

Abstract
Background
Poly (ADP-ribose) polymerase (PARP) inhibitors, which are among the most important breakthroughs in precision medicine, have played a crucial role in cancer treatment. Understanding the toxicity profiles of the different PARP inhibitors will improve strategic treatment in clinical practice.
Methods
PubMed, Cochrane Library, and Web of Science were systematically searched to include related studies published in English between January 2009 and February 2020. Only prospective, phase II and III randomized controlled trials were included. The following treatment groups were analyzed: niraparib, talazoparib, olaparib, rucaparib, conventional therapy (chemotherapy), one PARP inhibitor with one angiogenesis inhibitor, and placebo. Baseline data and adverse event data were extracted from the Bayesian random-effects network meta-analysis.
Results
Fourteen phase II and III randomized controlled trials (4,336 patients) were included. When considering grade 3–5 adverse events, olaparib may be a better choice (probability =57%), followed by conventional therapy (50%), talazoparib (45%), rucaparib (75%), niraparib (77%), and a PARP inhibitor with one angiogenesis inhibitor (94%). Niraparib and rucaparib had higher risks for hematological and gastrointestinal toxicities, respectively. Talazoparib was safer for gastrointestinal function. Constipation and neutropenia were less observed in olaparib, but the risks for anorexia increased. The combination of PARP inhibitor and angiogenesis inhibitor increased the risk of general, metabolic, and gastrointestinal disorders.
Conclusions
This network meta-analysis suggested that the toxicity spectrum of each PARP inhibitor is different. Olaparib had the best safety profile among all PARP inhibitors because of its mild toxicity and narrow spectrum. This study may guide clinicians and support further research.
Keywords: Poly (ADP-ribose) polymerase inhibitors (PARP inhibitors), network meta-analysis, adverse events, randomized controlled trials
Introduction
In recent years, a deeper understanding of the molecular characteristics of tumors has driven the development of precise treatment. The novel and promising poly (ADP-ribose) polymerase (PARP) inhibitors have led to breakthroughs in cancer treatment (1,2). They were designed to exploit synthetic lethality and kill cells that have homologous recombination deficiency (3). Great effectiveness has been observed in BRCA-mutant tumors or tumors with deficiencies in the homologous recombination repair pathway (4).
Currently, PARP inhibitors have been employed in the treatment of breast and ovarian cancer, either alone or in combination with other therapies (2,5). According to NCCN guidelines, HER2-negative metastatic breast cancer patients with germline BRCA1/2 mutations could receive treatment with olaparib or talazoparib. Ovarian cancer patients with germline or somatic BRCA1/2 mutations are recommended to receive PARP inhibitors alone or combined with bevacizumab. To enhance the anti-tumor effectiveness, clinical scientists are also investigating the most potent synergistic efficacy partner of PARP inhibitors. The efficacy and safety of the combination therapy, including PARP inhibitors plus angiogenesis inhibitors (6,7), immune checkpoint inhibitors (8-10), cyclin dependent kinase (CDK) 4/6 inhibitors (11,12), phosphoinositide 3-kinase (PI3K) inhibitors (13), protein kinase B (AKT) inhibitors (14), mammalian target of rapamycin (mTOR) inhibitors (15), WEE1 inhibitors (16) or mitogen-activated protein kinase (MEK) inhibitors (17) are being evaluated through various clinical trials.
To further explore the therapeutic potential of PARP inhibitors, clinical trials in other tumors have also been conducted. On one hand, PARP inhibitors have shown effectiveness in patients carrying BRCA1/2 mutations beyond breast or ovarian cancer, such as prostate cancer (18) and pancreatic cancer (19). On the other hand, numerous clinical trials have sought to extend the promising function of PARP inhibitors to tumors with BRCAness (3,20). For example, the concept of combining PARP inhibitors with targeted therapy involving homologous recombination repair genes, including ATM, RAD51, CHEK2 and PTEN, has brought novel insights in cancer treatment (21-23).
However, despite the great effectiveness of PARP inhibitors, their toxicity profiles are not yet understood. Because of differences in their mechanisms, the various PARP inhibitor drugs differ in terms of safety (24-26). The safety profile of the combination therapy with other drugs, such as an angiogenesis inhibitor, also remains to be determined.
Since the traditional meta-analysis is limited to pairwise comparisons, it is impossible to determine the relative advantages of candidate therapies that have not yet been directly compared. By introducing indirect comparison, network meta-analysis can not only solve this limitation, but also improve the accuracy by combining direct and indirect estimations (27).
Here, we conducted a network meta-analysis of randomized controlled trials to compare the relative safety of PARP inhibitors alone, the combination of PARP inhibitors and angiogenesis inhibitors, and conventional therapy. We reported the results of both dose- and drug-based meta-analyses and ranked these treatment options on the basis of safety. The subgroup analysis was carried out according to different specific adverse events and cancer type (ovarian cancer). We present the following article in accordance with the PRISMA reporting checklist (available at https://dx.doi.org/10.21037/atm-21-1883).
Methods
This network meta-analysis was conducted following the preferred reporting items for systematic reviews and meta-analysis (PRISMA) and the PRISMA extension statement for network meta-analysis (28). A protocol was created in the Prospective Register of Systematic Reviews (PROSPERO CRD42020198263).
Date sources and searches
The databases of PubMed, Cochrane Library, and Web of Science were used to search for related studies. Randomized controlled trials published in English between January 2009 and February 2020 were eligible. Key words used in the search strategy included cancer, carcinoma, neoplasm, poly (ADP-ribose) polymerase inhibitors (BRCA mutation, PARP inhibitors, PARPis, PARPi), and specific PARPi drug names (fluzoparib, niraparib, olaparib, rucaparib, talazoparib, iniparib, and veliparib). The search strategy will be described in detail in the supplementary materials. Two independent investigators (SN-B, YP-Y) evaluated the data of the related publications after the primary screening of all eligible studies.
Study selection
Only prospective, phase II and III randomized controlled trials that compared two or three treatments in the following group were included: niraparib, talazoparib, olaparib, rucaparib, conventional therapy (chemotherapy), one PARP inhibitor with one angiogenesis inhibitor, or placebo. Studies that compared different doses of one PARP inhibitor were also included. Eligible studies must have toxicity regarding adverse events of all grades. Conference abstracts, posters, and ongoing randomized controlled trials were excluded. Trials wherein PARP inhibitors were used as neoadjuvant or adjuvant treatments were also excluded.
Data extraction and quality assessment
The primary outcome was the difference in adverse events between different PARP inhibitors. The study name, study ID, first author, year of publication, cancer type, study design, number of patients, treatments, region, version of the Common Terminology Criteria for Adverse Events (CTCAE), the frequency of each specific grade 1–5 and grade 3 or higher adverse events, and follow-up time were extracted and summarized. The CTCAE was used to evaluate adverse events. Adverse events of any grade indicated complete toxicity while grade 3 or higher adverse events indicated severe toxicity. We evaluated the full text, reference lists of the related publications, and supplementary materials from clinicaltrials.gov and other available sources to obtain the latest and complete data. For each study, both original and updated information were extracted and reviewed in this network meta-analysis.
Two independent investigators (SNB, YJH) assessed the risk of bias in the randomized trials using the Cochrane Collaboration’s tool. The considered sources of bias included random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and others (29). Methodological quality was also evaluated using the modified Jadad quality scale (30). Scores ≥4 were considered to be of high quality (Supplementary materials, Table S1).
Data synthesis and statistical analysis
Direct and indirect data from all eligible trials, including odds ratios and 95% confidence intervals (CIs), were synthesized to evaluate the difference in safety between the various PARP inhibitors. The safety profiles of the PARP inhibitors were analyzed in both overview and detail, according to the number of general and specific adverse events. Both dose- and drug-based analyses compared and assessed grade 3 or higher adverse events. All reported P values were two-sided, and results with P<0.05 were considered significant. Odds ratios >1 represented a safety benefit for the control group.
To compare the different treatments, we generated a network diagram using STATA version 14.0 (StataCorp) (31). A Bayesian framework using Markov chain Monte Carlo methods in R version 4.0.2 and its appropriate packages was employed in both dose- and drug-based network meta-analyses (32). The drug based meta-analysis, wherein we combined different treatment arms with different doses of the same PARP inhibitor into one arm, was conducted after completing the dose-based meta-analysis. Studies that only had one treatment group after the combination were ruled out. To address inter-study heterogeneity, random effects and consistency models were employed to calculate odds ratios and 95% CIs (33). To achieve posterior distributions of model parameters, non-informative uniform and normal prior distributions were used. Besides, initial values of over-dispersion (with a scaling of 5) in four chains were also applied to fit the model (32). Overall, 100,000 iterations (including 100,000 tuning iterations) and a thinning interval of 10 for each chain were generated. The convergence of iterations was evaluated in accordance with the Brooks-Gelman-Rubin statistic (34). The safety of each treatment was assessed and ranked according to the odds ratio and posterior probabilities.
Consistency between direct and indirect evidence is imperative to achieve reliable results. The inconsistency of the entire network was assessed by node splitting analysis, wherein P<0.05 indicated significant inconsistency (35,36). The frequentist framework was also used in pairwise meta-analysis to estimate odds ratios and 95% CIs. We evaluated the inter-study heterogeneity using the χ2 and I2 tests. Statistical significance was set at P<0.05. The heterogeneity was considered to be low, moderate, and high when I2 value was estimated to be under 25%, 25–50%, and more than 50%, respectively (37). The fixed effects model was used when there was no significant heterogeneity. Otherwise, the random effects model was used. Results from the network (indirect) and pairwise (direct results) meta-analysis were compared to check for inconsistency.
Subgroup analyses were carried out according to different specific adverse events and cancer types. To ensure the stability and reliability of the network meta-analysis, we also performed a sensitivity analysis by controlling the source of this study, including phase III randomized controlled trials, studies using the current recommended dosage of PARP inhibitors, and studies explicitly reporting grade ≥3 adverse events.
Results
Systematic review and characteristics
The initial search yielded 1,080 potentially relevant studies, of which 121 reports were assessed in full text, and 14 randomized controlled trials were declared eligible (Figure 1) (6,19,38-49). In the dose based-network meta-analysis, 13 randomized controlled studies (6,19,38,39,41-49), representing nine treatments with different doses [niraparib 300 mg once daily; talazoparib 1 mg once daily; olaparib 200, 300 or 400 mg twice daily; rucaparib 600 mg twice daily; one PARP inhibitor with one angiogenesis inhibitor; conventional therapy (chemotherapy) and placebo], (n=4,219) were evaluated (Figure 2A). Twelve randomized controlled trials (6,19,38,39,41-48), which included seven treatments [niraparib, talazoparib, olaparib, rucaparib, one PARP inhibitor with one angiogenesis inhibitor, conventional therapy (chemotherapy), and placebo], (n=4,121) were assessed in the drug-based network meta-analysis (Figure 2B). The main characteristics of all studies are shown in Table 1. Nine studies (64.3%) were phase III trials and, five (35.7%) were phase II. Twelve (85.7%) were multinational trials. Thirteen (92.9%) studies were two-arm trials, and one (7.2%) included three arms. Cancer types investigated in these studies included breast, ovarian, prostate, and pancreatic cancers. The modified Jadad scores indicated that 14 studies were of high quality (Supplementary materials, Table S1).
Figure 1.
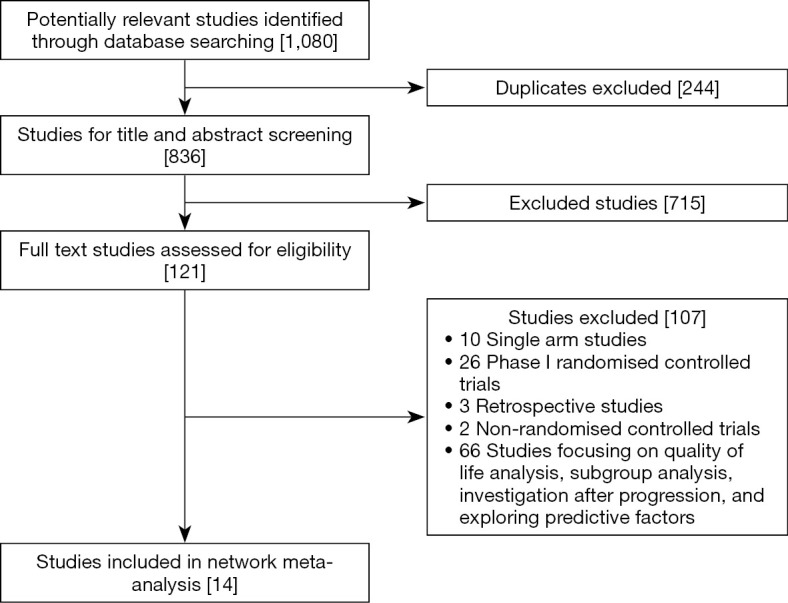
Flowchart of study selection.
Figure 2.

Network plots of comparisons for dose (A) and drug (B) based network meta-analyses. Each circular node represents a type of treatment. The circle size is proportional to the total number of patients. The width of lines is proportional to the number of studies performing head-to-head comparisons in the same study. PARPi, poly (ADP-ribose) polymerase inhibitor; AI, angiogenesis inhibitor.
Table 1. Baseline characteristics of 14 studies for Bayesian network meta-analysis by cancer type.
| First author, year | Study ID | Region | Trial phase | Total No. | Safety analysis No | Arm | Treatment (median follow-up time, months) | CTCAE version |
|---|---|---|---|---|---|---|---|---|
| Breast cancer | ||||||||
| Advanced HER2-negative breast cancer and a germline BRCA1/2 mutation | ||||||||
| Jennifer K. Litton, 2018 | EMBRACA | MN | III | 431 | 286 | 1 | Talazoparib 1 mg once daily (11.2) | 4.03 |
| 126 | 2 | ICC (capecitabine, eribulin, gemcitabine, or vinorelbine every 3 weeks) (11.2) | ||||||
| Mark Robson, 2017 | OlympiAD | MN | III | 302 | 205 | 1 | Olaparib 300 mg twice daily (14.5) | 4.0 |
| 91 | 2 | ICC (capecitabine, eribulin, or vinorelbine every 3 weeks) (14.1) | ||||||
| Ovarian cancer | ||||||||
| Measurable or evaluable high-grade serous or endometrioid platinum-sensitive recurrent ovarian cancer | ||||||||
| Mansoor Raza Mirza, 2019 | NSGO-AVANOVA2 | MN | II | 97 | 48 | 1 | Niraparib 300mg once daily plus bevacizumab 15mg/kg once every 3 weeks (16.9) | 4.0 |
| 49 | 2 | Niraparib 300mg once daily (16.9) | ||||||
| Joyce F. Liu, 2019* | NCT01116648 | USA | II | 90 | 46 | 1 | Olaparib 400 mg twice daily (46.0) | 4.0 |
| 44 | 2 | Olaparib 200 mg twice daily and cediranib 30 mg daily (46.0) | ||||||
| Robert L. Coleman, 2017 | ARIEL3 | MN | III | 564 | 372 | 1 | Rucaparib 600 mg twice daily (NR) | 4.03 |
| 189 | 2 | Placebo (NR) | ||||||
| Ovarian cancer that recurred within 12 months of prior platinum therapy and with confirmed germline BRCA1 or BRCA2 mutations | ||||||||
| Stan B. Kaye, 2011† | NCT00628251 | MN | II | 97 | 32 | 1 | Olaparib 200 mg twice daily (NR) | 3.0 |
| 32 | 2 | Olaparib 400 mg twice daily (NR) | ||||||
| 32 | 3 | Pegylated liposomal doxorubicin 50 mg/m2 intravenously every 28 days (NR) | ||||||
| Advanced ovarian cancer following response on front-line platinum-based chemotherapy | ||||||||
| A. González-Martín, 2019 | PRIMA | MN | III | 733 | 484 | 1 | Niraparib 300 mg once daily (13.8) | 4.03 |
| 244 | 2 | Placebo (13.8) | ||||||
| High-grade serous platinum-sensitive, recurrent ovarian cancer | ||||||||
| Mansoor R. Mirza, 2016 | ENGOT-OV16/NOVA | MN | III | 553 | 367 | 1 | Niraparib 300 mg once daily (16.9) | 4.02 |
| 179 | 2 | Placebo (16.9) | ||||||
| Jonathan Ledermann, 2012 | Study 19 NCT00753545 | MN | II | 265 | 136 | 1 | Olaparib 400 mg twice daily (78.0) | 3.0 |
| 128 | 2 | Placebo (78.0) | ||||||
| Newly diagnosed advanced high-grade serous or endometrioid platinum-sensitive ovarian cancer with BRCA1 or BRCA2 mutations | ||||||||
| K. Moore, 2018 | SOLO1 | MN | III | 391 | 260 | 1 | Olaparib 300 mg twice daily (40.7) | 4.0 |
| 130 | 2 | Placebo (41.2) | ||||||
| Advanced high-grade serous or endometrioid platinum-sensitive ovarian cancer with BRCA1 or BRCA2 mutations | ||||||||
| Eric Pujade-Lauraine, 2017 | SOLO2 | MN | III | 295 | 195 | 1 | Olaparib 300 mg twice daily (22.1) | 4.0 |
| 99 | 2 | Placebo (22.2) | ||||||
| Richard T. Penson, 2020 | SOLO3 | MN | III | 266 | 178 | 1 | Olaparib 300 mg twice daily (13.8) | 4.0 |
| 76 | 2 | ICC (pegylated liposomal doxorubicin, paclitaxel, gemcitabine, or topotecan) (3.9) | ||||||
| Prostate cancer | ||||||||
| Metastatic castration-resistant prostate cancer with DDR gene aberrations | ||||||||
| Joaquin Mateo, 2020† | TOPARP-B | UK | II | 98 | 49 | 1 | Olaparib 400 mg twice daily (24.8) | 4.02 |
| 49 | 2 | Olaparib 300 mg twice daily (24.8) | ||||||
| Pancreatic cancer | ||||||||
| Metastatic pancreatic adenocarcinoma with germline BRCA mutations that had not progressed during first-line platinum-based chemotherapy | ||||||||
| Talia Golan, 2019 | POLO | MN | III | 154 | 91 | 1 | Olaparib 300 mg twice daily (9.1) | 4.0 |
| 60 | 2 | Placebo (3.8) | ||||||
*, the study was excluded from the dose and drug based network meta-analysis; †, grade ≥3 adverse events were not available in the study by Stan B. Kaye-2011/Joaquin Mateo-2020, so serious adverse events were used. CTCAE, Common Terminology Criteria for Adverse Events; MN, multinational; NR, not reported; ICC, investigator’s choice chemotherapy.
Dose based network meta-analysis
To investigate the safety profiles of differing doses of one PARP inhibitor, a dose-based network meta-analysis was conducted. The results obtained in the consistency model indicated that, in terms of grade 3–5 adverse events, there was no significant difference between varied olaparib doses (Supplementary materials, Figure S1). The node splitting analysis also demonstrated that there was no significant inconsistency (all P>0.05; Supplementary materials, Table S2). Therefore, there was no significant difference in the safety profile between different doses of one PARP inhibitor, and this was an important prerequisite for the drug-based network meta-analysis.
Drug based network meta-analysis
The drug-based network meta-analysis compares the risks of grade 3–5 adverse events between niraparib, talazoparib, olaparib, rucaparib, a PARP inhibitor with one angiogenesis inhibitor, conventional therapy (chemotherapy), and placebo. The results are shown in Figure 3A. Logically, the placebo group was found to have the lowest risk of grade 3–5 adverse events. The combination of one PARP inhibitor and one angiogenesis inhibitor was shown to cause more toxicity than all other treatments except niraparib and rucaparib. Olaparib was significantly safer than niraparib and rucaparib, but the difference between talazoparib was not significant. Niraparib had a higher risk of adverse events than talazoparib and olaparib, but it was not significantly different compared to rucaparib. It was also indicated that conventional therapy (chemotherapy) was significantly safer than niraparib and rucaparib but was not different from the other two PARP inhibitors.
Figure 3.

Safety profile (A) and ranking histograms (B) according to the drug based network meta-analysis in the consistency model. Each cell of the safety profile contains the pooled odds ratios and 95% confidence intervals for grade 3–5 adverse events; significant results are in bold. Ranking histograms indicate the probability of the highest risk of grade 3–5 adverse events, the second highest, the third highest, and so on. PARPi, poly (ADP-ribose) polymerase inhibitor; AI, angiogenesis inhibitor.
The safety ranking for grade 3–5 adverse events, from the lowest to highest risk, was placebo, olaparib, conventional therapy (chemotherapy), talazoparib, rucaparib, niraparib, and one PARP inhibitor with one angiogenesis inhibitor (Figure 3B). Therefore, in clinical practice, when considering grade 3–5 adverse events, olaparib may be a better choice (probability =57%), followed by conventional therapy (50%), talazoparib (45%), rucaparib (75%), niraparib (77%), and a PARP inhibitor with one angiogenesis inhibitor (94%). The probabilities are detailed in the Supplementary materials, Table S3.
In addition to inducing synthetic lethality, PARP1 or PARP2 become trapped in DNA damage sites, leading to PARP-DNA complexes and interfering with DNA replication (25,50). The rank order of trapping potency is talazoparib >> olaparib = rucaparib > niraparib = veliparib in HeyA8 cells treated with 1 mmol/L methyl methanesulfonate. This difference is believed to be the main reason for the different recommended doses of existing PARP inhibitors, as PARP trapping appears to drive myelosuppresion (26).
The results of the three head-to-head comparisons are presented in Figure 4. Pairwise meta-analysis of two comparisons, which were olaparib and niraparib versus placebo, demonstrated that our network meta-analysis results were prominently consistent. No significant inter-study heterogeneity was found in any pairwise meta-analysis comparisons (I2=0%; P>0.05) (Supplementary materials, Figure S2).
Figure 4.

Forest plots depicting the direct and indirect results of head-to-head comparisons.
Subgroup analysis based on specific adverse events
Based on specific grade 1–5 adverse events, the subgroup network meta-analysis included nine to 13 studies (3,372 patients to 4,211 patients). We evaluated the risk of all treatment groups for specific adverse events, including hematological disorders (anemia, neutropenia and thrombocytopenia), gastrointestinal disorders (nausea, vomiting, constipation), metabolism disorders (anorexia), and general disorders (fatigue) (Table 2). In terms of the safety of the PARP inhibitor monotherapy, no meaningful results were observed between the different PARP inhibitors. However, olaparib was safer than niraparib for constipation and thrombocytopenia. Compared to the combination of PARP inhibitor and angiogenesis inhibitor, niraparib was safer for anorexia, and olaparib was safer for anorexia and constipation. No differences were found between the combination and talazoparib or rucaparib. Interestingly, compared to conventional therapy, the risk for nausea, vomiting, and anemia was increased in almost all PARP inhibitors. Only olaparib was found to be safer than conventional therapy for neutropenia.
Table 2. Effect of treatment on each specific grade 1-5 adverse event.
| Group | Fatigue | Anorexia | Nausea | Vomiting | Constipation | Anaemia | Neutropenia | Thrombocytopenia |
|---|---|---|---|---|---|---|---|---|
| No of studies | 13 | 10 | 13 | 13 | 13 | 13 | 10 | 9 |
| No of patients | 4,211 | 3,101 | 4,211 | 4,211 | 4,211 | 4,211 | 3,668 | 3,372 |
| Niraparib as control | ||||||||
| Talazoparib | 1.2 | 0.62 | 0.469 | 0.644 | 0.816 | 0.902 | 1.012 | 0.786 |
| Olaparib | 1.225 | 0.999 | 1.135 | 1.237 | 0.47* | 0.552 | 0.565 | 0.185* |
| Rucaparib | 1.617 | 0.88 | 1.3 | 1.438 | 0.654 | 1.064 | 0.825 | 0.637 |
| Conventional therapy | 0.884 | 0.65 | 0.437* | 0.581 | 0.787 | 0.176* | 1.431 | 0.157 |
| Placebo | 0.557* | 0.457 | 0.244* | 0.43* | 0.354* | 0.106* | 0.183* | 0.041* |
| PARPi + AI | 2.033 | 3.461* | 1.348 | 2.048 | 1.29 | 0.766 | 0.78 | 0.817 |
| Talazoparib as control | ||||||||
| Olaparib | 1.015 | 1.602 | 2.418 | 1.917 | 0.576 | 0.612 | 0.557 | 0.234 |
| Rucaparib | 1.343 | 1.42 | 2.774 | 2.238 | 0.8 | 1.191 | 0.816 | 0.817 |
| Conventional therapy | 0.734 | 1.047 | 0.933 | 0.9 | 0.961 | 0.196* | 1.413 | 0.202* |
| Placebo | 0.462 | 0.73 | 0.521 | 0.667 | 0.433 | 0.118* | 0.18* | 0.053* |
| PARPi + AI | 1.691 | 5.623 | 2.847 | 3.17 | 1.58 | 0.856 | 0.762 | 1.036 |
| Olaparib as control | ||||||||
| Rucaparib | 1.318 | 0.884 | 1.149 | 1.16 | 1.391 | 1.929 | 1.462 | 3.467 |
| Conventional therapy | 0.723 | 0.655 | 0.386* | 0.47* | 1.677 | 0.319* | 2.532* | 0.853 |
| Placebo | 0.454* | 0.456* | 0.215* | 0.348* | 0.751 | 0.193* | 0.324* | 0.224* |
| PARPi + AI | 1.663 | 3.486* | 1.186 | 1.655 | 2.739* | 1.389 | 1.369 | 4.385 |
| Rucaparib as control | ||||||||
| Conventional therapy | 0.547 | 0.741 | 0.336* | 0.403* | 1.199 | 0.164* | 1.729 | 0.246 |
| Placebo | 0.345* | 0.516 | 0.187* | 0.299* | 0.54 | 0.1* | 0.222* | 0.065* |
| PARPi + AI | 1.256 | 3.951 | 1.036 | 1.435 | 1.968 | 0.716 | 0.933 | 1.258 |
| Conventional therapy as control | ||||||||
| Placebo | 0.63 | 0.699 | 0.558 | 0.74 | 0.449* | 0.608 | 0.128* | 0.262 |
| PARPi + AI | 2.308 | 5.327 | 3.063* | 3.533* | 1.644 | 4.385* | 0.54 | 5.185 |
| Placebo as control | ||||||||
| PARPi + AI | 3.664* | 7.671* | 5.515* | 4.75* | 3.665* | 7.179* | 4.262* | 19.788* |
Values are odds ratios. Significant values are shown in *. PARPi: poly (ADP-ribose) polymerase inhibitor; AI, angiogenesis inhibitor.
We also assessed the ranking profile of all treatment groups (Figure 5). The combination of PARP inhibitor and angiogenesis inhibitor had the highest risk for causing general, metabolic, and gastrointestinal disorders. Niraparib had a higher risk of causing hematological toxicities, including anemia, thrombocytopenia, and neutropenia. The main adverse events of rucaparib were gastrointestinal disorders. Talazoparib was the safest PARP inhibitors for gastrointestinal function. Moreover, olaparib, which has a narrow and mild toxicity profile, had the lowest risk for constipation and neutropenia, but a higher risk for anorexia. Interestingly, the conventional therapy had the lowest risk for fatigue, nausea, vomiting, anemia, and thrombocytopenia, but the highest risk for neutropenia.
Figure 5.

Toxicity spectra and rankings in the subgroup analysis based on each specific grade 1-5 adverse event and cancer type. PARP inhibitor drugs are shown with a dark background. CT, conventional therapy; PARPi, poly (ADP-ribose) polymerase inhibitor; AI, angiogenesis inhibitor.
Subgroup analysis based on cancer type (ovarian cancer)
The approval of PARP inhibitors (olaparib, niraparib, and rucaparib) for recurrent epithelial ovarian cancer has emerged as a new and exciting treatment method. We therefore assessed the safety of different PARP inhibitors in ovarian cancer.
This subgroup analysis included nine studies (6,41-48), and only adverse events of grade 3 or higher were analyzed (Figure 6). This indicated that olaparib was safer than niraparib, rucaparib, and the combination of PARP inhibitor and angiogenesis inhibitor. Conventional therapy had lower risks for grade 3–5 adverse events than niraparib, rucaparib, and the combination of PARP inhibitor and angiogenesis inhibitor, but was not different from that of olaparib. No differences were observed between the combination of PARP inhibitor and angiogenesis inhibitor, and niraparib or rucaparib. The safety ranking, from the lowest to the highest risk, was placebo, conventional therapy, olaparib, rucaparib, niraparib, and one PARP inhibitor with one angiogenesis inhibitor. In conclusion, olaparib may be the safest option among the analyzed PARP inhibitors for the treating ovarian cancer.
Figure 6.
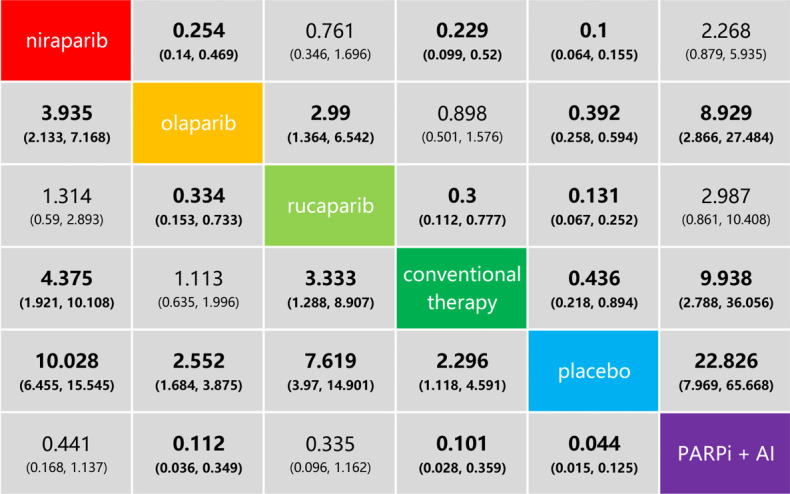
Safety profiles in the subgroup analysis based on ovarian cancer. Each cell of the safety profile contains the pooled odds ratios and 95% confidence intervals for grade 3–5 adverse events; significant results are in bold. PARPi, poly (ADP-ribose) polymerase inhibitor; AI, angiogenesis inhibitor.
Sensitivity analysis
To ensure the reliability of this study, we conducted a sensitivity analysis on three subgroups: phase III studies (19,38,39,42-44,46-48), studies explicitly reporting grade ≥3 adverse events (6,19,38,39,42-48), and studies using the current recommended dosage of PARP inhibitors (6,19,38,39,42-44,46-48). Similar results were observed in these three subgroups.
The individual sensitivity analysis of nine phase III studies suggested that olaparib was safer than conventional therapy and all other PARP inhibitors. This finding was consistent with the drug-based network meta-analysis. Talazoparib, rucaparib, and niraparib caused more risks for adverse events than the conventional therapy. The combination of a PARP inhibitor and an angiogenesis inhibitor was absent in this analysis. Two additional sensitivity analyses, based on 11 studies explicitly reporting grade ≥3 adverse events and 10 studies using the current recommended dosage of PARP inhibitors, showed that the safety ranking from the lowest to the highest risk, was placebo, olaparib, conventional therapy, talazoparib, rucaparib, niraparib, and one PARP inhibitor plus one angiogenesis inhibitor (Supplementary materials, Table S4).
Discussion
The successful development of PARP inhibitors has become a milestone in cancer treatment. Despite their promising effectiveness in clinical practice, the toxicity profiles of different PARP inhibitors need to be clarified.
To the best of our knowledge, there have been relatively few network meta-analyses comparing the toxicity outcomes of PARP inhibitors for the treatment of advanced cancers. In this network meta-analysis, we included 14 randomized controlled studies (4,336 patients) to compare the safety profile of placebo, niraparib, talazoparib, olaparib, rucaparib, conventional therapy (chemotherapy), and one PARP inhibitor with one angiogenesis inhibitor. Because of the strict inclusion criteria and a reasonable study design, our network meta-analysis features good transitivity, and results achieved in this study can be interpreted appropriately (51,52). The sensitivity analysis further confirmed the reliability of our results, ensuring the homogeneity and consistency of our study.
In the dose-based safety analysis, no significant differences were found between different doses of one PARP inhibitor. The general safety profile ranking, from lowest to highest, was placebo, olaparib, conventional therapy (chemotherapy), talazoparib, rucaparib, niraparib, and one PARP inhibitor with one angiogenesis inhibitor in the drug-based analysis.
In the subgroup analyses according to different specific adverse events, the toxicity spectrum of each PARP inhibitor was different. Niraparib and rucaparib had higher risks for hematological and gastrointestinal toxicities, respectively. Talazoparib was safer for gastrointestinal function. Constipation and neutropenia were less observed in olaparib, but the risks for anorexia increased. The combination of PARP inhibitor and angiogenesis inhibitor increased the risk of general, metabolic, and gastrointestinal disorders. In addition, the subgroup analysis based on cancer type revealed that olaparib may be a better choice for treating ovarian cancer. Moreover, similar results were reported in the sensitivity analyses. Therefore, olaparib is considered a safer drug for cancer treatment because of its mild toxicity and narrow toxicity spectrum.
Limitations
This study had some limitations. First, inevitable confounding factors, based on clinical trial data, such as the difference in follow-up time and cancer type, were shown in this network meta-analysis. Second, the difference in common comparisons (conventional therapy) may have influenced the general function of the network meta-analysis, especially the exchangeability, thus undermining the balance of this study. Third, placebo-controlled trials tended to recruit more patients with mild diseases. Finally, this study did not include too many clinical trials, and the subgroup analysis of cancer type was limited to ovarian cancer. Given that NCT01116648 did not provide the number of patients who had grade ≥3 adverse events, we excluded it when analyzing the dose- and drug-based network meta-analysis. Since our findings were based on clinical trials, further studies are warranted in the clinical setting.
Conclusions
In this network meta-analysis, different toxicity spectra were observed among the different PARP inhibitors. After comparing various PARP inhibitors, conventional therapy (chemotherapy), and the combination of PARP inhibitor and angiogenesis inhibitor, olaparib was found to be the safer choice. The integration of evidence from this network meta-analysis can improve the management of adverse events and modify prescriptions for PARP inhibitors in the clinical setting. Since there have been no randomized controlled trials comparing the safety profile of PARP inhibitors directly, this study provides a useful guide for clinicians and researchers. Further studies are needed to explore the relative characteristics of PARP inhibitors.
Supplementary
The article’s supplementary files as
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Funding: This work was financially supported by the National Key Research and Development Program of China (ZDZX2017ZL-01), High-level Innovation Team of Nanjing Medical University (JX102GSP201727), Wu Jieping Foundation (320.6750.17006), Key Medical Talents (ZDRCA2016023), 333 Project of Jiangsu Province (BRA2017534 and BRA2015470), the Collaborative Innovation Center for Tumor Individualization Focuses on Open Topics (JX21817902/008) and Project of China Key Research and Development Program Precision Medicine Research (2016YFC0905901).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Reporting Checklist: The authors have completed the PRISMA reporting checklist. Available at https://dx.doi.org/10.21037/atm-21-1883
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://dx.doi.org/10.21037/atm-21-1883). The authors have no conflicts of interest to declare.
References
- 1.Curtin NJ, Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov 2020;19:711-36. 10.1038/s41573-020-0076-6 [DOI] [PubMed] [Google Scholar]
- 2.Mateo J, Lord CJ, Serra V, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol 2019;30:1437-47. 10.1093/annonc/mdz192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152-8. 10.1126/science.aam7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tarsounas M, Sung P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol 2020;21:284-99. 10.1038/s41580-020-0218-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boussios S, Mikropoulos C, Samartzis E, et al. Wise Management of Ovarian Cancer: On the Cutting Edge. J Pers Med 2020;10:41. 10.3390/jpm10020041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirza MR, Åvall Lundqvist E, Birrer MJ, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol 2019;20:1409-19. 10.1016/S1470-2045(19)30515-7 [DOI] [PubMed] [Google Scholar]
- 7.Ray-Coquard I, Pautier P, Pignata S, et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N Engl J Med 2019;381:2416-28. 10.1056/NEJMoa1911361 [DOI] [PubMed] [Google Scholar]
- 8.Konstantinopoulos PA, Waggoner SE, Vidal GA, et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J Clin Oncol 2018;36:106. 10.1200/JCO.2018.36.15_suppl.106 [DOI] [Google Scholar]
- 9.Domchek SM, Postel-Vinay S, Im SA, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol 2020;21:1155-64. 10.1016/S1470-2045(20)30324-7 [DOI] [PubMed] [Google Scholar]
- 10.Drew Y, Penson RT, O'Malley DM, et al. 814MO Phase II study of olaparib (O) plus durvalumab (D) and bevacizumab (B) (MEDIOLA): Initial results in patients (pts) with non-germline BRCA-mutated (non-gBRCAm) platinum sensitive relapsed (PSR) ovarian cancer (OC). Ann Oncol 2020;31:S615-6. 10.1016/j.annonc.2020.08.953 [DOI] [Google Scholar]
- 11.Yi J, Liu C, Tao Z, et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019;43:225-37. 10.1016/j.ebiom.2019.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Chen L, Huang B, et al. Efficacy and mechanism of the combination of PARP and CDK4/6 inhibitors in the treatment of triple-negative breast cancer. J Exp Clin Cancer Res 2021;40:122. 10.1186/s13046-021-01930-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konstantinopoulos PA, Barry WT, Birrer M, et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol 2019;20:570-80. 10.1016/S1470-2045(18)30905-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yap TA, Kristeleit R, Michalarea V, et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov 2020;10:1528-43. 10.1158/2159-8290.CD-20-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westin SN, Litton JK, Williams RA, et al. Phase I trial of olaparib (PARP inhibitor) and vistusertib (mTORC1/2 inhibitor) in recurrent endometrial, ovarian and triple negative breast cancer. J Clin Oncol 2018;36:5504. 10.1200/JCO.2018.36.15_suppl.5504 [DOI] [Google Scholar]
- 16.Tutt A, Stephens C, Frewer P, et al. VIOLETTE: A randomized phase II study to assess the DNA damage response inhibitors AZD6738 or AZD1775 in combination with olaparib (Ola) versus Ola monotherapy in patients (pts) with metastatic, triple-negative breast cancer (TNBC). J Clin Oncol 2019;37:TPS1112. 10.1200/JCO.2019.37.15_suppl.TPS1112 [DOI] [Google Scholar]
- 17.Kurnit KC, Meric-Bernstam F, Hess K, et al. Abstract CT020: Phase I dose escalation of olaparib (PARP inhibitor) and selumetinib (MEK Inhibitor) combination in solid tumors with Ras pathway alterations. Cancer Res 2019;79:CT020. [Google Scholar]
- 18.Hussain M, Mateo J, Fizazi K, et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 2020;383:2345-57. 10.1056/NEJMoa2022485 [DOI] [PubMed] [Google Scholar]
- 19.Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med 2019;381:317-27. 10.1056/NEJMoa1903387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boussios S, Moschetta M, Karihtala P, et al. Development of new poly(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer: Quo Vadis? Ann Transl Med 2020;8:1706. 10.21037/atm.2020.03.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neeb A, Herranz N, Arce-Gallego S, et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur Urol 2021;79:200-11. 10.1016/j.eururo.2020.10.029 [DOI] [PubMed] [Google Scholar]
- 22.Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol 2018;29:1203-10. 10.1093/annonc/mdy099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Risdon EN, Chau CH, Price DK, et al. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021;26:e115-29. 10.1634/theoncologist.2020-0697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012;72:5588-99. 10.1158/0008-5472.CAN-12-2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaFargue CJ, Dal Molin GZ, Sood AK, et al. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol 2019;20:e15-28. 10.1016/S1470-2045(18)30786-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hopkins TA, Ainsworth WB, Ellis PA, et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol Cancer Res 2019;17:409-19. 10.1158/1541-7786.MCR-18-0138 [DOI] [PubMed] [Google Scholar]
- 27.Lu G, Ades AE. Combination of direct and indirect evidence in mixed treatment comparisons. Stat Med 2004;23:3105-24. 10.1002/sim.1875 [DOI] [PubMed] [Google Scholar]
- 28.Hutton B, Salanti G, Caldwell DM, et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann Intern Med 2015;162:777-84. 10.7326/M14-2385 [DOI] [PubMed] [Google Scholar]
- 29.Higgins JP, Altman DG, Gøtzsche PC, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011;343:d5928. 10.1136/bmj.d5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jadad AR, Moore RA, Carroll D, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary? Control Clin Trials 1996;17:1-12. 10.1016/0197-2456(95)00134-4 [DOI] [PubMed] [Google Scholar]
- 31.Chaimani A, Higgins JP, Mavridis D, et al. Graphical tools for network meta-analysis in STATA. PLoS One 2013;8:e76654. 10.1371/journal.pone.0076654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sutton AJ, Abrams KR. Bayesian methods in meta-analysis and evidence synthesis. Stat Methods Med Res 2001;10:277-303. 10.1177/096228020101000404 [DOI] [PubMed] [Google Scholar]
- 33.Mills EJ, Thorlund K, Ioannidis JP. Demystifying trial networks and network meta-analysis. BMJ 2013;346:f2914. 10.1136/bmj.f2914 [DOI] [PubMed] [Google Scholar]
- 34.Brooks SP, Gelman A. General Methods for Monitoring Convergence of Iterative Simulations. J Comput Graph Stat 1998;7:434-55. [Google Scholar]
- 35.Lu G, Ades AE. Assessing Evidence Inconsistency in Mixed Treatment Comparisons. J Am Stat Assoc 2006;101:447-59. 10.1198/016214505000001302 [DOI] [Google Scholar]
- 36.Dias S, Welton NJ, Caldwell DM, et al. Checking consistency in mixed treatment comparison meta-analysis. Stat Med 2010;29:932-44. 10.1002/sim.3767 [DOI] [PubMed] [Google Scholar]
- 37.Higgins JP, Thompson SG, Deeks JJ, et al. Measuring inconsistency in meta-analyses. BMJ 2003;327:557-60. 10.1136/bmj.327.7414.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Litton JK, Rugo HS, Ettl J, et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med 2018;379:753-63. 10.1056/NEJMoa1802905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robson M, Im SA, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med 2017;377:523-33. 10.1056/NEJMoa1706450 [DOI] [PubMed] [Google Scholar]
- 40.Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol 2014;15:1207-14. 10.1016/S1470-2045(14)70391-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaye SB, Lubinski J, Matulonis U, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol 2012;30:372-9. 10.1200/JCO.2011.36.9215 [DOI] [PubMed] [Google Scholar]
- 42.Penson RT, Valencia RV, Cibula D, et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J Clin Oncol 2020;38:1164-74. 10.1200/JCO.19.02745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.González-Martín A, Pothuri B, Vergote I, et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 2019;381:2391-402. 10.1056/NEJMoa1910962 [DOI] [PubMed] [Google Scholar]
- 44.Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med 2016;375:2154-64. 10.1056/NEJMoa1611310 [DOI] [PubMed] [Google Scholar]
- 45.Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 2012;366:1382-92. 10.1056/NEJMoa1105535 [DOI] [PubMed] [Google Scholar]
- 46.Moore K, Colombo N, Scambia G, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 2018;379:2495-505. 10.1056/NEJMoa1810858 [DOI] [PubMed] [Google Scholar]
- 47.Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1274-84. 10.1016/S1470-2045(17)30469-2 [DOI] [PubMed] [Google Scholar]
- 48.Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:1949-61. 10.1016/S0140-6736(17)32440-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 2020;21:162-74. 10.1016/S1470-2045(19)30684-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boussios S, Karihtala P, Moschetta M, et al. Veliparib in ovarian cancer: a new synthetically lethal therapeutic approach. Invest New Drugs 2020;38:181-93. 10.1007/s10637-019-00867-4 [DOI] [PubMed] [Google Scholar]
- 51.Gangadhar TC, Vonderheide RH. Mitigating the toxic effects of anticancer immunotherapy. Nat Rev Clin Oncol 2014;11:91-9. 10.1038/nrclinonc.2013.245 [DOI] [PubMed] [Google Scholar]
- 52.Ter Veer E, van Oijen MGH, van Laarhoven HWM. The Use of (Network) Meta-Analysis in Clinical Oncology. Front Oncol 2019;9:822. 10.3389/fonc.2019.00822 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as