


Abstract
Transcriptional regulation by Wnt signalling is primarily thought to be accomplished by a complex of β-catenin and TCF family transcription factors (TFs). Although numerous studies have suggested that additional TFs play roles in regulating Wnt target genes, their mechanisms of action have not been investigated in detail. We characterised a Wnt-responsive element (WRE) downstream of the Wnt target gene Axin2 and found that TCFs and Caudal type homeobox (CDX) proteins were required for its activation. Using a new separation-of-function TCF mutant, we found that WRE activity requires the formation of a TCF/CDX complex. Our systematic mutagenesis of this enhancer identified other sequences essential for activation by Wnt signalling, including several copies of a novel CAG DNA motif. Computational and experimental evidence indicates that the TCF/CDX/CAG mode of regulation is prevalent in multiple WREs. Put together, our results demonstrate the complex nature of cis- and trans- interactions required for signal-dependent enhancer activity.
INTRODUCTION
The Wnt/β-catenin signalling pathway is highly conserved across the animal kingdom, plays numerous essential roles in animal development, and is required for the homeostasis of many tissues in adult organisms (1,2). While it is well known that the Wnt pathway affects cell behaviour by transcriptionally regulating gene expression, many questions remain about how Wnt signalling controls gene expression in a cell-specific manner (3–6).
The prevalent model of transcriptional regulation by the Wnt pathway is centred upon the regulation of β-catenin protein levels and the activity of transcription factors (TFs) of the TCF/LEF family (TCFs). In the absence of Wnt signals, β-catenin protein levels are kept low in cells by a ‘destruction complex’, which targets β-catenin for proteasomal degradation. The binding of Wnt ligands to their extracellular receptor complexes inactivates the destruction complex, allowing β-catenin to accumulate (7). β-catenin then complexes with TCFs, which are bound to specific TCF binding sites in cis-regulatory regions named Wnt-responsive elements (WREs) (8). Subsequently, β-catenin recruits a variety of co-factors to regulate the transcription of Wnt target genes (9,10).
The view of TCFs as the major transcriptional effectors of Wnt signalling is supported by genetic studies in vertebrates and invertebrates (11,12). More recently, studies in Drosophila and mammalian cell culture systems have confirmed that the majority of Wnt target genes fail to be activated in the absence of TCFs (13–15). Functional TCF binding sites have been identified in many WREs, and synthetic reporters containing multimers of TCF sites are specifically activated by Wnt/β-catenin signalling (3,8). In light of this, TCFs are often considered necessary and sufficient for WRE activity. In addition to the High Mobility Group (HMG) domain that all TCFs possess which binds to TCF binding sites (16), invertebrate TCFs and some vertebrate TCF isoforms also contain an additional DNA-binding domain, the C-clamp (17,18). C-clamps recognize GC-rich motifs termed Helper sites (19,20), which are essential for activation of multiple Wnt targets in Drosophila, C. elegans and mammalian systems (19,21–23).
The idea that WRE activity is solely mediated by TCFs is inconsistent with broader studies of enhancers, which suggest that these cis-regulatory elements are primarily regulated by combinatorial TF activity. Enhancers tend to contain clusters of binding sites for multiple TFs that vary in their number and relative orientation (24,25). In this manner, several TFs can be recruited to an enhancer through their cognate binding sites. There is evidence of protein-protein interactions between many TFs known to co-regulate enhancers. This has led to the idea of enhancer regulation by TF collectives, groups of TFs formed by protein-protein and protein-DNA interactions that act together to regulate transcription (26). Although theoretical frameworks suggest that a model of gene regulation involving TFs working in distinct collectives to activate enhancers can explain the observed specificity of gene regulatory networks, there are few examples of studies which have directly identified and tested the role of TF-TF interactions on gene expression (25).
Consistent with the aforementioned view of enhancer structure, there are several lines of evidence that WREs are more complex than previously thought. While synthetic WREs consisting of high affinity TCF binding sites upstream of a minimal promoter (e.g. TOPflash) provide sensitive readouts for the Wnt pathway in cell culture, they do not faithfully recapitulate patterns of Wnt signalling at the organismal level (8,19). Reporters knocked into endogenous Wnt target genes such as Axin2 have proven to be better markers for Wnt signalling (27), suggesting that the endogenous WREs regulating them contain additional information and are not just collections of TCF binding sites. Similarly, recent studies in Xenopus found that β-catenin recruitment to chromatin was insufficient for the activation of nearby genes, leading the authors to suggest that additional TFs are required at WREs to act as spatio-temporal specificity cues (28,29).
Correlative experiments using chromatin immunoprecipitation (ChIP) based techniques have identified several TFs that show significant levels of chromatin co-occupancy with TCFs (6,30). Some of these TFs also directly interact with TCFs, suggesting that they could be part of a Wnt/TCF TF collective. For instance, TCF7L2 and CDX2 (Caudal type homeobox 2) show significant levels of chromatin co-occupancy in colorectal cancer cells (31), and protein-protein interactions have been observed between other members of the TCF and CDX families (32). Similarly, the TFs Sp5 and Sp8 interact with multiple TCFs and bind to several WREs in mouse embryonic stem cells (33). Co-occupancy at genomic locations and interactions between TCFs and other TFs have also been reported, including Smads and AP-1 (3), as well as TEADs (34). Together, these studies suggest the existence of multiple TCF-containing TF collectives which contribute to the high cell-type specificity seen in Wnt target genes. However, the importance of interactions between TCFs and other TFs has not been rigorously tested, and whether there is a TF ‘binding site grammar’ shared by WREs has not been systematically examined.
In this study, we have analysed a novel WRE located downstream of the Axin2 Wnt target gene in great detail. We found that in addition to TCF proteins, enhancer activity was regulated by CDX protein levels and that TCF7L2 and CDX1 are recruited to this WRE. We found evidence for the existence of a TCF7–CDX1 complex, and our experiments using a specific separation-of-function mutant of TCF7 support a model where TCF7–CDX1 complex formation is required for enhancer activity. Systematic scanning mutagenesis of the WRE revealed that as is typical for WREs, it contains four TCF binding sites that are absolutely required for activation. In addition, it identified other sequences required for WRE activity, including two sequence motifs resembling the CDX consensus sequence and a previously unidentified orphan DNA motif we refer to as the CAG site. Functional TCF, CDX and CAG sites are also present in a WRE linked to human cancers located upstream of the c-Myc oncogene. Computational analysis of chromatin bound by TCF7L2 and CDX2 revealed an enrichment of CAG motifs, suggesting that a TCF/CDX/CAG cassette exists in other WREs. Using synthetic reporters, we also saw that the CDX and CAG sites do not respond to Wnt/β-catenin signalling, but these motifs can potentiate the ability of TCF sites to respond to pathway activation in a cell-type specific manner. These results define a binding site grammar for a subset of WREs and provide a more detailed view of their structure beyond the simplistic model of WREs as clusters of TCF sites.
MATERIALS AND METHODS
Cell culture and transfection
All cell lines were grown at 37°C and 5% CO2. HEK293T (ATCC, CRL-3216) and HeLa (ATCC, CCL-2) were grown in DMEM (Dulbecco's modified Eagle's medium, Gibco, 11995065) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin/glutamine (PSG, Gibco, 10378016). LS174T cells (ATCC, CL-188) were cultured in MEM (minimum essential medium, Gibco, 11095080) containing 10% FBS and PSG. Cells were transfected using Lipofectamine 2000 (Invitrogen, 11668030) or PEI MAX (Polysciences, 24765–1) according to manufacturer instructions.
Plasmids
The CREAX fragment was amplified from human genomic DNA with the following primers: 5′-CTTGCTCGAGGGAACCCGCTGAATGGCTGG-3′, 5′-ACTCAGATCTCAACACAGCGCTCCTGTCCA-3′, and cloned into upstream of the minimal promoter in pGL4.23 (Promega, E841A) with XhoI and BglII. Mutations were made in the CREAX reporter using the Quickchange II Kit (Stratagene, 200518). The mutated regions are shown in Supplementary Figure S1 and details of the mutations are shown in Supplementary Tables S1 and S2. The TOPflash and Ax2 reporters (35) along with the β-catenin* (S33Y mutation, cloned into pcDNA3.1) expression plasmid were a kind gift from Dr Eric Fearon, University of Michigan. Expression constructs for shRNAs were generated by cloning in the appropriate oligonucleotides into the pSUPER vector (OligoEngine, VEC-PBS-0002) according to the manufacturer's instructions. Targeting sequences for the shRNA constructs are listed in Supplementary Table S3.
The TCF7 open reading frame (ORF) was amplified with PCR (primers: 5′-GAATTCGAGCACTGTCATCGGAAGGAAC-3′ and 5′-GGTACCATGCCGCAGCTGGACTCC-3) and cloned into pcDNA3.1 with KpnI and EcoRI. The CDX1 ORF was similarly amplified and cloned (primers: 5′-GGTACCATGTATGTGGGCTATGTGCTGGAC-3′ and 5′-GAATTCTGGCAGAAACTCCTCTTTCACAG-3′). The HA tag was generated by annealing two oligonucleotides (5′-AATTCTACCCATACGATGTTCCAGATTACGCTTACCCATACGATGTTCCAGATTACGCTTACCCATACGATGTTCCAGATTACGCTTAAT-3′ and 5′-CTAGATTAAGCGTAATCTGGAACATCGTATGGGTAAGCGTAATCTGGAACATCGTATGGGTAAGCGTAATCTGGAACATCGTATGGGTAG-3′) which were then cloned into protein expression plasmids using EcoRI and XbaI. Flag tags were added in a similar manner using different oligos (5′-AATTCGACTACAAGGATGACGATGACAAAGACTACAAGGATGACGATGACAAAGACTACAAGGATGACGATGACAAATAAT-3′ and 5′-CTAGATTATTTGTCATCGTCATCCTTGTAGTCTTTGTCATCGTCATCCTTGTAGTCTTTGTCATCGTCATCCTTGTAGTCG-3′). GST-CDX1 was generated by PCR cloning the CDX1 ORF into pGEX-6P-1 (Cytiva, 28-9546-48) using the following primers: 5′-CGCGGATCCATGTGCTGGACAAGGATTCG-3′ and 5′-ATAGTTTAGCGGCCGCTTATGGCAGAAACTCCTCTTTCACAG-3. His-TCF7 was PCR cloned into pET52b (Millipore, 71554–3) using the primers 5′-CGGGGTACCTCATGCCGCAGCTGGACTCC-3′ and 5′-ATAGTTTAGCGGCCGCGAGCACTGTCATCGGAAGGAAC-3′. Site-directed mutagenesis was performed using the primers 5′-GAAGAGGCGGTCGGAGGAAGAGCACCAAGAATCCAC-3′ and 5′-GTGGATTCTTGGTGCTCTTCCTCCGACCGCCTCTTC-3′ to generate HA- and His-tagged basic tail mutant versions of TCF7.
The synthetic TCF/CDX/CAG reporter was created by annealing the following oligos: 5′-GATCTCCAACAGTCACGGTACCTTTGATCTTGTAGTTTATGCGTACCAACAGTCACGGTACCTTTGATCTTGTAGTTTATGCGTACCAACAGTCACGGTACCTTTGATCTTGTAGTTTATGCC-3′ and 5′-TCGAGGCATAAACTACAAGATCAAAGGTACCGTGACTGTTGGTACGCATAAACTACAAGATCAAAGGTACCGTGACTGTTGGTACGCATAAACTACAAGATCAAAGGTACCGTGACTGTTGGA-3′, and cloning into pGL4.23 with BglII and XhoI. The other synthetic reporters were generated by site-directed mutagenesis of this reporter using primers shown in Supplementary Table S4.
Luciferase assays and analysis
Luciferase assays were performed with firefly-luciferase reporter plasmids and either β-galactosidase (LacZ) or renilla luciferase as an internal transfection control. Activities of LacZ and luciferase were measured using the Tropix Galacto-Star (Applied Biosystems, T1056) and Tropix Luc-Screen systems (Applied Bisosystems, T1035). Experiments with firefly and renilla luciferase were assayed using Promega's Dual-luciferase reporter assay system (Promega, E1910) as per the manufacturer's protocol. For each well assayed, the firefly/LacZ or firefly/renilla ratio was used as the measure of luciferase activity. All luciferase assays were done in triplicates except for the mutagenesis screen, which was done with duplicates of each data point. The mean and standard deviation of the firefly/LacZ or firefly/renilla ratios were calculated. Data from the luciferase assays are expressed in terms of relative luciferase activity (RLA) or fold activation. To calculate RLA, a basal condition was selected (specified in each figure). The mean and standard deviation of the ratios of all conditions were then proportionally expressed in terms of the mean of the ratios of the basal conditions. Fold activation was calculated as the quotient of the luciferase activity under the specified Wnt-activated condition to the activity without Wnt signalling. Both Wnt on and Wnt off conditions were done in triplicate with appropriate error propagation used to calculate the standard deviation in the quotient.
Measurement of transcript levels by RT-qPCR
Cells were lysed with TRIzol (Invitrogen, 15596026) and RNA extraction was performed using the Rneasy mini kit (Qiagen, 74104). Reverse transcription was performed using SuperScript III Reverse Transcriptase (Invitrogen, 18080093). Quantitative PCR was then performed on a CFX Connect Real-Time PCR Detection System (Bio-Rad, 1855201) using the Power SYBR Green PCR Master Mix (Applied Biosystems, 4368577). The mean and standard deviation of transcript levels were quantified using the Pffafl method (36) with Axin2 transcript levels normalised to G6PD transcript levels.
Gel shift assay
EMSA was performed as described previously (19) using a 6% native gel. GST-tagged CDX1 and 20 fmol biotinylated DNA probe (5′- Bio-GGCCAACAGTCACGGTACCTTTGATCTTGTAGTTTATGCGTACCAACAGTCACGGTACCTTTGATCTTGTAGTTTATGCGT-3′) were incubated with 50μg/ml poly (dI-dC), 0.05% NP-40, 5 mM MgCl2 and 2μl of 50% glycerol in the presence of binding buffer (10 mM Tris–HCl, pH 7.5, 50 mM KCl, 1 mM DTT) for 5 min on ice and 20 min at room temperature. For competition assays, unlabelled CDX binding probe (5′-TCTTGTAGTTTATGCGTACGTAGTTTATGCGTACC-3′) was incubated with the reaction mixture containing protein for 10 min prior to adding the labelled probe.
CAG site enrichment analysis
A list of CAG sites identified in the CREAX and c-Myc WREs was generated and converted into a MEME motif file using the sites2meme utility from the MEME suite (37). To calculate an appropriate p-value threshold to search for CAG sites across the genome, the FIMO utility (38) was run with the CAG site motif file to identify CAG sites in the c-Myc-335 WRE. The CAG site with the highest p-value in the c-Myc-335 WRE had a P-value of 0.003611, and this was set as the threshold.
Data files analysed came from a previously published study examining the binding of TCF7L2 and CDX2 using ChIP-chip (31). Raw data from the original study are publicly available online (GEO accession no. GSE22572). Files containing the coordinates of regions bound by TCF7L2 and CDX2 were kindly provided to us by Dr. Michael P. Verzi. The bedtools intersect program (39) was used with the ‘-wa’ option to generate a BED file containing the coordinates of the overlapping TCF7L2 and CDX2 peaks and with the ‘-v’ option to generate files containing locations of peaks bound exclusively by TCF7L2 or CDX2. The twoBitToFa utility from the UCSC genome browser (40) was then used to obtain the sequences of each of the regions bound by TCF7L2 and CDX2. FIMO was then run to identify CAG sites using the following command: ‘fimo –thresh 0.003611 CAG-sites-meme.txt tcf7l2-cdx2-common.fasta’. Shuffled versions of these sequences were generated using the fasta-shuffle-letters utility from the MEME suite: ‘fasta-shuffle-letters -dna tcf7l2-cdx2-common.fasta LS174T-shuffle-001.fasta’. This command was run 100 times to generate 100 different shuffles. Random genomic loci were selected for comparison using the shuffle functionality from bedtools as follows: ‘bedtools shuffle -i LS174T_TCF4_CDX2_intersect_wa.bed -g hg18.chrom.sizes -seed 1 -chrom > Random_1.bed’. 10 shuffled versions were generated by running the command with seed values from 1 to 10 and the resulting files were concatenated to generate a file with 1180 randomly selected sequences. The number of CAG sites in each of the original and shuffled peaks were tabulated and the number of sites per kb was calculated.
Antibodies
Western blots were probed with the following antibodies: Anti-Flag-HRP (Sigma-Aldrich, A8592), Anti-HA (Roche, 11867423001), Anti-His (Cytiva, 27-4710–01), and Anti-GST (Invitrogen, A-5800). Anti-Flag immunoprecipitations were carried out using magnetic beads pre-conjugated with the antibody (Millipore, M8823). Anti-TCF7L2 (Cell Signaling Technology, 2569S) antibodies were also used for ChIP.
Generation of a Flag-tagged cell line using CRISPR/Cas9 and CRISPRi
A guide RNA targeting a site immediately downstream of the human CDX1 stop codon (atgcccaccctgtgccccgg) was designed using CRISPOR (41). This was then cloned into PX458, a plasmid expressing Cas9 protein (42). pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138). The homology-directed repair template was designed based on the pFETCh_Donor plasmid from the CETCh-seq protocol (43). pFETCh_Donor (EMM0021) was a gift from Eric Mendenhall & Richard M. Myers (Addgene plasmid # 63934). Since HEK293T cells are G-418 resistant, the neomycin resistance gene in the plasmid was changed to puromycin resistance. The puromycin resistance gene from sgOpti (44) was PCR amplified using the following primers – 5′-atcgtttcgcatgacagaatacaaaccaaccgtgc-3′ and 5′-gtataagaagacatgatgttcgaatcaagcaccaggttttcgtgtc-3′. sgOpti was a gift from Eric Lander & David Sabatini (Addgene plasmid # 85681). pFETCh_Donor was digested with AleI-v2 and BstBI (NEB). The 3x-Flag-P2A cassette was amplified from pFETCh_Donor (primers: 5′-atactctgtaatcctactcaataaacgtgtcacg-3′, 5′-attctgtcatgcgaaacgatccaggtccagggttc-3′). The fragments were then joined by Gibson assembly to create pFETCh_Puro, in which the neomycin resistance cassette was replaced with puromycin resistance. HEK293T genomic DNA was purified using the Dneasy Blood & Tissue kit (Qiagen, 69504). Homology arms were amplified using the following primers—arm 1 with 5′- ctgacgtcgacggatcgggaTCCCCGACCTGCAGCCCA-3′ and 5′- CCGGAACCTCCTCCGCTC-3′ and arm 2 with 5′- ttcgaacatcCTCGGGTGCTGGGAGTGT-3′ and 5′- actgtgctggatatctgcagGCTCTGCTTGGTCCGAATAAAG-3′. The 3xFlag-P2A-Puromycin construct was amplified using the following primers: 5′- gggagcggaggaggttccggTGGAGGTGGTTCTGGAGATTAC-3′ and 5′- agcacccgagGATGTTCGAATCAAGCACC-3′. The pcDNA3.1 plasmid (Invitrogen, V79020) was digested with EcoRI-HF and BglII (NEB) and the homology arms and flag-tagging constructs were assembled into the plasmid using Gibson assembly to generate the repair template for genome editing.
HEK293T cells were co-transfected with the Cas9/gRNA expressing plasmid and the repair template. Puromycin selection was performed 48 hours post-transfection using 500 ng/ml puromycin. Over 150 distinct puromycin-resistant colonies of cells could be seen, and these were allowed to grow under selection to create a polyclonal cell population. Cells were subsequently grown with 250 ng/ml puromycin to maintain selection. Genomic DNA was purified using the Dneasy blood & tissue kit to confirm the presence of the edits. PCRs were set up with primers specific to the Flag construct (5′-caatgcctgtgaaagaggagtttctg-3′ and 5′-CAATCTTTCATAAAAAGGCAGATTTCG-3′) and primers flanking the insertion site, which would generate a 450 bp amplicon from the unedited locus (5′-tcttgcactctctctttcactctctcc-3′ and 5′-tttatccaacaggcttactgcacagat-3′). The sequence of the edited locus was confirmed by Sanger sequencing the product from the Flag-specific PCR.
For CRISPRi experiments, oligonucleotide sequences to express four gRNAs targeting the CREAX locus were identified using CRISPOR and cloned into sgOpti after digestion with Esp3I (NEB). The gRNA target sites and oligo sequences are listed in Supplementary Table S5. The dCas9-KRAB construct was expressed from pHR-UCOE-SFFV-dCas9-mCherry-ZIM3-KRAB (45), which was a gift from Mikko Taipale (Addgene plasmid # 154473; http://n2t.net/addgene:154473;RRID:Addgene_154473). Cells were transfected with the dCas9-KRAB construct and either the gRNA expression plasmids or sgOpti as negative control.
Chromatin immunoprecipitation (ChIP)
Cells were fixed in 0.75% formaldehyde at room temperature for 10 min. They were then incubated for 5 minutes with 125 mM glycine to quench the formaldehyde. Following this, they were rinsed thrice with PBS and resuspended in RIPA buffer (Sigma-Aldrich, R0278). The cell suspension was sonicated using a Covaris M220 focused ultrasonicator. Cell debris was pelleted by high-speed centrifugation. One fraction of the supernatant was saved as the input fraction and the rest was used for the immunoprecipitation (IP). The cell lysate was pre-cleared with 30 μl Protein A agarose beads (Millipore, 16-156) for 1 h at 4°C and then incubated overnight on a rotor with antibody coated beads. The beads were then washed once in low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 500 mM NaCl) and LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris–HCl pH 8.0). DNA was eluted by gentle shaking in elution buffer (1% SDS, 100 mM NaHCO3) for 15 min. Decrosslinking was performed by adding NaCl to a final concentration of 0.2 M. RNA was digested with Rnase A (Invitrogen, 12091021) followed by treatment with Proteinase K (Sigma-Aldrich, 3115879001). DNA fragments from the input and IP fractions were purified using a PCR purification kit (Qiagen, 28104) and measured with qPCR.
Three sets of primers were used for qPCR—one targeting the CREAX locus (5′-TCCATAGCCAACAGTCACGC-3′ and 5′-GCAATCCTGCCAGCAATCTC-3′) and one targeting a flanking region located 1.3 kb away from CREAX (Control 1: 5′-CGTCATCCTGCAACAAGCTG-3′ and 5′-TCTCCATCCACCCTGACCTT-3′). Quantitative PCR was then performed on a CFX Connect Real-Time PCR Detection System (Bio-Rad, 1855201) using the Power SYBR Green PCR Master Mix (Applied Biosystems, 4368577). The percent input was determined for each of 3 replicates and the mean and standard deviation were calculated.
Nuclear extract preparation and co-immunoprecipitation
Cells were rinsed with ice-cold PBS and then collected into 1 ml of cold PBS. Cells were pelleted and resuspended with 400 μl buffer A (10 mM HEPES pH7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.5% NP-40) supplemented with a protease inhibitor cocktail (Roche, 11873580001). This suspension was incubated on ice for 15 min followed by vigorous vortexing for 10 seconds to lyse cell membranes. Nuclei were pelleted by centrifugation. The pellet was resuspended in 100μl buffer B (20 mM HEPES pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2mM EDTA, 0.5 mM DTT) supplemented with protease inhibitor cocktail on ice for 20 minutes to lyse nuclei. Cell debris was removed by high-speed centrifugation and the supernatant was used for co-immunoprecipitation.
Three-fold diluted nuclear extracts were pre-cleared with 50 μl Protein-G agarose beads (Millipore, 16–266) for 1 h at 4°C and then incubated overnight on a rotor with 3 μl anti-FLAG antibody (Sigma, F3165). Each reaction was incubated with Protein-G agarose beads (50 μl) for one more hour. Beads were washed four times with 3-fold diluted buffer B. Proteins were eluted in 2x SDS sample buffer (0.125 M Tris–HCl pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol and 0.01% (w/v) Bromophenol blue) and analysed using western blotting.
Western blotting
Cell lysates or protein samples were lysed and denatured in hot 2× SDS sample buffer. Protein samples were separated by SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad, 162-0177) and blocked in 5% bovine serum albumin (BSA, Dot Scientific, DSA30075-100). Protein blots were incubated with the appropriate concentration of primary antibody diluted in 5% BSA overnight at 4°C. They were then washed thrice with Tris-buffered saline containing 1% Tween-20 (TBS-T). Blots were incubated with the secondary antibody diluted in 5% BSA for 1 h. After washing thrice in TBS-T, they were developed using a chemiluminescence substrate (Pierce, 32109) and imaged in a LI-COR Odyssey CLx imager. Images were processed using the GNU Image Manipulation Program.
Recombinant protein expression and purification
Plasmids expressing GST- and His- tagged proteins were transformed into BL21(DE3) competent cells (Thermo Scientific, EC0114) and grown in LB media at 37°C. Protein expression was induced by the addition of IPTG when the OD reached 0.6. After 3–4 h, cells were collected by centrifugation and resuspended in the appropriate lysis buffer. Cells were lysed using sonication and proteins were purified using standard protocols (46,47).
GST pull-down assays
5 μg each of the recombinant GST-tagged bait and His-tagged prey proteins were incubated for 1.5 h in 200 μl of pull-down buffer (20 mM Tris–HCl pH7.6, 150 mM NaCl, 1% Triton X-100) at 4°C. Glutathione Fast Flow Sepharose 4 beads (GE Healthcare, 17-5132-01) that had been pre-washed in binding buffer were then added to the binding reaction and it was incubated in a rotor for 2 h at 4°C. The supernatant was then removed and the beads were washed 5 times with 600 μl of binding buffer. Proteins were then eluted using 20 μl of 2× SDS sample buffer and analysed using western blotting.
RESULTS
Identification and characterisation of CREAX, a highly Wnt-regulated enhancer of Axin2
To test the features of the TF collective model in the context of Wnt signalling, we looked for highly responsive WREs which could be studied under cell culture conditions. Axin2 is a robustly activated Wnt target gene in several mammalian tissues (27,48–50) and cell lines (35,51,52). A recent β-catenin ChIP-seq study found two TCF-dependent β-catenin-bound regions near the Axin2 gene locus (13), one near the promoter and one located about 45 kb downstream of the TSS (Figure 1A). Previous studies found peaks of TCF7L2 binding at both loci (53). The promoter-proximal region, termed Ax2, was found to be responsive to Wnt signalling in a reporter assay in HEK293T cells (51). The distal enhancer, which we named CREAX (CDX Regulated Enhancer of Axin2), was shown to be active in the LS174T colorectal cancer cell line (54). We cloned both elements into pGL4.23 vectors upstream of a minimal TATA-box promoter and luciferase (luc) gene and examined their activity in HEK293T cells with and without Wnt signalling.
Figure 1.

Characterisation of CREAX (CDX Regulated Enhancer of Axin2), a highly Wnt-responsive enhancer near Axin2. (A) Position of CREAX (CDX Regulated Enhancer of Axin2) relative to the Axin2 gene locus. Blue boxes represent exons and red bars are regions bound by β-catenin in LS174T cells (Mokry et al., 2010). Known TCF7L2-bound regions in HEK293T cells are denoted by asterisks (Doumpas et al., 2018). Black bars denote regions cloned to generate the Ax2 and CREAX reporters. Positions are shown with respect to the hg18 assembly. (B) RT-qPCR measurement of Axin2 transcript levels showing an increase upon treatment with CHIR-99021 (CHIR) in HEK293T cells. (C) Activity levels of luciferase reporters of the previously identified Ax2 promoter-proximal WRE (Jho et al., 2002) and CREAX with CHIR treatment shows that CREAX is more sensitive to Wnt signalling. (D) CREAX-luciferase reporter activity increases in a dose-dependent manner to CHIR concentration in HEK293T cells. (E) CREAX-luciferase reporter activity increases in a dose-dependent manner upon transfection of increasing amounts of a plasmid expressing stabilised β-catenin containing the S33Y mutation (β-catenin*). (F) Cartoon showing positions of guide RNAs used to recruit the dCas9-KRAB construct to the CREAX locus for CRISPRi experiments. (G) RT-qPCR results showing that the increase in Axin2 transcript levels upon CHIR treatment is suppressed by the recruitment of dCas9-KRAB to the CREAX locus. (H) Activation of the CREAX-luciferase reporter by β-catenin* is not affected by orientation, meeting the classical definition of an enhancer. In (B-E,G, and H), data are presented as mean ± SD from three replicates (N = 3) for each condition. *P< 0.05; **P< 0.005; ***P< 0.001; ****P < 0.0001; n.s. P > 0.05.
Activating the Wnt/β-catenin pathway in HEK293T cells using the GSK3 inhibitor CHIR-99021 (CHIR) resulted in a 10-fold increase in Axin2 transcript levels as detected by RT-qPCR (Figure 1B). The same treatment caused a modest increase in activity of the Ax2-luciferase reporter but a much stronger increase in the CREAX-luciferase reporter (Figure 1C). CREAX-luc activity increased in a dose-dependent manner with increasing concentrations of CHIR (Figure 1D). Similarly, CREAX-luc showed dose-responsive activation when co-transfected with increasing amounts of a plasmid expressing β-catenin containing the stabilising S33Y mutation (β-catenin*, Figure 1E) which prevents it from being targeted by the destruction complex (7). We then performed a CRISPR inhibition assay (44) using a construct expressing a catalytically inactive Cas9 fused to the KRAB transcriptional repressor domain (dCas9-KRAB) to examine whether CREAX contributed to the Wnt-dependent upregulation of Axin2 mRNA. As previous studies using dCas9-based constructs to perturb gene expression have shown stronger and more robust responses to a mixture of multiple gRNAs targeting a locus than a single gRNA (45,55,56), we identified 4 gRNAs targeting the CREAX locus (Figure 1F) and pooled them for the assay. When co-transfected with the four gRNAs and the dCas9-KRAB construct, we found that CHIR treatment resulted in significantly lower Axin2 mRNA levels, suggesting that the CREAX locus is a regulator of Wnt-dependent Axin2 transcription (Figure 1G). Finally, we found that CREAX activity was not affected by reversing its orientation, meeting the standard requirement for an enhancer (Figure 1H). The high amplitude of response to Wnt/β-catenin signalling provided a strong starting point for a detailed analysis of the TFs and cis-regulatory sequences that regulate the activity of this WRE.
TCF and CDX family proteins bind to and regulate CREAX and a distal enhancer of c-Myc
To identify the TFs that regulate CREAX activity, we computationally scanned the sequence of the enhancer for putative binding sites of TFs known to co-localise with TCFs at WREs. We used motif information from the JASPAR database (57) along with the FIMO utility from the MEME suite (38) to perform this search. This preliminary analysis identified potential binding sites for TCF and CDX family TFs in CREAX. We found no sequences resembling Helper sites, the DNA motif recognized by TCF7 and TCF7L2 isoforms containing the C-clamp DNA binding domain (17,20,23). A similar scan of the Ax2 WRE sequence identified putative TCF binding sites, but none for CDX. To confirm a role for CDX proteins in regulating CREAX, we activated Wnt signalling in HEK293T cells with β-catenin* and examined the effect of RNAi against CDX and TCF TFs on CREAX-luc reporter activity. RNAi against CDX1 or the TCFs LEF1 and TCF7 reduced CREAX reporter activity (Figure 2A). Similarly, RNAi against LEF1 or CDX1 also reduced the upregulation of Axin2 mRNA levels upon CHIR treatment (Figure 2B).
Figure 2.

CDX and TCF family members directly bind to and regulate the activity of CREAX and a distal enhancer of c-Myc. (A) RNAi-mediated depletion of CDX1, LEF1, or TCF7 reduces the activation of CREAX-luciferase by β-catenin* in HEK293T cells. Cells were transfected with CREAX-luc, β-catenin* and CDX1, LEF1, or TCF7 shRNA expression plasmids. (B) RNAi-mediated depletion of CDX1 or LEF1 reduces the increase in Axin2 mRNA levels caused by CHIR treatment in HEK293T cells. (C) Activity of a reporter of the c-Myc-335 enhancer previously shown to be bound by TCF7L2 and CDX2 in LS174T cells is reduced by RNAi against TCF7L2, CDX1 or CDX2. Cells were transfected with the reporter and plasmids expressing shRNA targeting TCF7L2, CDX1 or CDX2. (D) CHIR-stimulated c-Myc-335 reporter activity is decreased by RNAi-mediated depletion of LEF1, TCF7l2 and CDX1 in HEK293T cells. (E) Cartoon showing a portion of the CDX1 gene locus in HEK293T cells edited to express C-terminally Flag-tagged CDX1. The different sets of primers used to validate the edits are shown with arrows. (F) Anti-Flag western blot showing the expression of Flag-tagged protein in the CDX1-flag cells. (G) PCR validation showing that the CDX1-flag cells are heterozygous for the CDX1-flag allele. (H) Cartoon showing positions of the amplicons used to measure the ChIP signals from the CREAX and control loci. (I) TCF7L2 ChIP-qPCR signals from the CREAX and control loci in Flag-CDX1 HEK293T cells with and without a 24 h CHIR treatment. ChIP signals were measured using primers shown in panel H and normalized to signal from input chromatin. (J) Flag ChIP-qPCR signals from CREAX and control loci in Flag-CDX1 HEK293T cells with and without a 24 hour CHIR treatment. In (A-D,I,J), data are shown as mean ± SD from three replicates (N = 3) for each condition. P-value comparisons are made with the control condition unless otherwise indicated. P< 0.05; **P< 0.005; ***P< 0.001; ****P < 0.0001; n.s. P > 0.05.
After examining previously studied WREs to find ones that might be co-regulated by TCF and CDX TFs, we identified c-Myc-335, a distal upstream enhancer of the c-Myc oncogene (31,58). This WRE had been of interest in cancer biology since it can contain a naturally occurring SNP (rs6983267) linked to increased colorectal and prostate cancer risk (59,60). This polymorphism was found to reside in a TCF binding site and increase its affinity to TCF7L2 (61–63). To test whether this WRE was regulated in a similar manner to CREAX, we constructed a c-Myc-335 luciferase reporter. We tested it in both HEK293T cells and in LS174T colorectal cancer cells, the latter of which have mutations in β-catenin that result in constitutively active Wnt signalling (64). In these cancer cells, RNAi against TCF7L2, CDX1 and CDX2 reduces the activity of the c-Myc-335 reporter (Figure 2C). Similarly, RNAi against LEF1, TCF7l2 or CDX1 reduced the activation of c-Myc-335-luc by β-catenin* in HEK293T cells (Figure 2D).
Most previous literature on the subject of TCF/CDX co-occupancy focuses on CDX2 in colorectal cancer cells (31,58). Since CREAX activity in HEK293T cells was sensitive to CDX1 protein levels, we were interested in whether CDX1 was also capable of co-occupying WREs along with TCFs. To this end we used ChIP-qPCR to test the binding of CDX1 to CREAX in HEK293T cells. Since we were unable to find ChIP-quality antibodies against CDX1, we generated a polyclonal HEK293T cell line expressing C-terminally flag-tagged CDX1 protein from the endogenous gene locus. To create this line, we adapted the previously reported CETCh-seq protocol (43) to generate a polyclonal HEK293T cell population using CRISPR/Cas9 mediated genome editing that express a Flag-tagged CDX1 protein. The edit replaced the endogenous stop codon with a cassette containing a 3xFlag epitope upstream of the coding sequences for the P2A self-cleaving peptide and the puromycin resistance gene (Figure 2E). After selecting for puromycin resistance, we obtained a population of edited HEK293T cells which expressed a flag-tagged CDX1 detectable on a western blot (Figure 2F). PCR using combinations of primers located inside and outside the Flag epitope showed that the polyclonal cell population contains a mixture of both the tagged and untagged alleles (Figure 2E,G).
Using the CDX1-Flag cell line, we performed ChIP using antibodies against TCF7L2 and the Flag tag (for CDX1). We conducted the experiments both under control conditions and after activating Wnt signalling using CHIR to examine chromatin state under conditions in which the enhancer was active. To detect the binding of TCF7L2 and CDX1, we used qPCR with primers located in the enhancer and compared the results to the signal from primers in a flanking region outside the enhancer (Figure 2H). Both TCF7L2 and CDX1 were enriched at the CREAX locus in the presence and absence of CHIR. However, while the CDX1 peak was stronger in the presence of CHIR, there was no significant change in the strength of the TCF7L2 signal from the enhancer (Figure 2I,J).
Direct TCF–CDX protein-protein interactions are required for enhancer function
Previous reports indicated that LEF1 and CDX1 could form a complex and showed that residues on the basic tail of LEF1 were required for this interaction (65). The basic tail is a short stretch of basic amino acid residues found in all TCFs located immediately C-terminal to the HMG domain (3) (Figure 3A). These residues are conserved in other TCFs (Figure 3B). To test the role of TCF–CDX interactions in enhancer activity, we attempted to generate TCF proteins with mutations preventing their interaction with CDX TFs. As the N-terminal portion of the basic tail is involved in contacting DNA (66), we focused on residues at the C-terminal end of the motif. We made charge-swap mutations in two residues (R350E, K352E) of a TCF7 isoform lacking a C-clamp to generate a variant we named BTmut (basic tail mutant). We transfected HEK293T cells with plasmids expressing Flag-tagged CDX1 and HA-tagged WT TCF7 or BTmut. We found that TCF7 robustly co-immunoprecipitated CDX1 and BTmut did so much more weakly (Figure 3C), suggesting that the mutations severely compromised the ability of BTmut to complex with CDX1. This result was also confirmed by a pulldown assay with recombinant GST-tagged CDX1 protein and His-tagged TCF7 or BTmut (Figure 3D). These results suggested that BTmut could be a useful tool for testing the importance of TCF–CDX interactions in WRE activation.
Figure 3.
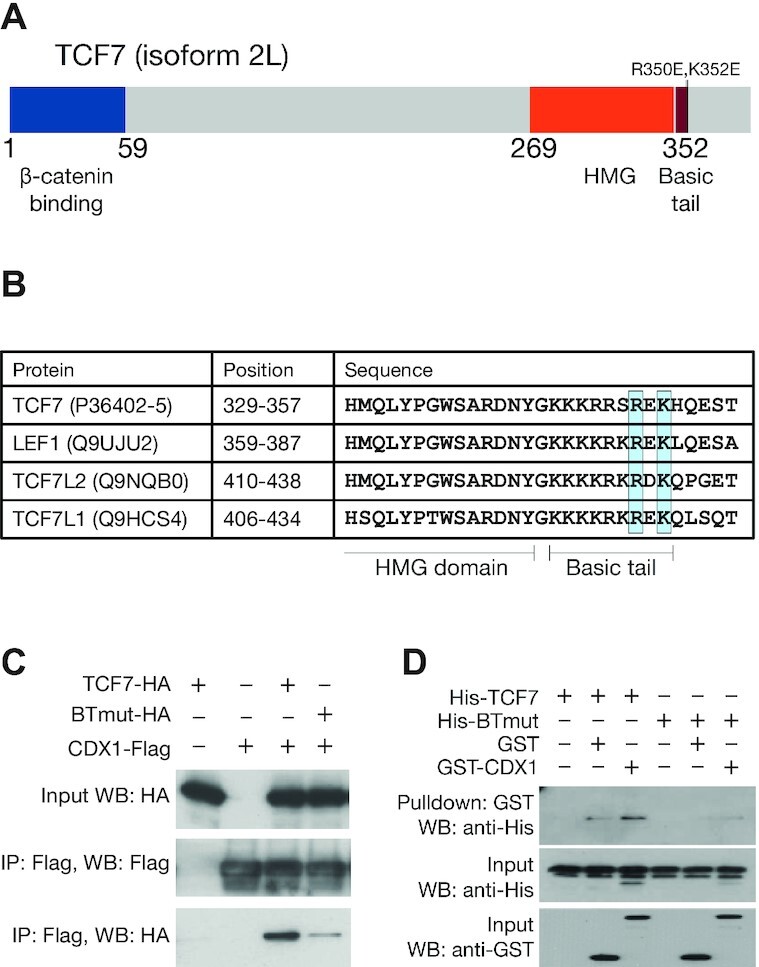
TCFs bind to CDX proteins through highly conserved residues in the basic tail. (A) Cartoon of TCF7 protein with functional domains highlighted. Positions of R350E and K352E, mutations which interfere with the TCF–CDX interaction, are highlighted. (B) Alignment of the C-terminal portion of the HMG domain and basic tail of the 4 human TCFs showing conservation of R350 and K352. Uniprot identifiers for the proteins are shown along with positions of the residues in the alignment. (C) Co-IP showing that the BTmut variant of TCF7 with 2 amino acid substitutions (R350E,K352E) shows attenuated binding to CDX1. HEK293T cells were transfected with CDX1-flag and HA-tagged WT or BTmut variants of TCF7. Cell lysates were subjected to an anti-Flag IP and examined using anti-HA and anti-Flag western blots (WB). Input samples were used as loading control. (D) GST pulldown showing direct binding between recombinant CDX1 and WT TCF7 but not BTmut. Purified His-tagged TCF7 or BTmut proteins were pulled down using glutathione beads with GST or GST-tagged CDX1 as bait. Precipitates were analysed on an anti-His WB.
For BTmut to be an informative reagent for probing the importance of TCF7–CDX1 binding in transcriptional regulation, we needed to ensure that the mutations in the basic tail did not interfere with other essential functions of TCF7. To this end, we compared the activity of WT and BTmut TCF7 proteins using the TOPflash reporter in HEK293T cells, a synthetic reporter containing 6 multimerised TCF sites (67). RNAi against LEF1 reduced the activation of TOPflash by β-catenin*. Consistent with previous work in the field which suggests that the 4 mammalian TCFs bind to very similar DNA motifs and can be functionally redundant (3,15), we found that in the case of TOPflash, the LEF1 RNAi effect could be rescued by the overexpression of TCF7 (Figure 4A).
Figure 4.

TCF–CDX interactions are important for the activation of natural WREs and Wnt target genes. (A) Luciferase assay showing that TOPflash activity can be driven by both WT TCF7 and BTmut. HEK293T cells were transfected with TOPflash reporter, β-catenin*, and plasmids expressing HA-tagged TCF7, BTmut, and shRNA against LEF1 as indicated. (B) Luciferase assay showing that BTmut cannot drive CREAX-luc activity. HEK293T cells were transfected similarly to the TOPflash experiment above. (C) RT-qPCR assay showing that LEF1 depletion by RNAi in HEK293T cells causes a reduction in Axin2 mRNA levels that can be rescued by overexpressing WT TCF7 but not BTmut. (D) Luciferase assay showing that CHIR-activated c-Myc—335 enhancer activity in HEK293T cells is reduced by LEF1 RNAi and can be rescued by overexpressing WT TCF7. BTmut is only capable of partially rescuing the RNAi effect. (E) Luciferase assay showing a qualitative difference in the ability of TCF7 and BTmut to drive c-Myc-335 reporter expression in LS174T cells. Cells were transfected with the c-Myc-335-luc reporter and the indicated shRNA and protein expressing plasmids. (F) Luciferase assay showing that the activation of CREAX-luc in HEK293T cells by β-catenin* is further increased by overexpressing WT TCF7 but not BTmut. Data are shown as mean ± SD from three replicates (N = 3) for each condition in all panels. P< 0.05; **P< 0.005; ***P< 0.001; ****P < 0.0001; n.s. P > 0.05.
BTmut was also similarly competent for rescuing TOPflash activity (Figure 4A), strongly suggesting that the BTmut protein still retained its ability to enter the nucleus and bind to DNA and β-catenin. Strikingly, in marked contrast to TOPflash, BTmut overexpression failed to rescue CREAX-luc activity (Figure 4B). These results support a model of TCF–CDX protein-protein interactions having an essential role in activating CREAX. Similarly, Axin2 mRNA levels lowered by LEF1 RNAi were rescued by WT TCF7 overexpression by not by BTmut, suggesting that TCF–CDX interactions were important for the Wnt-dependent expression of Axin2 (Figure 4C). In the case of the c-Myc-335 enhancer, BTmut only partially rescued the loss of activity caused by LEF1 RNAi while WT TCF7 completely rescued it (Figure 4D). In LS174T cells, we found that the effect of TCF7L2 RNAi on c-Myc-335 reporter activity could be fully rescued by TCF7 overexpression, while BTmut displayed limited rescuing activity (Figure 4E). Finally, overexpression of β-catenin* along with WT TCF7 in HEK293T cells caused a synergistic upregulation of CREAX-luc reporter activity that was not seen with BTmut (Figure 4F). In sum, these experiments provide strong evidence that TCF–CDX interactions play an essential role in activating multiple WREs.
Identification of TCF/CDX/CAG site clusters in CREAX
One of the key ideas of the TF collective model is that enhancers are bound by a variety of TFs, implying the existence of several distinct, functionally important regulatory motifs in these enhancers (26). We wondered whether there were factors in addition to TCF and CDX proteins that regulated CREAX activity. We sought to identify which portions of the CREAX sequence were important for Wnt-responsive activity by systematic mutagenesis of the CREAX-luc reporter plasmid. First, we looked for TCF-binding sites in the enhancer using the FIMO utility from the MEME suite (38) and a list of functionally validated TCF-binding sites (21). Based on this search, we flagged four potential TCF-binding sites identified by FIMO with a P-value threshold of <0.001 and designated these regions T1-T4. We then divided up the remaining regions in the enhancer into blocks of roughly 10 nucleotides each, splitting the 420 bp enhancer into 42 blocks. We then generated 42 constructs, each containing mutations in a different block and tested their ability to be activated by β-catenin* in HEK293T cells (see Supplementary Table S1 for details). In order to compare results across independent experiments, we performed each experiment with the WT CREAX-luc reporter as a control and expressed the fold activation of the mutant constructs as a percentage of the WT reporter.
We found that mutations in the four annotated TCF sites reduced the fold activation to at least 55% of the WT reporter. To identify additional regions of regulatory importance, we examined constructs which showed less than 55% of WT activation and those showing more than 100% of WT activity levels (red and blue lines, Figure 5A). Twenty eight out of 42 constructs fell outside these thresholds. Sixteen of them showed lower activity upon mutation, suggesting that they contained motifs required for activating the enhancer, while 12 mutant constructs showed higher activity, implying a repressive function (Figure 5B).
Figure 5.

Identification of multiple sets of recurring DNA motifs important for the activation of CREAX by Wnt signalling by a systematic mutagenesis screen. (A) Luciferase assay showing fold activation by β-catenin* of CREAX mutant constructs in HEK293T cells. Four TCF-binding sites were identified and mutated (labelled T1-T4), and the remaining nucleotides were mutated as indicated for non-overlapping screening mutagenesis. Mutations which decreased fold activation to at least 55% (blue line) or more than 100% of WT (red line) were considered hits from the screen. Bars represent a mean of two independent experiments (N = 2). (B) 28 of the 42 CREAX mutagenesis contructs showed significant changes in activation by β-catenin*. Of the 28, 16 showed reduced activity (suggesting that these regions play a role in enhancer activation) and 12 showed increased activity (suggesting a repressive effect). (C) The cartoon depicts the 420 bp CREAX fragment with the activating regions annotated. In addition to TCF binding sites (red), CREAX activity is regulated by multiple CDX binding sites (green) and CAG sites (teal), which are shown with their site logos.
To understand the requirements for the transcriptional activation of CREAX by the Wnt pathway, we examined the 16 sequence blocks containing activating information. In these, we looked for the existence of recurring motifs besides the TCF sites we had already flagged. Among the non-TCF site regions, we found 2 nearly identical sequences (TTTATGC and TTAATGC), which fit the consensus binding site for CDX family proteins (68). Additionally, we found five copies of a novel motif centred around the consensus sequence CAG, which we named ‘CAG sites’ (Figure 5C).
TCF, CDX, and CAG sites are functionally important for the activity of CREAX and the c-Myc-335 WRE
To confirm the role of TCF, CDX and CAG sites in CREAX activity, we generated reporter constructs with targeted mutations in the respective sites (see Supplementary Figure S1 and Table S2 for details). Strikingly, we found that abrogating the function of any of the three sets of motifs caused a severe reduction in CREAX reporter activation by β-catenin* (Figure 6A). To confirm that the putative CDX sites we had identified were specifically recognized by CDX protein, we performed gel-shift assays with recombinant GST-CDX1 and a probe containing a TTTATGC sequence. GST-CDX1 caused a gel mobility shift in the biotinylated probe, and the addition of excess unlabelled probe decreased the shift intensity in a manner consistent with competition (Figure 6B). These results provided support that the sites we identified were indeed CDX sites.
Figure 6.
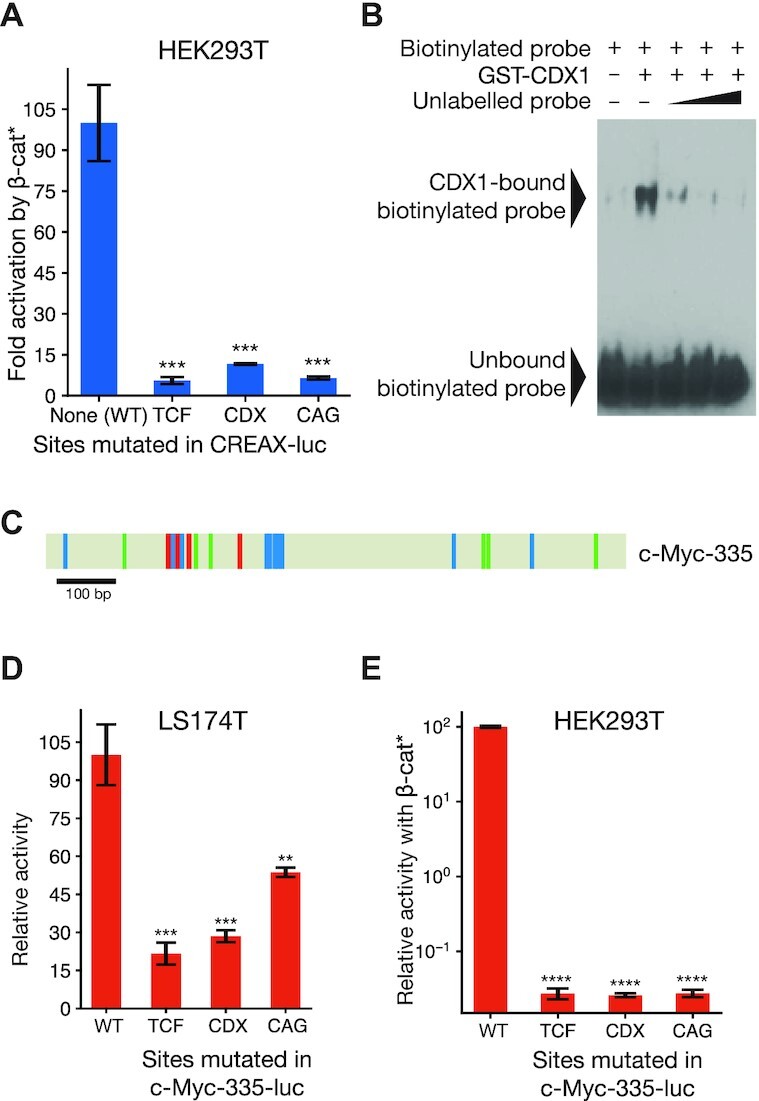
TCF and CDX binding sites and CAG sites are functionally important for CREAX and the c-Myc-335 enhancer. (A) Effect of mutating the 4 TCF sites, 2 CDX sites, or the 5 CAG sites on CREAX-luciferase reporter activity in HEK293T cells. Cells were transfected with the indicated reporters and an empty vector or plasmid expressing β-catenin* and the fold activation by β-catenin* was compared. (B) CDX1 can bind to the CDX sites in CREAX in vitro. A biotin-labelled probe containing CDX sites from CREAX binds to GST-tagged recombinant CDX1 in an EMSA. An unlabelled probe consisting of these CDX sites competes with the biotinylated probe for binding GST-CDX1. (C) Cartoon of the c-Myc-335 enhancer showing TCF binding sites (red), CDX binding sites (green), and CAG sites (teal). (D) Luciferase assay showing that the mutation of TCF, CDX, or CAG sites in the c-Myc-335 enhancer decreases its activity in LS174T cells. Cells were transfected with the indicated reporters and their relative activity was analysed by a luciferase assay. (E) Luciferase assay showing that the mutation of TCF, CDX, or CAG sites in the c-Myc-335 enhancer decreases its β-catenin*-driven activity in HEK293T cells. Data in (A, D, E) are shown as mean ± SD from three replicates (N = 3). P< 0.05; **P< 0.005; ***P< 0.001; ****P < 0.0001; n.s. P > 0.05.
Similarly, our examination of the c-Myc-335 enhancer identified four potential TCF binding sites, 6 CDX sites, and 10 CAG sites (Figure 6C; Supplementary Figure S1). To examine their role in WRE regulation, we constructed reporters with each class of motif mutated. As was the case for CREAX in HEK293T cells, we found that all three mutant reporters displayed a dramatic reduction in activity in LS174T cells (Figure 6D) and in HEK293T cells (Figure 6E).
CAG sites are enriched in regions bound by both TCF7L2 and CDX2
After establishing the functional importance of CAG sites in two WREs, we were interested in whether regulation by TCF, CDX and CAG sites was a feature of other WREs as well. Looking through the sequences of previously studied WREs, we identified TCF, CDX and CAG sites in the Wnt-responsive promoters of DEFA5 and DEFA6 (defensin alpha 5 and 6), which have been shown to be active in Paneth cells of the small intestine (69) (Supplementary Figure S2). To examine this at a larger scale, we analysed data from a previous ChIP-chip study that had identified 118 loci bound by both TCF7L2 and CDX2 in chromosomes 8, 11 and 12 of LS174T cells (31). We generated a position-weighted matrix using the 15 CAG sites from CREAX and c-Myc-335 (site logo in Figure 7A) and used it to look for CAG sites in the 118 doubly-bound loci using the FIMO utility.
Figure 7.

CAG sites are enriched at loci bound by TCF7L2 and CDX2. (A) Site logo of functionally validated CAG sites identified in the CREAX and c-Myc-335 WREs. (B) Distribution of CAG sites in regions bound by TCF7L2 and CDX2 in colorectal cancer cells from Verzi et al. Each data point shows the number of CAG sites per kb in a single ChIP-chip peak. A search for CAG sites was performed in 3 shuffled versions of each peak's sequence to see if there was an enrichment of the CAG site motif in these sites. (C) CAG sites are enriched in loci bound by TCF7L2 and CDX2. Actual number of CAG sites identified by FIMO are shown in the red scatter plot. The number of CAG sites was calculated in 100 shuffled variants of the sequence of each locus, and the blue line shows the average number of CAG sites in these variants. The shaded region represents the standard deviation in the number of CAG sites (N = 100). (D) 99 (83.9%) of loci bound by both TCF7L2 and CDX2 had more than the expected number of CAG sites. The CAG site count was more than one standard deviation above the mean in 66 of those loci. (E) Box plots showing the number of CAG sites in loci bound by both TCF7l2 and CDX2, only TCF7l2, only CDX2, and randomly selected genomic regions in colorectal cancer cells. P< 0.05; **P< 0.005; ***P< 0.001; ****P < 0.0001; n.s. P > 0.05.
The ChIP-chip peaks common to TCF7L2 and CDX2 range in length from 695 to 1594 bp with a mean length of 1090 bp. On average, we found that the common peaks contained 9.93 CAG sites per kb (Figure 7B). Since the identity of the TF(s) that bind to the CAG sites is currently unknown, we had to generate a negative control dataset to test whether they were significantly enriched in the TCF7L2 and CDX2 bound loci. Using an algorithm similar to that used in DREME and other motif scanning programs (37,70,71), we compared the number of CAG sites found in each peak to the number that would be expected due to random chance. For each peak, we shuffled the peak's sequence 100 times to generate comparison sequence of identical length and nucleotide composition. We then calculated the number of CAG sites in each of the 100 shuffled controls and computed the mean and standard deviation to estimate of the number of CAG sites expected from a random distribution (Figure 7C). Surprisingly, 99/118 (83.9%) common peaks had more CAG sites than expected from chance, with the number of CAG sites in 66 (55.9%) peaks exceeding the expectation value by more than one standard deviation. Only 5 loci contained significantly fewer CAG sites than expected (Figure 7D).
After determining that CAG sites were enriched in loci bound by both TCF7L2 and CDX2, we examined their prevalence in loci bound by just one or neither of the TFs. For each TCF7L2 and CDX2 bound loci, we randomly selected 10 regions of identical length on the same chromosome to generate a set of 1180 random DNA sequences. While the number of CAG sites/kb was 9.93 for the TCF7L2/CDX2 common peaks, it was 10.10 for regions bound by TCF7L2 but not CDX2, 8.89 for regions bound exclusively by CDX2, and 8.71 in the random DNA sequences. This represents a statistically significant enrichment of CAG sites in all regions bound by TCF7L2 irrespective of CDX2 binding. At the same time, CAG sites are not enriched in loci bound exclusively by CDX2 compared to the background levels in the genome (Figure 7E). Put together, our analysis suggests that CAG motifs are widespread in several WREs, a subset of which contain the tripartite TCF/CDX/CAG regulatory logic.
CDX and CAG sites sensitise WREs to Wnt signalling in a cell-type specific manner
Having identified the importance of TCF, CDX and CAG sites in WREs, we then attempted to generalise our findings using synthetic WREs based on the design principles of CREAX and c-Myc-335. We generated 5 reporters with different combinations of TCF, CDX and CAG sites. The first reporter contained three TCF binding sites alone, similar to the TOPflash reporter corresponding to the conventional model of WREs. The second and third reporters consisted of TCF and either CDX or CAG sites respectively. The fourth reporter contained a combination of CDX and CAG sites without TCF sites, and the fifth contained TCF, CDX, and CAG sites, reflecting the composition of CREAX and c-Myc-335 (Figure 8A). The spacing between the sites were kept constant to discount any effects caused by changes in relative positions between TF binding sites. We then tested all 5 reporters in three cell lines—HEK293T cells, HeLa cells and LS174T cells. HEK293T and HeLa cells have low basal levels of Wnt signalling, so we compared the fold activation shown by each reporter upon transfection with β-catenin* (Figure 8B, C). Since LS174T cells have a constitutively active Wnt pathway, we compared the relative activity of each reporter to the empty vector (Figure 8D).
Figure 8.
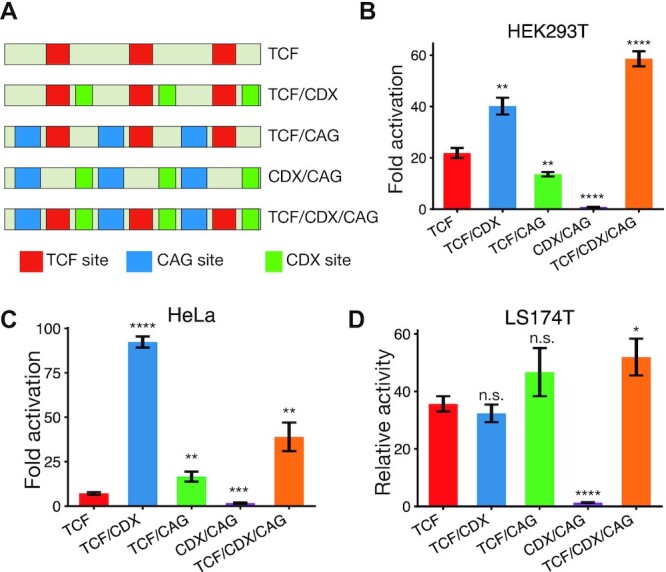
CDX and CAG sites sensitise WREs to Wnt signalling. (A) Cartoons of the five synthetic WREs with different combinations of TCF, CDX and CAG sites. Constructs shown were cloned upstream of a minimal promoter to generate luciferase reporters. (B–D) Relative activity levels of the synthetic WREs in HEK293T, LS174T and HeLa cells. HEK293T (B) and HeLa cells (C) were transfected with indicated reporters and an empty vector or β-catenin* expression plasmid. Fold activation by β-catenin* was compared. LS174T cells were transfected with indicated reporters, and activity was normalised to that of the empty vector with only the minimal promoter (D). Each bar represents mean ± SD from three replicates (N = 3) P< 0.05; **P< 0.005; ***P< 0.001; ****P < 0.0001; n.s. P > 0.05.
These experiments yielded three main conclusions. Firstly, we found that the TCF/CDX/CAG construct was more active than the construct containing only TCF sites in all 3 cell lines. From this we concluded that CDX and CAG sites play a general role in sensitising WREs to Wnt signalling. Secondly, the construct lacking TCF sites and containing only CDX and CAG sites did not show Wnt-responsive activity, supporting a model of them supporting WRE activation through TCFs. Thirdly, the best-performing WREs were different in each cell type – while the TCF/CDX/CAG was the clear winner in HEK293T cells, it showed a similar level of activity as the TCF/CAG reporter in LS174T cells. Similarly, while both the TCF/CAG and TCF/CDX/CAG were more sensitive than the TCF-only reporter in HeLa cells, the TCF/CDX construct was more than twice as sensitive as the TCF/CDX/CAG construct. When looked at simultaneously, the contributions of TCF, CDX, and CAG sites to WRE activity across cell types paint a picture of a TF collective containing TCF and CDX proteins whose activity is modulated by additional cell type specific factors.
DISCUSSION
The question of what makes certain regions of the genome act as enhancers has long been of interest in the field of gene regulation. It is widely accepted that enhancers contain clusters of binding sites for multiple TFs. However, understanding the ‘binding site grammar’ of enhancers—the rules governing the composition, distribution, and orientation of TF binding sites that allow them to drive expression under specific conditions—remains a challenge (25,72). The problem of enhancer composition is typically discussed using two overarching models, the enhanceosome and the flexible billboard. The enhanceosome is best exemplified by the Interferon-β promoter (73,74) and models enhancers as consisting of specific TF binding sites organised in a specific order and orientation. The flexible billboard model portrays enhancers as collections of TF binding sites which vary in their composition and arrangement. Enhancer activation occurs when a sufficient number of TF binding sites are occupied (24). While there is experimental evidence of some constraint in binding site order and orientation on some enhancers (75–77), other studies support the flexible billboard view (78,79). The TF collective model adds a further level of complexity to the billboard model by invoking protein-protein interactions between TFs as an additional means of recruiting TFs to enhancers (26). While computational efforts to understand this enhancer grammar have had some success incorporating aspects of all three aforementioned models (80), our results with WREs demonstrate the need for additional bottom up experimental approaches to understand the logic of enhancer composition.
Here, we report on two WREs whose binding site grammar is consistent with the billboard rubric of enhancer function. A combination of ChIP and binding site mutagenesis indicate that the CREAX and c-Myc-335 WREs are direct targets of TCF and CDX TFs (Figures 2 and 5). Systematic mutagenesis of CREAX revealed the presence of multiple copies of another motif, which we have termed CAG sites (Figure 5). These CAG sites are required for Wnt responsiveness of the CREAX and c-Myc-335 WREs (Figure 6). CAG motifs are also enriched in the vast majority of regions bound by TCF7L2 in a colorectal cell line (Figure 7). While TCF/CDX/CAG site-based regulation appears to define a significant class of WREs, the arrangement of the binding sites is highly variable. For example, in CREAX, the TCF and CAG sites are roughly evenly spread out, while in the c-Myc-335 enhancer, three out of four TCF sites and five of the CAG sites form separate clusters (Figures 5C and 6C). In the 118 sites bound by TCFL2 and CDX2, there is also a great flexibility of motif composition, with the density of CAG sites ranging from 1.3 to 21.3 sites per kb. While all these sites have not been functionally tested, the systematic analysis of CREAX supports a model where all the TCF, CDX and CAG sites contribute to activation by Wnt signalling (Figure 5A).
In addition to supporting the billboard model for WRE function, our data demonstrating the importance of TCF–CDX physical interactions also fits with the TF collective vision of enhancer activation. Our work extends previous findings demonstrating an interaction between the basic tail of TCFs and the homeodomain of CDXs (32). We found that mutation of two residues in the basic tail of TCF7 (BTmut), resulting in a decreased binding to CDX1, did not affect its ability to activate a TCF-site reporter. However, BTmut was dramatically crippled in supporting CREAX activation and showed a significant defect in regulating c-Myc-335 (Figure 4). The arginine and lysine residues that mediate TCF7–CDX1 interactions are conserved among all 4 mammalian TCFs as well as in several vertebrate TCFs (Supplementary Table S6), suggesting a broad role for TCF–CDX associations in gene regulation.
Our data provide compelling support for the importance of TCF–CDX interactions in WRE activation, but the precise mechanism remains to be elucidated. A previous study identified the recruitment of CDX1 and LEF1 to CDX1 regulatory DNA, and the absence of CDX binding sites in this sequence led the authors to surmise that TCFs could recruit CDX proteins to DNA (32). Another study found that the binding of TCF7L2 decreased upon CDX2 depletion by RNAi at loci bound by both TCF7L2 and CDX2, suggesting that CDX2 could recruit TCFs to chromatin (31). CDXs have also been reported to act as pioneer factors, making chromatin more accessible to binding by other TFs (81,82). However, since our data suggest that CREAX is bound by TCF and CDX proteins even in the absence of Wnt signalling, experiments in TCF or CDX knockout backgrounds will be required to distinguish between the various models. It is also possible that TCFs recruit CDXs to some loci with the reverse happening at others.
Our findings on how CDXs can directly co-regulate WREs could provide mechanistic insight into previous findings in vertebrate development. Wnt/β-catenin signalling, TCFs and CDXs have all been demonstrated to be important for axial patterning in mammalian development, and specifying posterior/caudal cell fates (83–85). CDXs are transcriptional targets of Wnt signalling (86,87) suggesting a hierarchy of action. However, consistent with our results, there is also evidence that CDXs and the Wnt pathway function at the same level of the axial patterning hierarchy (88,89). This suggests that TCF/β-catenin and CDX may also directly act on enhancers controlling posterior fate genes. Wnt signalling and CDXs are also known to intersect in other contexts such as the haematopoietic system (90) and primordial germ cells (91). Mice engineered to express the BTmut variant could be a powerful tool to test the importance of WRE activation by CDX proteins in these and other contexts.
Our synthetic enhancer experiments demonstrated that CDX and CAG sites contribute to WRE function across cell types (Figure 8). The addition of CDX sites increased WRE activity in HEK293T and HeLa cells, while it had no effect in LS174T cells. One explanation for this observation could be that while TCF and CDX TFs directly bind to these WREs in all three cell types, the TF collectives regulating these WREs contain additional cell type-specific TFs which contribute to enhancer activity without binding to DNA. A similar model has been proposed for TBX3 regulation of Wnt targets in mammalian limb development. Interestingly, a enhancer corresponding to CREAX is known to be bound by TBX3 and BCL9 in developing mouse forelimbs (92). Similarly, the addition of CAG sites increases WRE activity in LS174T and HeLa cells but decreases it in HEK293T cells. Since the identity of the CAG site binding protein is still unknown, this could imply the existence of multiple TFs capable of binding CAG sites expressed at different levels in various cell types. In addition to the cis-regulatory information that recruits TFs to an enhancer, the trans-regulatory environment of the cell can dictate the identity of the TF collective which binds to the enhancer. Transcriptional regulation supplementing the direct binding of TFs to DNA provides additional control over enhancer activity to produce the exquisite spatio-temporal specificity of transcription seen throughout animal development.
The systematic mutagenesis of the CREAX enhancer provides a dramatic illustration that naturally occurring WREs consist of more than clusters of TCF binding sites. Our identification of CDX and CAG sites in WREs refines our understanding of the binding site grammar underlying some WREs. It is important to note that the identified TCF, CDX and CAG sites only account for 11 of the 16 regions required for activation of CREAX (Figure 5C). In addition, there are 12 regions with repressive activity. These functional regions don’t contain consensus AP-1 sites, which have been implicated in WRE function (93). Nor do they contain the repressive motif recently identified in several WREs (94). Future experiments will explore how these sequences interact with the TCF/CDX/CAG cassette, to more fully understand the multiple layers of regulation in WREs.
DATA AVAILABILITY
Source data for all figures will be provided by the corresponding author upon request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Benjamin L. Allen for his helpful comments on the manuscript and Aaron Buckingham for contributing to the initial stages of this project.
Contributor Information
Aravinda-Bharathi Ramakrishnan, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109 USA.
Lisheng Chen, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109 USA.
Peter E Burby, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109 USA.
Ken M Cadigan, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109 USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institute of Health [R01 GM108468 to K.M.C]; University of Michigan Rogel Cancer Center [to K.M.C]; University of Michigan M-cubed [to K.M.C]; University of Michigan Department of Molecular, Cellular, and Developmental Biology [Biosciences fellowship to A.-B.R.]; Rackham Graduate School, University of Michigan [One-Term Dissertation Fellowship and a Graduate Student Research Grant to A.-B.R.]. Funding for open access charge: Chair's Research Funds (to K.M.C.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Clevers H., Nusse R.. Wnt/β-catenin signaling and disease. Cell. 2012; 149:1192–1205. [DOI] [PubMed] [Google Scholar]
- 2.Clevers H., Loh K.M., Nusse R.. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014; 346:1248012. [DOI] [PubMed] [Google Scholar]
- 3.Archbold H.C., Yang Y.X., Chen L., Cadigan K.M.. How do they do Wnt they do?: regulation of transcription by the Wnt/β-catenin pathway. Acta Physiol. 2012; 204:74–109. [DOI] [PubMed] [Google Scholar]
- 4.Lien W.-H., Fuchs E.. Wnt some lose some: transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 2014; 28:1517–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramakrishnan A.-B., Cadigan K.M.. Wnt target genes and where to find them. F1000Res. 2017; 6:746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Söderholm S., Cantù C.. The WNT/β-catenin dependent transcription: a tissue-specific business. WIREs Mechanisms of Disease. 2021; 13:e1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamos J.L., Weis W.I.. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2013; 5:a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barolo S.Transgenic Wnt/TCF pathway reporters: all you need is Lef?. Oncogene. 2006; 25:7505–7511. [DOI] [PubMed] [Google Scholar]
- 9.Cadigan K.M.Payre S.P.Chapter one - TCFs and Wnt/β-catenin signaling: more than one way to throw the switch. Current Topics in Developmental Biology, Transcriptional Switches During Development. 2012; 98:Academic Press; 1–34. [DOI] [PubMed] [Google Scholar]
- 10.Valenta T., Hausmann G., Basler K.. The many faces and functions of β-catenin. EMBO J. 2012; 31:2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Es J.H., Haegebarth A., Kujala P., Itzkovitz S., Koo B.-K., Boj S.F., Korving J., van den Born M., van Oudenaarden A., Robine S.et al.. A critical role for the wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell. Biol. 2012; 32:1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schweizer L., Nellen D., Basler K.. Requirement for Pangolin/dTCF in drosophila wingless signaling. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:5846–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doumpas N., Lampart F., Robinson M.D., Lentini A., Nestor C.E., Cantù C., Basler K.. TCF/LEF dependent and independent transcriptional regulation of Wnt/β-catenin target genes. EMBO J. 2019; 38:e98873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franz A., Shlyueva D., Brunner E., Stark A., Basler K.. Probing the canonicity of the Wnt/Wingless signaling pathway. PLos Genet. 2017; 13:e1006700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreira S., Polena E., Gordon V., Abdulla S., Mahendram S., Cao J., Blais A., Wood G.A., Dvorkin-Gheva A., Doble B.W.. A single TCF transcription factor, regardless of its activation capacity, is sufficient for effective trilineage differentiation of ESCs. Cell Rep. 2017; 20:2424–2438. [DOI] [PubMed] [Google Scholar]
- 16.Cadigan K.M., Waterman M.L.. TCF/LEFs and wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012; 4:a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atcha F.A., Syed A., Wu B., Hoverter N.P., Yokoyama N.N., Ting J.-H.T., Munguia J.E., Mangalam H.J., Marsh J.L., Waterman M.L.. A unique DNA binding domain converts T-Cell factors into strong wnt effectors. Mol. Cell. Biol. 2007; 27:8352–8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravindranath A.J., Cadigan K.M.. Structure-function analysis of the C-clamp of TCF/pangolin in Wnt/ß-catenin signaling. PLoS One. 2014; 9:e86180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang J.L., Chang M.V., Barolo S., Cadigan K.M.. Regulation of the feedback antagonist naked cuticle by Wingless signaling. Dev. Biol. 2008; 321:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoverter N.P., Zeller M.D., McQuade M.M., Garibaldi A., Busch A., Selwan E.M., Hertel K.J., Baldi P., Waterman M.L.. The TCF C-clamp DNA binding domain expands the Wnt transcriptome via alternative target recognition. Nucleic Acids Res. 2014; 42:13615–13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Archbold H.C., Broussard C., Chang M.V., Cadigan K.M.. Bipartite recognition of DNA by TCF/Pangolin is remarkably flexible and contributes to transcriptional responsiveness and tissue specificity of wingless signaling. PLoS Genet. 2014; 10:e1004591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhambhani C., Ravindranath A.J., Mentink R.A., Chang M.V., Betist M.C., Yang Y.X., Koushika S.P., Korswagen H.C., Cadigan K.M.. Distinct DNA binding sites contribute to the TCF transcriptional switch in C. elegans and drosophila. PLos Genet. 2014; 10:e1004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoverter N.P., Ting J.-H., Sundaresh S., Baldi P., Waterman M.L.. A WNT/p21 circuit directed by the C-Clamp, a Sequence-Specific DNA binding domain in TCFs. Mol. Cell. Biol. 2012; 32:3648–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnosti D.N., Kulkarni M.M.. Transcriptional enhancers: intelligent enhanceosomes or flexible billboards?. J. Cell. Biochem. 2005; 94:890–898. [DOI] [PubMed] [Google Scholar]
- 25.Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PubMed] [Google Scholar]
- 26.Spitz F., Furlong E.E.M.. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012; 13:613–626. [DOI] [PubMed] [Google Scholar]
- 27.van Amerongen R., Bowman A.N., Nusse R.. Developmental stage and time dictate the fate of Wnt/β-catenin-responsive stem cells in the mammary gland. Cell Stem Cell. 2012; 11:387–400. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura Y., Hoppler S.. Genome-wide analysis of canonical Wnt target gene regulation in Xenopus tropicalis challenges β-catenin paradigm. Genesis. 2017; 55:e22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura Y., Alves E., de P., Veenstra G.J.C., Hoppler S.. Tissue- and stage-specific Wnt target gene expression is controlled subsequent to β-catenin recruitment to cis-regulatory modules. Development. 2016; 143:1914–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhambhani C., Cadigan K.M., Hoppler S., Moon R.T.. Finding a needle in a genomic haystack. Wnt Signaling in Development and Disease. 2014; John Wiley & Sons, Inc; 73–87. [Google Scholar]
- 31.Verzi M.P., Hatzis P., Sulahian R., Philips J., Schuijers J., Shin H., Freed E., Lynch J.P., Dang D.T., Brown M.et al.. TCF4 and CDX2, major transcription factors for intestinal function, converge on the same cis-regulatory regions. Proc. Natl Acad. Sci. 2010; 107:15157–15162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Béland M., Pilon N., Houle M., Oh K., Sylvestre J.-R., Prinos P., Lohnes D.. Cdx1 autoregulation is governed by a novel Cdx1-LEF1 transcription complex. Mol. Cell. Biol. 2004; 24:5028–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy M.W., Chalamalasetty R.B., Thomas S., Garriock R.J., Jailwala P., Yamaguchi T.P.. Sp5 and Sp8 recruit β-catenin and Tcf1-Lef1 to select enhancers to activate Wnt target gene transcription. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:3545–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiao S., Li C., Hao Q., Miao H., Zhang L., Li L., Zhou Z.. VGLL4 targets a TCF4–TEAD4 complex to coregulate Wnt and Hippo signalling in colorectal cancer. Nat. Commun. 2017; 8:14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung J.Y., Kolligs F.T., Wu R., Zhai Y., Kuick R., Hanash S., Cho K.R., Fearon E.R.. Activation of AXIN2 Expression by β-Catenin-T Cell Factor. A feedback repressor pathway regulating Wnt signaling. J. Biol. Chem. 2002; 277:21657–21665. [DOI] [PubMed] [Google Scholar]
- 36.Pfaffl M.W.A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001; 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Concordet J.-P., Haeussler M.. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protocols. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savic D., Partridge E.C., Newberry K.M., Smith S.B., Meadows S.K., Roberts B.S., Mackiewicz M., Mendenhall E.M., Myers R.M.. CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. Genome Res. 2015; 25:1581–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fulco C.P., Munschauer M., Anyoha R., Munson G., Grossman S.R., Perez E.M., Kane M., Cleary B., Lander E.S., Engreitz J.M.. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science. 2016; 354:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alerasool N., Segal D., Lee H., Taipale M.. An efficient KRAB domain for CRISPRi applications in human cells. Nat. Methods. 2020; 17:1093–1096. [DOI] [PubMed] [Google Scholar]
- 46.Harper S., Speicher D.W.. Purification of proteins fused to glutathione S-tranferase. Methods Mol. Biol. 2011; 681:259–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nallamsetty S., Waugh D.S.. A generic protocol for the expression and purification of recombinant proteins in Escherichia coli using a combinatorial His 6 -maltose binding protein fusion tag. Nat. Protoc. 2007; 2:383–391. [DOI] [PubMed] [Google Scholar]
- 48.Lim X., Tan S.H., Yu K.L., Lim S.B.H., Nusse R.. Axin2 marks quiescent hair follicle bulge stem cells that are maintained by autocrine Wnt/β-catenin signaling. Proc. Natl Acad. Sci. U.S.A. 2016; 113:E1498–E1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang B., Zhao L., Fish M., Logan C.Y., Nusse R.. Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature. 2015; 524:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lustig B., Jerchow B., Sachs M., Weiler S., Pietsch T., Karsten U., Wetering M., Clevers H., Schlag P.M., Birchmeier W.et al.. Negative feedback loop of wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 2002; 22:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jho E., Zhang T., Domon C., Joo C.-K., Freund J.-N., Costantini F.. Wnt/β-Catenin/Tcf signaling induces the transcription of axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 2002; 22:1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van der Flier L.G., Sabates–Bellver J., Oving I., Haegebarth A., De Palo M., Anti M., Van Gijn M.E., Suijkerbuijk S., Van de Wetering M., Marra G.et al.. The intestinal Wnt/TCF signature. Gastroenterology. 2007; 132:628–632. [DOI] [PubMed] [Google Scholar]
- 53.Mokry M., Hatzis P., Bruijn E., Koster J., Versteeg R., Schuijers J., Wetering M., Guryev V., Clevers H., Cuppen E.. Efficient double fragmentation ChIP-seq provides nucleotide resolution protein-DNA binding profiles. PLoS One. 2010; 5:e15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hatzis P., Flier L.G., Driel M.A., Guryev V., Nielsen F., Denissov S., Nijman I.J., Koster J., Santo E.E., Welboren W.et al.. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 2008; 28:2732–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W., Rangarajan S., Shivalila C.S., Dadon D.B., Jaenisch R.. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013; 23:1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez-Pinera P., Kocak D.D., Vockley C.M., Adler A.F., Kabadi A.M., Polstein L.R., Thakore P.I., Glass K.A., Ousterout D.G., Leong K.W.et al.. RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nat. Methods. 2013; 10:973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D.et al.. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic. Acids. Res. 2020; 48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewis A., Freeman-Mills L., de la Calle-Mustienes E., Giráldez-Pérez R.M., Davis H., Jaeger E., Becker M., Hubner N.C., Nguyen L.N., Zeron-Medina J.et al.. A polymorphic enhancer near GREM1 influences bowel cancer risk through differential CDX2 and TCF7L2 binding. Cell Rep. 2014; 8:983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tomlinson I., Webb E., Carvajal-Carmona L., Broderick P., Kemp Z., Spain S., Penegar S., Chandler I., Gorman M., Wood W.et al.. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat. Genet. 2007; 39:984–988. [DOI] [PubMed] [Google Scholar]
- 60.Zanke B.W., Greenwood C.M., Rangrej J., Kustra R., Tenesa A., Farrington S.M., Prendergast J., Olschwang S., Chiang T., Crowdy E.et al.. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet. 2007; 39:989–994. [DOI] [PubMed] [Google Scholar]
- 61.Pomerantz M.M., Ahmadiyeh N., Jia L., Herman P., Verzi M.P., Doddapaneni H., Beckwith C.A., Chan J.A., Hills A., Davis M.et al.. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 2009; 41:882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tuupanen S., Turunen M., Lehtonen R., Hallikas O., Vanharanta S., Kivioja T., Björklund M., Wei G., Yan J., Niittymäki I.et al.. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009; 41:885–890. [DOI] [PubMed] [Google Scholar]
- 63.Wright J.B., Brown S.J., Cole M.D.. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell. Biol. 2010; 30:1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van de Wetering M., Sancho E., Verweij C., de Lau W., Oving I., Hurlstone A., van der Horn K., Batlle E., Coudreuse D., Haramis A.-P.et al.. The β-Catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002; 111:241–250. [DOI] [PubMed] [Google Scholar]
- 65.Béland M., Pilon N., Houle M., Oh K., Sylvestre J.-R., Prinos P., Lohnes D.. Cdx1 autoregulation is governed by a Novel Cdx1-LEF1 transcription complex. Mol. Cell. Biol. 2004; 24:5028–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giese K., Amsterdam A., Grosschedl R.. DNA-binding properties of the HMG domain of the lymphoid-specific transcriptional regulator LEF-1. Genes Dev. 1991; 5:2567–2578. [DOI] [PubMed] [Google Scholar]
- 67.Korinek V., Barker N., Morin P.J., van Wichen D., de Weger R., Kinzler K.W., Vogelstein B., Clevers H.. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997; 275:1784–1787. [DOI] [PubMed] [Google Scholar]
- 68.Berger M.F., Badis G., Gehrke A.R., Talukder S., Philippakis A.A., Peña-Castillo L., Alleyne T.M., Mnaimneh S., Botvinnik O.B., Chan E.T.et al.. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008; 133:1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wehkamp J., Wang G., Kübler I., Nuding S., Gregorieff A., Schnabel A., Kays R.J., Fellermann K., Burk O., Schwab M.et al.. The paneth cell α-Defensin deficiency of ileal crohn's disease is linked to Wnt/Tcf-4. J. Immunol. 2007; 179:3109–3118. [DOI] [PubMed] [Google Scholar]
- 70.Bailey T.L.DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011; 27:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bailey T.L.STREME: accurate and versatile sequence motif discovery. Bioinformatics. 2020; 10.1093/bioinformatics/btab203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wasserman W.W., Sandelin A.. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004; 5:276–287. [DOI] [PubMed] [Google Scholar]
- 73.Panne D., Maniatis T., Harrison S.C.. An atomic model of the interferon-β Enhanceosome. Cell. 2007; 129:1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thanos D., Maniatis T.. Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell. 1995; 83:1091–1100. [DOI] [PubMed] [Google Scholar]
- 75.Farley E.K., Olson K.M., Zhang W., Rokhsar D.S., Levine M.S.. Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:6508–6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jolma A., Yin Y., Nitta K.R., Dave K., Popov A., Taipale M., Enge M., Kivioja T., Morgunova E., Taipale J.. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015; 527:384–388. [DOI] [PubMed] [Google Scholar]
- 77.Swanson C.I., Evans N.C., Barolo S.. Structural rules and complex regulatory circuitry constrain expression of a notch- and egfr-regulated eye enhancer. Dev. Cell. 2010; 18:359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Junion G., Spivakov M., Girardot C., Braun M., Gustafson E.H., Birney E., Furlong E.E.M.. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012; 148:473–486. [DOI] [PubMed] [Google Scholar]
- 79.Smith R.P., Taher L., Patwardhan R.P., Kim M.J., Inoue F., Shendure J., Ovcharenko I., Ahituv N.. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 2013; 45:1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen L., Capra J.A.. Learning and interpreting the gene regulatory grammar in a deep learning framework. PLoS Comput. Biol. 2020; 16:e1008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumar N., Tsai Y.-H., Chen L., Zhou A., Banerjee K.K., Saxena M., Huang S., Toke N.H., Xing J., Shivdasani R.A.et al.. The lineage-specific transcription factor CDX2 navigates dynamic chromatin to control distinct stages of intestine development. Development. 2019; 146:dev172189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Verzi M.P., Shin H., Roman A.K.S., Liu X.S., Shivdasani R.A.. Intestinal master transcription factor CDX2 controls chromatin access for partner transcription factor binding. Mol. Cell. Biol. 2013; 33:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Galceran J., Fariñas I., Depew M.J., Clevers H., Grosschedl R.. Wnt3a-/–like phenotype and limb deficiency in Lef1(-/-)Tcf1(-/-) mice. Genes Dev. 1999; 13:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lohnes D.The Cdx1 homeodomain protein: an integrator of posterior signaling in the mouse. Bioessays. 2003; 25:971–980. [DOI] [PubMed] [Google Scholar]
- 85.Shimizu T., Bae Y.-K., Muraoka O., Hibi M.. Interaction of Wnt and caudal-related genes in zebrafish posterior body formation. Dev. Biol. 2005; 279:125–141. [DOI] [PubMed] [Google Scholar]
- 86.Pilon N., Oh K., Sylvestre J.-R., Bouchard N., Savory J., Lohnes D.. Cdx4 is a direct target of the canonical Wnt pathway. Dev. Biol. 2006; 289:55–63. [DOI] [PubMed] [Google Scholar]
- 87.Pilon N., Oh K., Sylvestre J.-R., Savory J.G.A., Lohnes D.. Wnt signaling is a key mediator of Cdx1 expression in vivo. Development. 2007; 134:2315–2323. [DOI] [PubMed] [Google Scholar]
- 88.Amin S., Neijts R., Simmini S., van Rooijen C., Tan S.C., Kester L., van Oudenaarden A., Creyghton M.P., Deschamps J.. Cdx and T Brachyury Co-activate growth signaling in the embryonic axial progenitor niche. Cell Rep. 2016; 17:3165–3177. [DOI] [PubMed] [Google Scholar]
- 89.Young T., Rowland J.E., van de Ven C., Bialecka M., Novoa A., Carapuco M., van Nes J., de Graaff W., Duluc I., Freund J.-N.et al.. Cdx and hox genes differentially regulate posterior axial growth in mammalian embryos. Dev. Cell. 2009; 17:516–526. [DOI] [PubMed] [Google Scholar]
- 90.Lengerke C., Schmitt S., Bowman T.V., Jang I.H., Maouche-Chretien L., McKinney-Freeman S., Davidson A.J., Hammerschmidt M., Rentzsch F., Green J.B.A.et al.. BMP and wnt specify hematopoietic fate by activation of the Cdx-hox pathway. Cell Stem Cell. 2008; 2:72–82. [DOI] [PubMed] [Google Scholar]
- 91.Bialecka M., Young T., Sousa Lopes S.C., ten Berge D., Sanders A., Beck F., Deschamps J.. Cdx2 contributes to the expansion of the early primordial germ cell population in the mouse. Dev. Biol. 2012; 371:227–234. [DOI] [PubMed] [Google Scholar]
- 92.Zimmerli D., Borrelli C., Jauregi-Miguel A., Söderholm S., Brütsch S., Doumpas N., Reichmuth J., Murphy-Seiler F., Aguet Mi., Basler K.et al.. TBX3 acts as tissue-specific component of the Wnt/β-catenin transcriptional complex. eLife. 2020; 9:e58123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yochum G.S., Cleland R., Goodman R.H.. A genome-wide screen for β-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell. Biol. 2008; 28:7368–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim K., Cho J., Hilzinger T.S., Nunns H., Liu A., Ryba B.E., Goentoro L.. Two-element transcriptional regulation in the canonical wnt pathway. Curr. Biol. 2017; 27:2357–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for all figures will be provided by the corresponding author upon request.