

SUMMARY
Pancreatic β-cell failure is the key pathogenic element of the complex metabolic deterioration in type 2 diabetes (T2D); its underlying pathomechanism is still elusive. Here, we identify pleckstrin homology domain leucine-rich repeat protein phosphatases 1 and 2 (PHLPP1/2) as phosphatases whose upregulation leads to β-cell failure in diabetes. PHLPP levels are highly elevated in metabolically stressed human and rodent diabetic β-cells. Sustained hyper-activation of mechanistic target of rapamycin complex 1 (mTORC1) is the primary mechanism of the PHLPP upregulation linking chronic metabolic stress to ultimate β-cell death. PHLPPs directly dephosphorylate and regulate activities of β-cell survival-dependent kinases AKT and MST1, constituting a regulatory triangle loop to control β-cell apoptosis. Genetic inhibition of PHLPPs markedly improves β-cell survival and function in experimental models of diabetes in vitro, in vivo, and in primary human T2D islets. Our study presents PHLPPs as targets for functional regenerative therapy of pancreatic β cells in diabetes.
In brief
Lupse et al. show that chronic metabolic stress and high sugar in diabetes leads to hyper-activation of the metabolism-control pathwaymTORC1 and initiation of a triangle loop of cellular survival control by the phosphatase PHLPP. PHLPP inhibits pro-survival AKT, activates pro-apoptotic MST1, and ultimately leads to β-cell death and dysfunction.
Graphical Abstract
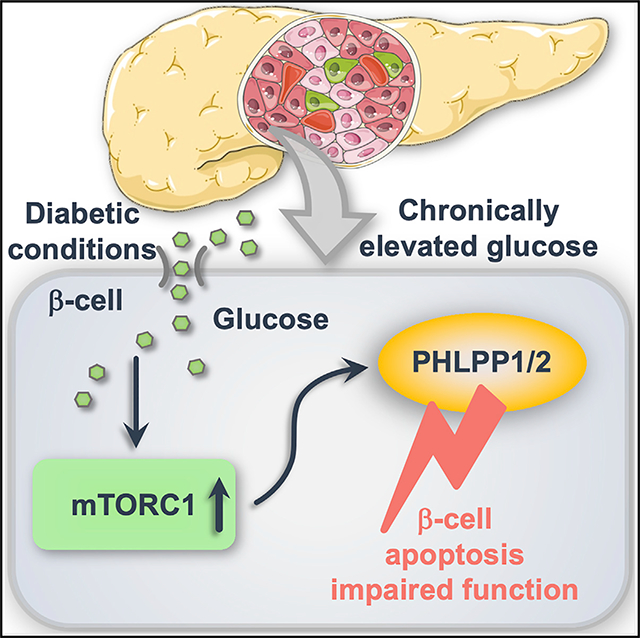
INTRODUCTION
Type 2 diabetes (T2D) is a heterogeneous multifactorial metabolic disease, characterized by insulin resistance and progressive loss of functional β-cell mass. Pancreatic β-cell failure finally results from decreased insulin secretory function and/or β-cell death (Alejandro et al., 2015; Ashcroft and Rorsman, 2012; Butler et al., 2003; Weir et al., 2020), hallmarks of T2D; however, underlying molecular mechanisms are still not fully characterized, and there is currently no β-cell-specific therapy for a cure (Donath et al., 2019). In addition to β-cell death and dysfunction, other mechanisms, such as β-cell dedifferentiation (Cinti et al., 2016; Jeffery and Harries, 2016; Talchai et al., 2012) and failure of adaptive expansion because of impaired regeneration (Aguayo-Mazzucato and Bonner-Weir, 2018; Tiwari et al., 2016), have been proposed as possible causes for β-cell failure in T2D.
The coordinated cellular stress response and enormous metabolic adaptation are necessary for normal β-cell insulin-secretory function, glucose homeostasis, and prevention of T2D; these are largely directed by the highly complex dynamics of signal transduction pathways. Perturbations in β-cell signaling have complex consequences leading to imbalanced and improper transcriptional and post-transcriptional alterations, metabolic deterioration, continuous decline in β-cell function and viability and the cumulative development of diabetic complications. Thus, comprehensive understanding of cell-fate decisions during stress and metabolic overload will provide new targets for the development of therapeutic approaches aiming at prevention and repair of β-cell failure in T2D.
Serine-threonine phosphatases (STPs) are important components of multiple cell signaling nodes and serve as potential targets for drug development. The pleckstrin homology (PH) domain leucine-rich repeat protein phosphatases (PHLPPs) enzymes are members of the protein phosphatase 2C (PP2C) grouped in the protein phosphatase metal-dependent (PPM) family of STP (Brognard and Newton, 2008). The PHLPP family includes two isozymes, PHLPP1 (also referred to as suprachiasmatic nucleus circadian oscillatory protein [SCOP]) and PHLPP2 (Grzechnik and Newton, 2016). PHLPP1/2 are ubiquitously expressed and involved in several cellular processes, such as proliferation, survival, stress response, inflammation, memory formation, and T cell development (Brognard and Newton, 2008; Chen et al., 2013; Cohen Katsenelson et al., 2019; Gao et al., 2005; Liu et al., 2009; Masubuchi et al., 2010; Patterson et al., 2011). Initial studies identified PHLPP as a hydrophobic motif phosphatase to antagonize pro-survival signaling pathways. It is well-established that PHLPP1/2, when activated as a cellular response to cytotoxic stress, mediate cell death through dephosphorylation of multiple substrates, such as AKT, PKC, p70S6K, and MST1 (Gao et al., 2005, 2008; Liu et al., 2011b; Qiao et al., 2010). Notably, PHLPP1 single-nucleotide polymorphism (SNP) has been recently associated with T2D (Turki et al., 2013; Yako et al., 2016). In line with that genetic association, PHLPP1 expression is elevated in liver and skeletal muscle of insulin-resistant rodents (Behera et al., 2018; Liu et al., 2012) as well as in adipose tissue and skeletal muscle of human obese individuals, including patients with T2D (Andreozzi et al., 2011), indicating an important role for PHLPP1 in diabetes progression. Consistently, PHLPP1 is an important regulator of AKT signaling in the heart: knockdown or genetic deletion of PHLPP1 enhances pro-survival AKT activity in cardiac myocytes and, in turn, provides protection against ischemic injury (Aviv and Kirshenbaum, 2010; Chen et al., 2013; Miyamoto et al., 2010). Similarly, PHLPP1 depletion is neuroprotective and increases AKT signaling and survival in hippocampal and striatal neurons (Jackson et al., 2013).
So far, the physiological role of PHLPP1/2 in the human β-cell—whether PHLPP1/2 are upregulated in T2D, whether such upregulation would trigger β-cell death and impaired insulin secretion, and whether PHLPP1/2 inhibition can rescue β-cells in diabetes—is not known. In the present work, we aimed to investigate molecular and cellular mechanisms of PHLPP1/2-induced β-cell failure and to test whether inhibition of PHLPP1/2 prevent β-cell destruction and diabetes in vivo.
RESULTS
PHLPP1/2 are upregulated by diabetogenic conditions and impair β-cell survival and function
To identify PHLPP1/2 upregulation and its correlation with β-cell apoptosis, we exposed isolated human islets and the classically used rat β-cell line INS-1E to a diabetic milieu in vitro (chronically elevated glucose concentrations). PHLPP1/2 were highly upregulated at the protein level in INS-1E cells (Figures 1A and 1B) and in primary isolated human islets (Figures 1C and 1D). Consistently, PHLPP1/2 levels were increased in islets of hyperglycemic high fat/high sucrose (HFD)-fed mice for 16 weeks (Figures 1E and 1F). β-cell-specific PHLPP upregulation was confirmed by double staining for PHLPP2 and insulin from paraffin-embedded sections from HFD in comparison with normal diet (ND)-fed control mice (Figure 1G). Similarly, PHLPP1/2 protein levels were also elevated in islets of another model of T2D, the obese diabetic leptin-receptor-deficient db/db mice (Figures 1H and 1I). These data show that PHLPP1/2 are markedly elevated by pro-diabetic conditions in vitro and in vivo in mouse models of T2D.
Figure 1. PHLPP1/2 is upregulated by diabetogenic conditions and impairs β-cell survival and function.
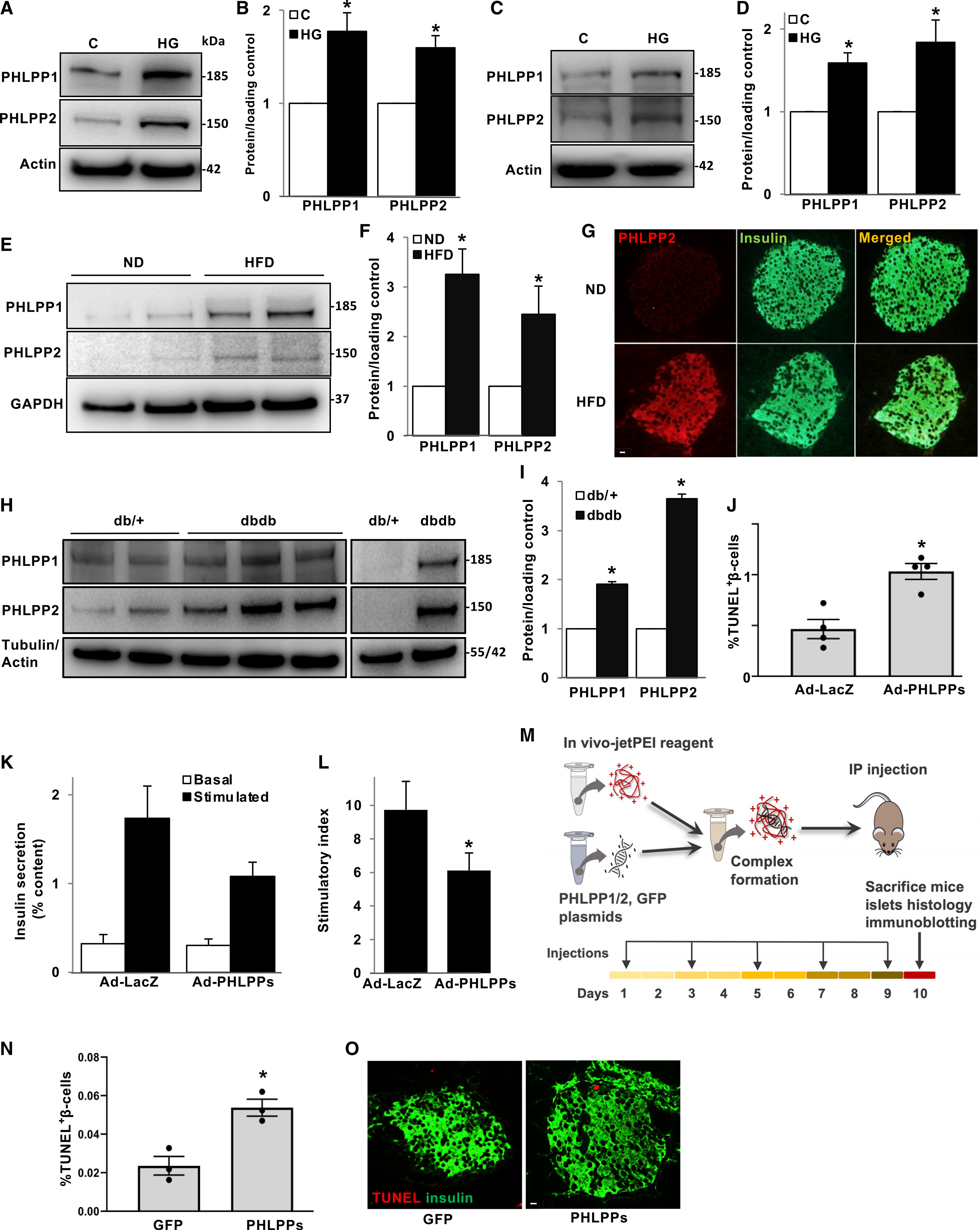
(A–D) Representative western blots (A and C) and quantitative densitometry analysis (B and D) of INS-1E cells (A and B; n = 6) or isolated human islets (C and D; n = 6) treated with high glucose (22 mM) for 2 days.
(E and F) Representative western blots (E) and quantitative densitometry analysis (F) of isolated islets from normal diet (ND) or high-fat/high-sucrose diet (HFD)-fed mice for 16 weeks (n = 8).
(G) Representative images shown double immunostaining for PHLPP2 in red and insulin in green in pancreatic sections from ND- and HFD-treated mice.
(H and I) Representative western blots (H) and quantitative densitometry analysis (I) of isolated islets from 10-week-old diabetic db/db mice and their heterozygous db/+ littermates (n = 5).
(J–L) Human islets transduced with LacZ control or PHLPP1 and PHLPP2 adenoviruses for 48 h. (J) Pooled TUNEL analysis (n = 4; an average of 18,718 β cells were counted from each treatment condition). (K) Insulin secretion during 1 h of incubation with 2.8 mM (basal) and 16.7 mM (stimulated) glucose, normalized to insulin content. (L) Insulin stimulatory index denotes the ratio of stimulated and basal (n = 5).
(M) Scheme of the in vivo experimental strategy. 8-week-old male C57BL/6 mice were intraperitoneally (i.p) administrated a mixture of in vivo jetPEI-PHLPP1/2 or −GFP control constructs, one of five injections on every alternate day, and sacrificed after 10 days, one day after the last injection.
(N and O) Pooled TUNEL analysis (N) and double staining for TUNEL (red) and insulin (green) (O) of isolated pancreases from GFP- or PHLPP1/2-transfected mice (n = 3; an average of 13,618 β cells were counted from each treatment condition).
Data are expressed as means ± SEM. *p < 0.05 compared with untreated or nondiabetic or LacZ or GFP control. White scale bars depict 10 μm.
To examine the role of PHLPP1/2 in β-cell death, we checked whether PHLPP1/2 overexpression alone is sufficient to promote β-cell apoptosis. PHLPP1/2 overexpression in INS-1E cells and human islets achieved by adenoviral gene transfer induced human (Figure 1J) and rodent (Figure S1A) β-cell apoptosis. In addition, overexpression of PHLPPs impaired glucose-stimulated insulin secretion (GSIS) in isolated human islets (Figures 1K and 1L) suggesting its detrimental role on both β-cell survival and insulin secretion.
To investigate whether PHLPPs directly induce β-cell death in vivo, we used the Polyplus-transfection reagent jetPEI, a polyethylenimine-based delivery system for safe and efficient introduction of nucleic acids into tissues in vivo as successfully reported previously for delivery into islets (Goyal et al., 2019; Kim et al., 2013). A solution of jetPEI carrier complexed with either HA-PHLPP1- and 2- or GFP (control)-expressing constructs was injected intraperitoneally (i.p.) into nondiabetic, wild-type (WT) mice. Mice were given a total of five injections every alternate day and sacrificed 1 day after the last injection (Figure 1M). PHLPP overexpression in islets was evaluated ex vivo. Immunohistochemistry of pancreatic sections as well as immunoblot analyses of isolated islets showed successful β-cell/islet upregulation of PHLPPs (Figures S1B and S1C). Chronic administration of PHLPPs significantly induced β-cell apoptosis, as compared with control GFP plasmids, represented by elevated TUNEL-positive β-cells (Figures 1N and 1O) as well as increased levels of caspase-3 cleavage (Figure S1C), a universal marker of apoptosis. In addition, our data show that there was no change in either proliferation or β-cell mass, suggesting that despite promoting apoptosis, a short time of 10-day PHLPP overexpression did not change β-cell mass (Figures S1D and S1E). Together, these data suggest that PHLPPs are highly elevated in diabetic β-cells and are harmful for β-cell survival.
PHLPP1/2 inhibit pro-survival AKT and activate pro-apoptotic MST1 signaling in pancreatic β-cells
To better understand the detrimental function of upregulated PHLPPs in β-cells, we analyzed the key down-stream substrates of PHLPPs. The first well established physiological substrate of PHLPP1/2 is AKT. PHLPP1 and PHLPP2 inactivate AKT through de-phosphorylation of AKT at Ser473 (Brognard et al., 2007; Gao et al., 2005). In light of the essential role of AKT in survival and adaptive growth of the pancreatic β cells (Elghazi and Bernal-Mizrachi, 2009; Yuan et al., 2018), we first sought to analyze the PHLPP-AKT axis that might coordinately control β-cell viability. PHLPP1/2 overexpression reduced AKT-Ser473 phosphorylation in human islets (Figure 2A) and in INS-1E cells (Figure 2B). Conversely, PHLPP1/2 knockdown in INS-1E cells enhanced phospho-AKT levels (Figure 2C), consistent with our observation in PHLPP1-deleted mouse embryonic fibroblasts (MEFs; Figure S2A) and isolated mouse islets (Figure 2D). Mitogens, such as insulin-like growth factor I (IGF-I) and insulin, exert their cell survival action primarily through phosphorylation and activation of AKT in the IRS-PI3K pathway (Trumper et al., 2000; Tuttle et al., 2001). In this line, we wondered whether modulation of PHLPP1/2 might alter insulin- or IGF-I-induced AKT phosphorylation by direct functional regulation of AKT. Indeed, ectopic overexpression of PHLPP1/2 alone or together diminished stimulated AKT phosphorylation in β cells (Figures S2B and S2C). Similar changes occurred in vivo; pancreatic islets isolated from regularly fed in vivo jetPEI-PHLPP1/2-transfected mice showed a decrease in AKT-Ser473 phosphorylation (Figure S2D).
Figure 2. PHLPP1/2 inhibits pro-survival AKT and activates pro-apoptotic MST1 signaling in pancreatic β-cells.
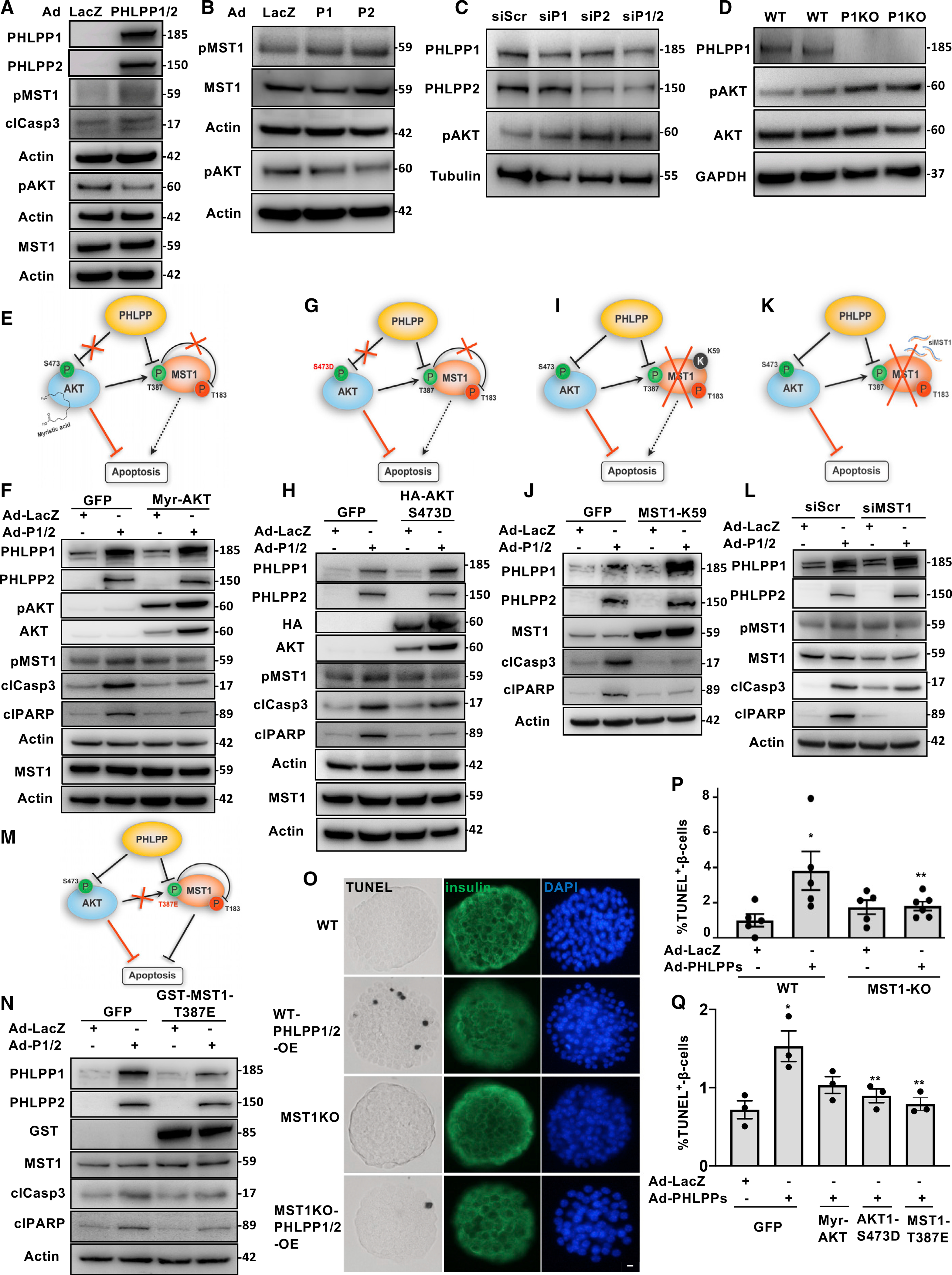
(A–D) Representative western blots of isolated human islets (A) and INS-1E cells (B) transduced with LacZ control or PHLPP1 and PHLPP2 adenoviruses for 48 h or (C) transfected with PHLPP1 and/or PHLPP2 siRNA or control siScr for 2 days. (D) Representative western blots of islets isolated from WT and PHLPP1-KO mice (A–D: n = 3).
(E–N) Schematic cartoons and representative western blots of INS-1E cells overexpressed with adenoviruses for LacZ (control) or PHLPP1/2 and transfected with GFP or siScr (control), Myr-AKT1 (E and F), HA-tagged AKT-S473D (G and H), kinase dead MST1-K59R (I and J), siRNA to MST1 (K and L), or MST1-T387E (M and N) plasmids (all n = 2).
(O and P) Representative images of triple staining for TUNEL (black), insulin (green) and DAPI (blue) (O; scale bar depicts 10 μm) and pooled TUNEL analysis (P) of isolated islets from MST1-KO mice and their WT littermates after transduction with adenoviruses for LacZ (control) or PHLPP1/2 (n = 5–6; an average of 15,301 β cells were counted from each treatment condition).
(Q) Pooled TUNEL analysis of isolated human islets overexpressed with adenoviruses for LacZ (control) or PHLPP1/2 and transfected with GFP (control) or Myr-AKT1 or HA-tagged AKT-S473D, or MST1-T387E plasmids (n = 3; an average of 14,034 β cells were counted from each treatment condition).
Data are expressed as means ± SEM. *p < 0.05 compared with LacZ control. **p < 0.05 MST1-KO-PHLPP or PHLPP-AKT-S473D or PHLPP- MST1-T387E compared with WT-PHLPP or PHLPP-GFP.
The second important PHLPP1/2 target is mammalian sterile 20-like kinase 1 (MST1); PHLPP1/2 directly bind and activate pro-apoptotic MST1 signaling by dephosphorylation at the auto-inhibitory MST1-Thr-387 site (Qiao et al., 2010). Because MST1 is a key regulator of β-cell survival in diabetes (Ardestani et al., 2014), we next investigated the potential upstream role of PHLPPs on MST1 regulation of β-cell apoptosis. PHLPP1/2 overexpression activated MST1 as manifested by increased phosphorylation at the MST1-Thr183-activating residue in human islets (Figure 2A) and INS-1E cells (Figure 2B). Because the PHLPP substrates AKT and MST1 mutually inhibit each other (Ardestani et al., 2014), several complementary experimental settings were designed to investigate the PHLPP-AKT-MST1 crosstalk in β cells in depth:
We started with overexpression of Myr-AKT1, a constitutively active form of AKT with a myristoylation sequence attached to the membrane, which is not sensitive to PHLPPs (Figure 2E). Myr-AKT1 counteracted PHLPP-induced MST1 activation and caspase-3 and PARP cleavage (apoptosis readouts) in β cells (Figure 2F), suggesting that the lack of PHLPP-induced AKT inhibition antagonizes MST1 activation and apoptosis.
In the second experiment, INS-1E cells were transfected with the phospho-mimetic mutant AKT1-S473D, in which serine 473 is permanently replaced with the phospho-mimetic amino acid aspartic acid. Thus, AKT can no longer be dephosphorylated by PHLPP and is constitutively active (Figure 2G). AKT1-S473D mutant suppressed MST1 activation and β-cell apoptosis triggered by PHLPP overexpression (Figure 2H), indicating the critical role of AKT-Ser473 phosphorylation in regulating AKT as well as MST1 activity and subsequent survival.
In the third experiment, the kinase-dead mutant of MST1 (K59R-MST1; Figure 2I) was overexpressed. The critical lysine in the ATP binding pocket of MST1 kinase is mutated so that it cannot receive any ATP and, thus, is inactive (Yamamoto et al., 2003). K59R-MST1 antagonized the pro-apoptotic effect of PHLPPs overexpression in β cells as shown by reduced caspase-3 and PARP cleavage (Figure 2J).
To further confirm whether MST1 hyperactivity has a role in β-cell apoptosis upon PHLPPs overexpression, small-interfering RNA (siRNA) was used to suppress MST1 expression in INS-1E cells (Figure 2K). Consistent with the dominant-negative results described in Figure 2J, MST1 silencing abrogated PHLPP-induced β-cell apoptosis, especially seen by the fully diminished PARP cleavage (Figure 2L).
Given that PHLPP-mediated dephosphorylation of MST1 at the Thr387 inhibitory site increases the activity of MST1 (Qiao et al., 2010), we examined whether MST1-Thr387 phosphorylation mediates the PHLPP-dependent regulation of MST1 in β-cells. INS-1E cells were transfected with the phospho-mimetic mutant MST1-T387E, in which threonine 387 is permanently replaced by the phospho-mimetic amino acid glutamic acid. Thus, MST1 is no longer dephosphorylated by PHLPP and MST1-T387E mimics the inhibitory phosphorylation of MST1 rendering the protein inactive (Figure 2M). Overexpression of the MST1-T387E mutant reduced PHLPP-induced β-cell apoptosis (Figure 2N), showing the important role of MST1-T387 dephosphorylation by PHLPP in the regulation of MST1 activation and β-cell death.
To further support the role of MST1 as the main mediator of PHLPP-induced apoptosis in β-cells, we isolated islets from MST1 knockout (MST1-KO) or WT littermate mice and found that MST1-KO islets were largely resistant to PHLPP1/2-induced apoptosis (Figures 2O, 2P, and S2E). We also tested whether decreased AKT or increased MST1 activities were responsible for PHLPP-induced β-cell death in human islets by direct introduction of PHLPP-insensitive AKT and MST1 mutants. Both AKT/MST1 mutants significantly abolished the number of TUNEL-positive apoptotic human β-cells upon PHLPPs overexpression confirming previous observations in rodent β-cells (Figures 2Q and S2F).
In summary, multiple gain- and loss-of-function experimental approaches targeting endogenous AKT and MST1 activities as well as PHLPP site-specific mutation analysis using phospho-mimetic mutants of AKT (AKT-S473D) and MST1 (MST1-T387E) showed that the critical kinases AKT and MST1 regulate the pro-apoptotic action of PHLPPs in β cells.
mTORC1 hyper-activation induces PHLPPs translation
Chronic exposure of INS-1E cells as well as human islets to elevated glucose concentrations upregulated PHLPPs levels (Figure 1) without changing PHLPP1/2 mRNA expression (Figures S3A and S3B) as well as PHLPP1/2 protein stability (Figure S3C), suggesting that high-glucose-induced PHLPP1/2 induction neither occurred at transcriptional nor post-translational levels. The mechanistic target of rapamycin complex 1 (mTORC1) signaling is a principal regulator of protein translation to control major cellular functions, such as metabolism, growth, and survival (Saxton and Sabatini, 2017). We have previously reported aberrant mTORC1 hyper-activation in diabetic islets (Yuan et al., 2017). To define whether mTORC1 regulates PHLPPs expression, mTORC1 signaling was inhibited by chemical inhibitors against mTORC1 (rapamycin) and S6K1, a major down-stream effector of mTORC1 (PF-4708671; S6K1i) (Pearce et al., 2010). Activation of mTORC1 was demonstrated by increased phosphorylation of its downstream target S6K1 at Thr389 (pS6K), and the direct S6K substrate ribosomal protein S6 at Ser235/236 (pS6), as well as eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) at Thr37/46 (p4E-BP1). In TSC2-KO MEFs, an experimental model of constitutive mTORC1 activation (Bachar et al., 2009), PHLPP1/2 were highly upregulated compared with WT MEFs, and blocking mTOR signaling by rapamycin resulted in a marked decrease of PHLPP1/2 protein expression in TSC2-KO, but not in WT, MEFs (Figure S3D). Likewise, in pancreatic β cells under glucotoxic conditions, mTORC1 inhibition by rapamycin resulted in decreased levels of pS6K1, pS6, and p4EBP1 (mTORC1 readouts) and blocked high-glucose-induced PHLPP1 and PHLPP2 upregulation in INS-1E cells (Figure 3A) and in isolated human islets (Figure 3B) providing direct evidence of the PHLPP regulation by mTORC1, which was upregulated upon chronic exposure to increased glucose concentrations (Figures 3A and 3B). Consistently, pharmacological inhibition of S6K1 by PF-4708671 suppressed high-glucose-induced PHLPPs induction in β cells (Figure S3E). In line with that observation, selective inhibition of endogenous mTORC1 by siRNA-mediated silencing of raptor, mTORC1’s critical subunit, counteracted mTORC1 signaling, reduced PHLPPs levels, and substantially protected INS-1E cells from high-glucose-induced MST1 activation and apoptosis (Figure 3C). Likewise, knockdown of S6K1 demonstrated that the depletion of mTORC1 down-stream signaling markedly reduced PHLPPs as well as MST1 activation and apoptosis (Figure 3D). This further corroborated hyper-activated mTORC1 as an up-stream regulator of PHLPP1/2 expression in the context of glucose-induced β-cell apoptosis. To further explore whether mTORC1-S6K signaling is a principal regulator of PHLPPs in β cells, we overexpressed the constitutively active form of S6K1 in INS-1E cells. Notably, sustained S6K1 overexpression effectively induced PHLPP1/2; this was accompanied by higher MST1 phosphorylation, recapitulating hallmarks of metabolically stressed β cells cultured under diabetes-associated glucotoxic conditions, including induction of PHLPP1/2 as well as activating MST1 (Figure 3E). Likewise, pharmacological induction of mTORC1 by the small-molecule mTOR activator 3-benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-one (3BDO) (Ge et al., 2014; Peng et al., 2014) or MHY1485 (Choi et al., 2012) upregulated PHLPPs, activated pro-apoptotic MST1, and impaired β-cell survival in INS1-E β-cells (Figures S3F–S3I).
Figure 3. mTORC1 hyper-activation induces PHLPPs translation.
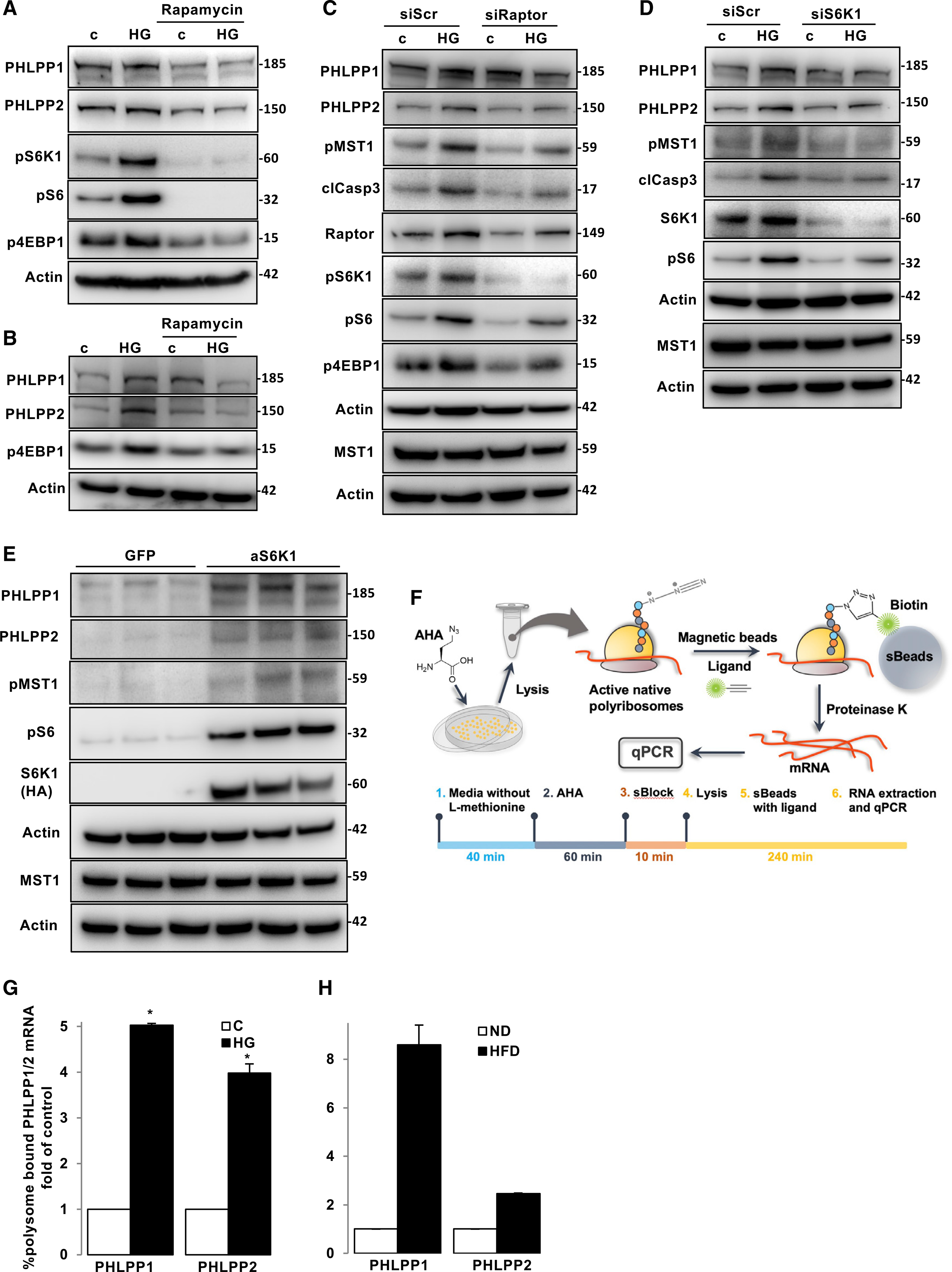
(A and B) Representative western blots of INS-1E cells (A) and isolated human islets (B) pre-treated with 100 nM rapamycin and cultured with 22.2 mM glucose for 2 (INS-1E) and 3 (human islets) days. n = 3.
(C and D) Representative western blots of INS-1E cells transfected with siS6K1 (C), siRaptor (D) or siScr and then exposed to 22.2 mM glucose for 2 days. n = 3
(E) Representative western blots of INS-1E cells transfected with active S6K1 or GFP control plasmids for 2 days. n = 3.
(F) Experimental strategy of the AHARIBO-based isolation of active polyribosomes and associated RNAs.
(G and H) qPCR measurement of PHLPP1 or PHLPP2 mRNA associated with polysomes of INS-1E cells treated with high glucose (G) (n = 3) or of isolated islets from mice fed for 16 weeks with a ND or HFD (H) (n = 2 independent experiments; each pooled from 8–10 mice/condition).
Data are expressed as means ± SEM. *p < 0.05 compared with untreated controls.
An elegant previous study used high-resolution transcriptome-scale ribosome profiling to show that subsets of mRNAs that are specifically regulated by mTORC1 at the translational level consist of established 5′ terminal oligopyrimidine (TOP) or previously unrecognized TOP-like motifs (Thoreen et al., 2012). Interestingly, the 5′ untranslated region (UTR) of PHLPP1 mRNA contains a TOP motif (5′-CTTCTCCCTTCTCC-3′) and PHLPP2 mRNA contains a TOP-like motif (5′-CCTTGCC-3′), proposing potential mTORC1-dependent regulation of PHLPPs at the translational level (Liu et al., 2011a; Wen et al., 2013). Because mTORC1 induces PHLPPs, we hypothesized that the expression of PHLPP1/2 under diabetic conditions is upregulated at the translational level. We have used AHARIBO, a minimally invasive, non-canonical amino acid tagging and isolation method of active polyribosomes (RiboMINATI). The protocol relies on the pulse incubation of cell cultures with l-azidohomoalanine (AHA). Then, a small molecule (sBlock) blocks the nascent peptide attached to the ribosome. A biotin tag is linked to the newly synthesized AHA-labeled proteins, and mRNAs associated to polysomes are analyzed by qPCR (Figure 3F). High-glucose-treated INS-1E cells (Figure 3G) and islets isolated from long-term HFD-fed diabetic mice (Figure 3H) displayed a higher percentage of PHLPP1/2 mRNAs bound to polysomes as compared with WT islets, suggesting a marked elevation in PHLPPs translation.
Altogether, these results indicate that PHLPP levels under diabetic conditions are regulated by mTORC1 at the translational level.
Loss of PHLPPs attenuated stress-induced β-cell injury in vitro and in vivo
To examine whether PHLPP1/2 upregulation is causative for β-cell apoptosis, islets isolated from WT and PHLPP1-KO mice were chronically exposed to glucolipotoxic conditions as well as the mixture of pro-inflammatory cytokines interleukin-1beta (IL-1β) and interferon gamma (IFN-γ). PHLPP1 deletion potently inhibited pro-inflammatory cytokine- as well as high-glucose/palmitate-induced β-cell death (Figure 4A). In addition, in human islets transfected with siRNAs directed to PHLPP1 and/or PHLPP2 before exposure to pro-diabetic stimuli (Figure S4A), apoptosis triggered by pro-inflammatory cytokines as well as by the mixture of high-glucose/palmitate was significantly abolished by the knockdown of PHLPP1 or PHLPP2 individually or together (Figure 4B).
Figure 4. Loss of PHLPPs attenuated stress-induced β-cell injury in vitro and in vivo.
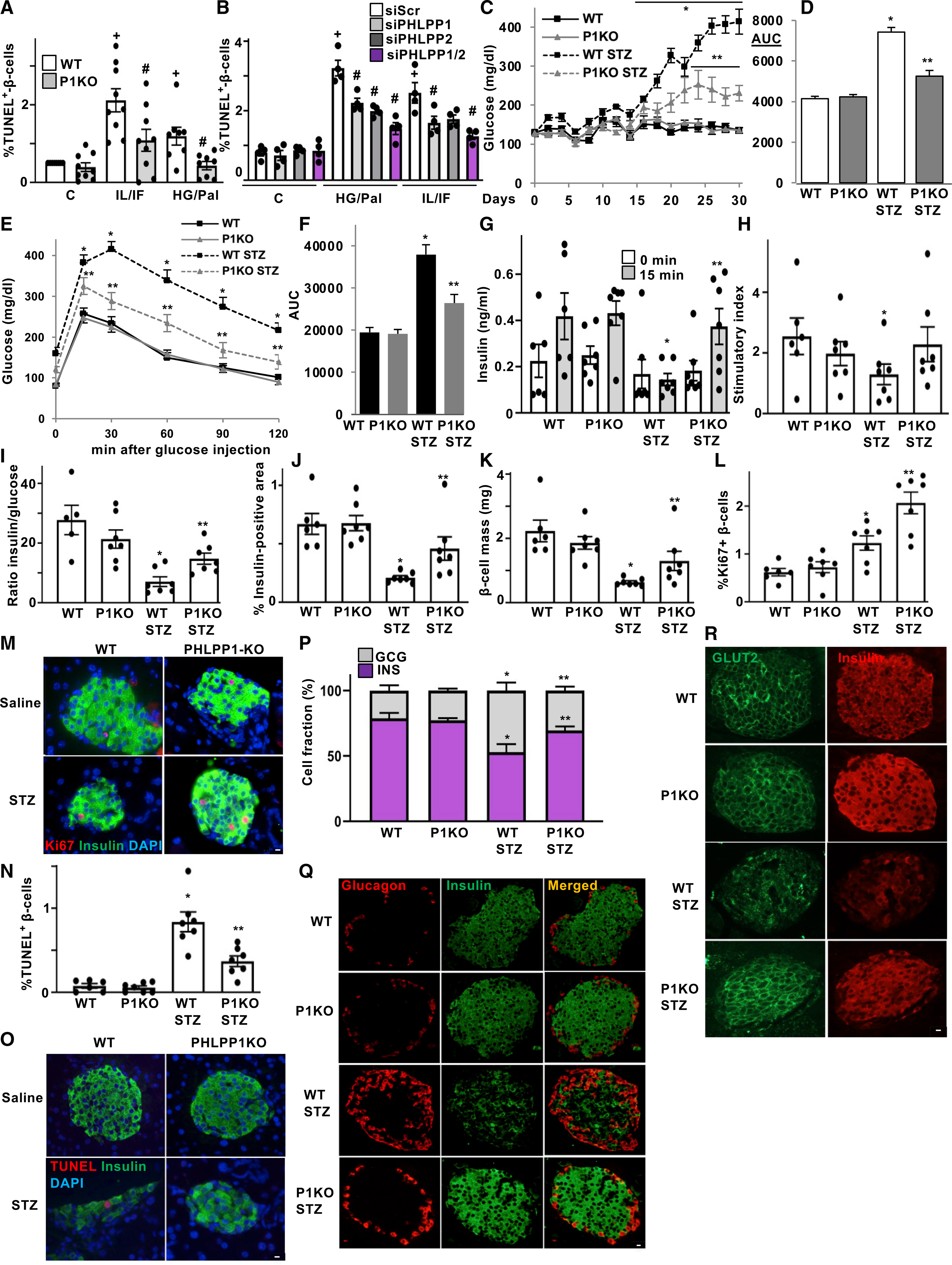
(A and B) TUNEL analysis of isolated islets from PHLPP1-KO mice and their WT littermates (A) and of isolated human islets transfected with PHLPP1 and/or PHLPP2 siRNA or control siScr (B) and then treated with 22.2 mM glucose plus 0.5 mM palmitate (HG/Pal) or the mixture of 2 ng/mL IL1β plus 1,000 U/mL IFN-γ (IL/IF) for 3 days. n = 4–9.
(C–R) PHLPP1-KO and WT control mice injected with streptozotocin (STZ; 40 mg per kg body weight) or saline for 5 consecutive days (n = 6–7). (C) Random-fed blood glucose measurements after first saline or STZ injection (day 0) over 30 days and (D) respective area-under-the curve (AUC) analyses. (E) i.p. glucose tolerance test (GTT) and (F) respective AUC analyses in PHLPP1-KO and WT mice. (G) Insulin levels during an i.p. GTT measured before (0 min) and 15 min after glucose injection and expressed (H) as the ratio of secreted insulin at 15 to 0 min (stimulatory index). (I) Ratio of secreted insulin and glucose calculated at the fed state. (J) Insulin-positive area and (K) β-cell mass (given as the percentage of insulin-positive to the entire pancreatic section area from 10 sections spanning the width of the pancreas). (L–O) Quantitative analyses and representative images from triple staining for Ki67 (L and M; an average of 11,609 β cells were counted from each treatment condition) or TUNEL (N and O; an average of 12,733 β cells were counted from each treatment condition), insulin, and DAPI; expressed as the percentage of TUNEL- or Ki67-positive β cells ± SEM (P and Q) Quantitative analyses (P) and representative images (Q) of the percentage of α cells (red) and β cells (green). (R) Representative double-staining for Glut2 (green), and insulin (red).
Data are expressed as means ± SEM. +p < 0.05 versus untreated control. #p < 0.05 PHLPP1-KO or siPHLPP1/2 versus WT or siScr at the same treatment conditions. *p < 0.05 WT-STZ compared with WT saline-injected mice, **p < 0.05 PHLPP1-KO-STZ versus WT-STZ mice. White scale bars depict 10 μm.
Because PHLPP1/2 silencing improved islet β-cell survival under diabetic conditions in vitro, we hypothesized that PHLPP1 deficiency might be beneficial against β-cell injury and diabetes development in vivo. PHLPP1-KO mice are viable, fertile, and showed no significant differences in basal glycemia, food intake, and body weight compared to WT control mice (Figures 5, S5A, and S5B). We tested whether PHLPP1-KO mice are protected from diabetes progression in the multiple low-dose streptozotocin (MLD-STZ) model of β-cell destruction and diabetes (Horwitz et al., 2018; Luo et al., 2019). MLD-STZ for 5 consecutive days induced progressive hyperglycemia and glucose intolerance rendering WT mice overtly diabetic, whereas blood glucose in PHLPP1-KO mice was robustly attenuated (Figures 4C and 4D), and glucose tolerance significantly improved at all time points (Figures 4E and 4F). In line with the impairment in glucose tolerance, glucose-induced insulin secretion was fully blunted in the MLD-STZ-treated WT mice, whereas PHLPP1-KO animals exhibited a marked restoration in insulin secretion 15 min after the glucose challenge; stimulatory index was unchanged compared with non-STZ injected mice, together with a significantly increased insulin-to-glucose ratio in PHLPP1-KO mice, compared with STZ-injected WT controls (Figures 4G–4I).
Figure 5. PHLPP1 deletion protects from HFD-induced diabetes.
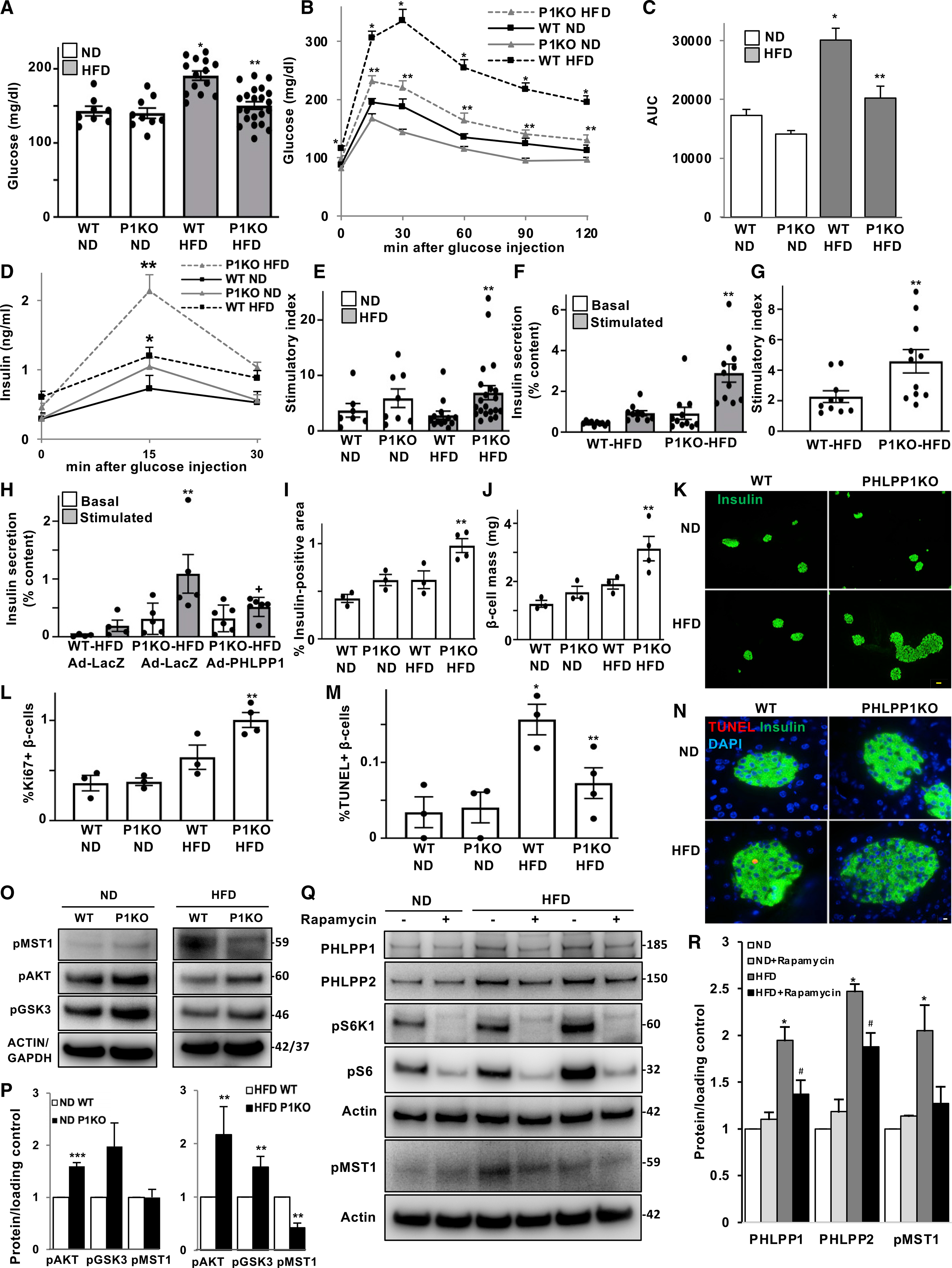
(A–C) PHLPP1-KO and WT control mice were fed an ND or an HFD (“Surwit”) for 17 weeks. (A) Random-fed blood glucose, (B) i.p. GTT, and (C) respective AUC analyses. n = 8–22.
(D and E) Insulin secretion during an i.p. GTT measured before (0 min), 15 and 30 min after glucose injection and expressed as (E) the ratio of secreted insulin at 15 to 0 min (stimulatory index) (n = 7–21).
(F and G) Islets were isolated from all four treatment groups, cultured overnight, and subjected to an in vitro GSIS (F). Insulin secretion during 1 h of incubation with 2.8 mM (basal) and 16.7 mM glucose (stimulated), normalized to insulin content, and (G) the stimulatory index denotes the ratio of stimulated to basal insulin secretion (n = 10–11).
(H) Islets isolated from HFD-fed groups transduced with LacZ control or PHLPP1 adenoviruses for 1 day and subjected to an in vitro GSIS (n = 4–6).
(I–K) Insulin-positive area (I) and β-cell mass (J) (given as percentage of the insulin-positive area to the entire pancreatic section from 10 sections spanning the width of the pancreas) and (K) respective representative images (n = 3–4; yellow scale bar depicts 50 μm).
(L–N) Quantitative analyses from double/triple staining for Ki67 (L) or TUNEL (M) (and representative images: N; white scale bar scale depicts 10 μm), insulin, and DAPI expressed as percentage of Ki67- or TUNEL-positive β cells (n = 3–4; an average of 7,648 [Ki67] or 9,009 [TUNEL] β cells were counted from each treatment condition).
(O and P) Representative western blots (O) and quantitative densitometry analysis (P) of isolated islets from WT and PHLPP1-KO mice fed an ND or an HFD (n = 3–4). Islet samples isolated from mice under ND or HFD were run on different gels. For each cohort, protein expression signal is normalized to the corresponding WT mice and quantitative densitometry analysis as a fold of the change is presented separately.
(Q and R) Representative western blots (Q) and quantitative densitometry analysis (R) of islets isolated from ND- and HFD-fed mice treated with 100 nM rapamycin (PHLPP1/PHLPP2, n = 7; pMST1, n = 3).
Data are expressed as means ± SEM. *p < 0.05 WT-HFD versus WT-ND mice, **p < 0.05 PHLPP1-KO-HFD versus WT-HFD mice, ***p < 0.05 PHLPP1-KO-ND versus WT-ND, +p < 0.05 HFD-PHLPP1-KO-AdPHLPP1 versus HFD-PHLPP1-KO-LacZ, #p < 0.05 rapamycin-HFD versus control-HFD.
Consistent with the metabolic improvements, β-cell volume and β-cell mass were significantly restored in PHLPP1-KO, compared with STZ-WT mice (Figures 4J and 4K). To determine whether the regeneration of the β-cell mass was a result of an increased β-cell number because of augmented β-cell replication and/or decreased β-cell apoptosis, we further assessed the effects of PHLPP1 ablation on β-cell survival and proliferation. Together with increased β-cell apoptosis in WT-STZ animals, β-cell proliferation—as represented by double-labeled Ki67/insulin-positive β-cells—was elevated, showing an enhanced compensatory capacity in response to STZ-induced β-cell injury (Figures 4L–4O). PHLPP1 deletion fostered β-cell proliferation (Figures 4L and M) and suppressed β-cell apoptosis (Figures 4N and 4O), compared with the STZ-WT group. Loss of PHLPP1 had no effect on basal β-cell mass and turnover (apoptosis/proliferation) in non-diabetic mice. These results suggest that PHLPP1 ablation restores β-cell mass predominantly as a result of reduced β-cell apoptosis and a demand for β-cell compensation. Islet cells from MLD-STZ-treated WT mice were architecturally disrupted, with fewer insulin-positive β cells and proportionally more glucagon-positive α cells compared with that of untreated WT mice (Figures 4P and 4Q). In contrast, the percentage of glucagon-positive α cells as well as insulin-positive β cells in MLD-STZ-injected PHLPP1-KO islets was similar to non-STZ-treated WT control mice and confined to the rim of the islets (Figures 4P and 4Q).
We next checked whether PHLPP1 deficiency could also restore the expression of several key markers of the glucose-sensing machinery as well as of insulin production. Immunostaining of pancreatic sections from STZ-treated mice showed a profound loss in both nuclear PDX1- and NKX6.1-positive cells. Many cells within the islets, which still express insulin, had lost their PDX1 or NKX6.1 expression. Such PDX1/NKX6.1 protein expression was markedly restored by PHLPP1 inhibition (Figures S4B and S4C). Consistently, although the expression of the PDX1 canonical down-stream target GLUT2 was reduced and its membrane localization disrupted in β cells of MLD-STZ-treated WT mice, GLUT2 expression as well as its membrane localization was largely preserved in MLD-STZ-treated PHLPP1-KO islets, compared with that of STZ-injected WT mice (Figure 4R). This was also confirmed in vitro in isolated mouse islets, in which PHLPP1 deletion restored the STZ-induced loss of GLUT2 expression (Figure S4D).
The combination of these metabolic and morphological data suggests that PHLPP1 ablation leads to enhanced β-cell mass and proliferation, reduced apoptosis, and restored insulin secretion and glucose tolerance in an in vivo model of β-cell destruction and diabetes.
PHLPP1 deletion protects from HFD-induced diabetes
To further characterize the physiological relevance of our findings in a second, diet-induced diabetes model, we sought to examine whether PHLPP1 is indispensable for the long-term β-cell compensatory response in the diet-induced obesity mouse model of HFD-induced diabetes. For this purpose, WT and PHLPP1-KO male mice were placed on either ND or HFD for 17 weeks, which led to chronic hyperglycemia, insulin resistance, and β-cell failure in WT mice (Ardestani et al., 2014; Collins et al., 2010). On an ND diet, PHLPP1-KO mice were normal, healthy, and indistinguishable from WT controls; systemic PHLPP1 deletion had no effect on weight gain or on food intake in either ND or HFD groups (Figures S5A and S5B). When maintained on a long-term HFD, WT mice developed mild hyperglycemia and drastic impairment in glucose tolerance, which was robustly attenuated in HFD-treated PHLPP1-KO mice (Figures 5A–5C). To assess whether these metabolic improvements were due to changes in insulin sensitivity, we performed an insulin tolerance test. Under the non-diabetogenic conditions of a chow diet, WT and PHLPP1-KO mice had a similar response to exogenous insulin. Under HFD conditions, PHLPP1-KO mice had a slightly better insulin tolerance compared with that of insulin-resistant WT mice (Figure S5C). However, this is unlikely to be the major mechanism for the robust systemic improvement of glucose tolerance in obese HFD-treated PHLPP1-KO mice. PHLPP1 deletion significantly enhanced insulin secretion as well as stimulatory index during an i.p. glucose challenge in HFD-fed mice (Figures 5D and 5E). To further assess whether the improvements in glucose homeostasis in PHLPP1-KO mice were directly linked to insulin secretion, we measured GSIS ex vivo in isolated islets from HFD-fed groups. Islets from PHLPP1-KO mice on an HFD exhibited substantially increased insulin secretion in response to glucose; also, the stimulatory index was highly improved compared with that of WT-HFD mice (Figures 5F and 5G), recapitulating the in vivo phenotype. We next asked a critical question: does reconstitution of the PHLPP1 reverse the enhancement in glucose responsiveness evident in HFD-treated PHLPP1-KO islets? PHLPP1 overexpression resulted in a reversal in GSIS in PHLPP1-reconstituted islets isolated from HFD-PHLPP1-KO mice compared with LacZ-transduced HFD-PHLPP1-KO counterparts suggesting a potential cell-autonomous action of PHLPP1 in the regulation of insulin secretion (Figure 5H).
Consistent with the improved insulin secretion in vivo and ex vivo, PHLPP1-KO mice displayed a greater compensatory response, i.e., β-cell volume and β-cell mass were significantly increased relative to WT control mice under the HFD diet (Figures 5I–5K). These findings implicate functional β-cell-mass restoration as a key factor for the metabolic benefits in the PHLPP1-KO mice. Similar to the STZ model of β-cell destruction (Figure 4), PHLPP1-KO mice showed significantly more β-cell proliferation (Figure 5L) and fewer β-cell apoptosis (Figures 5M and 5N), compared with that of WT-HFD mice.
In addition, western blot analysis of isolated islets from ND-treated WT and PHLPP1-KO mice showed PHLPP deletion increased phosphorylation of AKT and its downstream target GSK3, whereas MST1 phosphorylation remained unchanged (Figures 5O and 5P). Importantly, genetic inhibition of PHLPP1 resulted in suppressed MST1 activation and restored AKT activation in islets isolated from HFD-subjected mice (Figures 5O and 5P), confirming the regulatory MST1-AKT axis downstream of PHLPPs in vivo in diabetic islets. Because mTORC1 induced PHLPPs in vitro, we also investigated whether diabetes-induced PHLPP expression is regulated by mTORC1 in ND- or HFD-treated mouse islets. Similar to INS-1E cells and human islets cultured under high-glucose conditions, mTORC1 inhibition by rapamycin reduced the levels of both PHLPP isoforms in isolated islets from HFD-treated diabetic mice (Figures 5Q and 5R), further confirming mTORC1 signaling as an upstream regulator of PHLPPs. In addition, rapamycin did not significantly affect the PHLPP levels under ND control conditions (Figures 5Q and 5R).
Altogether, PHLPP1 genetic inhibition elicited a robust glucose-lowering response in obese hyperglycemic mice through—at least, in part—a coordinated increase in both β-cell mass and secretory function.
Genetic inhibition of PHLPP1/2 improves insulin secretion and β-cell survival in human islets from patients with T2D
To identify whether the observed PHLPP1/2 upregulation in research models of diabetes is relevant to human T2D and may contribute to its pathogenesis, we investigated islets from patients with T2D. Western blot analysis of human islets from patients with T2D exhibited significant upregulation of PHLPP1/2, compared with islets from non-diabetic individuals (Figures 6A and 6B). In line with the pattern seen under a pro-diabetic milieu in vitro, PHLPP1 and PHLPP2 mRNA abundance was unchanged in T2D human islets (Figure S6A), again indicating that a post-transcriptional mechanism was responsible for the increase in PHLPPs. Immunofluorescence staining confirmed increased protein expression of PHLPP2 in the insulin-positive area in islets from paraffin-embedded pancreas sections from autopsies of patients with T2D, in comparison to an only faint PHLPP2 expression in non-diabetic controls (Figure 6C). Increased β-cell apoptosis as a major pathological feature of diabetes was observed in human islets under the same experimental settings (Dharmadhikari et al., 2017). To understand the patho-physiological effect of increased PHLPPs in human T2D islets, siRNA-mediated knockdown was used to examine whether PHLPP inhibition restores β-cell function and survival (Figure S6B). Importantly, although genetic inhibition of PHLPP1 or PHLPP2 alone or together had no effect on insulin secretion in the nondiabetic cohort, their loss significantly improved GSIS in five independent human islet preparations isolated from organ donors with T2D (Figures 6D and 6E). In addition, β-cell viability was restored by genetic inhibition of PHLPPs in T2D islets (Figure 6F). Moreover, targeted inhibition of endogenous mTORC1 by siRNA-mediated silencing of raptor in isolated T2D islets substantially decreased PHLPPs (Figures 6G and 6H), further indicating the mTORC1-dependent PHLPPs upregulation in human T2D islets, in confirmation with the PHLPP reduction seen by mTORC1 inhibition in β cells and human islets under long-term high-glucose treatment (Figure 3). This shows a detrimental effect of abnormally upregulated PHLPPs on β-cell function and survival in human T2D islets and suggests that the higher protein expression of PHLPPs might be linked to the impaired insulin secretion and metabolic deterioration in human diabetes (Figure 7).
Figure 6. Genetic inhibition of PHLPP1/2 improved insulin secretion and β-cell survival in human islets from patients with T2D.
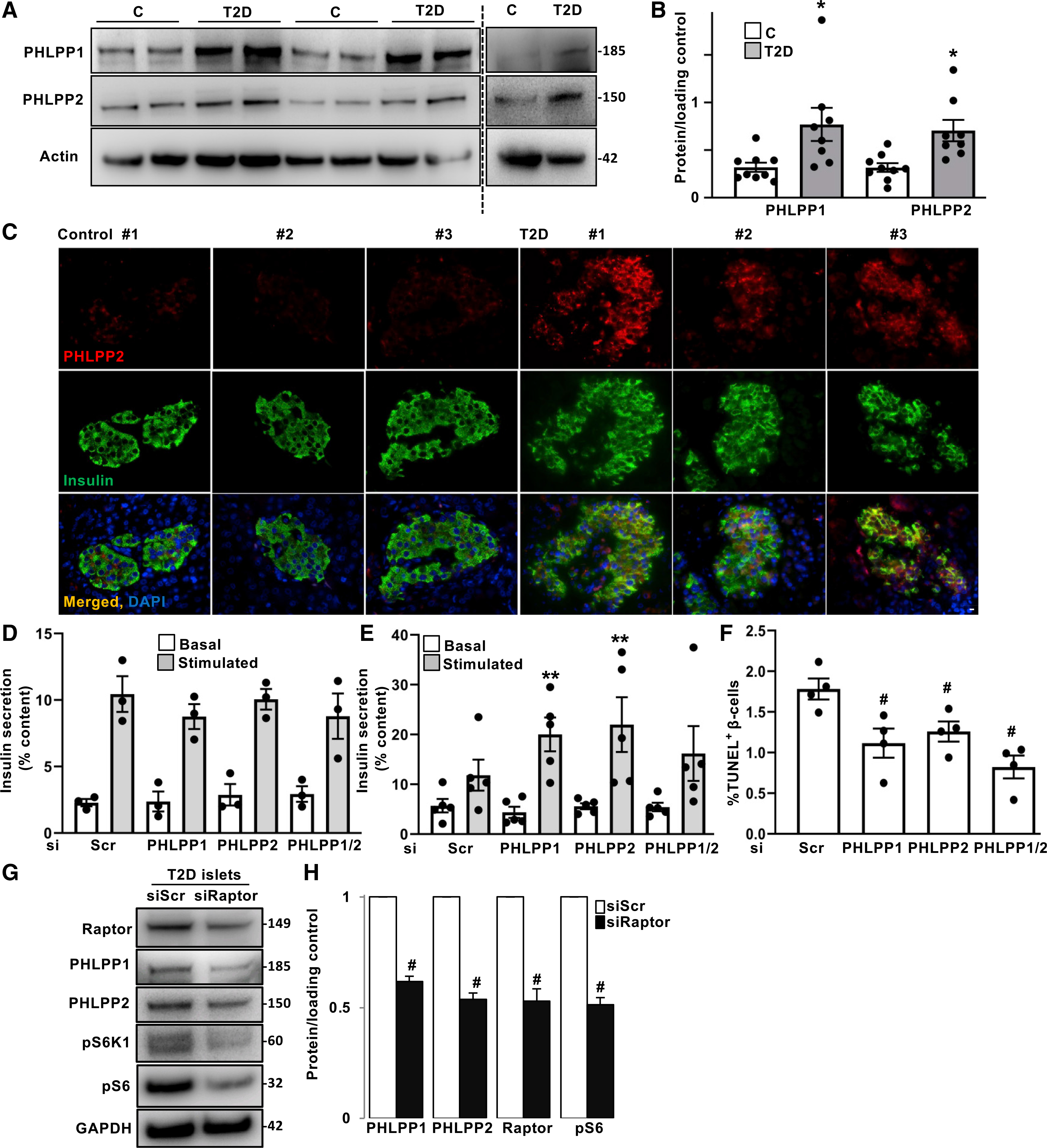
(A and B) Representative western blots (A) and quantitative densitometry analysis (B) of human isolated islets from non-diabetic controls (n = 9) and patients with T2D (n = 8).
(C) Representative images of double immunostaining for PHLPP2 in red and insulin in green of pancreatic autopsy sections from non-diabetic controls (n = 4) and patients with T2D (n = 4; scale bar depicts 10 μm).
(D–F) Isolated human islets from non-diabetic individuals (D) and patients with T2D (E and F) were transfected with PHLPP1 and/or PHLPP2 siRNA or control siScr for 2 days. (D and E) Insulin secretion during 1 h of incubation with 2.8 mM (basal) and 16.7 mM (stimulated) glucose, normalized to insulin content (n = 3 controls; n = 5 T2D; each from three independent replicates, respectively). (F) Pooled TUNEL analysis (n = 4; each from three independent replicates, an average of 2,515 β cells were counted from each treatment condition).
(G and H) Representative western blots (G) and quantitative densitometry analysis (H) of human isolated islets from patients with T2D transfected with raptor siRNA or control siScr for 2 days (n = 3).
Data are expressed as means ± SEM. *p < 0.05 T2D versus control islets. **p < 0.05 siPHLPP1/2-transfected stimulated versus siPHLPP1/2-transfected basal. #p < 0.05 siPHLPP1/2- or siRaptor-transfected compared with siScr-transfected T2D islets.
Figure 7. PHLPPing mTORC1 toward β-cell failure: Graphical summary of the results.
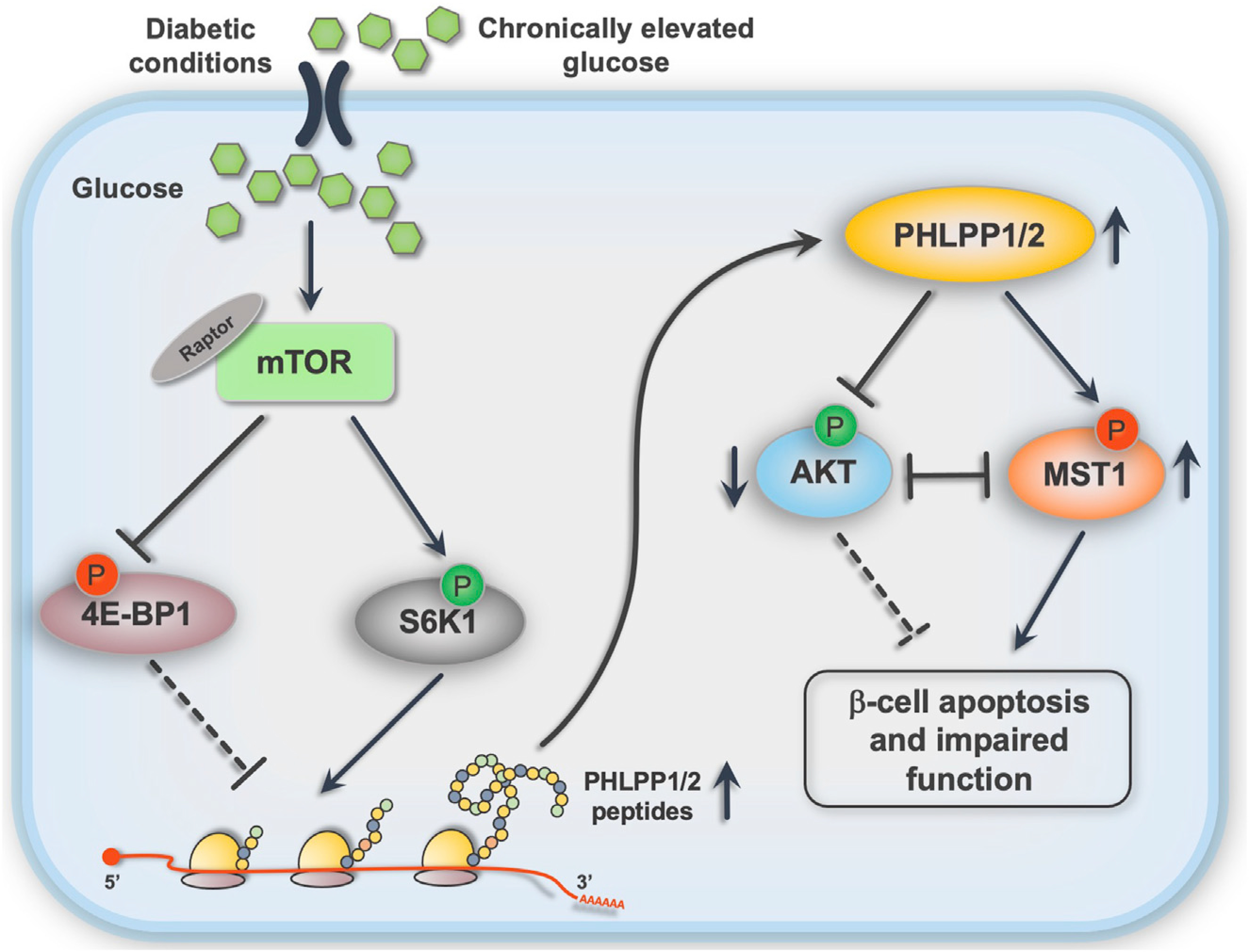
Chronic metabolic stress leads to hyper-activation of mTORC1, promoting the PHLPP translational machinery, which leads to the triangle loop of PHLPP activity, AKT inhibition, and MST1 activation and, ultimately, to β-cell death and dysfunction.
DISCUSSION
In the present study, we provide direct evidence for PHLPP protein upregulation in β-cells as an initiator path toward β-cell failure in diabetes, because (1) PHLPPs were highly upregulated in human islets and β-cells under glucotoxic conditions in vitro and in islets from diabetic mouse models and in patients with T2D; (2) PHLPP1/2 overexpression itself was sufficient to trigger β-cell death and dysfunction; (3) mechanistically, the apoptosis-inducing effects of PHLPP1/2 were mediated not only through the inactivation of AKT pro-survival signaling but also through the activation of pro-apoptotic MST1 kinase, two downstream PHLPP substrates (both of these downstream signals have been implicated in β-cell failure and diabetes before); (4) chronic hyper-activation of mTORC1 was identified as a primary mechanism of PHLPPs upregulation, linking metabolic stress to ultimate β-cell death; and (5) genetic PHLPP1 inhibition protected against glucose intolerance and defective insulin secretion and promoted β-cell survival, β-cell proliferation, and compensatory β-cell mass expansion in two mouse models of diabetes and restored β-cell function and survival in human T2D islets.
Protein phosphorylation/de-phosphorylation is a key biochemical component of intracellular signaling pathways and has a crucial role in the transduction of signals to ultimately decide the fate of cells (Duncan et al., 2010; Takeda et al., 2011). PHLPP phosphatases potently promote cell death by inhibiting proliferative pathways (O’Neill et al., 2013). AKT kinase, which was the first well-established, physiological substrate of PHLPPs, is the master pro-survival kinase in β-cells. Activation of the AKT signaling pathways downstream of mitogen receptors, such as insulin, insulin growth factors (IGF family), and phosphoinositide 3-kinase (PI3K) (Boucher et al., 2014; Taniguchi et al., 2006), has a pivotal role in controlling β-cell growth, proliferation, and apoptosis. AKT-mediated phosphorylation of multiple substrates positively regulates insulin transcription, insulin secretion, and β-cell growth and survival (Assmann et al., 2009; Bernal-Mizrachi et al., 2001; Tuttle et al., 2001). PHLPPs directly de-phosphorylate AKT and inhibit its intrinsic catalytic activity. Consequently, PHLPP-induced inactivation of AKT results in apoptosis and inhibition of cell proliferation (Brognard et al., 2007; Gao et al., 2005). Defective phosphorylation of AKT at Ser473 is an important biochemical hallmark of human and rodent diabetic β-cells (Kim et al., 2012; Shirakawa et al., 2017; Wang et al., 2010; Yuan et al., 2017), but mechanistically, a key upstream element responsible for defective AKT signaling in the β-cell has not yet been well described.
In addition, the second PHLPP downstream target in the β-cell found in this study, MST1 (Jung et al., 2014; Qiao et al., 2010), is crucial for β-cell survival because it acts as an essential apoptotic molecule in the presence of diabetic stimuli and is a common component in the diverse signaling pathways leading to impaired β-cell survival and function in diabetes (Ardestani et al., 2014, 2019). In-depth investigations of the PHLPP downstream pathway demonstrate that PHLPP-AKT-MST1 constitutes a stress-sensitive survival pathway. Under acute stress conditions, AKT promoted cell survival by inhibiting MST1, but prolonged, unresolved metabolic stress upregulated PHLPPs with two obvious functional outputs: (1) a decrease in AKT activity through direct AKT-Ser473 de-phosphorylation, and (2) an increase in MST1 activity through direct MST1-Thr387 de-phosphorylation, which leads, in turn, to auto-phosphorylation of MST1-Thr183 and subsequent MST1 activation to induce apoptosis. Both these mechanisms cumulatively amplified pro-apoptotic MST1 signaling. This Thr387 site is similar to the site that is phosphorylated by AKT to inactivate MST1 and terminate apoptosis (Jang et al., 2007). Thus, PHLPP, AKT, and MST1 form an auto-inhibitory triangle that regulates β-cell apoptosis in a tightly controlled manner. Our data are fully in line with a recently published report that shows upregulation of PHLPP1/2 in INS-1 cells in response to elevated glucose and their link to AKT (Hribal et al., 2020).
mTORC1 signaling is an instrumental pathway in nutrient sensing and the integration of metabolic, energetic, and hormonal stimuli to control cellular metabolism, survival, and anabolic growth (Gonzalez and Hall, 2017; Mossmann et al., 2018; Saxton and Sabatini, 2017). Although physiological mTORC1 activation is necessary for the maintenance of β-cell growth, homeostasis, metabolic compensation, and insulin secretion, its long-term, sustained, aberrant activation—as illustrated in β-cells from patients with T2D and later in rodent islets (Jaafar et al., 2019; Yuan et al., 2017)—can promote β-cell failure, underscoring the dual and complex action of mTORC1 signals in pancreatic β-cells (Ardestani et al., 2018). A prerequisite for rescuing the β-cell from chronic metabolic stress would be to unravel the molecular mechanisms/targets underlying the “pathogenic arm” of inappropriate hyper-activated mTORC1 seen in diabetic β-cells. That would require opening the narrow mTORC1-based therapeutic window and avoiding compromising important mTORC1 homeostatic signals for β-cell homeostasis. We have identified PHLPP1/2 as mediators of the mTORC1-directed β-cell switch under diabetic conditions. Although sustained hyper-activation of mTORC1 upregulated PHLPPs and promoted β-cell apoptosis in metabolically stressed β-cells, both genetic and pharmacological interception of mTORC1 blocked PHLPP1/2 upregulation in response to nutritional stress. These data suggest that PHLPP is an important element of pathogenic mTORC1 signaling and that mTORC1 stimulation is essential for PHLPP1/2 to act as detrimental signals in stressed β-cells. The mTORC1-PHLPP1/2 axis offers a mechanistic link between glucotoxicity and dysregulation of β-cell survival and function. Only indirectly addressed here, the inhibition of PHLPP as an mTORC1 target could restore active AKT levels by halting the mTORC1-PHLPP-AKT loop.
Although suppression of PHLPP would be a desirable approach for β-cell mass preservation or expansion, the effects of prolonged PHLPPs inhibition should not be underestimated because of their function as tumor suppressors, as a logical consequence of apoptosis inhibition. PHLPP1-KO mice develop normally with no anatomical defects, consistent with previously reported studies showing that PHLPP1-KO mice are viable and show no overt changes in growth, anatomy, or development (Chen et al., 2013; Masubuchi et al., 2010). In addition, mice with systemic deletion/inhibition of PHLPP1 show promising neuro-, cardio-, and intestine-protection as well as tissue regeneration in several pathological settings (Chen et al., 2013; Hwang et al., 2018; Jackson et al., 2018; Moc et al., 2015; Wen et al., 2015; Zhang et al., 2019, 2020). In our long-term mouse studies, no tumorigenic features were observed in PHLPP1-KO mice; they live to a relatively old age without development of tumors. This may be due to compensatory actions by the other PHLPP isoform PHLPP2 or by the other AKT phosphatase PTEN (Chen et al., 2011; Molina et al., 2012). Moreover, PHLPP1 heterozygous mice are fully viable and show no growth abnormality compared with that of WT mice (Chen et al., 2011), and relative PHLPP1 deficiency can activate AKT signaling as efficiently as full PHLPP1 deletion (Moc et al., 2015). Nevertheless, although normal physiological functions and life span in PHLPP1-KO animals are generally not affected, PHLPP1 deletion accelerates tumor development in a mouse model of cancer (Li et al., 2014). Obviously, PHLPPs act as tumor suppressors and, thus, control oncogenic pathways. Although their permanent inactivation could lead to cancer development, it should be equally apparent that PHLPPs, and, in general, many other tumor suppressors, such as PTEN (Wang et al., 2010; Yang et al., 2014; Zeng et al., 2013), MST1 (Ardestani et al., 2014), P53 (Kung and Murphy, 2016), or p27Kip1 (Uchida et al., 2005), are required for normal growth, compensatory proliferation, and regeneration of pancreatic β-cells, indicating that “regenerative pathways and oncogenic pathways are the same, differing only in their level, mechanism, and the duration of activation and safe regulatory mechanisms to turn on and off regenerative, and oncogenic pathways will need to be developed before regenerative approaches become accepted” (Wang et al., 2015). Pharmacological inhibition of PHLPPs—if proven to be selective with no or only little acceptable side effects—could recapitulate a moderate, but not absolute, PHLPP inhibition, which is unlikely to lead to uncontrolled cell proliferation and tumor development.
Another feature of PHLPP deletion observed in the study was the normalization of STZ-induced α-cell hyperplasia, which is a classical feature of diabetes (Cho et al., 2011; Dunning and Gerich, 2007; Moin and Butler, 2019; Yoon et al., 2003). The focus of this study was purely on β-cell survival mechanisms during diabetes progression; we cannot exclude a possibility that a β-to-ɑ transdifferentiation might occur in diabetes and could also be regulated by PHLPP.
There is a critical need to develop therapeutic interventions to restore and maintain insulin secretion and β-cell mass in patients with T2D. Our multi-model approach not only shows PHLPPs as key phosphatases regulating β-cell survival but also identifies PHLPP-related up- and down-stream signal transductions that are activated by a pro-diabetic condition. PHLPPs deficiency restored normoglycemia and β-cell function and survival in vivo and in vitro. The identification of PHLPPs as key player in β-cell failure may have potential therapeutic relevance for the preservation and/or restoration of functional β-cell mass and glucose homeostasis in patients with T2D.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary materials. All raw data are available upon reasonable request from the authors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kathrin Maedler (kmaedler@uni-bremen.de)
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data generated from this study are included in this paper. All raw data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell culture, treatment and islet isolation
Human islets were isolated from pancreases of nondiabetic organ donors or donors with type 2 diabetes (both from males and females) at Universities of Illinois at Chicago, Wisconsin, Lille or ProdoLabs and cultured on extra cellular matrix (ECM)-coated dishes (Novamed, Israel) or on Biocoat Collagen I coated dishes (#356400, Corning, ME, USA). The clonal rat β-cell line INS-1E was kindly provided by Claes Wollheim (Geneva & Lund University). Mouse embryonic fibroblasts (MEFs) isolated from Tuberous sclerosis complex 2 knock-out (MEF-TSC2-KO) and respective WT mice (generously provided to our lab by Gil Leibowitz, Hadassah-Hebrew University Medical Center, Jerusalem) or from PHLPP1 knock-out (MEF-PHLPP1-KO) and respective WT mice (generously provided to our lab by Alexandra Newton, UCSD) were cultured in complete DMEM (Sigma-Aldrich, MO, USA) medium at 25 mM glucose. Human islets were cultured in complete CMRL-1066 (Invitrogen, CA, USA) medium at 5.5 mM glucose and mouse islets and INS-1E cells in complete RPMI-1640 (Sigma-Aldrich, MO, USA) medium at 11.1 mM glucose as described previously (Ardestani et al., 2014). Mouse islets were isolated by pancreas perfusion with a Liberase TM (#05401119001; Roche, Switzerland) solution (Ardestani et al., 2019) according to the manufacturer’s instructions and digested at 37°C, followed by washing and handpicking. Human and mouse islets and INS-1E cells were exposed to complex diabetogenic conditions: 22.2 mM glucose, in combination with 0.5 mM palmitic acid, or the mixture of 2 ng/mL recombinant human IL-1β (R&D Systems, MN, USA) plus 1,000 U/ml recombinant human IFN-γ (PeProTech, NJ, USA) for 1–3 days. Palmitic acid was dissolved as described previously (Maedler et al., 2001). In some experiments, cells or primary islets were additionally cultured with 100 nM Rapamycin or 10 μM S6K1 selective inhibitor PF-4708671 (Calbiochem, CA, USA) for 1–2 days, 10 to 20 μM 3-Benzyl-5-((2-nitrophenoxy)methyl)-dihydrofuran-2(3H)-one (3BDO) (J&K Scientific, Belgium) for 3h, 25 μM MHY1485 (Selleck Chemicals, TX, USA) for 3h, 100 ng/ml IGF1 (#407251; Calbiochem, CA, USA), 100 nM recombinant human insulin, 50 μg/ml cycloheximide (CHX) or 1 mM streptozotocin (STZ) (all Sigma-Aldrich). All human islet experiments were performed in the islet biology laboratory, University of Bremen. Human islets were distributed by the two JDRF and NIH supported approved coordination programs in Europe (Islet for Basic Research program; European Consortium for Islet Transplantation ECIT) and in the US (Integrated Islet Distribution Program IIDP) (Hart and Powers, 2019). Autopsy pancreases from non-diabetic controls and from patients with T2D were obtained from the National Disease Research Interchange (NDRI).
Ethical approval for the use of human islets had been granted by the Ethics Committee of the University of Bremen. The study complied with all relevant ethical regulations for work with human cells for research purposes. Organ donors are not identifiable and anonymous, such approved experiments using human islet cells for research is covered by the NIH Exemption 4 (Regulation PHS 398).
Animals
For multiple low dose streptozotocin (MLD-STZ) experiments, 8- to 10-week old male PHLPP1-KO mice (RRID: MGI:5795609; generously provided by Alexandra Newton, UCSD) (Masubuchi et al., 2010) on a C57BL/6J genetic background and their WT littermates were injected with multiple low-dose STZ (40 mg/kg body weight), freshly dissolved in 50 mM sodium citrate buffer, for five consecutive days. For the high-fat, high-sucrose diet (HFD) experiments, 8-week-old male WT and PHLPP1-KO mice were fed a normal diet (ND, Harlan Teklad Rodent Diet 8604, containing 12.2, 57.6 and 30.2% calories from fat, carbohydrate and protein, respectively) or a HFD (Surwit Research Diets, New Brunswick, NJ, containing 58, 26 and 16% calories from fat, carbohydrate and protein, respectively) for 17 weeks. For both groups, random blood was obtained from the tail vein of nonfasted mice, and glucose was measured using a Glucometer (FreeStyle; Abbott, IL, USA). Heterozygous leptin receptor deficient mice on the C57BLKS/J background (Leprdb/+, db/+) were purchased from Jackson Laboratory, ME, USA. By breeding of these mice, we obtained diabetic Leprdb/db (db/db) as well as non-diabetic heterozygous Leprdb/+ (db/+) mice.
All mice used in the experiments were housed in a temperature-controlled room with a 12-h light-dark cycle and were allowed free access to food and water in agreement with NIH animal care guidelines, §8 German animal protection law, German animal welfare legislation and with the guidelines of the Society of Laboratory Animals (GV-SOLAS) and the Federation of Laboratory Animal Science Associations (FELASA).
Ethical approval for the mouse experiments had been granted by the Bremen Senate (Senator for Science, Health and consumer protection) and we have complied with all relevant ethical regulations for animal testing and research.
METHOD DETAILS
Glucose and insulin tolerance tests and insulin secretion
For intraperitoneal glucose tolerant tests (ipGTT), mice were fasted overnight for 12h and injected i.p. with glucose (B.Braun, Germany) at a dose of 1g/kg body weight. Blood samples were collected at time points 0, 15, 30, 60, 90, and 120 min for glucose measurements by using a Glucometer (FreeStyle; Abbott, IL, USA). For i.p. insulin tolerance tests (ipITT), mice were initially fasted for a period of 4 h followed by recombinant human insulin injection (Novo Nordisk, Denmark) at a dose of 0.75 U/kg body weight. Glucose concentration was determined with the Glucometer at time points 0, 15, 30, 60 and 90 min. Blood samples for insulin secretion was collected before (0 min) and after (15 and 30 min) i.p. injection of glucose (2g/kg body weight) and measured by using ultrasensitive mouse ELISA kit (ALPCO Diagnostics, NH, USA).
Plasmids and siRNAs
To knock down PHLPP1, PHLPP2, MST1, raptor and S6K1 SMARTpool technology from Dharmacon, CO, USA was used. A mix of ON-TARGETplus siRNAs directed against the following sequences: rat PHLPP1 (#L-094929–02) sequences CAGCUUGACCUGCGAGACA; GUGAAUAACUUCCGUGACA; UAAUAGUAGUCUCCGGAAA; GAAUGUACAAUGUCCGAAA, rat PHLPP2 (#L-104590–02) sequences ACAAAUGGGCUGAGCGCUU; UAGUCUGAGUCUUCGGAAA; GCAUCUAUAACGUCCGCAA; CCGUGGACCUCUCGUGUUA, human PHLPP1 (#L-019103–00) sequences GAAUGUAUAAUGUCCGUAA; GAUCUAAGGUUGAACGUAA; GGAAUCAACUGGUCACAUU; GAUAUUGGCCAUAAUCAAA, human PHLPP2 (#L-022586–01) sequences CCUAUAUUGUUAUGCGAGA; CCGUGGAUCUCUCGUGUUA; GAUCCAGUUUGUAGACCUA; UGCAACGACUUGACAGAAA, human Raptor (#L-004107–00) sequences UGGCUAGUCUGUUUCGAAA; CACGGAAGAUGUUCGACAA; AGAAGGGCAUUACGAGAUU; UGGAGAAGCGUGUCAGAUA, rat Raptor (#L-086862–02) sequences GAGCUUGACUCCAGUUCGA, GCUAGGAACCUGAACAAAU, GCACACAGCAUGGGUGGUA, GAAUCAUGAGGUGGUAUAA, rat MST1 (#L-093629–02) sequences CUCCGAAACAAGACGUUAA; CGGCAGAAAUACCGCUCCA; CGAGAUAUCAAGGCGGGAA; GGAUGGAGACUACGAGUUU, and rat S6K1 (#L-099323–02) sequences GGCCAGAGCACCUGCGUAU; ACAAAAGCAGAGCGGAAUA; GCGCCUGACUUCCGACACA; CGGAGAACAUCAUGCUUAA. An ON-TARGETplus nontargeting siRNA pool (Scramble; siScr) served as controls.
Following plasmids have been used: constitutively active form of AKT1; Myr-HA AKT1 was a gift from William Sellers (Addgene plasmid # 9008; http://addgene.org/9008; RRID: Addgene_9008) (Ramaswamy et al., 1999). Phospho-mimetic AKT1 mutant; pCDNA3-HA-AKT1 S473D was a gift from Wenyi Wei (Harvard Medical School, USA) (Liu et al., 2014). Kinase-dead MST1; pCMV-MST1-K59R was a gift from J. Sadoshima and Y. Maejima (Rutgers New Jersey Medical School, USA) (Yamamoto et al., 2003). Phospho-mimetic MST1 mutant; GST-MST1 T387E from Qi Qi and Keqiang Ye (Emory University School of Medicine, USA) (Jang et al., 2007). pcDNA3 HA-PHLPP1 full length was a gift from Alexandra Newton (Addgene plasmid # 37100; http://addgene.org/37100; RRID: Addgene_37100) (Warfel et al., 2011). pcDNA3-HA-PHLPP2 was a gift from Alexandra Newton (Addgene plasmid # 22403; http://addgene.org/22403; RRID: Addgene_22403) (Brognard et al., 2007). Constitutively active form of S6K1: pRK7-HA-S6K1-F5A-E389-R3A was a gift from John Blenis (Addgene plasmid # 8991; http://addgene.org/8991; RRID: Addgene_8991) (Schalm and Blenis, 2002). GFP plasmid was used as a control.
Transfection
To achieve silencing and overexpression in human islets and INS-1E cells previously described protocol was used (Ardestani et al., 2014). In brief, human islets were dispersed into smaller cell aggregates using accutase (PAA) to increase transfection efficiency and subsequently cultured on ECM- or Collagen I- coated dishes for 1–2 days. To deliver desired siRNA/DNA into dispersed isolated islets as well as INS-1E cells two different transfection methods were used. First, partially dispersed islets or INS-1E cells were pre-incubated in transfection Ca2+-KRH medium (KCl 4.74 mM, KH2PO4 1.19 mM, MgCl26H2O 1.19 mM, NaCl 119 mM, CaCl2 2.54 mM, NaHCO3 25 mM, HEPES 10 mM) for 1h. After that lipoplexes (#11668019; Lipofectamine 2000, Invitrogen, CA, USA)/siRNA ratio 1:20 pmol or lipoplexes/DNA ratio 2.5:1) were added to Ca2+-KRH medium for 6h to transfect the islets or INS-1E cells. After transfection, medium was replaced for fresh CMRL-1066 or RPMI-1640 medium containing 20% FCS. Second, jetPRIME transfection reagent (#114–75; Polyplus transfection, France) was mixed with jetPRIME buffer and siRNA/DNA according to manufacturer’s instructions. The jetPRIME-siRNA/DNA complexes were then added to complete CMRL-1066 or RPMI-1640 to transfect dispersed human islets or INS-1E cells. Efficient transfection was evaluated based on western blot, qPCR and fluorescent microscopy.
In vivo nucleic acid delivery
A commercially available cationic polymer transfection reagent in vivo-jetPEI (#201–50G; Polyplus transfection, France) was used to deliver HA-conjugated PHLPP1- and PHLPP2-expressing constructs via i.p. injection according to manufacturer’s instructions. Briefly, 50 μg of each PHLPP1 and PHLPP2 or 100 μg control GFP plasmids were diluted in 200 μL of 5% glucose solution and mixed with in vivo-jetPEI transfection reagent based on the recommended ionic balance (N/p = 6–8). For an optimum of PHLPP1/2 overexpression, the plasmid/jetPEI mixture was i.p. injected into C57BL/6J male mice (Jackson Laboratory) five times every alternate day for 10 days. Mice were sacrificed 24h after last injection and pancreas/islets isolated.
Adenovirus transduction
The adenoviruses control Ad-LacZ as well as Ad-h-PHLPP1 and Ad-h-PHLPP2 expressing human PHLPP1 and PHLPP2 were purchased from Vector Biolabs, PA, USA. For transduction, isolated human or mouse islets or INS-1E cells were first plated for 1 day followed by infection at a multiplicity of infection (MOI) of 20 (for INS-1E) or 100 (for human and mouse islets) for 4h in CMRL-1066 or RPMI-1640 medium without FCS. After 4h incubation, adenovirus was washed off with 1xPBS and replaced by fresh complete medium. Human islets or INS-1E cells were additionally incubated for 1–3 days.
Glucose-stimulated insulin secretion (GSIS)
Human and mouse islets were pre-incubated in Krebs-Ringer bicarbonate buffer (KRB) containing 2.8 mM glucose for 30 min followed by fresh KRB containing 2.8 mM glucose for 1h (basal) and additional 1h in KRB containing 16.7 mM glucose (stimulated). Islets were washed with 1xPBS and lysed with RIPA buffer for measuring total insulin content. Insulin was determined using human and mouse insulin ELISA (ALPCO Diagnostics, NH USA). Secreted insulin was normalized to insulin content.
Immunohistochemistry
Mouse pancreases were dissected and fixed in 4% formaldehyde at 4°C for 8h and dehydrated before embedding in paraffin. Human pancreatic sections obtained from autopsy from both male and female organ donors and mouse sections (both 2 μm) were deparaffinized, rehydrated and incubated overnight at 4°C with rabbit anti-Ki-67 (#M7249; Dako), rabbit anti-HA-tag (#2367;CST), rabbit anti-pAKT (#9272;CST), mouse anti-NKX6.1 (#F55A12; DSHB, University of Iowa, USA [Ben-Othman et al., 2017]), rabbit anti-PHLPP2 (#A300–661A, Bethyl, TX, USA), rabbit anti-PDX1 antibody (#47267; Abcam, UK), rabbit anti-GLUT2 antibody (#07–1402; Chemicon, CA, USA), rabbit anti-glucagon (#A0565; Dako) or for 2 h at room temperature with anti-insulin (#A0546; Dako), followed by fluorescein isothiocyanate (FITC)- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, PA, USA). Slides were mounted with Vectashield with 4′6-diamidino-2-phenylindole (DAPI) (Vector Labs, CA, USA). Pancreatic β-cell apoptosis was analyzed by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) technique according to the manufacturer’s instructions (In situ Cell Death Detection Kit, TMR red; Roche, Switzerland) and double stained for insulin. Fluorescence was analyzed using a Nikon MEA53200 (Nikon GmbH, Germany) microscope, and images were acquired using NIS-Elements software from Nikon.
Morphometric analysis
For morphometric data, ten sections (spanning the width of the pancreas) per mouse were analyzed. Pancreatic tissue area and insulin-positive area (VECTASTAIN ABC Kit; Vector Labs, USA) were determined by computer-assisted measurements by using a Nikon MEA53200 (Nikon GmbH, Germany) microscope, and images were acquired by using NIS-Elements software from Nikon. Mean percent β-cell fraction per pancreas was calculated as the ratio of insulin-positive and whole pancreatic tissue area. Pancreatic β-cell mass was obtained by multiplying the β-cell fraction by the weight of the pancreas (Ardestani et al., 2014).
Western blot analysis
Human or mouse islets and INS-1E cells were washed twice with ice-cold PBS and lysed with RIPA lysis buffer containing Protease and Phosphatase Inhibitors (Thermo Fisher Scientific (TFS), MA, USA). Protein concentrations were measured by the BCA protein assay (TFS). Proteins were separated by size on NuPAGE 4%–12% Bis-Tris gel (Invitrogen; CA, USA) and electrically transferred into PVDF membranes. Membranes were blocked at room temperature using mixture of 2.5% milk (Cell Signaling Technology (CST), MA, USA) and 2.5% BSA for 1h and incubated overnight at 4°C with rabbit anti-cleaved caspase-3 (#9664), rabbit anti-cleaved PARP (rat specific; #9545), rabbit anti-tubulin (#2146), rabbit anti-GAPDH (#2118), rabbit anti-β-actin (#4967), rabbit anti-GFP (#2956), rabbit anti-HA (#2367), rabbit anti-p4EBP1 (#2855), rabbit anti-pS6 (#4858), rabbit anti-pS6K (#9234), rabbit anti-Raptor (#2280), rabbit anti-MST1 (#3682), rabbit anti-AKT (#9272), rabbit anti-pAKT (#4058), rabbit anti-GST (#2625), rabbit anti-pGSK3 (#9336) (all CST), rabbit anti-PHLPP1 (RRID: AB_2750897 #22789-I-AP, Proteintech, IL, USA), rabbit anti-PHLPP2 (#A300–661A; Bethyl, TX, USA), rabbit anti-pMST1(T183) (#ab79199, Abcam, UK) and rabbit anti-GLUT2 antibody (#07–1402; Chemicon, CA, USA). All primary antibodies were used at 1:1000 dilution in 1xTris-buffered saline plus Tween-20 (1xTBS-T) containing 5% BSA. Additionally, membranes were incubated with horseradish-peroxidase-linked anti-rabbit (Jackson ImmunoResearch, PA, USA) and developed using Immobilon Western HRP chemiluminescence assay system (#WBKLS0500; Millipore, MA, USA). Analysis of the immunoblots was performed using Vision Works LS Image Acquisition and Analysis software Version 6.8 (UVP BioImaging Systems, CA, USA).
Protein degradation analysis
INS-1E cells left untreated or treated with high glucose at 22 mM. At 2 days after incubation, cells were treated with 50 μg/ml translation initiation inhibitor cycloheximide (CHX) to the medium at the times indicated and the lysates were subjected to western blotting.
qPCR analysis
Total RNA was isolated from cultured human or mouse islets or INS-1E cells using TriFast (PEQLAB Biotechnologie, Germany). cDNA synthesis (RevertAid reverse transcriptase, Thermo Fisher Scientific (TFS), MA, USA) and quantitative RT-PCR was performed as previously described (Ardestani et al., 2014). The Applied Biosystems StepOne Real-Time PCR system (Applied Biosystems, CA, USA) with TaqMan® Fast Universal PCR Master Mix for TaqMan assays (Applied Biosystems) were used for analysis. TaqMan® Gene Expression Assays were used for PHLPP1 (#Hs01597875_m1), PHLPP2 (#Hs00982295_m1), PPIA (#Hs99999904_m1), and TUBA1A (#Hs00362387_m1) for human, Phlpp1 (#Mm01295850_m1), Phlpp2 (#Mm01244267_m1), Ppia (#Mm03024003_g1), and Tuba1a (#Mm00846967_g1) for mouse, and Phlpp1 (#Rn00572211_m1), Phlpp2 (#Rn01431647_m1), Ppia (#Rn00690933_m1) and Tuba1a (#Rn01532518_g1) for rat. qPCR was performed and analyzed by the Applied Biosystems StepOne Real-Time PCR system. The ΔΔCT method was used to analyze the relative changes in gene expression.
Translatome analysis
Active polyribosomes with associated mRNAs and nascent peptides for translatome analysis were isolated after an adapted protocol of AHARIBO RNA (#AHA003-R; IMMAGINA Biotechnology, Italy) (Figure 3F). INS-1E cells were cultured till 80% confluency in complete RPMI-1640 medium supplemented with 22.2 mM glucose for 1h. Pancreatic islets from mice fed a ND or HFD were isolated and cultured in complete RPMI-1640 medium. Cells were exposed to L-methionine-free medium (Invitrogen, CA, USA) for 40 min to deplete methionine reserves, followed by 1h treatment with 0.5 mM L-azidohomoalanine (AHA). After AHA incorporation, translation was blocked using sBlock for 10 min and cells were lysed in 1% sodium deoxycholate, 5 U/ml DNase 1, sBlock, 1x proteinase and phosphatase inhibitor cocktail, 200 U/ml RiboLock RNase Inhibitor. 5% of the lysate was saved for input. The remaining lysate was mixed with pre-functionalized magnetic beads (magnetic beads+ biotinylated alkyne ligand) for a chemo-selective “click reaction” between an azide and an alkyne for the effective pull-down of active ribosome complexes. After pull-down, ribosome complexes were digested using proteinase K (VWR, PA, USA) for 75 min. Ribosome associated mRNAs were then extracted with Phenol:Chloroform:Isoamyl Alcohol (Sigma-Aldrich) and used for qPCR analysis. The modified version of the ΔΔCT method was used to calculate a fold change of Up/downregulation of target gene at the translational level.
QUANTIFICATION AND STATISTICAL ANALYSIS
At least three independent biological replica (referred to “n”) were used for human and mouse islets (from different donors/islet preparations from independent experiments), or INS-1E cells (independent experiments) or mice as reported in all figure legends, unless otherwise stated. Data are presented as means ± SEM. Mean differences were determined by Student’s t tests. p value < 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| rabbit anti-Ki-67 | Dako; now Agilent, Santa Clara, CA, USA | #M7249; RRID:AB_2250503 |
| rabbit anti-glucagon | Dako | #A0565; RRID:AB_10013726 |
| guinea pig anti-insulin | Dako | #A0546; RRID:AB_2617169 |
| mouse anti-NKX6.1 | DSHB, University of Iowa, USA | #F55A12; RRID:AB_532379 |
| rabbit anti-PHLPP2 | Bethyl, TX, USA | #A300-661A; RRID:AB_2299551 |
| rabbit anti-PDX1 | Abcam, UK | #47267; RRID:AB_777179 |
| rabbit anti-GLUT2 | Chemicon, CA, USA), | #07-1402; RRID:AB_1587076 |
| rabbit anti-HA-tag | Cell signaling technology (CST), Danvers, MA, USA | #2367; RRID:AB_10691311 |
| rabbit anti-pAKT | CST | #9272; RRID:AB_329827 |
| rabbit anti-cleaved caspase-3 | CST | #9664; RRID:AB_2070042 |
| rabbit anti-cleaved PARP | CST | rat specific; #9545; RRID:AB_2283565 |
| rabbit anti-tubulin | CST | #2146; RRID:AB_2210545 |
| rabbit anti-GAPDH | CST | #2118; RRID:AB_561053 |
| rabbit anti–β-actin | CST | #4967; RRID:AB_330288 |
| rabbit anti-GFP | CST | #2956; RRID:AB_1196615 |
| rabbit anti-p4EBP1 | CST | #2855; RRID:AB_560835 |
| rabbit anti-pS6 | CST | #4858; RRID:AB_916156 |
| rabbit anti-pS6K | CST | #9234; RRID:AB_2269803 |
| rabbit anti-Raptor | CST | #2280; RRID:AB_561245 |
| rabbit anti-MST1 | CST | #3682; RRID:AB_2144632 |
| rabbit anti-AKT | CST | #9272; RRID:AB_329827 |
| rabbit anti-pAKT | CST | #4058; RRID:AB_331168 |
| rabbit anti-GST | CST | #2625; RRID:AB_490796 |
| rabbit anti-pGSK3 | CST | #9336; RRID:AB_331405 |
| rabbit anti-PHLPPI | Proteintech, IL, USA | RRID: AB_2750897 #22789-I-AP |
| rabbit anti-pMST1(T183) | Abcam, UK | #ab79199; RRID:AB_2271183 |
| horseradish-peroxidase-linked anti-rabbit | Jackson Immuno Research, PA, USA | #111-035-003; RRID:AB_2313567 |
| horseradish-peroxidase-linked anti-mouse | Jackson | #115-035-003; RRID:AB_10015289 |
| Cy3-conjugated donkey anti-mouse | Jackson | #715-165-150; RRID:AB_2340813 |
| Cy3-conjugated anti-rabbit | Jackson | #711-165-152; RRID:AB_2307443 |
| FITC-conjugated donkey anti-guinea pig | Jackson | #706-096-148; RRID:AB_2340454 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| adenoviruses Ad-LacZ | Vector Biolabs, PA, USA | #1080 |
| Ad-h-PHLPP1 | Vector Biolabs | N/A |
| Ad-h-PHLPP2 | Vector Biolabs | #ADV-214159 |
|
| ||
| Biological samples | ||
|
| ||
| Human FFPE pancreatic sections from autopsy | this paper | N/A |
| Mouse FFPE pancreatic sections from autopsy | this paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Immobilon Western HRP Substrat | Millipore, MA, USA | #WBKLS0500 |
| Protease and Phosphatase Inhibitors | Thermo Fisher Scientific, USA | #78440 |
| RevertAid reverse transcriptase | #EP0451 | |
| S6K1 selective inhibitor PF-4708671 | Calbiochem, USA | #S2163 |
| Rapamycin | Calbiochem | #53123-88-9 |
| IGF1 | Calbiochem | #407251 |
| recombinant human IL-1β | R&D Systems, USA | #201-LB |
| recombinant human IFN-γ | PeProTech, USA | #300-02 |
| MHY1485 | Selleck Chemicals, USA | #S7811 |
| 3-Benzyl-5-((2-nitrophenoxy)methyl)-dihydrofuran-2(3H)-one (3BDO) | J&K Scientific, Belgium | #1914077 |
| recombinant human insulin | Sigma-Aldrich, USA | #91077C |
| cycloheximide | Sigma-Aldrich | #C4859-1ML |
| streptozotocin | Sigma-Aldrich | #S0130 |
| Phenol:Chloroform:Isoamyl Alcohol | Sigma-Aldrich | #77617 |
| Liberase TM | Roche, Switzerland | #05401119001 |
| jetPRIME® transfection reagent | Polyplus, France | #114-75 |
| in vivo-jetPEI | Polyplus | #201-50G |
| Vectashield with 4′6-diamidino-2-phenylindole (DAPI) | Vector Labs, USA | #H-1200-10 |
| TriFast | PEQLAB Biotechnologie, Germany | #30-2010 |
| Lipofectamine 2000 | Invitrogen, USA | #11668019 |
|
| ||
| Critical commercial assays | ||
|
| ||
| translatome analysis: AHARIBO RNA | IMMAGINA Biotechnology, Italy | #AHA003-R |
| Pierce BCA Protein Assay | Thermo Fisher Scientific, USA | #23225 |
| Insulin ELISA Assay | ALPCO Diagnostics, USA | #80-INSMSU-E01 |
| In situ Cell Death Detection Kit, TMR red | Roche, Switzerland | #12156792910 |
| VECTASTAIN ABC Kit | Vector Labs, USA | #PK-4000 |
|
| ||
| Deposited data | ||
|
| ||
| N/A | ||
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| rat β-cell line INS-1E | Laboratory of Claes Wollheim, University of Geneva | RRID: CVCL_0351 |
| Mouse embryonic fibroblasts (MEFs) isolated from Tuberous sclerosis complex 2 knock-out (MEF-TSC2-KO) and respective WT mice | Laboratory of Gil Leibowitz, Hadassah University | N/A |
| Mouse embryonic fibroblasts (MEFs) isolated from PHLPP1 knock-out (MEF-PHLPP1-KO) and respective WT mice | Laboratory of Alexandra Newton, UCSD | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| PHLPP1-KO mice | Laboratory of Alexandra Newton, UCSD | RRID: MGI:5795609 |
| leptin receptor deficient mice Leprdb/+(db/+), Leprdb/db (db/db) | Jackson Laboratory, ME, USA | #000642; BKS.Cg-Dock7m +/+ Leprdb/J |
| Isolated mouse islets | this paper | N/A |
| Isolated human islets | this paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| rat PHLPP1 sequences 5’CAGCUUGACCUGCGAGACA3’; 5’GUGAAUAACUUCCGUGACA3’; 5’UAAUAGUAGUCUCCGGAAA3’; 5’GAAUGUACAAUGUCCGAAA3’ | ON-TARGETplus siRNAs, Dharmacon, CO, USA | #L-094929-02 |
| rat PHLPP2 sequences 5’ACAAAUGGGCUGAGCGCUU3’; 5’UAGUCUGAGUCUUCGGAAA3’; 5’GCAUCUAUAACGUCCGCAA3’; 5’CCGUGGACCUCUCGUGUUA3’ | ON-TARGETplus | #L-104590-02 |
| human PHLPP1 sequences 5’GAAUGUAUAAUGUCCGUAA3’; 5’GAUCUAAGGUUGAACGUAA3’; 5’GGAAUCAACUGGUCACAUU3’; 5’GAUAUUGGCCAUAAUCAAA3’, | ON-TARGETplus | #L-019103-00 |
| human PHLPP2 sequences 5’CCUAUAUUGUUAUGCGAGA3’; 5’CCGUGGAUCUCUCGUGUUA3’; 5’GAUCCAGUUUGUAGACCUA3’; 5’UGCAACGACUUGACAGAAA3’ | ON-TARGETplus | #L-022586-01 |
| human Raptor sequences 5’UGGCUAGUCUGUUUCGAAA3’; 5’CACGGAAGAUGUUCGACAA3’; 5’AGAAGGGCAUUACGAGAUU3’; 5’UGGAGAAGCGUGUCAGAUA3’ | ON-TARGETplus | #L-004107-00 |
| rat Raptor sequences 5’GAGCUUGACUCCAGUUCGA3’, 5’GCUAGGAACCUGAACAAAU3’, 5’GCACACAGCAUGGGUGGUA3’, 5’GAAUCAUGAGGUGGUAUAA3’ | ON-TARGETplus | #L-086862-02 |
| rat MST1 sequences 5’CUCCGAAACAAGACGUUAA3’; 5’CGGCAGAAAUACCGCUCCA; 5’CGAGAUAUCAAGGCGGGAA3’; 5’GGAUGGAGACUACGAGUUU3’ | ON-TARGETplus | #L-093629-02 |
| rat S6K1 sequences 5’GGCCAGAGCACCUGCGUAU3’; 5’ACAAAAGCAGAGCGGAAUA3’; 5’GCGCCUGACUUCCGACACA3’; 5’CGGAGAACAUCAUGCUUAA3’ | ON-TARGETplus | #L-099323-02 |
| human PHLPP1 | TaqMan® Gene Expression Assays, Applied Biosystems, CA, USA | #Hs01597875_m1 |
| human PHLPP2 | Applied Biosystems | #Hs00982295_m1 |
| mouse Phlpp1 | Applied Biosystems | #Mm01295850_m1 |
| mouse Phlpp2 | Applied Biosystems | #Mm01244267_m1 |
| rat Phlpp1 | Applied Biosystems | #Rn00572211_m1 |
| rat Phlpp2 | Applied Biosystems | #Rn01431647_m1 |
| human TUBA1A | Applied Biosystems | #Hs00362387_m1 |
| mouse Ppia | Applied Biosystems | #Mm03024003_g1 |
| mouse Tuba1a | Applied Biosystems | #Mm00846967_g1 |
| rat Ppia | Applied Biosystems | #Rn00690933_m1 |
| human PPIA | Applied Biosystems | #Hs99999904_m1 |
| rat Tuba1a | Applied Biosystems | #Rn01532518_g1 |
|
| ||
| Recombinant DNA | ||
|
| ||
| Phospho-mimetic AKT1 mutant; pCDNA3-HA-AKT1 S473D | Laboratory of Wenyi Wei, Harvard Medical School Liu et al., 2014 | N/A |
| Kinase-dead MST1; pCMV-MST1-K59R | Laboratory of J. Sadoshima and Y. Maejima; Yamamoto et al., 2003 | N/A |
| Phospho-mimetic MST1 mutant; GST-MST1 T387E | Laboratory of Qi Qi and Keqiang Ye; Jang et al., 2007 | N/A |
| active form of AKT1; Myr-HA AKT1 | Ramaswamy et al., 1999 | William Sellers, Broad Institute of MIT http://n2t.net/addgene:9008; RRID: Addgene_9008 |
| pcDNA3 HA-PHLPP1 full length | Warfel et al., 2011 | Alexandra Newton, UCSD http://n2t.net/addgene:37100; RRID: Addgene_37100 |
| pcDNA3-HA-PHLPP2 | Brognard et al., 2007 | Alexandra Newton, UCSD http://n2t.net/addgene:22403; RRID: Addgene_22403 |
| Constitutively active form of S6K1; pRK7-HA-S6K1-F5A-E389-R3A | Schalm and Blenis, 2002 | John Blenis, Weill Cornell Medicine http://n2t.net/addgene:8991; RRID: Addgene_8991 |
|
| ||
| Software and algorithms | ||
|
| ||
| Vision Works LS Image Acquisition and Analysis software Version 6.8 | UVP BioImaging Systems, CA, USA | https://www.labortechnik.com/en/vision-works-ls/analysis-software |
| NIS-Elements software, v3.22.11 | Nikon GmbH, Germany | https://www.nikon.com/products/microscope-solutions/support/download/software/imgsfw/nis-f_v4600064.htm |
| GraphPad Prism v8.4.3 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
|
| ||
| Other | ||
|
| ||
| Biocoat Collagen I coated dishes | Corning, ME, USA | #356400 |
| CMRL-1066 | Invitrogen, USA | #11530037 |
| RPMI-1640 |
Sigma-Aldrich, MO, USA | #R8758 |
| DMEM high glucose | #D6429 | |
| The Applied Biosystems StepOne Real-Time PCR system | Applied Biosystems, USA | N/A |
| Nikon MEA53200 | Nikon GmbH, Germany | N/A |
| Glucometer FreeStyle Lite | Abbott, USA | N/A |
Highlights.
PHLPP1/2 are highly elevated in metabolically stressed β cells in diabetes
Metabolic-stress-induced mTORC1 hyper-activation leads to PHLPP upregulation
PHLPPs regulate β-cell survival-dependent kinases AKT and MST1
PHLPP inhibition restores glycemia, β-cell survival, and function
ACKNOWLEDGMENT
This work was supported by the German Research Foundation (DFG). Human pancreatic islets were kindly provided by the NIDDK-funded Integrated Islet Distribution Program (IIDP) at City of Hope, NIH grant no 2UC4DK098085, the JDRF-funded IIDP Islet Award Initiative, and through the ECIT Islet for Basic Research program supported by JDRF (JDRF award 31–2008-413). We thank J. Kerr-Conte and Francois Pattou (European Genomic Institute for Diabetes, Lille) and ProdoLabs for high-quality human islet isolations, Katrischa Hennekens (University of Bremen) for excellent technical assistance and animal care, and Petra Schilling (University of Bremen) for pancreas sectioning. PHLPP1-KO mice were kindly provided by Alexandra C. Newton (University of California at San Diego), MEF-TSC2-KO by Gil Leibowitz (Hadassah-Hebrew University Medical Center, Jerusalem), INS-1E cells by Claes Wollheim (Lund and Geneva Universities), and plasmids from William Sellers and Wenyi Wei (both Harvard Medical School, USA), J. Sadoshima and Y. Maejima (Rutgers New Jersey Medical School, USA), Alexandra Newton (UCSD), and John Blenis (Cornell University). Human pancreatic sections were provided from the National Disease Research Interchange (NDRI), supported by the NIH. The graphical abstract was created using smart servier medical art under https://creativecommons.org/
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109490.
REFERENCES
- Aguayo-Mazzucato C, and Bonner-Weir S (2018). Pancreatic β cell regeneration as a possible therapy for diabetes. Cell Metab. 27, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Méneur C, and Bernal-Mizrachi E (2015). Natural history of β-cell adaptation and failure in type 2 diabetes. Mol. Aspects Med. 42, 19–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreozzi F, Procopio C, Greco A, Mannino GC, Miele C, Raciti GA, Iadicicco C, Beguinot F, Pontiroli AE, Hribal ML, et al. (2011). Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 54, 1879–1887. [DOI] [PubMed] [Google Scholar]
- Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, Frogne T, Tao W, Oberholzer J, Pattou F, et al. (2014). MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 20, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardestani A, Lupse B, Kido Y, Leibowitz G, and Maedler K (2018). mTORC1 signaling: a double-edged sword in diabetic β cells. Cell Metab. 27, 314–331. [DOI] [PubMed] [Google Scholar]
- Ardestani A, Li S, Annamalai K, Lupse B, Geravandi S, Dobrowolski A, Yu S, Zhu S, Baguley TD, Surakattula M, et al. (2019). Neratinib protects pancreatic beta cells in diabetes. Nat. Commun. 10, 5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, and Rorsman P (2012). Diabetes mellitus and the β cell: the last ten years. Cell 148, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann A, Ueki K, Winnay JN, Kadowaki T, and Kulkarni RN (2009). Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol. Cell. Biol. 29, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviv Y, and Kirshenbaum LA (2010). Novel phosphatase PHLPP-1 regulates mitochondrial Akt activity and cardiac cell survival. Circ. Res. 107, 448–450. [DOI] [PubMed] [Google Scholar]
- Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, and Leibowitz G (2009). Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS ONE 4, e4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera S, Kapadia B, Kain V, Alamuru-Yellapragada NP, Murunikkara V, Kumar ST, Babu PP, Seshadri S, Shivarudraiah P, Hiriyan J, et al. (2018). ERK1/2 activated PHLPP1 induces skeletal muscle ER stress through the inhibition of a novel substrate AMPK. Biochim. Biophys. Acta Mol. Basis Dis. 1864 (5 Pt A), 1702–1716. [DOI] [PubMed] [Google Scholar]
- Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, Hadzic B, Druelle N, Napolitano T, Navarro-Sanz S, et al. (2017). Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell 168, 73–85 e11. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, and Permutt MA (2001). Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J. Clin. Invest. 108, 1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher J, Kleinridders A, and Kahn CR (2014). Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 6, a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brognard J, and Newton AC (2008). PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 19, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brognard J, Sierecki E, Gao T, and Newton AC (2007). PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931. [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, and Butler PC (2003). Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110. [DOI] [PubMed] [Google Scholar]
- Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O’Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, et al. (2011). Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 20, 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Van Winkle JA, Lyden PD, Brown JH, and Purcell NH (2013). PHLPP1 gene deletion protects the brain from ischemic injury. J. Cereb. Blood Flow Metab. 33, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Kim JW, Shin JA, Shin J, and Yoon KH (2011). β-cell mass in people with type 2 diabetes. J. Diabetes Investig. 2, 6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Park YJ, Park JY, Jeong HO, Kim DH, Ha YM, Kim JM, Song YM, Heo HS, Yu BP, et al. (2012). Inhibitory effect of mTOR activator MHY1485 on autophagy: suppression of lysosomal fusion. PLoS ONE 7, e43418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, Marselli L, Suleiman M, Ratner LE, Marchetti P, and Accili D (2016). Evidence of β-cell dedifferentiation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 101, 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Katsenelson K, Stender JD, Kawashima AT, Lordén G, Uchiyama S, Nizet V, Glass CK, and Newton AC (2019). PHLPP1 counter-regulates STAT1-mediated inflammatory signaling. eLife 8, e48609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Hoppa MB, Walker JN, Amisten S, Abdulkader F, Bengtsson M, Fearnside J, Ramracheya R, Toye AA, Zhang Q, et al. (2010). Progression of diet-induced diabetes in C57BL6J mice involves functional dissociation of Ca2+ channels from secretory vesicles. Diabetes 59, 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmadhikari G, Stolz K, Hauke M, Morgan NG, Varki A, de Koning E, Kelm S, and Maedler K (2017). Siglec-7 restores β-cell function and survival and reduces inflammation in pancreatic islets from patients with diabetes. Sci. Rep. 7, 45319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Dinarello CA, and Mandrup-Poulsen T (2019). Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 19, 734–746. [DOI] [PubMed] [Google Scholar]
- Duncan JS, Turowec JP, Vilk G, Li SS, Gloor GB, and Litchfield DW (2010). Regulation of cell proliferation and survival: convergence of protein kinases and caspases. Biochim. Biophys. Acta 1804, 505–510. [DOI] [PubMed] [Google Scholar]
- Dunning BE, and Gerich JE (2007). The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr. Rev. 28, 253–283. [DOI] [PubMed] [Google Scholar]
- Elghazi L, and Bernal-Mizrachi E (2009). Akt and PTEN: beta-cell mass and pancreas plasticity. Trends Endocrinol. Metab. 20, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, and Newton AC (2005). PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24. [DOI] [PubMed] [Google Scholar]
- Gao T, Brognard J, and Newton AC (2008). The phosphatase PHLPP controls the cellular levels of protein kinase C. J. Biol. Chem. 283, 6300–6311. [DOI] [PubMed] [Google Scholar]
- Ge D, Han L, Huang S, Peng N, Wang P, Jiang Z, Zhao J, Su L, Zhang S, Zhang Y, et al. (2014). Identification of a novel MTOR activator and discovery of a competing endogenous RNA regulating autophagy in vascular endothelial cells. Autophagy 10, 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González A, and Hall MN (2017). Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 36, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal N, Tiwary S, Kesharwani D, and Datta M (2019). Long non-coding RNA H19 inhibition promotes hyperglycemia in mice by upregulating hepatic FoxO1 levels and promoting gluconeogenesis. J. Mol. Med. (Berl.) 97, 115–126. [DOI] [PubMed] [Google Scholar]
- Grzechnik AT, and Newton AC (2016). PHLPPing through history: a decade in the life of PHLPP phosphatases. Biochem. Soc. Trans. 44, 1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart NJ, and Powers AC (2019). Use of human islets to understand islet biology and diabetes: progress, challenges and suggestions. Diabetologia 62, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz E, Krogvold L, Zhitomirsky S, Swisa A, Fischman M, Lax T, Dahan T, Hurvitz N, Weinberg-Corem N, Klochendler A, et al. (2018). β-cell dna damage response promotes islet inflammation in type 1 diabetes. Diabetes 67, 2305–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hribal ML, Mancuso E, Arcidiacono GP, Greco A, Musca D, Procopio T, Ruffo M, and Sesti G (2020). The Phosphatase PHLPP2 Plays a Key Role in the Regulation of Pancreatic Beta-Cell Survival. Int. J. Endocrinol. 2020, 1027386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SM, Feigenson M, Begun DL, Shull LC, Culley KL, Otero M, Goldring MB, Ta LE, Kakar S, Bradley EW, et al. (2018). Phlpp inhibitors block pain and cartilage degradation associated with osteoarthritis. J. Orthop. Res. 36, 1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaafar R, Tran S, Shah AN, Sun G, Valdearcos M, Marchetti P, Masini M, Swisa A, Giacometti S, Bernal-Mizrachi E, et al. (2019). mTORC1 to AMPK switching underlies β-cell metabolic plasticity during maturation and diabetes. J. Clin. Invest. 129, 4124–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Verrier JD, Drabek T, Janesko-Feldman K, Gillespie DG, Uray T, Dezfulian C, Clark RS, Bayir H, Jackson EK, and Kochanek PM (2013). Pharmacological inhibition of pleckstrin homology domain leucine-rich repeat protein phosphatase is neuroprotective: differential effects on astrocytes. J. Pharmacol. Exp. Ther. 347, 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Dixon CE, Janesko-Feldman K, Vagni V, Kotermanski SE, Jackson EK, and Kochanek PM (2018). Acute physiology and neurologic outcomes after brain injury in SCOP/PHLPP1 KO mice. Sci. Rep. 8, 7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SW, Yang SJ, Srinivasan S, and Ye K (2007). Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. J. Biol. Chem. 282, 30836–30844. [DOI] [PubMed] [Google Scholar]
- Jeffery N, and Harries LW (2016). β-cell differentiation status in type 2 diabetes. Diabetes Obes. Metab. 18, 1167–1175. [DOI] [PubMed] [Google Scholar]
- Jung S, Kang JG, Lee JH, Song KJ, Ko JH, and Kim YS (2014). PHLPP1 regulates contact inhibition by dephosphorylating Mst1 at the inhibitory site. Biochem. Biophys. Res. Commun. 443, 1263–1269. [DOI] [PubMed] [Google Scholar]
- Kim W, Lao Q, Shin YK, Carlson OD, Lee EK, Gorospe M, Kulkarni RN, and Egan JM (2012). Cannabinoids induce pancreatic β-cell death by directly inhibiting insulin receptor activation. Sci. Signal. 5, ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Park KJ, Kim GH, Jeong EA, Lee DY, Lee SS, Kim DJ, Roh GS, Song J, Ki SH, and Kim WH (2013). In vivo activating transcription factor 3 silencing ameliorates the AMPK compensatory effects for ER stress-mediated β-cell dysfunction during the progression of type-2 diabetes. Cell. Signal. 25, 2348–2361. [DOI] [PubMed] [Google Scholar]
- Kung CP, and Murphy ME (2016). The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 231, R61–R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Stevens PD, Liu J, Yang H, Wang W, Wang C, Zeng Z, Schmidt MD, Yang M, Lee EY, et al. (2014). PHLPP is a negative regulator of RAF1, which reduces colorectal cancer cell motility and prevents tumor progression in mice. Gastroenterology 146, 1301–1312e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Weiss HL, Rychahou P, Jackson LN, Evers BM, and Gao T (2009). Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene 28, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stevens PD, and Gao T (2011a). mTOR-dependent regulation of PHLPP expression controls the rapamycin sensitivity in cancer cells. J. Biol. Chem. 286, 6510–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stevens PD, Li X, Schmidt MD, and Gao T (2011b). PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol. Cell. Biol. 31, 4917–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhou L, Xiong K, Godlewski G, Mukhopadhyay B, Tam J, Yin S, Gao P, Shan X, Pickel J, et al. (2012). Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 142, 1218–1228.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. (2014). Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Soläng C, Mejia-Cordova M, Thorvaldson L, Blixt M, Sandler S, and Singh K (2019). Kinetics of immune cell responses in the multiple low-dose streptozotocin mouse model of type 1 diabetes. FASEB Bioadv. 1, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, and Donath MY (2001). Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50, 69–76. [DOI] [PubMed] [Google Scholar]
- Masubuchi S, Gao T, O’Neill A, Eckel-Mahan K, Newton AC, and Sassone-Corsi P (2010). Protein phosphatase PHLPP1 controls the light-induced resetting of the circadian clock. Proc. Natl. Acad. Sci. USA 107, 1642–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K, Castillo R, Glembotski CC, Sussman MA, Newton AC, and Brown JH (2010). PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ. Res. 107, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moc C, Taylor AE, Chesini GP, Zambrano CM, Barlow MS, Zhang X, Gustafsson AB, and Purcell NH (2015). Physiological activation of Akt by PHLPP1 deletion protects against pathological hypertrophy. Cardiovasc. Res. 105, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moin ASM, and Butler AE (2019). Alterations in beta cell identity in type 1 and type 2 diabetes. Curr. Diab. Rep. 19, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Agarwal NK, Morales FC, Hayashi Y, Aldape KD, Cote G, and Georgescu MM (2012). PTEN, NHERF1 and PHLPP form a tumor suppressor network that is disabled in glioblastoma. Oncogene 31, 1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossmann D, Park S, and Hall MN (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757. [DOI] [PubMed] [Google Scholar]
- O’Neill AK, Niederst MJ, and Newton AC (2013). Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 280, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SJ, Han JM, Garcia R, Assi K, Gao T, O’Neill A, Newton AC, and Levings MK (2011). Cutting edge: PHLPP regulates the development, function, and molecular signaling pathways of regulatory T cells. J. Immunol. 186, 5533–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, and Alessi DR (2010). Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 431, 245–255. [DOI] [PubMed] [Google Scholar]
- Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, and Miao J (2014). An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E−/− mice. Sci. Rep. 4, 5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W, Stein J, Stein GS, Iglehart JD, Shi Q, and Pardee AB (2010). Mst1 is an interacting protein that mediates PHLPPs’ induced apoptosis. Mol. Cell 38, 512–523. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, and Sellers WR (1999). Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA 96, 2110–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, and Blenis J (2002). Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12, 632–639. [DOI] [PubMed] [Google Scholar]
- Shirakawa J, Fernandez M, Takatani T, El Ouaamari A, Jungtrakoon P, Okawa ER, Zhang W, Yi P, Doria A, and Kulkarni RN (2017). Insulin signaling regulates the FoxM1/PLK1/CENP-A pathway to promote adaptive pancreatic β cell proliferation. Cell Metab. 25, 868–882.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Naguro I, Nishitoh H, Matsuzawa A, and Ichijo H (2011). Apoptosis signaling kinases: from stress response to health outcomes. Antioxid. Redox Signal. 15, 719–761. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, and Accili D (2012). Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, and Kahn CR (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S, Roel C, Tanwir M, Wills R, Perianayagam N, Wang P, and Fiaschi-Taesch NM (2016). Definition of a Skp2-c-Myc pathway to expand human beta-cells. Sci. Rep. 6, 28461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trümper K, Trümper A, Trusheim H, Arnold R, Göke B, and Hörsch D (2000). Integrative mitogenic role of protein kinase B/Akt in beta-cells. Ann. N Y Acad. Sci. 921, 242–250. [DOI] [PubMed] [Google Scholar]
- Turki A, Mahjoub T, Mtiraoui N, Abdelhedi M, Frih A, and Almawi WY (2013). Association of POL1, MALT1, MC4R, PHLPP and DSEL single nucleotide polymorphisms in chromosome 18q region with type 2 diabetes in Tunisians. Gene 527, 243–247. [DOI] [PubMed] [Google Scholar]
- Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, Polonsky KS, Naji A, and Birnbaum MJ (2001). Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat. Med. 7, 1133–1137. [DOI] [PubMed] [Google Scholar]
- Uchida T, Nakamura T, Hashimoto N, Matsuda T, Kotani K, Sakaue H, Kido Y, Hayashi Y, Nakayama KI, White MF, and Kasuga M (2005). Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat. Med. 11, 175–182. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu Y, Yan Lu S, Nguyen KT, Schroer SA, Suzuki A, Mak TW, Gaisano H, and Woo M (2010). Deletion of Pten in pancreatic ß-cells protects against deficient ß-cell mass and function in mouse models of type 2 diabetes. Diabetes 59, 3117–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, García-Ocaña A, and Stewart AF (2015). Diabetes mellitus–advances and challenges in human β-cell proliferation. Nat. Rev. Endocrinol. 11, 201–212. [DOI] [PubMed] [Google Scholar]
- Warfel NA, Niederst M, Stevens MW, Brennan PM, Frame MC, and Newton AC (2011). Mislocalization of the E3 ligase, β-transducin repeat-containing protein 1 (β-TrCP1), in glioblastoma uncouples negative feedback between the pleckstrin homology domain leucine-rich repeat protein phosphatase 1 (PHLPP1) and Akt. J. Biol. Chem. 286, 19777–19788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, Gaglia J, and Bonner-Weir S (2020). Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 8, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YA, Stevens PD, Gasser ML, Andrei R, and Gao T (2013). Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol. Cell. Biol. 33, 4594–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YA, Li X, Goretsky T, Weiss HL, Barrett TA, and Gao T (2015). Loss of PHLPP protects against colitis by inhibiting intestinal epithelial cell apoptosis. Biochim. Biophys. Acta 1852 (10 Pt A), 2013–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yako YY, Guewo-Fokeng M, Balti EV, Bouatia-Naji N, Matsha TE, Sobngwi E, Erasmus RT, Echouffo-Tcheugui JB, and Kengne AP (2016). Genetic risk of type 2 diabetes in populations of the African continent: a systematic review and meta-analyses. Diabetes Res. Clin. Pract. 114, 136–150. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, et al. (2003). Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J. Clin. Invest. 111, 1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KT, Bayan JA, Zeng N, Aggarwal R, He L, Peng Z, Kassa A, Kim M, Luo Z, Shi Z, et al. (2014). Adult-onset deletion of Pten increases islet mass and beta cell proliferation in mice. Diabetologia 57, 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, et al. (2003). Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J. Clin. Endocrinol. Metab. 88, 2300–2308. [DOI] [PubMed] [Google Scholar]
- Yuan T, Rafizadeh S, Gorrepati KD, Lupse B, Oberholzer J, Maedler K, and Ardestani A (2017). Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia 60, 668–678. [DOI] [PubMed] [Google Scholar]
- Yuan T, Lupse B, Maedler K, and Ardestani A (2018). mTORC2 signaling: a path for pancreatic β cell’s growth and function. J. Mol. Biol. 430, 904–918. [DOI] [PubMed] [Google Scholar]
- Zeng N, Yang KT, Bayan JA, He L, Aggarwal R, Stiles JW, Hou X, Medina V, Abad D, Palian BM, et al. (2013). PTEN controls β-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell 12, 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Smith MP, Zhou GK, Lai A, Hoy RC, Mroz V, Torre OM, Laudier DM, Bradley EW, Westendorf JJ, et al. (2019). Phlpp1 is associated with human intervertebral disc degeneration and its deficiency promotes healing after needle puncture injury in mice. Cell Death Dis. 10, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang X, Liu M, Liu D, Pan J, Tian J, Jin T, Xu Y, and An F (2020). Inhibition of PHLPP1 ameliorates cardiac dysfunction via activation of the PI3K/Akt/mTOR signalling pathway in diabetic cardiomyopathy. J. Cell. Mol. Med. 24, 4612–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated from this study are included in this paper. All raw data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.