

Abstract
Intrahepatic cholangiocarcinoma (iCCA) is a lethal malignant neoplasm with limited therapeutic options. Previous studies have found that Notch1 overexpression alone suffices to induce iCCA in the mouse, albeit after long latency. The current study found that activation of the Yes-associated protein (Yap) proto-oncogene occurs during Notch1-driven iCCA progression. After co-expressing activated Notch1 intracellular domain (Nicd) and Yap (YapS127A) in the mouse liver, rapid iCCA formation and progression occurred in Nicd/Yap mice. Mechanistically, an increased expression of amino acid transporters and activation of the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway was detected in Nicd/Yap mouse liver tumors. Significantly, the genetic deletion of Raptor, the major mTORC1 component, completely suppressed iCCA development in Nicd/Yap mice. Elevated expression of Notch1, YAP, amino acid transporters, and members of the mTORC1 pathway was also detected ubiquitously in a collection of human iCCA specimens. Their levels were associated with a poor patient outcome. This study demonstrates that Notch and YAP concomitant activation is frequent in human cholangiocarcinogenesis. Notch and YAP synergize to promote iCCA formation by activating the mTORC1 pathway.
Intrahepatic cholangiocarcinoma (iCCA) is a deadly tumor and the second most common primary liver cancer type.1,2 Most cases of iCCA are diagnosed at advanced stages of the disease, when limited therapeutic options are available. The prognosis of patients with iCCA is dismal because of the late diagnosis, high tumor recurrence rate, and resistance to chemotherapy.1 Unlike the progress achieved in hepatocellular carcinoma treatment3,4 in the last decade, a combination of gemcitabine and platin-based drugs remains the first-line treatment for advanced iCCA not eligible for locoregional therapies or surgical resection. However, with median and 5-year overall survival remaining relatively poor at approximately 28 months (range, 9 to 53 months) and 30% (range, 5% to 56%), respectively, the benefit of this combinatorial treatment is almost negligible.5,6 Therefore, the development of more effective therapeutic strategies against this lethal tumor is imperative.1,2 Recently, pemigatinib, a soluble inhibitor of fibroblast growth factor receptor (FGFR) 1, FGFR2, and FGFR3, received accelerated approval by the US Food and Drug Administration for the treatment of adults with previously treated, unresectable, locally advanced, or metastatic iCCA harboring FGFR2 fusions or other rearrangements.7 The approval of pemigatinib and the encouraging benefits of this drug for patients with iCCA indicate that successful therapeutic approaches against advanced iCCA can be achieved. Thus, considerable efforts need to be directed toward the identification of relevant molecular targets in this disease.8
Recent high-throughput genomic and transcriptomic studies have uncovered the genetic and epigenetic landscape of iCCA,9, 10, 11, 12, 13 providing novel insights into the pathways that lead to cholangiocarcinogenesis. In particular, the Notch and Hippo cascades have been identified as two key signaling pathways driving iCCA development and progression. The Notch family consists of four distinct receptors (Notch1 through 4) and two types of ligands (Jagged1/2 and DLL1/3/4).14,15 When the Notch signaling is activated, the Notch intracellular domain (NICD) is cleaved and released to the nucleus to initiate the expression of its downstream target genes.15,16 The Notch pathway plays a vital role in cholangiocyte differentiation, bile duct development,17,18 and cholangiocarcinogenesis.19,20 The overexpression of Notch receptors and ligands, such as Jagged1, has been reported in human iCCA.19,20 Moreover, using genomic data from a vast iCCA collection, a recent study identified a Notch1 signature in human iCCA, suggesting the use of γ-secretase inhibitors to treat this subgroup of patients.21 The oncogenic role of the Notch pathway in iCCA has been investigated and validated in animal models.19,20 Indeed, this study found that overexpression of Nicd1 alone suffices to induce iCCA over long latency, and it synergizes with activated AKT signaling to drive rapid iCCA formation in mice.22,23 Other groups subsequently reported similar results by overexpressing either Notch1 or the other Notch receptors.24, 25, 26, 27, 28
The Yes-associated protein (YAP) is a major downstream effector of the Hippo pathway. YAP acts as a transcription factor in regulating genes involved in cell proliferation and apoptosis, profoundly influencing organ size, cell renewal, and tumor development.29, 30, 31 Multiple studies have demonstrated the activation of YAP in human iCCA samples.32,33 In particular, YAP levels predicted poor prognosis in patients with iCCA,34,35 and YAP silencing in human iCCA cells resulted in growth inhibition.36,37 Moreover, YAP regulates genes that modulate proliferation, apoptosis, and angiogenesis in iCCA cells.38 A recent study found that overexpression of an activated form of YAP (YAPS127A) alone in the liver cannot induce histologic alterations in mice. In contrast, co-expression of YAPS127A with activated AKT promotes fast iCCA development in vivo.26,37,39
The mammalian target of rapamycin (mTOR) cascade is a pivotal player in regulating cell growth and metabolism in response to growth factors and nutrients via phosphorylation of its downstream effectors.40,41 Previous studies have demonstrated the critical function of the mTOR pathway in iCCA formation and progression.42, 43, 44 An earlier investigation found that mTOR inhibition restrains iCCA cell growth in vitro and possesses antitumor activities in the AKT/YAP iCCA mouse model in vivo.37 Notably, the pan-mTOR inhibitor MLN0128 combined with the CDK4/6 inhibitor palbociclib markedly suppressed AKT/YAP iCCA tumor growth.39
In the current study, the expression and molecular crosstalk between the YAP and Notch pathways using mouse models and human specimens were investigated. Co-expression of activated forms of YAP and Notch1 triggered rapid iCCA formation in a mTOR complex 1 (mTORC1)–dependent manner in mice. In human iCCA specimens, the YAP, Notch1, and mTOR pathways were coordinately activated, especially in the most biologically aggressive tumors. Thus, the Nicd/Yap model might represent a valuable tool for the study of human iCCA.
Materials and Methods
Mouse, Plasmids, and Hydrodynamic Tail Vein Injection
Wild-type FVB/N mice and Raptorfl/fl mice were from the Jackson Laboratory (Sacramento, CA). Mice were housed and monitored following protocols approved by the Committee for Animal Research at the University of California, San Francisco. The plasmids, including pT3EF1α-NICD (MYC tagged), pT3EF1α-YAPS127A, pCMV, pCMV-Cre (Cre), and pCMV–sleeping beauty transposase (SB), have been described previously in detail.26,37,45,46 Briefly, 20 μg of pT3EF1α-NICD and pT3EF1α-YAPS127A constructs and the SB plasmid at the ratio of 25:1 to all oncogenes were diluted in 2 mL of 0.9% sodium chloride solution and injected into the tail vein of 20-g FVB/N mice in approximately 5 seconds. Concerning Raptor ablation experiments, 20 μg of pT3EF1α-NICD and pT3EF1α-YAPS127A, with 60 μg of pCMV or Cre, and SB plasmids were injected into Raptorfl/fl mice. All plasmids were extracted using the Endotoxin-free Maxi prep kit (Sigma-Aldrich, St. Louis, MO) before injection. Mice were sacrificed at indicated time points or when mice became moribund or developed large abdominal masses.
Protein Extraction and Western Blot Analysis
Mouse liver tissues and human cell lines were homogenized in mammalian protein extraction reagent (catalog number 78503; Thermo Fisher Scientific, Waltham, MA) plus protease inhibitor cocktail (catalog number 78440; Thermo Fisher Scientific). Proteins were quantified with the BCA assay (catalog number 23225; Thermo Fisher Scientific). The antibodies used in this study are listed in Table 1.
Table 1.
Antibody List
| Antibody | Source | Catalog no. | Species | Application | Dilution | Retrieval buffer |
|---|---|---|---|---|---|---|
| p-AKTS473 | Cell Signaling Technology (Danvers, MA) | 9271 | Mouse | Western blot | 1:1000 | NA |
| Total AKT | Cell Signaling Technology | 9272 | Mouse | Western blot | 1:1000 | NA |
| mTOR | Cell Signaling Technology | 2983 | Mouse | Western blot | 1:1000 | NA |
| p-4EBP1 | Cell Signaling Technology | 9451 | Mouse | Western blot | 1:1000 | NA |
| 4EBP1 | Cell Signaling Technology | 9644 | Mouse | Western blot | 1:1000 | NA |
| p-RPS6 | Cell Signaling Technology | 4858 | Mouse | Western blot | 1:1000 | NA |
| RPS6 | Cell Signaling Technology | 2217 | Mouse | Western blot | 1:1000 | NA |
| p-ERK1/2 | Cell Signaling Technology | 9101 | Mouse | Western blot | 1:1000 | NA |
| Total ERK1/2 | Cell Signaling Technology | 9102 | Mouse | Western blot | 1:1000 | NA |
| YAP | Cell Signaling Technology | 14074 | Mouse | Western blot | 1:1000 | NA |
| Notch1 | Cell Signaling Technology | 4380 | Mouse | Western blot | 1:1000 | NA |
| MYC tagged | Cell Signaling Technology | 2278 | Mouse | Western blot | 1:1000 | NA |
| RICTOR | Cell Signaling Technology | 2114 | Mouse | Western blot | 1:500 | NA |
| RAPTOR | Cell Signaling Technology | 2280 | Mouse | Western blot | 1:1000 | NA |
| GAPDH | Cell Signaling Technology | 5174 | Mouse | Western blot | 1:3000 | NA |
| CK19 | Abcam (Cambridge, UK) | ab181604 | Mouse | Immunohistochemistry | 1:800 | Sodium citrate |
| HNF4α | Abcam | ab181604 | Mouse | Immunohistochemistry | 1:2000 | Tris-EDTA |
| SOX9 | Abcam | ab185230 | Mouse | Immunohistochemistry | 1:2000 | Sodium citrate |
| Ki-67 | Cell Signaling Technology | 12202 | Mouse | Immunohistochemistry | 1:150 | Sodium citrate |
| p-ERK1/2 | Cell Signaling Technology | 4370 | Mouse | Immunohistochemistry | 1:200 | Sodium citrate |
| p-mTOR Ser2448 | Cell Signaling Technology | 2976 | Mouse | Immunohistochemistry | 1:150 | Sodium citrate |
| MYC-tagged | Cell Signaling Technology | 2278 | Mouse | Immunohistochemistry | 1:100 | Sodium citrate |
| CK19 | Cell Signaling Technology | 12434 | Human | Immunohistochemistry | 1:500 | Sodium citrate |
| RAPTOR | Abcam | ab40768 | Human | Immunohistochemistry | 1:200 | Sodium citrate |
| p-4EBP1 | Cell Signaling Technology | 2855 | Human | Immunohistochemistry | 1:300 | Sodium citrate |
| Notch1 | Lifespan Biosciences (Seattle, WA) | LS-C114369 | Human | Immunohistochemistry | 1:100 | Sodium citrate |
| YAP | Cell Signaling Technology | 14074 | Human | Immunohistochemistry | 1:200 | Sodium citrate |
| p-YAP | Cell Signaling Technology | 13008 | Human | Immunohistochemistry | 1:300 | Sodium citrate |
| CTGF | Atlas Antibodies (Stockholm, Sweden) | AMAb91366 | Human | Immunohistochemistry | 1:100 | Sodium citrate |
CK, cytokeratin; CTGF, connective tissue growth factor; ERK, extracellular signal-regulated kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HNF4α, hepatocyte nuclear factor 4α; mTOR, mammalian target of rapamycin; NA, not applicable; p-, phosphorylated; MYC tagged, pT3EF1α-NICD; YAP, Yes-associated protein.
Histopathologic Analysis
Two experienced pathologists and liver experts (M.E. and S.R.) analyzed hematoxylin and eosin–stained liver sections based on the criteria established by Frith and Ward.47 Cholangiomas, alias biliary adenomas, are benign cholangiocellular lesions that compress the surrounding parenchyma but lack cytologic atypia, substantial mitotic/apoptotic activity, necrosis, infiltration/invasion, and fibrosis. When cystic, cholangiomas are referred to as cystic cholangiomas or cystadenomas. Cholangiocellular lesions characterized by cytologic atypia and extensive proliferation are defined as intracystic papillary neoplasms with intraepithelial neoplasia. If these neoplasms result from malignant transformation of benign cystadenomas, they are referred to as cystadenocarcinomas. Cystadenocarcinomas that invade the surrounding liver tissue resemble iCCA. They consist of atypical tumor cells that form ductules, glands, or solid sheets, cause local or vascular invasion, and have the potential for metastasis. In the absence of these features, severe cytologic alterations (nuclear pleomorphism, prominent nucleoli, basophilic cytoplasm, nuclear hyperchromasia, or high nuclear/cytoplasmic ratio), pronounced mitotic and apoptotic activity, necrosis, and desmoplastic stromal reaction or fibrosis indicate iCCA.
Immunohistochemistry
Liver samples were fixed in 10% formalin and embedded in paraffin. The hematoxylin and eosin staining was performed following the standard procedure, and immunohistochemistry was conducted as previously described.23,48,49 Briefly, 5-μm mouse or human liver sections were deparaffinized and rehydrated. Next, the slides went through antigen retrieval in sodium citrate buffer (pH 6.0) or Tris-EDTA (pH 9.0) for 10 minutes in the microwave. Subsequently, 10% goat serum and Avidin-Biotin (catalog number SP-2001; Vector Laboratories, Burlingame, CA) blocking steps were performed after quenching the endogenous peroxidase by incubating sections with 3% H2O2. Slides were incubated with primary antibodies (Table 1) overnight at 4°C. After washes, slides were incubated with a secondary antibody (catalog number B2770; Thermo Fisher Scientific) and Vectastain Elite ABC (catalog number PK-6100; Vector Laboratories) at room temperature. Finally, the slides were stained with DAB (catalog number SK-4105; Vector Laboratories).
RT-qPCR
Total RNA was extracted from frozen liver tissues and cells and transcribed to cDNA with Reverse Transcription Supermix (catalog number 1708841; Bio-Rad Laboratories, Hercules, CA). In mouse tissues, mRNA expression was determined by quantitative real-time RT-PCR (RT-qPCR) using the SYBR Green Master Mix (catalog number 1725124; Bio-Rad Laboratories) in a Quant Studio 6 Flex system (Applied Biosystems, Rockford, IL). Thermal cycling conditions included an initial hold period at 95°C for 10 minutes followed by a three-step PCR program of 95°C for 15 seconds, 60°C for 1 minute, and 72°C for 30 seconds for 40 cycles. Each gene expression was normalized to the normal mouse liver with 18S rRNA using the −ΔΔCt method. The mouse primer sequences are listed in Table 2. For human iCCA samples, gene expression assays for human NOTCH1 (Hs01062014_m1), YAP1 (Hs00902712_g1), GLS1 (Hs01005622_m1), HES1 (Hs00172878_m1), CTGF/CCN2 (Hs00170014_m1), SLC1A5 (Hs01056542_m1), SLC38A1 (Hs01562175_m1), RAPTOR (Hs00375332_m1), and GAPDH (Hs02786624_g1) genes were purchased from Applied Biosystems (Foster City, CA). Quantitative values for each gene were calculated by using the 7500 Analysis Software version 2.0.6 (Applied Biosystems) and expressed as number target (NT): NT = 2−ΔCt, wherein the ΔCt value of each sample was calculated by subtracting the mean Ct value of the target gene from the mean Ct value of the GAPDH gene.
Table 2.
Primers for Quantitative Real-Time RT-PCR
| Gene | Forward | Reverse |
|---|---|---|
| 18S rRna | 5′-CGGCTACCACATCCAAGGAA-3′ | 5′-GCTGGAATTACCGCGGCT-3′ |
| Hes1 | 5′-AAAGCCTATCATGGAGAAGAGGCG-3′ | 5′-GGAATGCCGGGAGCTATCTTTCTT-3′ |
| Hes5 | 5′-AAGCGCCCTTGCGAGGAAAC-3′ | 5′-GGTAGTTGTCGGTGAATTGGAC-3′ |
| HeyL | 5′-CAGCCCTTCGCAGATGCAA-3′ | 5′-CCAATCGTCGCAATTCAGAAAG-3′ |
| Hey1 | 5′-GCGCGGACGAGAATGGAAA-3′ | 5′-TCAGGTGATCCACAGTCATCTG-3′ |
| Hey2 | 5′-AAGCGCCCTTGTGAGGAAAC-3′ | 5′-GGTAGTTGTCGGTGAATTGGAC-3′ |
| Ctgf | 5′-GGGCCTCTTCTGCGATTTC-3′ | 5′-ATCCAGGCAAGTGCATTGGTA-3′ |
| Cyr61 | 5′-CTGCGCTAAACAACTCAACGA-3′ | 5′-GCAGATCCCTTTCAGAGCGG-3′ |
| Slc38a1 | 5′-AGCAACGACTCTAATGACTTCAC-3′ | 5′-CCTCCTACTCTCCCGATCTGA-3′ |
| Slc7a5 | 5′-CTACGCCTACATGCTGGAGG-3′ | 5′-GAGGGCCGAATGATGAGCAG-3′ |
| Slc1a5 | 5′-TTCGCTATCGTCTTTGGTGTG-3′ | 5′-ATGGTGGCATCATTGAAGGAG-3′ |
| Sox9 | 5′-ACTCTGGGCAAGCTCTGGAG-3′ | 5′-GGAAGGGTCTCTTCTCGCTCT-3′ |
| Ck19 | 5′-GGGGGTTCAGTACGCATTGG-3′ | 5′-GAGGACGAGGTCACGAAGC-3′ |
| Epcam | 5′-GCGGCTCAGAGAGACTGTG-3′ | 5′-CCAAGCATTTAGACGCCAGTTT-3′ |
Human Liver Tissue Specimens
Human iCCA samples (n = 132) were collected at the Medical University of Greifswald (Greifswald, Germany) and the Medical University of Regensburg (Regensburg, Germany). Institutional review board approval was obtained at the local ethical committees of the Medical University of Greifswald (approval BB 67/10) and the Medical University of Regensburg (approval 16-316-101). Informed consent was obtained from all individuals. Table 3 summarizes the available clinicopathologic data of the patients.
Table 3.
Clinicopathologic Features of Patients with Intrahepatic Cholangiocarcinoma (iCCA)
| Feature | n |
|---|---|
| Sex | |
| Male | 30 |
| Female | 20 |
| Age, y | |
| <60 | 14 |
| ≥60 | 36 |
| Cause | |
| HBV | 12 |
| HCV | 8 |
| Hepatolithiasis | 8 |
| PSC | 2 |
| NA | 20 |
| Liver cirrhosis | |
| Yes | 21 |
| No | 29 |
| Tumor differentiation | |
| Well | 22 |
| Moderately | 20 |
| Poorly | 8 |
| Tumor size, cm | |
| <5 | 35 |
| ≥5 | 15 |
| Tumor number | |
| Single | 13 |
| Multiple | 37 |
| Prognosis | |
| Better (≥3 years) | 14 |
| Poorer (<3 years) | 36 |
| Lymph node metastasis | |
| Yes | 18 |
| No | 32 |
| Lung metastasis | |
| Yes | 8 |
| No | 42 |
HBV, hepatitis B virus; HCV, hepatitis C virus; NA, not available; PSC, primary sclerosing cholangitis.
Cell Lines
Normal human cholangiocytes (NHC-SS and C324) were isolated from normal liver tissues collected from patients with local hepatic adenomas or colon metastasis. They were characterized for biliary markers and grown in fully supplemented Dulbecco’s modified Eagle’s medium/F-12 medium as previously described.46 HuccT1, RBE (Riken, Tsukuba, Japan), and KKUM-213 (Sekisui XenoTech LLC, Kansas City, KS) human iCCA cell lines were grown following the protocols provided by the companies.
Statistical Analysis
All data are presented as means ± SD. Statistical analysis was performed using a two-tailed unpaired t-test for comparisons between two groups. Low and high mRNA levels of the investigated genes in iCCA samples were recoded into binary variables (0/1) using the corresponding median value as the cutoff. Kaplan-Meier curves were used for survival analysis, and P values were assessed using the log-rank test (GraphPad Prism version 8.0; GraphPad Inc., San Diego, CA). P < 0.05 was considered to be statistically significant.
Results
Activation of Hippo/YAP, ERK/MAPK, and mTOR Pathways during Notch1-Driven Cholangiocarcinoma Progression in Mice
A previous study indicated for the first time that overexpression of an activated/cleaved form of Notch1 (Nicd1) in the liver of mice (referred to as Nicd) suffices to induce iCCA development, albeit after long latency.22 Multiple cysts lined by flat or cuboidal cytologically benign cells were detected as early as 20 weeks after hydrodynamic injection in these mice (Figure 1, A and C). These benign lesions, classified as cystadenomas, exhibited positive immunoreactivity for MYC-tagged Notch1 and the biliary marker cytokeratin 19 (CK19) (Figure 1, E and G). By 29 to 33 weeks after injection, cystadenomas were replaced by cystic lesions that consisted of micropapillary proliferation of atypical cells (cystadenocarcinomas) and solid and highly invasive iCCA, often coexisting in the same liver (Figure 1, B, D, F, and H). When looking at other pathways relevant in cholangiocarcinogenesis, including the YAP/Hippo, extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK), and mTOR cascades, low or absent immunoreactivity was found for nuclear YAP, phosphorylated/activated (p-)ERK1/2, and p-mTOR proteins in cystadenoma lesions (Figure 2, A, C, and E). Notably, the same proteins were instead strongly expressed in the advanced lesions (cystadenocarcinomas and iCCA) of Nicd mice (Figure 2, B, D, and F). Thus, activation of the YAP/Hippo, ERK/MAPK, and mTOR cascades occurs at the progression stage of Nicd-induced cholangiocarcinogenesis.
Figure 1.

Overview of Notch1 intracellular domain (Nicd)–induced cholangiocarcinogenesis. A–D: Hematoxylin and eosin (HE) staining of early [20 weeks post injection (w.p.i.), A and C] and late (32 w.p.i., B and D) cholangiocellular lesions developed in the mouse liver after hydrodynamic gene delivery of the activated/cleaved Nicd plasmid. Representative cystadenoma lesion (arrows) consisting of multiple cysts (A). Higher magnification image showing the cystic formations lined by a flat or cuboidal epithelium composed of cytologically benign cells (C). At later time points, invasive cystadenocarcinoma (CAK) and intrahepatic cholangiocarcinomas (iCCA), often co-expressing, were detected on the liver surface of Nicd-overexpressing mice (B). At high magnification, the iCCA appears to be composed of highly malignant cells and frequent mitoses (D). E–H: Both early and late lesions display positive immunoreactivity for the biliary marker cytokeratin 19 (CK19) (E) and the injected pT3EF1α-NICD (Myc-tagged) plasmid (H). Scale bars = 200 μm (A and B); 50 μm (C–H). Original magnification: ×40 (A and B); ×400 (C–H).
Figure 2.

Staining pattern of Hippo/Yes-associated protein (YAP), Ras/mitogen-activated protein kinase (MAPK), and mammalian target of rapamycin (mTOR) pathways in Notch1 intracellular domain (Nicd)–induced cholangiocarcinogenesis. Both early [20 weeks post injection (w.p.i,), A, C, and E] and late (32 w.p.i., B, D, and F) cholangiocellular lesions from Nicd mice depicted in Figure 1 are shown. Although no immunoreactivity for nuclear YAP, phosphorylated/activated extracellular signal-regulated kinase (pERK1/2), and phosphorylated/activated mTOR-Ser2448 was detected in early lesions (A, C, and E), the advanced lesions displayed robust immunoreactivity for the same antibodies (B, D, and F). The pERK staining was used as a surrogate marker of Ras/MAPK pathway activation. Scale bars = 50 μm; Original magnification, ×400.
Co-Expression of Activated Yap or NRas Accelerates Notch1 Driven Mouse Cholangiocarcinoma Development
The data reported above suggest that the YAP/Hippo, ERK/MAPK, and mTOR pathways might contribute to the malignant conversion and tumor progression of the early lesions induced by Nicd overexpression. To validate this hypothesis, this study overexpressed Nicd alone or in association with an oncogenic form of Ras (NRasV12) or an activated form of Yap (YAPS127A) in the mouse liver via hydrodynamic tail vein injection (Figure 3A). In accordance with previous data from the study laboratory, overexpression of YapS127A or NRasV12 oncogenes alone did not lead to any alteration in the liver parenchyma by 42 weeks after injection.33,37 In striking contrast, co-expression of Nicd with NRasV12 or YAPS127A in the mouse liver (referred to as Nicd/NRas and Nicd/Yap mice, respectively) resulted in accelerated iCCA development when compared with mice overexpressing Nicd alone. Specifically, Nicd/NRas mice exhibited a slight acceleration of cholangiocarcinogenesis and needed euthanasia because of a high tumor burden by 19 to 25 weeks after injection. A more substantial acceleration of tumor development was detected in Nicd/Yap mice, in which a high tumor burden occurred as early as 10 to 13 weeks after injection (Figure 3, B and C). Proliferation was most elevated in the tumor lesion from Nicd/Yap mice, intermediate in Nicd/NRas mice, and lowest in Nicd mice (Figure 3E), as assessed by Ki-67 immunohistochemistry. Nicd/Ras mouse lesions were virtually identical to those formed in Nicd mice at the histomorphologic level, consisting initially of cystadenomas and subsequently of cystadenocarcinomas and invasive iCCA (Figure 3D and Supplemental Figure S1). These data indicate that NRasV12 co-expression (and the consequent ERK/MAPK activation) slightly accelerates Notch1-induced cholangiocarcinogenesis without modifying the lesions' phenotype.
Figure 3.

Co-overexpression of activated forms of Notch1 intracellular domain (Nicd) and Ras (NRasV12) or Yes-associated protein (Yap) (YAPS127A) cooperates for the formation of intrahepatic cholangiocarcinoma (iCCA) in mice. A: Experimental study design. B: Survival curve of the various mouse models tested. C: Liver weight/body weight ratio of the mouse models investigated. D: Representative histopathologic features of cholangiocellular lesions developed by a Nicd/Ras mouse 23 weeks post injection (w.p.i.). The lesions are indistinguishable from those developing in Nicd mice. Indeed, both invasive cystadenocarcinomas (CAK) and intrahepatic cholangiocarcinoma (iCCA, also shown in the top middle panel at higher magnification) can be concomitantly observed. The lesions display immunoreactivity for the injected Nicd plasmid (Myc tagged), phosphorylated/activated extracellular signal-regulated kinase (a surrogate marker of Ras/mitogen-activated protein kinase pathway activation), and the biliary markers cytokeratin (CK) 19 and 7. E: Proliferation rate of the tumor lesions developed in Nicd, Nicd/Ras, and Nicd/Yap mice, as assessed by Ki-67 index. Representative Ki-67 staining in the various models is also shown. Data are expressed as means ± SD. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001. Scale bars = 100 μm. Original magnification: ×40 (D, top left panel); ×200 (D, all other panels, and E); HE, hematoxylin and eosin.
Morphologic and Molecular Aspects of Nicd/Yap-Driven Mouse Cholangiocarcinogenesis
Next, the histopathologic features of the lesions that developed in mouse livers co-expressing Nicd and YAPS127A oncogenes (Nicd/Yap mice) were analyzed in detail (Figure 4). In these mice, most lesions developed as small microcystic tumors with flat benign epithelia, resembling the initial liver lesions detected in Nicd and Nicd/Ras mice (Figure 4A). The stromal component of some of these lesions revealed the presence of inflammatory cells (Figure 4B), consisting of lymphocytes, neutrophils, and plasma cells (Supplemental Figure S2). These cells disappeared during carcinogenesis. Although most of these lesions increased in size without alteration of the cytology (Figure 4C), some of them evolved into an intracystic (yet noninvasive) micropapillary proliferation of atypical cells. They eventually became invasive, with the septa between the cysts infiltrated by invasive iCCA (Figure 4, D–G). Concomitantly with these lesions, a second type of lesions developed into iCCA without an intermediate step of cystic lesions. They were less frequent than the first type of lesions and consisted of malignant cells with atypical nuclei (Figure 4, H and I). As expected, both types of lesions exhibited intense immunoreactivity for the injected proto-oncogenes (MYC-tagged NICD and YAP) (Figure 5). Moreover, they were positive for the CK19 cholangiocellular maker and were proliferating, as suggested by positive immunolabeling for Ki-67 (Figure 6A). At the molecular level, real-time RT-qPCR analysis of Sox9, Epcam, and Ck19 revealed a high expression of these biliary epithelium markers in Nicd/Yap tumors when compared with healthy livers (Supplemental Figure S3). The same tumor cells stained negative for hepatocyte nuclear factor 4α (Figure 6A), confirming their cholangiocellular nature.
Figure 4.

Histopathologic characterization of cholangiocellular lesions developing in Notch1 intracellular domain (Nicd)/Yes-associated protein (Yap) (YapS127A) mice, as revealed by hematoxylin and eosin (HE) staining. A: Most of the lesions developed as small microcysts lined by a flat benign epithelium, often increasing in size but retaining their benign appearance. B and C: The stromal component of these lesions was rich in inflammatory cells, which disappeared along with tumor progression. D–F: Although most of these lesions increased in size without altering the cytologic features, some evolved into an intracystic micropapillary proliferation of atypical cells (boxed area in D) and became eventually invasive (F), with the septa between the cysts being infiltrated by invasive intrahepatic cholangiocarcinoma (iCCA). G: Often the invasive iCCA destroyed the cystic architecture of the more benign lesions. H and I: Concomitantly with these lesions, a second type of lesions developed into iCCA without an intermediate step of cystic lesions. These tumor lesions consisted of malignant cells with atypical nuclei. Scale bars: 50 μm (C and E); 100 μm (A, B, F, H, and I); 500 μm (G); 1000 μm (D). Original magnification: ×20 (D); ×40 (G); ×200 (A, B, F, H, and I); ×400 (C and E).
Figure 5.

Cholangiocellular lesions developed in Notch1 intracellular domain (Nicd)/Yes-associated protein (Yap) (YapS127A) mice robustly express the injected proto-oncogenes. Representative overview of the lesions formed in a Nicd/YapS127A mouse 8 weeks post injection (w.p.i.) exhibiting positive nuclear and/or cytoplasmic immunoreactivity for pT3EF1α-NICD (MYC-tagged) NICD and YAP proteins. Both the cystic lesions (C) and the intrahepatic cholangiocarcinoma lesions (iCCA) display similar, intense immunolabeling for the two injected proto-oncogenes. The boxed area in the top left panel is shown at higher magnification in the bottom row. Scale bars: 500 μm (top row); 100 μm (bottom row). Original magnification: ×40 (top row); ×200 (bottom row). HE, hematoxylin and eosin.
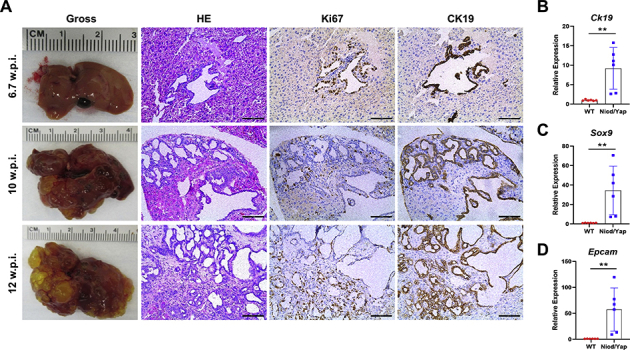
Figure 6.

Chronological and molecular characterization of the lesions developed in Notch1 intracellular domain (Nicd)/Yes-associated protein (Yap) (YapS127A) mice. A: Representative results of the gross and microscopic images of the tumor lesions at different weeks post injection (w.p.i.). Tumor lesions were positive for the cholangiocyte epithelial markers cytokeratin (CK) 19 and SOX9 and negative for the hepatocyte marker hepatocyte nuclear factor 4α (HNF4α), respectively. B: Relative expression of Yap downstream target genes (Cyr61 and Ccn2) in Nicd/Yap of end-stage tumors and wild-type (WT) livers. C: Relative expression of Notch downstream target genes (Hey1, Hey2, HeyL, Hes1, and Hes5) in Nicd/Yap of end-stage tumors and WT livers. Each gene expression was normalized to WT mouse livers with 18S rRNA using the −ΔΔCt method. Data are expressed as means ± SD. ∗P < 0.05, ∗∗P < 0.01 (t-test). Scale bars: 100 μm (second column); 50 μm (third, fourth, fifth, and sixth columns). Original magnification: ×200 (second column); ×400 (third, fourth, fifth, and sixth columns).
To ensure the activation of the Notch and Yap pathways in Nicd/Yap liver lesions, this study assessed the expression of the canonical downstream targets of these two signaling cascades by RT-qPCR. The expression of Cyr61 and Ctgf/Ccn2, two downstream effectors of Yap, were significantly higher in Nicd/Yap iCCA than in healthy livers (Figure 6B). Similarly, canonical Notch targets, including Hey1, Hey2, HeyL, Hes1, and Hes5, were also markedly up-regulated in Nicd/Yap tumors (Figure 6C). Altogether, the data indicate that cholangiocellular lesions, and not hepatocellular lesions, develop in Nicd/Yap mice. These lesions exhibit the activation of the Notch and Yap pathways.
Inactivation of mTORC1 Prevents Cholangiocarcinogenesis in Nicd/Yap Mice
YAP regulates glutamine metabolism via inducing the expression of amino acid transporters50,51 and glutaminase (GLS, alias GLS1).52 Thus, this study examined the expression of Slc1a5, Slc7a5, and Slc38a1 amino acid transporters and Gls1 in Nicd/Yap iCCA tissues. Notably, mouse iCCA samples displayed the up-regulation of all these genes (Figure 7, A and B). Because increased uptake of amino acids into tumor cells induces mTORC1,53,54 this study investigated the activation status of the mTOR pathway in Nicd/Yap iCCA lesions. Positive staining for p-mTOR confirmed mTOR activation in Nicd/Yap tumors (Figure 7C). Consistently, the two main downstream kinases of mTORC1 signaling, namely, S6 and 4-EBP1, were actively phosphorylated, and RAPTOR, the main component of mTORC1, was induced in Nicd/Yap cholangiocellular lesions. Furthermore, RICTOR, the unique subunit of mTORC2, and p-AKT, a major mTORC2 downstream effector, were up-regulated in Nicd/Yap lesions, implying the activation of mTORC1 and 2 in this model (Figure 7D).
Figure 7.

Activation of the mammalian target of rapamycin (mTOR) pathway in Notch1 intracellular domain (Nicd)/Yes-associated protein (Yap) (YapS127A) tumor lesions. A and B: Relative expression of amino acid transporters Slc1a5, Slc7a5, Slc38a1, and Gls1 in mouse wild-type (WT), normal livers, and Nicd/Yap tumor livers, as determined by quantitative real-time RT-PCR (RT-qPCR). C: Activation of the mTOR pathway in Nicd/Yap tumor lesions at various time points of carcinogenesis, as assessed by immunohistochemistry. D: Representative analysis of the mTOR pathway in WT and Nicd/YapS127A mouse livers, as evaluated by Western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was the loading control. Note the elevated levels also of activated/phosphorylated (p-) extracellular signal-regulated kinase (ERK1/2) proteins. The RT-qPCR relative expression of each gene was normalized to WT mouse livers with 18S rRNA using the −ΔΔCt method. Data are expressed as means ± SD. ∗P < 0.05, ∗∗P < 0.01. Scale bars = 50 μm. Original magnification, ×400. NL, normal liver.
Following these intriguing findings, whether mTORC1 is dispensable or required for Nicd/Yap-driven tumor development was tested. To answer this question, Raptor was deleted in the mouse liver using the Cre/LoxP system.55 In brief, Cre or control pCMV plasmids were co-injected with Nicd and YAPS127A into Raptorfl/fl mice via hydrodynamic tail vein injection (Figure 8A). As previously reported, the co-injection of Cre with given oncogenes allows the deletion of the floxed alleles while retaining the overexpression of the injected oncogenes in the same set of mouse hepatocytes.56 Consistent with previous studies, control group mice developed a lethal liver tumor burden between 10 and 19 weeks after injection.37,39 In striking contrast, no tumor lesions occurred in Nicd/Yap/Cre livers up to >40 weeks after injection (Figure 8B). The pCMV group exhibited massive liver tumors with higher liver weight and liver/body ratio than the Cre group (Figure 8, C and D). Histologically, Nicd/Yap/pCMV mice displayed iCCA lesions expressing the cholangiocellular markers CK19, SOX9, and EPCAM (not shown) but not hepatocyte nuclear factor 4α (Figure 8E). In contrast, Nicd/Yap/Cre livers were microscopically completely normal, indistinguishable from livers from uninjected mice. No Myc-tagged(+) hepatocytes could be detected in Nicd/Yap/Cre liver tissues, presumably because of the elimination of the transfected hepatocytes by spontaneous or immune-induced apoptosis (Figure 8E). These results support the complete suppression of Nicd/Yap cholangiocarcinogenesis in the absence of mTORC1.
Figure 8.

Notch1 intracellular domain (Nicd)/Yes-associated protein (Yap) (YapS127A) intrahepatic cholangiocarcinoma (iCCA) tumor formation depends on the mammalian target of rapamycin complex 1 (mTORC1) pathway in mice. A: Experimental study design. B: Survival curve of Raptorfl/fl mice bearing Nicd/YapS127A tumors. Ablation of Raptor [Nicd/Yap/pCMV-Cre (Cre)] completely prevented tumor formation compared with the control group (Nicd/Yap/pCMV). C and D: Liver weight and liver/body ratio of the pCMV and Cre mouse groups. E: Representative gross image, hematoxylin and eosin (HE), and immunohistochemistry of the pCMV and Cre mice. ∗∗P < 0.01. Scale bars: 100 μm (second column); 50 μm (third, fourth, fifth, and sixth columns). Original magnification: ×200 (second column); ×400 (third, fourth, fifth, and sixth columns).
To rule out the possibility that co-injection of Cre affected Nicd/Yap gene delivery and efficiency of the hepatocytic transformation, wild-type FVB/N mice (n = 3) were co-injected with Nicd/Yap/Cre plasmids. Mice were harvested at 4 to 6 weeks after injection. Histologic examination revealed the presence of iCCA lesions in all three mice injected (Supplemental Figure S4). The results demonstrate that Nicd/Yap/Cre injection readily induces cholangiocarcinogenesis in wild-type mice. Overall, the present data indicate that activated Yap and Notch synergistically induce iCCA development via the mTORC1 pathway. Suppression of mTORC1 is sufficient to abolish liver carcinogenesis in Nicd/Yap mice.
Coordinated Activation of Notch, YAP, and mTORC1 Pathways in Human iCCA Samples
Notch, YAP, and mTORC1 signaling cascades are implicated in human iCCA development and progression.19, 20, 21,32,33,42 To examine the relevance of these pathways and their eventual interplay in cholangiocarcinogenesis, this study investigated the activation status of Notch, YAP, and mTORC1 signaling in a human iCCA collection. By immunohistochemistry, this study analyzed in a group of 132 tumor samples and respective nontumorous counterparts the staining pattern of Notch1, YAP, p-YAP at Ser127, RAPTOR, and p-4EBP1 (Figure 9). In nontumorous surrounding liver tissues, weak or moderate nuclear positive staining for Notch1, YAP, and p-4EBP1 was limited to biliary cells (used as an internal positive control for the staining). In contrast, hepatocytes had absent or faint nuclear and cytoplasmic immunolabeling for these proteins. Cytoplasmic immunoreactivity for RAPTOR protein was moderate in hepatocytes and normal biliary cells. Importantly, normal hepatocytes and biliary cells exhibited moderate and robust immunolabeling for p-YAP, respectively. Tumors displayed weak or absent staining for p-YAP instead (Figure 9). YAP phosphorylation at the Ser127 residue indicates inactivated form of YAP.29 Consistent with YAP inactivation, immunoreactivity for the YAP target connective tissue growth factor (CTGF) was absent in hepatocytes and normal cholangiocytes but moderate in iCCA lesions (Supplemental Figure S5A). Pronounced nuclear and/or cytoplasmic immunoreactivity for Notch1, YAP, RAPTOR, p-4EBP1, and CTGF was detected in most iCCA specimens [112 of 132 (84.8%), 130 of 132 (98.5%), 106 of 132 (80.3%), 98 of 132 (74.2%), and 115 of 132 (87.12%), respectively], whereas p-YAP staining was ubiquitously [132 of 132 (100%)] low in tumor specimens. To further validate these observations in human iCCA cell lines, this study analyzed the levels of YAP, Notch, and mTORC1 cascades in human iCCA cell lines and normal cholangiocyte cell lines. Total and activated (cleaved) levels of Notch1 protein were higher in human HucctT1, RBE, and KKUM-213 iCCA cell lines than in NHC-SS and C324 human normal cholangiocyte cell lines (Supplemental Figure S5B). Although no significant differences in YAP total levels were observed in iCCA and normal cholangiocyte cell lines, the levels of p-YAP (Ser127) were higher in normal cholangiocytes than in iCCA cell lines. This finding implies an efficient YAP-mediated inactivation in normal cholangiocyte cell lines, which is significantly impaired in iCCA cells. Activated p-mTOR levels were highest in the iCCA cell lines (Supplemental Figure S5B). Finally, levels of Notch1 (HES1, HEY1), YAP (CYR61), and mTOR (SLC38A1, SLC1A5) target genes were significantly higher in iCCA cell lines, whereas RAPTOR mRNA was most elevated in HuccT1 and KKUM-213 cell lines (Supplemental Figure S5C).
Figure 9.

Coordinated activation of Yes-associated protein (YAP), Notch1, and mammalian target of rapamycin (mTOR) in human intrahepatic cholangiocarcinoma (iCCA) samples. Representative immunohistochemical pattern of YAP, phosphorylated/inactivated (p-) YAP at Ser127 (S127), Notch1, RAPTOR, and p-4EBP1 staining in sections from nontumorous surrounding livers (ST) and two human iCCAs (iCCA1 and iCCA2). In ST, faint to moderate nuclear positive staining for Notch1, YAP, and p-4EBP1 was limited to biliary cells, whereas hepatocytes exhibited absent or weak nuclear and cytoplasmic immunolabeling for the same proteins. Immunoreactivity for RAPTOR was moderate in nonneoplastic biliary cells and hepatocytes. In striking contrast, both tumors displayed strong immunoreactivity for nuclear and cytoplasmic YAP and Notch1, RAPTOR, and p-4EBP1. Intense immunoreactivity for p-YAP (S127) was detected in the nonneoplastic liver, especially in biliary cells; levels of p-YAP were low in tumors. Cytokeratin 19 (CK19) staining was used as a biliary marker. Arrows indicate representative biliary cells or tracts. Scale bars = 100 μm. Original magnification, ×200. HE, hematoxylin and eosin.
Subsequently, the levels of NOTCH1, YAP1, HES1, CCN2/CTGF, RAPTOR, SLC1A5, SLC38A1, and GLS1 genes were evaluated in a subset of the iCCA sample collection (n = 50), for which the clinicopathologic data were available, via RT-qPCR. All the genes tested were significantly higher in iCCA than in corresponding nontumorous surrounding liver tissues (Figure 10). Moreover, a significant, positive correlation occurred between the mRNA levels of the various genes (Supplemental Figures S6 and S7). Kaplan-Meier and linear regression analysis also indicated that NOTCH1-, YAP1-, HES1-, CCN2/CTGF-, RAPTOR-, SLC1A5-, and GLS1-high patients had a worse survival outcome, whereas the same relationship was not significant when evaluating SLC38A1 levels. However, a SLC38A1 mRNA level >75th percentile in iCCA specimens was also associated with significantly shorter patient survival (Figure 11 and Supplemental Tables S1–S5). In The Cancer Genome Atlas cholangiocarcinoma data set, this study detected a significantly increased RAPTOR, MLST8, and mTOR mRNA expression in iCCA than in the nonneoplastic counterparts (Supplemental Figure S8). Thus, The Cancer Genome Atlas data support a crucial role of mTORC1 in cholangiocarcinogenesis. Overall, the present findings reveal the coordinated and almost ubiquitous up-regulation of the Notch, Hippo/YAP, and mTORC1 pathways in human iCCA.
Figure 10.

Notch1, Yes-associated protein (YAP) 1, and related downstream effectors are overexpressed in human intrahepatic cholangiocarcinoma (iCCA) specimens. Quantitative real-time RT-PCR analysis of NOTCH1, YAP1, HES1, CCN2/CTGF, RAPTOR, SLC1A5, SLC38A1, and GLS1 mRNA levels in a collection of human iCCA and corresponding nontumorous surrounding liver tissues (ST). Quantitative values were calculated using the PE Biosystems analysis software and expressed as number target (N target). N target = 2−ΔCt, wherein the ΔCt value of each sample was calculated by subtracting the mean Ct value of the gene of interest from the mean Ct value of the GAPDH gene. n = 50 each in blue and red boxes. ∗∗∗∗P < 0.0001 (t-test) compared with ST.
Figure 11.

NOTCH1, YAP1, HES1, CCN2/CTGF, RAPTOR, SLC1A5, SLC38A1, and GLS1 mRNA levels correlate with an adverse outcome in human intrahepatic cholangiocarcinoma (iCCA). A–G: Kaplan-Meier survival curves of human iCCA with high and low levels of the investigated genes, showing the unfavorable outcome of patients with elevated expression of these genes. H: Although SLC38A1 levels did not reach statistical significance, a SLC38A1 mRNA expression >75th percentile in iCCA specimens was also associated with significantly shorter patient survival.
Discussion
Despite the recent advancements in identifying the genetic and epigenetic changes occurring in cholangiocarcinogenesis, the molecular pathogenesis of iCCA remains poorly delineated. In particular, additional studies are necessary to unravel the specific role(s) of the signaling pathways involved in this aggressive neoplasm. The present study investigated Notch signaling and its possible crosstalk with other molecular cascades. The study found that activation of the MAPK/ERK and Hippo/YAP pathways occurs in the late stage of Notch-driven cholangiocarcinogenesis in Nicd-overexpressing mice. Notably, simultaneous activation of Notch1 with oncogenic forms of NRas or Yap accelerated iCCA development in double-injected mice. The accelerated carcinogenesis was histologically associated with an earlier malignant conversion of the benign cystic lesions into cystadenocarcinomas and invasive iCCA. However, although all liver iCCA lesions developing in Nicd/Ras derived from the conversion of benign cystic lesions, this intermediate step was not present in some Nicd/Yap lesions. Indeed, some of these lesions consisted of invasive iCCA from the beginning, similar to those observed in Akt/Nicd mice.22 Thus, the present data indicate that Yap, but not Nras, partly modifies the histopathologic features of the cholangiocellular lesions induced by Notch1 overexpression in the mouse liver. Furthermore, tumor development was significantly faster in Nicd/Yap than in Nicd/Ras mice, implying more productive cooperation between Notch1 and Hippo/YAP pathways than between Notch1 and Ras/MAPK, at least in mouse iCCA.
YAP and transcriptional co-activator with PDZ-binding motif (TAZ) are the two oncogenic effectors downstream of Hippo kinases. The functional involvement of YAP in hepatic tumorigenesis has been extensively characterized. Indeed, YAP contributes to the development and progression of the major types of primary liver cancer, including hepatocellular carcinoma, hepatoblastoma, and iCCA.57 This study and others have found that YAP cooperates with different oncogenic signals, leading to distinct primary liver tumors. For instance, this study found that Yap synergizes with activated Notch to promote iCCA formation. When Yap and oncogenic forms of β-catenin are co-expressed in the mouse liver, hepatoblastoma develops instead.33 These animal models are unique tools to delineate the mechanisms whereby YAP influences lineage commitment and contributes to tumorigenesis. This study found at the molecular level that Yap activates the mTORC1 pathway in iCCA. In hepatocellular carcinoma, YAP seems instead to be mainly involved in cell-cycle progression and DNA replication.58 Concerning the YAP paralog TAZ, a few investigations have been conducted in liver cancer to date. Whether TAZ and YAP have distinct or redundant functions in iCCA remains to be defined. In particular, it would be interesting to determine whether active Taz can also cooperate with Nicd to induce iCCA formation in mice and, if so, whether the tumor development also requires functional mTORC1 signaling.
Furthermore, the present investigation has revealed an increased expression of Slc1a5, Slc7a5, and Slc38a1 amino acid transporters as well as Gls1 in Nicd/Yap iCCA lesions. These transporters and GLS1, known YAP transcriptional targets, induce mTORC1 activation.50, 51, 52 Moreover, in addition to activating the mTOR pathway, increased amino acid uptake into tumor cells might exert other effects.59 For instance, all these transporters contribute to glutamine transport. Together with the observed increased expression of GLS1 in iCCA, the current findings suggest augmented glutamine catabolism in iCCA. Glutamine can contribute to tumor progression by shuttling into the tricarboxylic acid cycle cycle for ATP generation. It is also critical for regulating cellular redox balance.60,61 The increased amino acid transporter expression in iCCA thus supports further investigation on these molecules for iCCA treatment.62 In addition, these transporters could allow the delivery of tumor-targeting drugs.63 Currently, the studies on amino acid transporters in iCCA development are scanty. Additional investigations are required to illustrate their function(s) in iCCA initiation and progression. The Nicd/Yap mouse iCCA model provides an excellent preclinical tool to address these questions in vivo.
Mounting evidence indicates that the mTOR plays a central role during tumor development. Targeting the mTOR pathway has been suggested to be a potentially effective therapeutic strategy against liver tumors, including iCCA.41 A previous study found that inhibition of mTORC1 via Raptor silencing hinders iCCA cell growth in vitro and in vivo.37 The Nicd/Yap iCCA model might represent an excellent system to address these issues in vivo. Importantly, this study discovered that MLN0128, a second-generation dual mTORC1/mTORC2 inhibitor, effectively suppressed tumor growth in mouse iCCA driven by Akt/Yap oncogenes.37 Because mTOR is a direct downstream effector of activated Akt signaling, the results observed in Akt/Yap mice were predictable. In contrast, in the Nicd/Yap model, activation of the mTOR signaling is not the consequence of the ectopically overexpressed Akt oncogene. Thus, this model might recapitulate more faithfully human iCCA subtypes exhibiting activated mTOR activation at physiologic levels. Testing whether pan-mTOR inhibitors can hamper iCCA progression in Nicd/Yap mice is important. The results can provide further evidence of the usefulness of mTOR inhibitors for the treatment of human iCCA.
Footnotes
Supported by NIH grants R01CA190606 (X.C.), R01CA239251 (X.C.), and P30DK026743 (University of California, San Francisco Liver Center); National Science Foundation for Young Scientists of China grant 81402308 (B.F.); Science Foundation of Peking University Cancer Hospital grant 2021-24 (B.F.); and Beijing University of Chinese Medicine New Faculty Start-up Fund grant 2021-JYB-XJSJJ029 (J.W.).
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2021.05.017.
Contributor Information
Biao Fan, Email: fanbiao1986@163.com.
Jingxiao Wang, Email: 201801022@bucm.edu.cn.
Supplemental Data
Supplemental Figure S1.

Representative histopathologic features of cholangiocellular lesions developing in Notch intracellular domain (Nicd) and Nicd/NRas mice. The lesions formed in Nicd mice (A–C) are indistinguishable from those developing in Nicd/NRas mice (D–F). Indeed, large cystic lesions (cyst), demarcated by a thin epithelium (B and E), and intrahepatic cholangiocarcinoma (iCCA) can be simultaneously detected in both mouse models. Scale bars: 500 μm (A and D); 100 μm (B, C, E, and F). Original magnification: ×40 (A and D); ×200 (B, C, E, and F). HE, hematoxylin and eosin; w.p.i., weeks post injection.
Supplemental Figure S2.
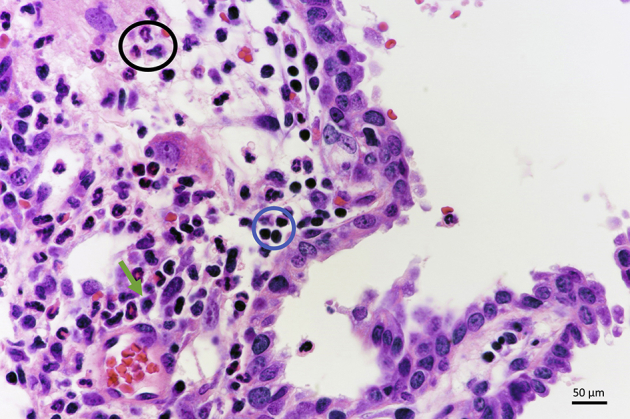
Notch intracellular domain (Nicd)/Yes-associated protein (Yap) mice exhibit an inflammatory infiltrate in some of the lesions. A representative example is shown in the image; the inflammatory infiltrate is mainly composed of neutrophils (black circle), lymphocytes (blue circle), and plasma cells (green arrow). Hematoxylin and eosin staining. Scale bar = 50 μm. Original magnification, ×400.
Supplemental Figure S3.
Chronological and molecular characterization of the lesions developed in Notch intracellular domain (Nicd)/Yes-associated protein (Yap) S127A mice. A: Representative results of the gross and microscopic images of the tumor lesions at different weeks after injection. The immunohistochemistry results indicate the positive immunoreactivity for the proliferation marker Ki-67 and the cholangiocyte marker cytokeratin 19 (CK19). B–D: Quantitative real-time RT-PCR shows up-regulation of the Ck19, Sox9, and Epcam genes in the Nicd/Yap mice of end-stage tumors compared with wild-type (WT) mice. Each gene expression was normalized to the WT mouse liver with 18S rRNA using the −ΔΔCt method. Data are expressed as means ± SD. ∗∗P < 0.01 (t-test). Scale bars = 100 μm. Original magnification, ×200. w.p.i., weeks post injection.
Supplemental Figure S4.
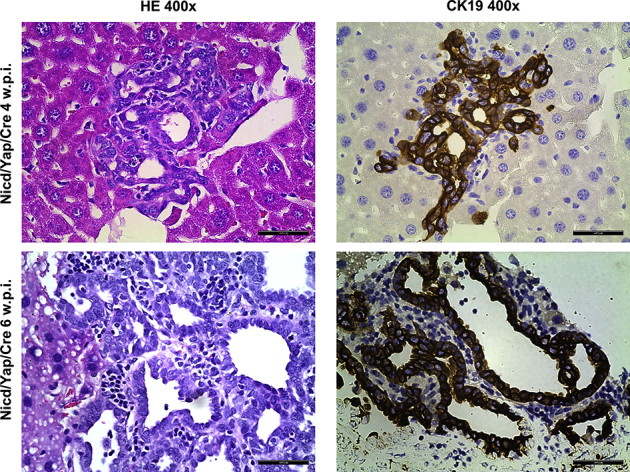
Co-expression of pCMV-Cre (Cre) does not affect Notch intracellular domain (Nicd)/Yes-associated protein (Yap)–driven iCCA formation in mice. Representative liver histologic results of two FVB/N mice 4 and 6 weeks after the hydrodynamic injection of Nicd/Yap/Cre plasmids. Cholangiocellular lesions can already be appreciated in this mouse's liver parenchyma, as indicated by hematoxylin and eosin (HE) staining and positive immunoreactivity for the cytokeratin 19 (CK19) cholangiocellular marker. Scale bars = 50 μm. Original magnification, ×400. w.p.i., weeks post injection; WT, wild type.
Supplemental Figure S5.

Activation of Yes-associated protein (YAP), Notch1, and mammalian target of rapamycin (mTOR) in human intrahepatic cholangiocarcinoma (iCCA) samples. A: Representative immunohistochemical pattern of connective tissue growth factor (CTGF), a YAP target, in sections from the nontumorous surrounding liver (ST) and two human iCCAs (iCCA1 and iCCA2; the same tumor samples are stained with other antibodies in Figure 8). In ST, no staining was detected for CTGF in normal hepatocytes and biliary cells (arrow). Both tumors exhibited strong immunoreactivity for cytoplasmic CTGF. B: Levels of Notch1, YAP, phosphorylated/inactivated YAP (S127), and phosphorylated/activated mTOR (S2448) in normal cholangiocyte (NHC-SS and C324) and iCCA (HuccT1, RBE, and KKUM-213) cell lines as determined by Western blot analysis. β-Actin was used as a loading control. C: Quantitative real-time RT-PCR analysis of Notch1 (HES1, HEY1), YAP (CYR61), and mTOR (SLC38A1, SLC1A5, and RAPTOR) mRNA levels in normal cholangiocyte (NHC-SS and C324) and iCCA (HuccT1, RBE, and KKUM-213) cell lines. Gray bars indicate normal cholangiocyte cell lines, and black bars indicate iCCA cell lines. Quantitative values were calculated using the PE Biosystems analysis software and expressed as number target (N target). N target = 2−ΔCt, wherein the ΔCt value of each sample was calculated by subtracting the mean Ct value of the gene of interest from the mean Ct value of the GAPDH gene. Scale bars: 50 μm (A, left panel); 100 μm (A, middle and right panels). Original magnification: ×400 (A, left panel); ×200 (A, middle and right panels).
Supplemental Figure S6.

mRNA levels of Notch1 correlated with YAP1, HES1, CCN2/CTGF, RAPTOR, SLC1A5, SLC38A1, and GLS1 in human intrahepatic cholangiocarcinoma (iCCA) samples. Linear regression analysis showing a highly statistically significant correlation between Notch1 expression and that of the other genes.
Supplemental Figure S7.

Expression levels of YAP1 correlated with Notch1, HES1, CCN2/CTGF, RAPTOR, SLC1A5, SLC38A1, and GLS1 in human intrahepatic cholangiocarcinoma (iCCA) specimens. Linear regression analysis showing a highly statistically significant correlation between YAP1 expression and that of the other genes.
Elevated mRNA expression of RPTOR (RAPTOR), MLST8, and MTOR genes in The Cancer Genome Atlas (TCGA) cholangiocarcinoma (CHOL) data set. These mRNA expression data were extracted from the TCGA CHOL data set web server (http://ualcan.path.uab.edu/index.html, last accessed May 20, 2021).
References
- 1.Razumilava N., Gores G.J. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizvi S., Khan S.A., Hallemeier C.L., Kelley R.K., Gores G.J. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018;15:95–111. doi: 10.1038/nrclinonc.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kudo M., Finn R.S., Qin S., Han K.H., Ikeda K., Piscaglia F., Baron A., Park J.W., Han G., Jassem J., Blanc J.F., Vogel A., Komov D., Evans T.R.J., Lopez C., Dutcus C., Guo M., Saito K., Kraljevic S., Tamai T., Ren M., Cheng A.L. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 4.Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., Li D., Verret W., Xu D.Z., Hernandez S., Liu J., Huang C., Mulla S., Wang Y., Lim H.Y., Zhu A.X., Cheng A.L., IMbrave150 Investigators Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 5.Valle J., Wasan H., Palmer D.H., Cunningham D., Anthoney A., Maraveyas A., Madhusudan S., Iveson T., Hughes S., Pereira S.P., Roughton M., Bridgewater J., ABC-02 Trial Investigators Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 6.Mavros M.N., Economopoulos K.P., Alexiou V.G., Pawlik T.M. Treatment and prognosis for patients with intrahepatic cholangiocarcinoma: systematic review and meta-analysis. JAMA Surg. 2014;149:565–574. doi: 10.1001/jamasurg.2013.5137. [DOI] [PubMed] [Google Scholar]
- 7.Hoy S.M. Pemigatinib: first approval. Drugs. 2020;80:923–929. doi: 10.1007/s40265-020-01330-y. [DOI] [PubMed] [Google Scholar]
- 8.Abou-Alfa G.K., Sahai V., Hollebecque A., Vaccaro G., Melisi D., Al-Rajabi R., Paulson A.S., Borad M.J., Gallinson D., Murphy A.G., Oh D.Y., Dotan E., Catenacci D.V., Van Cutsem E., Ji T., Lihou C.F., Zhen H., Feliz L., Vogel A. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21:671–684. doi: 10.1016/S1470-2045(20)30109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farshidfar F., Zheng S., Gingras M.C., Newton Y., Shih J., Robertson A.G. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;18:2780–2794. doi: 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowery M.A., Ptashkin R., Jordan E., Berger M.F., Zehir A., Capanu M., Kemeny N.E., O'Reilly E.M., El-Dika I., Jarnagin W.R., Harding J.J., D'Angelica M.I., Cercek A., Hechtman J.F., Solit D.B., Schultz N., Hyman D.M., Klimstra D.S., Saltz L.B., Abou-Alfa G.K. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res. 2018;24:4154–4161. doi: 10.1158/1078-0432.CCR-18-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goeppert B., Konermann C., Schmidt C.R., Bogatyrova O., Geiselhart L., Ernst C., Gu L., Becker N., Zucknick M., Mehrabi A., Hafezi M., Klauschen F., Stenzinger A., Warth A., Breuhahn K., Renner M., Weichert W., Schirmacher P., Plass C., Weichenhan D. Global alterations of DNA methylation in cholangiocarcinoma target the Wnt signaling pathway. Hepatology. 2014;59:544–554. doi: 10.1002/hep.26721. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y., Deng X., Li Q., Wang F., Miao L., Jiang Q. Emerging roles of long noncoding RNAs in cholangiocarcinoma: advances and challenges. Cancer Commun (Lond) 2020;40:655–680. doi: 10.1002/cac2.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C., Zhang B., Meng D., Ge C. Comprehensive analysis of DNA methylation and gene expression profiles in cholangiocarcinoma. Cancer Cell Int. 2019;19:352. doi: 10.1186/s12935-019-1080-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bray S.J. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17:722–735. doi: 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- 15.Kovall R.A., Gebelein B., Sprinzak D., Kopan R. The canonical notch signaling pathway: structural and biochemical insights into shape, sugar, and force. Dev Cell. 2017;41:228–241. doi: 10.1016/j.devcel.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie G., Karaca G., Swiderska-Syn M., Michelotti G.A., Kruger L., Chen Y., Premont R.T., Choi S.S., Diehl A.M. Crosstalk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology. 2013;58:1801–1813. doi: 10.1002/hep.26511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Razumilava N., Gores G.J. Notch-driven carcinogenesis: the merging of hepatocellular cancer and cholangiocarcinoma into a common molecular liver cancer subtype. J Hepatol. 2013;58:1244–1245. doi: 10.1016/j.jhep.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jors S., Jeliazkova P., Ringelhan M., Thalhammer J., Durl S., Ferrer J., Sander M., Heikenwalder M., Schmid R.M., Siveke J.T., Geisler F. Lineage fate of ductular reactions in liver injury and carcinogenesis. J Clin Invest. 2015;125:2445–2457. doi: 10.1172/JCI78585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cigliano A., Wang J., Chen X., Calvisi D.F. Role of the Notch signaling in cholangiocarcinoma. Expert Opin Ther Targets. 2017;21:471–483. doi: 10.1080/14728222.2017.1310842. [DOI] [PubMed] [Google Scholar]
- 20.Rauff B., Malik A., Bhatti Y.A., Chudhary S.A., Qadri I., Rafiq S. Notch signalling pathway in development of cholangiocarcinoma. World J Gastrointest Oncol. 2020;12:957–974. doi: 10.4251/wjgo.v12.i9.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Rourke C.J., Matter M.S., Nepal C., Caetano-Oliveira R., Ton P.T., Factor V.M., Andersen J.B. Identification of a pan-gamma-secretase inhibitor response signature for notch-driven cholangiocarcinoma. Hepatology. 2020;71:196–213. doi: 10.1002/hep.30816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan B., Malato Y., Calvisi D.F., Naqvi S., Razumilava N., Ribback S., Gores G.J., Dombrowski F., Evert M., Chen X., Willenbring H. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911–2915. doi: 10.1172/JCI63212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L., Che L., Tharp K.M., Park H.M., Pilo M.G., Cao D., Cigliano A., Latte G., Xu Z., Ribback S., Dombrowski F., Evert M., Gores G.J., Stahl A., Calvisi D.F., Chen X. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology. 2016;63:1900–1913. doi: 10.1002/hep.28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zender S., Nickeleit I., Wuestefeld T., Sorensen I., Dauch D., Bozko P., El-Khatib M., Geffers R., Bektas H., Manns M.P., Gossler A., Wilkens L., Plentz R., Zender L., Malek N.P. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell. 2016;30:353–356. doi: 10.1016/j.ccell.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Guest R.V., Boulter L., Dwyer B.J., Kendall T.J., Man T.Y., Minnis-Lyons S.E., Lu W.Y., Robson A.J., Gonzalez S.F., Raven A., Wojtacha D., Morton J.P., Komuta M., Roskams T., Wigmore S.J., Sansom O.J., Forbes S.J. Notch3 drives development and progression of cholangiocarcinoma. Proc Natl Acad Sci U S A. 2016;113:12250–12255. doi: 10.1073/pnas.1600067113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J., Dong M., Xu Z., Song X., Zhang S., Qiao Y., Che L., Gordan J., Hu K., Liu Y., Calvisi D.F., Chen X. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene. 2018;37:3229–3242. doi: 10.1038/s41388-018-0188-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitchen P., Lee K.Y., Clark D., Lau N., Lertsuwan J., Sawasdichai A., Satayavivad J., Oltean S., Afford S., Gaston K., Jayaraman P.S. A runaway PRH/HHEX-Notch3-positive feedback loop drives cholangiocarcinoma and determines response to CDK4/6 inhibition. Cancer Res. 2020;80:757–770. doi: 10.1158/0008-5472.CAN-19-0942. [DOI] [PubMed] [Google Scholar]
- 28.Matsumori T., Kodama Y., Takai A., Shiokawa M., Nishikawa Y., Matsumoto T., Takeda H., Marui S., Okada H., Hirano T., Kuwada T., Sogabe Y., Kakiuchi N., Tomono T., Mima A., Morita T., Ueda T., Tsuda M., Yamauchi Y., Kuriyama K., Sakuma Y., Ota Y., Maruno T., Uza N., Marusawa H., Kageyama R., Chiba T., Seno H. Hes1 is essential in proliferating ductal cell-mediated development of intrahepatic cholangiocarcinoma. Cancer Res. 2020;80:5305–5316. doi: 10.1158/0008-5472.CAN-20-1161. [DOI] [PubMed] [Google Scholar]
- 29.Hansen C.G., Moroishi T., Guan K.L., YAP and T.A.Z. a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Misra J.R., Irvine K.D. The Hippo signaling network and its biological functions. Annu Rev Genet. 2018;52:65–87. doi: 10.1146/annurev-genet-120417-031621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moya I.M., Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20:211–226. doi: 10.1038/s41580-018-0086-y. [DOI] [PubMed] [Google Scholar]
- 32.Li H., Wolfe A., Septer S., Edwards G., Zhong X., Abdulkarim A.B., Ranganathan S., Apte U. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 2012;32:38–47. doi: 10.1111/j.1478-3231.2011.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tao J., Calvisi D.F., Ranganathan S., Cigliano A., Zhou L., Singh S., Jiang L., Fan B., Terracciano L., Armeanu-Ebinger S., Ribback S., Dombrowski F., Evert M., Chen X., Monga S.P.S. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014;147:690–701. doi: 10.1053/j.gastro.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimachi K., Nishio M., Aishima S., Kuroda Y., Iguchi T., Komatsu H., Hirata H., Sakimura S., Eguchi H., Bekki Y., Takenaka K., Maehara Y., Suzuki A., Mimori K. Altered expression of Hippo signaling pathway molecules in intrahepatic cholangiocarcinoma. Oncology. 2017;93:67–74. doi: 10.1159/000463390. [DOI] [PubMed] [Google Scholar]
- 35.Sugiura K., Mishima T., Takano S., Yoshitomi H., Furukawa K., Takayashiki T., Kuboki S., Takada M., Miyazaki M., Ohtsuka M. The expression of yes-associated protein (YAP) maintains putative cancer stemness and is associated with poor prognosis in intrahepatic cholangiocarcinoma. Am J Pathol. 2019;189:1863–1877. doi: 10.1016/j.ajpath.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 36.Marti P., Stein C., Blumer T., Abraham Y., Dill M.T., Pikiolek M., Orsini V., Jurisic G., Megel P., Makowska Z., Agarinis C., Tornillo L., Bouwmeester T., Ruffner H., Bauer A., Parker C.N., Schmelzle T., Terracciano L.M., Heim M.H., Tchorz J.S. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62:1497–1510. doi: 10.1002/hep.27992. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S., Song X., Cao D., Xu Z., Fan B., Che L., Hu J., Chen B., Dong M., Pilo M.G., Cigliano A., Evert K., Ribback S., Dombrowski F., Pascale R.M., Cossu A., Vidili G., Porcu A., Simile M.M., Pes G.M., Giannelli G., Gordan J., Wei L., Evert M., Cong W., Calvisi D.F., Chen X. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol. 2017;67:1194–1203. doi: 10.1016/j.jhep.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Werneburg N., Gores G.J., Smoot R.L. The Hippo Pathway and YAP signaling: emerging concepts in regulation, signaling, and experimental targeting strategies with implications for hepatobiliary malignancies. Gene Expr. 2020;20:67–74. doi: 10.3727/105221619X15617324583639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song X., Liu X., Wang H., Wang J., Qiao Y., Cigliano A., Utpatel K., Ribback S., Pilo M.G., Serra M., Gordan J.D., Che L., Zhang S., Cossu A., Porcu A., Pascale R.M., Dombrowski F., Hu H., Calvisi D.F., Evert M., Chen X. Combined CDK4/6 and Pan-mTOR inhibition is synergistic against intrahepatic cholangiocarcinoma. Clin Cancer Res. 2019;25:403–413. doi: 10.1158/1078-0432.CCR-18-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saxton R.A., Sabatini D.M. mTOR Signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu X., Paliogiannis P., Calvisi D.F., Chen X. Role of the mammalian target of rapamycin pathway in liver cancer: from molecular genetics to targeted therapies. Hepatology. 2021;73 Suppl 1:49–61. doi: 10.1002/hep.31310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung J.Y., Hong S.M., Choi B.Y., Cho H., Yu E., Hewitt S.M. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin Cancer Res. 2009;15:660–667. doi: 10.1158/1078-0432.CCR-08-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corti F., Nichetti F., Raimondi A., Niger M., Prinzi N., Torchio M., Tamborini E., Perrone F., Pruneri G., Di Bartolomeo M., de Braud F., Pusceddu S. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: a review of current evidences and future perspectives. Cancer Treat Rev. 2019;72:45–55. doi: 10.1016/j.ctrv.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 44.Montal R., Sia D., Montironi C., Leow W.Q., Esteban-Fabro R., Pinyol R., Torres-Martin M., Bassaganyas L., Moeini A., Peix J., Cabellos L., Maeda M., Villacorta-Martin C., Tabrizian P., Rodriguez-Carunchio L., Castellano G., Sempoux C., Minguez B., Pawlik T.M., Labgaa I., Roberts L.R., Sole M., Fiel M.I., Thung S., Fuster J., Roayaie S., Villanueva A., Schwartz M., Llovet J.M. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J Hepatol. 2020;73:315–327. doi: 10.1016/j.jhep.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X., Calvisi D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184:912–923. doi: 10.1016/j.ajpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urribarri A.D., Munoz-Garrido P., Perugorria M.J., Erice O., Merino-Azpitarte M., Arbelaiz A., Lozano E., Hijona E., Jimenez-Aguero R., Fernandez-Barrena M.G., Jimeno J.P., Marzioni M., Marin J.J., Masyuk T.V., LaRusso N.F., Prieto J., Bujanda L., Banales J.M. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut. 2014;63:1658–1667. doi: 10.1136/gutjnl-2013-305281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frith C.H., Ward J.M. A morphologic classification of proliferative and neoplastic hepatic lesions in mice. J Environ Pathol Toxicol. 1979;3:329–351. [PubMed] [Google Scholar]
- 48.Wang C., Cigliano A., Jiang L., Li X., Fan B., Pilo M.G., Liu Y., Gui B., Sini M., Smith J.W., Dombrowski F., Calvisi D.F., Evert M., Chen X. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and N-Ras protooncogenes in mice. Hepatology. 2015;61:200–213. doi: 10.1002/hep.27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y., Xu M., Liu P., Liang B., Qian M., Wang H., Song X., Nyshadham P., Che L., Calvisi D.F., Li F., Lin S., Chen X. Mammalian target of rapamycin complex 2 signaling is required for liver regeneration in a cholestatic liver injury murine model. Am J Pathol. 2020;190:1414–1426. doi: 10.1016/j.ajpath.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park Y.Y., Sohn B.H., Johnson R.L., Kang M.H., Kim S.B., Shim J.J., Mangala L.S., Kim J.H., Yoo J.E., Rodriguez-Aguayo C., Pradeep S., Hwang J.E., Jang H.J., Lee H.S., Rupaimoole R., Lopez-Berestein G., Jeong W., Park I.S., Park Y.N., Sood A.K., Mills G.B., Lee J.S. Yes-associated protein 1 and transcriptional co-activator with PDZ-binding motif activate the mammalian target of rapamycin complex 1 pathway by regulating amino acid transporters in hepatocellular carcinoma. Hepatology. 2016;63:159–172. doi: 10.1002/hep.28223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu P., Calvisi D.F., Kiss A., Cigliano A., Schaff Z., Che L., Ribback S., Dombrowski F., Zhao D., Chen X. Central role of mTORC1 downstream of YAP/TAZ in hepatoblastoma development. Oncotarget. 2017;8:73433–73447. doi: 10.18632/oncotarget.20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bertero T., Oldham W.M., Cottrill K.A., Pisano S., Vanderpool R.R., Yu Q., Zhao J., Tai Y., Tang Y., Zhang Y.Y., Rehman S., Sugahara M., Qi Z., Gorcsan J., 3rd, Vargas S.O., Saggar R., Saggar R., Wallace W.D., Ross D.J., Haley K.J., Waxman A.B., Parikh V.N., De Marco T., Hsue P.Y., Morris A., Simon M.A., Norris K.A., Gaggioli C., Loscalzo J., Fessel J., Chan S.Y. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. 2016;126:3313–3335. doi: 10.1172/JCI86387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicklin P., Bergman P., Zhang B., Triantafellow E., Wang H., Nyfeler B., Yang H., Hild M., Kung C., Wilson C., Myer V.E., MacKeigan J.P., Porter J.A., Wang Y.K., Cantley L.C., Finan P.M., Murphy L.O. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimobayashi M., Hall M.N. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016;26:7–20. doi: 10.1038/cr.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang C., Che L., Hu J., Zhang S., Jiang L., Latte G., Demartis M.I., Tao J., Gui B., Pilo M.G., Ribback S., Dombrowski F., Evert M., Calvisi D.F., Chen X. Activated mutant forms of PIK3CA cooperate with RasV12 or c-Met to induce liver tumour formation in mice via AKT2/mTORC1 cascade. Liver Int. 2016;36:1176–1186. doi: 10.1111/liv.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiao Y., Wang J., Karagoz E., Liang B., Song X., Shang R., Evert K., Xu M., Che L., Evert M., Calvisi D.F., Tao J., Wang B., Monga S.P., Chen X. Axis inhibition protein 1 (Axin1) deletion-induced hepatocarcinogenesis requires intact beta-catenin but not Notch cascade in mice. Hepatology. 2019;70:2003–2017. doi: 10.1002/hep.30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang S., Zhou D. Role of the transcriptional co-activators YAP/TAZ in liver cancer. Curr Opin Cell Biol. 2019;61:64–71. doi: 10.1016/j.ceb.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Wang H., Wang J., Zhang S., Jia J., Liu X., Zhang J., Wang P., Song X., Che L., Liu K., Ribback S., Cigliano A., Evert M., Wu H., Calvisi D.F., Zeng Y., Chen X. Distinct and overlapping roles of hippo effectors YAP and TAZ during human and mouse hepatocarcinogenesis. Cell Mol Gastroenterol Hepatol. 2020;11:1095–1117. doi: 10.1016/j.jcmgh.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scalise M., Console L., Rovella F., Galluccio M., Pochini L., Indiveri C. Membrane transporters for amino acids as players of cancer metabolic rewiring. Cells. 2020;9:2028. doi: 10.3390/cells9092028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shah R., Chen S. Metabolic signaling cascades prompted by glutaminolysis in cancer. Cancers (Basel) 2020;12:2624. doi: 10.3390/cancers12092624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mates J.M., Campos-Sandoval J.A., Santos-Jimenez J.L., Marquez J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019;467:29–39. doi: 10.1016/j.canlet.2019.09.011. [DOI] [PubMed] [Google Scholar]
- 62.Cormerais Y., Vucetic M., Pouyssegur J. Targeting amino acids transporters (SLCs) to starve cancer cells to death. Biochem Biophys Res Commun. 2019;520:691–693. doi: 10.1016/j.bbrc.2019.10.173. [DOI] [PubMed] [Google Scholar]
- 63.Zhang L., Sui C., Yang W., Luo Q. Amino acid transporters: emerging roles in drug delivery for tumor-targeting therapy. Asian J Pharm Sci. 2020;15:192–206. doi: 10.1016/j.ajps.2019.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elevated mRNA expression of RPTOR (RAPTOR), MLST8, and MTOR genes in The Cancer Genome Atlas (TCGA) cholangiocarcinoma (CHOL) data set. These mRNA expression data were extracted from the TCGA CHOL data set web server (http://ualcan.path.uab.edu/index.html, last accessed May 20, 2021).