

SUMMARY
Background
Genome-wide association studies (GWASs) in Parkinson’s disease (PD) have increased the scope of biological knowledge about the disease over the past decade. We sought to use the largest aggregate of GWAS data to identify novel risk loci and gain further insight into disease etiology.
Methods
We performed the largest meta-GWAS of PD to date, involving the analysis of 7.8M SNPs in 37.7K cases, 18.6K UK Biobank proxy-cases (having a first degree relative with PD), and 1.4M controls. We carried out a meta-analysis of this GWAS data to nominate novel loci. We then evaluated heritable risk estimates and predictive models using this data. We also utilized large gene expression and methylation resources to examine possible functional consequences as well as tissue, cell type and biological pathway enrichments for the identified risk factors. Additionally we examined shared genetic risk between PD and other phenotypes of interest via genetic correlations followed by Mendelian randomization.
Findings
We identified 90 independent genome-wide significant risk signals across 78 genomic regions, including 38 novel independent risk signals in 37 loci. These 90 variants explained 16–36% of the heritable risk of PD depending on prevalence. Integrating methylation and expression data within a Mendelian randomization framework identified putatively associated genes at 70 risk signals underlying GWAS loci for follow-up functional studies. Tissue-specific expression enrichment analyses suggested PD loci were heavily brain-enriched, with specific neuronal cell types being implicated from single cell data. We found significant genetic correlations with brain volumes, smoking status, and educational attainment. Mendelian randomization between cognitive performance and PD risk showed a robust association.
Interpretation
These data provide the most comprehensive understanding of the genetic architecture of PD to date by revealing many additional PD risk loci, providing a biological context for these risk factors, and demonstrating that a considerable genetic component of this disease remains unidentified.
INTRODUCTION
Parkinson’s disease is a neurodegenerative disorder, affecting approximately 1 million individuals in the United States alone (5). PD patients suffer from a combination of progressive motor and non-motor symptoms affecting daily function and quality of life. The prevalence of PD is projected to double in some age groups by 2030, creating a substantial burden on healthcare systems (5).
Early investigations into the role of genetic factors in PD focused on the identification of rare mutations underlying familial disease, (6,7) over the past decade there has been a growing appreciation for the contribution of genetics in sporadic disease(1,8). Genetic studies of sporadic PD have altered the foundational view of disease etiology.
We executed a series of experiments to explore the genetics of PD (summarized in Figure 1). We performed the largest-to-date GWAS for PD, including 7.8M SNPs, 37.7K cases, 18.6K UK Biobank (UKB) “proxy-cases” (individuals without PD that have a family history of PD) and 1.4M controls. We identified mechanistic candidate genes for PD, providing valuable therapeutic targets. We assessed the function of these potential risk genes via Mendelian randomization, expression enrichment, and protein-protein interaction network analysis. We estimated PD heritability, developed a polygenic risk score that predicted a substantial proportion of this heritability, and leveraged these results to inform future studies. Finally, we identified candidate PD biomarkers and risk factors using genetic correlation and Mendelian randomization.
Figure 1: Workflow and rationale summary.
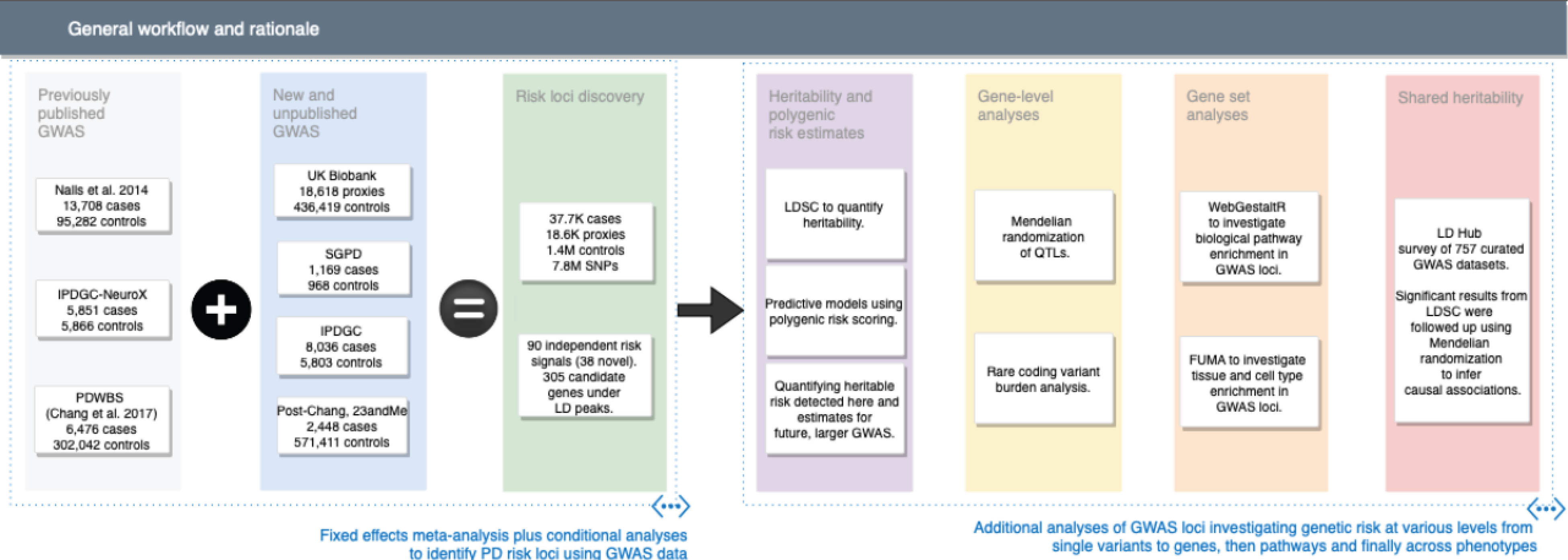
This figure describes study design and rationale behind the analyses included in this report.
SUMMARY OF METHODS
GWAS Study design and risk locus discovery
Three sources of data were used for discovery analyses, these include three previously published studies, 13 new datasets, and proxy-case data from the UK BioBank (UKB). Previous studies include summary statistics published in Nalls et al. 2014, GWAS summary statistics from the 23andMe Web-Based Study of Parkinson’s Disease (PDWBS) in Chang et al. 2017, and the publicly available NeuroX dataset from the International Parkinson’s Disease Genomics Consortium (IPDGC) previously used previously as a replication sample. These cohorts have been reported in detail (1,9). We included 13 new case-control sample series for meta-analyses through either publicly available data or collaborations (please see Supplementary Table S1 for details regarding these studies). All samples from the 13 new datasets underwent similar standardized quality control for inclusion, mirroring that of previous studies. We attempted to generate summary statistics for GWAS meta-analyses as uniformly as possible. This analysis utilized fixed-effects meta-analyses as implemented in METAL to combine summary statistics across all sources (10).
Conditional-joint analysis to nominate variants of interest
To nominate variants of interest, we employed a conditional and joint analysis strategy (COJO, http://cnsgenomics.com/software/gcta/) to algorithmically identify variants that best account for the heritable variation within and across loci (11). Additional analyses described below were utilized to further scrutinize putative associated variants and account for possible differential linkage disequilibrium (LD) signatures, including using the massive single site reference data from 23andMe in more conditional analyses. If a variant nominated during the COJO phase of analysis was greater than 1Mb from any of the genome-wide significant loci nominated in Chang et al. 2017, we considered this to be novel. We defined nominated risk variants as from a single locus if they were within +/− 250kb of each other. We instituted two filters after fixed-effects and COJO analyses, excluding variants that 1) had a random-effects P value across all datasets > 4.67E-04 and 2) a conditional analysis P > 4.67E-04 using participant level 23andMe genotype data. Please see Supplementary Table S2 summarizing all variants nominated plus the Methods and Results Supplement.
Refining heritability estimates and determining extant genetic risk
We used the R package PRSice2 for risk profiling (12), this carries out polygenic risk score (PRS) profiling in the standard weighted allele dose manner (1,9,13–15). In addition, PRSice incorporates permutation testing where case and control labels are swapped in the withheld samples to generate an empirical P. This workflow identifies the best P thresholds for variant inclusion while carrying out LD pruning. In many cases this best P threshold for PRS construction does not meet what is commonly regarded as genome-wide signfiicance.
A two stage design was also employed, training on the largest single array study (NeuroX-dbGaP) and then tested on the second largest study (HBS) using the same array. These two targeted array studies were chosen for three reasons: precedent in the previous publications where the NeuroX-dbGaP dataset was used in PRS; direct genotyping of larger effect rare variants in GBA and LRRK2; participant level genotypes for these datasets are publicly available.
To calculate heritability in clinically defined PD datasets, we used LD score regression (LDSC) employing the LD references for Europeans provided with the software (16). This workflow was also repeated on a per cohort level (see Supplementary Appendix).
Functional causal inferences via Quantitative Trait Loci (QTL)
We used MR to test whether changes in DNA methylation and/or RNA expression of genes physically proximal to significant PD risk loci were causally related to PD risk. To nominate genes of interest for MR analyses, we took our putative 90 loci in the large LD reference used for the COJO phase of analysis and identified SNPs in LD with our SNPs at an r2 > 0.5 within +/− 1MB (Supplementary Table S5). MR was used by integrating discovery phase summary statistics with quantitative trait locus (QTL) association summary statistics across well-curated methylation and expression datasets. We used the curated versions of Qi et al., 2018 brain methylation and expression summary statistics (multi-study and multi-tissue meta-analysis), as well as a specific focus on substantia nigra data (GTEx), we made use of the blood expression data from Võsa et al. 2018 (eQTLGen, all available here http://cnsgenomics.com/software/smr/#Overview and here http://www.eqtlgen.org) (17–21). For all QTL analyses, we utilized the multi-SNP summary-based Mendelian randomization (SMR) method as a framework to carry out MR. All MR effect estimates are reported on the scale of a standard deviation increase in the exposure variable relating to a similar change in PD risk. Simply, these MR analyses compare the local polygenic risk of an exposure (methylation or expression) to similar polygenic risk in an outcome (PD), inferring causal associations under the assumption that there is no intermediate confounder associated with both parameters and that the association is not simply due to LD.
To further investigate expression enrichment across cell types in PD, we integrated GWAS summary statistics with expression and network data from the FUMA webserver (https://fuma.ctglab.nl/, version 1.3.1) (22).
Rare coding variant burden tests
A uniformly quality controlled and imputed dataset from the IPDGC was used to carry out burden tests for all rarer coding variants successfully imputed in an average of 85% of the sample series (17,188 cases and 22,875 controls). These analyses include all variants at a hard call threshold of imputation quality > 0.8. After annotation with annovar, we had a total of 37,503 exonic coding variants (nonsynonymous, stop or splicing) at MAF < 5% and a subset of 29,016 at MAF < 1% (23). For inclusion in this phase, a gene must have contained at least 2 coding variants. After assembling this subset of 113 testable genes, we used the optimized sequence kernel association test to generate summary statistics at maximum MAFs of 1% and 5% (24).
LD score regression and causal inference
To investigate correlations of PD genetics with that of multiple traits and diseases, we employed bivariate LDSC (16). These analyses were carried out using data from the 757 GWAS available via LD Hub and biomarker GWAS summary statistics on c-reactive protein and cytokine measures; LD Hub was accessed on June 20th, 2018 (version 1.2.0) (25–27). P values from the bivariate LDSC were adjusted for FDR to account for multiple testing. Traits showing significant genetic correlations with PD were analyzed using MR methods. We excluded the UKB data when a nominated trait was from summary statistics derived from the UKB or if the UKB was included as part of a meta-analysis.
When complete GWAS summary statistics were available for traits of interest (relating to smoking and education), we used the more powerful bi-directional generalized summary-data-based Mendelian Randomization (GSMR). We analyzed GWAS summary statistics for smoking initialization (453,693 records from a self-report survey with 208,988 regular smokers and 244,705 never regular smokers) and current smoking (CS) within the UKB, CS contrasted 47,419 current smokers versus 244,705 never regular smokers. The same analysis was carried out incorporating recent GWAS data regarding educational attainment (N = 766,345) from self report in the UK and cognitive performance (N = 257,828) as measured by the g composite score (28). These were analyzed using methods to mirror that of the UKB PD GWAS dataset. Combined left and right putamen volume from a T2 magnetic resonance imaging GWAS available from Oxford Brain Imaging Genetics (BIG) Server (accessed December 28th, 2018) (29). All MR analyses included GWAS on the scale of tens of thousands of samples and overcame the considerable power demands of the methodology.
For additional quality control, methods details and ancillary results, see the Methods Supplement.
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
RESULTS
To maximize our power for locus discovery we used a single stage design, meta-analyzing all available GWAS summary statistics. Supporting this design, we found strong genetic correlations using PD cases ascertained by clinicians compared to 23andMe self-reported cases (genetic correlation from LDSC (rG) = 0.85, SE = 0.06) and UKB proxy cases (rG = 0.84, SE = 0.134).
We identified a total of 90 independent genome-wide significant association signals through our analyses of 37,688 cases, 18,618 UKB proxy-cases and 1,417,791 controls at 7,784,415 SNPs (Figure 2, Table 1, Supplementary Appendices, Table S1, Table S2). Of these, 38 signals are new and more than 1MB from loci described previously (1) (Table S3).
Figure 2: Manhattan plot.
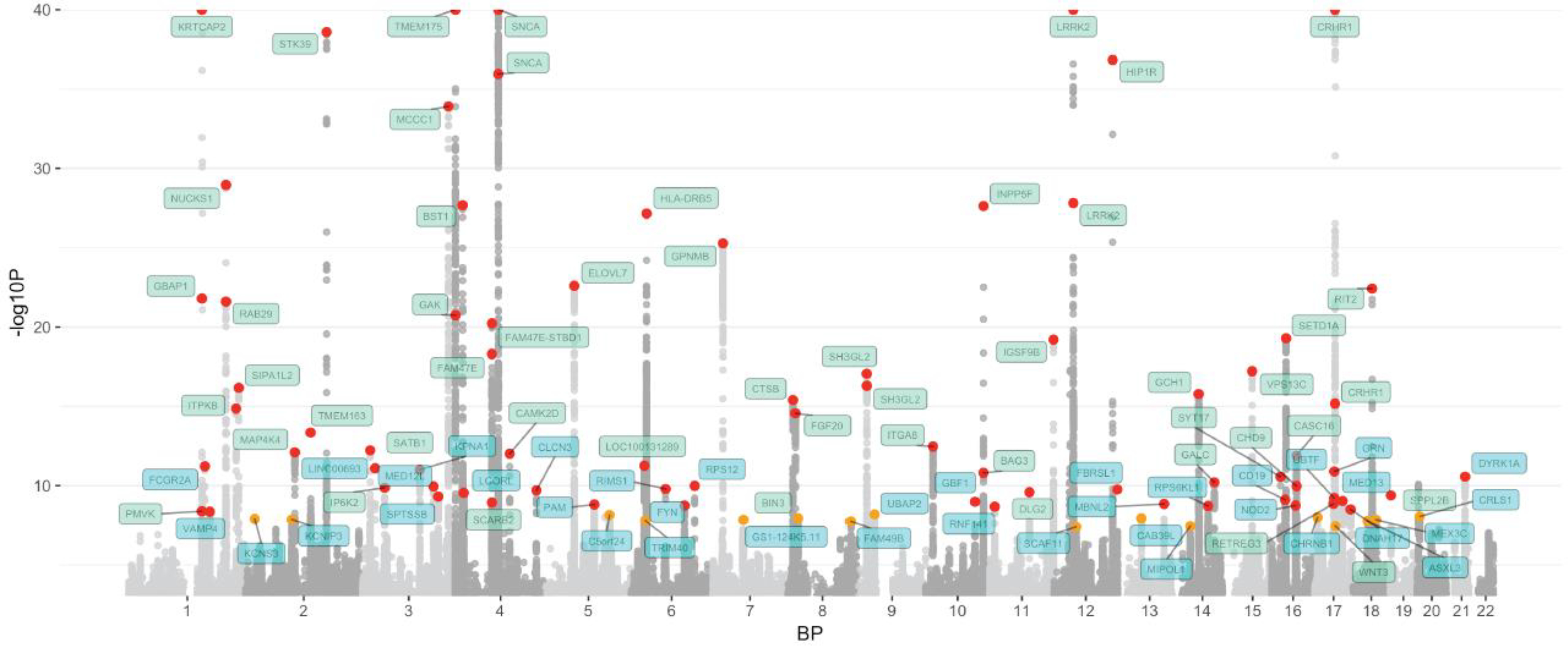
The nearest gene to each of the 90 significant variants are labeled in green for previously-identified loci and in blue for novel loci. −log10 P values were capped at 40. Variant points are color coded red and orange, with orange representing significant variants at P 5E-08 and 5E-9 and red representing significant variants at P < 5E-9. The X axis represents the base pair position of variants from smallest to largest per chromosome (1–22).
Table 1.
Novel loci associated with Parkinson’s disease.
| SNP | CHR | BP | Nearest Gene | Effect allele | Other allele | Effect allele frequen cy | OR | Low 95% CI | High 95% CI | Beta | SE | P, fixed-effects | P, COJO | P, conditional | P, random-effects | I2, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs6658353 | 1 | 161469054 | FCGR2A | c | g | 0.501 | 1.07 | 1.05 | 1.09 | 0.06 5 | 0.009 | 6.10E–12 | 4.69E12 | 1.38E–05 | 3.71E–05 | 40.2 |
| rs11578699 | 1 | 171719769 | VAMP4 | t | c | 0.195 | 0.93 | 0.91 | 0.95 | −0.070 | 0.012 | 4.47E–09 | 4.45E09 | 2.63E–03 | 1.09E–07 | 5.1 |
| rs76116224 | 2 | 18147848 | KCNS3 | a | t | 0.904 | 1.12 | 1.08 | 1.16 | 0.11 0 | 0.019 | 1.27E–08 | 1.27E08 | 3.75E–07 | 1.27E–08 | 0 |
| rs2042477 | 2 | 96000943 | KCNIP3 | a | t | 0.242 | 0.94 | 0.92 | 0.96 | −0.066 | 0.012 | 1.38E–08 | 1.48E08 | 3.49E–05 | 1.38E–08 | 0 |
| rs6808178 | 3 | 28705690 | LINC00693 | t | c | 0.379 | 1.07 | 1.05 | 1.09 | 0.06 6 | 0.010 | 8.09E–12 | 7.18E12 | 8.84E–05 | 8.09E–12 | 0 |
| rs55961674 | 3 | 122196892 | KPNA1 | t | c | 0.172 | 1.09 | 1.06 | 1.12 | 0.086 | 0.013 | 9.98E–12 | 8.30E12 | 2.80E–06 | 9.98E–12 | 0 |
| rs11707416 | 3 | 151108965 | MED12L | a | t | 0.367 | 0.94 | 0.92 | 0.96 | −0.063 | 0.010 | 1.13E–10 | 1.02E10 | 2.66E–04 | 1.77E–07 | 10.9 |
| rs1450522 | 3 | 161077630 | SPTSSB | a | g | 0.674 | 0.94 | 0.92 | 0.96 | −0.062 | 0.010 | 5.01E–10 | 4.90E10 | 3.51E–04 | 2.27E–05 | 24.6 |
| rs34025766 | 4 | 17968811 | LCORL | a | t | 0.159 | 0.92 | 0.90 | 0.94 | −0.084 | 0.013 | 2.87E–10 | 2.82E10 | 7.43E–06 | 2.87E–10 | 0 |
| rs62333164 | 4 | 170583157 | CLCN3 | a | g | 0.326 | 0.94 | 0.92 | 0.96 | −0.064 | 0.010 | 2.00E–10 | 1.77E10 | 5.10E–05 | 2.17E–05 | 21.3 |
| rs26431 | 5 | 102365794 | PAM | c | g | 0.703 | 1.06 | 1.04 | 1.09 | 0.062 | 0.010 | 1.57E–09 | 1.65E09 | 6.00E–03 | 2.36E–07 | 7.9 |
| rs11950533 | 5 | 134199105 | C5orf24 | a | c | 0.102 | 0.91 | 0.88 | 0.94 | −0.092 | 0.016 | 7.16E–09 | 6.73E09 | 5.08E–04 | 2.68E–08 | 1.9 |
| rs9261484 | 6 | 30108683 | TRIM40 | t | c | 0.245 | 0.94 | 0.92 | 0.96 | −0.064 | 0.011 | 1.62E–08 | 1.43E08 | 1.26E–06 | 1.62E–08 | 0 |
| rs12528068 | 6 | 72487762 | RIMS1 | t | c | 0.284 | 1.07 | 1.05 | 1.09 | 0.066 | 0.010 | 1.63E–10 | 1.79E10 | 9.80E–06 | 1.63E–10 | 0 |
| rs997368 | 6 | 112243291 | FYN | a | g | 0.805 | 1.07 | 1.05 | 1.10 | 0.071 | 0.012 | 1.84E–09 | 1.97E09 | 2.61E–05 | 1.84E–09 | 0 |
| rs75859381 | 6 | 133210361 | RPS12 | t | c | 0.967 | 0.80 | 0.75 | 0.86 | −0.221 | 0.034 | 1.04E–10 | 9.67E11 | 1.09E–06 | 1.04E–10 | 0 |
| rs76949143 | 7 | 66009851 | GS1-124K5.11 | a | t | 0.051 | 0.87 | 0.82 | 0.91 | −0.143 | 0.025 | 1.43E–08 | 1.51E08 | 5.47E–09 | 2.04E–06 | 12.3 |
| rs2086641 | 8 | 130901909 | FAM49B | t | c | 0.723 | 0.94 | 0.92 | 0.96 | −0.061 | 0.011 | 1.81E–08 | 1.57E08 | 6.07E–06 | 1.81E–08 | 0 |
| rs6476434 | 9 | 34046391 | UBAP2 | t | c | 0.734 | 0.94 | 0.92 | 0.96 | −0.062 | 0.011 | 6.58E–09 | 6.56E09 | 2.74E–04 | 6.58E–09 | 0 |
| rs10748818 | 10 | 104015279 | GBF1 | a | g | 0.851 | 0.92 | 0.90 | 0.95 | −0.079 | 0.013 | 1.05E–09 | 1.23E09 | 7.47E–06 | 1.05E–09 | 0 |
| rs7938782 | 11 | 10558777 | RNF141 | a | g | 0.878 | 1.09 | 1.06 | 1.12 | 0.087 | 0.015 | 2.12E–09 | 1.97E09 | 2.17E–07 | 2.12E–09 | 0 |
| rs7134559 | 12 | 46419086 | SCAF11 | t | c | 0.404 | 0.95 | 0.93 | 0.97 | −0.054 | 0.010 | 3.96E–08 | 3.80E08 | 1.69E–02 | 1.84E–05 | 25.2 |
| rs11610045 | 12 | 133063768 | FBRSL1 | a | g | 0.490 | 1.06 | 1.04 | 1.08 | 0.060 | 0.009 | 1.77E–10 | 1.62E10 | 3.57E–05 | 8.79E–07 | 19.5 |
| rs9568188 | 13 | 49927732 | CAB39L | t | c | 0.740 | 1.06 | 1.04 | 1.09 | 0.062 | 0.011 | 1.15E–08 | 1.11E08 | 4.29E–06 | 2.46E–04 | 21.4 |
| rs4771268 | 13 | 97865021 | MBNL2 | t | c | 0.230 | 1.07 | 1.05 | 1.09 | 0.068 | 0.011 | 1.45E–09 | 1.67E09 | 1.41E–04 | 1.45E–09 | 0 |
| rs12147950 | 14 | 37989270 | MIPOL1 | t | c | 0.438 | 0.95 | 0.93 | 0.97 | −0.053 | 0.010 | 3.54E–08 | 3.58E08 | 1.06E–03 | 3.54E–08 | 0 |
| rs3742785 | 14 | 75373034 | RPS6KL1 | a | c | 0.787 | 1.07 | 1.05 | 1.10 | 0.071 | 0.012 | 1.92E–09 | 2.08E09 | 2.22E–06 | 8.18E–06 | 24.8 |
| rs2904880 | 16 | 28944396 | CD19 | c | g | 0.309 | 0.94 | 0.92 | 0.96 | −0.065 | 0.011 | 7.87E–10 | 8.68E10 | 1.39E–05 | 7.87E–10 | 0 |
| rs6500328 | 16 | 50736656 | NOD2 | a | g | 0.599 | 1.06 | 1.04 | 1.08 | 0.059 | 0.010 | 1.82E–09 | 1.53E09 | 1.43E–03 | 1.82E–09 | 0 |
| rs12600861 | 17 | 7355621 | CHRNB1 | a | c | 0.648 | 0.95 | 0.93 | 0.96 | −0.057 | 0.010 | 1.01E–08 | 1.15E08 | 5.10E–03 | 1.01E–08 | 0 |
| rs2269906 | 17 | 42294337 | UBTF | a | c | 0.653 | 1.07 | 1.04 | 1.09 | 0.063 | 0.010 | 6.24E–10 | 8.63E09 | 1.17E–05 | 6.24E–10 | 0 |
| rs850738 | 17 | 42434630 | FAM171A2 | a | g | 0.606 | 0.93 | 0.91 | 0.95 | −0.071 | 0.011 | 1.29E–11 | 3.55E10 | 4.18E–04 | 2.17E–07 | 17 |
| rs61169879 | 17 | 59917366 | BRIP1 | t | c | 0.164 | 1.09 | 1.06 | 1.11 | 0.082 | 0.013 | 9.28E–10 | 9.40E10 | 9.07E–07 | 6.21E–06 | 16.4 |
| rs666463 | 17 | 76425480 | DNAH17 | a | t | 0.833 | 1.08 | 1.05 | 1.11 | 0.076 | 0.013 | 3.20E–09 | 2.90E09 | 1.62E–05 | 4.17E–04 | 41 |
| rs1941685 | 18 | 31304318 | ASXL3 | t | g | 0.498 | 1.05 | 1.04 | 1.07 | 0.053 | 0.009 | 1.69E–08 | 1.61E08 | 1.64E–08 | 1.69E–08 | 0 |
| rs8087969 | 18 | 48683589 | MEX3C | t | g | 0.550 | 0.94 | 0.93 | 0.96 | −0.058 | 0.010 | 1.41E–08 | 1.46E08 | 1.09E–04 | 1.41E–08 | 0 |
| rs77351827 | 20 | 6006041 | CRLS1 | t | c | 0.128 | 1.08 | 1.05 | 1.11 | 0.080 | 0.014 | 8.87E–09 | 7.94E09 | 1.84E–05 | 4.38E–07 | 11.2 |
| rs2248244 | 21 | 38852361 | DYRK1A | a | g | 0.283 | 1.07 | 1.05 | 1.10 | 0.071 | 0.011 | 2.74E–11 | 2.51E11 | 6.31E–05 | 8.78E–06 | 34.3 |
Summary statistics for 38 novel genome-wide significant PD variants. Columns include single nucleotide polymorphism ID (SNP), chromosome (CHR), base pair position (BP) based on hg19 build, nearest gene annotation for the variant, effect allele designation and frequency, as well as metrics for the odds ratio (OR), regression coefficient (beta), and standard error of the beta for the SNP from fixed-effects meta-analysis as well as the index of heterogeneity (I2). We also include four P values from: fixed-effects meta-analyses, random-effects meta-analyses, standard conditional analyses in 23andMe, and a conditional joint analysis approach (COJO).
We detected 10 loci containing more than one independent risk signal (22 risk SNPs in total across these loci), of which nine had been identified by previous GWAS, including multi-signal loci in the vicinity of GBA, NUCKS1/RAB29, GAK/TMEM175, SNCA and LRRK2. The novel multi-signal locus comprised independent risk variants rs2269906 (UBTF/GRN) and rs850738 (FAM171A2). Detailed summary statistics on all nominated loci can be found in Table S2, including variants filtered out during additional quality control.
To quantify how much of the genetic liability we have explained and what direction to take with future PD GWAS we generated updated heritability estimates and PRS. Using LDSC on a meta-analysis of all 11 clinically-ascertained datasets from our GWAS and estimated the liability-scale heritability of PD as 0.22 (95% CI 0.18 – 0.26), only slightly lower than a previous estimate derived using GCTA (0.27, 95% CI 0.17 – 0.38) (2,16,30). LDSC is known to be more conservative than GCTA, however, our LDSC heritability estimate does fall within the 95% confidence interval of the GCTA estimate.
To determine the proportion of SNP-based heritability explained by our PD GWAS results using PRS, we used a two-stage design, with variant selection and training in the NeuroX-dbGaP dataset (5,851 cases and 5,866 controls) and then validation in the Harvard Biomarker Study (HBS, 527 cases and 472 controls). Using equations from Wray et al. 2010 and our current heritability estimates, the 88 variant PRS explained a minimum 16% of the genetic liability of PD assuming a global prevalence of 0.5% (2,31). The 1805 variant PRS explained roughly 26% of PD heritability. In a high-risk population with a prevalence of 2%, the 1805 variant PRS explained a maximum 36% of PD heritable risk (2,31) (Table S4).
We then attempted to quantify strata of risk in our more inclusive PRS. Compared to individuals with PRS values in the lowest quartile, the PD odds ratio for individuals with PRS values in the highest quartile was 3.74 (95% CI = 3.35 – 4.18) in the NeuroX-dbGaP cohort and 6.25 (95% CI = 4.26 – 9.28) in the HBS cohort (Table 2, Figure 3, Figure S1).
Table 2.
Summary of genetic predictive model performance.
| Study | Max P threshold | pseudo R2 from PRS* | Beta | SE | P | OR, highest quartile PRS | 95% CI, highest quartile PRS | N SNPs | N samples | AUC | 95% CI (DeLong) | Sensitivity | Specificity | Positive predictive value (PPV) | Negative predictive value (NPV) | Balanced accuracy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Training dataset: IPDGC - Neurox | 1.35E–03 | 0.029 | 0.553 | 0.022 | 8.99E–135 | 3.74 | 3.35 – 4.18 | 1809 | 11,243 | 0.640 | 0.630 – 0.650 | 0.569 | 0.632 | 0.591 | 0.611 | 0.601 |
| Test dataset: HBS | 4.00E–02 | 0.054 | 0.709 | 0.072 | 8.28E–23 | 6.25 | 4.26 – 9.28 | 1805 | 999 | 0.692 | 0.660 – 0.725 | 0.628 | 0.686 | 0.691 | 0.623 | 0.657 |
These are estimates of performance for predictive models including single study estimates, estimates from meta-analyses across studies, as well as a two stage design. Here the best P value threshold column denotes the filtering value for SNP inclusion to achieve the maximal pseudo (Nagelkerke's) R2. The odds ratio (OR) colum is the exponent of the regression coefficient (beta) from logistic regression of the polygenic risk score (PRS) on case status, with the standard error (SE) representing the precision of these estimates. These same metrics are derived across array types and datasets using random-effects meta-analyses. The area under the curve (AUC) is included as the most common metric for predictive model performance. In the table
denotes R2 approximation adjusted for an estimated prevalence of 0.5%, equivalent to roughly half of the unadjusted R2 estimates for the PRS. All calculations and reported statistics include only the PRS and no other parameters after adjusting for principal components 1-5, age and sex at variant selection in the NeuroX-dbGaP dataset.
Figure 3: Predictive model details.
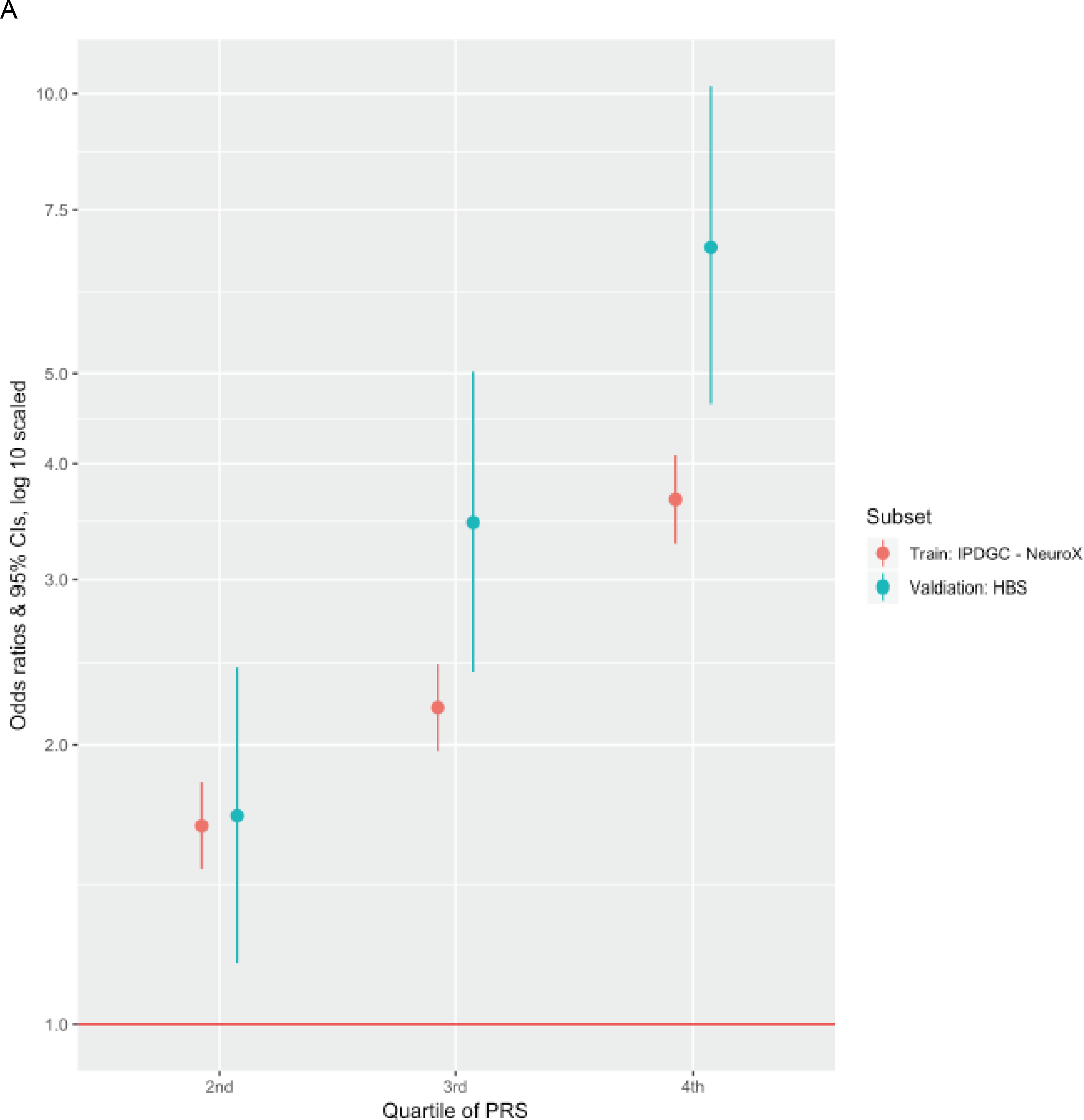
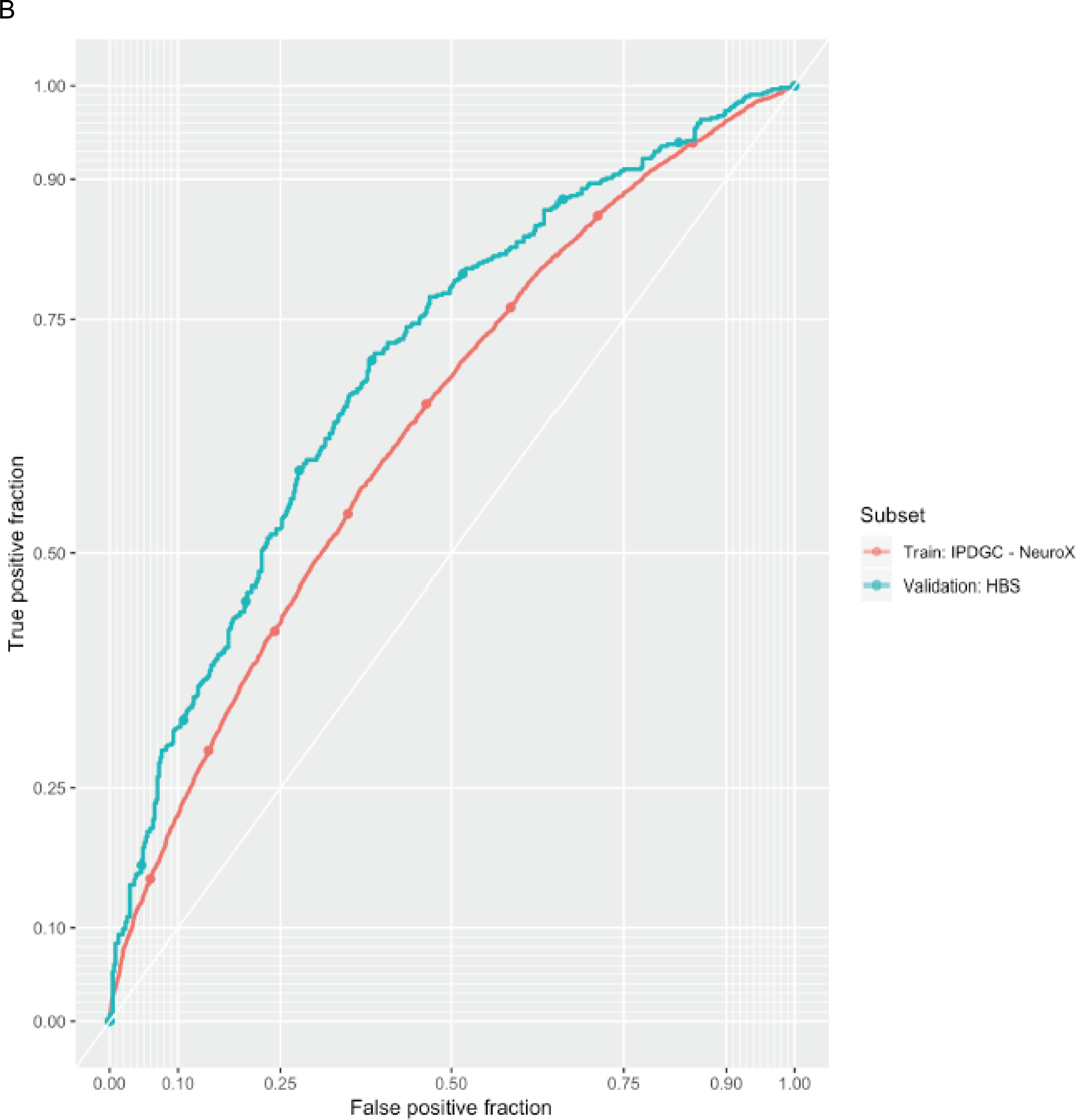
A. The odds ratio of developing PD for each quartile of polygenic risk score (PRS) compared to the lowest quartile of genetic risk. B. PRS receiver-operator curves for the more inclusive 1805 variant PRS in the validation dataset as well as in the corresponding training dataset that was used for PRS thresholding and SNP selection.
Variants in the range of 5E-08 < P < 1.35E-03 (used in the 1805 variant PRS) were rarer and had smaller effect estimates than variants reaching genome-wide significance. These sub-significant variants had a median minor allele frequency of 21.3% and a median effect estimate (absolute value of the log odds ratio of the SNP parameter from regresion) of 0.047. Genome-wide significant risk variants were more common with a median minor allele frequency of 25.1%, and had a median effect estimate of 0.081. Here we assume that the lower minor allele frequencies and smaller effect size estimates are typical and representative of variants contributing to our more inclusive PRS and represent future GWAS hits. We performed power calculations to forecast the number of additional PD cases needed to achieve genome-wide significance at 80% power for a variant with a minor allele frequency of 21.3% and an effect estimate of 0.047 (32). Assuming that future data is well-harmonized with current data and that disease prevalence is 0.5%, we estimated that we would need a total of ~99K cases, ~2.3 times more than this work for these to reach genome-wide significance. These variants already contribute towards the current increases in AUC when considering the 1805 variant PRS outperforms the 88 variant PRS. Expanding future studies to this size will invariably identify new loci and improve the AUC for a genetic predictor in PD (maximum potential AUC estimated at 85% using the equations from Wray et al. 2010) (31).
There were 305 genes within the 78 GWAS loci. We sought to identify the likely causal gene(s) in each locus using large QTL datasets and summary-data-based Mendelian randomization (Table 3, Table S5, Table S6) (33). This method allows for functional inferences between two datasets to be made in an analogous framework to a randomized controlled trial, treating the genotype as the randomizing factor.
Table 3.
Summary of significant functional inferences from QTL associations via Mendelian randomization for nominated genes of interest.
| Gene | Probe | CHR | Probe, BP | Top SNP, BP | Top SNP | N SNPs | QTL reference | Effect | SE | P | Bonferroni adjusted P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| VAMP4 | ENSG00000117533 | 1 | 171,690,343 | 171,717,417 | rs10913587 | 98 | Võsa et al. 2018 - blood expression | −0.272 | 0.05 | 5.67E07 | 1.19E–04 |
| KCNIP3 | ENSG00000115041 | 2 | 96,007,438 | 95,989,766 | rs3772034 | 14 | Qi et al. 2018 - brain expression | −0.161 | 0.04 | 1.12E05 | 1.15E–03 |
| MAP4K4 | ENSG00000071054 | 2 | 102,410,880 | 102,338,377 | rs6733355 | 3 | Võsa et al. 2018 - blood expression | 1.119 | 0.24 | 2.32E06 | 4.87E–04 |
| TMEM163 | ENSG00000152128 | 2 | 135,344,950 | 135,248,544 | rs598668 | 28 | Qi et al. 2018 - brain expression | 0.074 | 0.02 | 3.55E07 | 3.65E–05 |
| KPNA1 | ENSG00000114030 | 3 | 122,187,294 | 122,201,610 | rs73190142 | 110 | Võsa et al. 2018 - blood expression | 0.310 | 0.05 | 1.56E06 | 3.28E–04 |
| GAK | ENSG00000178950 | 4 | 884,612 | 906,131 | rs11248057 | 1 | Qi et al. 2018 - brain expression | 0.508 | 0.10 | 7.47E07 | 7.69E–05 |
| CAMK2D | ENSG00000145349 | 4 | 114,527,635 | 114,730,260 | rs115671064 | 146 | Võsa et al. 2018 - blood expression | −0.006 | 0.05 | 5.74E06 | 1.21E–03 |
| PAM | ENSG00000145730 | 5 | 102,228,247 | 102,118,633 | rs2432162 | 679 | Võsa et al. 2018 - blood expression | −0.031 | 0.01 | 2.08E06 | 4.36E–04 |
| LOC100131289 | cg21339923 | 6 | 27,636,378 | 27,636,378 | rs78149975 | 2 | Qi et al. 2018 - brain methylation | −0.094 | 0.02 | 1.53E06 | 3.06E–04 |
| TRIM40 | cg01641092 | 6 | 30,094,300 | 30,094,315 | rs9261443 | 8 | Qi et al. 2018 - brain methylation | 0.072 | 0.01 | 6.15E06 | 1.23E–03 |
| HLA-DRB5 | cg26036029 | 6 | 32,552,443 | 32,570,311 | rs34039593 | 8 | Qi et al. 2018 - brain methylation | −0.153 | 0.02 | 7.53E10 | 1.51E–07 |
| GPNMB | ENSG00000136235 | 7 | 23,295,156 | 23,294,668 | rs858274 | 74 | Qi et al. 2018 - brain expression | 0.090 | 0.01 | 2.73E21 | 2.81E–19 |
| CTSB | ENSG00000164733 | 8 | 11,713,495 | 11,699,279 | rs4631423 | 33 | Qi et al. 2018 - brain expression | −0.150 | 0.04 | 4.37E09 | 4.50E–07 |
| BIN3 | ENSG00000147439 | 8 | 22,502,296 | 22,456,517 | rs71513892 | 32 | Qi et al. 2018 - brain expression | 0.046 | 0.01 | 1.43E06 | 1.48E–04 |
| SH3GL2 | ENSG00000107295 | 9 | 17,688,103 | 17,684,784 | rs10756899 | 15 | Qi et al. 2018 - brain expression | 0.252 | 0.05 | 5.83E08 | 6.00E–06 |
| ITGA8 | ENSG00000077943 | 10 | 15,659,036 | 15,548,925 | rs7910668 | 6 | Qi et al. 2018 - brain expression | −0.201 | 0.05 | 6.13E05 | 6.32E–03 |
| RNF141 | ENSG00000110315 | 11 | 10,548,001 | 10,553,355 | rs4910153 | 120 | Võsa et al. 2018 - blood expression | −0.054 | 0.05 | 6.25E07 | 1.31E–04 |
| IGSF9B | cg25790212 | 11 | 133,800,774 | 133,800,477 | rs11223626 | 1 | Qi et al. 2018 - brain methylation | −0.172 | 0.04 | 3.24E06 | 6.48E–04 |
| FBRSL1 | cg03621470 | 12 | 133,137,479 | 133,138,334 | rs10781619 | 16 | Qi et al. 2018 - brain methylation | −0.057 | 0.01 | 6.35E05 | 1.27E–02 |
| CAB39L | ENSG00000102547 | 13 | 49,950,524 | 49,918,175 | rs35214871 | 30 | Qi et al. 2018 - brain expression | 0.097 | 0.02 | 3.51E08 | 3.62E–06 |
| GCH1 | ENSG00000131979 | 14 | 55,339,148 | 55,348,837 | rs3825611 | 6 | Qi et al. 2018 - brain expression | 0.113 | 0.03 | 2.76E04 | 2.85E–02 |
| SYT17 | ENSG00000103528 | 16 | 19,229,472 | 19,273,554 | rs727747 | 4 | Qi et al. 2018 - brain expression | 0.177 | 0.05 | 1.54E04 | 1.58E–02 |
| SETD1A | ENSG00000099381 | 16 | 30,982,526 | 30,950,352 | rs7206511 | 34 | Võsa et al. 2018 - blood expression | −0.710 | 0.09 | 2.75E13 | 5.77E–11 |
| CHRNB1 | ENSG00000170175 | 17 | 7,354,703 | 7,373,595 | rs60488855 | 18 | Qi et al. 2018 - brain expression | 0.115 | 0.03 | 1.67E05 | 1.72E–03 |
| UBTF | ENSG00000108312 | 17 | 42,290,697 | 42,297,631 | rs113844752 | 34 | Võsa et al. 2018 - blood expression | −0.466 | 0.09 | 5.68E06 | 1.19E–03 |
| MAPT | ENSG00000186868 | 17 | 44,038,724 | 44,862,347 | rs199502 | 6 | Qi et al. 2018 - brain expression | 0.265 | 0.03 | 7.13E24 | 7.35E–22 |
| WNT3 | ENSG00000108379.5 | 17 | 44,875,148 | 44,908,263 | rs9904865 | 2 | GTEx v7 - substantia nigra brain expression | −0.082 | 0.02 | 4.01E06 | 4.81E–05 |
| DNAH17 | cg09006072 | 17 | 76,425,972 | 76,427,732 | rs589582 | 3 | Qi et al. 2018 - brain methylation | 0.100 | 0.02 | 2.44E05 | 4.88E–03 |
| MEX3C | ENSG00000176624 | 18 | 48,722,797 | 48,731,131 | rs12458916 | 40 | Võsa et al. 2018 - blood expression | −0.291 | 0.05 | 5.28E05 | 1.11E–02 |
Multi-SNP eQTL Mendelian randomization results focusing only on the most significant association per nearest gene to the PD risk variant of interest after Bonferroni correction. If a locus was significantly associated with both brain and blood QTLs after multiple test correction, we opted to show the most signficant brain tissue derived association here after filtering for possible polygenicity (HEIDI P > 0.01). Effects with significant HEIDI P values may indicate a possible effect complicated by LD and are less likely to be a true causal association. All tested QTL summary statistics can be found in Supplementary Table S6. Effect estimates represent the change in PD odds ratio per one standard deviation increase in gene expression or methylation.
Of the 305 genes under linkage disequilibrium (LD) peaks around our risk variants of interest, 237 were possibly associated with at least one QTL in public reference datasets and were therefore testable via SMR (Methods Supplement, Table S6). The expression or methylation of 151 of these 237 genes (63.7%) was significantly associated with a possible causal change in PD risk.
Of the 90 PD GWAS risk variants, 70 were in loci containing at least one of these putatively causal genes after multiple test correction (Table 3). For 53 out of these 70 PD GWAS hits (75.7%), the gene nearest to the most significant SNP was a putatively causal gene (Table S2). Most loci tested contained multiple putatively causal genes. Interestingly, the nearest putatively causal gene to the rs850738/FAM171A2 GWAS risk signal is GRN, a gene known to be associated with frontotemporal dementia (FTD) (34). Mutations in GRN have also been shown to be connected with another lysosomal storage disorder, neuronal ceroid lipofuscinosis (35).
As an orthogonal approach for nominating genes under GWAS peaks we carried out rare coding variant burden analyses. We performed kernel-based burden tests on the 113 genes out of the 305 under our GWAS peaks that contained two or more rare coding variants (MAF< 5% or MAF < 1%). After Bonferroni correction for 113 genes, we identified 7 significant genes: LRRK2, GBA, CATSPER3 (rs11950533/C5orf24 locus), LAMB2 (rs12497850/IP6K2 locus), LOC442028 (rs2042477/KCNIP3 locus), NFKB2 (rs10748818/GBF1 locus), and SCARB2 (rs6825004 locus). These results suggest that some of the risk associated with these loci may be due to rare coding variants or that these are pleomorphic risk loci. The LRRK2 and NFKB2 associations at MAF < 1% remained significant after correcting for all ~20,000 genes in the human genome (P = 2.15E-10 and P = 4.02E-07, Table S7, Table S5).
We tested whether genes of interest were enriched in 10,651 biological pathways(from gene ontology annotations) using Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA) (22,36). We found 10 significantly enriched pathways (FDR-adjusted P < 0.05, Table S8), including four related to vacuolar function and three related to known drug targets (calcium transporters: ikeda_mir1_targets_dn and ikeda_mir30_targets_up, kinase signaling: kim_pten_targets_dn). At least three candidate genes within novel loci are involved in lysosomal storage disorders (GUSB, GRN, and NEU1), a pathway of keen interest in PD (37). Our GWAS results also include candidate genes VAMP4 and NOD2 from the endocytic pathway (38).
To determine the tissues and cell types most relevant to PD etiology using FUMA (22,36) we tested whether the genes highlighted by our PD GWAS were enriched for expression in 53 tissues from across the body. We found 13 significant tissues, all of which were brain-derived (Figure S2A), in contrast to what has been seen in Alzheimer’s disease which shows a strong bias towards blood, spleen, lungs and microglial enrichments (39). To further disentangle the enrichment in brain tissues, we tested whether our PD GWAS genes were enriched for expression in 88 brain cell types using single cell RNA sequencing reference data from mouse brains (http://dropviz.org)(40). After FDR correction we found seven significant brain cell types, all of which were neuronal (Figure S2B). The strongest enrichment was for neurons in the substantia nigra (SN) at P = 1.0E-06, with additional significant results at P < 5.0E-4 for the globus pallidus (GP), thalamus (TH), posterior cortex (PC), frontal cortex (FC), hippocampus (HC) and entopeduncular nucleus (ENT).
Next, we used cross-trait genetic correlation and MR to identify possible PD biomarkers and risk factors by comparing with 757 other GWAS datasets curated by LD hub (41). We found four significant genetic correlations (FDR-adjusted P < 0.05, Table S10) including positive correlations with intracranial volume and putamen volume (42), and negative correlations with current tobacco use and “academic qualifications: National Vocational Qualifications (NVQ) or Higher National Diploma (HND) or Higher National Certificate (HNC) or equivalent” (43). The negative association with one’s academic qualifications suggests that individuals without a college education may be at less risk of PD. The correlation between PD and smoking status may not be independent from the correlation between PD and education as smoking status and years of education were significantly correlated (44).
We used MR to assess whether there was evidence of a causal relationship between PD and five phenotypes related to academic qualification, smoking, and brain volumes described above (Figure S4). Cognitive performance had a large, significant causal effect on PD risk (MR effect = 0.213, SE = 0.041, Bonferroni-adjusted P = 8.00E-07), while PD risk did not have a significant causal effect on cognitive performance (Bonferroni-adjusted P = 0.125). Educational attainment also had a significant causal effect on PD risk (MR effect = 0.162, SE = 0.040, Bonferroni-adjusted P = 2.06E-04), but PD risk also had a weak but significant causal effect on educational attainment (MR effect = 0.007, SE = 0.002, Bonferroni-adjusted P = 7.45E-3). There was no significant causal relationship between PD and current smoking status (forward analysis: MR effect = −0.069, SE = 0.031, Bonferroni-adjusted P = 0.125; reverse analysis: MR effect = 0.004, SE = 0.010, Bonferroni-adjusted P = 1). Smoking initiation (the act of ever starting smoking) did not have a causal effect on PD risk (MR effect = −0.063, SE = 0.034, Bonferroni-adjusted P = 0.315), whereas PD had a small, but significantly positive causal effect on smoking initiation (MR effect = 0.027, SE = 0.006, Bonferroni-adjusted P = 1.62E-05). Intracranial volume could not be tested because its GWAS did not contain any genome-wide significant risk variants. There was no significant causal relationship between PD and putamen volume (P > 0.05 in both the forward and reverse directions).
DISCUSSION
Our work marks a significant step forward in our understanding of the genetic architecture of PD and provides a genetic reference set for the broader research community. We identified 90 independent common genetic risk factors for PD, nearly doubling the number of known PD risk variants. We re-evaluated the cumulative contribution of genetic risk variants, both genome-wide significant and not-yet discovered, in order to refine our estimates of heritable PD risk. We also nominated likely genes at each locus for further follow-up using QTL analyses and rare variant burden analyses. Our work has highlighted the pathways, tissues, and cell types involved in PD etiology. Finally, we identified intracranial and putaminal volume as potential future PD biomarkers, and cognitive performance as a PD risk factor. Altogether, the data presented here has significantly expanded the resources available for future investigations into potential PD interventions.
We were able to explain 16%−36% of PD heritability, the range being directly related to prevalence estimates varying (0.5% to 2%). Power estimates suggest that expansions of case numbers to 99K cases will continue to reveal additional insights into PD genetics. While these risk variants will have relatively small effects and/or be quite rare, they will help to further expand our knowledge of the genes and pathways that drive PD risk.
Population-wide screening for individuals who are likely to develop PD is currently not feasible using our 1805 variant PRS alone. There would be roughly 14 false positives per true positive assuming a prevalence of 0.5%. While large-scale genome sequencing and non-linear machine learning methods will likely improve these predictive models, we have previously shown that we will need to incorporate other data sources (e.g. smell tests, family history, age, sex) in order to generate algorithms that have more possible value in population-wide screening (4).
Evaluating these results in the larger context of pathway, tissue, and cellular functionality revealed that genes near PD risk variants showed enrichment for expression in the brain, contrasting with previous work in Alzheimer’s disease. Strikingly, we showed that the expression enrichment of genes at PD loci occurred exclusively in neuronal cell types. We also found that PD genes were enriched in chemical signaling pathways and pathways involving the response to a stressor. We believe that this contrast, in which the pathway enrichment analyses suggest at least some immune component to PD and the expression enrichment analysis does not suggest any significant immune related tissue component should be viewed with a critical eye. In particular, the marginal P values of most immune related pathways in our recent analyses after multiple test correction reinforce this moderate view. These observations may be informative for disease modeling efforts, highlighting the importance of disease modeling in neurons and possibly incorporating a cellular stress component. This information can help inform and focus stem cell derived therapeutic development efforts that are currently underway.
We found four phenotypes that were genetically correlated with PD. Putamen and intracranial volumes may prove to be valuable in future PD biomarker studies. Our bi-directional GSMR results suggest a complex etiological connection between smoking initiation and PD that will require further follow-up. One of the implications of this work is that PD trials of nicotine or other smoking-related compound(s) may be less likely to succeed. The strong causal effect of cognitive performance on PD is supported by observational studies (45).
While this study marks major progress in assessing genetic risk factors for PD, there remains a great deal to be done. No defined external validation dataset was used, which may be seen as a limitation. Also, external replication of the novel associations we present will be difficult simply due to the sample sizes needed. Simulations have suggested that without replication variants with P values between 5E-08 and 5E-9 should be interpreted with greater caution (46,47). We found 16 risk variants in this range, including two known variants near WNT3 (proximal to the MAPT locus) and BIN3. To a degree, the fact that we filtered our variants with a secondary random-effects meta-analysis may make our 90 PD GWAS hits somewhat more robust due to the conservative nature of random-effects.
This study focused on PD risk in individuals of European ancestry. Adding datasets from non-European populations would be helpful to further improve our granularity in association testing and ability to fine-map loci through integration of more variable LD signatures while also evaluating population specific associations. Also, risk predictions may not generalize across populations in some cases and ancestry specific PRS should be investigated. Additionally, large ancestry-specific PD LD reference panels, such as those for Ashkenazi Jewish patients, will help us further unravel the genetic architecture of loci such as GBA and LRRK2. This may be particularly crucial at these loci where LD patterns may be variable within European populations, accentuating the possible influence of LD reference series on conditional analyses in some cases (48). Finally, our work utilized state-of-the-art QTL datasets to nominate candidate genes, but many QTL associations are hampered by both small sample size and low cis-SNP density. Larger QTL studies and PD-specific network data from large scale cellular screens would allow us to build a more robust functional inference framework.
As the field moves forward there are some critical next steps that should be prioritized. First, allowing researchers to share participant-level data in a secure environment would facilitate inclusiveness and uniformity in analyses while maintaining the confidentiality of study participants. Our work suggests that GWASes of increasing size will continue to provide useful biological insights into PD. In addition to studies of the genetics of PD risk, studies of disease onset, progression, and subtype will be important and will require large series of well-characterized patients(49). We also believe that work across diverse populations is important, not only to be able to best serve these populations but also to aid in fine mapping of loci. Notably, the use of genome sequencing technologies could further improve discovery by capturing rare variants and structural variants, but with the caveat that very large sample sizes will be required. While there is still much left to do, we believe that our current work represents a significant step forward and that the results and data will serve as a foundational resource for the community to pursue this next phase of PD research.
DATA ACCESS
GWAS summary statistics for 23andMe datasets (post-Chang and data included in Chang et al. 2017 and Nalls et al. 2014) will be made available through 23andMe to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Please visit research.23andme.com/collaborate/#publication for more information and to apply to access the data. An immediately accessible version of the summary statistics is available here excluding Nalls et al. 2014, 23andMe post-Chang et al. 2017 and Web-Based Study of Parkinson’s Disease (PDWBS) but including all analyzed SNPs. After applying with 23andMe, the full summary statistics including all analyzed SNPs and samples in this GWAS meta-analysis will be accessible to the approved researcher(s). Underlying participant level IPDGC data is available to potential collaborators, please contact ipdgc.contact@gmail.com.
Supplementary Material
RESEARCH IN CONTEXT.
Evidence before this study
Previous studies such as Chang et al. 2017 and its predecessors have utilized GWAS methods to discover 42 independent risk loci associated with PD (1). Some of these loci harboring common risk variants also include rare variants implicated in familial PD risk such as SNCA, LRRK2 or GBA. Earlier studies like Keller et al. 2012 have attempted to quantify how much heritable risk is captured by common variation that can be easily imputed using commercial genotyping arrays and estimate the amount of risk explained by GWAS (2). As far back as Nalls et al. 2011, GWAS studies of PD have integrated expression and methylation datasets to evaluate possible candidate genes for follow-up at PD loci (3). Many epidemiological and observational studies have attempted to assess risk of PD and various exposures like smoking, caffeine or occupational hazards, with a mixed track record of success at validating putative associations.
Added value of this study
The primary deliverable of this study was increasing the count of independent common genetic risk factors for PD to 90. We added 38 novel risk variants not identified as genome-wide significant in previous reports. We refined heritability estimates and genetic risk predictions suggesting that common genetic variants account for approximately 22% of PD risk on the liability scale, with a range of 16–36% of that risk being explained by GWAS loci in this study. These updated risk predictions also suggested that polygenic risk scoring can be used to achieve an area under the curve of near 70%, although this prediction uses many more variants than just the 90 independent risk factors identified in this report. Of the 90 risk variants we have characterized here, we have nominated at least one possible candidate gene for follow-up functional studies in 70 of these genomic regions by mining recently available expression and methylation reference datasets on a scale not possible just a few years ago. We have additionally mined single cell RNA sequencing data from mice to identify tissue-specific signatures of enrichment relating to PD genetic risk, showing a major focus on neuronal cell types. We also utilized the massive amount of publicly available GWAS results to survey genetic correlations between PD and other phenotypes showing significant correlations with smoking, education and brain morphology. Subsequent analyses using Mendelian randomization (MR) methods showed that there are likely causal links between increased cognitive performance and PD risk on a genetic level.
Implications of all available evidence
First and foremost, this study increased the scope of our knowledge of PD genetics by adding 38 novel risk factors, directly broadening our knowledge base of disease etiology. Using updated heritability estimates and risk predictions, we took preliminary steps down the long path to early detection. In future studies, combining genetic and clinico-demographic risk factors may lead to earlier detection and refined diagnostics, which may help improve clinical trials (4). The generation of copious amounts of public summary statistics created by this effort relating to both the GWAS and subsequent analyses of gene expression and methylation patterns may be of use to investigators planning follow-up functional studies in stem cells or other cellular screens, allowing them to prioritize targets more efficiently using our data as additional evidence. We hope our findings may have some downstream clinical impact in the future such as improved patient stratification for clinical trials and genetically informed drug targets.
ACKNOWLEDGEMENTS
See supplemental materials (Text S2).
Disclosures and conflicts of interest
Dr. Nalls reports that this work was carried out under a consulting contract with NIH, he also consults for Lysosomal Therapeutics Inc, Neuron23 Inc and Illumina Inc. Dr. Heilbron reports other support from 23andMe, during the conduct of the study outside the submitted work. Dr. Chang reports other support from Genentech outside the submitted work. Ms. Tan reports grants from Parkinson’s UK during the conduct of the study. Dr. Noyce reports grants from Parkinson’s UK, grants from Virginia Kieley benefaction, grants and non-financial support from GE Healthcare, personal fees from Profile, Bial and Britannia, outside the submitted work. Dr. von Coelln reports grants from American Brain Foundation, grants from Michael and Eugenia Brin Foundation, during the conduct of the study. Dr. Pihlstrøm reports grants from Southeastern Regional Health Authority, Norway, during the conduct of the study. Dr. Siitonen reports grants from Sigrid Juselius Foundation, during the conduct of the study. Dr. Scholz is a scientific advisory council member for the Lewy Body Dementia Association. Dr. Corvol reports grants from French Ministry of Health, during the conduct of the study; grants from Sanofi, personal fees from Ever Pharma, personal fees from Denali, personal fees from Biogen, personal fees from Air Liquide, personal fees from BrainEver, personal fees from Theranexus, outside the submitted work. Dr. L Shulman reports grants from NIH, outside the submitted work. Dr. Tienari reports no conflicts of interest. In addition, Dr. Tienari has a patent c9orf72 in the diagnosis and treatment of neurodegenerative disease pending. Dr. Toft reports grants from Research Council of Norway, grants from Regional Health Authority South-Eastern Norway, grants from Michael J. Fox Foundation, personal fees from Roche, outside the submitted work. Dr. Andreassen reports grants from Research Council of Norway, grants from KG Jebsen Stiftelsen, during the conduct of the study; personal fees from Lundbeck, outside the submitted work; In addition, Dr. Andreassen has a patent PCT/US2014/011014 pending. Dr. Bhangale reports other from Genentech, outside the submitted work. Dr. BRICE reports grants from France Parkinson + FRC, grants from ANR - EPIG - Agence nationale de recherche, grants from ANR - JPND - Agence nationale de recherche, grants from RDS (Roger de Spoelberch Foundation), grants from France Alzheimer, grants from ENP -Ecole des neurosciences Paris, grants from Institut de France, grants from CHU de Nimes, grants from ERA NET, grants from ANR - EPIG, grants from APHP, outside the submitted work. Dr. Gan-Or reports personal fees from Lysosomal Therapeutics Inc, personal fees from Idorsia, personal fees from Inception Sciences, personal fees from Denali, personal fees from Prevail Therapeutics, outside the submitted work. Dr. Gasser reports grants from The Michael J Fox Foundation for Parkinsońs Research, personal fees from UCB Pharma, other from “Joint Programming for Neurodegenerative Diseases” program, funded by the European Commission, personal fees from Novartis, personal fees from Teva, personal fees from MedUpdate, outside the submitted work; In addition, Dr. Gasser has a patent Patent Number: EP1802749 (A2) KASPP (LRRK2) gene, its production and use for the detection and treatment of neurodegenerative disorders issued. Dr. Heutink is a consultant for NEURON23, inc. Dr. J Shulman reports grants from Burroughs Wellcome Fund during the conduct of the study; grants from National Institutes of Health and personal fees from Helis Medical Foundation, outside the submitted work. Dr. Hinds reports other from 23andMe, Inc., outside the submitted work. Dr. Morris reports grants from Parkinson’s UK, grants from Medical Research Council, grants from Cure Parkinson’s Trust, during the conduct of the study; grants from PSP Association, grants from CBD Solutions, personal fees from Teva, personal fees from Boehringer Ingelheim, personal fees from GSK, grants from Drake Foundation, personal fees from Biogen, personal fees from UCB, personal fees from Biohaven, outside the submitted work. Dr. Graham reports other from Genentech, during the conduct of the study; other from Genentech, outside the submitted work.
Footnotes
All others have no disclosures or potential conflicts of interest.
Full consortia membership (PubMed indexed) is available in the supplemental materials (Text S1).
WORKS CITED
- 1.Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 2017October;49(10):1511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keller MF, Saad M, Bras J, Bettella F, Nicolaou N, Simón-Sánchez J, et al. Using genome-wide complex trait analysis to quantify “missing heritability” in Parkinson’s disease. Hum Mol Genet 2012November15;21(22):4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin U-M, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011February19;377(9766):641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nalls MA, McLean CY, Rick J, Eberly S, Hutten SJ, Gwinn K, et al. Diagnosis of Parkinson’s disease on the basis of clinical and genetic classification: a population-based modelling study. Lancet Neurol 2015October;14(10):1002–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007January30;68(5):384–6. [DOI] [PubMed] [Google Scholar]
- 6.Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science. 1996November15;274(5290):1197–9. [DOI] [PubMed] [Google Scholar]
- 7.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003October31;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 8.Fung H-C, Scholz S, Matarin M, Simón-Sánchez J, Hernandez D, Britton A, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 2006November;5(11):911–6. [DOI] [PubMed] [Google Scholar]
- 9.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014September;46(9):989–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010September1;26(17):2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ANthropometric Traits (GIANT) Consortium, DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 2012March18;44(4):369–75, S1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics. 2014;31(9):1466–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin U-M, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011February19;377(9766):641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International Parkinson’s Disease Genomics Consortium (IPDGC), Wellcome Trust Case Control Consortium 2 (WTCCC2). A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet 2011June;7(6):e1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nalls MA, Escott-Price V, Williams NM, Lubbe S, Keller MF, Morris HR, et al. Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov Disord 2015May;30(6):850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015March;47(3):291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013June;45(6):580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun 2018June11;9(1):2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016May;48(5):481–7. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 2018March2;9(1):918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis [Internet]. bioRxiv 2018. [cited 2018 Oct 25]. p. 447367. Available from: https://www.biorxiv.org/content/early/2018/10/19/447367.abstract [Google Scholar]
- 22.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017November28;8(1):1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 2015;10(10):1556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 2012August10;91(2):224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LD Hub [Internet]. [cited 2018 Jun 20]. Available from: http://ldsc.broadinstitute.org/ldhub/
- 26.Prins BP, Abbasi A, Wong A, Vaez A, Nolte I, Franceschini N, et al. Investigating the Causal Relationship of C-Reactive Protein with 32 Complex Somatic and Psychiatric Outcomes: A Large-Scale Cross-Consortium Mendelian Randomization Study. PLoS Med 2016June;13(6):e1001976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahola-Olli AV, Würtz P, Havulinna AS, Aalto K, Pitkänen N, Lehtimäki T, et al. Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am J Hum Genet 2017January5;100(1):40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet [Internet]. 2018July23; Available from: 10.1038/s41588-018-0147-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature. 2018October;562(7726):210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011January7;88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wray NR, Yang J, Goddard ME, Visscher PM. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet 2010February26;6(2):e1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 2006February;38(2):209–13. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016May;48(5):481–7. [DOI] [PubMed] [Google Scholar]
- 34.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006August24;442(7105):920–4. [DOI] [PubMed] [Google Scholar]
- 35.Smith KR et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. - PubMed - NCBI [Internet]. [cited 2019 Jan 10]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22608501 [DOI] [PMC free article] [PubMed]
- 36.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017July3;45(W1):W130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 2017December1;140(12):3191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bandres-Ciga S, Saez-Atienzar S, Bonet-Ponce L, Billingsley K, Vitale D, Blauwendraat C, et al. The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson’s disease. Mov Disord 2019April;34(4):460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet [Internet]. 2019January7; Available from: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saunders A et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. - PubMed - NCBI [Internet]. [cited 2018 Oct 31]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30096299 [DOI] [PMC free article] [PubMed]
- 41.LD Hub [Internet]. [cited 2018 Jun 26]. Available from: http://ldsc.broadinstitute.org
- 42.Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivières S, Jahanshad N, et al. Common genetic variants influence human subcortical brain structures. Nature. 2015April9;520(7546):224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapid GWAS of thousands of phenotypes for 337,000 samples in the UK Biobank [Internet]. Neale lab. [cited 2018 Jun 24]. Available from: http://www.nealelab.is/blog/2017/7/19/rapid-gwas-of-thousands-of-phenotypes-for-337000-samples-in-the-uk-biobank [Google Scholar]
- 44.Bulik-Sullivan B, ReproGen Consortium, Finucane HK, Anttila V, Gusev A, Day FR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valdés EG, Andel R, Sieurin J, Feldman AL, Edwards JD, Långström N, et al. Occupational complexity and risk of Parkinson’s disease. PLoS One. 2014September8;9(9):e106676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Y, Zheng Z, Visscher PM, Yang J. Quantifying the mapping precision of genome-wide association studies using whole-genome sequencing data. Genome Biol 2017May16;18(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pulit SL, de With SAJ, de Bakker PIW. Resetting the bar: Statistical significance in whole-genome sequencing-based association studies of global populations. Genet Epidemiol 2017February;41(2):145–51. [DOI] [PubMed] [Google Scholar]
- 48.Rivas MA, Avila BE, Koskela J, Huang H, Stevens C, Pirinen M, et al. Insights into the genetic epidemiology of Crohn’s and rare diseases in the Ashkenazi Jewish population. PLoS Genet 2018May;14(5):e1007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwaki H, Blauwendraat C, Leonard HL, Kim JJ, Liu G, Maple-Grødem J, et al. Genome-wide association study of Parkinson’s disease progression biomarkers in 12 longitudinal patients’ cohorts [Internet]. bioRxiv. 2019. [cited 2019 Apr 25]. p. 585836. Available from: 10.1101/585836v1.abstract [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.