

Abstract
Objective
Parkinson's disease (PD) is a complex neurodegenerative disorder. Men are on average ~ 1.5 times more likely to develop PD compared to women with European ancestry. Over the years, genomewide association studies (GWAS) have identified numerous genetic risk factors for PD, however, it is unclear whether genetics contribute to disease etiology in a sex‐specific manner.
Methods
In an effort to study sex‐specific genetic factors associated with PD, we explored 2 large genetic datasets from the International Parkinson's Disease Genomics Consortium and the UK Biobank consisting of 13,020 male PD cases, 7,936 paternal proxy cases, 89,660 male controls, 7,947 female PD cases, 5,473 maternal proxy cases, and 90,662 female controls. We performed GWAS meta‐analyses to identify distinct patterns of genetic risk contributing to disease in male versus female PD cases.
Results
In total, 19 genomewide significant regions were identified and no sex‐specific effects were observed. A high genetic correlation between the male and female PD GWAS were identified (rg = 0.877) and heritability estimates were identical between male and female PD cases (~ 20%).
Interpretation
We did not detect any significant genetic differences between male or female PD cases. Our study does not support the notion that common genetic variation on the autosomes could explain the difference in prevalence of PD between males and females cases at least when considering the current sample size under study. Further studies are warranted to investigate the genetic architecture of PD explained by X and Y chromosomes and further evaluate environmental effects that could potentially contribute to PD etiology in male versus female patients. ANN NEUROL 2021;90:41–48
Parkinson's disease (PD) is an age‐related, progressive neurodegenerative disorder. On average, men are ~ 1.5 times more likely to develop PD compared to women in European ancestry cohorts.1 The reasons for the increased risk in men (relative to women) is not well understood. Possible explanations might include different degrees of exposure to environmental risk factors (such as pesticides and heavy metals), putative risk and protective factors (head trauma, caffeine, and urate), the influence of sex‐specific hormones, differential aging, cardiovascular risk, and life expectancy, or potential genetic factors, either linked or independent of these other factors.2, 3, 4, 5
There are also differences in the clinical presentation of PD by sex, female patients are more likely to experience dyskinesia and a slower decline in performance of activities of daily living, whereas it has been shown that male patients have a higher risk of developing cognitive impairment.6 Symptoms that present into the earliest phases of PD also differ by sex; rapid eye movement (REM) sleep behavior disorder (RBD) is much more common in men, and depression and anxiety appear to be more common in women.7, 8 PD is a genetically complex disease, with a substantial genetic component explained by rare and common variants.9 Several large case‐control genomewide association studies (GWAS) have been performed, the most recent of which identified 92 risk signals across 80 loci.10, 11 Over the last 20 years, we have gained a great deal of insight into the genetic architecture and etiology of PD and this now serves as the basis for several therapeutic approaches. However, the interplay between sex and genetics in PD has not been broadly investigated and it is currently unknown whether the genetic risk varies between men and women. Here, we investigate whether there is a difference in the genetic architecture of autosomal risk according to genetic sex using multiple large case‐control cohorts.
Methods
International Parkinson's Disease Genomics Consortium Data
Genotyping data, all derived from Illumina platform‐based genotyping, was obtained from members of the International Parkinson's Disease Genomics Consortium (IPDGC), collaborators, and publicly available datasets (Supplementary Table S2). All PD cases were diagnosed using standard UK Brain bank or the Movement Disorder Society (MDS) criteria.12, 13 Control participants were excluded if they had any known neurological disease. All datasets underwent quality control separately, both on individual‐level data and variant‐level data, before imputation was performed, as previously described.11, 14 Quality control steps included: relatedness filtering (PIHAT > 0.125, at the level of first cousin one random individual was removed from each pair), removal of genetic ancestry outliers departing 6 standard deviations from the European CEU/TSI HapMap3 populations, removal of samples with call rates < 95% and whose genetically determined sex from X‐chromosome did not match that from clinical data, as well as samples exhibiting excess heterozygosity estimated by an F‐statistic > ± 0.15. The quality control process and underlying scripts for filtering can be found at https://github.com/neurogenetics/GWAS-pipeline. Filtered genotype data was imputed using the Michigan imputation server with the Haplotype Reference Consortium reference panel r1.1 2016 under default settings with phasing using the EAGLE option.15, 16 For GWAS analyses, variants passing the post‐imputation quality criteria of R2 > 0.3 and minor allele frequency (MAF) > 1% were included. Data were split into male and female datasets based on genetic sex. Sex specific case‐control GWAS were performed using RVTESTS (version 20190205) under default settings17 using logistic regression on genotype dosages adjusted for the following covariates: age at onset for cases and age of last examination for controls (for a small subset age was not available and missing values were imputed with the mean value using ‐ imputeCov), principal components (PCs) 1 to 5 to account for population stratification, and dataset origin. Age was not included in 3 datasets due to missing data (MF, VANCE) or co‐linearity (FINLAND). PCs were calculated from non‐imputed genotype data using FlashPCA (version 2.0).18
UK Biobank Data
Imputed UK Biobank (UKB) genotype data (version 3) was downloaded (April 2018) under application number 33601.19, 20 PD cases were identified using data fields 42032 and 42033. “Proxy” PD cases were included as part of the analyses, considering individuals who reported a parent affected with PD (paternal PD = data field 20107 and maternal PD = data field 20110) because it has been previously shown to share genetic risk with PD cases.11 Controls were set as people with no report of PD, and no parent affected with PD, and with an age of recruitment over 60 (data field 21022). Covariates were obtained from the data fields: age of recruitment (data field 21022) and Townsend index (data field 189). Individuals were filtered for relatedness (PIHAT > 0.125, at the level of first cousin one random individual was removed from each pair) based on the pre‐imputed genotype data using Genomewide Complex Trait Analysis (GCTA).21 Only European ancestry individuals were included from data field 22006. Imputed genotypes were converted to PLINK2.pgen files using PLINK2 (version v2.00a2LM)22 and filtered for missingness (removing samples with variant missingness > 0.1 and MAF < 0.01), Hardy–Weinberg equilibrium of p ≥ 1E‐6 and imputation quality (R2 > 0.8). GWAS was performed using PLINK2 logistic regression with covariates, including age of recruitment, Townsend index, and 5 PCs generated by FlashPCA to account for population substructure.18 Four GWAS were performed in the UKB data (1) male PD cases versus male controls, (2) female PD cases versus female controls, (3) subjects with a father affected with PD versus controls, and (4) subjects with a mother affected with PD versus controls (Supplementary Table S2). Proxy conversion was performed as previously described.23
Additional Analyses
Post GWAS quality control was applied to remove variants with an MAF < 1%, unrealistic beta values for GWAS (> 5 or < 5), and multi‐allelic variants. Sex‐specific meta‐analyses were performed using METAL version 2018‐08‐28 under default settings.24 Post GWAS meta‐analysis, the following filtering steps were further applied: variants present in at least 13 out of the 19 datasets and displaying an I2 heterogeneity value of < 80 were included. Mirror Manhattan plot generated with both filtered datasets using the Hudson package (https://github.com/anastasia-lucas/hudson). Linkage Disequilibrium Score regression (LDSC) was performed to calculate the genetic correlation between summary statistics.25 To assess differences in the magnitude of associations between men's and women's heterogeneity tests were performed. Additionally, genetic heritability was calculated excluding the UKB proxies for men and women separately. All figures and statistical calculations were created and performed using R (version 4.0.3) or Python (version 3.7). The genetic risk score was estimated using IPDGC data, and11 as the reference dataset to define risk‐weighted alleles. Locus numbering was obtained as previously described.26 Large effect size variants from the GBA and LRRK2 regions were excluded (rs76763715, rs35749011, rs34637584, and rs114138760).
Results
Initial Data Overview
After quality control, we included 13,020 male PD cases, 7,936 paternal proxies, 89,660 male controls, 7,947 female PD cases, 5,473 maternal proxies, and 90,662 female controls totaling 214,698 individuals (Supplementary Table S2). The odds ratio for PD in men versus women (excluding proxies) was 1.72 (95% confidence interval [CI] = 1.66–1.78). Meta‐analyses at MAF > 1%, variant present at least 13 out of the 19 datasets and displaying an I2 heterogeneity value of < 80 resulted in 7,153,507 variants passing quality control for men and 7,141,404 variants for women. No evidence of genomic inflation was observed, with Lambda 1000 and LDSC intercept values of 1.0013 and 0.9548 (SE = 0.0077) for male PD GWAS and 1.0008 and 0.9516 (SE = 0.0077) for female PD GWAS. In total, 14 and 13 genomic regions reached genomewide significance in the male and female GWAS meta‐analyses respectively, of which 8 were identified in both and 11 were only genomewide significant in either GWAS. However, all these 11 genomic regions show significant signals at p > 1E‐4, similar effect sizes and overlapping 95% CIs of regression coefficients (Fig 1). As expected and previously reported in the largest PD GWAS meta‐analysis,11 the SNCA and MAPT loci were the main significant hits in both the male and female specific GWAS (see Fig 1), where the top SNCA variant was rs356182; (PD_male: p = 3.47E‐44, beta = 0.256, SE = 0.0184; and PD_female: p = 1.41E‐25, beta = 0.219, SE = 0.0209) and top MAPT variant was rs75010486; (PD_male: p = 1.48E‐32, beta = 0.244, SE = 0.0206; and PD_female: p = 5.02E‐29, beta = 0.268, SE = 0.0239). Where the SNCA locus represents the association between SNCA gene expression and PD risk and the MAPT locus the association between MAPT haplotypes H1/H2 and PD risk.
FIGURE 1.
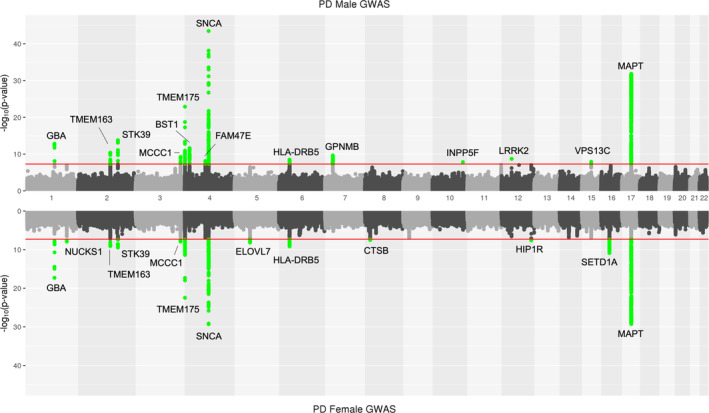
Mirror Manhattan plot of male and female specific Parkinson's disease GWAS. On the top male PD GWAS, and bottom female PD GWAS. The red line indicates the ‐log10 p value genomewide significant threshold of 5E‐8. Green dots indicate variants passing genomewide significance. Note that green highlighted signals do not represent male or female specific Parkinson's disease GWAS signals and that for each region of interest nominal significance is identified in the opposite Manhattan plot. Signals are annotated based on the closest gene from ref. 11. GWAS = genomewide association studies; PD = Parkinson's disease. [Color figure can be viewed at www.annalsofneurology.org]
Comparing Parkinson's Disease Risk Signals Between Men and Women
To identify potential genetic differences in risk for PD, we performed four main analyses: (1) a genomewide LDSC correlation between male and female specific summary statistics, (2) an analysis of whether the known cumulative genetic risk score was different between men and women, (3) an assessment of whether the known PD risk variants from ref. 11 were affecting men and women differently, and (4) an investigation for potential novel sex‐specific associations in male and female specific summary statistics. The genomewide correlation using LDSC resulted in a very high genetic correlation between the male and female PD GWASes (rg = 0.877, SE = 0.0699) which was highly significant at a p = 4.24E‐36. This shows that on an autosomal genetic level, PD risk is similar between men and women as it is similar among self‐reported, clinically diagnosed, and family history defined cases with genetic correlations at 84% or more as shown in ref. 11 when comparing IPDGC (clinically diagnosed) summary statistics with 23andMe data (mostly self‐reported; rg = 0.85) or UK Biobank (rg = 0.84; family history defined cases). Heritability estimates (excluding UKB proxies) were similar between males and females (h2_male = 0.2077, SE = 0.0238, and h2_female = 0.1857, SE = 0.0304) as expected based on previous estimates.
Next, we tested whether there was a difference of cumulative genetic risk score, based on known GWAS hits and using weights from ref. 11 between male and female PD cases using the IPDGC data. Each cohort was analyzed separately and results were meta‐analyzed. Meta‐analysis showed that there was no difference between the cumulative genetic risk score in men and women with PD, p = 0.572 (Fig 2). Subsequently, we investigated whether any of the known GWAS risk signals were associated with PD differently between male and female cases. Out of the 90 independent risk signals from the most recent PD GWAS,11 85 were present in the sex‐specific GWAS summary statistics. An almost perfect correlation was observed between the effect sizes of the sex‐specific GWAS for these 85 variants (Pearson correlation R2 > 0.95; Fig 3A, Supplementary Table S3). Similarly, high correlations were found comparing effect sizes between male PD GWAS and ref. 11 (> 0.98) and female PD GWAS and ref. 11 (> 0.96). Minor differences were observed in some instances. For example, the GALC genomic region (locus 58, rs979812) showed slight differences in magnitude of effect in men versus women; PD_male p = 0.06462, beta = 0.031, SE = 0.0168; PD_female p = 3.07E‐06, beta = 0.0895, SE = 0.0192, and p_difference = 0.0218. However, these were not significant following multiple test corrections (Supplementary Table S3).
FIGURE 2.
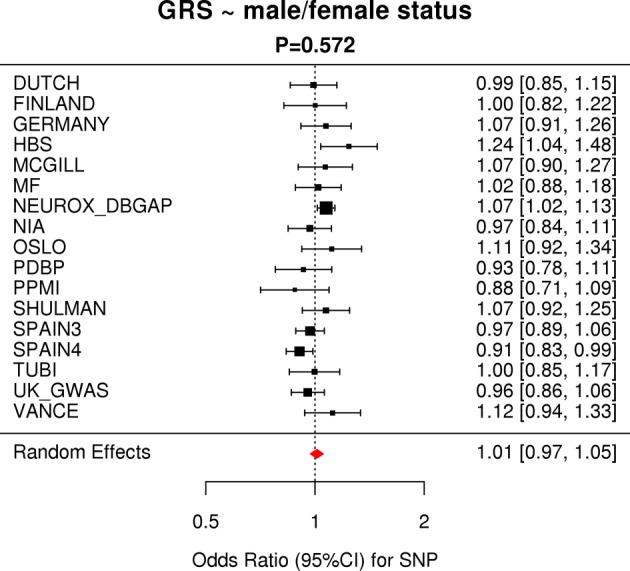
Meta‐analysis of the genetic risk score versus male/female status shows no difference between “genetic load” of Parkinson's disease associated risk. Red diamond indicates the effect estimate (odds ratio) and 95% confidence interval of the aggregate result. CI = confidence interval; GRS = genetic risk score; SNP = single nucleotide polymorphism. [Color figure can be viewed at www.annalsofneurology.org]
FIGURE 3.

(A) Beta‐beta plot of11 genome wide significant risk signals. Very high correlation (Pearson correlation R2 > 0.95) is observed between effect sizes from the male and female specific GWAS. Additional details can be found in Supplementary Table S2. (B) Effect sizes of the male PD GWAS hits passing genomewide significance plotted versus matching female PD GWAS effect sizes. (C) Effect sizes of the female PD GWAS hits passing genome wide significance plotted versus matching male PD GWAS effect sizes. GWAS = genomewide association studies; PD = Parkinson's disease. [Color figure can be viewed at www.annalsofneurology.org]
Finally, we investigated the full summary statistics to identify potential new sex specific hits that have not been identified yet as PD GWAS risk signals. All variants passing p < 5.0E‐8 from the male PD GWAS were extracted and compared to the summary statistics of the female PD GWAS and vice versa. When plotting the effect sizes of these variants, no differences were observed between male PD GWAS and female PD GWAS effect sizes of variants passing p < 5.0E‐8 (see Fig 3B, C).
Discussion
Men are ~ 1.5 times more likely to develop PD compared to women in European ancestry. This is similar to dementia with Lewy bodies (DLB), which is also more common in men, but in contrast with Alzheimer's disease (AD) in which women are almost 1.5 to 2 times more likely to develop disease.27 Where on the phenotype level slight differences are observed between men and women in PD, DLB, and AD and overall men have a faster progression/cognitive decline and shorter lifespan after diagnosis.6,27 Sex‐dependent autosomal effects on clinical progression of AD have been observed28 and data on sex‐dependent autosomal effects for other neurodegenerative diseases is lacking. Here, we assessed whether an autosomal genetic difference explains these differences by performing GWAS using several large case‐control datasets and separating these by men and women. Overall, based on the results presented here, we could not identify that autosomal genetics contributes to the difference of observed prevalence between men and women. As expected, the results from the sex‐specific GWAS are highly similar to previous PD GWAS and in particular to the PD GWAS from 2011 with a very similar sample size (15 K cases).27
The question of what actually causes the difference in prevalence will need to be further evaluated. One possibility is genetic variation on the sex chromosomes. Chromosome X contains ~ 850 genes and chromosome Y ~ 70 genes and account for ~ 5% and ~ 2% of the human genome size, respectively. Although there is ongoing work in this area, thus far, no association has been found that can explain the significant over‐representation of male PD patients. In the most recent X chromosome association study, one replicated genomewide signal was identified and no differences were identified between male and female effect size.28 Given that chromosome Y is only present in men, it could be a good candidate for increased risk. Currently, no large genetic association studies have been performed to investigate if certain chromosome Y haplotypes are over‐represented in cases versus controls. There are reports stating that certain Y chromosome genes show male‐specific effects for potential dopaminergic loss.29 Besides the differences in sex chromosomes in men and women, there are several other differences not directly assessed here that may likely contribute to sex differences in PD, including (1) differences in gene expression in cells and tissues on both autosomes and sex chromosomes,30, 31 (2) hormone production, and (3) the environment including for example smoking behavior.32 All these possibilities need to be studied further with a specific focus on PD.
As for any GWAS study, there are limitations. First, due to the study design, we can only investigate common variants that are present in the imputation panels, meaning that we cannot investigate rare variants and structural variation.33, 34 Second, given that the majority of the data included in this study was also used in the discovery of the known 90 risk variants from ref. 11 and sex was used as a covariate in that analysis, it is not surprising that there is a high correlation of effect sizes between the male and female specific GWAS results from Figure 3A due to circularity. However, by using a more unbiased approach in Figure 3B and C no differences in effect sizes were identified for any genomewide significant hits from the sex‐specific GWAS. Third, although we included a very large number of cases and controls, there could be small effect size variants that play a role in disease, not currently detected due to lack of statistical power.
Overall, by combining our manuscript with Guen et al. we provide evidence that there are no male or female specific PD GWAS hits and that the difference in prevalence of PD between men and differences females cannot be explained by common genetics when considering the current sample size under study. Further studies are warranted to investigate the genetic architecture of PD explained by the sex chromosomes and further evaluate environmental effects that could potentially contribute to PD etiology in men versus differences.
Author Contributions
C.B., M.A.N., and A.B.S. contributed to conception and design of the study. C.B., H.I., M.B.M., and M.A.N. contributed to acquisition and analysis of data. C.B., M.A.N., A.B.S., A.J.N., and S.B.C. contributed to drafting the text or preparing the figures.
Potential Conflicts of Interest
The authors declared no conflict of interest.
Data Availability
All summary statistics are available at https://pdgenetics.org/resources. Four summary statistics tables are made available: (1) male PD GWAS (all data), (2) female PD GWAS (all data), (3) male PD GWAS (no UKB proxy data), and (4) female PD GWAS (no UKB proxy data). All code used has been made available on GitHub: https://github.com/neurogenetics/Autosomal-sex-differences-PDv2.
Supporting information
TABLE S1. IPDGC consortium members and affiliations
TABLE S2. Overview of included data
TABLE S3. Summary statistics of the genome wide signals from 11
Acknowledgments
The authors would like to thank all of the subjects who donated their time and biological samples to be a part of this study. We also would like to thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). See for a complete overview of members, acknowledgments, and funding http://pdgenetics.org/partners. This work was supported in part by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institute on Aging (NIA), and the National Institute of Environmental Health Sciences both part of the National Institutes of Health, Department of Health and Human Services; project numbers 1ZIA‐NS003154, Z01‐AG000949‐02, and Z01‐ES101986. In addition, this work was supported by the Department of Defense (award W81XWH‐09‐2‐0128), and The Michael J. Fox Foundation for Parkinson's Research. This work was supported by National Institutes of Health grants R01NS037167, R01CA141668, and P50NS071674, American Parkinson Disease Association (APDA); Barnes Jewish Hospital Foundation; Greater St Louis Chapter of the APDA. The KORA (Cooperative Research in the Region of Augsburg) research platform was started and financed by the Forschungszentrum für Umwelt und Gesundheit, which is funded by the German Federal Ministry of Education, Science, Research, and Technology and by the State of Bavaria. This study was also funded by the German Federal Ministry of Education and Research (BMBF) under the funding code 031A430A, the EU Joint Programme ‐ Neurodegenerative Diseases Research (JPND) project under the aegis of JPND ‐www.jpnd.eu ‐ through Germany, BMBF, funding code 01ED1406 and iMed ‐ the Helmholtz Initiative on Personalized Medicine. This study utilized the high‐performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD, USA (http://biowulf.nih.gov), and DNA panels, samples, and clinical data from the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository. People who contributed samples are acknowledged in descriptions of every panel on the repository website. We thank P. Tienari (Molecular Neurology Programme, Biomedicum, University of Helsinki), T. Peuralinna (Department of Neurology, Helsinki University Central Hospital), L. Myllykangas (Folkhalsan Institute of Genetics and Department of Pathology, University of Helsinki), and R. Sulkava (Department of Public Health and General Practice Division of Geriatrics, University of Eastern Finland) for the Finnish controls (Vantaa85+ GWAS data). This study was also funded by the Sigrid Juselius Foundation (KM). We used genomewide association data generated by the UK control individuals from the 1958 Birth Cohort and National Blood Service. UK population control data was made available through WTCCC1. As with previous IPDGC efforts, this study makes use of data generated by the Wellcome Trust Case‐Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under awards 076113, 085475, and 090355. This study was also supported by Parkinson's UK (grants 8047 and J‐0804). Sequencing and genotyping done in McGill University was supported by grants from the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) program and Parkinson's Society Canada. The access to part of the participants at McGill has been made possible thanks to the Quebec Parkinson's Network (http://rpq-qpn.ca/en). We thank the Quebec Parkinson's Network (http://rpq-qpn.org) and its members. Harvard NeuroDiscovery Biomarker Study (HBS) is a collaboration of HBS investigators and funded through philanthropy and NIH and Non‐NIH funding sources. The HBS Investigators have not participated in reviewing the data analysis or content of the manuscript. PPMI – a public‐private partnership – is funded by the Michael J. Fox Foundation for Parkinson's Research and funding partners, the full names of all of the PPMI funding partners can be found at www.ppmi-info.org/fundingpartners. The PPMI Investigators have not participated in reviewing the data analysis or content of the manuscript. For up‐to‐date information on the study, visit www.ppmi-info.org. Parkinson's Disease Biomarker Program (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript.
[Correction added on August 12, 2021 after first online publication: Wellcome Trust 090355 085475 076113 and Medical Research Council G1100643 G0700943 were removed from Acknowledgments section.]
Contributor Information
Cornelis Blauwendraat, Email: cornelis.blauwendraat@nih.gov.
and the International Parkinson's Disease Genomics Consortium (IPDGC):
Alastair J. Noyce, Rauan Kaiyrzhanov, Ben Middlehurst, Demis A. Kia, Manuela Tan, Henry Houlden, Catherine S. Storm, Huw R. Morris, Helene Plun‐Favreau, Peter Holmans, John Hardy, Daniah Trabzuni, John Quinn, Vivien Bubb, Kin Y. Mok, Kerri J. Kinghorn, Nicholas W. Wood, Patrick Lewis, Sebastian R. Schreglmann, Ruth Lovering, Lea R'Bibo, Claudia Manzoni, Mie Rizig, Mina Ryten, Sebastian Guelfi, Valentina Escott‐Price, Viorica Chelban, Thomas Foltynie, Nigel Williams, Karen E. Morrison, Carl Clarke, Kirsten Harvey, Benjamin M. Jacobs, Alexis Brice, Fabrice Danjou, Suzanne Lesage, Jean‐Christophe Corvol, Maria Martinez, Claudia Schulte, Kathrin Brockmann, Javier Simón‐Sánchez, Peter Heutink, Patrizia Rizzu, Manu Sharma, Thomas Gasser, Susanne A. Schneider, Mark R. Cookson, Sara Bandres‐Ciga, Cornelis Blauwendraat, David W. Craig, Kimberley Billingsley, Mary B. Makarious, Derek P. Narendra, Faraz Faghri, J. Raphael Gibbs, Dena G. Hernandez, Kendall Van Keuren‐Jensen, Joshua M. Shulman, Hirotaka Iwaki, Hampton L. Leonard, Mike A. Nalls, Laurie Robak, Jose Bras, Rita Guerreiro, Steven Lubbe, Timothy Troycoco, Steven Finkbeiner, Niccolo E. Mencacci, Codrin Lungu, Andrew B. Singleton, Sonja W. Scholz, Xylena Reed, Ryan J. Uitti, Owen A. Ross, Francis P. Grenn, Anni Moore, Roy N. Alcalay, Zbigniew K. Wszolek, Ziv Gan‐Or, Guy A. Rouleau, Lynne Krohn, Kheireddin Mufti, Jacobus J. van Hilten, Johan Marinus, Astrid D. Adarmes‐Gómez, Miquel Aguilar, Ignacio Alvarez, Victoria Alvarez, Francisco Javier Barrero, Jesús Alberto Bergareche Yarza, Inmaculada Bernal‐Bernal, Marta Blazquez, Marta Bonilla‐Toribio, Juan A. Botía, María Teresa Boungiorno, Dolores Buiza‐Rueda, Ana Cámara, Fátima Carrillo, Mario Carrión‐Claro, Debora Cerdan, Jordi Clarimón, Yaroslau Compta, Monica Diez‐Fairen, Oriol Dols‐Icardo, Jacinto Duarte, Raquel Duran, Francisco Escamilla‐Sevilla, Mario Ezquerra, Cici Feliz, Manel Fernández, Rubén Fernández‐Santiago, Ciara Garcia, Pedro García‐Ruiz, Pilar Gómez‐Garre, Maria Jose Gomez Heredia, Isabel Gonzalez‐Aramburu, Ana Gorostidi Pagola, Janet Hoenicka, Jon Infante, Silvia Jesús, Adriano Jimenez‐Escrig, Jaime Kulisevsky, Miguel A. Labrador‐Espinosa, Jose Luis Lopez‐Sendon, Adolfo López de Munain Arregui, Daniel Macias, Irene Martínez Torres, Juan Marín, Maria Jose Marti, Juan Carlos Martínez‐Castrillo, Carlota Méndez‐del‐Barrio, Manuel Menéndez González, Marina Mata, Adolfo Mínguez, Pablo Mir, Elisabet Mondragon Rezola, Esteban Muñoz, Javier Pagonabarraga, Pau Pastor, Francisco Perez Errazquin, Teresa Periñán‐Tocino, Javier Ruiz‐Martínez, Clara Ruz, Antonio Sanchez Rodriguez, María Sierra, Esther Suarez‐Sanmartin, Cesar Tabernero, Juan Pablo Tartari, Cristina Tejera‐Parrado, Eduard Tolosa, Francesc Valldeoriola, Laura Vargas‐González, Lydia Vela, Francisco Vives, Alexander Zimprich, Lasse Pihlstrom, Mathias Toft, Pille Taba, Sulev Koks, Sharon Hassin‐Baer, Kari Majamaa, Ari Siitonen, Pentti Tienari, Njideka U. Okubadejo, Oluwadamilola O. Ojo, Rauan Kaiyrzhanov, Chingiz Shashkin, Nazira Zharkinbekova, Vadim Akhmetzhanov, Gulnaz Kaishybayeva, Altynay Karimova, Talgat Khaibullin, and Timothy L. Lynch
References
- 1.Moisan F, Kab S, Mohamed F, et al. Parkinson disease male‐to‐female ratios increase with age: French Nationwide study and meta‐analysis. J Neurol Neurosurg Psychiatry 2016;87:952–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascherio A, Weisskopf MG, O'Reilly EJ, et al. Coffee consumption, gender, and Parkinson's disease mortality in the cancer prevention study II cohort: the modifying effects of estrogen. Am J Epidemiol 2004;160:977–984. [DOI] [PubMed] [Google Scholar]
- 3.Gao X, O'Reilly ÉJ, Schwarzschild MA, Ascherio A. Prospective study of plasma urate and risk of Parkinson disease in men and women. Neurology 2016;86:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nandipati S, Litvan I. Environmental exposures and Parkinson's disease. Int J Environ Res Public Health 2016;13:881. 10.3390/ijerph13090881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor KSM, Cook JA, Counsell CE. Heterogeneity in male to female risk for Parkinson's disease. J Neurol Neurosurg Psychiatry 2007;78:905–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwaki H, Blauwendraat C, Leonard HL, et al. Differences in the presentation and progression of Parkinson's disease by sex. Mov Disord 2020;36:106–117. 10.1002/mds.28312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Postuma RB, Iranzo A, Hu M, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain 2019;142:744–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiba M, Bower JH, Maraganore DM, et al. Anxiety disorders and depressive disorders preceding Parkinson's disease: a case‐control study. Mov Disord 2000;15:669–677. [DOI] [PubMed] [Google Scholar]
- 9.Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol 2020;19:170–178. 10.1016/s1474-4422(19)30287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foo JN, Chew EGY, Chung SJ, et al. Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: a genome‐wide association study. JAMA Neurol 2020;77:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol 2019;18:1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibb WR, Lees AJ. A comparison of clinical and pathological features of young‐ and old‐onset Parkinson's disease. Neurology 1988;38:1402–1406. [DOI] [PubMed] [Google Scholar]
- 13.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591–1601. [DOI] [PubMed] [Google Scholar]
- 14.Blauwendraat C, Heilbron K, Vallerga CL, et al. Parkinson's disease age at onset genome‐wide association study: defining heritability, genetic loci, and α‐Synuclein mechanisms. Mov Disord 2019;34:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das S, Forer L, Schönherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhan X, Hu Y, Li B, et al. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 2016;32:1423–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abraham G, Qiu Y, Inouye M. FlashPCA2: principal component analysis of biobank‐scale genotype datasets. Bioinformatics 2017;33:2776–2778. [DOI] [PubMed] [Google Scholar]
- 19.Bycroft C, Freeman C, Petkova D, et al. The UKbiobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sudlow C, Gallacher J, Allen N, et al. UKbiobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Hong Lee S, Goddard ME, Visscher PM. GCTA: a tool for genome‐wide complex trait analysis. Am J Hum Genet 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CC, Chow CC, Tellier LC, et al. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu JZ, Erlich Y, Pickrell JK. Case‐control association mapping by proxy using family history of disease. Nat Genet 2017;49:325–331. [DOI] [PubMed] [Google Scholar]
- 24.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of Genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulik‐Sullivan BK, Loh P‐R, Finucane HK, et al. LD score regression distinguishes confounding from Polygenicity in genome‐wide association studies. Nat Genet 2015;47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grenn FP, Kim JJ, Makarious MB, et al. The Parkinson's disease genome‐wide Association Study Locus Browser. Mov Disord 2020;35:2056–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Podcasy JL, Epperson CN. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin Neurosci. 2016;18437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan CC, Banks SJ, Thompson WK. Sex‐dependent autosomal effects on clinical progression of Alzheimer's disease. Brain. 2020;1432272–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.International Parkinson's Disease Genomics Consortium (IPDGC), and Wellcome Trust Case Control Consortium 2 (WTCCC2) . A Two‐Stage Meta‐Analysis Identifies Several New Loci for Parkinson's Disease. PLoS Genet 2011;7:e1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Guen Y, Le Guen Y, Napolioni V, et al. Common x‐chromosome variants are associated with parkinson's disease risk. Ann Neurol 2021. 10.1002/ana.26051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, Pinares‐Garcia P, Loke H, et al. Sex‐specific neuroprotection by inhibition of the Y‐chromosome gene, in experimental Parkinson's disease. Proc Natl Acad Sci USA 2019;116:16577–16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliva M, Muñoz‐Aguirre M, Kim‐Hellmuth S, et al. The impact of sex on gene expression across human tissues. Science 2020;369:eaba3066. 10.1126/science.aba3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trabzuni D, Ramasamy A, Imran S, et al. Widespread sex differences in gene expression and splicing in the adult human brain. Nat Commun 2013;4:2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heilbron K, Jensen MP, Bandres‐Ciga S, et al. Unhealthy behaviours and parkinson's disease: a mendelian randomisation study. medRxiv 2020. 10.1101/2020.03.25.20039230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. IPDGC consortium members and affiliations
TABLE S2. Overview of included data
TABLE S3. Summary statistics of the genome wide signals from 11
Data Availability Statement
All summary statistics are available at https://pdgenetics.org/resources. Four summary statistics tables are made available: (1) male PD GWAS (all data), (2) female PD GWAS (all data), (3) male PD GWAS (no UKB proxy data), and (4) female PD GWAS (no UKB proxy data). All code used has been made available on GitHub: https://github.com/neurogenetics/Autosomal-sex-differences-PDv2.