


Abstract
With the dramatic development of single-cell RNA sequencing (scRNA-seq) technologies, the systematic decoding of cell-cell communication has received great research interest. To date, several in-silico methods have been developed, but most of them lack the ability to predict the communication pathways connecting the insides and outsides of cells. Here, we developed CellCall, a toolkit to infer inter- and intracellular communication pathways by integrating paired ligand-receptor and transcription factor (TF) activity. Moreover, CellCall uses an embedded pathway activity analysis method to identify the significantly activated pathways involved in intercellular crosstalk between certain cell types. Additionally, CellCall offers a rich suite of visualization options (Circos plot, Sankey plot, bubble plot, ridge plot, etc.) to present the analysis results. Case studies on scRNA-seq datasets of human testicular cells and the tumor immune microenvironment demonstrated the reliable and unique functionality of CellCall in intercellular communication analysis and internal TF activity exploration, which were further validated experimentally. Comparative analysis of CellCall and other tools indicated that CellCall was more accurate and offered more functions. In summary, CellCall provides a sophisticated and practical tool allowing researchers to decipher intercellular communication and related internal regulatory signals based on scRNA-seq data. CellCall is freely available at https://github.com/ShellyCoder/cellcall.
INTRODUCTION
In multicellular organisms, intercellular communication allows multiple cells to coordinate with one another to form tissues, organs or systems and then accomplish various biological tasks (1–3), and aberrant loss or gain of extracellular recognition functions can contribute to various diseases (4,5). Despite advances in technology, a comprehensive understanding of intercellular communication, the associated internal signal cascades, and its highly integrated and extremely dynamic nature remain largely mysterious (6). Recently, with the rapid development of single-cell RNA sequencing (scRNA-seq) technologies, the systematic deciphering of the intercellular crosstalk mediated by ligand-receptor (L–R) interactions quickly became a research focus (7–10). For example, analysis of intercellular communication in the alveolar niche revealed functional pathways that mediate the growth and self-renewal of alveolar type 2 progenitor cells, including IL-6/Stat3, Bmp, and Fgf signaling (9). Hepatocyte-derived VEGFA has been found to activate PLVAP in tumor endothelial cells and likely promote oncofetal reprogramming of the tumor immune microenvironment (TIME) (10). In a sense, scRNA-seq technologies have greatly facilitated the development of intercellular communication research and have become an essential bioinformatics pipeline for scRNA-seq processing.
To date, many algorithms have been developed to infer cell-cell communications, such as CellPhoneDB (11), Nichenet (12), SingleCellSignalR (13), NATMI (14) CellChat (15) and RNA-Magnet (16). Most of these methods infer intercellular communication based only on the expression intensity and/or specificity of L–R pairs, such as their coexpression (sum, mean or product) (11,17), differential expression (18,19) and expression correlation (20). In addition, some of these methods introduced thresholds for the inferred communication scores by evaluating the specificity of paired L–R, with methodologies such as differential expressed analysis of ligands/receptors and nonparametric tests (permutation test and rank-sum test) (11,15,18). However, there are certain limitations for inferring intercellular communication based on the expression intensity/specificity of paired L–R. First, the expression of some receptors is not closely correlated with cell-cell communication because they usually exhibit a constant/stable expression level in cells (21), and mRNAs encoding some surface receptors usually show low abundance, which likely leads to these receptors not being detected at the single-cell level (16,22). More importantly, cell-cell communication includes not only intercellular signaling but also the intracellular transmission and amplification of the signal through specific signaling pathways, generally culminating in altered activity of downstream transcription factors (TFs) and gene regulatory networks (GRNs) (12,23). To address these problems, several methods have taken intracellular signaling into account, including Nichenet (12) and SoptSC (24). Nichenet was applied to predict ligand-target gene (TG) links between cells by combining their expression data with prior knowledge on extracellular signaling and downstream GRNs (22). The integrated network (including L–R interactions, intracellular signaling and gene regulatory interactions) embedded in Nichenet was collected from multiple sources with patchy data quality (some interactions were predicted). Nichenet could not provide definitive lines of communication connecting the outside and inside of the cells. SoptSC predicts intercellular communication by combining the expression of L–R interactions and the genes in its downstream signal transduction pathway but does not collect intracellular signaling pathway data.
Here, to infer the pathways connecting the inside and outside of the cells, we developed CellCall, a toolkit to infer intercellular communication networks and internal regulatory signals by integrating intracellular and intercellular signaling. (i) CellCall collects ligand–receptor–transcript factor (L–R–TF) axis datasets based on KEGG pathways. (ii) According to prior knowledge of L–R–TF interactions, CellCall infers intercellular communication by combining the expression of ligands/receptors and downstream TF activities for certain L–R pairs. (iii) CellCall embeds a pathway activity analysis method to identify the crucial pathways involved in communications between certain cell types. (iv) CellCall offers a rich suite of visualization options (Circos plot, Sankey plot, bubble plot, ridge plot, etc.) to intuitively present the analysis results. In addition, we demonstrated the overall features of CellCall by applying it to case studies on scRNA-seq datasets of human testicular cells and TIME, evaluated the reliability and functionality of CellCall predictions by immunofluorescence assays, and performed comparison analysis with other tools.
MATERIALS AND METHODS
Collection of L–R–TF axis and TF–TG interaction data
The L–R–TF axis dataset was extracted from the KEGG pathway analysis using the following steps. (1) We collected human L–R interactions from the NATMI (14), Cellinker (25), CellTalkDB (26), CellChat (15) and STRING v11 databases (only literature or experimental supported data were collected) (27); ligand/receptor complexes were included among the L–R interactions. (2) We extracted the TFs downstream of the L–R interaction from the KEGG pathway. Only the L–R interaction and downstream TFs in the same branch of a given pathway were identified as an L–R–TF axis. A total of 19 144 human L–R–TF axes were obtained.
Human TF–TG interactions were collected from TRANSFAC (28), JASPAR (29), RegNetwork (30), Pathway Commons (31), TRRUST (32), Ontogenet (33), ReMap (34), EVEX (35), HTRIDB (36), CHEA (37), ENCODE (38) and MOTIFMAP (39). A total of 587,248 human experimental supported interactions were collected. Moreover, a total of 12 069 mouse L–R–TF axes and 554 207 TF–TG interactions were obtained by the orthology majority-voting scheme described in Cellinker (25).
Inferring intercellular communication
To infer the cell-cell communications between different cell types, Sk is defined as the communication score of an L–R interaction k between cell types i and j, which is evaluated by integrating the L2 norm of the L–R interaction 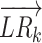 and the activity score of the downstream TF TFk. The formula is as follows:
and the activity score of the downstream TF TFk. The formula is as follows:
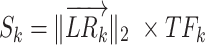 |
(1) |
where TFk is the activity score of the TF downstream of L–R interaction k. 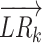 is a two-dimensional vector represented by the normalized expression value (normalized by the softmax function) of the ligand and receptor for L–R interaction k:
is a two-dimensional vector represented by the normalized expression value (normalized by the softmax function) of the ligand and receptor for L–R interaction k:
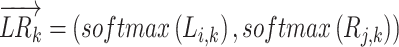 |
(2) |
where Li,k is the mean expression value of the ligand in cell type i and Rj,k is the mean expression value of the receptor in cell j. In addition, to prevent the influence of dropout of the scRNA-seq data, users can also choose the quantile expression value (25%, 50% and 75%) of the ligand/receptor to represent Li,k and Rj,k.
If the ligand is a complex containing n subunits, L is defined as the geometric mean of the expression value of all subunits:
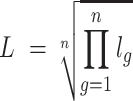 |
(3) |
where lg is the expression value of subunit g in the ligand complex.
Similar to the case for ligands, if a receptor is a complex containing n subunits, R is defined as the geometric mean of the expression value of all subunits:
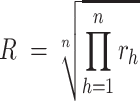 |
(4) |
where rh is the expression value of subunit h in the receptor complex.
The TF activity score TFk was evaluated according to the expression of the TF regulon. According to SCENIC (40), a regulon is defined as the set of TGs of a TF that are coexpressed with the TF in the single-cell expression profile. The formula is as follows:
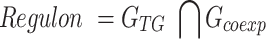 |
(5) |
where GTG is the gene set of all TGs for a TF, and Gcoexp is the gene set of all coexpressed genes of a TF. Gene coexpression was calculated by Spearman's rank correlation coefficient (P < 0.05, |R| > 0.1).
Then, TFk is the gene set enrichment analysis (GSEA) enrichment score (ES) (41) for the regulon. The formula is as follows:
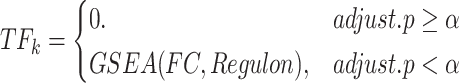 |
(6) |
where FC is the fold change (receiver cells / other cells) of all TGs in Regulon, and adjust.p is the significance level of GSEA. If adjust.p is lower than the threshold 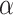 (default as 0.05), then TFk is equal to the ES of GSEA; otherwise, TFk is equal to 0.
(default as 0.05), then TFk is equal to the ES of GSEA; otherwise, TFk is equal to 0.
If there are n TFs (n > 1) downstream of L–R interaction k, the activity score TFk is defined as the weighted sum of all TFs. The formula is as follows:
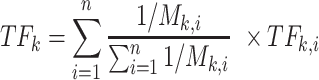 |
(7) |
where M is the shortest step from TFk,i to receptor k in a pathway.
Pathway activity analysis
CellCall embeds a pathway activity analysis method to help explore the main pathways involved in communication between certain cells. First, CellCall quantifies the activity of pathway i based on the Jaccard similarity coefficient. The formula of the pathway activity score nPASi is as follows:
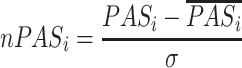 |
(8) |
where nPASi is the z-score normalized PASi, and the PASi was calculated as follows:
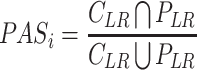 |
(9) |
where CLR is the L–R interaction between certain cell types inferred by intercellular communication analysis. PLR is the L–R interaction in the pathway.
CellCall also estimated the significance of pathway activity by hypergeometric testing. The formula is as follows:
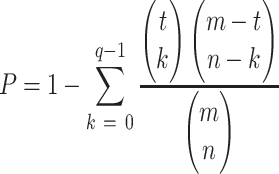 |
(10) |
where m is the number of all L–R interactions and t is the number of L–R interactions inferred by intercellular communication analysis between two cell types. n is the number of L–R interactions in a pathway. q is the overlap of t and n.
Data collection and processing of scRNA-seq datasets
The scRNA-seq data of 2532 human testicular cells were collected from our previous study (GSE106487) (Supplementary Table S1) (42), and the expression levels were normalized by log2[TPM/10 + 1] (transcripts per million, TPM). The 10 processed TIME scRNA-seq datasets were collected from the TISCH database (Supplementary Table S2) (43). A standardized analysis workflow based on MAESTRO v1.1.0 (44) was applied for quality control, batch effect removal, cell clustering and cell type annotation based on the expression matrix, with the expression in each cell scaled to 10 000.
Immunofluorescence of the human testicular cells
Human seminiferous tubules were fixed with 4% paraformaldehyde and stored in 70% ethanol. Then, fixed samples were embedded in paraffin and sliced into 5-μm sections by microtome (Leica RM2235). Immunostaining was performed as described previously (42). Deparaffined and rehydrated sections were washed in PBS for 5 min after antigen retrieval and then blocked with 5% BSA at room temperature for 1 h. Subsequently, the sections were incubated with primary antibodies at 4°C for more than 8 hours and secondary antibodies (Jackson ImmunoResearch, Alexa 488-, Alexa 594-) for 1 h at room temperature. Nuclei were counterstained with 10 μg/ml Hoechst 33342 for 15 min at room temperature. All images were captured with a ZEISS LSM880 confocal microscope. The experiments performed in this study were approved by the Third Affiliated Hospital of Guangzhou Medical University (2017-055). And informed consents to every donor in our study have been signed.
The following primary antibodies were used: mouse anti-γH2AX (Abcam, ab26350), mouse anti-FGFR3 (Santa Cruz, sc-13121), mouse anti-BMPR1B (Abcam, ab1565836), mouse anti-DDX4 (Abcam, ab27591), rabbit anti-INHBB (Abcam, ab69286), mouse anti-ACVR2B (R&D Systems, MAB3392), rabbit anti-pSMAD2 (Cell Signaling Technology, #18338), rabbit anti-GDF5 (Abcam, ab93855), rabbit anti-pSMAD1 (Abcam, ab226821) and rabbit anti-ACVR1B (Abcam, ab109300).
Comparison of CellCall with CellPhoneDB, CellChat, iTALK and SingleCellSignalR
All the methods were applied to infer intercellular communications using their own default parameters. An scRNA-seq dataset of human testicular cells was adopted as the testing data (from STs to SSCs). Meanwhile, the 1,141 L–R interactions from the dataset embedded in CellCall were used for the other four methods. The Wilcoxon rank-sum test was used to detect the differentially expressed genes analysis for iTALK (P < 0.05, |log2(FC)| > 1). The rules of literature curation are as follows: (i) the ligand and/or receptor has been mentioned to be expressed in testicular cells; (ii) the receptor has been reported to be located in SSCs and (iii) the L–R interaction has been reported to be related to spermatogenesis.
Statistical analysis
Spearman correlation tests, survival analyses and Fisher's exact tests were performed in R. The correlations between TFs and target genes were evaluated using Spearman correlation analysis in the R package ‘psych’. Kaplan–Meier, log-rank test and univariate Cox regression in the R package ‘survival’ were used to assess the relationships between TF expression and survival time. Hypergeometric tests and Fisher's exact tests were used in the R package ‘stats’, enrichment analysis was used in the R package ‘clusterProfiler’. The receiver operating characteristic (ROC) curve was used in the R package ‘pROC’.
RESULTS
Overview of CellCall
The core algorithm of CellCall and the underlying intercellular communication model are shown in Figure 1. Basically, the schematic diagram and biological model of intercellular communication can be described as follows: cell signals are transmitted from sender cells to receiver cells through intercellular L–R interactions, and then the signals are transduced to the receiver cell interior through a specific signaling pathway, generally culminating in changes in the activity of downstream TFs and GRNs (Figure 1A and B). According to this biological model, we built a statistical model for intercellular communication consisting of two parts, an L–R pair (intercellular signaling) and a regulon (intracellular signaling) (Figure 1C). The L–R pair was defined as a two-dimensional vector represented by the expression value of the ligand and receptor. The regulon was defined as the set of TGs for a TF that were co-expressed with the TF. Then, the cell-cell communication score of an L–R pair is calculated by integrating intercellular signaling (expression of ligand and receptor) and intracellular signaling (activity score of downstream TFs). Notably, the activity status (active or inactive) and score of downstream regulons were estimated by gene set enrichment analysis (GSEA). When multiple regulons could be activated by one L–R interaction, the activity score was defined as the weighted sum of all activated regulons. Ultimately, CellCall was able to not only quantify intercellular communication for certain L–R pairs but also infer its internal regulatory signaling as reflected by TF activity. In addition, CellCall embeds a pathway activity analysis method to help explore the main pathways involved in intercellular crosstalk among certain cells (see method for details) (Figure 1D).
Figure 1.

Overview of CellCall. (A) The schematic diagram of intercellular communication. (B) The biological model of intercellular communication. (C) The statistical model of intercellular communication. (D) CellCall provides a variety of functions including intercellular communication analysis and pathway activity analysis and offers a rich suite of visualization tools to intuitively present the results of the analysis, including heatmap, Circos plot, bubble plot, Sankey plot, TF enrichment plot and ridge plot.
The accuracy of the prior knowledge of L–R interactions and downstream TFs is crucial for inferring meaningful intercellular communications (15). Therefore, we extracted the data for the L–R–TF axis construction from the KEGG database, and only the L–R interactions and downstream TFs in the same branch of a pathway were identified as an L–R–TF axis. Experimentally supported TF–TG interactions were collected from multiple databases. The software package CellCall was implemented in R. In addition to intercellular communication analysis and pathway activity analysis, CellCall also offers a rich suite of visualization tools to intuitively present the results of the analysis, including heatmap, Circos plot, bubble plot, Sankey plot, TF enrichment plot and ridge plot (see Figure 1D). The usage of these visualization kits is demonstrated in the case studies as follows.
Inferring cell-cell communication between the human spermatogenic niche and germ cells
The testicular niche plays important roles in spermatogenesis through a complex intercellular signal transduction cascade (45,46). Hence, we applied CellCall to an scRNA-seq dataset of human testicular cells from our previous study (42) (see Figure 2A and Supplementary Table S1). As Sertoli (ST) cells are the only somatic cells located in the seminiferous tubule that could support the development of germ cells and act as a spermatogenic niche (47,48), we analyzed intercellular signaling from Sertoli cells to 14 other types of germ cells, including spermatogonial stem cells (SSCs), differentiating spermatogonia (Diff.ing SPG), differentiated spermatogonia (Diff.ed SPG), three consecutive stages of leptotene spermatocytes (L1, L2 and L3), zygotene (Z), pachytene (P), diplotene (D), spermatocyte 7 (SPC7) and spermatids at four stages (S1, S2, S3 and S4). As seen in Figure 2B and Supplementary Figure S1, SSCs were the prominent receiver of signals from Sertoli cells compared to other germ cell types. Pathway activity analysis showed that intercellular signaling from Sertoli cells to SSCs was enriched mainly in the Notch signaling pathway, Hippo signaling pathway, MAPK signaling pathway, PI3K-Akt signaling pathway and human cytomegalovirus infection pathway (Figure 2C). These pathways have been reported to be critical for spermatogenesis (49–51).
Figure 2.

Case study of the application of CellCall on human testicular cells. (A) UMAP of 2532 human testicular cells. (B) Circos plot of intercellular communication from Sertoli cells to other germ cells. (C) Pathway activity analysis of intercellular communications from Sertoli cells to other germ cells. (D) The intercellular communications from Sertoli cells to other germ cells (normalized score greater than 0.5). (E) Sankey plot of intercellular communications from Sertoli cells to SSCs.
A total of 47 intercellular communication pathways from Sertoli cells to SSCs were identified (Figure 2D and Supplementary Figure S1), most of which (41/47, see Supplementary Table S3 for details) have been implicated in spermatogenesis or SSC differentiation. For instance, FGF1-FGFR1/3 signaling is required for the maintenance of SSCs (52–55). BMP8B-BMPR/1A/1B/2 signaling could regulate the proliferation and differentiation of spermatogonia by activating SMAD1/2/3/5/8 (56,57). Activin signaling (INHBB-ACVR2A/1B/2B) can influence germ cell development through the phosphorylation of SMAD2/3(58). EphB/ephrin-B signaling (EFNB1/2-EPHB4/6) may modulate spermatogenesis and spermiation (59). Further analysis of the TFs downstream of these intercellular communications revealed that most of these TFs were related to spermatogenesis (see Figure 2E). For example, HES1, RBPJ and SMAD1/2 are intracellular effectors of the Notch signaling pathway, which plays critical roles in SSC proliferation and differentiation (60,61). FOXOs are crucial effectors of the PI3K/Akt signaling pathway in SSCs and are required for both SSC homeostasis and the initiation of spermatogenesis (62). Enrichment analysis of these TFs indicated that all TFs were obviously activated (see Figure 3A), and most fold change (FC) values of TGs were larger than 1 (Figure 3B).
Figure 3.

Enrichment analysis of downstream TFs and immunofluorescence of the INHBB-ACVR2A/B-pSMAD2 axes. (A) Enrichment analysis of the six top TFs in SSCs. (B) Ridge plot of the density distribution of FC of TGs for the six TFs. (C) Dual immunofluorescence of VASA (red) and INHBB (green) in adult human testicular paraffin sections. (D) Dual immunofluorescence of ACVR2B (red) and INHBB (green) in adult human testicular paraffin sections. (E) Dual immunofluorescence of ACVR2B (red) and pSMAD2 (green) in adult human testicular paraffin sections. (F) Dual immunofluorescence of FGFR3 (red) and ACVR1B (green) in adult human testicular paraffin sections. Triangles and circles indicate SSCs, and arrows indicate Sertoli cells. The scale bars represent 10 μm.
To confirm the intercellular communications inferred by CellCall, an immunostaining assay was performed to colocalize the expression of the INHBB-ACVR2A/B-SMAD2 axes between STs and SSCs. The results showed that ACVR2B (red) costained with INHBB around FGFR3 + SSCs in adult human testicular paraffin sections (Figure 3C, D and Supplementary Figure S2), and ACVR2B costained with phosphorylated SMAD2 (pSMAD2) in FGFR3 + SSCs (Figure 3E). In addition, ACVR1B was also identified to be expressed in FGFR3 + SSCs (Figure 3F).
Inferring cell-cell communication among germ cells
Previous studies have mainly focused on crosstalk between the spermatogenic niche and germ cells. However, recent studies have implied that intercellular communication among different germ cells also plays roles in spermatogenesis and deserves further investigation (63). Hence, we applied CellCall to infer the candidate intercellular communications between SSCs and other differential germ cells. As shown in Figure 4A, B and Supplementary Figure S1, intercellular communication from P to SSCs may play critical roles in crosstalk between SSCs and other differential germ cells. As shown in the Sankey plot, TFs downstream of the intercellular communication from P to SSCs, such as HES1, SMAD1/9, TCF7 and ID4, have also been reported to be involved in spermatogenesis (60,64) (see Figure 4C). We also confirmed a communication axis from P to SSCs (GDF5-BMPR1B-SMAD1) by immunostaining (Figure 4D). The results showed that BMPR1B+ (receptor) and pSMAD1 + SSCs costained with GDF5+ (ligand) spermatocytes in adult human testicular sections (see Figure 4E). GDF5-BMPR1B signaling has been reported to play important roles in chondrogenesis and osteogenesis (65). The potential roles and mechanisms of this novel communication in spermatogenesis deserve further elucidation.
Figure 4.

Analysis of intercellular communication from other germ cells to SSCs. (A) Circos plot of intercellular communication from other germ cells to SSCs. (B) The intercellular communications from other germ cells to SSCs (normalized score greater than 0.5). (C) Sankey plot of intercellular communications from P to SSCs. (D) The expression of GDF5, BMPR1B and SMAD1 across different cell types. (E) Dual immunofluorescence of γH2AX (red) and GDF5 (green) in adult human testicular paraffin sections. Dual immunofluorescence of BMPR1B (red) and GDF5 or pSMAD1 (green) in adult human testicular paraffin sections. Yellow triangles indicate P, white triangles and circles indicate SSCs. The scale bars represent 10 μm.
Inferring intercellular communication among immune cells in the TIME
Increasing studies have shown that intercellular crosstalk between immune cells in the tumor niche is involved in the linkage of inflammation, immunity and tumorigenesis, which is crucial to tumor development (66). In this study, we applied CellCall to 10 TIME scRNA-seq datasets (see Supplementary Table S2), which comprised 6 types of cancer, including two liver hepatocellular carcinoma (LIHC) datasets, one non-Hodgkin lymphoma (NHL) dataset, two nonsmall cell lung cancer (NSCLC) datasets, one kidney renal clear cell carcinoma (KRIC) dataset, three colorectal cancer (CRC) datasets and one breast invasive carcinoma (BRCA) dataset. All datasets included both tumor and normal samples. First, intercellular communication among six immune cell types, namely, B cells (B), conventional CD4 T cells (CD4Tconv), CD8 T cells (CD8 T), exhausted CD8 T cells (CD8Tex), monocytes/macrophages (Mono/Macro) and natural killer (NK) cells, was analyzed by CellCall. As shown in Figure 5A and Supplementary Figure S3, compared to other cell types, Mono/Macro cells received significantly more signals from other immune cells across all datasets, indicating the dominant role of Mono/Macro in the intercellular crosstalk of immune cells in the TIME (67,68). Then, to investigate the difference in intercellular communication between normal and tumor tissues, we identified tumor-specific intercellular communications (i.e. those occurring only in tumor samples) for all datasets. Seven common tumor-specific intercellular communications identified in more than four datasets were obtained (see Figure 5B), all of which mainly involved intercellular communication from other cells to Mono/Macro, including CCL3/4/5-CCR1/5 and TNF–TNFRSF1B signaling (see Figure 5C). Many studies have revealed that C–C motif chemokines (CCL3/4/5) secreted in the TIME play important roles in monocyte/macrophage differentiation, activation, polarization and recruitment by binding specific C–-C motif chemokine receptors (CCR1/5) (69). TNF–TNFRSF1B signaling has been reported to be a central negative regulator of M2 tumor-associated macrophages (70).
Figure 5.

Case study of the application of CellCall in the TIME. (A) Circos plot of intercellular communication among immune cells in the NSCLC dataset (GSE139555). (B) Common tumor-specific intercellular communications in 10 TIME datasets. (C) Intercellular crosstalk from other immune cells to Mono/Macro cells in the TIME. (D) Sankey plot of seven common tumor-specific intercellular communications and downstream TFs. (E) Association of TF expression with patient survival in the TCGA pan-cancer data. (F) Kaplan–Meier curves of selected TFs. Statistical significance and hazard ratios were calculated using multivariate Cox regression.
Moreover, we also investigated the TFs activated downstream of these communications. Most of the activated TFs involved in cancer progression and the TIME (Figure 5D) (71,72), such as the NFκB family (NFKB1, NFKBIA, NFKBIB and RELA) and STAT family (STAT1, STAT2, STAT3 and STAT5B) occupy central roles in M1 and M2 macrophage polarization (71). To further certify the functionality of these TFs in cancer and the capacity of CellCall, we investigated the association of the expression of the top 10 TFs with patient survival using TCGA pancancer data. As shown in Figure 5E and Supplementary Figure S4, all TFs significantly affected the overall survival of patients with different cancers. For example, STAT5B had positive effects on long-term survival in most cancers, such as LIHC and KIRC (Figure 5E and F). However, IKBKB had opposite effects on survival in different cancer types, such as BLCA versus LGG (Figure 5E and F). These results suggested that CellCall is able to effectively infer the crucial intercellular communications of the TIME and identify the underlying intracellular processes affected by intercellular crosstalk.
Comparison of CellCall with other tools
We systematically compare the general features of CellCall with those of nine other tools in three aspects: data, approach and visualization. As shown in Table 1, CellCall, Nichenet and SingleCellSignalR collected intracellular signaling data in addition to L–R interactions. CellCall collected 19 144 L–R–TF axes from KEGG pathway analysis. Nichenet built an integrated network including ligand receptors, intracellular signaling and GRNs from multiple data sources with patchy data quality (some interactions were predicted). Although SingleCellSignalR collected intracellular signaling data from KEGG pathways and Reactome (73), it did not consider intracellular signaling in predicting intercellular communication. Conversely, SoptSC accounted for intracellular signaling but did not include intracellular signaling data. The remaining tools include L–R interactions from known databases and the literature (ranging from 380 to 3251 entries). In addition, CellCall CellPhoneDB, CellChat (15) and ICELLNET (74) contain ligand/receptor complex information, which should not be ignored because many ligands/receptors act as multisubunit complexes (25). CellCall and Nichenet scored intercellular communication based on L/R/TG expression. SoptSC is based on L/R/downstream gene expression, but the remaining tools are based on L/R expression only. Meanwhile, CellCall, CellPhoneDB, CellChat, iTALK (18) and SingleCellSignalR offer thresholds for communication scores. CellPhoneDB and CellChat evaluate the statistical significance of the communication by permutation tests. iTALK identifies positive L–R communication based on the differential expression of ligands/receptors. SingleCellSignalR uses ad hoc benchmarking to generate a customized threshold for different scRNA-seq datasets. In contrast, CellCall filters the intercellular communication scores according to the activity status of the downstream TFs. Moreover, CellCall uses an embedded pathway activity analysis method to help explore the pathways involved in crosstalk between certain cells. In terms of visualization, CellCall provides a rich suite of display options to intuitively present the analysis results, including heatmap, Circos plot, bubble plot, Sankey plot, TF enrichment plot and ridge plot, providing a greater abundance of visualization choices than is available in other tools.
Table 1.
Feature comparison of CellCall with other tools
| CellCall | CellPhoneDB | SingleCellSignalR | CellChat | Nichenet | NATMI | iTALK | RNA-Magnet | SoptSC | ICELLNET | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Data | Data size | 19 144 (L–R–TF) | 1396 | 3251 | 1939 | 12 019 | 2187 | 2649 | 2000 | N | 380 |
| Literature supported L–R interactions | 1141 | 398 | 1483 | 891 | 0 | 2187 | 0 | 1618 | N | 379 | |
| Intracellular signaling | KEGG | N | KEGG/ Reactome | N | multiple sources | N | N | N | N | N | |
| Complex | Y | Y | N | Y | N | N | N | N | N | Y | |
| Approach | Scoring based on expression | L/R/TG | L/R | L/R | L/R | L/R/TG | L/R | L/R | L/R | L/R/ Downstream gene | L/R |
| Score threshold | TF activities | Permutation test | ad hoc benchmark | Permutation test | N | N | Differentially expressed | N | N | N | |
| Pathway activity analysis | Y | N | N | N | N | N | N | N | N | N | |
| Visualization | Heatmap | Y | N | N | Y | Y | Y | N | Y | N | Y |
| Circos plot | Y | N | Y | Y | N | Y | Y | N | Y | N | |
| Bubble plot | Y | Y | N | Y | N | N | N | N | N | Y | |
| Sankey plot | Y | N | N | Y | N | N | N | N | N | N | |
| Ridge plot | Y | N | N | N | N | N | N | N | N | N | |
| TF enrichment plot | Y | N | N | N | N | N | N | N | N | N |
Next, we compared the performance of CellCall with that of four other tools that offered thresholds for communication scores (CellPhoneDB, CellChat, iTALK and SingleCellSignalR) on a dataset of human testicular cells (42). As shown in Figure 6A, CellCall, CellPhoneDB, CellChat, iTALK and SingleCellSignalR identified 47, 54, 42, 63 and 70 intercellular communications from Sertoli cells to SSCs by their own default cut-offs, respectively (see Supplementary Table S3 and Supplementary Materials for details). In the curated literature related to these intercellular communications, over 87% of CellCall-identified intercellular communications (41/47) have been reported to be involved in spermatogenesis (see Figure 6B and Supplementary Table S3). This high literature support rate is superior or comparable to those of CellPhoneDB (81.48%, Fisher's exact test P = 0.396), CellChat (78.57%, Fisher's exact test P = 0.586), iTALK (55.56%, Fisher's exact test P = 3.7e–04) and SingleCellSignalR (31.43%, Fisher's exact test P = 1.5e–09). Then we used ROC curves to compare these methods, as the results shown in Figure 6C, CellCall achieved the highest area under curve of receiver operating characteristic (AUC) (CellCall: 0.731, CellPhoneDB: 0.725, CellChat: 0.680, iTALK: 0.691, SingleCellSignalR: 0.586). And the results of these methods with their own optimal cut-point values (the black dots on the curves, which defined by Youden Index) are shown in Figure 6D. These results suggest that CellCall might infer intercellular communication with greater accuracy than these existing methods.
Figure 6.

Comparison of the performance of CellCall with CellPhoneDB, CellChat, iTALK and SingleCellSignalR on the scRNA-seq dataset of human testicular cells (from ST cells to SSCs). (A) UpSetR plot of results from the five tools with their own default cut-offs. (B) Comparison of the literature support rates of CellCall and four other tools (Fisher's exact test). (C) The ROC curves of these methods. (D) UpSetR plot of results from the five tools with their own optimal cut-point values (the black dots on the curves, which defined by Youden Index).
DISCUSSION
Cells rely on intercellular signal transduction to recognize and respond to cues in their environment (25). This process not only promotes the proper functioning of individual cells but also allows communication and coordination among groups of cells to carry out tasks no single cell could accomplish on its own (75). Hence, investigation of intercellular communications could facilitate understanding of the fundamental basis of biological activities and help to reveal the pathogenic mechanisms of diseases (76,77). Here, we developed CellCall, a toolkit for intercellular communication analysis, by integrating paired L–R and internal TF activities. Case studies on scRNA-seq data of human testicular cells and TIME suggest that CellCall could effectively infer candidate intercellular communications and internal signaling under physiological and pathological conditions. In addition, comparative analysis with other tools indicated that CellCall has superior accuracy and richer functionality.
A case study on human testicular cells indicated prominent signal crosstalk between ST cells and SSCs. Most of the inferred intercellular communications (41/47) from ST cells to SSCs have been implicated in spermatogenesis or SSC differentiation (53,54,56,58,59), and some of them were confirmed by immunofluorescence assays. Intercellular communication analysis also indicated that intercellular crosstalk among germ cells may play roles in spermatogenesis, and a communication axis from P to SSCs (GDF5-BMPR1B-SMAD1) was confirmed by immunostaining. BMPR1B has been reported to receive extracellular signals from BMPs such as BMP4/8A/8B and then regulate the differentiation of SSCs by activating SMAD1/5/8 (78). GDF5 is also a BMP, and GDF5-BMPR1B signaling has been reported to play an important role in chondrogenesis and osteogenesis (65). Therefore, the potential role and mechanism of this communication in spermatogenesis deserves further elucidation. Furthermore, a case study on multiple TIME datasets indicated the dominant role of the Mono/Macro cell population in the intercellular crosstalk of immune cells in the TIME (67,68), and all of the inferred common tumor-specific intercellular communications and internal TFs have been reported to be involved in monocyte/macrophage differentiation, activation, polarization and recruitment (69,70,79). Survival analysis of these TFs using TCGA pancancer data showed that all the identified TFs were significantly associated with the overall survival of patients with different cancers. All these results indicate the reliable and distinct function of CellCall in intercellular communication analysis and internal TF activity exploration.
A systematic comparison of its general features with those of other tools show that CellCall has certain advantages for intercellular communication analysis. CellCall includes high-quality inter- and intracellular signaling data from KEGG pathways and considers ligand/receptor complex information. In terms of approach, CellCall infers intercellular communication by combining ligand/receptor expression and the downstream TF activities for a given L–R pair. The algorithm not only takes into account intracellular signaling but also offers a threshold for intercellular communication scores by evaluating downstream TF activities, which is more reasonable than assessing just the expression intensity and/or specificity of the L–R pair. In addition, CellCall uses an embedded pathway activity analysis method to identify the crucial pathways involved in crosstalk between certain cells. In terms of visualization, CellCall offers the most abundant visualization options among the available tools. Furthermore, a comparative analysis of the performance of CellCall and four other tools on the dataset of human testicular cells suggested that CellCall has superior accuracy in deciphering intercellular communication. All these results indicate that CellCall is a valuable and powerful tool for intercellular communication analysis and internal signal cascade exploration.
Although CellCall has special advantages and characteristics in comparison with other methods, it still has some limitations. First, all existing tools, including CellCall, infer intercellular communication based on scRNA-seq technologies (which contain only the gene expression values in single cells), meaning that CellCall considers only the downstream GRNs (regulation of gene expression) of L–R interactions. However, this type of L–R–TF model is only one part of the full picture of intercellular signal transduction. Some signaling pathways do not fit this model; for example, intracellular signaling related to autophagy involves multiple protein modifications, interactions, and enzyme reactions rather than changes in gene expression. Meanwhile, all the existing tools, including CellCall, are focused on only those L–R pairs in which both partners are proteinaceous and ignore other nonpeptidic molecules (such as small molecules, carbohydrates, lipids and nucleic acid ligands) (80). However, these limitations will be remedied by the appearance of new single-cell technologies for monitoring metabolites and protein modifications in the future.
In this study, we presented CellCall, a toolkit for intercellular communication analysis that integrates intracellular and intercellular signaling. The case study on scRNA-seq of human testicular cells and TIME suggest the reliable and unique function of CellCall in intercellular communication analysis and internal TF activity exploration. Meanwhile, comparative analysis with other tools indicated that CellCall was more accurate and multifunctional and is a useful and powerful method for deciphering intercellular communication. In summary, CellCall provides an elaborate and practical tool enabling researchers to infer intercellular communication and internal signaling based on scRNA-seq data. We believe that this proposed method will promote intercellular communication research and accelerate the development of related algorithms for scRNA-seq data.
DATA AVAILABILITY
The R package of CellCall is freely available at https://github.com/ShellyCoder/cellcall.
Supplementary Material
ACKNOWLEDGEMENTS
We thank professor Zhili Rong, Meng Zhao and Hao Lin for constructive comments of this research. We thank Dr. Xinyan Yang for preprocessing the scRNA-seq data of human testicular cells.
Contributor Information
Yang Zhang, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Tianyuan Liu, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Xuesong Hu, State Key Laboratory of Organ Failure Research, Department of Developmental Biology, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Mei Wang, State Key Laboratory of Organ Failure Research, Department of Developmental Biology, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Jing Wang, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Bohao Zou, Department of Statistics, University of California Davis, Davis, CA, USA.
Puwen Tan, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Tianyu Cui, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Yiying Dou, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Lin Ning, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Yan huang, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Shuan Rao, Department of Thoracic Surgery, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China.
Dong Wang, Department of Bioinformatics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
Xiaoyang Zhao, State Key Laboratory of Organ Failure Research, Department of Developmental Biology, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China; Guangdong Key Laboratory of Construction and Detection in Tissue Engineering, Southern Medical University, Guangzhou 510515, China; Department of Gynecology, Zhujiang Hospital, Southern Medical University, Guangzhou 510280, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Project of China [2019YFA0801800, 2017YFA0105001]; National Natural Science Foundation of China [82070109, 81770104, 62002153]; Guangdong Basic and Applied Basic Research Foundation [2019A1515010784, 2019A1515110701], China Postdoctoral Science Foundation [2020M682623, 2020M682785]; Guangzhou Science and Technology project key project topic [201904020031]. Funding for open access charge: National Key Research and Development Project of China [2019YFA0801800].
Conflict of interest statement. None declared.
REFERENCES
- 1.Douam F., Gaska J.M., Winer B.Y., Ding Q., Schaewen M.v., Ploss A.. Genetic dissection of the host tropism of human-tropic pathogens. Annu. Rev. Genet. 2015; 49:21–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scadden D.T.Nice neighborhood: emerging concepts of the stem cell niche. Cell. 2014; 157:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y., Wang C., Miao Z., Bi X., Wu D., Jin N., Wang L., Wu H., Qian K., Li C.et al.. ViRBase: a resource for virus-host ncRNA-associated interactions. Nucleic Acids Res. 2015; 43:D578–D582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massagué J., Obenauf A.C.. Metastatic colonization by circulating tumour cells. Nature. 2016; 529:298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seliger B.Basis of PD1/PD-L1 therapies. J. Clin. Med. 2019; 8:2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Handly L.N., Yao J., Wollman R.. Signal transduction at the single-cell level: approaches to study the dynamic nature of signaling networks. J. Mol. Biol. 2016; 428:3669–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramilowski J.A., Goldberg T., Harshbarger J., Kloppmann E., Lizio M., Satagopam V.P., Itoh M., Kawaji H., Carninci P., Rost B.et al.. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015; 6:7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong X., Kuang H., Ansari S., Liu T., Gong J., Wang S., Zhao X.Y., Ji Y., Li C., Guo L.et al.. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell. 2019; 75:644–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zepp J.A., Zacharias W.J., Frank D.B., Cavanaugh C.A., Zhou S., Morley M.P., Morrisey E.E.. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell. 2017; 170:1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma A., Seow J.J.W., Dutertre C.A., Pai R., Blériot C., Mishra A., Wong R.M.M., Singh G.S.N., Sudhagar S., Khalilnezhad S.et al.. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in Hepatocellular carcinoma. Cell. 2020; 183:377–394. [DOI] [PubMed] [Google Scholar]
- 11.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R.. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020; 15:1484–1506. [DOI] [PubMed] [Google Scholar]
- 12.Browaeys R., Saelens W., Saeys Y.. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020; 17:159–162. [DOI] [PubMed] [Google Scholar]
- 13.Cabello-Aguilar S., Alame M., Kon-Sun-Tack F., Fau C., Lacroix M., Colinge J.. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020; 48:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou R., Denisenko E., Ong H.T., Ramilowski J.A., Forrest A.R.R.. Predicting cell-to-cell communication networks using NATMI. Nat. Commun. 2020; 11:5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.-H., Myung P., Plikus M.V., Nie Q.. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021; 12:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baccin C., Al-Sabah J., Velten L., Helbling P.M., Grünschläger F., Hernández-Malmierca P., Nombela-Arrieta C., Steinmetz L.M., Trumpp A., Haas S.. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 2020; 22:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramilowski J.A., Goldberg T., Harshbarger J., Kloppmann E., Lizio M., Satagopam V.P., Itoh M., Kawaji H., Carninci P., Rost B.et al.. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun. 2015; 6:7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., Wang R., Zhang S., Song S., Jiang C., Han G., Wang M., Ajani J., Futreal A., Wang L.. iTALK: an R package to characterize and illustrate intercellular communication. 2019; bioRxiv doi:04 January 2019, preprint: not peer reviewed 10.1101/507871. [DOI]
- 19.Cillo A.R., Kürten C.H.L., Tabib T., Qi Z., Onkar S., Wang T., Liu A., Duvvuri U., Kim S., Soose R.J.et al.. Immune landscape of viral- and carcinogen-driven head and neck cancer. Immunity. 2020; 52:183–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen M., Giladi A., Gorki A.-D., Solodkin D.G., Zada M., Hladik A., Miklosi A., Salame T.-M., Halpern K.B., David E.et al.. Lung single-cell signaling interaction map reveals basophil role in macrophage imprinting. Cell. 2018; 175:1031–1044. [DOI] [PubMed] [Google Scholar]
- 21.Zinn K., Özkan E.. Neural immunoglobulin superfamily interaction networks. Curr. Opin. Neurobiol. 2017; 45:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armingol E., Officer A., Harismendy O., Lewis N.E.. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 2021; 22:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Türei D., Valdeolivas A., Gul L., Palacio-Escat N., Klein M., Ivanova O., Ölbei M., Gábor A., Theis F., Módos D.et al.. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Molecular systems biology. 2020; 17:e9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S., Karikomi M., MacLean A.L., Nie Q.. Cell lineage and communication network inference via optimization for single-cell transcriptomics. Nucleic Acids Res. 2019; 47:e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Liu T., Wang J., Zou B., Li L., Yao L., Chen K., Ning L., Wu B., Zhao X.et al.. Cellinker: a platform of ligand–receptor interactions for intercellular communication analysis. Bioinformatics. 2021; 10.1093/bioinformatics/btab036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao X., Liao J., Li C., Lu X., Cheng J., Fan X.. CellTalkDB: a manually curated database of ligand–receptor interactions in humans and mice. Brief. Bioinform. 2020; 22: 10.1093/bib/bbaa269. [DOI] [PubMed] [Google Scholar]
- 27.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P.et al.. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018; 47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K.et al.. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006; 34:D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H.et al.. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014; 42:D142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z.P., Wu C., Miao H., Wu H.. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database. 2015; 2015: 10.1093/database/bav095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerami E.G., Gross B.E., Demir E., Rodchenkov I., Babur O., Anwar N., Schultz N., Bader G.D., Sander C.. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011; 39:D685–D690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han H., Shim H., Shin D., Shim J.E., Ko Y., Shin J., Kim H., Cho A., Kim E., Lee T.et al.. TRRUST: a reference database of human transcriptional regulatory interactions. Sci. Rep. 2015; 5:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jojic V., Shay T., Sylvia K., Zuk O., Sun X., Kang J., Regev A., Koller D., Best A.J., Knell J.et al.. Identification of transcriptional regulators in the mouse immune system. Nat. Immunol. 2013; 14:633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Griffon A., Barbier Q., Dalino J., van Helden J., Spicuglia S., Ballester B.. Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape. Nucleic Acids Res. 2015; 43:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Landeghem S., Hakala K., Rönnqvist S., Salakoski T., Van de Peer Y., Ginter F.. Exploring biomolecular literature with EVEX: connecting genes through events, homology, and indirect associations. Advances in Bioinformatics. 2012; 2012:582765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bovolenta L.A., Acencio M.L., Lemke N.. HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics. 2012; 13:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lachmann A., Xu H., Krishnan J., Berger S.I., Mazloom A.R., Ma’ayan A.. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010; 26:2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The ENCODE (ENCyclopedia Of DNA Elements) project. Science. 2004; 306:636–640. [DOI] [PubMed] [Google Scholar]
- 39.Xie X., Rigor P., Baldi P.. MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics. 2009; 25:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J.et al.. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang M., Liu X., Chang G., Chen Y., An G., Yan L., Gao S., Xu Y., Cui Y., Dong J.et al.. Single-Cell RNA sequencing analysis reveals sequential cell fate transition during human spermatogenesis. Cell Stem Cell. 2018; 23:599–614. [DOI] [PubMed] [Google Scholar]
- 43.Sun D., Wang J., Han Y., Dong X., Ge J., Zheng R., Shi X., Wang B., Li Z., Ren P.et al.. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2020; 49:D1420–D1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C., Sun D., Huang X., Wan C., Li Z., Han Y., Qin Q., Fan J., Qiu X., Xie Y.et al.. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020; 21:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao L., Yao C., Xing X., Jing T., Li P., Zhu Z., Yang C., Zhai J., Tian R., Chen H.et al.. Single-cell analysis of developing and azoospermia human testicles reveals central role of Sertoli cells. Nat. Commun. 2020; 11:5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shami A.N., Zheng X., Munyoki S.K., Ma Q., Manske G.L., Green C.D., Sukhwani M., Orwig K.E., Li J.Z., Hammoud S.S.. Single-cell RNA sequencing of human, Macaque, and mouse testes uncovers conserved and divergent features of mammalian spermatogenesis. Dev. Cell. 2020; 54:529–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shinohara T., Orwig K.E., Avarbock M.R., Brinster R.L.. Restoration of spermatogenesis in infertile mice by sertoli cell transplantation1. Biol. Reprod. 2003; 68:1064–1071. [DOI] [PubMed] [Google Scholar]
- 48.Savvulidi F., Ptacek M., Savvulidi Vargova K., Stadnik L.. Manipulation of spermatogonial stem cells in livestock species. J. Anim. Sci. Biotechnol. 2019; 10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao X., Mruk D.D., Cheng C.Y.. Intercellular adhesion molecules (ICAMs) and spermatogenesis. Hum. Reprod. Update. 2013; 19:167–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dirami G., Ravindranath N., Achi M.V., Dym M.. Expression of Notch pathway components in spermatogonia and Sertoli cells of neonatal mice. J. Androl. 2001; 22:944–952. [DOI] [PubMed] [Google Scholar]
- 51.Ni F.-D., Hao S.-L., Yang W.-X.. Multiple signaling pathways in Sertoli cells: recent findings in spermatogenesis. Cell Death. Dis. 2019; 10:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Kopylow K., Staege H., Schulze W., Will H., Kirchhoff C.. Fibroblast growth factor receptor 3 is highly expressed in rarely dividing human type A spermatogonia. Histochem. Cell Biol. 2012; 138:759–772. [DOI] [PubMed] [Google Scholar]
- 53.Winge S.B., Nielsen J., Jørgensen A., Owczarek S., Ewen K.A., Nielsen J.E., Juul A., Berezin V., Rajpert-De Meyts E.. Biglycan is a novel binding partner of fibroblast growth factor receptor 3c (FGFR3c) in the human testis. Mol. Cell. Endocrinol. 2015; 399:235–243. [DOI] [PubMed] [Google Scholar]
- 54.Chen L., Li X., Wang Y., Song T., Li H., Xie L., Li L., Chen X., Ma L., Chen Y.et al.. Fibroblast growth factor 1 promotes rat stem leydig cell development. Front. Endocrinol. 2019; 10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cancilla B., Risbridger G.P.. Differential localization of fibroblast growth factor receptor-1, -2, -3, and -4 in fetal, immature, and adult rat testes. Biol. Reprod. 1998; 58:1138–1145. [DOI] [PubMed] [Google Scholar]
- 56.Wu F.-J., Lin T.-Y., Sung L.-Y., Chang W.-F., Wu P.-C., Luo C.-W.. BMP8A sustains spermatogenesis by activating both SMAD1/5/8 and SMAD2/3 in spermatogonia. Sci. Signal. 2017; 10:eaal1910. [DOI] [PubMed] [Google Scholar]
- 57.Lochab A.K., Extavour C.G.. Bone Morphogenetic Protein (BMP) signaling in animal reproductive system development and function. Dev. Biol. 2017; 427:258–269. [DOI] [PubMed] [Google Scholar]
- 58.Wijayarathna R., de Kretser D.M.. Activins in reproductive biology and beyond. Hum. Reprod. Update. 2016; 22:342–357. [DOI] [PubMed] [Google Scholar]
- 59.Gofur M.R., Ogawa K.. Compartments with predominant ephrin-B1 and EphB2/B4 expression are present alternately along the excurrent duct system in the adult mouse testis and epididymis. Andrology. 2019; 7:888–901. [DOI] [PubMed] [Google Scholar]
- 60.Garcia T.X., Farmaha J.K., Kow S., Hofmann M.C.. RBPJ in mouse Sertoli cells is required for proper regulation of the testis stem cell niche. Development. 2014; 141:4468–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcia T.X., Parekh P., Gandhi P., Sinha K., Hofmann M.C.. The NOTCH ligand JAG1 regulates GDNF expression in Sertoli cells. Stem Cells Dev. 2017; 26:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goertz M.J., Wu Z., Gallardo T.D., Hamra F.K., Castrillon D.H.. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J. Clin. Invest. 2011; 121:3456–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Endo T., Freinkman E., de Rooij D.G., Page D.C.. Periodic production of retinoic acid by meiotic and somatic cells coordinates four transitions in mouse spermatogenesis. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E10132–E10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Helsel A.R., Yang Q.-E., Oatley M.J., Lord T., Sablitzky F., Oatley J.M.. ID4 levels dictate the stem cell state in mouse spermatogonia. Development. 2017; 144:624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mang T., Kleinschmidt-Doerr K., Ploeger F., Schoenemann A., Lindemann S., Gigout A.. BMPR1A is necessary for chondrogenesis and osteogenesis, whereas BMPR1B prevents hypertrophic differentiation. J. Cell Sci. 2020; 133:jcs246934. [DOI] [PubMed] [Google Scholar]
- 66.Brücher B.L.D.M., Jamall I.S.. Cell-cell communication in the tumor microenvironment, carcinogenesis, and anticancer treatment. Cell. Physiol. Biochem. 2014; 34:213–243. [DOI] [PubMed] [Google Scholar]
- 67.DeNardo D.G., Ruffell B.. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019; 19:369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang J., Li D., Cang H., Guo B.. Crosstalk between cancer and immune cells: role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019; 8:4709–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M.. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004; 25:677–686. [DOI] [PubMed] [Google Scholar]
- 70.Kratochvill F., Neale G., Haverkamp J.M., Van de Velde L.A., Smith A.M., Kawauchi D., McEvoy J., Roussel M.F., Dyer M.A., Qualls J.E.et al.. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015; 12:1902–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Genard G., Lucas S., Michiels C.. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front. Immunol. 2017; 8:828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li L., Han L., Sun F., Zhou J., Ohaegbulam K.C., Tang X., Zang X., Steinbrecher K.A., Qu Z., Xiao G.. NF-κB RelA renders tumor-associated macrophages resistant to and capable of directly suppressing CD8(+) T cells for tumor promotion. Oncoimmunology. 2018; 7:e1435250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R.et al.. The reactome pathway knowledgebase. Nucleic Acids Res. 2020; 48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noël F., Massenet-Regad L., Carmi-Levy I., Cappuccio A., Grandclaudon M., Trichot C., Kieffer Y., Mechta-Grigoriou F., Soumelis V.. ICELLNET: a transcriptome-based framework to dissect intercellular communication. Nature communications. 2020; 12:1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wojtowicz W.M., Vielmetter J., Fernandes R.A., Siepe D.H., Eastman C.L., Chisholm G.B., Cox S., Klock H., Anderson P.W., Rue S.M.et al.. A human IgSF cell-surface interactome reveals a complex network of protein-protein interactions. Cell. 2020; 182:1027–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumar M.P., Du J., Lagoudas G., Jiao Y., Sawyer A., Drummond D.C., Lauffenburger D.A., Raue A.. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018; 25:1458–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang J., Guan M., Wang Q., Zhang J., Zhou T., Sun X.. Single-cell transcriptome-based multilayer network biomarker for predicting prognosis and therapeutic response of gliomas. Brief. Bioinform. 2020; 21:1080–1097. [DOI] [PubMed] [Google Scholar]
- 78.Li Y., Zhang Y., Zhang X., Sun J., Hao J.. BMP4/Smad signaling pathway induces the differentiation of mouse spermatogonial stem cells via upregulation of Sohlh2. Anat. Rec. 2014; 297:749–757. [DOI] [PubMed] [Google Scholar]
- 79.Rückerl D., Seoane P.I.. The M2 triangle: gp130 binding cytokines drive macrophages to promote tumor growth. Immunol. Cell Biol. 2018; 96:243–245. [DOI] [PubMed] [Google Scholar]
- 80.Armstrong J.F., Faccenda E., Harding S.D., Pawson A.J., Southan C., Sharman J.L., Campo B., Cavanagh D.R., Alexander S.P.H., Davenport A.P.et al.. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 2020; 48:D1006–D1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R package of CellCall is freely available at https://github.com/ShellyCoder/cellcall.