056838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon H., Im G.I. Autophagy in osteoarthritis. Connect Tissue Res. 2017;58:497–508. doi: 10.1080/03008207.2016.1240790. [DOI] [PubMed] [Google Scholar]
- 34.Rabanal-Ruiz Y., Otten E.G., Korolchuk V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017;61:565–584. doi: 10.1042/EBC20170027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwenofu O.H., Lackman R.D., Staddon A.P., Goodwin D.G., Haupt H.M., Brooks J.S. Phospho-S6 ribosomal protein: a potential new predictive sarcoma marker for targeted mTOR therapy. Mod Pathol. 2008;21:231–237. doi: 10.1038/modpathol.3800995. [DOI] [PubMed] [Google Scholar]
- 36.Rim Y.A., Nam Y., Ju J.H. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int J Mol Sci. 2020;21:2358. doi: 10.3390/ijms21072358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Block J.A., Shakoor N. Lower limb osteoarthritis: biomechanical alterations and implications for therapy. Curr Opin Rheumatol. 2010;22:544–550. doi: 10.1097/BOR.0b013e32833bd81f. [DOI] [PubMed] [Google Scholar]
- 38.Zamli Z., Sharif M. Chondrocyte apoptosis: a cause or consequence of osteoarthritis? Int J Rheum Dis. 2011;14:159–166. doi: 10.1111/j.1756-185X.2011.01618.x. [DOI] [PubMed] [Google Scholar]
- 39.Lotz M., Loeser R.F. Effects of aging on articular cartilage homeostasis. Bone. 2012;51:241–248. doi: 10.1016/j.bone.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wohl G.R., Shymkiw R.C., Matyas J.R., Kloiber R., Zernicke R.F. Periarticular cancellous bone changes following anterior cruciate ligament injury. J Appl Physiol (1985) 2001;91:336–342. doi: 10.1152/jappl.2001.91.1.336. [DOI] [PubMed] [Google Scholar]
- 41.Grogan S.P., D'Lima D.D. Joint aging and chondrocyte cell death. Int J Clin Rheumtol. 2010;5:199–214. doi: 10.2217/ijr.10.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Favre J., Jolles B.M. Gait analysis of patients with knee osteoarthritis highlights a pathological mechanical pathway and provides a basis for therapeutic interventions. EFORT Open Rev. 2016;1:368–374. doi: 10.1302/2058-5241.1.000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henriksen M., Graven-Nielsen T., Aaboe J., Andriacchi T.P., Bliddal H. Gait changes in patients with knee osteoarthritis are replicated by experimental knee pain. Arthritis Care Res (Hoboken) 2010;62:501–509. doi: 10.1002/acr.20033. [DOI] [PubMed] [Google Scholar]
- 44.de Sa Coutinho D., Pacheco M.T., Frozza R.L., Bernardi A. Anti-inflammatory effects of resveratrol: mechanistic insights. Int J Mol Sci. 2018;19:1812. doi: 10.3390/ijms19061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenjian S., Moini A., Yamini N., Kashani L., Faridmojtahedi M., Bahramrezaie M., Khodarahmian M., Amidi F. Resveratrol treatment in patients with polycystic ovary syndrome decreased pro-inflammatory and endoplasmic reticulum stress markers. Am J Reprod Immunol. 2020;83:e13186. doi: 10.1111/aji.13186. [DOI] [PubMed] [Google Scholar]
- 46.Posey K.L., Coustry F., Veerisetty A.C., Hossain M., Alcorn J.L., Hecht J.T. Antioxidant and anti-inflammatory agents mitigate pathology in a mouse model of pseudoachondroplasia. Hum Mol Genet. 2015;24:3918–3928. doi: 10.1093/hmg/ddv122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie Y.K., Zhou X., Yuan H.T., Qiu J., Xin D.Q., Chu X.L., Wang D.C., Wang Z. Resveratrol reduces brain injury after subarachnoid hemorrhage by inhibiting oxidative stress and endoplasmic reticulum stress. Neural Regen Res. 2019;14:1734–1742. doi: 10.4103/1673-5374.257529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan D., Liu X.M., Fang Z., Du L.L., Chang J., Lin S.H. Protective effect of resveratrol on kidney in rats with diabetic nephropathy and its effect on endoplasmic reticulum stress. Eur Rev Med Pharmacol Sci. 2018;22:1485–1493. doi: 10.26355/eurrev_201803_14497. [DOI] [PubMed] [Google Scholar]
- 49.Gaballah H.H., Zakaria S.S., Elbatsh M.M., Tahoon N.M. Modulatory effects of resveratrol on endoplasmic reticulum stress-associated apoptosis and oxido-inflammatory markers in a rat model of rotenone-induced Parkinson's disease. Chem Biol Interact. 2016;251:10–16. doi: 10.1016/j.cbi.2016.03.023. [DOI] [PubMed] [Google Scholar]
- 50.Li Y., Luo B., Zhang L., Ma M., Guo X. [Resveratrol attenuates endoplasmic reticulum stress and alveolar epithelial apoptosis in a rat model of chronic obstructive pulmonary disease] Zhonghua Jie He He Hu Xi Za Zhi. 2014;37:30–35. [PubMed] [Google Scholar]
- 51.Yamamoto K., Okano H., Miyagawa W., Visse R., Shitomi Y., Santamaria S., Dudhia J., Troeberg L., Strickland D.K., Hirohata S., Nagase H. MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 2016;56:57–73. doi: 10.1016/j.matbio.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park D., Jeong H., Lee M.N., Koh A., Kwon O., Yang Y.R., Noh J., Suh P.-G., Park H., Ryu S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep. 2016;6:21772. doi: 10.1038/srep21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gu H., Li K., Li X., Yu X., Wang W., Ding L., Liu L. Oral resveratrol prevents osteoarthritis progression in C57BL/6J mice fed a high-fat diet. Nutrients. 2016;8:233. doi: 10.3390/nu8040233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang M., Li X., Yu X., Liu X., Xu X., He J., Gu H., Liu L. Oral administration of resveratrol alleviates osteoarthritis pathology in C57BL/6J mice model induced by a high-fat diet. Mediators Inflamm. 2017;2017:7659023. doi: 10.1155/2017/7659023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li W., Cai L., Zhang Y., Cui L., Shen G. Intra-articular resveratrol injection prevents osteoarthritis progression in a mouse model by activating SIRT1 and thereby silencing HIF-2alpha. J Orthop Res. 2015;33:1061–1070. doi: 10.1002/jor.22859. [DOI] [PubMed] [Google Scholar]
- 56.Thaung Zaw J.J., Howe P.R.C., Wong R.H.X. Long-term resveratrol supplementation improves pain perception, menopausal symptoms, and overall well-being in postmenopausal women: findings from a 24-month randomized, controlled, crossover trial. Menopause. 2020;28:40–49. doi: 10.1097/GME.0000000000001643. [DOI] [PubMed] [Google Scholar]
- 57.Wong R.H.X., Evans H.M., Howe P.R.C. Resveratrol supplementation reduces pain experience by postmenopausal women. Menopause. 2017;24:916–922. doi: 10.1097/GME.0000000000000861. [DOI] [PubMed] [Google Scholar]
- 58.Hussain S.A., Marouf B.H., Ali Z.S., Ahmmad R.S. Efficacy and safety of co-administration of resveratrol with meloxicam in patients with knee osteoarthritis: a pilot interventional study. Clin Interv Aging. 2018;13:1621–1630. doi: 10.2147/CIA.S172758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marouf B.H., Hussain S.A., Ali Z.S., Ahmmad R.S. Resveratrol supplementation reduces pain and inflammation in knee osteoarthritis patients treated with meloxicam: a randomized placebo-controlled study. J Med Food. 2018:1253–1259. doi: 10.1089/jmf.2017.4176. [DOI] [PubMed] [Google Scholar]
- 60.Ma P., Yue L., Yang H., Fan Y., Bai J., Li S., Yuan J., Zhang Z., Yao C., Lin M., Hou Q. Chondroprotective and anti-inflammatory effects of amurensin H by regulating TLR4/Syk/NF-kappaB signals. J Cell Mol Med. 2020;24:1958–1968. doi: 10.1111/jcmm.14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen C., Savouret J.F., Widerak M., Corvol M.T., Rannou F. Resveratrol, potential therapeutic interest in joint disorders: a critical narrative review. Nutrients. 2017;9:45. doi: 10.3390/nu9010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Latruffe N., Lancon A., Frazzi R., Aires V., Delmas D., Michaille J.J., Djouadi F., Bastin J., Cherkaoui-Malki M. Exploring new ways of regulation by resveratrol involving miRNAs, with emphasis on inflammation. Ann N Y Acad Sci. 2015;1348:97–106. doi: 10.1111/nyas.12819. [DOI] [PubMed] [Google Scholar]
- 63.Deng Z., Li Y., Liu H., Xiao S., Li L., Tian J., Cheng C., Zhang G., Zhang F. The role of sirtuin 1 and its activator, resveratrol in osteoarthritis. Biosci Rep. 2019;39 doi: 10.1042/BSR20190189. BSR20190189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dvir-Ginzberg M., Mobasheri A., Kumar A. Erratum to: the role of sirtuins in cartilage homeostasis and osteoarthritis. Curr Rheumatol Rep. 2016;18:48. doi: 10.1007/s11926-016-0606-8. [DOI] [PubMed] [Google Scholar]
- 65.Dvir-Ginzberg M., Mobasheri A., Kumar A. The role of sirtuins in cartilage homeostasis and osteoarthritis. Curr Rheumatol Rep. 2016;18:43. doi: 10.1007/s11926-016-0591-y. [DOI] [PubMed] [Google Scholar]
- 66.Wang J., Gao J.S., Chen J.W., Li F., Tian J. Effect of resveratrol on cartilage protection and apoptosis inhibition in experimental osteoarthritis of rabbit. Rheumatol Int. 2012;32:1541–1548. doi: 10.1007/s00296-010-1720-y. [DOI] [PubMed] [Google Scholar]
- 67.Wang Z.M., Chen Y.C., Wang D.P. Resveratrol, a natural antioxidant, protects monosodium iodoacetate-induced osteoarthritic pain in rats. Biomed Pharmacother. 2016;83:763–770. doi: 10.1016/j.biopha.2016.06.050. [DOI] [PubMed] [Google Scholar]
- 68.Qin N., Wei L., Li W., Yang W., Cai L., Qian Z., Wu S. Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J Pharmacol Sci. 2017;134:166–174. doi: 10.1016/j.jphs.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 69.Wei Y., Jia J., Jin X., Tong W., Tian H. Resveratrol ameliorates inflammatory damage and protects against osteoarthritis in a rat model of osteoarthritis. Mol Med Rep. 2018;17:1493–1498. doi: 10.3892/mmr.2017.8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ebrahim H.A., Alzamil N.M., Al-Ani B., Haidara M.A., Kamar S.S., Dawood A.F. Suppression of knee joint osteoarthritis induced secondary to type 2 diabetes mellitus in rats by resveratrol: role of glycated haemoglobin and hyperlipidaemia and biomarkers of inflammation and oxidative stress. Arch Physiol Biochem. 2020 doi: 10.1080/13813455.2020.1771378. [Epub ahead of print] doi: 10.1080/13813455.2020.1771378. [DOI] [PubMed] [Google Scholar]
- 71.Yuce P., Hosgor H., Rencber S.F., Yazir Y. Effects of intra-articular resveratrol injections on cartilage destruction and synovial inflammation in experimental temporomandibular joint osteoarthritis. J Oral Maxillofac Surg. 2021;79:344.e1–344.e12. doi: 10.1016/j.joms.2020.09.015. [DOI] [PubMed] [Google Scholar]
- 72.Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem. 2012;151:217–219. doi: 10.1093/jb/mvr143. [DOI] [PubMed] [Google Scholar]
- 73.Ansari M.Y., Haqqi T.M. Interleukin-1beta induced stress granules sequester COX-2 mRNA and regulates its stability and translation in human OA chondrocytes. Sci Rep. 2016;6:27611. doi: 10.1038/srep27611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duan R., Xie H., Liu Z.Z. The role of autophagy in osteoarthritis. Front Cell Dev Biol. 2020;8:608388. doi: 10.3389/fcell.2020.608388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buckwalter J.A., Anderson D.D., Brown T.D., Tochigi Y., Martin J.A. The roles of mechanical stresses in the pathogenesis of osteoarthritis: implications for treatment of joint injuries. Cartilage. 2013;4:286–294. doi: 10.1177/1947603513495889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allen K.D., Griffin T.M., Rodriguiz R.M., Wetsel W.C., Kraus V.B., Huebner J.L., Boyd L.M., Setton L.A. Decreased physical function and increased pain sensitivity in mice deficient for type IX collagen. Arthritis Rheum. 2009;60:2684–2693. doi: 10.1002/art.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pirker W., Katzenschlager R. Gait disorders in adults and the elderly: a clinical guide. Wien Klin Wochenschr. 2017;129:81–95. doi: 10.1007/s00508-016-1096-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pasold J., Engelmann R., Keller J., Joost S., Marshall R.P., Frerich B., Muller-Hilke B. High bone mass in the STR/ort mouse results from increased bone formation and impaired bone resorption and is associated with extramedullary hematopoiesis. J Bone Miner Metab. 2013;31:71–81. doi: 10.1007/s00774-012-0394-9. [DOI] [PubMed] [Google Scholar]
- 79.Wang Y., Fan X., Xing L., Tian F. Wnt signaling: a promising target for osteoarthritis therapy. Cell Commun Signal. 2019;17:97. doi: 10.1186/s12964-019-0411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rowe M.A., Harper L.R., McNulty M.A., Lau A.G., Carlson C.S., Leng L., Bucala R.J., Miller R.A., Loeser R.F. Reduced osteoarthritis severity in aged mice with deletion of macrophage migration inhibitory factor. Arthritis Rheumatol. 2017;69:352–361. doi: 10.1002/art.39844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCoy A.M. Animal models of osteoarthritis: comparisons and key considerations. Vet Pathol. 2015;52:803–818. doi: 10.1177/0300985815588611. [DOI] [PubMed] [Google Scholar]
- 82.Wallace I.J., Worthington S., Felson D.T., Jurmain R.D., Wren K.T., Maijanen H., Woods R.J., Lieberman D.E. Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc Natl Acad Sci U S A. 2017;114:9332–9336. doi: 10.1073/pnas.1703856114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martinez G., Duran-Aniotz C., Cabral-Miranda F., Vivar J.P., Hetz C. Endoplasmic reticulum proteostasis impairment in aging. Aging Cell. 2017;16:615–623. doi: 10.1111/acel.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Naidoo N. ER and aging-protein folding and the ER stress response. Ageing Res Rev. 2009;8:150–159. doi: 10.1016/j.arr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 85.Kawasaki N., Asada R., Saito A., Kanemoto S., Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. doi: 10.1038/srep00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H., Zhang X.Y., Wu T.J., Cheng W., Liu X., Jiang T.T., Wen J., Li J., Ma Q.L., Hua Z.C. Endoplasmic reticulum stress regulates rat mandibular cartilage thinning under compressive mechanical stress. J Biol Chem. 2013;288:18172–18183. doi: 10.1074/jbc.M112.407296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.