

Abstract
The nutrient-sensing mammalian target of rapamycin (mTOR) is integral to cell fate decisions after T cell activation. Sustained mTORC1 activity favors the generation of terminally differentiated effector T cells instead of follicular helper and memory T cells. This is particularly pertinent for T cell responses of older adults, who have sustained mTORC1 activation in spite of dysfunctional lysosomes. Here, we show that lysosome-deficient T cells rely on late endosomes rather than lysosomes as an mTORC1 activation platform, where mTORC1 is activated by sensing cytosolic amino acids. T cells from older adults have an increased expression of the plasma membrane leucine transporter SLC7A5 to provide a cytosolic amino acid source. Hence, SLC7A5 as well as VPS39 deficiency (a member of the HOPS complex promoting early to late endosome conversion) substantially reduced mTORC1 activities in T cells from older but not young individuals. Late endosomal mTORC1 is independent of the negative feedback loop involving mTORC1-induced inactivation of the transcription factor TFEB that controls expression of lysosomal genes. The resulting sustained mTORC1 activation impaired lysosome function and prevented lysosomal degradation of PD-1 in CD4+ T cells from older adults, thereby inhibiting their proliferative responses. VPS39 silencing of human T cells improved their expansion to pertussis as well as to SARS-CoV-2 peptides in vitro. Furthermore, adoptive transfer of CD4+ Vps39-deficient LCMV-specific SMARTA cells improved germinal center responses, CD8+ memory T cell generation and recall responses to infection. Thus, curtailing late endosomal mTORC1 activity is a promising strategy to enhance T cell immunity.
One-sentence Summary
Sustained activation of mTORC1 at late endosomes in primary CD4+ T cell responses of older individuals impairs T helper cell function.
Introduction
Aging is associated with a decline in adaptive immunity, resulting in decreased efficacy of vaccination and increased morbidity from infections with newly as well as previously encountered pathogens (1, 2). Most noticeable are the increased susceptibility during the annual influenza epidemic (3) and, most recently, the increased risk for developing severe disease following SARS-CoV-2 infection (4–6). Although the increased morbidity is clearly multifactorial, age-related changes in CD4+ T cell survival and differentiation have been identified that contribute to the immune decline in older individuals (7, 8). CD4+ T helper cells are pivotal for mounting a protective immune response after vaccination or infections (9). Generation of CD4+ T follicular helper (TFH) cells is essential for germinal center (GC) formation, class switching and affinity maturation (10). Moreover, CD4+ T cells are required for effective CD8+ T cell memory and recall responses (11–13). Recent studies have shown a T cell-intrinsic bias towards short-lived effector cells differentiation in CD4+ T cell responses of older adults at the expense of TFH and memory precursor cells (8). One important mechanism causing this bias is a sustained activation of the mechanistic target of rapamycin complex 1 (mTORC1) (8).
mTORC1 plays an important role in regulating T cell responses by coordinating cell growth and cellular metabolism with environmental inputs, in particular nutrient resources, and facilitating the switch toward anabolic metabolism that is required for cell proliferation and effector cell differentiation (14–16). Complete block of mTORC1 activities by either a treatment with high dose of rapamycin or a genetic deletion of RHEB (an upstream activator of mTORC1) dramatically reduced the number of antigen-specific effector and memory precursor T cells at peak responses after primary infection (17, 18). RHEB-deficient memory CD8+ T cells even failed to respond to secondary immunization (18). However, overactivation of mTORC1 by genetic depletion of TSC1 (an upstream inhibitor of mTORC1) reduced the numbers of memory precursor CD8+ T cells at peak responses after primary infection instead of promoting them (19–21). Indeed, moderate inhibition of mTORC1 by pharmacological compounds produced increased clonal expansion of antigen-specific T cells after primary responses and enhanced memory recall responses after antigen rechallenge in several infection models (22–24). These data led to the conclusion that mTORC1 inhibition promotes long-term memory over short-lived effector CD8+ T cell differentiation (17). Taken together, fine-tuning of mTORC1 activity is important for generating protective primary and recall T cell responses, in particular for older adults, who have sustained mTORC1 activity after T cell activation resulting in the preferential generation of short-lived effector T cells (8).
mTORC1 translocates to the lysosomal membrane, where it is activated in response to amino acid signaling. In turn, mTORC1 suppresses lysosomal activity by phosphorylating TFEB (25, 26). We previously found that T cells from older adults had lower lysosomal gene expression and proteolytic activities due to reduced TFEB transcription (27). In parallel, mTORC1 activity was more sustained in activated T cells from older adults (8). How lysosome-deficient aged T cells maintain mTORC1 activity remains unresolved. The late endosome is an alternative platform for mTORC1 activation (28–30). Compared to the lysosome, late endosomes have no proteolytic activity and therefore no amino acid efflux (31). They originate by homotypic fusion and vacuole protein sorting (HOPS) complex-mediated conversion from early endosomes (32). Inhibiting early to late endosomal conversion by silencing Vam6/Vps39-like protein (VPS39, a key member of the HOPS complex) led to the formation of hybrid endosomal compartments and attenuated mTORC1 activity (29, 33). Conversely, inhibition of lysosomal activities induced an intracellular expansion of late endosomes (34). Consistent with this observation, an increased number of late endosomes is seen in responses of T cells from older individuals that fail to replenish their lysosomes (27).
Here, we describe that in activated CD4+ T cells from older adults, mTORC1 activation relies on late endosomes rather than lysosomes as an activation platform and the plasma membrane leucine transporter SLC7A5 as an amino acid source. SLC7A5 as well as VPS39 deficiency reduced mTORC1 activities in T cells from older but not young adults. Sustained activation of late endosomal mTORC1 suppressed lysosomal biogenesis by phosphorylating TFEB, thereby preventing lysosomal degradation of PD-1 and impairing T cell expansion. In vivo, inhibition of late endosome formation by silencing Vps39 in LCMV-specific CD4+ SMARTA cells enhanced T cell expansion and improved memory cell transition after LCMV infection, leading to enhanced CD4+ T cell helper responses. Thus, abrogation of late endosomal mTORC1 may be a promising strategy to boost immunity in older individuals.
Results
Lysosome-independent activation of mTORC1 in naïve CD4+ T cell responses.
mTORC1 is recruited to lysosomal membranes where it is activated by released amino acids. However, we found that after 5 days of in vitro stimulation with anti-CD3/anti-CD28 beads naïve CD4+ T cells from older (65–85 years) adults had impaired lysosomal activity in parallel to enhanced mTORC1 activity, as shown by reduced fluorescence of cleaved de-quenched bovine serum albumin (DQ-BSA) and increased S6K1 protein phosphorylation, the downstream effector of mTORC1 activity (Fig. 1A and fig. S1A and B). To examine this apparent conundrum, we investigated the effects of lysosome inhibition on mTORC1 activity; chloroquine and Bafilomycin A1, two lysosomal acidification inhibitors, were used for pharmacological inhibition at nontoxic doses (fig. S1C); TFEB silencing was used as a genetic intervention. All interventions reduced lysosomal activities but surprisingly neither reduced S6K1 (Fig. 1B and C and fig. S1D to F) nor AKT phosphorylation (fig. S1G), the downstream effector of the growth-factor signaling arm of mTORC1 pathway (35).
Fig. 1. Lysosome-independent activation of mTORC1 in naïve CD4+ T cell responses.
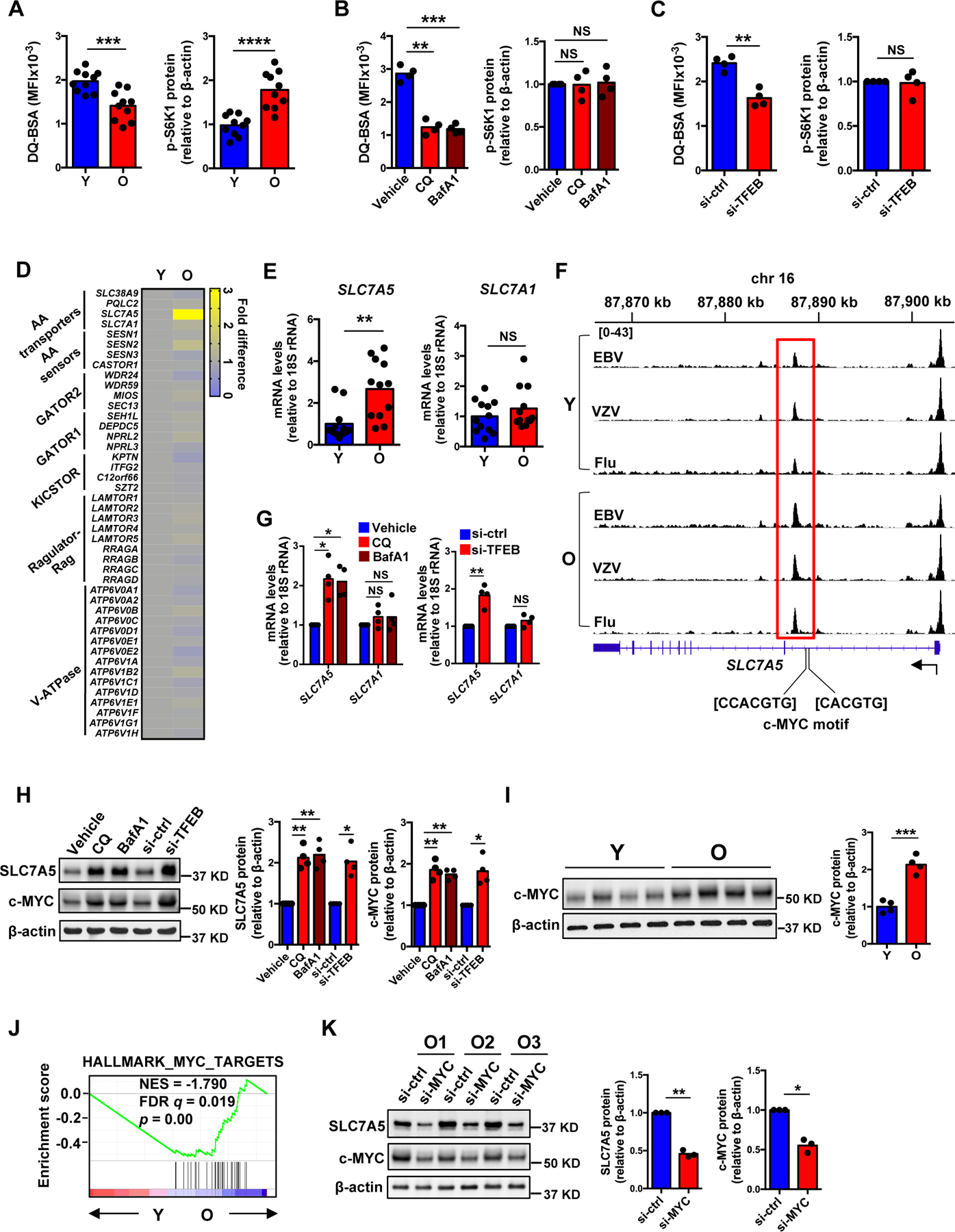
(A) Naïve CD4+ T cells were activated with anti-CD3/anti-CD28 beads for 5 days followed by DQ-BSA treatment for 6 hours. Fluorescence of cleaved DQ-BSA was analyzed by flow cytometry to determine lysosomal activities (left). Phospho-S6K1 (Thr389) protein expression was determined by Western blotting (right). Results are from ten young (Y, 20–35 years) and ten older (O, 65–85 years) healthy individuals. Intensities of p-S6K1 protein expression were normalized to β-actin and are shown relative to the mean from young individuals. (B) Naïve CD4+ T cells from young individuals were activated for 5 days with the last 2 days in the presence of vehicle, chloroquine (CQ) or bafilomycin A1 (BafA1). Lysosomal activities (left) and mTORC1 activities (right) were determined as in (A). (C) Naïve CD4+ T cells from young individuals were transfected with control or TFEB siRNA and activated for 5 days. Lysosomal activities (left) and mTORC1 activities (right) were determined. (D) Heat map showing expression differences of genes involved in the amino acid signaling arm of the mTORC1 pathway, comparing the transcriptome of day 5-activated naïve CD4+ T cells from older and young adults (re-analyzed from (36)). (E) SLC7A5 and SLC7A1 transcripts from day 5-activated naïve CD4+ T cells from twelve 20–35 year-old and twelve 65–85 year-old healthy adults. Results are expressed relative to the mean from young individuals. (F) Chromatin accessibility at SLC7A5 gene in human Epstein-Barr (EBV), varicella-zoster (VZV) and influenza (Flu) virus-specific CD4+ T cells from young and older adults. Averaged tracks (young n=3, older n=4) at SLC7A5 show increased peak in highlighted (red) region. (G) SLC7A5 and SLC7A1 transcripts in samples from (B) and (C). (H) SLC7A5 and c-MYC protein expression in samples from (B) and (C). (I) c-MYC protein expression in samples from (A). (J) GSEA comparing fold transcript differences in young compared with older naïve CD4+ T cells on day 5 after stimulation (re-analyzed from (36)) with that of “HALLMARK_MYC_TARGETS”. (K) SLC7A5 and c-MYC protein expression in day 5-stimulated naïve CD4+ T cells from three older individuals transfected with control or MYC siRNA. Statistical significance by two-tailed unpaired t test (A, E and I) or two-tailed paired t-test (B, C, G, H and K). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; NS, not significant.
We then set out to investigate the mechanism of how mTORC1 activity is increased even under the lysosome-deficient conditions in T cell responses of older adults. Because lysosome deficiency likely affects lysosomal efflux of amino acids, we examined expression of genes encoding the amino acid signaling arm of the mTORC1 pathway using data from our recent RNA-seq study comparing naïve CD4+ T cell responses from young and older adults after activation (36). SLC7A5 that encodes a plasma membrane leucine transporter is selectively more expressed in T cells from older adults while other genes involved in the nutrient arm do not change with age (Fig. 1D). Increased SLC7A5 transcription was confirmed in an independent cohort of day 5-activated naïve CD4+ T cells from twelve young and twelve older individuals. In contrast, no difference was seen for SLC7A1 that encodes the arginine transporter (Fig. 1E). SLC7A5 expression increased with activation in naïve CD4+ T cells from young and older donors, but expression was more sustained beyond day 3 in older naïve CD4+ T cells (fig. S2A–B). This difference persisted and was still detected for memory CD4+ T cells (fig. S2C). Accordingly, ATAC-seq studies showed increased chromatin accessibility of SLC7A5 in older memory CD4+ T cells in three virus-specific (EBV, VZV and influenza) systems compared to young cells (Fig. 1F). Consistent with increased SLC7A5 expression in lysosome-deficient aged T cells, pharmacological inhibition of lysosome activity or genetic silencing of TFEB upregulated SLC7A5 transcript and protein expression (Fig. 1G and H and fig. S1H). These data suggested that the up-regulation of SLC7A5 transcription in day 5-stimulated naïve CD4+ T cells from older adults was mechanistically related to their reduced lysosome activity.
Upstream of the differentially opened chromatin sites of SLC7A5 are two c-MYC binding motifs (Fig. 1F). c-MYC is the major driver of SLC7A5 transcription in T cell responses (37). c-MYC protein expression was increased after lysosomal inhibition or TFEB silencing (Fig. 1H), indicating a lysosome-dependent degradation of c-MYC protein. Consistently, day 5-activated aged naïve CD4+ T cells had higher c-MYC protein level compared to young cells (Fig. 1I). Global gene expression profiles obtained by RNA-seq supported the notion that day 5-activated older naïve CD4+ T cells had higher c-MYC activity. Gene set enrichment analysis (GSEA) showed that age-associated transcriptional signatures of day 5-activated naïve CD4+ T cells were correlated with the expression pattern of genes in the Hallmark_MYC_target pathway (Fig. 1J). To confirm that the increased SLC7A5 expression is related to higher c-MYC protein expression, we performed c-MYC silencing in naïve CD4+ T cells from older adults and found reduced SLC7A5 protein expression (Fig. 1K). Taken together, these data showed that lysosome deficiency stabilized c-MYC protein, thereby increasing SLC7A5 expression in day 5-stimulated naïve CD4+ T cells from older adults.
SLC7A5-dependent late endosomal mTORC1 activation in naïve CD4+ T cell responses.
To determine whether SLC7A5 activity accounts for the sustained mTORC1 activity, we treated T cell cultures from young and older adults with the specific SLC7A5 inhibitor JPH203. mTORC1 activities were monitored by Western blotting of S6K1 phosphorylation or by flow cytometry of S6RP phosphorylation on days 3 and 5 after activation. In titration experiments, a dose of 5 μM JPH203 was not cytotoxic (fig. S1C) and inhibited mTORC1 activity by about 50% (fig. S2D). On day 3, SLC7A5 inhibition reduced mTORC1 activity in cells from both young and older adults (Fig. 2A and B). On day 5, mTORC1 activity was already attenuated in T cell responses of young adults. In contrast, mTORC1 activity remained high in older activated T cells; the excess activity was sensitive to SLC7A5 inhibition (Fig. 2A and B). To exclude off-target effects of the inhibitor, we performed genetic SLC7A5 silencing and overexpression. Data observed were consistent with those obtained by JPH203 treatment (fig. S2E–H). Taken together, activated CD4+ T cells from older adults likely sustained uptake of extracellular amino acids through SLC7A5 to maintain mTORC1 activity in spite of reduced lysosomal function.
Fig. 2. SLC7A5-dependent late endosomal mTORC1 activation in naïve CD4+ T cell responses.
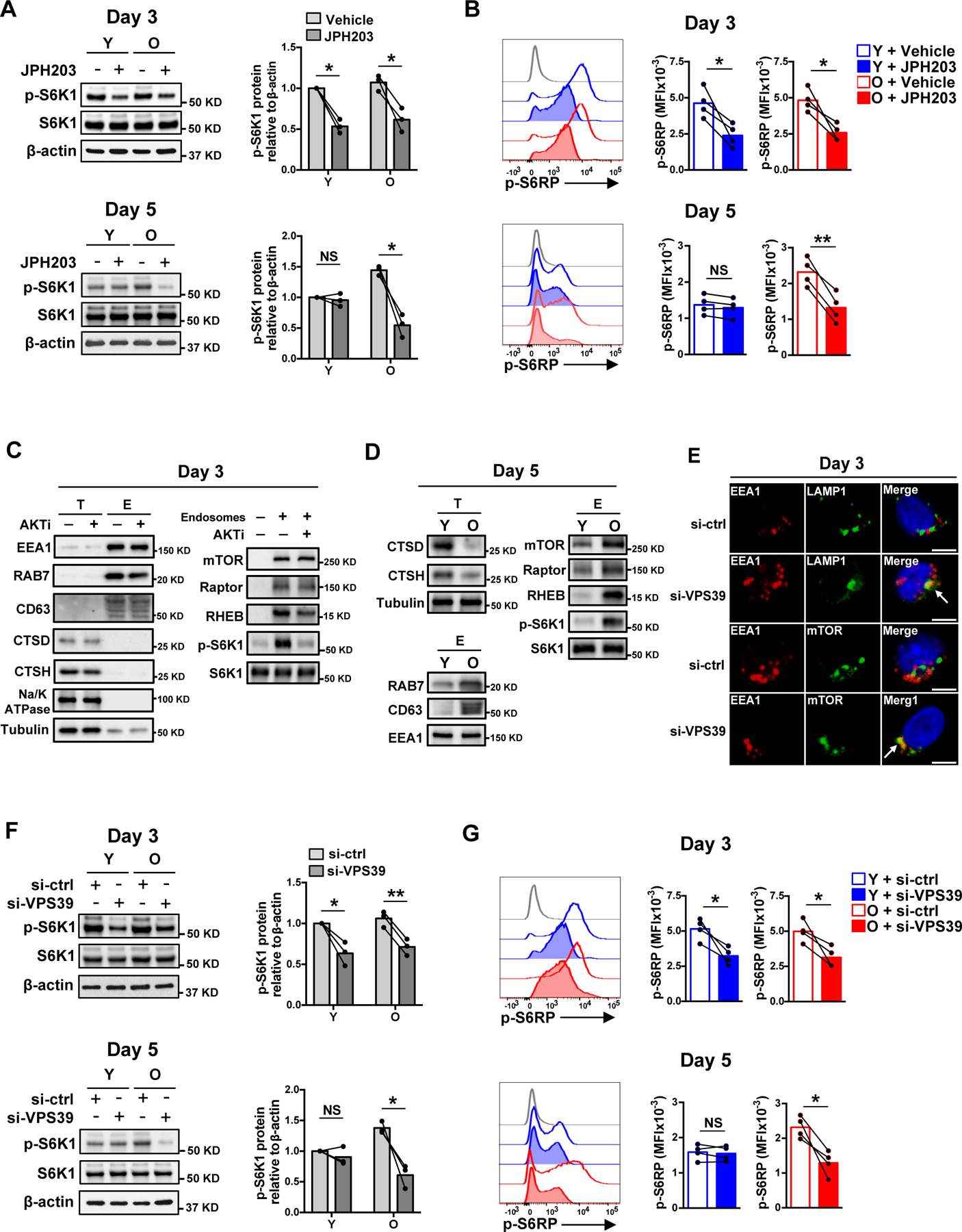
(A and B) Naïve CD4+ T cells from young and older healthy individual were activated with anti-CD3/anti-CD28 beads for 3 days in the presence of vehicle or SLC7A5 inhibitor JPH203 (upper panel). Alternatively, cells were activated with anti-CD3/anti-CD28 beads for 5 days with the last 2 days in the presence of vehicle or JPH203 (lower panel). mTORC1 activities were determined either by Western blotting of p-S6K1 (A) or by flow cytometry of intracellular p-S6RP (S235/S236) (B). Data are shown as one representative experiment (left) and cumulative data of three or four experiments (right). (C) Naïve CD4+ T cells were activated for 3 days followed by treatment or not with an AKT inhibitor for 2 hours prior to harvesting. Endosomes were isolated and analyzed for in vitro mTORC1 kinase activity toward S6K1. Total cell lysates (T) and endosome isolates (E) were analyzed by immunoblotting for indicated proteins. Data are shown as one representative of three experiments. (D) In vitro mTORC1 kinase activity of endosome isolates in day 5-stimulated naïve CD4+ T cells from one young and one older individual. Data are shown as one representative of three experiments. (E) Cells were stained with anti-EEA1, anti-LAMP1 and anti-mTOR. Confocal images representative of two independent experiments are shown. Scale bar, 5 μm. (F and G) mTORC1 activities in day 3- and day 5-stimulated naïve CD4+ T cells from young and older individuals after control or VPS39 silencing. The gray histogram represents isotype control. Comparison by two-tailed paired t test (A, B, F and G). *p < 0.05, **p < 0.01; NS, not significant.
We recently described that activated T cells from older adults failed to rebuild their lysosomes and expand the late endosomal compartments (27). To investigate whether these late endosomes provide an alternative platform for mTORC1 signaling in T cells, we isolated endosome fractions from naïve CD4+ T cells on day 3 after stimulation when mTORC1 activity peaks in both young and older individuals. The endosome isolates were enriched for early and late endosome markers (EEA1, RAB7 and CD63) and depleted of the lysosome markers Cathepsin D and Cathepsin H, and of the plasma membrane marker Na/K ATPase, indicating high purity of the endosome extracts (Fig. 2C). These endosome isolates contained mTOR, Regulatory-associated protein of mTOR (Raptor) and Ras homolog enriched in brain (RHEB) that are essential for substrate recruitment and kinase activity of mTORC1 (Fig. 2C). In an in vitro kinase assay, these endosome isolates exhibited mTORC1 kinase activity toward the substrate S6K1 that was not seen in endosomes extracted from cells treated with an AKT inhibitor (Fig. 2C). Day 5-stimulated naïve CD4+ T cells from older individuals had reduced lysosomal cathepsins, similar early endosome contents (EEA1) and increased late endosomal compartments, with increased late endosomal mTOR, Raptor and RHEB protein levels and higher mTORC1 kinase activity as shown by increased S6K1 phosphorylation in the in vitro kinase assay (Fig. 2D). Taken together, these data indicate that mTORC1 is activated on late endosomes and that late endosomal compartments are expanded in stimulated naïve CD4+ T cells from older adults accounting for the sustained mTORC1 activity.
We then performed VPS39 silencing to examine whether inhibition of late endosome maturation prevents mTORC1 activation. Confocal imaging confirmed that VPS39 silencing inhibited late endosome maturation and produced hybrid endosomal compartments that contain both the early endosome marker EEA1 and the late endosome/lysosome marker LAMP1 (Fig. 2E). These hybrid compartments co-localized with mTOR (Fig. 2E); however, S6K1 and S6RP phosphorylation were reduced after VPS39 silencing, documenting that these hybrid structures do not support mTORC1 activation (Fig. 2F and G). T cells from young and older adults exhibited reduced mTORC1 activities after VPS39 silencing on day 3 after T cell stimulation. On day 5, an effect of silencing was only observed in T cells from older adults (Fig. 2F and G), suggesting that the sustained mTORC1 signaling observed with age depended on the formation of late endosomes. VPS39 has also been implicated in regulating TGF-β/SMAD signaling (38); however, we did not observe changes of SMAD2/3 phosphorylation or reporter activities of a SMAD reporter in T cells after VPS39 silencing (fig. S3A–C), supporting the notion that the observed reduced mTORC1 activity is due to its impact on endosome maturation and not SMAD signaling.
Sustained activation of late endosomal mTORC1 suppresses lysosomal activities in naïve CD4+ T cell responses.
Our data so far demonstrated that CD4+ T cells from older adults sustained mTORC1 activity on late endosomes that relied on the presence of SLC7A5 and VPS39. mTORC1 phosphorylates TFEB, thereby preventing its transcriptional activity and inhibiting lysosome function (25). mTORC1 inhibition downregulated TFEB phosphorylation while upregulating TFEB mRNA and protein levels due to reduced AKT-mediated FOXO1 protein degradation (fig. S4A–C). Consistent with this notion, mTORC1 inhibition by Torin 1 partially restored the TFEB-dependent transcription of lysosomal cathepsin genes (CTSB, CTSD, CTSH and CTSS) (27) and lysosomal proteolytic activities in older, but not young, day 5-activated T cells (Fig. 3A and B). Inhibition of late endosomal mTORC1 in activated T cells from older adults by either VPS39 silencing or SLC7A5 inhibition had similar effects, while the already higher lysosomal activity in T cells from young adults could not be further increased (Fig. 3C–E). However, SLC7A5 inhibition could counteract the effects of lysosome inhibition in young T cells. Lysosomal cathepsins and lysosomal activities were increased in CQ and JPH203-cotreated young cells compared to young T cells treated with CQ alone (Fig. 3F). Together, these data demonstrate that sustained activation of mTORC1 in late endosomes suppressed lysosomal activities in day 5-stimulated naïve CD4+ T cells from older adults.
Fig. 3. Sustained activation of late endosomal mTORC1 suppresses lysosomal activities in naïve CD4+ T cell responses.
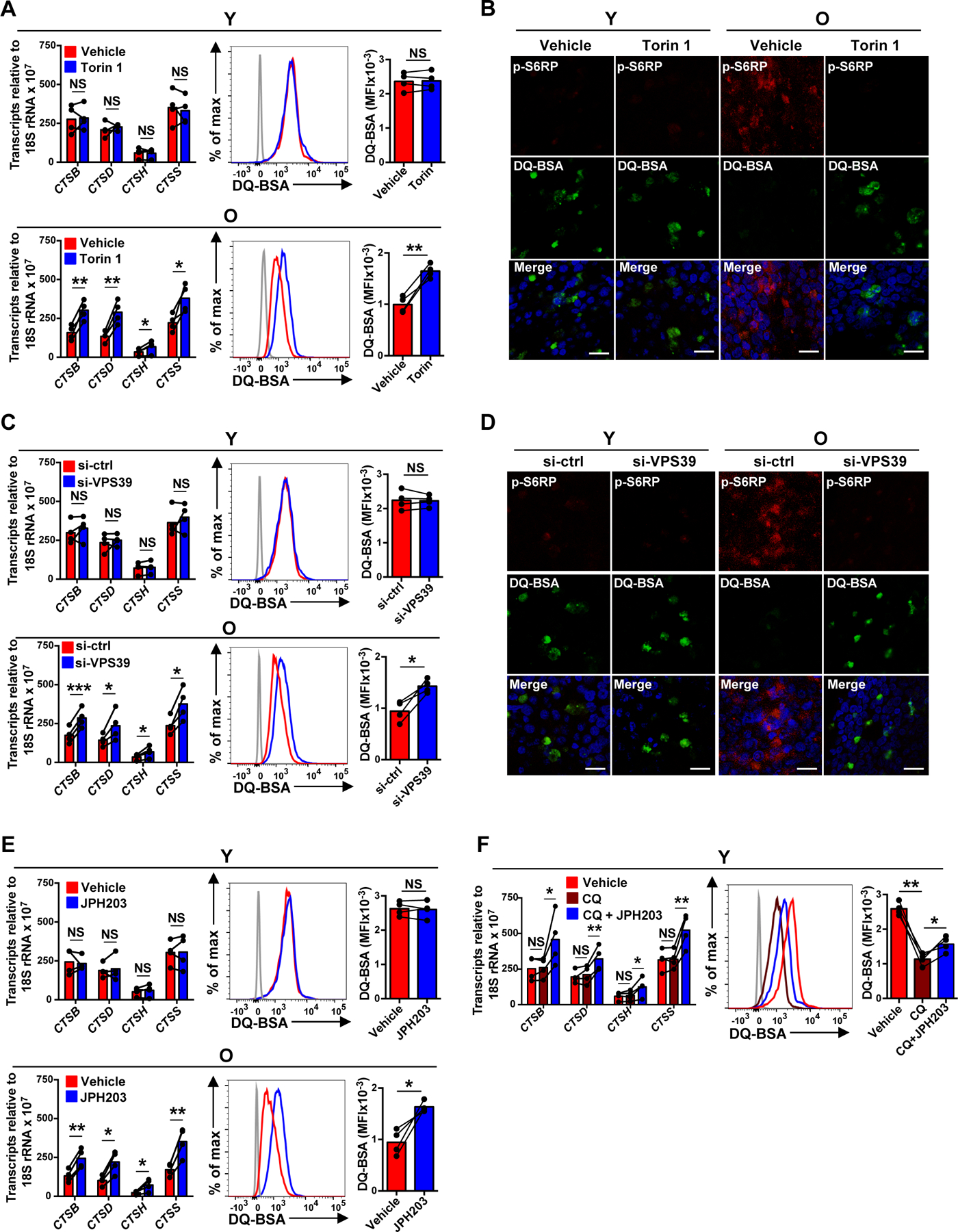
(A–F) Naïve CD4+ T cells from young (Y) and older (O) individuals were activated for 5 days. Indicated inhibitor or vehicle control was added for the last 2 days of culture (A–B and E–F). Alternatively, cells were transfected with control or VPS39 siRNA and then activated with anti-CD3/anti-CD28 beads for 5 days (C–D). (A, C, E and F) Lysosomal cathepsins expressions were determined by qRT-PCR (left); results are normalized to control samples using 18S rRNA as internal control; mean ± SD of four experiments. Lysosomal activities were determined by flow cytometry-based analysis of cells treated with 5 μg/mL of DQ-BSA for 6 hours. Results are shown as representative histograms (middle) and cumulative data from four experiments (right). The gray histogram represents DQ-BSA free samples. (B and D) Cells were treated with DQ-BSA (green) and stained with anti-pS6RP (red). Confocal images representative of two independent experiments show an inverse relationship between mTORC1 and lysosomal activity. Scale bar, 20 μm. Comparison by two-tailed paired t test (A, C, E and F). *p < 0.05, **p < 0.01; NS, not significant.
Sustained activation of late endosomal mTORC1 prevents PD-1 from lysosomal degradation.
We previously observed that in the course of T cell proliferation after activation, FOXO1 promoted the generation of lysosomes through induction of TFEB transcription (27). Moreover, Foxo1-deficient mouse CD4+ T cell showed up to fourfold increase of PD-1 protein levels after antigen priming in vivo (39), raising the possibility that lysosomal activity regulates PD-1 protein expression. Lysosome inhibition by TFEB silencing in T cells from young adults increased cell surface protein expression of PD-1 while transcript levels remained unchanged (Fig. 4A). In contrast, stimulation of lysosomal activities following late endosomal mTORC1 inhibition in T cells from older adults by either Torin 1 treatment, SLC7A5 inhibition or VPS39 silencing reduced cell surface protein expression without effects on transcripts of PD-1 (Fig. 4B–D). This effect was not due to the changes of intracellular versus cell surface trafficking routes of PD-1 since VPS39 silencing equally reduced cell surface and intracellular PD-1 protein expression (Fig. 4E–G). Assessment of PD-1 half-life in vitro in day 5-stimulated older naïve CD4+ T cells confirmed that VPS39-silenced cells underwent enhanced PD-1 protein degradation than control-silenced cells (Fig. 4H). These data suggest that mTORC1 at late endosomes stabilizes PD-1 protein by preventing lysosomal degradation of PD-1. In addition to PD-1, several molecules of functional relevance are targets of lysosomal degradation. CTLA-4, another inhibitory receptor, was also reduced after enhancing lysosomal activities by late endosomal mTORC1 inhibition (fig. S5C).
Fig. 4. Sustained activation of late endosomal mTORC1 prevents PD-1 from lysosomal degradation.
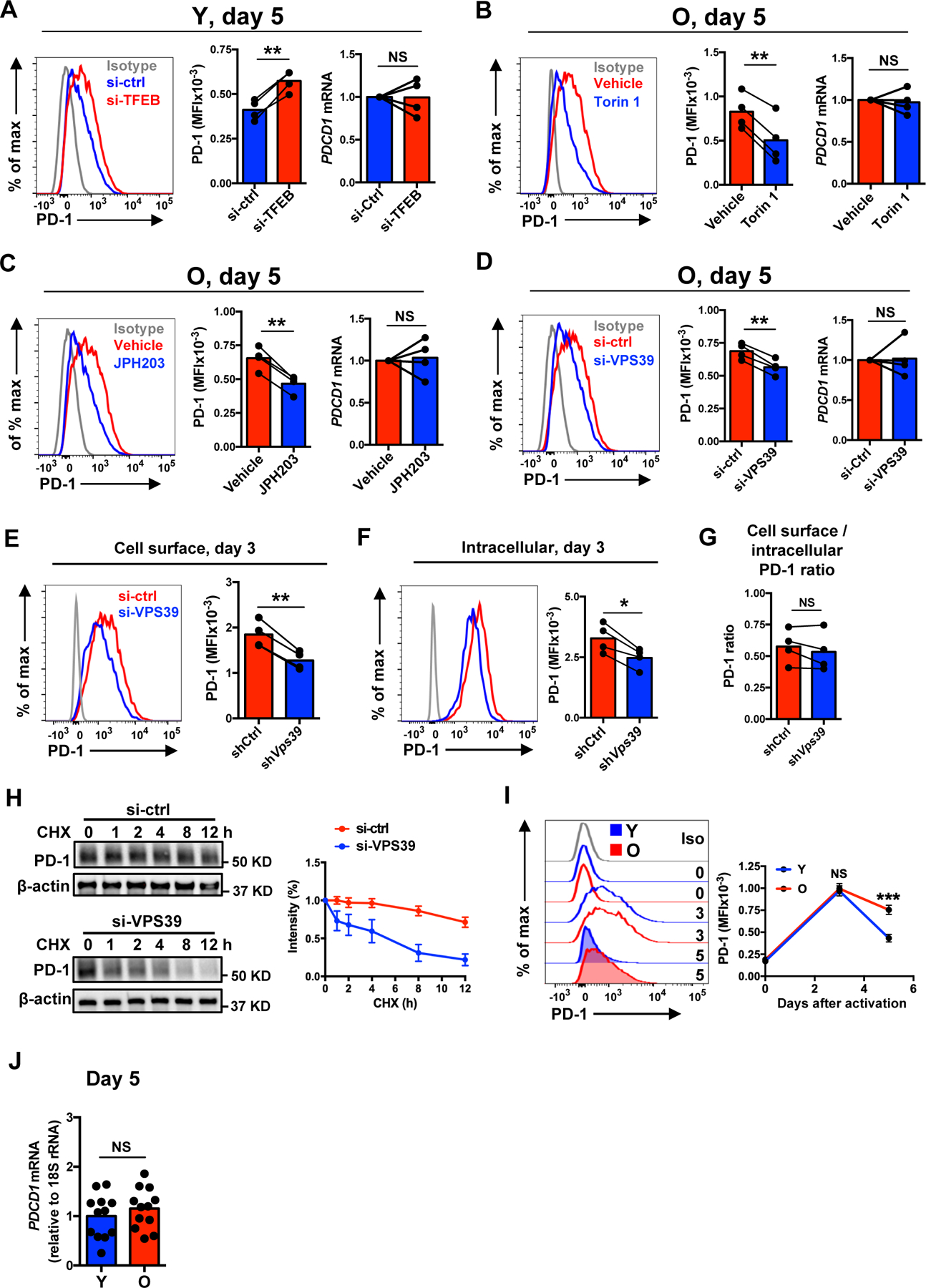
(A–D) Naïve CD4+ T cells from young and older individuals were activated with anti-CD3/anti-CD28 beads for 5 days with the last 2 days in the presence of vehicle or indicated inhibitor (B and C). Alternatively, cells were transfected with control or silencing RNA and activated for 5 days (A and D). Representative histograms showing cell surface protein expression of PD-1 (left) and cumulative data of cell surface protein expression (middle) and gene expression (right) of PD-1. (E–G) Cell surface expression (E), intracellular expression (F) and cell surface/intracellular PD-1 expression ratio (G) after control or VPS39 silencing in day 3-stimulated naïve CD4+ T cells from older individuals. (H) Control or VPS39-silenced, day 5-stimulated naïve CD4+ T cells from older individuals were treated with 5 μg/ml cycloheximide (CHX) to inhibit de novo PD-1 synthesis. Total PD-1 protein normalized to β-actin expression are shown as relative to non-treatment. Mean ± SEM of three experiments. (I) Longitudinal analysis of cell surface protein expression of PD-1 in naïve CD4+ T cells from ten young and ten older individuals. Mean ± SEM. (J) PD-1 gene expression comparison between day 5-stimulated young and older naïve CD4+ T cells. The gray histogram represents isotype control. Comparison by two-tailed paired (A–G) or two-tailed unpaired t test (I and J). *p < 0.05, **p < 0.01, ***p < 0.001; NS, not significant.
To determine whether PD-1 protein regulation changes with age, we monitored PD-1 expression at days 0, 3 and 5 after stimulation by flow cytometry. Consistent with the changes of the kinetics of mTORC1 activity with age (8), PD-1 protein expression peaked at day 3 after activation in both young and older activated naïve CD4+ T cells to then subside in activated T cells from young but not older adults. There were no age-related differences of protein expression of PD-1 at days 0 or 3, but activated T cells from older adults had more PD-1 protein at day 5 while no difference in PDCD1 transcription (Fig. 4I, J). No age-related difference was seen for CTLA-4 expression, which occurred early in a T cell response and before the observed age-related differences in mTORC1 activity (fig. S6).
Sustained activation of late endosomal mTORC1 impairs expansion of naïve CD4+ T cells from older adults.
Failure to timely degrade PD-1 protein raises the possibility that lysosome-deficient T cells receive an increased inhibitory signal suppressing cell expansion. To test this hypothesis, we stimulated naïve CD4+ T cells with anti-CD3/anti-CD28 beads in the presence or absence of PD-L1-Fc and anti-human IgG to cross-link PD-1 (40). VPS39 silencing induced increased proliferation and cell recovery in the presence of PD-1 stimulation (Fig. 5A). In contrast, VPS39 silencing only caused a minor increase in CD4+ T cell proliferation in the absence of PD-1 crosslinking that was not sufficient to increase cell numbers (Fig. 5A). These data support a model in which late endosomal mTORC1 activity regulates T cell expansion through lysosomal degradation of PD-1. Consistent with their sustained mTORC1 activation on late endosomes and reduced lysosomal activity and therefore degradation of PD-1, naïve CD4+ T cells from older adults had reduced potential to proliferate and to expand in the presence of PD-1 crosslinking (Fig. 5B). In the absence of exogenous PD-L1-Fc, age-related proliferation differences were not observed supporting the notion that the proliferative defect was related to increased PD-1 expression in T cells from older adults (Fig. 5B).
Fig. 5. Sustained activation of late endosomal mTORC1 impairs expansion of naïve CD4+ T cells from older adults.
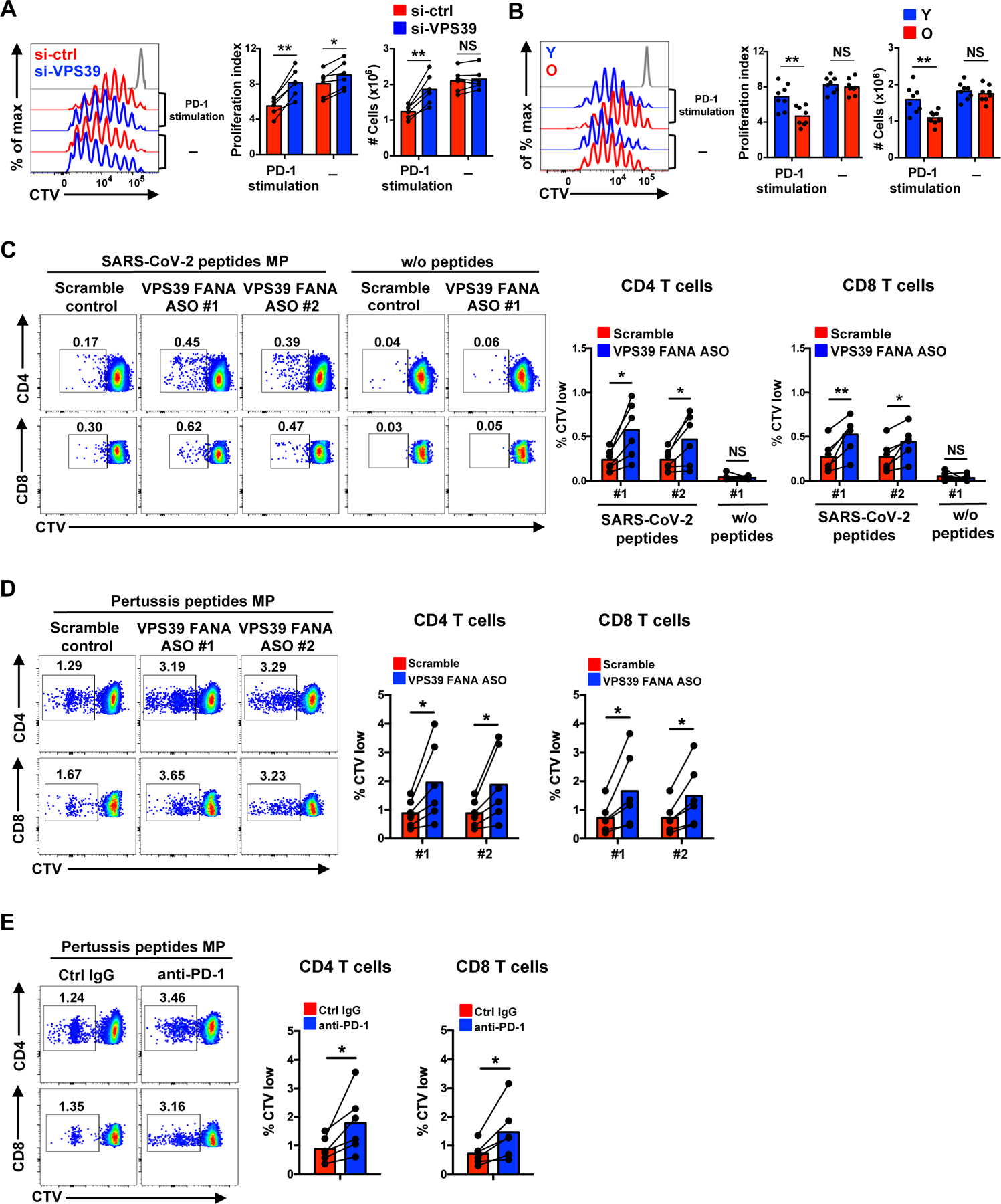
(A) Naïve CD4+ T cells from older individuals labeled with CellTrace Violet (CTV) were transfected with control or VPS39 siRNA and stimulated with anti-CD3/anti-CD28 beads for 5 days in the presence or absence of PD-L1-Fc and anti-human IgG Fc antibody to crosslink PD-1. Representative histograms (left), summary data of proliferation indices (middle) and cell numbers per culture (right). (B) CTV-labeled naïve CD4+ T cells from eight young and eight older individuals were activated by anti-CD3/anti-CD28 beads for 6 days with the last 3 day in the presence or absence of PD-1 crosslinking. Representative histograms (left), summary data of proliferation indices (middle) and cell numbers per culture (right). The gray histogram represents unstimulated cells. (C) CTV-labeled PBMCs from SARS-CoV-2 unexposed healthy individuals were cultured with SARS-CoV-2 peptide megapools for CD4+ and CD8+ T cells for 8 days. VPS39 silencing FANA ASO or scramble control were added to the culture on day 0. Representative flow plots of the frequencies of CTV low CD4+ and CD8+ T cells for indicated conditions and summary data from six individuals. (D and E) Pertussis peptide megapool responses of PBMC from six healthy individuals under the conditions of VPS39 genetic silencing (D) and PD-1 blockade (E). Comparison by two-tailed paired (A and C–E) or two-tailed unpaired t test (B). *p < 0.05, **p < 0.01, ***p < 0.001; NS, not significant.
A similar effect of endosomal differentiation interference was also seen for antigen-specific T cell responses. In these experiments, CTV-labeled PBMCs from healthy individuals were stimulated with a pool containing both HLA class II peptide megapool covering the entire SARS-CoV-2 orfeome together with HLA class I peptide megapools covering half of the entire orfeome. To silence VPS39 in human T cells, we added FANA antisense oligonucleotide (FANA ASO) that has the advantage of easy self-delivery without the need of transfection regents (41). Two VPS39 FANA ASO clones (#1 and #2) were added to replicate cultures at day 0. Frequencies of divided T cells were determined on day 8. Among eleven SARS-CoV-2-unexposed individuals tested, CD4+ and CD8+ T cell responses to SARS-CoV-2 peptide pool were observed in six individuals. VPS39 FANA ASO clone #1 and #2 significantly increased the SARS-CoV-2-reactive CD4+ and CD8+ T cell expansion compared to scrambled control FANA ASO in those individuals who showed a response (Fig. 5C). A similar increase of proliferating cells was not seen for PBMCs cultured in the absence of antigenic peptides (Fig. 5C). SARS-CoV-2 responses in unexposed individuals have been shown to derive from existing memory T cells (42). To examine whether VPS39 silencing improved human memory T cell responses more generally, we stimulated control- and VPS39-silenced T cells with pertussis peptide pools. VPS39–silencing augmented pertussis-specific responses (Fig. 5D) to a similar extent as anti-PD-1 blockade (Fig. 5E). These data indicate that VPS39-silencing improved memory T cell responses in general, potentially by interfering with the PD-1 checkpoint.
Inhibition of late endosomal mTORC1 promotes primary CD4+ T cell responses after LCMV infection in vivo.
To determine whether impaired expansion due to lysosome dysfunction is alsoseen with antigen-specific T cell responses in vivo, we used the LCMV infection mouse model. We retrovirally transduced SMARTA CD4+ T cells with shRNA specific for Tfeb (shTfeb) or control shRNA (shCtrl) and transferred the cells into 5- to 8-week-old B6 mice followed by acute LCMV infection. Tfeb silencing induced an increased PD-1 expression and reduced SMARTA CD4+ T cell expansion at peak responses (Fig. 6A–C).
Fig. 6. Inhibition of late endosomal mTORC1 promotes primary CD4+ T cell responses after LCMV infection in vivo.
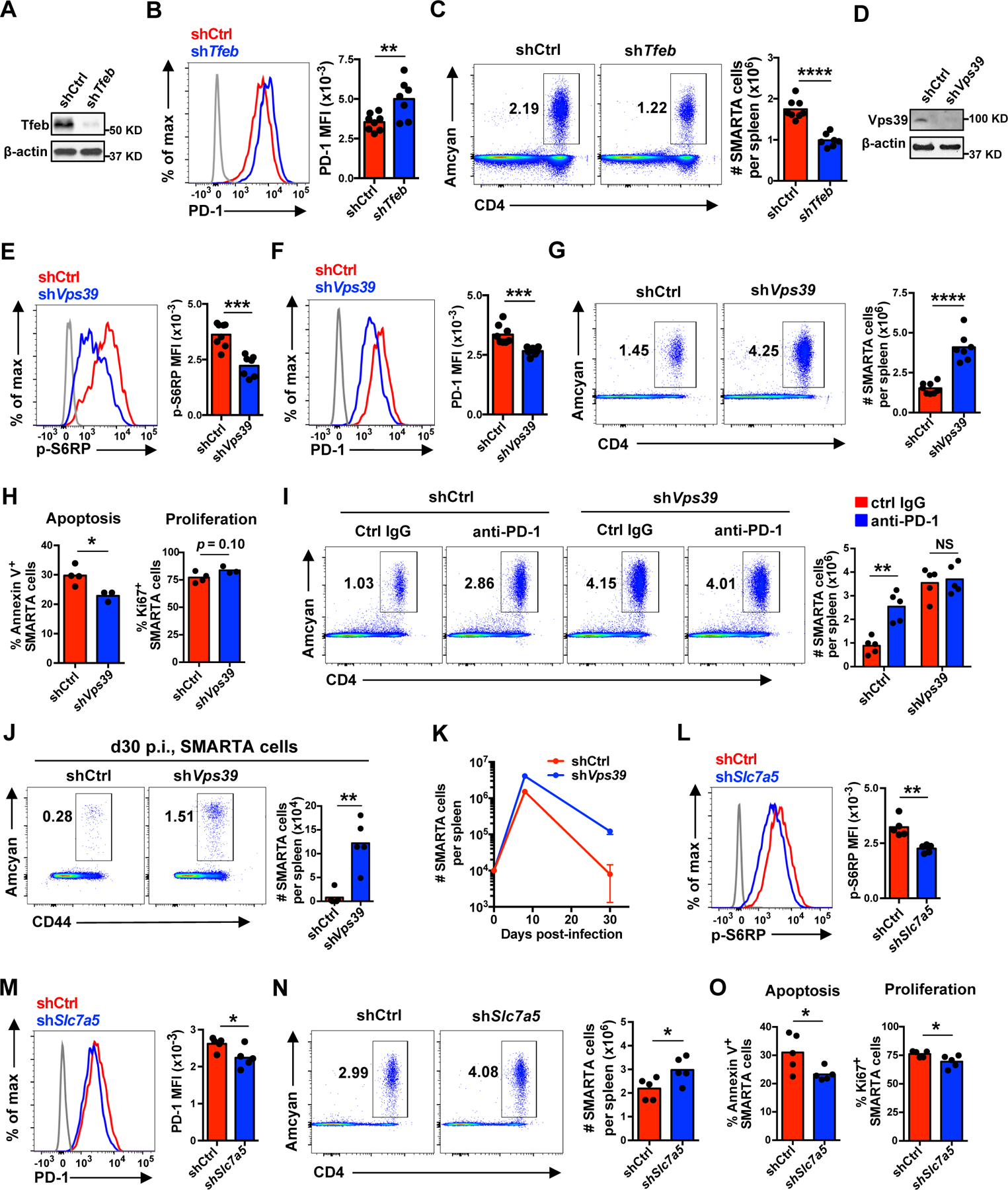
(A–C) 1 × 104 Tfeb shRNA or control shRNA retrovirally transduced Amcyan+ LCMV-specific naïve SMARTA TCR transgenic CD4+ T cells were adoptively transferred into CD45.2+ naïve recipients followed by infection with LCMV Armstrong. On day 8 post infection, spleens were harvested and analyzed. FACS plots are gated on CD4+ Amcyan+ SMARTA cells. (A) Tfeb protein expression in transduced cells before adoptive transfer. (B) PD-1 expression on day 8 p.i. (C) SMARTA CD4+ T cell numbers per spleen on day 8 p.i. (D–K) Analysis of T cells responses as described in (A–C) after adoptive transfer of Vps39 shRNA transduced SMARTA CD4+ T cells. (D) Vps39 protein expression in transduced cells before adoptive transfer. (E) Phosphorylation of S6RP on day 8 p.i. (F) PD-1 expression on day 8 p.i. (G) SMARTA CD4+ T cell numbers per spleen on day 8 p.i. (H) Cell apoptosis and proliferation of transferred SMARTA CD4+ T cells on day 8 p.i. (I) Mice infected with LCMV after adoptive transfer of transduced cells were additionally treated with anti-PD-1 antibody (29F.1A12) or control IgG on days 0, 3 and 6 post infection. SMARTA CD4+ T cell numbers were determined on day 8 after LCMV infection. (J) SMARTA CD4+ T cell numbers in the spleen at day 30 after infection. (K) Longitudinal analysis of SMARTA cells in the spleen of LCMV-infected B6 mice. (L–O) 1 × 104 Slc7a5 shRNA or control shRNA retrovirally transduced Amcyan+ LCMV-specific naïve SMARTA CD4+ T cells were adoptively transferred into CD45.2+ naïve recipients as in (A–C). Phosphorylation of S6RP (L), PD-1 expression (M), SMARTA cell numbers per spleen (N), and cell apoptosis and proliferation (O) of transduced SMARTA CD4+ T cells on day 8 post LCMV infection. Data are pooled from two independent experiments with 3–4 mice per group (B–C and E–G), representative of two independent experiments with 3–5 mice per group (A, D and H–K) or one experiment with 5 mice per group (L–O). Statistical significance by two-tailed unpaired t test (B–C, E–H and J–O) or one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test (I). The gray histograms represent naïve cells. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; NS, not significant.
To determine whether LCMV-specific CD4+ T cell responses can be boosted by inhibiting late endosomal mTORC1 in vivo, we transferred SMARTA CD4+ T cells transduced with shRNA specific for Vps39 (shVps39) or control shRNA (shCtrl) into B6 mice followed by acute LCMV infection. Vps39-silenced SMARTA CD4+ T cells had reduced mTORC1 activities, decreased PD-1 expression, enhanced lysosomal activities and up to a threefold increase in cell numbers in the spleen at peak responses on day 8 (Fig. 6D–G and fig. S5A). Vps39-silenced, expanded SMARTA CD4+ T cells were functional and produced similar levels of cytokines as control-silenced cells after peptide restimulation ex vivo (fig. S5B). A decrease of the Annexin V+ population indicated reduced apoptosis as a mechanism of the increased SMARTA CD4+ T cell recovery after Vps39 silencing, with an additional trend for increased cell proliferation as shown by Ki67+ population (Fig. 6H). To further examine whether the increased SMARTA CD4+ T cell expansion is at least in part due to reduced PD-1 expression, we blocked the PD-1/PD-L1 pathway by injecting mice with anti-PD-1 blocking antibody. PD-1 blockade promoted control-silenced SMARTA CD4+ T cell expansion at peak responses after LCMV infection, but did not further improve Vps39-silenced SMARTA CD4+ T cell expansion (Fig. 6I). At day 30 after infection, Vps39-silenced SMARTA CD4+ T cells showed over 15-fold increase in cell numbers with less contraction during the effector/memory transition than control-silenced SMARTA cells (Fig. 6J and K), consistent with the previously reported role of mTORC1 inhibition in promoting memory generation (17). Vps39 silencing by a second shRNA reproduced the data (fig. S5D and E), excluding the possibility of off-targets effects. Late endosomal mTORC1 inhibition by partial Slc7a5 silencing in SMARTA CD4+ T cells (fig. S5G) also produced similar effects to Vps39 silencing, although to a lesser extent. mTORC1 activities and PD-1 protein expression were both reduced after Slc7a5 silencing, while SMARTA cell number were slightly increased (Fig. 6L–N). Again, this increase of cell number was mostly due to reduced cell apoptosis, while proliferation was even slightly decreased (Fig. 6O). Taken together, these data show that inhibition of late endosomal mTORC1 promotes virus-specific CD4+ T cell expansion and memory generation after primary acute LCMV infection.
Inhibition of late endosomal mTORC1 augments CD4+ T cell helper responses in vivo.
The robust expansion at primary peak responses and the reduced contraction during memory transition of Vps39-silenced SMARTA CD4+ T cells prompted us to further examine whether antibody and memory CD8+ T cell responses in LCMV-infected host mice were affected. Relative frequencies of CXCR5hi PD-1hi germinal center (GC) within adoptively transferred SMARTA CD4+ T cells remained unchanged (Fig. 7A), suggesting that Vps39 silencing did not bias differentiation. However, the number of GC SMARTA TFH cells on day 8 was substantially increased after Vps39 silencing (Fig. 7A). Consequently, Fas+GL-7+ GC B cell numbers (Fig. 7B), CD138+IgD− plasma cell numbers (Fig. 7C) and LCMV-specific serum IgG titer (Fig. 7D) were increased in mice receiving Vps39-silenced SMARTA CD4+ T cells compared to mice receiving control-silenced cells. At day 30 after infection, the number of endogenous GP33 tetramer-positive CD8+ T memory cells increased by 2-fold in mice receiving Vps39-silenced SMARTA CD4+ cells compared to mice receiving control-silenced cells (Fig. 7E and fig. S5F). After rechallenge with Listeria monocytogenes expressing the GP33 epitope (Lm-GP33), memory CD8+ T cells underwent increased fold expansion and appeared to have superior memory cell quality as indicated by an increased capacity to produce IFNγ on a per-cell basis (Fig. 7F–H).
Fig. 7. Inhibition of late endosomal mTORC1 augments CD4+ T cell helper responses in vivo.
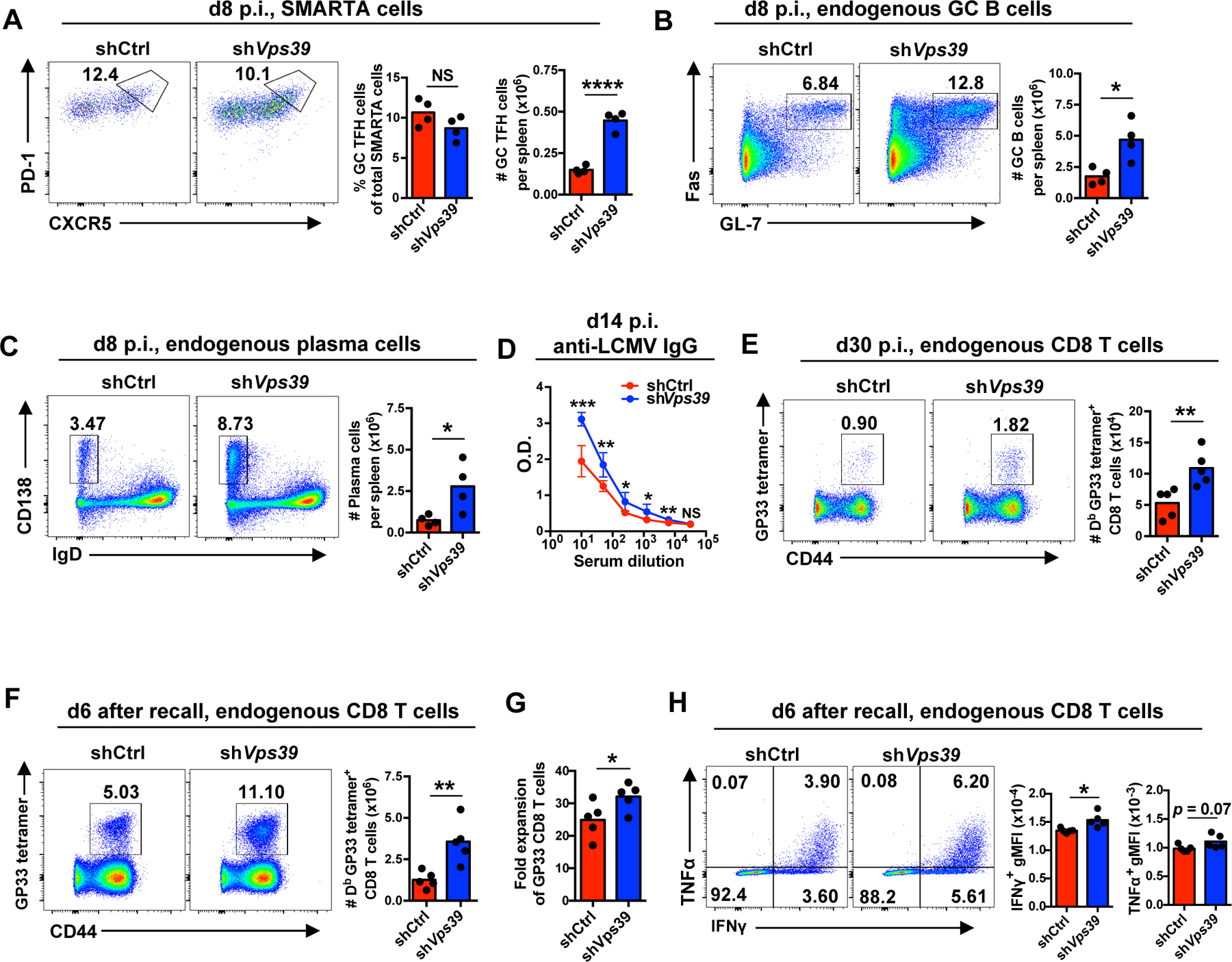
1 × 104 Vps39 shRNA or control shRNA retrovirally transduced Amcyan+ naïve SMARTA CD4+ T cells were adoptively transferred into CD45.2+ naïve recipient followed by LCMV infection. On days 8 (A–C) and 30 (E) post infection, spleens were harvested and analyzed. Alternatively, on day 30, immune mice were rechallenged with Lm-gp33 for 6 days (F–H). (A) Number of germinal center (GC) SMARTA TFH cells on day 8 after infection. (B and C) Number of endogenous Fas+ GL-7+ GC B cells (B) and CD138+ IgD− plasma cells (C) in host mice on day 8 after infection. Representative contour plots gated on B220+ CD4− B cells (left) and summary data (right). (D) Anti-LCMV nucleoprotein IgG titers in serum on day 14 after infection. n = 5 mice per group. (E) Numbers of Db LCMV GP33–41 (GP33) tetramer+ cells gated on endogenous CD8+ T cells in the spleen of host mice on day 30 after infection. (F) Numbers of endogenous Db LCMV GP33 tetramer+ CD8+ T cells in the spleen on day 6 after Lm-GP33 challenge. (G) Fold expansion of Db GP33 tetramer+ memory CD8+ T cells upon Lm-GP33 on day 6. (H) Cytokine production by CD8+ T cells harvested on day 6 after infection and restimulated with GP33 peptide in vitro. Data are representative of two independent experiments with 4–5 mice per group (A–E) or one experiment with 5 mice per group (F–H). Statistical significance by two-tailed unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; NS, not significant.
To examine whether these changes were due to reduced PD-1 expression in the adoptively transferred cells, we blocked the PD-1/PD-L1 pathway by injecting mice with anti-PD-1 blocking antibody. PD-1 blockade increased the numbers of transferred GC SMARTA TFH cells, endogenous GC B cells and plasma cells in mice receiving control-silenced cells while it did not further increase these numbers in mice receiving Vps39-silenced cells (fig. S7A–C), consistent with the lower expression of PD-1 on Vps39-silenced cells.
Discussion
Generation of immune memory, in the form of pathogen-neutralizing antibodies or pathogen-specific memory T cells, is at the core of successful vaccination. mTORC1 activation has been shown to be one of the major determinants in T cell fate decisions, favoring short-lived effector instead of TFH and memory T cells (17, 43). Previous studies have shown that mTORC1 activation is sustained for a longer time in a T cell response of older adults, contributing to the impaired vaccination efficacy (8). Here we show that mTORC1 signaling occurs at the late endosome and not at the lysosome in older T cells. This has important consequences for the regulation of mTORC1 activity as it changes a negative regulatory feedback to a forward loop that propagates lysosomal dysfunction. mTORC1 phosphorylates TFEB, thereby inhibiting transcription of lysosomal genes and impairing proteolytic activity that is required for further activation of mTORC1 at the lysosome. In contrast, reduced lysosomal activity in T cells from older adults induces the expansion of late endosomes and the expression of the leucine transporter SLC7A5, the latter by failing to degrade c-MYC. Together, increased late endosome mass and increased SLC7A5 activity support further mTORC1 activation leading to further lysosome dysfunction. Progressively dysfunctional lysosomes fail to degrade PD-1, resulting in its increased and sustained cell surface expression and inhibition of proliferation. This cycle can be broken by inhibiting or silencing SLC7A5 or VPS39 that restore TFH generation in T cells from older adults and augments the generation of germinal center and CD8+ memory response in the LCMV model of infection.
Lysosomal activation of mTORC1 is controlled during cell proliferation by the cross-regulation of lysosomal and mTORC1 activities (25). When lysosomal activity is deficient and amino acid efflux is low, mTORC1-dependent phosphorylation of TFEB is reduced, resulting in the translocation of TFEB into the nucleus, where it stimulates lysosomal gene transcription and restores lysosomal activities; adequate lysosomal activities trigger the mTORC1-dependent phosphorylation and cytoplasm retention of TFEB and the termination of its transcriptional induction of lysosomal genes (25, 44). This feedback loop appears to be dysregulated in lysosome-deficient aged T cells as they fail to down-regulate mTORC1 to restore TFEB-dependent lysosomal genes expression; instead they show enhanced late endosomal mTORC1 activity with even more reduced lysosomal genes expression compared to the young cells.
Late endosomes are acidic organelles that, in contrast to lysosomes, do not have proteolytic activity and amino acid recycling ability (31). They function as temporary storage tanks for proteins that are sorted into the lysosomal degradation pathway after fusion with lysosomes or secreted as exosomes. Late endosome turnover is regulated downstream by lysosomal degradation and upstream by VPS39-mediated biogenesis from early endosomes (31, 32). Inhibition of lysosomal activities induces an intracellular expansion of late endosomes (34). We have recently described that activated T cells from older adults are deficient in rebuilding functional lysosomes as consequence of FOXO1 degradation and therefore expand this late endosomal compartment, which provides an alternative platform for mTORC1 activation. Although late endosomes do not degrade protein to recycle amino acids for mTORC1 signaling, the increased plasma membrane leucine transporter SLC7A5 induced by lysosome inhibition facilitated the uptake of extracellular amino acids as an alternative amino acid source for late endosomal mTORC1 activation. In addition to late endosomes, mTORC1 can be activated on the surface of Golgi via recruitment to the Golgi membrane, involving RAB1A and Golgi-resident RHEB (45). Recruitment of mTORC1 in mitochondria membrane by co-targeting Raptor and RHEB to mitochondria can also allow its activation (46). These sites of lysosome-independent mTORC1 signaling may contribute to a sustained high level of mTORC1 activities, as we saw in T cells from older adults in spite of impaired lysosomes, but they likely lack the positive feedback loop occurring downstream of late endosomal mTORC1.
One additional component in this network is activated AKT that is more sustained in naïve T cell responses of older adults due to the increased expression of miR-21 and the repression of its target phosphatase and tensin homolog (PTEN) (8). In addition to phosphorylating tuberous sclerosis complex 2 (TSC2) and PRAS40, both negative regulators of mTOR activity, AKT also phosphorylates and inactivates FOXO1 that is needed for generation of new lysosomes. We recently described that FOXO1 promotes lysosomal activities through induction of TFEB transcription in CD4+ T cells (27). This function was impaired in T cell responses of older adults due to increased degradation of FOXO1 after T cell stimulation. Sustained mTORC1 activation at the late endosomes further inhibits lysosome activity that plays an important role in promoting effector T cell survival, mainly through the autophagy/lysosome pathway (47, 48).
We show that one pathway by which lysosome dysfunction in older adults affects T cell responses is a failure to curtail activation-induced PD-1 expression. FOXO1-deficient mouse CD4+ T cells, in part recapitulating FOXO1-deficiency in activated T cells from older adults, had an up to four-fold increase of PD-1 protein levels and a major defect in T cell expansion after antigen priming in vivo (39). Moreover, PD-1 is degraded by lysosomes (49). Indeed, proliferation of T cells from older adults, which was intact after polyclonal stimulation, was impaired by increased cross-linking of PD-1 with PD-L1 compared to that of T cells from young adults.
Silencing of VPS39 to inhibit late endosome generation and activation of mTORC1 promoted T cell expansion to anti-CD3 bead stimulation in the presence of PD-L1 stimulating PD-1. We observed a similarly enhanced expansion of VPS39-deficient T cells in response to stimulation with SARS-CoV-2 and pertussis peptides in vitro that resembled the response achieved by PD-1 blockade. In the acute LCMV infection model, adoptive transfer of VPS39-deficient LCMV-specific CD4+ T cells resulted in increased clonal expansion, germinal center formation and improved CD8+ T-cell recall responses. These results with VPS39-silenced T cells resembled published studies, where the PD-1/PD-L1 pathway blockade enhanced both effector function and frequency of memory precursor of antigen-specific CD8+ T cells and reduced viral load in acute LCMV infection (50). In an immunization model, PD-1 blockade promoted the generation of follicular helper T cells that are specialized in promoting cognate B cell responses (40, 51). Thus, targeting VPS39 could serve as a strategy to boost immue responses to different types of virus infections and to establish the improved immune memory in older individuals.
Inhibition of late endosomal mTORC1 may serve as a strategy in compensating for age-related defects in T cell differentiation and the generation of TFH and memory cells (7, 8, 52). In addition to VPS39, there are five other protein components of HOPS complex (VPS11, VPS16, VPS18, VPS33 and VPS41) that also play a role in mediating early to late endosome conversion and could be targeted (53). In addition, partial inhibition of SLC7A5 to compensate for the physiological differences in expression in CD4+ T cells from young and older adults could also be explored. However, complete inhibition will likely be detrimental because knockout of Slc7a5 profoundly impaired T cell activation and expansion (54). Based on the early studies in the mouse that mTORC1 inhibition by low dose of rapamycin produced an enhanced primary and memory recall CD8+ T cell response (17) as well as promoted follicular helper cell over Th1 cell generation (43), clinical studies have been started. Treatment with low dose combination of a catalytic (BEZ235) plus an allosteric (RAD001) mTOR inhibitor enhanced the immune response to the influenza vaccine and reduced the percentage of CD4+ and CD8+ T lymphocytes expressing PD-1 in older individuals (55). However, a subsequent study by resTORbio was not successful, missing the primary endpoint (www.ClinicalTrials.gov; Identifier: NCT04139915). It should be noted that the design of the study was to show an overall improvement in immune health. The vaccine response was not specifically targeted, in fact, there was a wash out period before the vaccination. Targeting mTORC1 activation on late endosomes in the days subsequent to the initiation of the T cell activation rather than mTORC1 globally may be more advantageous to stimulate T cell responses and enhance vaccine responses functions in general and in particular in older individuals.
Materials and Methods
Study design
The aim of this study was to examine the mechanism by which activated naïve CD4+ T cells from older adults exhibit increased and sustained mTORC1 activation in spite of lysosomal dysfunction, and to identify targets for intervention other than direct mTORC1 inhibition to improve T follicular helper cell responses and memory T cell generation. We used purified naïve CD4+ T cells collected from young (20–35 years old) and older (65–85 years old) healthy individuals to perform in vitro signaling and functional studies after polyclonal activation. Pharmacological inhibition as well as gene expression silencing were applied to interrogate lysosomal and endosomal pathways in the context of age. In vitro human data were validated in vivo in a mouse model of LCMV infection by adoptively transferring antigen-specific CD4+ T cells that had been genetically manipulated. Mice were randomly assigned to control versus sample groups. Data analysis was conducted in an unblinded manner. Sampling and experimental replicates were specified in figure legends. No outliers were removed.
Study population and cell isolation
PBMC were collected from 53 young (20–35 years old) and 64 older (65–85 years old) healthy individuals of both gender with no history of autoimmune disease or cancer and no uncontrolled renal disease, diabetes mellitus, or cardiovascular disease. 98 of them were de-identified samples purchased from the Stanford Blood Center (Palo Alto, CA, USA) from donors younger than 35 years (43 donors) or older than 65 years (55 donors). 19 samples were from individuals recruited from the local area. The study was in accordance with the Declaration of Helsinki, approved by the Stanford Institutional Review Board, and all participants gave informed written consent. Naïve CD4+ T cells were purified with human CD4+ T cell enrichment cocktail (15062, STEMCELL Technologies), followed by negative selection with anti-CD45RO magnetic beads (19555, STEMCELL Technologies).
Cell culture
Isolated human naïve CD4+ T cells were activated with Dynabeads Human T-Activator CD3/CD28 (11132D, Thermo Fisher Scientific) in RPMI 1640 (Sigma) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and streptomycin (Thermo Fisher Scientific). Mouse T cells were activated in plates coated with 8 µg/ml anti-CD3 Ab (16–0032-82, eBioscience) and 8 µg/ml anti-CD28 Ab (16–0281-82, eBioscience) in culture medium supplemented with 10 ng/ml IL-2 (21212, Peprotech).
Western blotting
Cells were lysed in RIPA buffer containing PMSF and protease and phosphatase inhibitors (sc-24948, Santa Cruz Biotechnology) for 30 minutes on ice. Proteins were separated on denaturing 4%–15% SDS-PAGE (4561086, Bio-Rad), transferred onto nitrocellulose membrane (1704270, Bio-Rad) and probed with antibodies to SLC7A5 (5347S), c-MYC (E5Q6W, 18583S), β-actin (13E5, 4970S), S6K1 (49D7, 2708S), pS6K1 Kinase (Thr389) (108D2, 9234S), EEA1 (C45B10, 3288S), RAB7 (D95F2, 9367S), Cathepsin D (2284S), Na/K ATPase (3010S), Tubulin (11H10, 2125S), mTOR (7C10, 2983S), Raptor (24C12, 2280S), RHEB (E1G1R, 13879S), PD-1 (D4W2J, 86163S, all Cell Signaling Technology), CD63 (ab68418), Cathepsin H (ab185935) and VPS39 (ab224671, all Abcam). Membranes were developed using HRP-conjugated secondary antibodies (Cell Signaling Technology) and Chemiluminescent Western Blot Detection Substrate (Thermo Fisher Scientific).
Flow cytometry
For cell surface staining, cells were incubated with fluorescently conjugated antibodies in PBS containing 2% FBS at 4 °C for 30 minutes. For intracellular cytokine assays, cells were stimulated with 10 μM LCMV GP66–77 DIYKGVYQFKSV or 0.2 μM LCMV GP33–41 KAVYNFATC (Anaspec) in the presence of Brefeldin A (GolgiPlug, BD Biosciences) for 4 hours at 37 °C. Cells were then incubated with antibodies to cell surface molecules, permeabilized with Cytofix/Cytoperm kit (BD Biosciences) and stained with fluorescently labeled antibodies specific for the indicated cytokines. For pS6RP (S235/S236) staining, cells were treated with FOXP3 Fix/Perm Buffer Set (421403, Biolegend) before incubation with fluorescently conjugated antibodies at room temperature for 60 minutes. Dead cells were excluded from the analysis using LIVE/DEAD Fixable Aqua (eBioscience). Staining for flow cytometry was done with monoclonal antibodies against: CD4 (anti-human: RPA-T4; anti-mouse: RM4–5), CD8 (anti-human: RPA-T8; anti-mouse: 53–6.7), CD44 (IM7), B220 (RA3–6B2), Fas (Jo2), GL-7 (GL7), CD138 (281–2), IgD (11–26), CD62L (MEL-14), CD127 (SB/199), KLRG-1 (2F1), pS6RP (S235/S236, N7–548, all BD Bioscience), PD-1 (anti-human: A17188B; anti-mouse: 29F.1A12), CTLA-4 (UC10–4B9), IFN-γ (XMG1.2), TNF-α (MP6-XT22) and IL-2 (JES6–5H4, all Biolegend). Mouse CXCR5 was stained with biotin-conjugated anti-CXCR5 (2G8, BD Bioscience) followed by APC-streptavidin binding (BD Bioscience). Db GP33–41 KAVYNFATC (GP33) tetramer was obtained from the NIH tetramer core facility (Atlanta, GA). To stain LCMV-specific CD8+ T cells, cells were incubated with Db GP33 tetramers along with cell surface antibodies at 4 °C for 30 minutes in antibody staining buffer. Cells were analyzed on an LSRII or LSR Fortessa (BD Biosciences). Flow cytometry data were analyzed using FlowJo (TreeStar).
RNA isolation and quantitative RT-PCR
Total RNA was isolated using the RNeasy Plus Mini Kit (74134, QIAGEN) and converted to cDNA using the SuperScript VILO cDNA Synthesis Kit (11754, Invitrogen). Quantitative RT-PCR was performed on the ABI 7900HT system (Applied Biosystems) using Power SYBR Green PCR Master Mix (4368706, Thermo Fisher Scientific), according to the manufacturer’s instructions. Oligonucleotide primer sets are shown in Table S1.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) software from the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp) was used to determine the enrichment of gene sets in T cells from young (20–35 years) or older (65–85 years) adults. The datasets describing age-associated differences in activated CD4+ T cells were obtained from SRA database under accession number SRP158502 (36).
Transfection
Naïve CD4+ T cells were transfected with either SMARTpool negative control siRNA, SMARTpool TFEB siRNA, SMARTpool MYC siRNA or SMARTpool VPS39 siRNA (all from Dharmacon) using the Amaxa Nucleofector system and P3 primary cell Nucleofector Kit (Lonza). Cells were rested for 2 hours, washed and activated by dynabeads for 5 days. Cells were then harvested and analyzed.
Pharmacologic inhibition
The SLC7A5 inhibitor JPH203 (S8667, Selleckchem) was used at a dose of 5 μM for all experiments. Lysosome inhibitors Chloroquine (PHR1258–1G, Sigma-Aldrich) and Bafilomycin A1 (11038, Cayman CHEMICAL) were used at doses of 20 μM and 10 nM, respectively. The mTORC1 inhibitor Torin 1 (4247, Tocris) was used at a dose of 100 nM. The protein synthesis inhibitor cycloheximide (C4859, Sigma-Aldrich) was used at a dose of 5 μg/ml.
Endosome isolation and in vitro mTORC1 kinase activity assay
Endosomes were isolated from naïve CD4+ T cells on day 3 after activation when mTORC1 activity peaked, by using an endosome isolation kit (ED-028, Invent Biotechnologies) according to manufacturer’s instructions. In some experiments, cells were pre-treated for 2 hours with 1 μM AKT inhibitor (MK-2206 2HCl, Selleckchem) prior to harvesting. Briefly, 2 × 107 cells were suspended in lysis buffer A (supplemented with protease inhibitor cocktail) and cell extracts were filtered to remove intact cells, larger organelles and plasma membranes. The flow-through cell lysates were mixed with the supplied precipitation buffer B followed by centrifugation. After centrifugation, endosomes were enriched in pellets. Supernatants were discarded, and the endosome pellets were resuspended in RIPA lysis buffer (sc-24948, Santa Cruz Biotechnology). Protein concentrations were measured using BCA Protein Assay Kit (23227, Pierce) and equal amounts of protein were loaded for immunoblotting analysis of indicated markers. Alternatively, the endosome pellets were resuspended in mTORC1 kinase buffer (25 mM HEPES, 50 mM KCl, 10 mM MgCl2, 20% glycerol, 4 mM MnCl2 and 250 μM ATP) prepared according to a previous study (56) and divided into aliquots of 10 μl each containing 10 μg endosomes. 100 ng of S6K1 Human Recombinant Protein (TP317324, Origene) diluted in 5 μl of mTORC1 kinase buffer was added as the substrate, and the Kinase assays were performed at 30°C for 20 min in a final volume of 15 μl. Reactions were stopped by adding 5 μl of 4 x sample buffer and loaded for immunoblotting analysis.
Confocal microscopy
Cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with primary antibodies to LAMP1 (D2D11, #9091), mTOR (7C10, #2983), pS6RP (S235/236) (D57.2.2E, #4858) and EEA1 (E9Q6G, #48453, all Cell Signaling Technology) at 4°C overnight. Incubation with secondary antibodies was performed at room temperature for 2 hours using Alexa Fluor 488–conjugated AffiniPure Donkey anti-Rabbit immunoglobulin G (IgG) or Cy3-conjugated AffiniPure Donkey anti-Mouse IgG (Jackson Immuno Research Laboratories). The images were analyzed using an LSM 710 microscope system with ZEN 2010 software (Carl Zeiss) and a 63× oil immersion objective (Carl Zeiss).
DQ-BSA lysosomal activity assay
Cells were treated with 5 μg/mL of DQ-BSA (D12050, Thermo Fisher Scientific) diluted in prewarmed medium and incubated at 37 °C for 6 h. Cells were then briefly washed once with ice-cold PBS containing 2% FBS and kept on ice. Fluorescence of cleaved DQ-BSA was analyzed by flow cytometry.
Cell proliferation assay in vitro
Freshly purified naïve CD4+ T cells were labeled with CellTrace Violet (CTV; Thermo Fisher Scientific) and stimulated with anti-CD3/anti-CD28 beads in the presence of recombinant human PD-L1/B7-H1 Fc chimera (156-B7, R&D systems; 5 ug/ml) crosslinked by goat anti-human IgG Fc polyclonal antibody (G-102-C, R&D systems; 5 ug/ml). At day 5–6, cells were harvested and analyzed by flow cytometry.
SARS-CoV-2 and pertussis-reactive CD4+ and CD8+ T cell responses
PBMCs were isolated by density gradient centrifugation using Lymphoprep (STEMCELL Technologies) from pheresis samples of SARS-CoV-2 unexposed healthy donors. The HLA class II peptide megapool covering the entire SARS-CoV-2 orfeome and the HLA class I peptide megapool covering half of the entire orfeome were described previously (57, 58). PBMCs were labeled with CellTrace Violet (CTV; Thermo Fisher Scientific) and cultured with either the mixture of SARS-CoV-2 class II megapool (1 ug/ml) and class I megapool (1 ug/ml) or pertussis megapool (1 ug/ml) in RPMI 1640 media (Sigma) supplemented with 5% human AB serum (Sigma), 1 ug/ml anti-CD28 (ebioscience) and 100 U/ml penicillin and streptomycin (Thermo Fisher Scientific). VPS39-silencing FANA ASO (FANA antisense oligonucleotide, 1 uM, AUM Biotech), scrambled control FANA ASO (1 uM, AUM Biotech), anti-PD-1 blocking antibody (A17188B, 1 ug/ml) or control IgG (1 ug/ml) were added to the culture on day 0. Peptide-reactive CD4+ and CD8+ T cell responses were measured on day 8.
Mice, adoptive transfers and LCMV infection
Naïve CD4+ T cells specific to the GP66–77 epitope of LCMV obtained from 5- to 8-week-old SMARTA TCR transgenic mice (CD45.1, a kind gift from Rafi Ahmed at Emory University) were activated in plates coated with anti-CD3 Ab and anti-CD28 Ab. Cells were transduced with a retroviral vector expressing either scrambled control RNA, Tfeb shRNA (5’-CGGCAGTACTATGACTATGAT-3’), Vps39 shRNA (5’-AGTGAGCATGTGCTGAAGAAG-3’) or Slc7a5 shRNA (5’- CGCAATATCACGCTGCTCAAC-3’) on days 1 and 2 after activation. On day 6 after activation, retrovirus-transduced Amcyan-positive SMARTA cells were isolated and 1 × 104 transduced cells were intravenously (i.v.) transferred to 5- to 8-week-old female C57BL/6 (CD45.2) mice (Jackson Laboratory). On day 3 post-transfer, mice were infected intraperitoneally (i.p.) with 2 × 105 PFU of LCMV Armstrong. On day 8 post-infection, spleens were harvested and analyzed. For PD-1 Blockade: anti–PD-1 (29F.1A12, #BE0273, Bio X Cell; 200 μg, i.p.) or control IgG (2A3, #BE0089, Bio X Cell; 200 μg, i.p.) was administered to LCMV-infected B6 mice on days 0, 3 and 6 post infection. On day 8 post-infection, spleens were harvested and analyzed. For recall responses of CD8+ memory T cells, recombinant Listeria monocytogenes expressing the LCMV glycoprotein 33–41 epitope (Lm-gp33) were grown to log phase in BHI broth. Concentrations were determined by measuring the O.D. at 600 nm (O.D. of 1 = 1 × 109 CFU/ml). LCMV-immune mice were injected i.v. with 2 × 105 colony forming units (CFU) for recall responses. All animal experiments were approved by the Stanford Administrative Panel on Laboratory Animal Care Committee.
ELISA
Serum was collected from mice on day 14 after LCMV (Armstrong) infection. LCMV-specific IgG was measured using an anti-LCMV-NP IgG ELISA kit (AE-300200–1, Alpha Diagnostic International) according to the manual. Briefly, ELISA plates coated with LCMV-VP1 antigen were pre-washed and incubated with 5-fold serial diluted serum for 60 min. Plates were then washed and incubated with anti-mouse IgG HRP for 30 min. After washing, bound antibody was detected by adding TMB substrate and the reaction was stopped with stop solution. Plates were read for absorbance at 450 nm.
Statistical analysis
Statistical analysis was performed using the Prism 7.0 software (GraphPad Software Inc.). Paired or unpaired two-tailed Student’s t-tests were used for comparing two groups. One-way ANOVA with Tukey’s post hoc test was used for multi-group comparisons. p < 0.05 was considered statistically significant. Statistical details and significance levels can be found in the figure legends.
Supplementary Material
Data file S1. Raw data file (Excel spreadsheet).
Supplementary Materials
Supplementary Materials and Methods
Fig. S1. Activated naïve CD4+ T cells from older adults lose lysosomal function while maintaining mTORC1 activity.
Fig. S2. SLC7A5-dependent mTORC1 activation in naïve CD4+ T cell responses from older adults.
Fig. S3. VPS39 silencing does not affect TGF-β/SMAD signaling in naïve CD4+ T cell responses.
Fig. S4. Effects of mTORC1 inhibition on TFEB expression and phosphorylation.
Fig. S5. Inhibition of late endosomal mTORC1 promotes virus-specific CD4+ T cell expansion without affecting their cytokine production.
Fig. S6. CTLA-4 cell surface expression does not change with age.
Fig. S7. Improved CD4+ T cell helper responses after Vps39 silencing depends on reduced PD-1 expression.
Fig. S8. Key gating strategies used in this study.
Table S1. Oligonucleotide primer sets used in this study.
Acknowledgements:
We thank the Palo Alto Veterans Administration Flow Cytometry Core (B. Carter) for assistance with flow cytometry and cell sorting; Dr. R. Ahmed (Emory University) for providing SMARTA mice and LCMV-Armstrong; the NIH tetramer core facility (Atlanta, GA) for providing tetramers; Dr. Xin Yang (Stanford University) for assistance with I.V. injection.
Funding:
This work was supported by the National Institutes of Health R01 AR042527, R01 HL117913, R01 AI108906, R01 HL142068, and P01 HL129941 to C.M.W., R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191 to J.J.G., National Institutes of Health contract Nr. 75N9301900065 to A.S. and D.W. and with resources and the use of facilities at the Palo Alto Veterans Administration Healthcare System. Tetramers were provided by the NIH Tetramer Core Facility supported by contract HHSN272201300006C from NIAID. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Peptide pools utilized in this paper are available from A.S. (alex@lji.org) under a material transfer agreement (MTA) from La Jolla Institute for Immunology.
References:
- 1.Nikolich-Zugich J, The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol 19, 10–19 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Haynes L, Swain SL, Why aging T cells fail: implications for vaccination. Immunity 24, 663–666 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K, Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, Cereda D, Coluccello A, Foti G, Fumagalli R, Iotti G, Latronico N, Lorini L, Merler S, Natalini G, Piatti A, Ranieri MV, Scandroglio AM, Storti E, Cecconi M, Pesenti A, Network C-LI, Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA, (2020). [DOI] [PMC free article] [PubMed]
- 5.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B, Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang JJ, Dong X, Cao YY, Yuan YD, Yang YB, Yan YQ, Akdis CA, Gao YD, Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 75, 1730–1741 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Fang F, Yu M, Cavanagh MM, Hutter Saunders J, Qi Q, Ye Z, Le Saux S, Sultan W, Turgano E, Dekker CL, Tian L, Weyand CM, Goronzy JJ, Expression of CD39 on Activated T Cells Impairs their Survival in Older Individuals. Cell Rep 14, 1218–1231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim C, Hu B, Jadhav RR, Jin J, Zhang H, Cavanagh MM, Akondy RS, Ahmed R, Weyand CM, Goronzy JJ, Activation of miR-21-Regulated Pathways in Immune Aging Selects against Signatures Characteristic of Memory T Cells. Cell Rep 25, 2148–2162 e2145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laidlaw BJ, Craft JE, Kaech SM, The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol 16, 102–111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crotty S, T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 50, 1132–1148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matloubian M, Concepcion RJ, Ahmed R, CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68, 8056–8063 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP, CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421, 852–856 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Shedlock DJ, Shen H, Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300, 337–339 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Saxton RA, Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Zeng H, Chi H, mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol 46, 103–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, Guan KL, mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol 21, 63–71 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R, mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, Delgoffe GM, Powell JD, mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest 125, 2090–2108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang K, Neale G, Green DR, He W, Chi H, The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol 12, 888–897 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishna S, Yang J, Wang H, Qiu Y, Zhong XP, Role of tumor suppressor TSC1 in regulating antigen-specific primary and memory CD8 T cell responses to bacterial infection. Infect Immun 82, 3045–3057 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shrestha S, Yang K, Wei J, Karmaus PW, Neale G, Chi H, Tsc1 promotes the differentiation of memory CD8+ T cells via orchestrating the transcriptional and metabolic programs. Proc Natl Acad Sci U S A 111, 14858–14863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrer IR, Wagener ME, Robertson JM, Turner AP, Araki K, Ahmed R, Kirk AD, Larsen CP, Ford ML, Cutting edge: Rapamycin augments pathogen-specific but not graft-reactive CD8+ T cell responses. J Immunol 185, 2004–2008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E, Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest 124, 188–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Rao R, Vazzana J, Goedegebuure P, Odunsi K, Gillanders W, Shrikant PA, Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. J Immunol 188, 3080–3087 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM, The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5, ra42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Napolitano G, Di Malta C, Esposito A, de Araujo MEG, Pece S, Bertalot G, Matarese M, Benedetti V, Zampelli A, Stasyk T, Siciliano D, Venuta A, Cesana M, Vilardo C, Nusco E, Monfregola J, Calcagni A, Di Fiore PP, Huber LA, Ballabio A, A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature, (2020). [DOI] [PMC free article] [PubMed]
- 27.Jin J, Li X, Hu B, Kim C, Cao W, Zhang H, Weyand CM, Goronzy JJ, FOXO1 deficiency impairs proteostasis in aged T cells. Sci Adv 6, eaba1808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marat AL, Wallroth A, Lo WT, Muller R, Norata GD, Falasca M, Schultz C, Haucke V, mTORC1 activity repression by late endosomal phosphatidylinositol 3,4-bisphosphate. Science 356, 968–972 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Flinn RJ, Yan Y, Goswami S, Parker PJ, Backer JM, The late endosome is essential for mTORC1 signaling. Mol Biol Cell 21, 833–841 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flinn RJ, Backer JM, mTORC1 signals from late endosomes: taking a TOR of the endocytic system. Cell Cycle 9, 1869–1870 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piper RC, Katzmann DJ, Biogenesis and function of multivesicular bodies. Annu Rev Cell Dev Biol 23, 519–547 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rink J, Ghigo E, Kalaidzidis Y, Zerial M, Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Binda M, Peli-Gulli MP, Bonfils G, Panchaud N, Urban J, Sturgill TW, Loewith R, De Virgilio C, The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell 35, 563–573 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Dobrowolski R, Vick P, Ploper D, Gumper I, Snitkin H, Sabatini DD, De Robertis EM, Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep 2, 1316–1328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu GY, Sabatini DM, mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21, 183–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu B, Li G, Ye Z, Gustafson CE, Tian L, Weyand CM, Goronzy JJ, Transcription factor networks in aged naive CD4 T cells bias lineage differentiation. Aging Cell 18, e12957 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchingo JM, Sinclair LV, Howden AJ, Cantrell DA, Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. Elife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Felici A, Wurthner JU, Parks WT, Giam LR, Reiss M, Karpova TS, McNally JG, Roberts AB, TLP, a novel modulator of TGF-beta signaling, has opposite effects on Smad2- and Smad3-dependent signaling. EMBO J 22, 4465–4477 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, Lin YC, Yang E, Goldrath AW, Li MO, Cantrell DA, Hedrick SM, ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity 42, 239–251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H, PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity 49, 264–274 e264 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guimond A, Viau E, Aube P, Renzi PM, Paquet L, Ferrari N, Advantageous toxicity profile of inhaled antisense oligonucleotides following chronic dosing in non-human primates. Pulm Pharmacol Ther 21, 845–854 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Mateus J, Grifoni A, Tarke A, Sidney J, Ramirez SI, Dan JM, Burger ZC, Rawlings SA, Smith DM, Phillips E, Mallal S, Lammers M, Rubiro P, Quiambao L, Sutherland A, Yu ED, da Silva Antunes R, Greenbaum J, Frazier A, Markmann AJ, Premkumar L, de Silva A, Peters B, Crotty S, Sette A, Weiskopf D, Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 370, 89–94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, Laidlaw BJ, Araki K, Ahmed R, Kaech SM, Craft J, The Interleukin-2-mTORc1 Kinase Axis Defines the Signaling, Differentiation, and Metabolism of T Helper 1 and Follicular B Helper T Cells. Immunity 43, 690–702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, Eastes AN, Davis O, De Cegli R, Zampelli A, Di Giovannantonio LG, Nusco E, Platt N, Guida A, Ogmundsdottir MH, Lanfrancone L, Perera RM, Zoncu R, Pelicci PG, Settembre C, Ballabio A, Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356, 1188–1192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas JD, Zhang YJ, Wei YH, Cho JH, Morris LE, Wang HY, Zheng XF, Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 26, 754–769 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawrence RE, Cho KF, Rappold R, Thrun A, Tofaute M, Kim DJ, Moldavski O, Hurley JH, Zoncu R, A nutrient-induced affinity switch controls mTORC1 activation by its Rag GTPase-Ragulator lysosomal scaffold. Nat Cell Biol 20, 1052–1063 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, Green DR, Jones DP, Virgin HW, Ahmed R, Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol 15, 1152–1161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, Simon AK, Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, He Q, Shen H, Xia A, Tian W, Yu W, Sun B, TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol 71, 731–741 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, Sharpe AH, Freeman GJ, Irving BA, Ahmed R, Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A 115, 4749–4754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hams E, McCarron MJ, Amu S, Yagita H, Azuma M, Chen L, Fallon PG, Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol 186, 5648–5655 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Cao W, Fang F, Gould T, Li X, Kim C, Gustafson C, Lambert S, Weyand CM, Goronzy JJ, Ecto-NTPDase CD39 is a negative checkpoint that inhibits follicular helper cell generation. J Clin Invest 130, 3422–3436 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balderhaar HJ, Ungermann C, CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J Cell Sci 126, 1307–1316 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA, Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14, 500–508 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB, mTOR inhibition improves immune function in the elderly. Sci Transl Med 6, 268ra179 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM, mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A, A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 27, 671–680 e672 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, Rawlings SA, Sutherland A, Premkumar L, Jadi RS, Marrama D, de Silva AM, Frazier A, Carlin AF, Greenbaum JA, Peters B, Krammer F, Smith DM, Crotty S, Sette A, Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 181, 1489–1501 e1415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Raw data file (Excel spreadsheet).
Supplementary Materials
Supplementary Materials and Methods
Fig. S1. Activated naïve CD4+ T cells from older adults lose lysosomal function while maintaining mTORC1 activity.
Fig. S2. SLC7A5-dependent mTORC1 activation in naïve CD4+ T cell responses from older adults.
Fig. S3. VPS39 silencing does not affect TGF-β/SMAD signaling in naïve CD4+ T cell responses.
Fig. S4. Effects of mTORC1 inhibition on TFEB expression and phosphorylation.
Fig. S5. Inhibition of late endosomal mTORC1 promotes virus-specific CD4+ T cell expansion without affecting their cytokine production.
Fig. S6. CTLA-4 cell surface expression does not change with age.
Fig. S7. Improved CD4+ T cell helper responses after Vps39 silencing depends on reduced PD-1 expression.
Fig. S8. Key gating strategies used in this study.
Table S1. Oligonucleotide primer sets used in this study.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Peptide pools utilized in this paper are available from A.S. (alex@lji.org) under a material transfer agreement (MTA) from La Jolla Institute for Immunology.