


Abstract
A key regulatory process during Drosophila development is the localized suppression of the hunchback mRNA translation at the posterior, which gives rise to a hunchback gradient governing the formation of the anterior-posterior body axis. This suppression is achieved by a concerted action of Brain Tumour (Brat), Pumilio (Pum) and Nanos. Each protein is necessary for proper Drosophila development. The RNA contacts have been elucidated for the proteins individually in several atomic-resolution structures. However, the interplay of all three proteins during RNA suppression remains a long-standing open question. Here, we characterize the quaternary complex of the RNA-binding domains of Brat, Pum and Nanos with hunchback mRNA by combining NMR spectroscopy, SANS/SAXS, XL/MS with MD simulations and ITC assays. The quaternary hunchback mRNA suppression complex comprising the RNA binding domains is flexible with unoccupied nucleotides functioning as a flexible linker between the Brat and Pum-Nanos moieties of the complex. Moreover, the presence of the Pum-HD/Nanos-ZnF complex has no effect on the equilibrium RNA binding affinity of the Brat RNA binding domain. This is in accordance with previous studies, which showed that Brat can suppress mRNA independently and is distributed uniformly throughout the embryo.
INTRODUCTION
One of the key processes during Drosophila development is the formation of body axes in the embryo. This is achieved by RNA localization and spatially restricted translation (1), which results in protein gradients along which the axes are established (2). Proper anterior-posterior axis formation is governed by localization of two maternally transcribed genes. The first, bicoid mRNA, localizes to the anterior pole of the oocyte (3), whereas the second, nanos mRNA, localizes to the posterior pole of the oocyte (4). Translation of bicoid and nanos mRNA then results in opposing gradients of Bicoid and Nanos proteins, which in turn control translation of another maternally supplied mRNA—hunchback mRNA (5–8). Hunchback mRNA is distributed uniformly and Nanos suppresses the mRNA translation at the posterior, whereas Bicoid activates it at the anterior. The resulting anterior-posterior gradient of Hunchback then ensures proper development of abdominal and thorax structures (9,10).
Nanos is thus the trans-acting molecule in the posterior suppression of hunchback mRNA translation. The related cis-acting elements are located within the 3′ untranslated region (UTR) of the mRNA and are called Nanos Response Elements (NREs) (11). There are two NREs (NRE1 and NRE2), that each contain two conserved sequences called BoxA (upstream) and BoxB (downstream) (Figure 1A). Nanos contains a zinc finger domain (Nanos-ZnF, Figure 1B), that binds the NREs (12), but requires the trans-acting molecules Pumilio and Brain Tumor (Brat) for suppression of hunchback mRNA translation (13,14). Pumilio and Nanos have been long established as trans-acting elements in the suppression of hunchback mRNA translation and a recent study revealed the underlying mechanism of their concerted recognition of hunchback mRNA (Figure 1B) (15). Pumilio features a Pumilio homology domain (Pum-HD), which is a sequence-specific single-stranded RNA binding domain (RBD), that binds to a site partially overlapping with BoxB of NREs (16–19). In contrast, no specific motif in hunchback mRNA is recognized by Nanos-ZnF, which binds the RNA nevertheless with high affinity, but presumably with low sequence specificity (12,20). However, Nanos-ZnF and Pum-HD together with hunchback mRNA form a high affinity ternary protein-RNA complex (15). Ternary complex formation triggers RNA specificity in Nanos-ZnF for bases adjacent to the binding site of Pum-HD and further enhances the affinity of Pum-HD for its binding site. While localization of Nanos at the posterior ensures the spatial restriction for hunchback mRNA suppression, Pumilio conveys specificity for hunchback mRNA binding. Thus, the concerted recognition of the RNA by Pumilio and Nanos simultaneously allows the spatially controlled suppression of hunchback mRNA during Drosophila development.
Figure 1.

The molecules involved in suppression of hunchback mRNA translation and their interactions. (A) Two cis-acting elements controlling the suppression are located within the 3′ UTR and called Nanos Response Element 1 and 2 (NRE1 and NRE2). The original definitions for BoxA and BoxB are underlined, however, we rather refer to the cognate RNA motifs throughout: BoxA: UUGUUG for Brat-NHL and BoxB: UGUACAUA for Pum-HD. The trans-acting proteins used in this study are: (B) the HD domain of Pumilio (Pum-HD, grey) and the ZnF domain of Nanos (Nanos-ZnF, pink, NED: Nanos effector domain, the resulting complex is shown below (PDB ID: 5KL1)) and (C) the NHL domain of Brain tumour (Brat-NHL, green, B: B-box domain, CC: coiled-coil domain, PDB ID: 4ZLR). (D) A list of RNAs used in this study for ITC experiments and complex formation. The shading of the RNA sequences indicates the binding site of the domain matching the colour based on the previously published structures of Brat-NHL-RNA complex (22) and Pum-HD-Nanos-ZnF-RNA complex (15) shown in (B) and (C). RNA comprising NRE2 indicated in the lower part of the left panel (NRE2 RNA), the protein domains and the published high-resolution structures (B and C) were used in this study for experimental measurements or computational modelling. (E) NRE2 was shown to form a stem loop (22), which is melted by addition of Pum-HD. Here we show by 1H-2D NOESY (right panel) that the Pum-HD domain is even able to disrupt base pairing in a larger NRE1 + NRE2 construct, as imino NOEs disappear with increasing temperature and addition of Pum-HD. (F) Scheme of the experimental ITC setup. RNA is always in the cell and protein in the syringe. Preformed RNA-protein complexes are in the cell. All values shown in ITC plots are the dissociation constants derived from fitting the individual experiment and the resulting fitting error. Brat-NHL-RNA (BoxA and NRE2) interaction studied by ITC. NRE2 binds slightly stronger, likely due to avidity effects. The measurement reveals a 1:1 interaction with a Kd of around 1 μM. Replicates are listed in Table 1 and shown in Supplementary Figure S1 (also for following panels). (G) ITC experiments investigating RNA binding of Pum-HD by testing different RNA lengths. Pum-HD binds with low nanomolar affinity to its cognate sequence BoxB. It does not bind to BoxA or extension of BoxA. However, a weak second binding is upstream of the BoxB motif. (H) ITC of proteins only, showing that they do not interact in absence of RNA. (I) ITC of Brat-NHL titrated to preformed protein-RNA complexes. The affinity of Brat-NHL to NRE2 does not increase in presence of Pum-HD, Nanos-ZnF or both, indicating that Brat-NHL does not bind RNA cooperatively with the other two proteins (Supplementary Figure S2E).
Brain Tumour has been identified as another trans-acting molecule in the suppression of hunchback mRNA translation more recently (14). The NCL-1, HT2A, and LIN-41 domain of Brat (Brat-NHL) was shown to interact specifically with a motif in BoxA of NREs (Figure 1C) and Brat mutants induce similar developmental defects in flies as Pumilio or Nanos mutants (21–23). Like Pumilio, Brat is uniformly distributed throughout the embryo (14), but how it contributes to the suppression of hunchback mRNA by Pumilio and Nanos remains unclear (24), despite a wealth of available experimental data on the relationship of Brat-NHL, Pum-HD and Nanos-ZnF. First, published data suggest that Brat, Pumilio and Nanos form a quaternary complex with hunchback mRNA in order to suppress its translation: in yeast four-hybrid and pull-down assays Brat-NHL, Pum-HD and Nanos-ZnF were all necessary to form a stable protein-RNA complex (14). In line with that, Pum-HD was reported to increase the affinity of Brat-NHL for hunchback mRNA (22). This would explain why Brat mutations induce phenotypes similar to those of Pumilio and Nanos mutants. Moreover, a vast majority of these mutants indirectly implied quaternary complex formation, as they abrogated complex formation in yeast four-hybrid assays. However, it became clear later that the point mutations substituted residues necessary for RNA binding of Brat-NHL (22), so the observed effect is likely due to abrogated RNA binding and not necessarily due to abrogation of quaternary complex formation. Furthermore, gene reporter assays in Dmel2 cells show that removing Brat-NHL has little effect on suppression of a reporter gene in the presence of Pumilio and Nanos, and that, conversely, Brat can suppress the mRNA translation independently in their absence (22,23). In summary, it remains unclear whether Brat, Pumilio and Nanos suppress hunchback mRNA translation cooperatively in a single quaternary protein-RNA complex or whether Brat suppresses hunchback mRNA independently of Pumilio and Nanos. To determine if the suppression activities are cooperative or independent, a combination of both functional and biophysical insights into the process is required. Since there is already a wealth of functional data, we obtained the biophysical results needed to fill the existing gap in our understanding of hunchback mRNA translation suppression.
We carried out an extensive structural, biophysical, and computational investigation of Pum-HD, Brat-NHL, Nanos-ZnF and their complex with hunchback mRNA NRE2 (hb complex). First, we analysed the influence of Pum-HD on the RNA binding of Brat-NHL using isothermal titration calorimetry (ITC) and showed that they do not bind RNA cooperatively. Moreover, Brat-NHL and Nanos-ZnF do not show any signs of interaction in vitro in the absence or presence of RNA and cooperativity in RNA binding could not be observed between Brat-NHL and the Pum-HD/Nanos-ZnF complex. Nevertheless, Brat-NHL, Pum-HD and Nanos-ZnF assemble all on a single NRE2 in a stable stoichiometric quaternary protein-RNA complex. However, our data reveals that the complex, in context of a single NRE2 and isolated RNA binding domains, is largely flexible and that Brat-NHL in the complex moves independently of Pum-HD and Nanos-ZnF. To investigate the extent of flexibility of the complex we used unrestrained molecular dynamics (MD) simulation validated against cross-linking/mass spectrometry, small-angle X-ray and neutron scattering data. Collectively, our results describe a complex, in which the unoccupied nucleotides of the NRE2 RNA function as a flexible linker between the Brat-NHL and Pum-HD-Nanos-ZnF moieties of the complex, reminiscent of beads on a string. Moreover, this study highlights the importance of combining a multitude of structure analysis methods with molecular dynamics simulations to obtain reliable atomistic ensembles of dynamic protein-RNA complexes.
MATERIALS AND METHODS
Protein expression and purification
The Pum-HD and Brat-NHL constructs used in this study comprise residues 1093–1426 and 756–1037 of the respective proteins (Figure 1B and C), which are connected by a short linker containing a Usp2cc cleavage site to a His6-Ubiquitin tag in pHUE plasmid (25,26). The expression was done as previously described (23), but Escherichia coli BL-21(DE3) Rosetta cells were used. The cells were then lysed first by incubation for 20 min on ice in lysis buffer (50 mM Tris, 1 M NaCl, 5% glycerol, 10 mM imidazole, pH 8.0) with 1 mg/ml lysozyme, 1 μg/ml DNase I, 2 μg/ml Rnase A and protease inhibitor tablets (Roche), and then by sonication at 4°C. The lysate was then cleared by centrifugation (18 000 g, 4°C, 1 h) and purified using a HisTrap HP 5 ml column (GE Healthcare) and a 10–100 mM imidazole linear gradient followed by elution with 250 mM imidazole, which were created by mixing the lysis buffer with 50 mM Tris, 1 M NaCl, 5% glycerol, 1 M imidazole, pH 8.0. Pooled fractions of the proteins were then cleaved by His6-tagged Usp2cc at 4°C overnight during dialysis in dialysis buffer (50 mM Tris, 150 mM NaCl, 1 mM DTT, pH 7.4). Pum-HD was then separated from the tag by reverse affinity chromatography and in the final step Pum-HD was further purified by size-exclusion chromatography (SEC) using HiLoad 16/600 Superdex 75 column (GE Healthcare) in 50 mM Tris, 150 mM NaCl, 1 mM DTT, pH 7.4 buffer. For Brat-NHL serial reverse affinity and heparin chromatography (HiTrap HP Heparin 5ml column - GE Healthcare) followed the affinity chromatography. The sample was loaded on the columns connected in series after equilibrating the columns in dialysis buffer. Brat-NHL was eluted using a 150 mM to 1.5 M NaCl gradient. The Nanos-ZnF plasmid used in this study contains residues 301–392 of Nanos (Figure 1B) connected via a linker with SenP2 cleavage site to a His6-SUMO tag in pETM11-SUMO3GFP vector (EMBL Protein Expression and Purification Core Facility). Nanos-ZnF was expressed by transforming the plasmid to E. coli BL-21(DE3) Rosetta cells, growing the cells at 37°C while shaking until OD600 of 0.6–1.0, followed by addition of IPTG to 0.3 mM final concentration and further overnight incubation at 16°C while shaking. Nanos-ZnF was then purified as described above for Brat-NHL, except that in the first affinity chromatography step the basic lysis buffer was 50 mM Tris, 500 mM NaCl, 50 μM ZnSO4, 10 mM imidazole, pH 8.0 and the tag was cleaved off using His6-tagged SenP2.
Isotopic labelling
To obtain diversely isotopically labelled proteins, the cells were grown in M9 minimal media and the expression and purification followed the standard protocols outlined above unless stated otherwise. In order to obtain 15N-labelled Nanos-ZnF and Brat-NHL, the expression was done using 15NH4Cl as the sole nitrogen source. In order to obtain various degrees of 2H-labelling, cells were grown as follows: a 5 ml overnight culture of H2O M9 minimal media with adequate isotopes was spun down, resuspended in 100 ml of D2O M9 minimal media with adequate isotopes, grown to an OD600 of ∼0.6 at 37°C while shaking, then diluted to 500 ml final volume with D2O M9 minimal medium. The perdeuterated Brat-NHL and Pum-HD for SANS measurements were obtained by expressing the proteins in D2O M9 minimal media with 2H-glucose as the sole carbon source. For NMR relaxation and 1H,13C HMQC experiments, Brat-NHL was expressed either uniformly 2H-, 13C- and 15N-labelled (for free form and RNA-bound measurements) or uniformly 2H- and 15N-labelled (in the hb complex) and in both cases with Cδ1 of isoleucines and one of the methyl groups of valines and leucines 1H,13C labelled, and the other methyl groups 2H-, 12C-labelled. The uniform labelling was achieved by expression in D2O M9 minimal media with 15NH4Cl as the sole nitrogen source, and 2H- or 2H,13C-glucose as the sole carbon souce, and adding 2-keto-3,3-d2-1,2,3,4-13C-butyrate and 2-keto-3-methyl-d3-3-d1-1,2,3,4-13C-butyrate one hour prior induction as described previously (27).
Hb complex formation
To form the hb complex, the purified Pum-HD, Brat-NHL and Nanos-ZnF proteins were incubated with a 23 nucleotide-long NRE2 RNA, encompassing the NRE2 in the 3′ UTR of hunchback mRNA of the sequence 5′ UUGUUGUCGAAAAUUGUACAUAA 3′ (Microsynth, Figure 1A). The complex was formed by incubating NRE2 RNA with Pum-HD, Nanos-ZnF and Brat-NHL in a 1:1:1:2 molar ratio, respectively, to account for the lower affinity of Brat-NHL towards the RNA. Pum-HD, Nanos-ZnF and NRE2 RNA were diluted to 10 μM, whereas Brat-NHL was diluted to 20 μM. Pum-HD and Nanos-ZnF were then mixed with the RNA, incubated on ice for 15 minutes prior to addition of Brat-NHL and incubation overnight at 4°C. The mixture was then concentrated by reducing the volume from 48 ml to 1 ml using a 3 kDa cutoff concentrator and the hb complex was purified by SEC using a HiLoad 16/600 Superdex 200 pg column (GE Healthcare). The identity of the hb complex peak was confirmed by SDS-PAGE, UV absorption measurement at 260 nm and size-exclusion chromatography-coupled multiangle laser light scattering (SEC-MALLS).
Size exclusion chromatography-multi-angle laser light scattering (SEC-MALLS)
100 μl of hb complex (3.0 mg/ml) were injected onto a Superdex 200 10/300 GL gel-filtration column (Cytiva) in 50 mM Tris, 150 mM NaCl, 1 mM DTT, pH 7.4 buffer at room temperature. The column was coupled to a MALS system (MiniDAWN and Optilab, Wyatt Technology). Data were analysed using the Astra 7 software (Wyatt Technology). Measurements were performed in duplicates.
Isothermal titration calorimetry
All isothermal titration calorimetry (ITC) measurements were done on a MicroCal PEAQ-ITC instrument (Malvern) at 20°C in 50 mM Tris, 150 mM NaCl, 0.5 mM TCEP, pH 7.4 buffer. Diluted RNAs were snap-cooled before the measurements by incubating at 65°C, shaking for 5 minutes and quickly cooling on ice for 20 minutes. The proteins were then concentrated and both the protein and RNA solution were degassed. The diluted RNA solution was then added to the cell and the concentrated protein solution was titrated to the RNA from the syringe. Each titration comprised either 13–24 injections always with an 0.4 μl initial injection followed by 1.5 or 3 μl injections. The number and volume of injections was selected to optimize the signal with respect to enthalpy change. The sample was stirred at 750 rpm, instrument feedback was set to high, the reference power was set to 10 μcal/s and the delays were set to 60 second initial delay followed by 150 second delays. The specifics about injections and concentrations in individual titrations are listed in Table 1.
Table 1.
Summary of ITC measurements. All measurements were performed at 20°C. BoxB refers to the cognate RNA motif of Pum-HD (UGU ACA UA) and BoxA refers to the cognate RNA motif of Brat-NHL (UUG UUG) not to the original definition outlined in Figure 1A
| Exp. name | Replicates | Conc. syringe (μM) | Conc. cell (μM) | N (sites) | N (error) | Kd (μM) | Kd (μM, error) | ΔHa (kcal/mol) | –T ΔSa (kcal/mol) | Injection no. | Injection vol. (μl) | Shown in figure |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pum-HD + BoxB | 3 | 50 (Pum-HD) | 1) 2.5 (BoxB) 2) & 3) 5 (BoxB) | 1.1, 1.1, 1.1 | 0.01, 0.013, 0.005 | 0.002, 0.0066, 0.0081 | 0.002, 0.004, 0.0017 | 46.0 ± 3.0 | 34.0 | 24 | 1.5 | 1, S1 |
| Pum-HD + BoxA | 2 | 300 (Pum-HD) | 30 (BoxA) | No binding (or too weak to be assessed by ITC) | 18 | 2 | 1, S1 | |||||
| Pum-HD + BoxAextb | 2 | 300 (Pum-HD) | 30 (BoxA) | No binding (or too weak to be assessed by ITC) | 18 | 2 | 1, S1 | |||||
| Pum-HD + NRE2 minus BoxBc | 2 | 300 (Pum-HD) | 15 (NRE2 minus BoxB) | 1.1, 1.2 | 0.008, 0.028 | 1.2, 1.0 | 0.08, 0.02 | -134.5 ± 13.5 | 100.5 | 18/24 | 2 | 1, S1 |
| Pum-HD + NRE2 minus BoxAd | 2 | 50 (Pum-HD) | 5 (NRE2 minus BoxA) | N1 = 1.1, 1.0 | N1 = 0.005, 0.01 | Kd1 = 0.01, 0.011 | Kd1 = 0.0003, 0.0006 | Site 1: –21.5 ± 0.7 | Site 1: 10.7 | 23 | 1.5 | |
| N2 = 0.9, 1.1 | N2 = 0.006, 0.01 | Kd2 = 0.32, 0.55 | Kd2 = 0.03, 0.02 | Site 2: −78.3 ± 8.1 | Site 2: 69.1 | |||||||
| Pum-HD + NRE2 | 2 | 300 (Pum-HD) | 20 (NRE2) | N1 = 0.9, 1.0 | N1 = 0.01, 0.002 | Kd1 = 0.002, 0.015 | Kd1 = 0.0008 | Site 1: –13.5 ± 0.3 | Site 1: 6.8 | 15/24 | 2 | 1, S1 |
| N2 = 1.4, 1.4 | N2 = 0.01, 0.01 | Kd2 = 2.0,1.3 | Kd2 = 0.1, 0.003 | Site 2: –16.3 ± 0.5 | Site 2: 10.9 | 2 | ||||||
| Brat-NHL + BoxA | 3 | 300 (Brat-NHL) | 15–30 (BoxA) | 0.8, 0.9, 1.1 | 0.03, 0.02, 0.02 | 1.1, 1.0, 0.8 | 0.3, 0.3, 0.2 | -15.5 ± 0.8 | 7.4 | 18 | 2 | 1, S1 |
| Brat-NHL + NRE2 | 3 | 300–375 (Brat-NHL) | 25–30 (NRE2) | 1.1, 1.1, 1.0 | 0.02, 0.02, 0.03 | 0.37, 0.32, 1.2 | 0.1, 0.1, 0.2 | -7.7 ± 0.45 | 0.4 | 18 | 2 | 1, S1 |
| NanosZnF + NRE2 | 7 | 250 (NanosZnF) | 20–25 (NRE2) | No binding (or too weak to be assessed by ITC) | 18 | 2 | 1, S1 | |||||
| Brat-NHL + NRE2/Pum-HD | 4 | 250–300 (Brat-NHL) | 20–30 (NRE2/Pum-HD) | 1.0, 1.0, 1.0, 1.1 | 0.02–0.03 | 3.3, 2.9, 0.9, 1.2 | 0.1–1.4 | –9.1 ± 0.34 | 1.0 | 18 | 2 | 1, S1 |
| Brat-NHL + NRE2/ NanosZnF | 5 | 250–300 (Brat-NHL) | 7–30 (NRE2/ NanosZnF) | 1.0, 1.0, 1.0, 1.1 | 0.02–0.04 | 5.4, 2.3, 0.9, 0.4, 0.2 | 0.1–0.4 | –10.1 ± 0.6 | 1.3 | 18 | 2 | 1, S1 |
| Brat-NHL + NRE2/ NanosZnF/ Pum-HD | 6 | 150–375 (Brat-NHL) | 10–25 (NRE2/ NanosZnF/ Pum-HD) | 1.0, 1.1, 0.9, 0.8, 0.7, 0.9 | 0.02–0.07 | 1.4, 3.5, 5.9, 3.8, 1.6, 2.8 | 0.2–0.5 | -48.5 ± 10.3 | 24.5 | 14 | 3 | 1, S1 |
| Brat-NHL + Pum-HD | 2 | 467 (Brat-NHL) | 30 (Pum-HD) | No binding (or too weak to be assessed by ITC) | 13/18 | 3 | 1, S1 | |||||
| Brat-NHL + NanosZnF | 3 | 375 (Brat-NHL) | 25 (NanosZnF) | No binding (or too weak to be assessed by ITC) | 13 | 3 | 1, S1 | |||||
aΔH (enthalpy change) and –TΔS (entropy change) is given as average over all replicates.
bBoxAext (UUG UUG UC) has two more bases to the 3′ compared to BoxA (UUG UUG).
cNRE2 minus BoxB is the same sequence as the 23-mer NRE2 except that the cognate Pum-HD motif has been removed, resulting in the following sequence: UUG UUG UCG AAA AU.
dNRE2 minus BoxA is the same sequence as the 23-mer NRE2 except that the cognate Pum-HD motif has been removed, resulting in the following sequence: UCG AAA AUU GUA CAU AA (All used sequences are shown in Figure 1D).
Small-angle scattering
Small-angle X-ray scattering (SAXS) data were acquired at the P12 beamline at DESY, Hamburg, Germany using SEC coupled online to the beamline and a MALLS instrument in parallel. The measurement was done in a 50 mM Tris, 150 mM NaCl, 1 mM DTT, 3% glycerol, pH 7.4 buffer at 25°C. An Agilent BioInert HPLC/FPLC system equipped with Superdex 200 Increase 10/300 GL column (GE Healthcare) was used for the SEC. The hb complex at 7.2 mg/ml concentration was injected and run at 0.6 ml/min flowrate. SAXS measurements were then done over a 1 second exposure period per frame using X-rays with 1.23 Å wavelength and Pilatus 6M at 3 m detector distance. The scattering curve was then obtained by averaging frames of the SEC peak with a consistent radius of gyration (Rg) and subtracting averaged buffer signal from frames in a baseline region of the SEC run before and after the hb complex peak.
For small-angle neutron scattering (SANS) measurements, samples of the hb complex with varying subunit-selective perdeuteration of the protein components were measured at concentrations ranging from 3.7 to 5 mg/ml in 200 μl of 50 mM Tris, 150 mM NaCl, 1 mM DTT, pH 7.4 buffer in Hellma® 100QS quartz cuvette at 20°C. The samples were generally measured at three different percentages of D2O content in the buffer—at 0% D2O, at D2O percentage close to 1H protein match point (∼42%) and at D2O percentage close to the 1H RNA match point (∼63%). Match points of each sample were calculated using SASSIE-web (https://sassie- web.chem.utk.edu/sassie2/).
The following samples were measured as standards: a boron/cadmium sample for 5 min, an empty cell for 10 minutes, and 0%, 42% and 63% D2O buffers each for 20 minutes. The samples of the hb complex were then measured for 60 minutes each. Transmission of each sample was measured to precisely determine the D2O content of the buffer. The measurements were done at the D22 instrument at ILL, Grenoble, France using a neutron wavelength of 6 Å, laterally shifted detector and a detector and collimator distance of 4 m, respectively. The samples are listed in detail in Table 2. The SANS data have been aqcuired at ILL as part of the beam time allocation group BAG-8-36.
Table 2.
List of samples for small-angle X-ray and neutron scattering measurements
| Labelling | ||||||
|---|---|---|---|---|---|---|
| Sample | Brat-NHL | Pum-HD | Nanos-ZnF | NRE2 RNA | Concentration | Buffer D2O % |
| SAXS | 1H | 1H | 1H | 1H | n/a | 0% |
| 1B1P1N 0% D2O | 1H | 1H | 1H | 3.7 mg/ml | 0% | |
| 1B1P1N 66% D2O | 1H | 1H | 1H | 3.7 mg/ml | 66% | |
| 1B2P2N 0% D2O | 1H | 2H | 2H | 5.0 mg/ml | 0% | |
| 1B2P2N 41% D2O | 1H | 2H | 2H | 4.3 mg/ml | 41% | |
| 2B2P1N 0% D2O | 2H | 2H | 1H | 4.0 mg/ml | 0% | |
| 2B2P1N 33% D2O | 2H | 2H | 1H | 4.0 mg/ml | 33% | |
| 2B2P1N 62% D2O | 2H | 2H | 1H | 4.0 mg/ml | 62% | |
All experimental scattering curves were buffer subtracted and initial points with beamstop shadow were removed. The data was processed and analysed using various software from the ATSAS package (28), specifically PRIMUS (29), CRYSON (30), CRYSOL (31) or EOM (32) as well as using ScÅtter (33).
Cross-linking/mass-spectrometry
Cross-linking of the hb complex was done by adapting a previously published protocol (34). The complex was cross-linked via lysine amines by a mixture of 1H- and 2H- labelled disuccinimidyl suberate (DSS) in 20 mM HEPES, 150 mM NaCl, 1 mM DTT, pH 7.4 buffer. First, 1.5 mg/ml hb complex was cross-linked by 0.2 and 1 mM DSS. The cross-linking was done once using 0.2 mM DSS and in three biological replicates using 1 mM DSS. The reaction was incubated while shaking at 37°C for 30 min and then quenched by addition of NH4HCO3 to 50 mM final concentration. The cross-linked complex was then digested by incubating with LysC (Wako) at 1:100 protease:protein ratio at 37°C for 3.5 h and with Trypsin at 1:50 protease:protein ratio at 37°C overnight. The digested peptides were then desalted using OASIS® HLB μElution Plate and enriched by SEC in 30% (v/v) acetonitrile (ACN) and 0.1% (v/v) trifluoro acetic acid (TFA) using a Superdex Peptide PC 3.2/30 column (GE Healthcare). After evaporating to dryness and dissolving in 4% (v/v) ACN in 1% (v/v) formic acid (FA) the samples were analysed by liquid chromatography–coupled tandem mass spectrometry (LC–MS/MS) using a nanoAcquity UPLC system (Waters) connected online to an LTQ-Orbitrap Velos Pro instrument (Thermo). LC was done using a BEH300 C18 nanoAcquity UPLC column (Waters) with a 3% to 85% (v/v) gradient of ACN in 0.1% (v/v) FA. The MS/MS was done using a top-20 strategy - by first acquiring survey MS scans in m/z range of 375–1600 m/z in the Orbitrap (resolution of 30 000) and fragmenting the top 20 of the most abundant ions per full scan by collision-induced dissociation (CID, with 40% normalized collision energy) and analysing them in the LTQ. Charge states 1, 2 and unknown were rejected to focus the acquisition on larger cross-linked peptides and dynamic exclusion was set to 60 s. Ion target values for full scans and for MS/MS scans were 1,000,000 (for 500 ms max fill time) and 10 000 (for 50 ms max fill time), respectively. The LC–MS/MS was done in two technical duplicates. Cross-linking of 1.5 mg/ml complex with 0.2 and 1 mM DSS produced a high number of multimeric intradomain cross-links, so eventually the cross-linking was repeated in two biological replicates using 0.95 mg/ml hb complex and 0.5 mM DSS. The cross-linking, digestion and peptide separation was done as described above, but the mass spectrometry analysis was done using UltiMate™ 3000 RSLCnano system (Thermo Fisher Scientific) directly coupled to an Orbitrap Fusion Lumos (Thermo Fisher Scientific). Dried peptides were dissolved in 4% (v/v) ACN in 1% (v/v) FA and then LC was done using μ-Precolumn C18 PepMap 100 trapping cartridge and nanoEase MZ HSS T3 column in 0.05% (v/v) TFA with a 2% to 85% (v/v) gradient of ACN in 0.1% (v/v) FA. Full scans were acquired with an m/z range of 375–1600 m/z at 120 000 resolution. For peptide fragment spectra, the quadrupole window was set to 0.8 m/z and the peptides were fragmented by CID (35% normalized collision energy). Charge states 3–7 were selected and dynamic exclusion was set to 60 s. Ion target values for full scans and for MS/MS scans were 200 000 (for 250 ms max fill time) and 20 000 (for 100 ms max fill time), respectively. The spectra were assigned using xQuest and the posterior probabilities were calculated using xProphet (34). The results were then filtered using the following parameters: FDR of 0.05, min delta score of 0.95, –4 to 7 ppm tolerance window and id-score higher than 25. The two biological replicates of 0.95 mg/ml hb complex cross-linked at 0.5 mM DSS yielded 20 unique interdomain cross-links (Supplementary Table S2).
NMR spectroscopy
All of the NMR experiments were measured on a Bruker Avance III NMR spectrometer operating at a magnetic field strength corresponding to an 800 MHz proton Larmor frequency, equipped with a Bruker TXI cryo-probe head. The measurements were done at 25°C in 50 mM Tris, 150 mM NaCl, 1 mM DTT, pH 7.4 buffer. All experiments except Brat-NHL backbone assignment experiments were recorded using apodization weighted sampling (35). Backbone resonance assignment of 2H-, 15N-, 13C-labelled Brat-NHL was achieved to a completion of 77% (excluding prolines) using TROSY-based 1H,15N-HSQC, HNCA, CBCA(CO)NH and HNCACB triple resonance experiments (36–38). NMR interaction studies were performed as follows: 260 μM 15N Nanos-ZnF was titrated by unlabelled Brat-NHL to a molar ratio of 1:1.5, with an intermediate step at 1:1 molar ratio. 170 μM 15N Brat-NHL was titrated by unlabelled Nanos-ZnF to a 1:1 molar ratio with no intermediate steps. The experiments were monitored by recording a 1H,15N-HSQC at each step. The methyl-HMQC spectra comparison of Brat-NHL was obtained by recording a 1H,13C-HMQC. For free Brat-NHL, methyl-HMQC spectra and 15N T1 and T2 relaxation experiments were measured on a 915 μM 1H/2H-methyl ILV 2H,13C,15N-labelled Brat-NHL sample (ILV: isoleucine, leucine, valine). The following delays were used for the T1: 20, 50, 100, 150, 400, 500, 650, 800, 1000 and 1200 ms, with the 20 and 150 ms delays measured in duplicates. 12.5, 25, 37.5, 50, 75, 125 and 150 ms delays were used for T2 experiment and the 25 ms delay was measured twice. For Brat-NHL bound to RNA, the methyl-HMQC spectra were acquired on a 325 μM 1:1 mixure of 1H/2H-methyl ILV 2H,13C,15N-labelled Brat-NHL with NRE2 RNA. For Brat-NHL in the hb complex, the methyl-HMQC, 15N T1 and T2 relaxation measurements were measured on a 65 μM hb complex reconstituted using 2H-labelled Pum-HD, unlabelled Nanos-ZnF, 1H/2H,13C/12C-methyl ILV 2H,15N labelled Brat-NHL and NRE2 RNA. For the relaxation experiments the following delays were used: 20, 150, 400, 800 and 1200 ms, with the 150 ms delay measured in duplicates for the T1 experiment and 12.5, 25, 50, 75 and 125 ms, with the 25 ms delay measured in duplicates for the T2 experiment.
All spectra were processed using NMRPipe (39) and analysed using CARA (http://cara.nmr.ch), CcpNmr Analysis (40), or Sparky (41). Peak fitting, error estimation and exponential fitting for the relaxation experiments was done using PINT (42,43). The experimental rotational correlation times were calculated according to Equation (1),
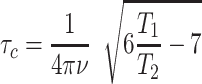 |
(1) |
where ν is the Larmor frequency in Hz, T1 is the 15N spin-lattice relaxation time and T2 is the 15N spin-spin relaxation time (44). The theoretical rotational correlation times were calculated from atomic structures using the ELM module in ROTDIF 3 (45,46).
Rigid body modelling
The computational modelling of the hb complex was done with Crystallography & NMR System (CNS) software (47,48) adapting a previously published protocol (49,50). The starting model of the complex was generated from the published structures of Brat NHL-RNA complex (PDB: 4ZLR) (22) and Pum HD-Nanos ZnF-RNA complex (PDB: 5KL1) (15) by threading the individual complexes on a single NRE2 RNA containing both binding sites and the connecting nucleotides. A pool of models of the complex was then generated by modified protocols within ARIA (51). First, the conformation of the NRE2 RNA nucleotides, which are not bound in either of the published RNA complexes, was randomized. The conformation of the proteins and the RNA elucidated in the published structures were then kept fixed throughout the modelling. The randomized structures were minimized unrestrained or with XL/MS derived distances employing standard simulated annealing protocols within ARIA. The pool of unrestrained models was then generated by 40,000 MD steps of 6 fs each, whereas the pool of models with XL/MS as distance restraints was generated by 120,000 MD steps of 2 fs each. Unrestrained modelling generated 5,055 models, modelling with XL/MS data generated 4572 models. The XL/MS data were put in as 31 Å lysine-lysine Cα–Cα upper distance limits with log harmonic potential (52). Each model was then fitted against the experimental data using CRYSON (30) and CRYSOL (31), then for each modelling an ensemble of best fitting models was selected according to a criterion of χ2 values of the fits of the model to each experimental curve. In case of unrestrained modelling the criterion was 0.23 quantile of the lowest χ2 values, whereas for modelling with XL/MS data the criterion used was 0.3 quantile of the lowest χ2 values.
All-atom molecular dynamics simulations
Eight different conformations of the protein-RNA complex were taken from the rigid-body modelling and henceforth used as starting conformations for all-atom explicit-solvent simulations. MD simulations were carried out with the Gromacs software (53), version 2019.6. Interactions of the protein and the RNA were described with the Amber99sbws force-field, and the TIP4P-2005s water model was used (54). Each of the eight initial conformations was placed in a dodecahedral simulation box, where the distance between the protein to the box edges was at least 2.0 nm. The boxes were filled with 184,665 water molecules, and 17 sodium ions were added to neutralize the systems. In total, the simulation systems contained 751 094 atoms. The energy of each simulation system was minimized within 400 steps with the steepest descent algorithm. Subsequently, the systems were equilibrated for 100 ps with harmonic position restraints applied to the backbone atoms of the proteins (force constant 1000 kJ mol−1nm−2). Finally, each of the eight replicas was simulated for 110 ns without any restraints. The temperature was kept at 298 K using velocity rescaling (τ = 0.1 ps) (55). The pressure was controlled at 1 bar with the Berendsen (τ = 1 ps) (56) and with the Parrinello-Rahman barostat (τ = 5 ps) (57) during equilibration and production simulations, respectively. The geometry of water molecules was constrained with the SETTLE algorithm (58), and LINCS was used to constrain all other bond lengths (59). Hydrogen atoms were modeled as virtual sites, allowing a time step of 4 fs. Lennard-Jones potentials with a cut-off at 1nm were used to describe dispersive interactions and short-range repulsion. Electrostatic interactions were computed with the smooth particle-mesh Ewald method (60,61). Visual inspection of the simulations revealed that the RNA–protein contacts were stable throughout the simulations.
Explicit-solvent SAXS/SANS calculations
The SAXS and SANS calculations were performed with an in-house modification of Gromacs 2018.8, as also implemented by our webserver WAXSiS (62–64). The implementation and tutorials are available at https://biophys.uni-saarland.de/software.html. Simulation frames from the time interval between 30 and 110 ns were used for SAXS/SANS calculations. A spatial envelope was built around all solute frames from all eight replicas of the protein-RNA complex. Solvent atoms inside the envelope contributed to the calculated SAXS/SANS curves. The distance between the protein-RNA complex and the envelope surface was at least 1.0 nm, such that all water atoms of the hydration shell were included. The buffer subtraction was carried out using 783 simulations frames of a pure-water simulation box, which was simulated for 110 ns and which was large enough to enclose the envelope. The orientational average was carried out using 1700 q-vectors for each absolute value of q, and the solvent electron density was corrected to the experimental value of 334 e/nm3, as described previously (62). During SANS calculations, the perdeuteration conditions and D2O concentrations were taken according to the experimental conditions. Here, we assigned the mean neutron scattering length to all potentially deuterated hydrogen atoms, as described previously (65). This protocol leads to a constant offset in the SANS curves that is absorbed into a fitting parameter (see below). To compare the experimental with the calculated SAXS/SANS curves, we fitted the experimental curve via Iexp,fit(q) = f⋅(Isam(q) – α⋅Ibuf(q) + c) by minimizing the chi-square with respect to the calculated curve. Here, the factor f accounts for the overall scale, and the offset c takes the uncertainties from the buffer subtraction and the incoherent scattering in SANS experiments into account. No fitting parameters owing to the hydration layer or excluded solvent were used, implying that also the radius of gyration was not adjusted by the fitting parameters.
RESULTS
Brat-NHL and Pum-HD/Nanos-ZnF bind NRE2 independently
We employed ITC to test whether Brat-NHL binds RNA cooperatively with Pum-HD, Nanos-ZnF or both. We focused on the relevant region of NRE2, which includes the earlier identified RNA binding motifs BoxA and BoxB (Figure 1A), which are supposedly recognized by Brat-NHL (Figure 1B) and Pum-HD/Nanos-ZnF (Figure 1C), respectively. However, later studies showed that the cognate motifs for both domains within NRE2 are shifted, being UUGUUG for Brat-NHL (22) and UGUACAUA for Pum-HD (66) to which we refer to in the following also as BoxA and BoxB (Figure 1D). The longest RNA used in this study is a 23-mer which we refer to as NRE2. This RNA consists of BoxA and BoxB with the additional eight bases in between, which harbour the AAU motif bound by Nanos-ZnF in the presence of Pum-HD (Figure 1D). The remaining five bases are presumably unbound. The 23-mer NRE2 has been shown previously to form a small stem-loop, covering parts of BoxA and BoxB by base pairing and unfolds upon addition of Pum-HD (22). Using 1H-2D NOESY experiments we can confirm that an extended RNA comprising NRE1 and NRE2 forms a stem-loop and that Pum-HD can likewise unwind this structure upon binding (Figure 1E). The imino-NOE cross peaks clearly indicate base pairing and are weakened upon temperature increase and vanish upon addition of Pum-HD, demonstrating that Pum-HD unfolds RNA secondary structure. We then performed a series of ITC experiments, where the RNA is provided in the cell, whether alone or as preformed protein-RNA complexes, and the titrant in the syringe is always one of the three proteins (Figure 1F). First, we confirmed that Brat-NHL binds to BoxA with low micromolar affinity (around 1 μM, Figure 1F and Supplementary Figure S1A). The affinity increases slightly by around two-fold to the 23-mer NRE2, likely due to avidity effects as the three bases 3′ to the BoxA motif (UCG) would permit sliding to an almost optimal BoxA motif downstream (UUGUCG as in NRE1, Figure 1F and Supplementary Figure S1B). This is in agreement with a previous study, where EMSA showed a Kd of around 1 μM between Brat-NHL and the 23-mer NRE2 RNA (22). In the same study, an RNA optimized for crystallization bound with a Kd of 40 nM. This increased affinity results from avidity effects, as a poly(U) sequence following the UUGUUG motif allows the NHL domain to slide to adjacent bases up to equimolar amounts of protein and RNA. Upon addition of excess of Brat-NHL, two NHL domains are bound to one RNA molecule. Next, we titrated Pum-HD to NRE2 and observed two binding events, where the tighter binding is difficult to resolve due to the high affinity (around 2 nM, Figure 1G). The second weaker binding event has a dissociation constant of around 2 μM. To test, whether NRE2 indeed consists of two binding sites for Pum-HD, we divided the RNA into several fragments. First, we could confirm, that the stronger binding site is BoxB, as has been demonstrated earlier (66) (around 8 nM, Figure 1G and Supplementary Figure S1C). Interestingly, Pum-HD does not bind at all to BoxA or to an extended BoxA (BoxAext, Figure 1G and Supplementary Figure S1D), but it does bind to an NRE2 sequence shortened by the cognate Pum-HD motif BoxB (NRE2-BoxB) with low micromolar affinity (around 1 μM, Figure 1G and Supplementary Figure S1D). This corresponds to the second weaker binding event observed with NRE2. Consequently, titrating to an RNA, which corresponds to NRE2 without BoxA, we again observed two binding events, where the first is in the low nanomolar range (around 10 nM), corresponding to Pum-HD binding to the BoxB motif and a second weaker event in the low micromolar range of Pum-HD binding to bases located between BoxA and BoxB (UCGAAAAU, Figure 1G and Supplementary Figure S1E). This segment also contains the Nanos-ZnF binding motif (AAU, Figure 1D). However, Nanos-ZnF does not bind NRE2 in absence of Pum-HD or Brat-NHL (Figure 1H and Supplementary Figure S1F), confirming the dependency of Nanos-ZnF on Pum-HD for RNA binding (15).
Prior to assessing cooperativity of Brat-NHL with Pum-HD and/or Nanos-ZnF for RNA binding, we also tested whether direct protein-protein interactions exist in absence of RNA. Neither titrating Brat-NHL to Pum-HD nor to Nanos-ZnF lead to any heat changes (Figure 1H and Supplementary Figure S1F).
Finally, we preformed NRE2-Pum-HD, NRE2-Nanos-ZnF and NRE2-NanosZnF/Pum-HD complexes in the cell to test whether the affinity of Brat-NHL towards protein-bound RNA increases in comparison to free NRE2 (Figure 1I and Supplementary Figure S1G–I). No change in affinity could be observed and it remains at low micromolar affinity across all experiments. These data clearly demonstrate that Brat-NHL binds RNA independently of Pum-HD and Nanos-ZnF.
Of note, titrating Brat-NHL to NRE2 or the preformed NRE2-Nanos-ZnF complex resulted in positive enthalpy changes at later titration points as opposed to all other titrations, including Brat-NHL titrated to the preformed NRE2-Pum-HD complex. We speculate that this is due to opening of the stem loop of NRE2 only at higher concentrations of Brat-NHL, as the majority of the Brat-NHL binding motif is accessible also in the base paired state. Nanos-ZnF on the contrary does not bind to this RNA in absence of other proteins, whereas Pum-HD opens the base pairs upon binding.
Although we could not observe any cooperativity between Brat-NHL and Pum-HD and/or Nanos-ZnF in RNA binding by ITC, we tested for a possible weak interaction between Nanos-ZnF and Brat-NHL by NMR spectroscopy. Typically, NMR titrations are applicable to a very broad range of interaction affinities from tight to weak or transient interactions, so we selected this method to avoid missing a potential interaction by probing a limited Kd range. We expressed both Brat-NHL and Nanos-ZnF 15N-labelled as well as unlabelled and then titrated either 15N-labelled protein by their unlabelled counterpart, while monitoring the titration by 1H,15N HSQC spectra (Figure 2). Neither of the two NMR titrations revealed any signs of interaction. We did not observe any changes of peak positions, that would be indicative of changes of the local chemical environment accompanying an interaction, or changes in peak intensity, reflecting changes of molecular tumbling upon complex formation. It should be noted that the Nanos-ZnF construct is one third of the molecular weight of the Brat-NHL construct (32 kDa), so a 4-fold increase of the apparent molecular weight upon complex formation should change molecular tumbling of Nanos-ZnF to a degree that obvious changes of peak intensities in NMR should be observed.
Figure 2.

Brat-NHL and Nanos-ZnF do not interact in the absence of RNA. NMR titrations were performed to test if Brat-NHL and Nanos-ZnF interact in vitro in the absence of RNA. (A) Overlay of the 1H,15N HSQC spectra of 15N labelled Nanos-ZnF in the free form and with unlabelled Brat-NHL added to 1:1.5 Nanos-ZnF:Brat-NHL molar ratio. (B) Overlay of the 1H,15N HSQC spectra of 15N labelled Brat-NHL in the free form and with unlabelled Nanos-ZnF added to 1:1 Brat-NHL:Nanos-ZnF molar ratio. The spectrum of the respective free proteins is always rendered with a lower contour limit, so it is visible that two spectra are overlaid and no chemical shift perturbations or signal intensity decrease is observed in either of the experiments.
Pum-HD, Nanos-ZnF and Brat-NHL assemble on a single NRE2 RNA
To ensure that all three proteins and hunchback mRNA form a quaternary hb complex, NRE2 RNA was incubated with Pum-HD, Nanos-ZnF and Brat-NHL in a 1:1:1:2 ratio, and purified as described in the methods section. The expected molecular weight of an equimolar quaternary complex of the NRE2 RNA with the three domains is approximately 87.8 kDa. The SEC chromatogram showed a major peak with a maximum at an elution volume corresponding to approximately 77 kDa based on the molecular weight standard (Gel filtration markers kit for MWs 12–200 kDa, Sigma, Figure 3A). The peak contains all three protein domains, as visible by SDS-PAGE (Figure 3B), and the NRE2 RNA, which is evidenced by a high ratio between UV absorption at wavelengths 260 and 280 nm (∼1.8). The peak was then analysed by SEC-MALLS to gain further insights into the stoichiometry of the complex. Notably, MALLS reveals a monodisperse particle with a molecular weight of 85.8 ± 0.5 kDa (Figure 3C, D). Collectively, these results confirm that the four components form a stable 1:1:1:1 hb complex of Pum HD, Nanos ZnF, Brat NHL and NRE2 RNA.
Figure 3.

Reconstitution of the hb complex and its NMR characterization. To reconstitute the hb complex, purified Pum-HD, Nanos-ZnF and Brat-NHL were mixed with NRE2 RNA as described in Material and Methods. Size-exclusion chromatography (SEC) was then used to purify the complex (A). The grey shaded area indicates the analysed fractions of the hb complex peak. (B) SDS-PAGE of the selected fractions from SEC to confirm that the major peak consists of the hb complex (B). (C) SEC-MALLS-based mass determination (D) confirmed the composition of the hb complex. The lines in (C) show the following: Molecular weight (MW, black), refractive index (RI, blue), light scattering (LS, red) and UV in green. Mw/Mn in (D) reflects the polydispersity of the sample and a value of 1 corresponds to a monodisperse sample. The MW estimated from SEC is based on a molecular weight standard (Gel filtration markers kit for MWs 12–200 kDa, Sigma). (E) Overlay of the methyl region of 1H,13C HMQC spectra of Brat-NHL in its free form (Free Brat-NHL) and Brat-NHL with NRE2 RNA (at 1:1 stoichiometric ratio). Red arrows are used to highlight larger chemical shift perturbations. (F) Overlay of the methyl region of 1H,13C HMQC spectra of Brat-NHL with NRE2 RNA (at 1:1 stoichiometric ratio) and of Brat-NHL in the hb complex (Brat-NHL in the complex). No chemical shift perturbations are seen between these two states indicating that no further contacts are formed between Brat-NHL and Pum-HD-Nanos-ZnF.
Having confirmed the presence of a biochemically stable quaternary protein-RNA complex, we then used NMR spectroscopy in order to probe whether transient contacts can be observed between Brat-NHL-NRE2 and Pum-HD/Nanos-ZnF-NRE2 moieties within the hb complex. As the size of the complex exceeds the sensitivity of standard biomolecular NMR, we used a sample of the complex containing unlabelled Nanos-ZnF and RNA, 2H-labelled Pum-HD and 1H,13C-methyl-ILV, 2H, 15N-labelled Brat-NHL (Brat-NHL in the complex) and recorded 1H,13C TROSY-HMQC spectra to compensate for line broadening by increased transverse relaxation due to slow molecular tumbling. We compared the methyl region from samples of free and RNA-bound 1H,13C-methyl-ILV, 2H,13C,15N-labelled Brat-NHL and of NRE2 RNA-bound Brat-NHL and Brat-NHL within the hb complex (Figure 3E, F). All samples would only provide signal from Brat-NHL, but would report on the residue-wise chemical environment of Brat-NHL in its free form, bound to RNA and in the hb complex. A comparison of the spectra of free and RNA-bound Brat-NHL shows obvious chemical shift perturbations (CSPs), confirming the interaction between Brat-NHL and RNA (Figure 3E). However, a comparison of the spectra of RNA-bound Brat-NHL and Brat-NHL in the complex reveals no further CSPs (Figure 3F), suggesting that Brat-NHL does not form any additional interaction surfaces in the hb complex with another protein or RNA component. This further confirms the absence of additional protein-protein contacts between Brat-NHL and Pum-HD/Nanos-ZnF or altered Brat-NHL-RNA contacts when part of the complex. This also suggests that both moieties (Brat-NHL and Pum-HD/Nanos-ZnF) may behave like beads on a string, which would allow independent movement of both parts with respect to each other.
Structural characterization of the hb complex
Although Brat-NHL binds RNA independently of Nanos-ZnF/Pum-HD, we nevertheless set out to obtain a structural model of the hb complex. This would provide important information whether preferred orientations between the proteins exist or which conformation space would be sterically excluded. Initially, we set out to investigate the hb complex using X-ray crystallography. We tested an extensive set of commercial crystallization screens covering various crystallization conditions (PEGs, salts, PEG smears, alternative polymer precipitants, etc.) at a wide range of hb complex concentrations and multiple temperatures. In addition, we designed and tested custom screens around the published crystallization conditions of Pum-HD, Nanos-ZnF and Brat-NHL. The extensive testing did not yield any diffracting crystals of the hb complex. Lastly, carrier-driven crystallization using a Pum-HD fused to an MBP-tag also failed, likely because the modification of Pum-HD interfered with complex formation. We then adopted an alternative integrative approach to obtain a structural model of the hb complex, which combines small-angle X-ray and neutron scattering (SAXS/SANS), NMR spectroscopy, cross-linking/mass-spectrometry (XL/MS), available crystal structures of Brat-NHL-RNA complex and Pum-HD-Nanos-ZnF-RNA complex (15,22), as well as molecular dynamics simulations.
First, SAXS data indicate a structured hb complex with an Rg of ∼37.4 Å according to Guinier analysis (Figure 4A, Supplementary Figure S2A, Supplementary Table S1). Indirect Fourier transformation results in an asymmetric distance distribution function (P(r)) with a maximum at ∼30 Å that smears out through an additional peak to a maximum particle dimension (Dmax) of ∼130 Å (Figure 4B). This is smaller than the sum of Dmax of the components (Supplementary Figure S2B). The P(r) of the Brat-NHL-RNA complex is a single approximately symmetrical peak with a maximum at ∼25 Å and a Dmax of ∼50 Å. The P(r) of the Pum-HD-Nanos-ZnF-RNA complex shows a more complex distribution with a maximum at ∼22 Å and a long, smeared tail with an additional peak and a Dmax of ∼90 Å. To learn whether the system possesses some degree of flexibility, the SAXS data were also plotted as a dimensionless Kratky plot (Figure 4C). This plot reveals approximately a Gaussian peak, which is typical for a folded globular biomolecule. However, the position of the maximum of the peak deviates from a position expected for an ideal globular folded protein indicating either a deviation from a globular shape or some degree of flexibility. In SANS measurements, subunit-selective perdeuteration combined with varying D2O concentration in the buffer is used during contrast matching to determine further structural parameters (e.g. centre-of-mass distances between the subunits) (65), which are useful in data-driven structure modelling of protein-RNA complexes (50,67–69). Here, we used several differentially subunit-selectively perdeuterated hb complex samples (Table 2). We obtained in total seven SANS scattering curves on a fully protonated hb complex, hb complex with perdeuterated Pum-HD and Nanos-ZnF, and hb complex with perdeuterated Brat-NHL and Pum-HD (Figure 4A).
Figure 4.

Characterization of the hb complex by small-angle scattering (SAS). (A) Scattering curves of the hb complex. The curve labelled hb complex SAXS corresponds to the scattering curve obtained for fully protonated hb complex using small-angle X-ray scattering (SAXS) coupled online to SEC. The remaining curves correspond to the scattering curves of hb complex obtained using small-angle neutron scattering (SANS). Each curve was measured on a distinct sample of the hb complex, that varied in perdeuteration of the components of the complex (Table 1). The perdeuteration is always indicated in the label - 1 stands for a fully protonated component, whereas 2 indicates a perdeuterated complex. B, P and N stands for Brat-NHL, Pum-HD and Nanos-ZnF, respectively. NRE2 RNA was always protonated and all curves shown here were measured at varying concentrations of D2O in the buffer as stated in the legend. (B) Distance distribution function of the hb complex obtained from indirect Fourier transformation of the SAXS scattering curve. (C) Normalized Kratky plot of the SAXS scattering curve. The indicated ideal maximum corresponds to an expected maximum for a folded globular protein (103). (D) Distribution of χ2 values of fits of the pool of models generated to model hb complex based on SAS data. To model the hb complex 5055 random models were generated and theoretical scattering curves of each model at each condition were then back-calculated and fitted against the experimental data using CRYSOL and CRYSON. The best fitting models were selected as the models in the top 0.22 quantile of χ2 values of fits of each curve and are highlighted in the distribution as either the purple or cyan dots according to whether they fall into the major or minor cluster. (E) Ensemble of hb complex models fitting the SAS data best. The models fall into two clusters representing two distinct conformations. The first, major cluster is shown with Brat-NHL in shades of purple and contains eleven models with Brat-NHL close to the C-terminus of Nanos-ZnF. The second, minor cluster is shown with Brat-NHL in shades of cyan and includes four models with Brat-NHL close to the N-terminus of Nanos-ZnF. All models in the ensembles are always superimposed on Pum-HD and Nanos-ZnF.
To search for a potential structure of the hb complex described by the experimental SAXS/SANS data, we generated a pool of ∼5000 models by unrestrained molecular dynamics (MD) in CNS (47,70) using NRE2 RNA, the structure of the Brat–NHL–RNA complex (PDB ID: 4ZLR) (22) and Pum-HD-Nanos-ZnF-RNA complex (PDB ID: 5KL1) (15). During structure calculation the unbound NRE2 RNA nucleotides (7–11) were randomized and the known structures preserved, including protein-RNA contacts. The pool of structures covers a large conformational space, which we then aimed to reduce comparing the back-calculated scattering curve of each model with the experimental data using CRYSOL and CRYSON (30,31). For each experimental curve we set a cut-off criterion of 0.23 quantile of lowest χ2 values of the fit between the experimental and back-calculated scattering (Figure 4D, Supplementary Figure S3A, C), and then searched for models that fulfil the criterion for each experimental curve (SAXS and SANS). This yielded an ensemble of fifteen models, which fall into two clusters representing two alternative conformations, that seem to fit the data equally well (Figure 4E). These two alternative conformations were present even when a stricter cut-off criterion was chosen. Two distinct conformations fitting the data equally well could be a consequence of either the general low resolution of the method or conformational heterogeneity of the complex. The low resolution would allow two distinct conformations to fit well, if the shapes of the conformations are roughly symmetric (this is an intrinsic limitation of the methods and has been extensively described elsewhere (71–73)). In case of conformational heterogeneity, the measured scattering curve is a linear sum of the scattering of individual conformations weighted by their population. In case of equally populated conformations, fitting each conformation individually to the experimental data would result in equally good fits. To resolve this, we acquired more data using XL/MS.
The cross-linking reaction described in the methods section was optimized to 0.5 mM DSS on 0.95 mg/ml hb complex. DSS cross-links the terminal side-chain amines of lysines and the linker arm allows distances of up to 11.4 Å to be cross-linked, resulting in an allowed distance between the Cα of linked residues of up to 24 Å. A tolerance of 2–6 Å is usually assumed as it could be shown that even larger distances are sometimes cross-linked (74). For structure calculations, distances between 26–30 Å are therefore used for Cα–Cα pairs. Our experiment yielded twenty inter-molecular cross-links with an id-score higher than 25 (Figure 5A, Supplementary Table S2). Eight of the cross-links were between Pum-HD and Nanos-ZnF, leaving twelve cross-links between Brat-NHL and Pum-HD or Nanos-ZnF. These were then used as lysine–lysine distance restraints with a log-harmonic potential in the same MD protocol as described above to generate another pool of ∼4500 models. The pool of models was then reduced as above, but with a cut-off criterion of 0.3 quantile of lowest χ2 for each curve (Figure 5B, Supplementary Figure S3B, C). Finally, an ensemble was obtained from the restricted pool by selecting only the models in 0.1 quantile of the lowest energy of XL/MS distance restraints violations. The ensemble comprised three models in a conformation similar to the conformation of the minor cluster described above, except that Brat-NHL was rotated by about 45° (Figure 5C, D). However, even the model with the least violations of XL/MS distance restraints satisfies a maximum of four of the twelve cross-links simultaneously (Figure 5C, model C) and visual inspection of other models revealed that some cross-links can only be satisfied exclusively. Such results would be expected for a flexible system, where there is no fixed position of Brat-NHL relative to the Pum-HD-Nanos-ZnF moiety.
Figure 5.

Modelling of the hb complex by combined use of SAS/cross-linking mass spectrometry and SAS-driven MD. (A) Cross-links of the hb complex obtained from the XL/MS experiments. Inter-molecular cross-links are indicated by lines in shades of orange. Grey loops indicate intra-molecular cross-links. Only cross-links with an id-score higher than 25 are shown. (B) Distribution of χ2 values of fits of the pool of models generated to model hb complex based on SAS and XL/MS data. To model the hb complex 4572 models were generated and theoretical scattering curves of each model at each condition were then back-calculated and fitted against the experimental data using CRYSOL and CRYSON (30,31). The models in the top 0.30 quantile of χ2 values of each curve were selected and then this pool was further restricted to only the models in the top 0.10 quantile of the lowest distance restraint violation energy. The selected ensemble is highlighted in the distribution as magenta dots. (C) Three hb complex models derived using a combination of the SAS and XL/MS data. All three adopting a similar conformation with Brat-NHL close to the N-terminus of Nanos-ZnF. Satisfied cross-links are indicated in green lines and unsatisfied in blue. (D) χ2 values against all SAS data for each model shown in C). In the last row the number of satisfied cross-links is shown. The last column indicates the range of χ2 values for all models of the total ensemble. (E) hb complex models obtained by all-atom MD simulations, of which Brat-NHL domains are shown as yellow spheres representing the centre-of-mass of the domain. This is overlayed with representatives of the ensemble filtered against SAS data alone (see Figure 4E) and filtered against SAS and XL/MS (see C)). All models in the ensembles are always superimposed on Pum-HD and Nanos-ZnF.
To test whether a continuous structural ensemble is compatible with the SAXS/SANS data, and to obtain an atomistic model of the overall complex in solution, we used all-atom MD simulations with the Amber99sbws/TIP4P-2005s force field, which has been refined to balance protein-protein versus protein-water interactions (54). To improve the sampling of the conformational space, we simulated 8 replicas for 110 ns, where each replica started from a different conformation taken from the rigid-body simulations. No bias or restraint was applied. In the simulations, the conformational space adopted by Brat-NHL relative to Pum-HD-Nanos-ZnF is characterized by an arch-shaped distribution, in which Brat-NHL (i) forms only occasional contacts with Pum-HD-Nanos-ZnF and (ii) takes various rotational states relative to Pum-HD (Figure 5E). SAXS and SANS curves were computed from a total of 6408 simulation frames using explicit-solvent calculations, taking atomistic representations for the hydration layer and excluded solvent into account. The experimental curves Iexp(q) were fitted to the calculated curve via Iexp,fit(q) = f⋅(Isam(q) – α⋅Ibuf(q) + c), but no parameters related to the hydration layer or excluded solvent were adjusted. Remarkably, we found nearly quantitative agreement between the calculated and experimental SAXS curves (reduced χ2 = 2.18) and reasonable agreement for the SANS curves (Supplementary Figure S3D, E), without the need to reweight the ensemble or couple the simulation to the experimental data, highlighting the quality of the applied force field and suggesting that the accessible conformational space was reasonably well sampled. For comparison, we also used the established ensemble optimization method (EOM) (32,75). EOM selected an ensemble of 7 models (Supplementary Figure S4) using the SAXS curve and the pool of random models generated previously for the modelling using only SAS data. The EOM ensemble has an Rflex of 88.3% (Rflex of the pool: 89.5%) and Rσ of 5.26, which shows that restricting a pool of approximately 5000 randomly generated models to an ensemble of 7 models with the use of experimental SAXS data does not reduce the represented conformational space at all. These results suggest that the hb complex adopts a continuous, heterogenous ensemble in solution.
To further validate this assumption experimentally, we used the perdeuterated, 15N,13C-labelled samples from Figure 3D and E in 1H,15N T1 and T2 relaxation experiments. The relaxation experiments were acquired on free Brat-NHL and on Brat-NHL in the complex. Brat NHL is a disc-shaped ∼32 kDa β-propeller domain, whereas the measured molecular weight of the hb complex is ∼85 kDa. Thus, forming the hb complex should be accompanied by a drastic change of relaxation properties of Brat-NHL, if the complex tumbles as one entity. However, the acquired relaxation parameters reveal that neither R1 (1/T1) nor R2 (1/T2) changes drastically between free Brat-NHL and Brat-NHL in the complex (Figure 6A and B). We used these values to calculate the rotational correlation time (τc), which is the time it takes the average molecule to rotate one radian in solution, and so it reports on the overall tumbling of the molecule and allows apparent molecular weight estimation (assuming isotropic tumbling). Brat-NHL has a theoretically predicted τc of 15.3 ns, whereas the predicted τc of the hb complex is 54 ns (with a standard deviation of 1.5 ns, assuming the whole complex tumbles jointly). Both values were predicted using ROTDIF 3 (45,46). The τc of the hb complex was calculated as a median value of the fifteen models generated by modelling using only the small-angle scattering data (described above). The experimentally measured τc of free Brat-NHL of 19.0 ± 4.3 ns matches the predicted value well. Strikingly, the measured τc of Brat-NHL in the complex was 15.6 ± 3.8 ns, which is similar to the value of free Brat-NHL and is far below the predicted value for a rigid hb complex (Figure 5C). The elevated τc of free Brat-NHL compared to Brat-NHL in the complex may result from weak unspecific multimerization of free Brat-NHL. Nevertheless, these τc values confirm that the hb complex is largely flexible and that Brat-NHL tumbles independently of the Pum-HD-Nanos-ZnF moiety. In such a scenario the measured small-angle scattering curve would be an average over all conformations and the cross-links would arise from stochastic encounters of the domains possibly yielding conflicting data. Additionally, we would not expect any further cooperativity or interaction upon recruitment of Brat-NHL to the complex.
Figure 6.

Flexibility of the hb complex. (A) 15N Spin-lattice relaxation rate (R1) of Brat-NHL. R1 was measured for both Brat-NHL in its free form (Free Brat-NHL) and in the hb complex with all the other components of the complex unlabelled (Brat-NHL in the complex). Values related to free Brat-NHL are displayed in green, whereas values related to Brat-NHL in the complex are displayed in orange. Measured values are plotted as dots with error bars, horizontal full line shows a median and the shaded horizontal bar indicates a range of two standard deviations of the median. (B) 15N Spin–spin relaxation rates (R2) of Brat-NHL. R2 was measured on the same samples as R1, and labelling corresponds to (A). (C) Rotational correlation time (τc) of Brat-NHL. The left panel of the plot indicates experimentally determined τc values calculated based on measured R1 and R2 values. The right panel shows theoretically expected τc values calculated from the crystal structure of Brat-NHL or an average of theoretically expected τc calculated for the ensemble of fifteen models of the hb complex modelled using the small-angle scattering data (Figure 4E).
Taken all together our data clearly show that a stable quaternary 1:1:1:1 complex of Pum-HD, Nanos-ZnF, Brat-NHL and NRE2 RNA forms. However, the RNA binding of Brat-NHL is completely independent of Pum-HD and Nanos-ZnF and no additional interaction between Brat-NHL and the Pum-HD-Nanos-ZnF moiety is observed. The resulting hb complex is flexible and the flexibility is presumably only limited by steric restrictions of the conformation of unbound nucleotides between the respective protein binding sites in NRE2 RNA.
DISCUSSION
Protein–RNA complexes play key roles at any stage of gene expression and underlying molecular mechanisms of their function have been elucidated for large machines like ribosomes, RNA polymerases or the spliceosome (76–78). However, >1000 proteins were identified to bind mRNA (79,80). In these cases our understanding of molecular mechanisms of the function of those proteins is mostly limited to insights from structures of individual RNA binding proteins (RBPs) bound to a short cognate RNA (81). However, these RBPs do not function in isolation. Only a few studies showed examples of cooperative RNA-recognition by multiple RBPs resulting in increased affinity and specificity (82,83). However, other mechanisms may be possible and detailed mechanistic understanding of complexes beyond those of individual RBPs is essential to formulate general molecular mechanisms governing the mRNA interactome. Shedding light into simultaneous RNA-recognition by multiple RBPs is often made difficult by the transient and dynamic nature of those complexes. Addressing this challenge often requires an integrative approach combining multiple methods (84). In this study, we used such an approach to obtain results allowing us to provide proper interpretation of previously published data and ultimately clarify our understanding of the structure and dynamics of the quaternary protein-RNA complex that suppresses hunchback mRNA translation.
In case of Pumilio and Nanos there is now sufficient evidence to conclude that these two proteins indeed act jointly (15), but how Brat-NHL fits into the picture remained unclear. Several conclusions can be drawn from our results. First, our binding assays reconcile the published results on RNA-binding by Pum-HD and Brat-NHL (Figures 1, 2, Table 1 and Supplementary Figure S1). Pum-HD was confirmed to bind BoxB with high affinity (16) and Brat-NHL binds with low micromolar affinity to BoxA (Figure 1F, G). Interestingly, Pum-HD also binds to nucleotides located in between BoxA and BoxB, part of which is bound by Nanos-ZnF within the hb complex (Figure 1G). Our binding assays show that this secondary binding of Pum-HD is of lower affinity and comparable to the binding of Brat-NHL to BoxA (∼1 μM Kd).
Furthermore, we show that incubating the RNA with Pum-HD, Nanos-ZnF or both does not enhance the Brat-NHL-RNA interaction (Figure 1H). The previously reported enhancement was based purely on EMSA experiments, which are not quantitative equilibrium assays (22). Moreover, the reported effect was very mild and likely in the error range of the method. We further confirm that Pum-HD indeed unfolds the secondary structure of the RNA (Figure 1E). However, we find no evidence that it enhances the Brat–NHL–RNA interaction and find no reasons to assume so, particularly considering that the reported Brat-NHL binding site lies predominantly outside of the secondary structure of the RNA (Figure 1A). In conclusion, we show that Pum-HD and Nanos-ZnF have no significant effect on the binding of Brat–NHL to the RNA, suggesting that Pum-HD/Nanos-ZnF and Brat-NHL recognize the hunchback mRNA neither competitively nor cooperatively, but independently. Although the hb complex forms, we did not detect any interaction by NMR between the two independent moieties of this complex (Figures 2, 3 and 6).
The binding sites of Brat-NHL and Pum-HD with Nanos-ZnF are separated by four nucleotides, so if there is no cooperativity in the RNA-binding and Pum-HD unfolds the secondary structure of the RNA, there should be no constraints on these four nucleotides. The unconstrained nucleotides should then give the hb complex flexibility allowing free movement of the Pum-HD-Nanos-ZnF and Brat-NHL moieties. We therefore derived an atomistic conformationally free ensemble from unbiased all-atom MD simulations, which quantitatively agreed with the SAXS, SANS and XL-MS data, suggesting that the hb complex is indeed largely flexible (Figure 5E). Our NMR relaxation measurements then confirm the flexibility, as we see no difference between the rotational correlation time of Brat-NHL in its free form and in the hb complex (Figure 6).
To put our findings into context with a previously established model and mutational analysis, we provided a table with mutations reported to have a phenotypic effect and abolish complex formation (Table 3). All mutations are located in the RNA binding domains used in this study, so they do not give insight into the importance of regions outside these boundaries on complex formation. Of note, until Loedige et al. and Weidmann et al. published their crystal structures of Brat-NHL-UUGUUG and the Pum-HD-Nanos-ZnF-RNA complex (15,22), it was assumed that Brat-NHL does not bind RNA directly but Pumilio. Edwards et al. (85) substituted three Brat-NHL residues (Y829, R847, R875) to alanines, which resulted in the absence of abdominal segmentation and they concluded that these mutations interfere with a direct Pum-HD-Brat-NHL interaction inferred from yeast-four-hybrid assays. However, the later crystal structure revealed that all three residues are directly involved in RNA binding and can therefore not contact Pum-HD. All three residues are located on the top surface of Brat-NHL and Edwards et al. provided a structural model of a Pum-HD-Brat-NHL complex based on their mutational analysis (85). The location of Brat-NHL with respect to Pum-HD is similar to our model, as it interacts with Pum-HD’s Puf repeat 8 with its top surface. In our model, the top surface is also oriented towards this end of Pum-HD, however, in between is the RNA and Nanos-ZnF. Another Brat-NHL mutation, H802L, causing a neoplastic larval brain is also located on the top surface and may interfere with RNA binding rather than the suggested interference of interaction with another unknown protein factor (14,86). Two further mutations (G774D and G860D) causing the same phenotypes were suggested to interfere with binding of Pumilio or another protein factor (14,86,87). These mutations are located on the side and bottom surface, distal from RNA and Pum-HD. Thus, these residues may interfere with another protein or instead decrease protein stability. Four mutations on Pum-HD have been reported to cause a defect in abdominal segmentation (G1330D, G1330Y, C1365R and T1366D), which was explained by disrupting a direct Brat interaction (14). However, all mutations are located on the Puf repeat 7, which has been shown later to be involved in the interaction with Nanos-ZnF (15). These could also cause Pumilio to unfold, as the Puf repeat interactions are important for the integrity of this domain. The Nanos-ZnF mutations M379K, causing defects in abdominal segmentation was also thought to affect a direct Brat interaction (14), but it was similarly shown later that this mutation interferes with the Nanos-ZnF-Pum-HD interaction (15). All these mutations are therefore not evidence of Brat-NHL-Pum-HD or Brat-NHL-Nanos-ZnF contacts and therefore do not contradict the validity of our ensemble model.
Table 3.
Previously tested point mutations of each protein component within the hb complex, their phenotype and effect on complex formation
| Protein | Mutation | Phenotype | Suggested molecular reasona | Actual effect on complex formationb | Reference |
|---|---|---|---|---|---|
| Brat-NHL | Y829A, R847A, R875A | No abdominal segmentation | Interferes with Pum binding (Yeast-four-hybrid assay) | Interferes with direct RNA binding | (85) |
| H802L | Neoplastic larval brain No abdominal segmentation | Interferes with another protein factor | May affect RNA binding indirectly | (14,86) | |
| G774D | No abdominal segmentation | Interferes with Pum binding | Not clear, mutations are on the side and bottom surface distal from Pum-HD/Nanos-ZnF | (14,87) | |
| G860D | Neoplastic larval brain | Interferes with another protein factor | (86) | ||
| Pum-HD | G1330D, G1330Y | No abdominal segmentation | Interferes with direct Brat interaction, no translation repression/ would clash with NHL E782 and D1021. | Not clear, located on Puf repeat 7, remote from Nanos and Brat | (14) |
| C1365R, T1366D | No abdominal segmentation | Interferes with direct Brat interaction, no translation repression | Not clear, located on Puf repeat 7, remote from Nanos and Brat | (14) | |
| Nanos-ZnF | M379K | No abdominal segmentation | Interferes with direct Brat interaction, no translation repression | Interferes with Pum binding instead | (14) |
| F321A, N325A, Y352A, T366A, K368Q, Y369A | Reduced translation repression | Interferes with Nos-RNA interaction within Pum-Nanos-RNA complex | Correct | (15) |
aThis column shows the molecular explanation of the phenotype with regards to hb complex formation as stated in the original publication (last column).
In summary our data clearly show that the RBD of Brat, and RBDs of Pumilio and Nanos, interact with hunchback RNA independently. Considering that the proteins are able to suppress translation independently (21–24,88,89), this could indicate that the suppression of hunchback mRNA by Brat is functionally separate of the suppression by Pumilio with Nanos. The independent action of both entities is to a certain degree in disagreement with an earlier study, where Brat-NHL binding was concluded to be dependent on direct and joint interactions with Pum-HD and Nanos-ZnF (14). However, Sonoda and Wharton used a Nanos-ZnF construct considerably longer (288–401) than in this study (301–392). The former is not suitable for biophysical studies as it shows two bands on a gel (14). Nonetheless, the possibility remains that residues responsible for cooperativity are located in these extensions. Also, the RNA used in the study was longer and Brat binding might be thus dependent on Pum/Nos binding to remove secondary structure in which the Brat binding motif could be hidden (22). On the other hand, a later study could show that Brat is able to repress translation independent of at least Pum (23). Furthermore, studies could show on a transcriptome-wide level that Pumilio and Brat have less joint mRNA targets than mRNAs, which are bound by both proteins individually (21,24,89). This suggests that Pumilio and Brat largely function independently. Of course, with regards to hunchback mRNA it remains to be seen if regions outside of the RNA binding domains tested here may contribute to cooperative RNA binding. The affinity of Brat-NHL for UUGUUG with a dissociation constant of 1 μM is rather weak. However, it is in the range of or even stronger than other classical RBDs (e.g. RRMs, CSDs etc.) (90,91). Also, single RBDs rarely act alone, but are often part of larger mRNP complexes, which achieve a much stronger binding by cooperativity, thus efficiently repressing translation (15,82,92). Similarly, Brat-NHL, although independent of Pum-HD/Nanos-ZnF could cooperatively bind together with another set of RBPs. Direct interactions with components of the microRNA pathway (93) or the deadenylation machinery (94–99) have been suggested in Drosophila and human. This could be yet another option of achieving a cooperative effect in RNA binding and efficient translational control. The translation of the maternal hunchback mRNA is suppressed from early embryogenesis to maternal-to-zygotic transition (MZT) (100). During MZT the maternally supplied mRNA is degraded by maternal and zygotic activities, so the embryo switches from a maternal to a zygotic genome (101). mRNA decay is often coupled to translation suppression and in fact Brat-mediated translation suppression was shown to control the maternal activity in RNA decay during MZT (21). Brat is distributed uniformly throughout the embryo (14), but Nanos is expressed in a gradient (4). If hunchback mRNA translation suppression by Brat and by the Pumilio-Nanos pair are separate, the suppression by Pumilio and Nanos might serve to establish the gradient of Hunchback in early embryogenesis, while the suppression by Brat might serve to control the maternal activity in RNA decay during MZT.
Considering all of these findings, there are two major questions to answer in order to gain a definitive and full understanding of the mechanism of hunchback mRNA translation suppression. First, it is crucial to extend this biophysical investigation ultimately to full-length proteins and RNA covering both NREs to test if Brat and Pumilio with Nanos still bind the RNA independently, especially considering that Brat has a dimeric coiled-coil domain (102). Secondly, functional experiments are necessary to decipher if the suppression activities of Brat and Nanos are separate. The suppression of hunchback mRNA translation is one of the best studied cases of localized translation suppression during development, so elucidating its mechanism fully will not only provide insights into the function of the specific trans-acting molecules, but will also give clues about general mechanisms of localized translation suppression during development.
DATA AVAILABILITY
All data will be made available upon request, including the structure ensemble of the complex model. The SAXS data has been submitted to the SASBDB with following identifier: SASDKR7.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the EMBL/DESY Hamburg PETRA-III (P12 beamline), and the ILL (D22 beamline) local contact for support (Anne Martel). This work was supported by an Emmy-Noether Fellowship (HE 7291_1) and the Priority Program SPP1935 (EP37/3-1, 3-2) of the Deutsche Forschungsgemeinschaft (DFG) and the EMBL.
Notes
Present address: Jakub Macošek, Biozentrum, University of Basel, Basel 4056, Switzerland.
Present address: Inga Loedige, IRI Life Sciences, Humboldt University, Berlin 10115, Germany.
Present address: Miloš Ivanović, Department of Biochemistry, University of Zurich, 8057 Zurich, Switzerland.
Contributor Information
Jakub Macošek, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Bernd Simon, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Johanna-Barbara Linse, Theoretical Physics and Center for Biophysics, Saarland University, Saarbrücken 66123, Germany.
Pravin Kumar Ankush Jagtap, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Sophie L Winter, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Jaelle Foot, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Karine Lapouge, Protein Expression and Purification Core Facility, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Kathryn Perez, Protein Expression and Purification Core Facility, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Mandy Rettel, Proteomics Core Facility, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Miloš T Ivanović, Theoretical Physics and Center for Biophysics, Saarland University, Saarbrücken 66123, Germany.
Pawel Masiewicz, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Brice Murciano, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Mikhail M Savitski, Proteomics Core Facility, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Inga Loedige, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany.
Jochen S Hub, Theoretical Physics and Center for Biophysics, Saarland University, Saarbrücken 66123, Germany.
Frank Gabel, Institut Biologie Structurale, University Grenoble Alpes, CEA, CNRS, Grenoble 38044, France.
Janosch Hennig, Structural and Computational Biology Unit, European Molecular Biology Laboratory Heidelberg, Heidelberg 69117, Germany; Chair of Biochemistry IV, Biophysical Chemistry, University of Bayreuth, Universitätsstrasse 30, 95447 Bayreuth, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Emmy-Noether Fellowship and a priority program (to J.H.); Deutsche Forschungsgemeinschaft (DFG) [HE 7291_1, EP37/3-1, 3-2]. Funding for open access charge: Deutsche Forschungsgemeinschaft; J.B.L. and J.S.H. were supported by the DFG [HU 1971/3-1].
Conflict of interest statement. None declared.
REFERENCES
- 1.Johnstone O., Lasko P.. Translational regulation and RNA localization in Drosophila oocytes and embryos. Annu. Rev. Genet. 2001; 35:365–406. [DOI] [PubMed] [Google Scholar]
- 2.Nüsslein-Volhard C., Frohnhöfer H.G., Lehmann R.. Determination of anteroposterior polarity in Drosophila. Science. 1987; 238:1675–1681. [DOI] [PubMed] [Google Scholar]
- 3.Berleth T., Burri M., Thoma G., Bopp D., Richstein S., Frigerio G., Noll M., Nüsslein-Volhard C.. The role of localization of bicoid RNA in organizing the anterior pattern of the Drosophila embryo. EMBO J. 1988; 7:1749–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C., Lehmann R.. Nanos is the localized posterior determinant in Drosophila. Cell. 1991; 66:637–647. [DOI] [PubMed] [Google Scholar]
- 5.Driever W., Nüsslein-Volhard C.. The bicoid protein is a positive regulator of hunchback transcription in the early Drosophila embryo. Nature. 1989; 337:138–143. [DOI] [PubMed] [Google Scholar]
- 6.Hülskamp M., Schröder C., Pfeifle C., Jäckle H., Tautz D. Posterior segmentation of the Drosophila embryo in the absence of a maternal posterior organizer gene. Nature. 1989; 338:629–632. [DOI] [PubMed] [Google Scholar]
- 7.Irish V., Lehmann R., Akam M.. The Drosophila posterior-group gene nanos functions by repressing hunchback activity. Nature. 1989; 338:646–648. [DOI] [PubMed] [Google Scholar]
- 8.Schröder C., Tautz D., Seifert E., Jäckle H.. Differential regulation of the two transcripts from the Drosophila gap segmentation gene hunchback. EMBO J. 1988; 7:2881–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann R., Nüsslein-Volhard C.. hunchback, a gene required for segmentation of an anterior and posterior region of the Drosophila embryo. Dev. Biol. 1987; 119:402–417. [DOI] [PubMed] [Google Scholar]
- 10.Struhl G., Johnston P., Lawrence P.A.. Control of Drosophila body pattern by the hunchback morphogen gradient. Cell. 1992; 69:237–249. [DOI] [PubMed] [Google Scholar]
- 11.Wharton R.P., Struhl G.. RNA regulatory elements mediate control of Drosophila body pattern by the posterior morphogen nanos. Cell. 1991; 67:955–967. [DOI] [PubMed] [Google Scholar]
- 12.Curtis D., Treiber D.K., Tao F., Zamore P.D., Williamson J.R., Lehmann R.. A CCHC metal-binding domain in Nanos is essential for translational regulation. EMBO J. 1997; 16:834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murata Y., Wharton R.P.. Binding of pumilio to maternal hunchback mRNA is required for posterior patterning in Drosophila embryos. Cell. 1995; 80:747–756. [DOI] [PubMed] [Google Scholar]
- 14.Sonoda J., Wharton R.P.. Drosophila brain tumor is a translational repressor. Genes Dev. 2001; 15:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weidmann C.A., Qiu C., Arvola R.M., Lou T.F., Killingsworth J., Campbell Z.T., Tanaka Hall T.M., Goldstrohm A.C.. Drosophila Nanos acts as a molecular clamp that modulates the RNA-binding and repression activities of Pumilio. Elife. 2016; 5:e17096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta Y.K., Lee T.H., Edwards T.A., Escalante C.R., Kadyrova L.Y., Wharton R.P., Aggarwal A.K.. Co-occupancy of two Pumilio molecules on a single hunchback NRE. RNA. 2009; 15:1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wharton R.P., Sonoda J., Lee T., Patterson M., Murata Y.. The Pumilio RNA-binding domain is also a translational regulator. Mol. Cell. 1998; 1:863–872. [DOI] [PubMed] [Google Scholar]
- 18.Zamore P.D., Bartel D.P., Lehmann R., Williamson J.R.. The PUMILIO-RNA interaction: a single RNA-binding domain monomer recognizes a bipartite target sequence. Biochemistry. 1999; 38:596–604. [DOI] [PubMed] [Google Scholar]
- 19.Zamore P.D., Williamson J.R., Lehmann R.. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA. 1997; 3:1421–1433. [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto H., Hara K., Hishiki A., Kawaguchi S., Shichijo N., Nakamura K., Unzai S., Tamaru Y., Shimizu T., Sato M.. Crystal structure of zinc-finger domain of Nanos and its functional implications. EMBO Rep. 2010; 11:848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laver J.D., Li X., Ray D., Cook K.B., Hahn N.A., Nabeel-Shah S., Kekis M., Luo H., Marsolais A.J., Fung K.Y.et al.. Brain tumor is a sequence-specific RNA-binding protein that directs maternal mRNA clearance during the Drosophila maternal-to-zygotic transition. Genome Biol. 2015; 16:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loedige I., Jakob L., Treiber T., Ray D., Stotz M., Treiber N., Hennig J., Cook K.B., Morris Q., Hughes T.R.et al.. The crystal structure of the NHL domain in complex with RNA reveals the molecular basis of drosophila brain-tumor-mediated gene regulation. Cell Rep. 2015; 13:1206–1220. [DOI] [PubMed] [Google Scholar]
- 23.Loedige I., Stotz M., Qamar S., Kramer K., Hennig J., Schubert T., Loffler P., Langst G., Merkl R., Urlaub H.et al.. The NHL domain of BRAT is an RNA-binding domain that directly contacts the hunchback mRNA for regulation. Genes Dev. 2014; 28:749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arvola R.M., Weidmann C.A., Tanaka Hall T.M., Goldstrohm A.C.. Combinatorial control of messenger RNAs by Pumilio, Nanos and Brain Tumor Proteins. RNA Biol. 2017; 14:1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catanzariti A.M., Soboleva T.A., Jans D.A., Board P.G., Baker R.T.. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004; 13:1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker R.T., Catanzariti A.M., Karunasekara Y., Soboleva T.A., Sharwood R., Whitney S., Board P.G.. Using deubiquitylating enzymes as research tools. Methods Enzymol. 2005; 398:540–554. [DOI] [PubMed] [Google Scholar]
- 27.Tugarinov V., Kay L.E.. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J. Am. Chem. Soc. 2003; 125:13868–13878. [DOI] [PubMed] [Google Scholar]
- 28.Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M.et al.. A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017; 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konarev P., Volkov V., Sokolova A., Koch M., Svergun D. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003; 36:1277–1282. [Google Scholar]
- 30.Svergun D.I., Richard S., Koch M.H., Sayers Z., Kuprin S., Zaccai G.. Protein hydration in solution: experimental observation by x-ray and neutron scattering. Proc. Natl. Acad. Sci. USA. 1998; 95:2267–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svergun D., Barberato C., Koch M.. CRYSOL - a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995; 28:768–773. [Google Scholar]
- 32.Tria G., Mertens H.D., Kachala M., Svergun D.I.. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2015; 2:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forster S., Apostol L., Bras W.. Scatter: software for the analysis of nano- and mesoscale small-angle scattering. J. Appl. Crystallogr. 2010; 43:639–646. [Google Scholar]
- 34.Leitner A., Walzthoeni T., Aebersold R.. Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protoc. 2014; 9:120–137. [DOI] [PubMed] [Google Scholar]
- 35.Simon B., Köstler H.. Improving the sensitivity of FT-NMR spectroscopy by apodization weighted sampling. J. Biomol. NMR. 2019; 73:155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K.. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:13585–13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pervushin K., Riek R., Wider G., Wüthrich K.. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sattler M., Schleucher J., Griesinger C.. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999; 34:93–158. [Google Scholar]
- 39.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 40.Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D.. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005; 59:687–696. [DOI] [PubMed] [Google Scholar]
- 41.Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niklasson M., Otten R., Ahlner A., Andresen C., Schlagnitweit J., Petzold K., Lundström P.. Comprehensive analysis of NMR data using advanced line shape fitting. J. Biomol. NMR. 2017; 69:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahlner A., Carlsson M., Jonsson B.H., Lundström P.. PINT: a software for integration of peak volumes and extraction of relaxation rates. J. Biomol. NMR. 2013; 56:191–202. [DOI] [PubMed] [Google Scholar]
- 44.Fushman D., Weisemann R., Thüring H., Rüterjans H.. Backbone dynamics of ribonuclease T1 and its complex with 2′GMP studied by two-dimensional heteronuclear NMR spectroscopy. J. Biomol. NMR. 1994; 4:61–78. [DOI] [PubMed] [Google Scholar]
- 45.Ryabov Y.E., Geraghty C., Varshney A., Fushman D. An efficient computational method for predicting rotational diffusion tensors of globular proteins using an ellipsoid representation. J. Am. Chem. Soc. 2006; 128:15432–15444. [DOI] [PubMed] [Google Scholar]
- 46.Berlin K., Longhini A., Dayie T.K., Fushman D. Deriving quantitative dynamics information for proteins and RNAs using ROTDIF with a graphical user interface. J. Biomol. NMR. 2013; 57:333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brunger A.T.Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007; 2:2728–2733. [DOI] [PubMed] [Google Scholar]
- 48.Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S.et al.. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 1998; 54:905–921. [DOI] [PubMed] [Google Scholar]
- 49.Simon B., Madl T., Mackereth C.D., Nilges M., Sattler M.. An efficient protocol for NMR-spectroscopy-based structure determination of protein complexes in solution. Angew. Chem. Int. Ed. Engl. 2010; 49:1967–1970. [DOI] [PubMed] [Google Scholar]
- 50.Lapinaite A., Simon B., Skjaerven L., Rakwalska-Bange M., Gabel F., Carlomagno T.. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature. 2013; 502:519–523. [DOI] [PubMed] [Google Scholar]
- 51.Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T.E., Nilges M.. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007; 23:381–382. [DOI] [PubMed] [Google Scholar]
- 52.Nilges M., Bernard A., Bardiaux B., Malliavin T., Habeck M., Rieping W.. Accurate NMR structures through minimization of an extended hybrid energy. Structure. 2008; 16:1305–1312. [DOI] [PubMed] [Google Scholar]
- 53.Abraham M., Murtola T., Schulz R., Páll S., Smith J., Hess B., Lindahl E.. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1-2:19–25. [Google Scholar]
- 54.Best R.B., Zheng W., Mittal J.. Balanced protein–water interactions improve properties of disordered proteins and non-specific protein association. J. Chem. Theory Comput. 2014; 10:5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bussi G., Donadio D., Parrinello M.. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007; 126:014101. [DOI] [PubMed] [Google Scholar]
- 56.Berendsen H., Postma J., Vangunsteren W., Dinola A., Haak J.. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984; 81:3684–3690. [Google Scholar]
- 57.Parrinello M., Rahman A.. Polymorphic transitions in single-crystals - a new molecular-dynamics method. J. Appl. Phys. 1981; 52:7182–7190. [Google Scholar]
- 58.Miyamoto S., Kollman P.. Settle - an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992; 13:952–962. [Google Scholar]
- 59.Hess B.P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008; 4:116–122. [DOI] [PubMed] [Google Scholar]
- 60.Essmann U., Perera L., Berkowitz M., Darden T., Lee H., Pedersen L. A smooth particle mesh ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 61.Darden T., York D., Pedersen L.. Particle mesh ewald - an N.LOG(N) method for ewald sums in large systems. J. Chem. Phys. 1993; 98:10089–10092. [Google Scholar]
- 62.Knight C.J., Hub J.S.. WAXSiS: a web server for the calculation of SAXS/WAXS curves based on explicit-solvent molecular dynamics. Nucleic Acids Res. 2015; 43:W225–W230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen P.C., Shevchuk R., Strnad F.M., Lorenz C., Karge L., Gilles R., Stadler A.M., Hennig J., Hub J.S.. Combined small-angle X-ray and neutron scattering restraints in molecular dynamics simulations. J. Chem. Theory Comput. 2019; 15:4687–4698. [DOI] [PubMed] [Google Scholar]
- 64.Chen P.C., Hub J.S.. Validating solution ensembles from molecular dynamics simulation by wide-angle X-ray scattering data. Biophys. J. 2014; 107:435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacrot B.The study of biological structures by neutron scattering from solution. Rep. Prog. Phys. 1976; 39:911–953. [Google Scholar]
- 66.Wang X., McLachlan J., Zamore P.D., Hall T.M.. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002; 110:501–512. [DOI] [PubMed] [Google Scholar]
- 67.Sonntag M., Jagtap P.K.A., Simon B., Appavou M.S., Geerlof A., Stehle R., Gabel F., Hennig J., Sattler M.. Segmental, domain-selective perdeuteration and small-angle neutron scattering for structural analysis of multi-domain proteins. Angew. Chem. Int. Ed. Engl. 2017; 56:9322–9325. [DOI] [PubMed] [Google Scholar]
- 68.Hennig J., Wang I., Sonntag M., Gabel F., Sattler M.. Combining NMR and small angle X-ray and neutron scattering in the structural analysis of a ternary protein-RNA complex. J. Biomol. NMR. 2013; 56:17–30. [DOI] [PubMed] [Google Scholar]
- 69.Capel M.S., Engelman D.M., Freeborn B.R., Kjeldgaard M., Langer J.A., Ramakrishnan V., Schindler D.G., Schneider D.K., Schoenborn B.P., Sillers I.Y.. A complete mapping of the proteins in the small ribosomal subunit of Escherichia coli. Science. 1987; 238:1403–1406. [DOI] [PubMed] [Google Scholar]
- 70.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S.et al.. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 1998; 54:905–921. [DOI] [PubMed] [Google Scholar]
- 71.Gabel F.A simple procedure to evaluate the efficiency of bio-macromolecular rigid-body refinement by small-angle scattering. Eur. Biophys. J. 2012; 41:1–11. [DOI] [PubMed] [Google Scholar]
- 72.Gabel F., Simon B., Nilges M., Petoukhov M., Svergun D., Sattler M.. A structure refinement protocol combining NMR residual dipolar couplings and small angle scattering restraints. J. Biomol. NMR. 2008; 41:199–208. [DOI] [PubMed] [Google Scholar]
- 73.Gabel F., Simon B., Sattler M.. A target function for quaternary structural refinement from small angle scattering and NMR orientational restraints. Eur. Biophys. J. 2006; 35:313–327. [DOI] [PubMed] [Google Scholar]
- 74.Merkley E.D., Rysavy S., Kahraman A., Hafen R.P., Daggett V., Adkins J.N.. Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 2014; 23:747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bernadó P., Mylonas E., Petoukhov M.V., Blackledge M., Svergun D.I.. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007; 129:5656–5664. [DOI] [PubMed] [Google Scholar]
- 76.Cramer P., Armache K.J., Baumli S., Benkert S., Brueckner F., Buchen C., Damsma G.E., Dengl S., Geiger S.R., Jasiak A.J.et al.. Structure of eukaryotic RNA polymerases. Annu. Rev. Biophys. 2008; 37:337–352. [DOI] [PubMed] [Google Scholar]
- 77.Yusupova G., Yusupov M.. Crystal structure of eukaryotic ribosome and its complexes with inhibitors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017; 372:20160184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilkinson M.E., Charenton C., Nagai K.. RNA splicing by the spliceosome. Annu. Rev. Biochem. 2020; 89:359–388. [DOI] [PubMed] [Google Scholar]
- 79.Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 81.Chen Y., Varani G.. Protein families and RNA recognition. FEBS J. 2005; 272:2088–2097. [DOI] [PubMed] [Google Scholar]
- 82.Hennig J., Militti C., Popowicz G.M., Wang I., Sonntag M., Geerlof A., Gabel F., Gebauer F., Sattler M.. Structural basis for the assembly of the Sxl-Unr translation regulatory complex. Nature. 2014; 515:287–290. [DOI] [PubMed] [Google Scholar]
- 83.Hennig J., Gebauer F., Sattler M.. Breaking the protein-RNA recognition code. Cell Cycle. 2014; 13:3619–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dimitrova-Paternoga L., Jagtap P.K.A., Chen P.C., Hennig J.. Integrative structural biology of Protein-RNA complexes. Structure. 2020; 28:6–28. [DOI] [PubMed] [Google Scholar]
- 85.Edwards T.A., Wilkinson B.D., Wharton R.P., Aggarwal A.K.. Model of the brain tumor-Pumilio translation repressor complex. Genes Dev. 2003; 17:2508–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arama E., Dickman D., Kimchie Z., Shearn A., Lev Z.. Mutations in the beta-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene. 2000; 19:3706–3716. [DOI] [PubMed] [Google Scholar]
- 87.Harris R.E., Pargett M., Sutcliffe C., Umulis D., Ashe H.L.. Brat promotes stem cell differentiation via control of a bistable switch that restricts BMP signaling. Dev. Cell. 2011; 20:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weidmann C.A., Goldstrohm A.C.. Drosophila Pumilio protein contains multiple autonomous repression domains that regulate mRNAs independently of Nanos and brain tumor. Mol. Cell. Biol. 2012; 32:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gerber A.P., Luschnig S., Krasnow M.A., Brown P.O., Herschlag D. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:4487–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Daubner G.M., Cléry A., Allain F.H.. RRM-RNA recognition: NMR or crystallography…and new findings. Curr. Opin. Struct. Biol. 2013; 23:100–108. [DOI] [PubMed] [Google Scholar]
- 91.Corley M., Burns M.C., Yeo G.W.. How RNA-Binding proteins interact with RNA: Molecules and mechanisms. Mol. Cell. 2020; 78:9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hennig J., Sattler M.. Deciphering the protein-RNA recognition code: combining large-scale quantitative methods with structural biology. Bioessays. 2015; 37:899–908. [DOI] [PubMed] [Google Scholar]
- 93.Neumüller R.A., Betschinger J., Fischer A., Bushati N., Poernbacher I., Mechtler K., Cohen S.M., Knoblich J.A.. Mei-P26 regulates microRNAs and cell growth in the Drosophila ovarian stem cell lineage. Nature. 2008; 454:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Komori H., Golden K.L., Kobayashi T., Kageyama R., Lee C.Y.. Multilayered gene control drives timely exit from the stem cell state in uncommitted progenitors during. Genes Dev. 2018; 32:1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wreden C., Verrotti A.C., Schisa J.A., Lieberfarb M.E., Strickland S.. Nanos and pumilio establish embryonic polarity in Drosophila by promoting posterior deadenylation of hunchback mRNA. Development. 1997; 124:3015–3023. [DOI] [PubMed] [Google Scholar]
- 96.Van Etten J., Schagat T.L., Hrit J., Weidmann C.A., Brumbaugh J., Coon J.J., Goldstrohm A.C.. Human Pumilio proteins recruit multiple deadenylases to efficiently repress messenger RNAs. J. Biol. Chem. 2012; 287:36370–36383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weidmann C.A., Raynard N.A., Blewett N.H., Van Etten J., Goldstrohm A.C.. The RNA binding domain of Pumilio antagonizes poly-adenosine binding protein and accelerates deadenylation. RNA. 2014; 20:1298–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Enwerem I.I.I., Elrod N.D., Chang C.T., Lin A., Ji P., Bohn J.A., Levdansky Y., Wagner E.J., Valkov E., Goldstrohm A.C.. Human Pumilio proteins directly bind the CCR4-NOT deadenylase complex to regulate the transcriptome. RNA. 2021; 27:445–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arvola R.M., Chang C.T., Buytendorp J.P., Levdansky Y., Valkov E., Freddolino P.L., Goldstrohm A.C.. Unique repression domains of Pumilio utilize deadenylation and decapping factors to accelerate destruction of target mRNAs. Nucleic Acids Res. 2020; 48:1843–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tautz D., Pfeifle C.. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989; 98:81–85. [DOI] [PubMed] [Google Scholar]
- 101.Tadros W., Lipshitz H.D.. The maternal-to-zygotic transition: a play in two acts. Development. 2009; 136:3033–3042. [DOI] [PubMed] [Google Scholar]
- 102.Liu C., Shan Z., Diao J., Wen W., Wang W.. Crystal structure of the coiled-coil domain of Drosophila TRIM protein Brat. Proteins. 2019; 87:706–710. [DOI] [PubMed] [Google Scholar]
- 103.Receveur-Brechot V., Durand D. How random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 2012; 13:55–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data will be made available upon request, including the structure ensemble of the complex model. The SAXS data has been submitted to the SASBDB with following identifier: SASDKR7.