


Abstract
Dysfunction of Tumour Suppressor Genes (TSGs) is a common feature in carcinogenesis. Epigenetic abnormalities including DNA hypermethylation or aberrant histone modifications in promoter regions have been described for interpreting TSG inactivation. However, in many instances, how TSGs are silenced in tumours are largely unknown. Given that miRNA with low expression in tumours is another recognized signature, we hypothesize that low expression of miRNA may reduce the activity of TSG related enhancers and further lead to inactivation of TSG during cancer development. Here, we reported that low expression of miRNA in cancer as a recognized signature leads to loss of function of TSGs in breast cancer. In 157 paired breast cancer and adjacent normal samples, tumour suppressor gene GPER1 and miR-339 are both downregulated in Luminal A/B and Triple Negative Breast Cancer subtypes. Mechanistic investigations revealed that miR-339 upregulates GPER1 expression in breast cancer cells by switching on the GPER1 enhancer, which can be blocked by enhancer deletion through the CRISPR/Cas9 system. Collectively, our findings reveal novel mechanistic insights into TSG dysfunction in cancer development, and provide evidence that reactivation of TSG by enhancer switching may be a promising alternative strategy for clinical breast cancer treatment.
INTRODUCTION
Tumour suppressor genes (TSGs) refer to genes that protect normal cells from malignant transformation. Since the discovery of the first TSG termed Rb (retinoblastoma susceptibility gene), a myriad of genes has been described as TSGs (1–3). Based on the ‘two-hit’ hypothesis, these TSGs take part in cell-cycle control, apoptosis, DNA damage repair and senescence (4–6). Loss of function or inactivation of TSGs is being recognized as common factors contributing to carcinogenesis in most cancers (7–9).
Despite cancer being generally considered a result of genetic abnormalities, accumulating evidence has indicated that epigenetic aberrations exert deep and ubiquitous functions in carcinogenesis (10–14). The leading epigenetic machinery for gene silencing in cancers involves promoter-specific DNA hypermethylation, abnormal histone modifications and posttranscriptional regulation by non-coding RNAs (15–19). Particularly, while a good deal of evidence demonstrates that DNA hypermethylation leads to TSG silencing in tumour development, it is nonetheless less prevalent than we are led to believe, suggesting that the whole story of TSG dysfunction is far from elucidation. For example, the well-known TSG TP53 is not methylated in head and neck cancer (20), as well as RB1 in breast, gastric, colorectal, and pancreatic cancers (21,22). Repressive histone modifications like H3K9me3 and H3K27me3 can only partially account for TSG inactivation, such as DAXX (23), KLF6 (24), EZH2 (25) and KDM6A (26). Since the regulation of eukaryotic gene expression is a knowingly complicated process (27), other uncovered players must exist behind TSG inactivation or loss of function. Given that enhancers are crucial cis-regulatory elements far superior in number compared to promoters in the mammalian genome (28–30), it is of great interest to investigate whether enhancer switching might play a role in TSG dysfunction in cancer.
Enhancers, with the length of about 50–1500nt, typically serve as transcription factor binding platforms to increase gene transcription levels independent of their orientation and locations (29,31). Histone markers such as H3K27ac and H3K4me1 are applied to identify and classify enhancer regions. More specifically, enhancers enriched with H3K4me1 are only taken as poised enhancers, whereas H3K27ac can be detected for ‘active’ enhancer to be distinguished from poised enhancers (32,33). However, the process in which enhancer elements are triggered to become active is still unclear. While analysing the regulatory function of enhancers, we surprisingly discovered that there is an ocean of miRNA genome loci overlapped within the active enhancer regions, that is, these annotated human miRNA precursor loci are within the peaks of H3K27ac modification. We termed these enhancer-overlapped miRNAs as NamiRNAs (Nuclear activating miRNAs) due to their function as an enhancer trigger. We further proposed a NamiRNA−enhancer−target gene activation network to strengthen our understanding of the crosstalk between NamiRNAs and enhancers in regulating gene transcription (34–36). Herein, the expression levels of NamiRNAs can positively endow their corresponding enhancers with an active or inactive status, like an on/off switch, thus up-regulating or down-regulating targeted genes (35). Later, other groups also found the interactions between these two elements, and discovered that the processing of these special miRNAs is controlled by enhancer constituents, all in support of our previous results (37). However, the lack of evidence validating how enhancers and miRNAs interplay in regulating TSG transcription prompted us to explore the underlying crosstalk between enhancers and miRNAs for loss of function of TSGs in cancers.
Breast cancer, a heterogeneous cancer with diversely clinical and morphological characteristics, is the leading cause of cancer-related death in women worldwide (38). According to the presence or absence of molecular markers for oestrogen or progesterone receptors (ER or PR) and human epidermal growth factor 2 (ERBB2; formerly HER2), it is classically categorized into four major subtypes: Luminal A, Luminal B (oestrogen and progesterone receptor-positive, about 70% of patients), HER2+ (HER2-overexpressing, about 15%∼20%), and triple-negative (tumours lacking all three standard molecular markers; about 15%) (39). It has been reported that hundreds of tumour suppressor genes are downregulated in breast cancer (40,41). For instance, the expression of GPER1 is lower in breast cancer patients compared to normal tissues, confirming that GPER1 establishes a pattern of tumour suppressor (42,43), which can inhibit breast cancer growth and proliferation (44,45). Similar results are found in ovarian cancer (46). However, the means by which GPER1 becomes downregulated in breast cancer or interplays with enhancer and NamiRNA needs further investigation.
Herein, we discovered that enhancer marker H3K27ac establishes a distinct pattern between breast cancer and normal cells for most of the tumour suppressor genes, but no obvious discrepancy of DNA methylation status in promoters, implying that DNA methylation is not a key player generally in breast cancer development. Meanwhile, we noticed that there are many annotated miRNAs overlapping with enhancer regions as NamiRNAs, many of which show lower expression and feature as tumour suppressors. Through bioinformatics analysis, we screened out miR-339 as a potential NamiRNA in breast cancer and GPER1 as one of the target genes regulated by miR-339. Interestingly, both miR-339 and GPER1 are down-regulated in Luminal A, Luminal B, and TNBC breast cancer subtypes compared to HER2-overexpressing subtype, and miR-339 could inhibit the proliferation and growth of breast cancer LM2-4175 and T47D cells, supporting that miR-339 could be used for breast cancer re-classification and a potential target for treatment. We further demonstrated that miR-339 up-regulated GPER1 by targeting the corresponding enhancer and relies on the integrity of the enhancer element. Finally, we validated miR-339 as a potential suppressor of breast tumour growth in mouse xenograft models by targeting GPER1. Our findings shed light on understanding enhancer-mediated TSGs loss of function in breast cancer development, in which NamiRNAs serve as a key regulator, and showcase miR-339 as a promising inhibitor for the clinical treatment of breast cancer.
MATERIALS AND METHODS
Cell lines and plasmids
Human breast cancer cell lines T47D (Luminal A subtype of breast cancer), MDA-MB-231 derived LM2-4175 (Triple Negative subtype of breast cancer) and human embryonic kidney HEK293T (HEK293T) cells were routinely maintained in Dulbecco's modified Eagle's medium (DMEM, HyClone) in a humidified atmosphere of 5% CO2 at 37°C, with routine detection of morphology and mycoplasma. The culture was supplemented with 10% fetal bovine serum (HyClone) and 1% penicillin/streptomycin (HyClone). All cells were expanded less than 6 months after resuscitation. MDA-MB-231–derived LM2-4175 cells were kindly provided by Guohong Hu (University of Chinese Academy of Sciences, Shanghai, China).
Fragments of pre-miR-339 (94 bp) and pri-miR-339 (336bp) were amplified from genomic DNAs in HEK293T cells and cloned into the vector pSUPER-retro-GFP/Neo and lentiviral vector pCDH-CMV-MCS-EF1-copGFP respectively, to generate miR-339 expression vectors for transient and stable transfection, respectively. The recombinant lentivirus was harvested at 72 h after transfection and then used to infect LM2-4175 and T47D cells. Cells were selected through flow cytometry (BD Biosciences) with strong GFP-positive. Human GPER1 short hairpin RNAs (shRNAs) and related shControl in pGLVH1/GFP + Puro vector were purchased from GenePharma (D02001).
Chemical reagents and antibodies
Inhibitors of miR-339 and the negative control RNA duplex (NC) were purchased from RiboBio (Guangzhou, China). The transfection of plasmids and miRNA inhibitors were performed using the Hieff Trans™ Liposomal Transfection Reagent (Yeasen, China) in line with the manufacturer's instructions.
Tissue samples
Paired breast cancer samples were collected from a cohort of 157 patients with malignant breast cancers who underwent initial surgery or surgical biopsies at Fudan University Shanghai Cancer Center (Shanghai, China) during the year 2018 to 2019. Another 29 ovarian tissue samples (23 ovarian cancer tissues and 6 normal tissues) and 13 endometrial tissues (10 cancer tissues and 3 normal tissues) were obtained from Jinshan Hospital of Fudan University. Studies involving human tumour tissues were carried out under the protocol approved by the Ethics Committee at Fudan University, and after approval by an institutional review obtained informed written consent from all patients. Tissue sections were frozen in liquid nitrogen immediately and stored at −80°C before usage.
RNA extraction, qRT-PCR and absolute qPCR
Total RNA fractions were isolated using TRIzol reagent (Invitrogen, 15 596 018). With the amount of 1 μg, the total RNA was reversely transcribed into cDNA using PrimeScript RT reagent Kit with gDNA Eraser (Takara). RNA purity and concentration were evaluated by using NanoDrop ND-2000 (Thermo Scientific). qRT-PCR was performed on LightCycler 480 II Real-time PCR system (Roche) using a SYBR Green qRT-PCR master mix kit (TIANGEN) according to the manufacturer's procedure. For qRT-PCR assay, standard vector with 2837 bp was diluted to 50 ng/μl, and its copy number was 1.61 × 1010 calculated by formula 6.02 × 1023 × concentration (50 ng/μl) × 10–9 / (2837 × 660). Next, the standard was diluted into four different gradient concentrations to abtain the standard curve. All primer sequences were listed in Supplementary Table S1. The 2−ΔΔCt method was applied to detect the relative expression of each gene.
Western blotting
Cultured cells were lysed in SDS lysis buffer with protease inhibitors (Roche) and centrifuged at 13 000 rpm for 15 min at 4°C. Protein cell lysates were loaded on a 4–12% gradient SDS-polyacrylamide gel (Life technologies) and then transferred onto PVDF membranes (Millipore, Billerica, MA). The immunoblots were probed with a primary antibody against GPER1 (1/2000 dilution), AGO2 (mouse anti-integrin, 1/1000 dilution) and GAPDH in 5% milk, followed by horseradish peroxide (HRP)-conjugated donkey anti-rabbit or anti-mouse secondary antibodies (1/2000 dilution; Amersham), and immunostaining was detected with an enhanced chemiluminescent system (ECL).
CRISPR/Cas9 system
An epiCRISPR/Cas 9 vector with GFP-positive was constructed with insertions of two guide RNAs to transfect HEK293T cells and thus delete the targeted enhancer regions (deleted 44 bp). Guide RNAs 5′-GGCCGCTCTCCCTGTCCTCC-3′ and 5′-GCTCTGTCGTCGAGGCGCTC-3′ for epiCRISPR/Cas9 vector construction were designed to carry out the deletion experiment in the enhancer region according to Zhang lab's public instructions (https://zlab.bio/guide-design-resources). The transfected cells were sent to Sanger Sequencing to determine if the constructed plasmid effectively deleted the target sequence. Transfected cells were separated through flow cytometry to 96-well plate, cultured in 5% CO2 at 37°C for 2 weeks, and then transferred into 24-well plate to obtain monoclonal cells. PCR products of the monoclonal cells were sent to Sanger sequence to screen the cells with efficient deletions.
ChIP and ChIP-qPCR
After 3 days of transfecting lentivirus of miR-339, the cells were washed twice using PBS and fixed in 1% formaldehyde for 15 min at room temperature. After the formaldehyde was quenched by 0.125 M glycine solution for 10 min, the cells were harvested and resuspended in lysis buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.5% NP-40,10 mM KCl) containing protease inhibitor cocktail (Roche). Nucleus extracts were gained and dissolved in nuclear lysis buffer (50 mM Tris–HCl pH 8.1, 0.3% SDS, 10 mM EDTA and 1× cocktail). Following sonication, the cell extract was incubated with antibody against H3K27ac at 4°C for 16 h and Protein A Dynabeads (Invitrogen). Qiagen DNA purification kit was then used for further immunoprecipitated DNA purification. ChIP template DNA was assayed in 10 μl qPCR reactions with a LightCycler 96 System (Roche) according to the protocol using chromatin region-specific primers listed in Supplemental Table S1.
Dual luciferase reporter assays
The predicted enhancer region containing miR-339 locus and corresponding mutated sites were amplified using PCR from HEK293T cells and inserted into pGL3-promoter plasmid for enhancer activity assays. The constructed plasmids were cotransfected with the Renilla luciferase reporter vector pRL-SV40 into HEK293T cells. Then the cell extracts were prepared after transfection for 48 hours and assayed using a Dual Luciferase Reporter Assay System (Promega) according to the manufacturer's procedures. Renilla luciferase was used to normalize for transfection efficiency, and the ratio of firefly/Renilla luciferase activities defined the relative activity of the enhancer region. The assay was repeated three times in duplicate.
CCK8 assays, colony formation and transwell assays
Cells transfected with miR-339 expression vectors, shGPER1 knock-down vectors, and corresponding control vectors were cultured in 96-well plates with 6000 cells in each well and live cells (CCK8) were detected daily at 0, 24, 48 and 72 h, respectively. The OD value at 0 h actually was detected at 6 h since it takes about 6 hours for the breast cancer cells to adhere to the glassware wall. All CCK8 assays were performed in triplicates to evaluate cell viability.
After incubation for 72 h, 4175 and T47D cells were transfected with miR-339 lentivirus or control and shGPER1 or control shRNA. Single-cell suspensions of transfected 4175 and T47D cells were seeded at 1000 cells/well in 6-well plates and cultured for 10–14 days to form colonies. Cell colonies were stained with 0.25% crystal violet on day 14, imaged, counted and reported as the number of colonies.
Similarly, transfected 4175 and T47D cells were seeded at 20 000 cells/well upper chamber (BD Biosciences) plated in 24-well plates, adding with serum-free DMEM. In the lower compartment, 20% FBS–DMEM was added as the chemoattractant. Cells were incubated for 24 h, then fixed with 100% methanol for 15min and stained with 0.1% crystal violet solution. Cells migrated to the lower surface were counted under a microscope.
Animals and in vivo assays
Breast cancer cells 4175 transfected with lentiviral constructs with overexpression miR-339 or knock-down GPER1 and related controls were harvested and selected through flow cytometry to gain stable transfection cells. Approximately 5 × 106 4175 cells were injected subcutaneously in the flank of each 6-week-old female Balb/c nude mouse, respectively. The volume of initiated-tumours were measured after 4 weeks’ feeding. The tumour size was calculated by using (X2Y)/2, where X = tumour width and Y = tumour length. After 8 weeks, mice were sacrificed to gain the tumours for further analyses. The standard procedure for histopathologic analyses was performed. All animal experiments were approved by the Fudan Committee on Animal Care and in compliance with ethical guidelines and procedures.
IHC assay
The tumour samples of mice were soaked in 10% neutral formalin for 72 h. Subsequently, paraffin embedding and sectioning were performed to further staining assay after dehydration in accordance with routine procedures. For immunohistochemistry assay, 5 μm tumour sections embedded in the paraffin were mounted on polylysine-coated glass slides. They were incubated at 60°C for 24 h and then defatted in xylene and followed by rehydration with ethanol. Then, the slides were immersed in 3% H2O2 for 30 min to quench endogenous peroxidase activity, rinsed, and incubated with the primary antibody (anti-G-protein coupled receptor 30 (1:50) overnight at 4°C. Subsequently, the slides were detected by using EnVision Detection Systems (Peroxidase/DAB, Rabbit/Mouse; DAKO, K5007), then counterstained with hematoxylin staining and photographed under the Olympus BX53 fluorescence microscope (Tokyo, Japan).
Next-generation sequencing and NamiRNA candidates screening analyses
For high-throughput mRNA and miRNA sequencing, total RNA containing small RNAs was extracted from ∼1 × 107 cells for each sample using TRIzol reagent according to the manufacturer's instructions (Invitrogen, 15 596 018). RNA concentration and quality were measured using Nanodrop 2000 (Thermo Scientific) and the Bioanalyzer 2100 (Agilent Technologies) for further library preparation. The RNA-seq analysis sequencing reads were aligned to the human reference genome hg38 using Tophat2 (http://ccb.jhu.edu/software/tophat). Read counts were calculated by using featureCounts (http://subread.sourceforge.net). And different expression genes (DEGs) and statistical analysis were performed with DESeq2 (version 3.12) in R (version 4.0). Fold change >1.5, P < 0.05.The heatmaps were created using the ComplexHeatmap (Bioconductor project). A total of 20 ng prepared DNA templates were used to build libraries for ChIP-seq. ChIP-Seq data analysis sequencing reads were aligned to human genome assembly hg38 using Bowtie2 (version 2.2.5). Then duplicate reads were removed with Samtools (version 0.1.19). MACS14 (https://github.com/taoliu/MACS/) was used to call the H3K27ac peaks with default setting, comparing with pCDH plasmid transferred H3K27ac ChIP-Seq. The miRNA target region was predicted by using miRanda (version 3.3a) among all the upregulated peak regions. The parameters of miRanda were optimized by comparing the difference in the up-regulated H3K27ac peaks group and the random peaks group as setting: score cutoff ≥120, energy cutoff ≤−20 kcal/mol.
Human microRNA annotations were downloaded from miRBase database (http://www.mirbase.org/). Enhancer region were downloaded from enhanceratlas 2.0 database (http://enhanceratlas.org/). If the miRNA transcriptional locus was overlapped with the enhancer regions, the miRNA was chosen as NamiRNA candidates, referring to our previous work (34).
For breast cancer samples, expression data were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/). Available DNA methylation data (450k) of TSGs for breast cancer (BC) and kidney renal clear cell carcinoma (KIRC) were downloaded from TCGA database. The difference between the mean methylation levels of paired samples >5% followed by Wilcoxon Rank Sum Test was considered to be significant. Then, the promoter sites modified by H3K27ac were removed (including TSS ± 1 kb region). GSE78011 and GSE174002 can be gained from the National Center for Biotechnology Information's Gene Expression Omnibus.
Statistical analysis
All values are presented as calculated means ± standard deviations with triplicated experiments unless otherwise noted. Statistical analyses between two samples were obtained by two-tailed Student t-test. If one-Anova method was performed among three samples, it will be indicated in the figures. **** means P < 0.0001, *** means P < 0.001, ** means P < 0.01 and * means P < 0.05. Values of P < 0.05 were taken as significant. All analyses were performed with GraphPad Prism (Version7.0, GraphPad Software, Inc.).
RESULTS
Lower expression pattern of TSGs in breast cancer without promoter DNA hypermethylation
Epigenetic dysregulation and DNA hypermethylation in promoter regions are considered critical in the inactivation or loss of function of TSGs (47–49). Therefore, we performed a comprehensive gene profiling, of which 1217 breast cancer and normal control tissues were downloaded from the TCGA database to acquire their expression patterns by bioinformatics analysis in combination with text retrieval in PubMed. We discovered that a total of 622 TSGs may be involved in breast cancer, like MAP3K8, DUSP6, NF1, CDH1, PTEN and GPER1. Next, we classified them according to up-regulated or down-regulated ones in breast cancer samples and found that, among the total 622 TSGs, there were 383 downregulated genes in breast cancer (Figure 1A, Supplementary Table S2). As expected, Gene Ontology (GO) analyses for these TSGs showed that they are significantly linked to typically cancer-associated p53 and Wnt signaling pathways (Supplementary Figure S1A) (4,50). To find out how these 383 TSGs become downregulated in breast cancer, we comprehensively analysed the methylation statuses of these TSGs’ promoter in breast cancer. Only 274 TSGs’ methylation data were available, based on a rank-sum test by profiling 878 breast cancer and corresponding normal tissue samples. Herein the differential ratio within ±5% was identified no methylation difference. Unexpectedly, the analysis results as seen in Cluster 1 of Figure 1B revealed that only 62 genes (about 22.6%) of the total downregulated TSGs, exhibited relative DNA hypermethylation status in promoter regions; yet 187 (about 68.2%) downregulated TSGs like PTCH1, MTUS1 and GPER1 showed hypomethylation but with no methylation difference in Cluster 2 (Figure 1B), and the remained 25 TSGs are DNA hypermethylated in promoter regions in both breast cancer and normal controls but without methylation difference (Cluster 3, Figure 1B).
Figure 1.
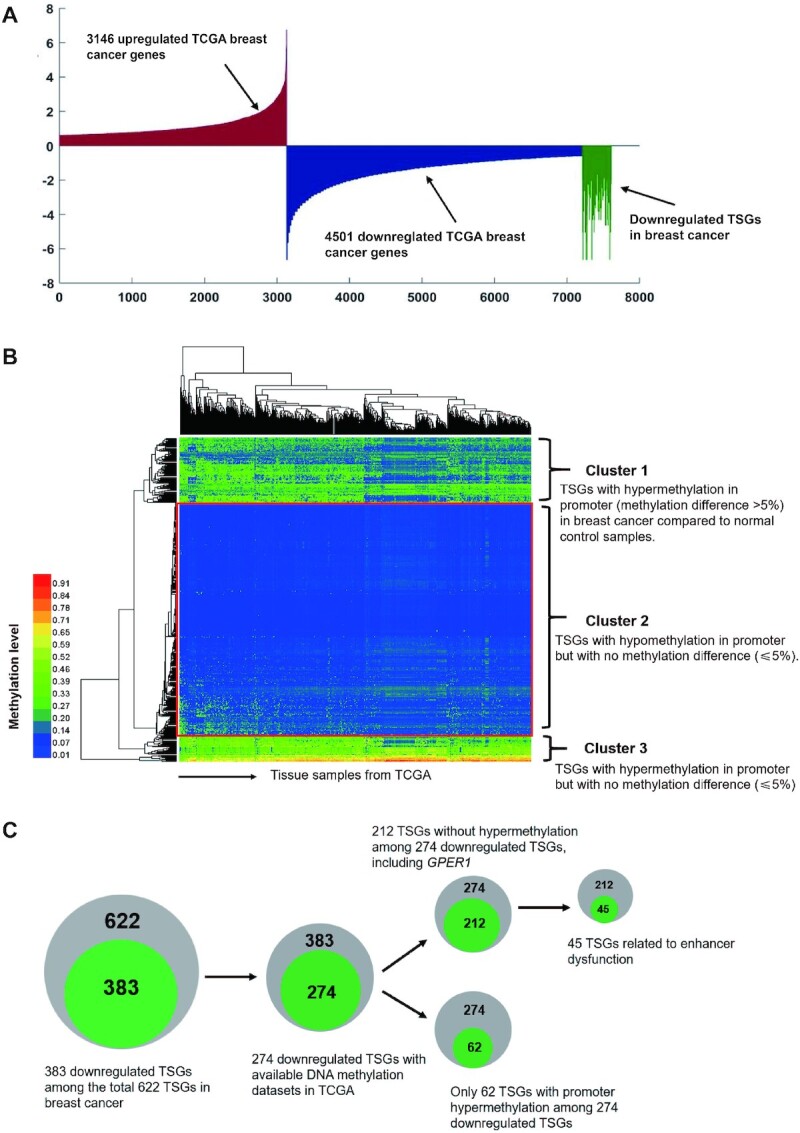
Downregulated TSGs in breast cancer without promoter DNA hypermethylation. (A) 383 TSGs downregulated in breast cancer by analysis of all up-regulated (3146) or down-regulated (4501) genes in breast cancer samples compared to normal controls. (B) Heatmap of promoter methylation status of TSGs in 878 TCGA breast cancer samples. The different DNA methylation level within ± 5% was identified no difference. Cluster 1 represents 62 genes (about 22.6%) of the total downregulated TSGs are hypermethylated in promoter regions in breast cancer; Cluster 2, as seen in red box, representing 187 TSGs (about 68.2%) with hypomethylation but without methylation difference between breast cancer and normal controls; Cluster 3 represents 25 TSGs are DNA hypermethylated in promoter in both breast cancer and normal controls but without methylation difference (within 5%). (C) Schematic diagram for the profiling processes of TSGs. Among the total 622 TSGs in breast cancer in TCGA database, there are 383 downregulated TSGs. 274 TSGs with available methylation data are obtained in the 383 downregulated TSGs, in which 187 TSGs are DNA hypomethylated but with no methylation difference (within 5%). Among these 187 TSGs, there is a number of 45 genes, accounting for about 24.1%, of which the downregulation is related to enhancer alteration.
In order to figure out how broad the patterns are seen in other tumour types, we investigate the DNA methylation states of 491 downregulated TSGs in kidney renal clear cell carcinoma (KIRC) from TSGene database (https://bioinfo.uth.edu/TSGene/). Available DNA methylation data of 464 TSGs and clinical information of patients with kidney renal clear cell carcinoma (KIRC) were downloaded from TCGA database. As seen in Supplementary Figure S1B, 358 downregulated TSGs (accounting for 77.1% of the total downregulated TSGs) showed hypomethylation in Cluster 3, and only 42 TSGs (about 9.1%), exhibited relative DNA hypermethylation status in promoter regions in Cluster 2; yet, the remained 64 TSGs (about 13.8%) are DNA hypermethylated in promoter regions in both cancer and normal controls but without methylation difference in Cluster 1. Taken together, these results suggest that promoter hypermethylation of TSG genes is not always the case in causing TSG downregulation.
To elucidate these abnormal regulation patterns, we focused our attention on another cis-regulatory element, the enhancers. By collecting the ChIP-seq profile from ENCODE database, we analysed the enrichment of H3K27ac, an active enhancer marker around these 187 TSGs with no significant difference for promoter DNA methylation in breast tumours compared to normal counterparts. As predicted, we found that there were 45 TSGs with significantly decreased H3K27ac peaks such as GPER1, supporting that the inactivation of enhancers is involved in downregulating TSG expression in breast cancer (Supplementary Table S3). All these profiling processes are presented in Figure 1C. Based on these results, we next explored how enhancer disfunction is involved in TSG alteration.
NamiRNAs act as key regulator in breast cancer leading to TSG lower expressions
Female hormone-related cancers like breast, ovarian, uterus and endometrial cancers pose serious risk to women's health worldwide. Therefore, there is an urgent need to explore potential initiative processes and treatment strategies to mitigate these malignancies. Since TSGs are usually downregulated in breast cancer, we set breast cancer as an example to investigate the underlying mechanism of TSGs downregulation or inactivation during tumourigenesis. Based on our previous work (34), it is worth mentioning that massive miRNA DNA loci were found overlapping within the enhancer region. These enhancer-overlapped miRNAs can positively regulate their target genes through enhancers, termed as nuclear activating miRNAs (NamiRNAs). Therefore, we pulled out a list of NamiRNA candidates in breast cancer based on text mining (Supplementary Table S4). Most of them have been reported to be tumour suppressors or display tumour suppressor features, such as miR-107 (51), miR-375 (52), miR-326 (53), miR-339 (54) and miR-638 (55). Among these NamamiRNA candidates, we randomly selected five NamiRNAs to perform absolute qPCR to testify their expression levels by comparing their copy numbers in breast cancer cell line 4175 and normal breast cell line MCF-10A. The result showed the copy numbers of these five NamiRNAs are indeed smaller in breast cancer cell line 4175 than those in the normal breast cell line MCF-10A (Supplementary Figure S2A). In addition, miR-339 and miR-1248 have the lowest copy numbers in breast cancer cell line 4175. We chose miR-339 as priority since it is clearly reported to exert suppressive functions on cancer development by positively impacting the p53-governed response in cancerous cells (56) as well as, by playing a role in controlling breast cancer hallmarks like proliferation, invasion, metastasis and apoptosis (57). Hence, miR-339 was screened as a specific example for us to explore the connection between enhancer and NamiRNA.
On account that NamiRNA positively regulates the target gene through enhancer and miR-339 is downregulated in breast cancer, we wanted to find miR-339′s target among the downregulated genes in the TCGA database. Firstly, we predicted potential target genes of miR-339 according to the seed sequence that overlapped with enhancer elements, leading us to a total of 10168 positively related target genes of miR-339 in genome-wide. Having recognized 383 tumour suppressor genes that are lower expressed in 878 TCGA breast cancer samples, we wanted to know how many TSGs are among these 10126 potential target genes of miR-339. Accordingly, we found that 332 TSGs downregulated in breast cancer (Figure 2A) were positively related to miR-339 regulation (Supplementary Table S5), including YAP1, DUSP6 and GPER1. Further KEGG pathway analysis of these 332 TSGs revealed that they were indeed involved in p53 signal pathway and cancer related pathways (Figure 2B).
Figure 2.

Down-regulated GPER1 and miR-339 in four different breast cancer subtypes. (A) 332 TSGs positively related to miR-339 regulation among the 383 downregulated TSGs. The green circle represents the number of 332 target genes positively related to miR-339, and orange circle represents 383 downregulated TSGs in breast cancer samples from TCGA database. (B) The 322 targeted genes of miR-339 associated to p53 pathway and pathways in cancer by KEGG pathway profiling. (C−F) GPER1, and miR-339 were both down-regulated in Luminal A (C, about 83.7% down-regulated, Correlation coefficient = 0.745), Luminal B (D, about 88.9% down-regulated, Correlation coefficient = 0.745), and TNBC (E, about 79.4% down-regulated, correlation coefficient = 0.808) breast cancer subtypes, except for the HER2-positive subgroup (F, only 42.8% down-regulated, correlation coefficient = 0.68) where they still exist a positive correlation between miR-339 and GPER1. TNBC means triple negative breast cancer subtype.
Breast cancer is an estrogen related carcinoma. Estrogen functions on regulating cell response mainly through binding the two classical receptors ER α And ER β. Except for these two classical estrogen receptors, GPER1, a membrane-bound G-protein-coupled oestrogen receptor, also known as GPR30 is a newly discovered estrogen receptor in recent years. It is involved in the rapid activation of ERK1/2 and CAMP mediated by estrogen, which plays inhibitory role in hormone sensitive malignant breast cancer (44). Moreover, it was reported that GPER1 functions as a tumour suppressor in coleorectal cancer (58) and the downregulation of GPER1 is correlated to the poor clinical outcome of breast cancer (42). According to our previous findings (34), NamiRNAs overlapped with enhancer are inclined to activate their neighboring genes. Among those TSGs, GPER1 is the only one located on chromosome 7 with miR-339, in the 60kb downstream. Hence, GPER1 is naturally selected as a prior target candidate of miR-339.
Further, in order to validate the positive relationship between miR-339 and its target genes, we performed RT-PCR assay to detect whether the expression level of its targeted genes (GPER1 and another five randomly selected target genes including CDKN1C, DUSP6, G0S2, STAT5A and MAP3K8) would increase after transfecting miR-339 in 4175 breast cancer cells. The result showed these target genes are all upregulated after transfecting miR-339 in 4175 cells (Supplementary Figure S2B), indicating miR-339 indeed plays positive regulation on targeted TSGs in breast cancer cells.
To further validate this concern, we performed RNA-seq high throughput analysis using 4175 cells with transfecting miR-339 lentivirus and empty control in triplicate. Interestingly, we observed a total of 127 genes are upregulated (fold change > 1.5, P < 0.05) after transfecting miR-339 (Supplementary Table S6), among which there is eight TSGs including G0S2, GPER1 and MAP3K8 (Right panel of Supplementary Figure S2C), which are consistent with the results verified by qPCR assay in Supplementary Figure S2B. Next, we figured out 91 genes are regulated by miR-339 through targeting enhancer among the total 127 upregulated genes (Supplementary Table S7), while other 36 ones are only regulated by enhancer without miR-339, indicating genes with NamiRNA are likely to be differentially regulated when compared to genes with the exact same properties, like with H3K27ac enrichment, minus the presence of NamiRNAs. Furthermore, we profiled whether these 91 upregulated genes with different targeting sites of miR-339 in enhancer. The results showed most upregulated genes with multiple targeting sites of miR-339 in enhancer, however, the number of targeting sites in enhancer is not correlated to the fold changes of these genes (Supplementary Table S7, correlation coefficient = −0.08672). Thereafter, with combination of the high throughput profilings and qPCR results, we utilized miR-339 and GPER1 as the prototypes to further investigate how NamiRNAs regulate their target TSGs with enhancers during breast cancer development and progression.
The behaviour of tumour suppressor genes varies in different breast cancer subtypes (59,60). For instance, TSG APC is downregulated in TNBC, yet upregulated in the Luminal B subtype (60). Given this, we wanted to further investigate the potential effects of miR-339 and its positive regulated TSG GPER1 in particular subtypes of breast cancer. We collected 157 pairs of breast tumour tissues and matched normal tissues (Luminal A, B, HER2-positive and TNBC are 43, 45, 35 and 34, respectively), and then examined the expression levels of miR-339 and its target gene GPER1 by RT-PCR. Results showed that both GPER1 and miR-339 were significantly down-regulated in most of three subtypes of Luminal A (about 83.7%), Luminal B (about 88.9%) and TNBC (about 79.4%), but not in HER2 positive subtypes (only 42.8%, Figure 2C–F and Supplementary Figure S3A, B). Of note, there is a positive correlation between the expressions of miR-339 and GPER1 in all these 4 different subgroups (Figure 2C–F). These results confirmed that miR-339 indeed positively regulated GPER1 in breast tumour samples, leading us to further seek their potential roles on breast cancer development. Moreover, we performed transcriptional mRNA detections via RT-PCR in ovarian and endometrial cancer tissue samples, as all these cancers are oestrogen-related and GPER1 is an oestrogen-related receptor. Accordingly, both miR-339 and GPER1 were downregulated in ovarian and endometrial cancer tissues compared to the controls, further validating the expression level of miR-339 positively correlates with that of GPER1 in these hormone-associated cancers (Supplementary Figure S3C, D). These findings provide us new insight on miRNA positively regulated genes in oestrogen-related cancer development. Furthermore, the cis-regulator miR-339 might be a new target for breast cancer treatment by reactivating GPER1, which gave us a clue to design gain-of-function and loss-of-function assays to validate this expectation.
MiR-339 specifically activates GPER1 to repress the proliferation of breast cancer cells
To elucidate the regulating effects of miR-339 on GPER1 in breast cancer, we transfected miR-339 and empty control lentivirus into 4175 (TNBC cell line) and T47D (Luminal A breast cancer cell line), and found that miR-339 was upregulated in 4175 and T47D cells transfected with miR-339 by 10.6- and 3.5-fold, respectively, compared to cells transfected with empty controls. As expected, GPER1 was accordingly upregulated by about 3.8- and 2.8-fold in 4175 and T47D miR-339 groups (Figure 3A, D). Conversely, transfection of miR-339-inhibitors into 4175 and T47D cells led to significant down-regulation of miR-339 and GPER1 (Figure 3B, E), and western blotting assay exhibited the consistent results (Figure 3C, F). These results demonstrated that miR-339 was able to activate TSG GPER1 expression in breast cancer cells.
Figure 3.

MiR-339 reactivates TSG GPER1 and inhibits proliferation of 4175 and T47D breast cancer cells. (A, B) MiR-339 reactivated its targeted TSG GPER1 expression (A) but downregulated GPER1 by transfecting miR-339 inhibitors (B) in 4175 cells through RT-PCR detection. (C) MiR-339 and its inhibitors respectively reactivated and downregulated the GPER1 protein expression levels in 4175 cells by western blot assay. (D–F) the expression of targeted TSG GPER1, was reactivated by miR-339 (D) and downregulated by miR-339 inhibitors in T47D cells (E) through RT-PCR detection and Western blot assay (F). (G, H) MiR-339 repressed the proliferation cells (G) and colony formation ability (H) of 4175 cells. (I) MiR-339 did not impact on breast cancer cell migration ability. (J, K) GPER1 knock-down by shGPER1 increased the proliferation (J) and colony formation ability (K) in 4175 cells. (L) GPER1 showed no impact on breast cancer cell migration ability. Data are presented as Mean ± SD of experiments conducted in triplicate. ****P < 0.0001, **P < 0.01, *P < 0.05. “ns” is representative of no significance.
Another crucial question we wanted to ask is whether miR-339 and GPER1 could affect the biological behaviour of breast cancer cells. Therefore, we detected the proliferation ability alterations of 4175 and T47D miR-339 groups and their corresponding controls by CCK8 and colony formation assays. The CCK8 assay showed that proliferation rate of both 4175 and T47D with miR-339 groups was significantly lower than that of the controls at 72 and 96 h (Figure 3G, Supplementary Figure S4A). For colony formation assay, a total number of 113 stained colonies were calculated in 4175 cells with transfecting miR-339 lentivirus, which is significantly less than the control group with 181 colonies after incubation for 14 days (Figure 3H). Similarly, a total number of 170 stained colonies were calculated in T47D cells with transfecting miR-339 lentivirus, which is discernibly less than the control group with a number of 263 colonies (Supplementary Figure S4B). Since metastasis is another feature of breast cancer, we performed transwell migration assays to test whether miR-339 can play an inhibitory function on breast cancer cell migrating abilities on top of exerting a repressive effect on their proliferation ability. However, transwell migration assay showed there was no significant difference for migration abilities between miR-339 transfected and control groups in 4175 and T47D cells (Figure 3I, Supplementary Figure S4C), suggesting that miR-339 exerted no effect on migrating ability of breast cancer cells.
Given that miR-339 can repress the proliferation of breast cancer cells, we wondered whether GPER1, as the target TSG of miR-339, can also inhibit their proliferation as well. Therefore, we knocked down GPER1 in 4175 and T47D breast cancer cell lines by transfecting shGPER1 lentivirus to discern whether GPER1 plays an inhibitory function on breast cancer cells. Subsequently, GPER1 expression was successfully downregulated in 4175 and T47D shGPER1 groups compared to their corresponding groups by RT-PCR detection (Supplementary Figure S4D). Furthermore, The CCK8 assay results showed GPER1 knock-down in 4175 and T47D shGPER1 groups led to an obvious increase in their proliferation ability at 72 and 96 h (Figure 3J, Supplementary Figure S4E), indicating that GPER1 may play an inhibitory function on breast cancer cells. Meanwhile, the colony formation assay showed GPER1 knock-down in both 4175 and T47D shGPER1 groups contributed to significantly more colony numbers compared to the controls (Figure 3K, Supplementary Figure S4F). In detail, a total number of 221 stained colonies were calculated in the 4175 shGPER1 group, while only 167 colonies in the corresponding control group. For T47D breast cancer cells, the stained colony number was 231 in shGPER1 group, yet in the control group they merely formed 173 stained colonies. We detected their migrating ability alterations as well. There was no difference between GPER1 knock-down and control groups (Figure 3L, Supplementary Figure S4G). These results indicate that GPER1 can indeed repress the proliferation and growth of breast cancer cells, yet has no impact on their migrating abilities.
To further confirm that TSG GPER1 plays an inhibitory function on breast cancer cells as modulated by miR-339, we subsequently performed a rescue assay by using 4175 and T47D shGPER1 groups. Specifically, we transfected miR-339 lentivirus and the empty control lentivirus into 4175 and T47D shGPER1 groups, respectively. Then, we confirmed that miR-339 was successfully expressed in 4175 and T47D shGPER1 groups compared to the control groups by RT-PCR assay (Supplementary Figure S5A). Here we defined 4175 shGPER1 + miR-339, T47D shGPER1 + miR-339 as the experimental group, and 4175 shGPER1 + empty control, T47D shGPER1 + empty control as the control group. Next, CCK8 and colony formation assays were performed to investigate their proliferation ability alterations. Results from CCK8 assays showed that the proliferation rate of both 4175 shGPER1 + miR-339 and T47D shGPER1 + miR-339 experimental groups were significantly lower than the controls at 72 and 96 h (Supplementary Figure S5B-C). Meanwhile, 156 and 158 stained colonies were formed after incubation for 14 days in 4175 and T47D experimental groups by colony formation assay, which is significantly smaller in the colony number than the control groups, indicating that transfected miR-339 can repress breast cancer cell proliferation by reactivating TSG GPER1 expression (Supplementary Figure S5D, E). All these results show that miR-339 is capable of inhibiting breast cancer cell growth and proliferation via targeting GPER1, which may provide a new strategy for breast cancer treatment.
MiR-339 activation depends on the enhancer integrity
Our observation that miR-339 could activate the tumour suppressor GPER1 expressions in breast cancer cell lines led us to further explore the underlying mechanism of how miR-339 modulates GPER1 positively as a cis-regulator via enhancer. Naturally, we explored the interactions between NamiRNA-339 and its corresponding enhancer elements to understand how they together affect TSG expression.
Firstly, we examined whether the genome sequence containing miR-339 locus can function as an enhancer. To test the potential enhancer activity of the miR-339 genome locus, a DNA fragment (588bp) containing miR-339 genome locus in the upstream 60 kb of GPER1 was amplified and inserted into pGL3 vector, named as pGL3-miR339. Then, pGL3-miR339 and pGL3-Control vector were transfected to HEK293T cells using lipofectamine, respectively. We then detected both the Luciferase and Renilla activities in HEK293T cells by Dual Luciferase Reporter Assay Kit (Promega) at 48h after transfection. As Expected, the activity of the reporter gene increased after pGL3-miR339 transfection compared to the control group with pGL3-Control vector transfection, demonstrating that the DNA fragment containing miR-339 genome locus can serve as an enhancer (Figure 4A).
Figure 4.

MiR-339 reactivates TSG GPER1 depended on Enhancer elements and AGO2 protein. (A, B) MiR-339 genome locus acts as an enhancer by Dual Luciferase Reporter Assay. (A) The activity of reporter gene was activated by pGL3-miR339 in HEK293T cells. (B, a, b) The ratio of Luc+/Rluc + was remarkably higher for the experimental group with co-transfecting pGL3-miR339 and pSR-miR339 than that of the control group with co-transfecting pGL3-miR339 and pSUPER empty plasmids; (b–d) The luciferase activity was reduced by either mutating enhancer elements (mut-pGL3-miR339) or mutating seed sequence of miR-339 (mut-pSR-miR339); (e) The luciferase activity was rescued by mutating both the enhancer and seed sequence of miR-339 with a matched complementary base pair. (C, D) The enrichment of H3K27ac in enhancer region containing miR-339 DNA locus by ChIP-qPCR assay (C) and H3K27ac ChIP-seq analyses (D). (E, F) The activation was abolished by deletion of the miR-339 enhancer region in HEK293T cells by CRISPR/Cas9. schematic diagram (E) shows a deletion of 44bp on enhancer region by CRISPR/Cas9 system, while miR-339 (F) failed to reactivate GPER1 expression. (G, H) AGO2 is essential to the activating function of miR-339 on GPER1 expression. GPER1 was no longer activated by miR-339 in RT-PCR (G) and western blot assays (H) by knocking down AGO2 in HEK293T cells. (I) AGO2 was recruited to miR-339 locus when transfected miR-339 by ChIP-qPCR assay. Data are presented as Mean ± SD of experiments conducted in triplicate. (C, F, G) One-ANOVA test, followed by Tukey's test. ****P <0.0001, ***P <0.001, **P <0.01, *P <0.05. “ns” is representative of no significance.
Secondly, we were interested in seeing whether miR-339 activates the reporter gene by interacting with the enhancer region. For this purpose, we initially amplified pre-miR-339 (94bp) and inserted this fragment into plasmid pSUPER-GFP/NEO to obtain new plasmid pSR-miR339. Transfection of pSR-miR339 increased miR-339 expression compared to the control group as confirmed by RT-PCR assay, shown in Supplementary Figure S5F. Then, we co-transfected pSR-miR339 and pGL3-miR339 into HEK293T cells and harvested these cells to detect luciferase activity by calculating the ratio of Luc+/Rluc+. The result showed that the ratio of Luc+/Rluc+ for the experimental group was remarkably higher than that in the control group with co-transfecting pGL3-miR339 and pSUPER empty plasmids, suggesting that miR-339 was able to activate the reporter gene by interacting with the enhancer (Figure 4B, a, b).
Thirdly, we were eager to learn how miR-339 interacts with enhancers, and in turn, plays an activating function. Since the seed sequence of miRNAs plays a critical role in its regulatory function (34), we mutated the seed sequence of miR-339 by constructing the plasmid mut-pSR-miR339 and its corresponding complementary sites in the enhancer region by constructing plasmid mut-pGL3-miR339. To detect if miR-339 interacts with the enhancer region via the seed sequence, we co-transfected pSR-miR339 and mut-pGL3-miR339, mut-pSR-miR339 and pGL3-miR339 respectively, any of which will have mismatch sites due to mutants in miR-339 or its target enhancer. As expected, the luciferase activity was down-regulated (Figure 4B, a–d), indicating that the miR-339 activating function needs to bind to the enhancer region through the seed sequence. Once the seed sequence of miR-339 or the complementary sequence on the enhancer region was mutated, the binding sites could form the mismatch and become impaired, which eventually contributes to a decrease of luciferase activity compared to the control group. Given that miR-339 can bind to its corresponding enhancer region to exhibit a positive regulatory function, this begs the question: can luciferase activity be rescued by mutating the binding sites in a complementary base-pair manner? To clear this point, we detected the luciferase activity after co-transfecting the two mutated plasmids mut-pGL3-miR339 and mut-pSR-miR339 and found that the luciferase activity was rescued (Figure 4B, c–e). Notably, these two plasmids mutated the seed sequence binding region of the enhancer and seed sequence of miR-339 in a base complementary fashion. Overall, these data indicate that miR-339 interacts with its targeted enhancer region positively in a base-pair match manner.
Fourthly, we aimed to see whether miR-339 can contribute to an increase in the activity of GPER1 enhancer. For this purpose, we carried out H3K27ac ChIP-qPCR since the histone modification of H3K27ac is frequently enriched as a surrogate marker for the enhancer. Expectedly, the enrichment of H3K27ac in the enhancer region 60kb upstream of GPER1, which contains the miR-339 genome locus when transfected with miR-339, is recognizably at higher level than that of the control group and knockout group (Figure 4C). Besides, ChIP-seq analyses confirmed the results in Figure 4D, supporting that miR-339 could raise enhancer activity. Knowing that miR-339 was able to upregulate enhancer activity, could enhancers also influence the regulatory function of miR-339 on GPER1?
In this final stage of our inquiry, we delved into identifying the effect of enhancer integrity on the regulatory function of miR-339 by deleting the enhancer region through CRISPR/Cas9 system. After we transfected miR-339 in HEK293T cells for 48 h, GPER1 expression level perceptibly increased approximately 2 folds by RT-PCR detection (Supplementary Figure S5F). To see whether miR-339 would no longer activate GPER1 after removing its enhancer region in HEK293T cells, we deleted the corresponding enhancer region in HEK293T cells by CRISPR/Cas9 system (Figure 4E). Subsequently, we transfected miR-339 in these HEK293T cells with the deleted enhancer region (44 bp), wherein miR-339 and GPER1 expression levels were determined by RT-PCR assays, respectively. When miR-339 was successfully expressed (Figure 4F), we saw that GPER1 could no longer be activated after the deletion of the enhancer region; in turn, it was significantly down-regulated (Figure 4F). Overall, these results demonstrate that enhancer integrity should exert a critical influence on GPER1 activation by miR-339, coinciding with the observations in the dual luciferase reporter assay.
AGO2 binding orchestrates the positive regulatory function of miR-339
AGO2 is a core protein for miRNA processing, which modulates the maturation of precursor miRNAs or enhance the stability of miRNAs (61). To determine whether AGO2 is involved in the miR-339 activating function, we constructed the shRNA expressed-plasmid shAGO2 to suppress AGO2 expression (shCtrl as the control plasmid). Then the shAGO2 plasmids and control plasmids were respectively co-transfected with miR-339 expression plasmids into HEK293T cells as shCtrl + miR-339 group and shAGO2 + miR-339 group. RT-PCR results confirmed that mRNA expression level of AGO2 was obviously downregulated in the shAGO2 + miR-339 group compared to the shCtrl and shCtrl + miR-339 groups, and meanwhile, miR-339 can be detected at a higher level in the shCtrl + miR-339 group and the shAGO2 + miR-339 group than in the shCtrl control group (Figure 4G). Subsequently, the changes of GPER1 expression after knocking down AGO2 was determined through RT-PCR and western blotting assays. Notably, GPER1 was no longer activated by miR-339 in the shAGO2 + miR-339 as it was in the shCtrl + miR-339 group, suggesting that the transcriptional activation of GPER1 gene by miR-339 depended on the AGO2 expression (Figure 4G, H). We hypothesized that miR-339 could recruit the AGO2 protein to GPER1 enhancer during its positive regulating processes. To test this theory, we performed an AGO2 ChIP-qPCR in HEK293T cells with upregulated miR-339, and detected whether AGO2 can be recruited to the miR-339 genomic locus overlapped with the enhancer region. The result showed that there was significantly more enrichment of AGO2 in the miR-339 locus than in the control group (Figure 4I), supporting that AGO2 was a vital player in GPER1 reactivation by miR-339.
MiR-339 acts as a potential suppressor for breast cancer due to TSG activation
Having demonstrated that miR-339 can activate TSG GPER1 in breast cancer, we then investigated whether miR-339 can act as a potential inhibitor for breast cancer treatment. Therefore, we would like to explore the effects of miR-339 and GPER1 on tumour growth in subcutaneous tumour models using 4175 breast cancer cell lines. The above-mentioned 4175 cells with stable expression of miR-339 were used in mouse models as the experiment group, and transfecting empty lentivirus as the control group. Accordingly, we injected these lentivirus-transfected 4175 cells into the flanks of nude mice to further validate effects of miR-339 on tumour growth. Expectedly, the growth rate of tumours in experiment group was reduced on average than in the control group (Figure 5A). In addition, the volume and weight of the sacrificed mice tumours were calculated. We observed that those miR-339-transfected cells formed significantly smaller tumours in mice compared to the control group (Figure 5B, C). These results together indicate that miR-339 was able to inhibit tumour cell proliferation.
Figure 5.

MiR-339 acts as an inhibitor by using mouse xenograft breast cancer models. (A) MiR-339 inhibited tumour growth by activating GPER1 via measuring the volume of tumours in mice by days. The nude mice formed the first measurable tumour marked as d0, and then tumours in mice were measured in the next 4 weeks until sacrifice. (B, C) MiR-339 inhibits the tumour growth by measuring weight and volume, which was counteracted if knockdown GPER1. The weight (B), the tumour virtual size (C, Left panel), and the calculated tumour volume (C, Right panel) of tumours in the sacrificed mice. (D, E) The alteration of Ki67 staining within breast cancer cell from xenograft mouse models. Ki67 staining decreased with miR-339 expression and increased after knocking down GPER1 by microscope (D) and by image J software calculation (E). Data are presented as mean ± SD of experiments conducted in triplicate (one-Anova test, followed by Tukey's test). ****P < 0.0001, **P < 0.01, *P < 0.05.
Since GPER1 is the activating target of miR-339 and function as a TSG for suppressing tumour cell proliferation, we wonder whether knockdown of GPER1 could reverse the effects of miR-339 in the tumour growth. Therefore, we knocked down GPER1 by transfecting shGPER1 in 4175 cells with stable expression of miR-339 and then injected these cells into the flanks of nude mice. Indeed, GPER1-knock-down obviously led to reverse the tumourigenicity by increasing the tumour volume and tumour weight (Figure 5A–C), further supporting that miR-339 function through activating the TSG GPER1 in breast cancer. Next, IHC staining of Ki67 was applied to detect the proliferating cells. Clearly, we found that there was less Ki67 positive cell when miR-339 was upregulated, and meanwhile, when we knockdown the miR-339 activating target GPER1, Ki67 positive cell was increased accordingly (Figure 5D, E). These findings in xenograft mouse models support that miR-339 and its target GPER1 together establish tumour suppressor features in breast cancer, and loss function of either will promote the cell growth in tumourigenesis. Therefore, miR-339 exhibits potential application in providing new treatment strategies for breast cancer, especially TNBC.
DISCUSSION
TSG promoter methylation and enhancer regulation in breast cancer
TSGs are frequently and aberrantly expressed during the process of tumourigenesis. Since the discovery of the first TSG by Knudson, encoding retinoblastoma protein (RB), TSGs have been linked with the famous ‘two-hit’ hypothesis and thought to play a role in carcinogenesis (1). In recent years, a myriad of evidence has gradually demonstrated that the low expression of tumour suppressor genes is closely related to epigenetic regulatory factors beyond gene mutation. Conventionally, the main epigenetic reason for TSG downregulation was thought to be ascribed to hypermethylation of their promoters, and sometimes to aberrant histone modifications and non-coding RNA dysregulations (13,14). Here, we found that ∼68.2% of the downregulated TSGs in breast cancer has no significant DNA hypermethylation in promoter regions compared with the normal control group, while the active enhancer marker H3K27ac on their enhancer regions display a significantly distinct enrichment pattern, implying that the abnormal H3K27ac modification of enhancers may partially explain the low expression of tumour suppressor genes. Our results support that enhancers are involved in TSG expression patterns: the sequence containing miR-339 genomic locus with a length of 588bp exhibited enhancer activity by dual luciferase reporter gene assay; in turn, a deletion of 44 bp in enhancer region of GPER1 leads to its downregulation. Meanwhile, our results were also supported by the finding that the lowly expressed tumour suppressor gene CST6 and BRMS1 were hypermethylated at promoters only in 17.9% and 32.1% of breast cancerous samples, respectively, which is in accordance with the idea that promoter hypermethylation is not essential to altered TSG expression cancerous tissues (62); furthermore, deletion of the enhancer sequence through CRISPR/Cas9 system can downregulate the expression of TSG CDKN1A, indicating that the low expression of the tumour suppressor gene may be linked with enhancer inactivation (63), yet the mechanism was unclear.
Enhancer switching on/off by NamiRNA
What was particularly striking about our results in our previous work was that there are a great number of miRNA genomic loci overlapped within the enhancer regions, which were nominated as NamiRNAs (Nuclear activating miRNAs) since they play activating functions in the nucleus and form a regulatory network with enhancers to co-regulate gene expression (34). Crucially, we revealed that NamiRNA miR-339 can reactivate its targeted TSG GPER1 via enhancer in our current work, and through this network, it is able to suppress the proliferation of breast cancer cells, demonstrating that miR-339 may act as a potential breast cancer inhibitor. Of note, ChIP-seq data exhibited that enrichment of the enhancer marker H3K27ac became significantly higher after overexpressing miR-339, implying that the activity of enhancers can be increased by NamiRNAs. Enhancer switching on or off depended on NamiRNAs, once the expression of NamiRNA was aberrantly downregulated, the corresponding enhancer would switch off and thus lead to inactivation of TSGs and vice versa. Moreover, low expression of miRNAs is a feature of tumours, as shown by miRNAs in normal tissues exhibiting higher expression levels than that in tumours (64–66). For instance, let-7 family can negatively regulate expression of MYC and RAS family oncogene to inhibit tumour development, and low expression of let-7 miRNA was observed in different cancers (67); the expression of miR-199a/199b in liver cells was downregulated, which was related to the occurrence and development of liver cancer (68). Most lowly expressed miRNAs in cancers display tumour suppressor features, is it possible that these miRNAs can play activating functions like NamiRNAs? As we speculate, these miRNAs like miR-339, which harbour tumour suppressor gene features, are upregulated in normal cells and downregulated in tumour cells; and these altered expressed miRNAs eventually contribute to targeted tumour suppressor gene inactivation, so as to promote the occurrence and development of tumour cells, among which enhancer activity is critical to the regulatory functions of these miRNAs (Figure 6).
Figure 6.
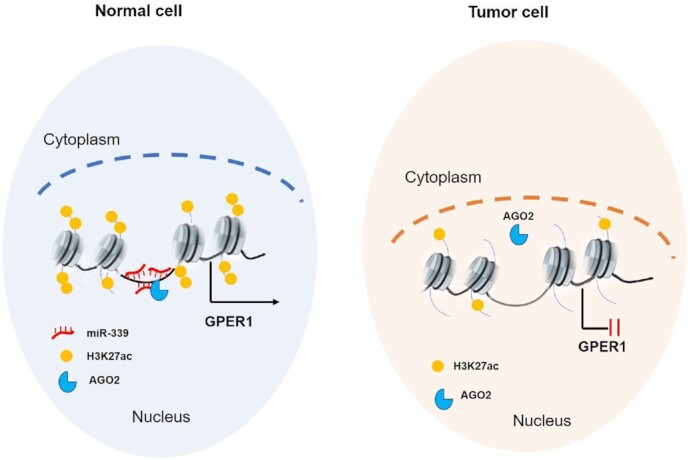
The model illustrated that how tumour suppressor gene GPER1 is silenced in breast cancer. In normal breast cell, NamiRNA miR-339 binds to the enhancer region colocalized with AGO2 to activate tumour suppressor gene GPER1, which protects the normal cell from transformation into malignant ones. However, when the expression of miR-339 is downregulated to reduce the enhancer activity , the corresponding TSG GPER1 would be silenced and failed to play its repressive functions on tumourigenesis.
MiR-339 activates GPER1 for repressing proliferation
miR-339, located on chromosome 7 of the human genome, has been reported to inhibit the proliferation of different types of cancers, like breast cancer (54), lung cancer (69), gastric cancer (70) and melanoma (71). In our work, we collected a total of 157 breast cancer tissue samples with different subtypes, and confirmed that the expression of miR-339 was downregulated in Luminal A/B and TNBC breast cancer subtypes, but there was no significant difference in HER2-positive breast cancer compared to corresponding adjacent tissues by RT-QPCR assay. Since almost only HER2-positive breast cancer subtype can be effectively treated by Herceptin (Trastuzumab Injection), yet other three subtypes face different problems on clinical treatments. Our results indicate miR-339 might bring a good therapeutic target for Luminal A/B and TNBC breast cancer subtypes. Moreover, our results demonstrate that miR-339 in breast cancer cells can activate GPER1 in T47D and 4175 breast cancer cells, thereafter the proliferation of these breast cancer cells was repressed significantly. Consistently, GPER1 expression was also significantly lower in ovarian cancer cells SOV-3 and OVCAR-3 than that in the normal control group, which function as a tumour suppressor gene to inhibit the proliferation of the ovarian cancer cells (72); meanwhile, more evidence found GPER1 can inhibit the growth of breast cancer cells and was even correlated to poor prognosis (73). Intriguingly, the in vivo assay in mice further verified that miR-339 and GPER1 can suppress the proliferation of breast cancer cells, supporting that miR-339 can act as a potential inhibitor and provide a new clinical strategy for breast cancer treatment, especially for the TNBC.
In summary, our current work unraveled the co-regulation of enhancers and NamiRNAs and their collective role on TSG inactivation in breast cancer. Furthermore, miR-339 was able to inhibit the proliferation of breast cancer cells by reactivating the expression of target TSG GPER1 through enhancers, implying that miR-339 can be used as a potential therapeutic target for breast cancer, especially for TNBC. Additionally, it is believed that the activating regulation of NamiRNAs and enhancers can also be used to explain the molecular mechanism of breast cancer development. Future research should aim to explore the interaction between enhancers and NamiRNAs in these fields to broaden our understanding of NamiRNAs’ regulatory role in various physiological and pathological processes.
DATA AVAILABILITY
The accession number for the sequencing data generated in this paper is GEO: GSE78011 and GSE174002. For breast cancer and kidney renal clear cell carcinoma samples, expression data were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/).
Supplementary Material
ACKNOWLEDGEMENTS
We sincerely appreciate Shuai Yang, Daoping Ru, Jin Li, Huaibing Luo and Feizhen Wu from Fudan University and Dongen Ju from Fourth Military Medical University for their critical comments in manuscript preparation. And we also thank Yue Yu for her kind assistance with language editing.
Author contributions: W.Y. conceived the topic and designed the study. W.Y., D.L. and H.W. guided the experimental work, while Y.L. conducted main work of the molecular experiments; F.Z., L.C., Y.T., M.L., X.R. and X.P. prepared the transfected cells; Q.L., Y.H. and B.Z. collected the clinical samples; F.Z. and Y.L. conducted the animal assays; Y.L, S.D., Q.Z. and D.Z. collected the experimental data; W.L.(Wei Li), S.W. and W.L.(Wenxuan Li) analysed the bioinformatic data; Y.L. wrote this paper; W.Y., D.L. and H.W. revised the report. All authors approved the final version.
Contributor Information
Ying Liang, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Qi Lu, Department of Gynaecology, Jinshan Hospital of Fudan University, Shanghai 201508, P. R. China.
Wei Li, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Dapeng Zhang, State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, P. R. China.
Fanglin Zhang, Fudan University Shanghai Cancer Center and Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Qingping Zou, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Lu Chen, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Ying Tong, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Mengxing Liu, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Shaoxuan Wang, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Wenxuan Li, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Xiaoguang Ren, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Peng Xu, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Zhicong Yang, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Shihua Dong, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Baolong Zhang, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Yanni Huang, Fudan University Shanghai Cancer Center and Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Daqiang Li, Fudan University Shanghai Cancer Center and Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
Hailin Wang, State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, P. R. China.
Wenqiang Yu, Shanghai Public Health Clinical Center and Department of General Surgery, Huashan Hospital, Cancer Metastasis Institute and Laboratory of RNA Epigenetics, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, P. R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2018YFC1005004]; Major Special Projects of Basic Research of Shanghai Science and Technology Commission [18JC1411101]; National Natural Science Foundation of China [31872814, 31671308]; Shanghai Science and Technology Commission [18JC1411104, 20Y11914100]; National Natural Science Foundation of China [22021003]; Strategic Priority Research Program of the Chinese Academy of Sciences [XDPB2004]. Funding for open access charge: Shanghai Science and Technology Commission [18JC1411104, 20Y11914100], Jinshan Hospital of Fudan University.
Conflict of interest statement. Wenqiang Yu and Ying Liang are listed as inventors on patents’ application related to this work; no other relationships or activities that could appear to have influenced the submitted work.
REFERENCES
- 1.Knudson A.G. JrMutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U.S.A. 1971; 68:820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knudson A.G.Antioncogenes and human cancer. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:10914–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macleod K.Tumor suppressor genes. Curr. Opin. Genet. Dev. 2000; 10:81–93. [DOI] [PubMed] [Google Scholar]
- 4.Aubrey B.J., Strasser A., Kelly G.L.. Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb. Perspect. Med. 2016; 6:a026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatzistergos K.E., Williams A.R., Dykxhoorn D., Bellio M.A., Yu W., Hare J.M.. Tumor suppressors RB1 and CDKN2a cooperatively regulate cell-cycle progression and differentiation during cardiomyocyte development and repair. Circ. Res. 2019; 124:1184–1197. [DOI] [PubMed] [Google Scholar]
- 6.Zhao W., Wiese C., Kwon Y., Hromas R., Sung P.. The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu. Rev. Biochem. 2019; 88:221–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baretta Z., Mocellin S., Goldin E., Olopade O.I., Huo D.Z.. Effect of BRCA germline mutations on breast cancer prognosis: a systematic review and meta-analysis. Medicine (Baltimore). 2016; 95:e4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Philpott C., Tovell H., Frayling I.M., Cooper D.N., Upadhyaya M.. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genomics. 2017; 11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giacomelli A.O., Yang X.P., Lintner R.E., McFarland J.M., Duby M., Kim J., Howard T.P., Takeda D.Y., Ly S.H., Kim E.et al.. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018; 50:1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Law C.T., Wei L., Tsang F.H., Chan C.Y., Xu I.M., Lai R.K., Ho D.W., Lee J.M., Wong C.C., Ng I.O.et al.. HELLS regulates chromatin remodeling and epigenetic silencing of multiple tumor suppressor genes in human hepatocellular carcinoma. Hepatology. 2019; 69:2013–2030. [DOI] [PubMed] [Google Scholar]
- 11.Llinas-Arias P., Esteller M.. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: an update. Open Biol. 2017; 7:170152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okugawa Y., Grady W.M., Goel A.. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 2015; 149:1204–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones P.A., Baylin S.B.. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002; 3:415–428. [DOI] [PubMed] [Google Scholar]
- 14.Bird A.DNA methylation patterns and epigenetic memory. Genes Dev. 2002; 16:6–21. [DOI] [PubMed] [Google Scholar]
- 15.Rhee I., Bachman K.E., Park B.H., Jair K.W., Yen R.W.C., Schuebel K.E., Cui H.M., Feinberg A.P., Lengauer C., Kinzler K.W.et al.. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002; 416:552–556. [DOI] [PubMed] [Google Scholar]
- 16.Teneng I., Tellez C.S., Picchi M.A., Klinge D.M., Yingling C.M., Snider A.M., Liu Y., Belinsky S.A.. Global identification of genes targeted by DNMT3b for epigenetic silencing in lung cancer. Oncogene. 2015; 34:621–630. [DOI] [PubMed] [Google Scholar]
- 17.Li P., Shan J.X., Chen X.H., Zhang D., Su L.P., Huang X.Y., Yu B.Q., Zhi Q.M., Li C.L., Wang Y.Q.et al.. Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res. 2015; 25:588–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai Y., Tsai H.C., Yen R.W.C., Zhang Y.W., Kong X.Q., Wang W., Xia L.M., Baylin S.B.. Critical threshold levels of DNA methyltransferase 1 are required to maintain DNA methylation across the genome in human cancer cells. Genome Res. 2017; 27:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhawan A., Scott J.G., Harris A.L., Buffa F.M.. Pan-cancer characterisation of microRNA across cancer hallmarks reveals microRNA-mediated downregulation of tumour suppressors. Nat. Commun. 2018; 9:5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wichmann G., Rosolowski M., Krohn K., Kreuz M., Boehm A., Reiche A., Scharrer U., Halama D., Bertolini J., Bauer U.et al.. The role of HPV RNA transcription, immune response-related gene expression and disruptive TP53 mutations in diagnostic and prognostic profiling of head and neck cancer. Int. J. Cancer. 2015; 137:2846–2857. [DOI] [PubMed] [Google Scholar]
- 21.Wang L.H., Wu C.F., Rajasekaran N., Shin Y.K.. Loss of tumor suppressor gene function in human cancer: An overview. Cell. Physiol. Biochem. 2018; 51:2647–2693. [DOI] [PubMed] [Google Scholar]
- 22.Berge E.O., Knappskog S., Lillehaug J.R., Lonning P.E.. Alterations of the retinoblastoma gene in metastatic breast cancer. Clin. Exp. Metastasis. 2011; 28:319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueda H., Akiyama Y., Shimada S., Mogushi K., Serizawa M., Matsumura S., Mitsunori Y., Aihara A., Ban D., Ochiai T.et al.. Tumor suppressor functions of DAXX through histone H3.3/H3K9me3 pathway in pancreatic NETs. Endocr. Relat. Cancer. 2018; 25:619–631. [DOI] [PubMed] [Google Scholar]
- 24.Keung E.Z., Rai K.. H3K9me3-mediated repression of KLF6: discovering a novel tumor suppressor in liposarcoma using a systematic epigenomic approach. Mol. Cell. Oncol. 2016; 3:e1093691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu Y., Chen J., Pang B., Li C., Zhao J., Shen K.. EZH2-induced H3K27me3 is associated with epigenetic repression of the ARHI tumor-suppressor gene in ovarian cancer. Cell Biochem. Biophys. 2015; 71:105–112. [DOI] [PubMed] [Google Scholar]
- 26.Ler L.D., Ghosh S., Chai X., Thike A.A., Heng H.L., Siew E.Y., Dey S., Koh L.K., Lim J.Q., Lim W.K.et al.. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 2017; 9:eaai8312. [DOI] [PubMed] [Google Scholar]
- 27.Cinghu S., Yang P.Y., Kosak J.P., Conway A.E., Kumar D., Oldfield A.J., Adelman K., Jothi R.. Intragenic enhancers attenuate host gene expression. Mol. Cell. 2017; 68:104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson R., Sandelin A., Danko C.G.. A unified architecture of transcriptional regulatory elements. Trends Genet. 2015; 31:426–433. [DOI] [PubMed] [Google Scholar]
- 29.Bulger M., Groudine M.. Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev. Biol. 2010; 339:250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kowalczyk M.S., Hughes J.R., Garrick D., Lynch M.D., Sharpe J.A., Sloane-Stanley J.A., McGowan S.J., De Gobbi M., Hosseini M., Vernimmen D.et al.. Intragenic enhancers act as alternative promoters. Mol. Cell. 2012; 45:447–458. [DOI] [PubMed] [Google Scholar]
- 31.Bulger M., Groudine M.. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011; 144:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ko J.Y., Oh S., Yoo K.H.. Functional enhancers as master regulators of tissue-specific gene regulation and cancer development. Mol. Cells. 2017; 40:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A.et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao M., Li J., Li W., Wang Y., Wu F.Z., Xi Y.P., Zhang L., Ding C., Luo H.B., Li Y.et al.. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017; 14:1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang Y., Xu P., Zou Q., Luo H., Yu W.. An epigenetic perspective on tumorigenesis: loss of cell identity, enhancer switching, and NamiRNA network. Semin. Cancer Biol. 2019; 57:1–9. [DOI] [PubMed] [Google Scholar]
- 36.Liang Y., Zou Q., Yu W.. Steering against wind: a new network of NamiRNAs and enhancers. Genomics Proteomics Bioinformatics. 2017; 15:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki H.I., Young R.A., Sharp P.A.. Super-enhancer-mediated RNA processing revealed by integrative microRNA network analysis. Cell. 2017; 168:1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller K.D., Nogueira L., Mariotto A.B., Rowland J.H., Yabroff K.R., Alfano C.M., Jemal A., Kramer J.L., Siegel R.L.. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019; 69:363–385. [DOI] [PubMed] [Google Scholar]
- 39.Perou C.M., Sorlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A.et al.. Molecular portraits of human breast tumours. Nature. 2000; 406:747–752. [DOI] [PubMed] [Google Scholar]
- 40.Zhao M., Sun J., Zhao Z.. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 2013; 41:D970–D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao M., Kim P., Mitra R., Zhao J., Zhao Z.. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016; 44:D1023–D1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ignatov T., Weißenborn C., Poehlmann A., Lemke A., Semczuk A., Roessner A., Costa S.D., Kalinski T., Ignatov A.. GPER-1 expression decreases during breast cancer tumorigenesis. Cancer Invest. 2013; 31:309–315. [DOI] [PubMed] [Google Scholar]
- 43.Poola I., Abraham J., Liu A., Marshalleck J.J., Dewitty R.L.. The cell surface estrogen receptor, G protein-coupled receptor 30 (GPR30), is markedly down regulated during breast tumorigenesis. Breast Cancer (Auckl). 2008; 1:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weissenborn C., Ignatov T., Poehlmann A., Wege A.K., Costa S.D., Zenclussen A.C., Ignatov A.. GPER functions as a tumor suppressor in MCF-7 and SK-BR-3 breast cancer cells. J. Cancer Res. Clin. Oncol. 2014; 140:663–671. [DOI] [PubMed] [Google Scholar]
- 45.Ariazi E.A., Brailoiu E., Yerrum S., Shupp H.A., Slifker M.J., Cunliffe H.E., Black M.A., Donato A.L., Arterburn J.B., Oprea T.I.et al.. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 2010; 70:1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang C., Lv X., He C., Hua G., Tsai M.Y., Davis J.S.. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death. Dis. 2013; 4:e869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thienpont B., Steinbacher J., Zhao H., D’Anna F., Kuchnio A., Ploumakis A., Ghesquiere B., Van Dyck L., Boeckx B., Schoonjans L.et al.. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016; 537:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang S., Wang R.H., Akagi K., Kim K.A., Martin B.K., Cavallone L.Kathleen Cuningham Foundation Consortium for Research into Familial Breast, C. Kathleen Cuningham Foundation Consortium for Research into Familial Breast, C. Haines D.C., Basik M., Mai P.et al.. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat. Med. 2011; 17:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J., Haanpaa M.K., Gruber J.J., Jager N., Ford J.M., Snyder M.P.. High-Resolution bisulfite-sequencing of peripheral blood DNA methylation in early-onset and familial risk breast cancer patients. Clin. Cancer Res. 2019; 25:5301–5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherr C.J.Principles of tumor suppression. Cell. 2004; 116:235–246. [DOI] [PubMed] [Google Scholar]
- 51.Datta J., Smith A., Lang J.C., Islam M., Dutt D., Teknos T.N., Pan Q.. microRNA-107 functions as a candidate tumor-suppressor gene in head and neck squamous cell carcinoma by downregulation of protein kinase C epsilon. Oncogene. 2012; 31:4045–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding L., Xu Y.J., Zhang W., Deng Y.J., Si M.S., Du Y., Yao H.M., Liu X.Y., Ke Y.H., Si J.M.et al.. MiR-375 frequently downregulated in gastric cancer inhibits cell proliferation by targeting JAK2. Cell Res. 2010; 20:784–793. [DOI] [PubMed] [Google Scholar]
- 53.Nawaz Z., Patil V., Paul Y., Hegde A.S., Arivazhagan A., Santosh V., Somasundaram K.. PI3 kinase pathway regulated miRNome in glioblastoma: identification of miR-326 as a tumour suppressor miRNA. Mol. Cancer. 2016; 15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu Z.S., Wu Q., Wang C.Q., Wang X.N., Wang Y., Zhao J.J., Mao S.S., Zhang G.H., Zhang N., Xu X.C.. MiR-339-5p inhibits breast cancer cell migration and invasion in vitro and may be a potential biomarker for breast cancer prognosis. BMC Cancer. 2010; 10:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma K.L., Pan X.R., Fan P.S., He Y.H., Gu J., Wang W., Zhang T.Y., Li Z.H., Luo X.Y.. Loss of miR-638 in vitro promotes cell invasion and a mesenchymal-like transition by influencing SOX2 expression in colorectal carcinoma cells. Mol. Cancer. 2014; 13:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jansson M.D., Damas N.D., Lees M., Jacobsen A., Lund A.H.. miR-339-5p regulates the p53 tumor-suppressor pathway by targeting MDM2. Oncogene. 2015; 34:1908–1918. [DOI] [PubMed] [Google Scholar]
- 57.Bertoli G., Cava C., Castiglioni I.. MicroRNAs: new biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics. 2015; 5:1122–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Q., Chen Z.J., Jiang G.M., Zhou Y., Yang X.L., Huang H.B., Liu H.L., Du J., Wang H.S.. Epigenetic down regulation of G protein-coupled estrogen receptor (GPER) functions as a tumor suppressor in colorectal cancer. Mol. Cancer. 2017; 16:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brinkman A.B., Nik-Zainal S., Simmer F., Rodriguez-Gonzalez F.G., Smid M., Alexandrov L.B., Butler A., Martin S., Davies H., Glodzik D.et al.. Partially methylated domains are hypervariable in breast cancer and fuel widespread CpG island hypermethylation. Nat. Commun. 2019; 10:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smid M., Wang Y., Zhang Y., Sieuwerts A.M., Yu J., Klijn J.G., Foekens J.A., Martens J.W.. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008; 68:3108–3114. [DOI] [PubMed] [Google Scholar]
- 61.Salomon W.E., Jolly S.M., Moore M.J., Zamore P.D., Serebrov V.J.C.. Single-molecule imaging reveals that argonaute reshapes the binding properties of its nucleic acid guides. Cell. 2015; 162:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chimonidou M., Strati A., Tzitzira A., Sotiropoulou G., Malamos N., Georgoulias V., Lianidou E.S.. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin. Chem. 2011; 57:1169–1177. [DOI] [PubMed] [Google Scholar]
- 63.Korkmaz G., Lopes R., Ugalde A.P., Nevedomskaya E., Han R., Myacheva K., Zwart W., Elkon R., Agami R.. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016; 34:192–198. [DOI] [PubMed] [Google Scholar]
- 64.Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A.et al.. MicroRNA expression profiles classify human cancers. Nature. 2005; 435:834–838. [DOI] [PubMed] [Google Scholar]
- 65.Zhu M., Yi M., Kim C.H., Deng C., Li Y., Medina D., Stephens R.M., Green J.E.. Integrated miRNA and mRNA expression profiling of mouse mammary tumor models identifies miRNA signatures associated with mammary tumor lineage. Genome Biol. 2011; 12:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Persson M., Andren Y., Mark J., Horlings H.M., Persson F., Stenman G.. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18740–18744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Akao Y., Nakagawa Y., Naoe T.. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol. Pharm. Bull. 2006; 29:903–906. [DOI] [PubMed] [Google Scholar]
- 68.Preusse M., Theis F.J., Mueller N.S.. miTALOS v2: analyzing tissue specific microrna function. PLoS One. 2016; 11:e0151771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ren H., Zhang Y., Zhu H.. MiR-339 depresses cell proliferation via directly targeting S-phase kinase-associated protein 2 mRNA in lung cancer. Thorac Cancer. 2018; 9:408–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen B., Zhang Y., Yu S., Yuan Y., Zhong Y., Lu J., Feng J.. MicroRNA-339, an epigenetic modulating target is involved in human gastric carcinogenesis through targeting NOVA1. FEBS Lett. 2015; 589:3205–3211. [DOI] [PubMed] [Google Scholar]
- 71.Weber C.E., Luo C., Hotz-Wagenblatt A., Gardyan A., Kordass T., Holland-Letz T., Osen W., Eichmuller S.B.. miR-339-3p Is a tumor suppressor in melanoma. Cancer Res. 2016; 76:3562–3571. [DOI] [PubMed] [Google Scholar]
- 72.Ignatov T., Modl S., Thulig M., Weissenborn C., Treeck O., Ortmann O., Zenclussen A., Costa S.D., Kalinski T., Ignatov A.. GPER-1 acts as a tumor suppressor in ovarian cancer. J Ovarian Res. 2013; 6:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weissenborn C., Ignatov T., Poehlmann A., Wege A.K., Costa S.D., Zenclussen A.C., Ignatov A.. GPER functions as a tumor suppressor in MCF-7 and SK-BR-3 breast cancer cells. J Cancer Res Clin. 2014; 140:663–671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data generated in this paper is GEO: GSE78011 and GSE174002. For breast cancer and kidney renal clear cell carcinoma samples, expression data were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/).