

Abstract
A disintegrin and metalloproteinase domain-15 (ADAM15) is expressed by cells implicated in the pathogenesis of chronic obstructive pulmonary disease (COPD), but its contributions to COPD are unknown. To address this gap, ADAM15 levels were measured in samples from cigarette smoke (CS)-versus air-exposed wild-type (WT) mice. CS-induced COPD-like disease was compared in CS-exposed WT, Adam15−/−, and Adam15 bone marrow chimeric mice. CS exposure increased Adam15 expression in lung macrophages and CD8+ T cells and to a lesser extent in airway epithelial cells in WT mice. CS-exposed Adam15−/− mice had greater emphysema, small airway fibrosis, and lung inflammation (macrophages and CD8+ T cells) than WT mice. Adam15 bone marrow chimera studies revealed that Adam15 deficiency in leukocytes led to exaggerated pulmonary inflammation and COPD-like disease in mice. Adam15 deficiency in CD8+ T cells was required for the exaggerated pulmonary inflammation and COPD-like disease in CS-exposed Adam15−/− mice (as assessed by genetically deleting CD8+ T cells in Adam15−/− mice). Adam15 deficiency increased pulmonary inflammation by rendering CD8+ T cells and macrophages resistant to CS-induced activation of the mitochondrial apoptosis pathway by preserving mTOR signaling and intracellular Mcl-1 levels in these cells. These results strongly link ADAM15 deficiency to the pathogenesis of COPD.
Keywords: macrophage, CD8+ T cell, mammalian target of rapamycin, myeloid cell leukemia-1 (Mcl-1), mitochondria
Introduction
COPD is the 3rd leading cause of death worldwide1. COPD is characterized by airflow limitation that is poorly reversible and associated with an abnormal pulmonary inflammatory response to noxious particles or gases including those contained in cigarette smoke (CS)2.
Airflow limitation in COPD is caused both by pulmonary emphysema and small airway disease3. Inhaling CS induces alveolar septal cell apoptosis4 and stimulates the recruitment of leukocytes in the lung. Proteinases released by CS-activated leukocytes and epithelial cells degrade extracellular matrix (ECM) proteins within the alveolar walls to promote emphysema development2.
Prior studies of proteinases in COPD have focused on the activities of matrix metalloproteinases (MMPs) and serine and cysteine proteinases2,5,6. Since the 1960s, proteinases were thought to exclusively promote the development of COPD6,7. However, this dogma was recently challenged when a disintegrin and metalloproteinase domain-8 (Adam8) was identified as the first proteinase that protects mice from developing CS-induced COPD-like disease8. ADAMs are type I transmembrane proteinases with multiple domains including: 1) a pro-domain which maintains latency of the metalloproteinase (MP) domain; 2) a MP domain which can shed proteins from cell surfaces; 3) disintegrin and cysteine-rich domains which can regulate cell adhesion and migration; 4) a surface-anchoring transmembrane domain; and 5) a cytoplasmic domain which can regulate intracellular signaling9–11. Whether other ADAMs regulate CS-induced COPD-like disease in mice is unclear.
ADAM15 has potential to contribute to the pathogenesis of COPD. Human ADAM15 and murine Adam15 are expressed by cells implicated in the pathogenesis of COPD including leukocytes, epithelial cells, and fibroblasts2. Although ADAM15 has minimal ECM protein-degrading activity12, it has activities that could be relevant to the pathogenesis of COPD including shedding growth factor receptors13, and regulating cellular adhesion and migration12–14. However, ADAM15’s contributions to COPD have not been explored. We quantified and localized ADAM15 expression in the lungs of CS-versus air-exposed WT mice and compared COPD-like lung disease in CS-exposed WT versus Adam15−/− mice and Adam15 bone marrow (BM) chimeric mice. Our results show that Adam15 deficiency promotes exaggerated COPD-like disease in CS-exposed mice by inciting an excessive pulmonary inflammatory response by increasing the survival of CD8+ T cells and macrophages by inhibiting CS-induced activation of the mitochondrial apoptosis pathway by abrogating CS-induced inactivation of mTOR-Mcl-1 signaling in these cells.
Results
Adam15 expression is increased in the lungs of CS-exposed WT mice:
Exposing WT mice to CS for 1 month increased lung Adam15 transcript and protein levels (Fig. 1A–1B); levels increased further after 2 months of CS exposure but did not increase further after 6 months of CS exposure. Adam15 immuno-staining was detected in alveolar macrophages (AMs), CD8+ T cells, bronchial and alveolar epithelial cells, and α-smooth muscle actin (α-SMA)-positive cells around the small airways in WT mice that were exposed to CS for 6 months (Fig. E1).
Figure 1: A Disintegrin and A Metalloproteinase Domain 15 (Adam15) expression is induced in the lungs of wild-type (WT) mice exposed to cigarette smoke (CS) and is upregulated on the surface of macrophages, monocytes and CD8+ T cells exposed to CS extract (CSE) in vitro.
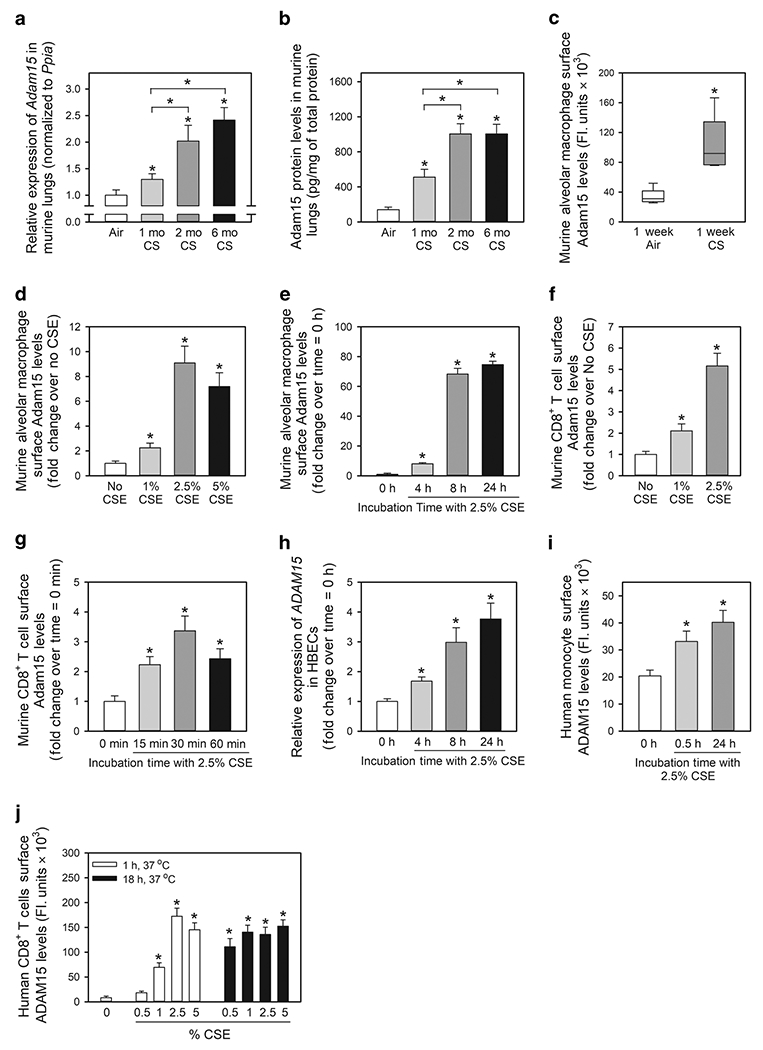
In A and B, Adam15 steady state mRNA and protein levels were measured in the lungs of C57BL/6 WT mice that were exposed to air or CS for up to 6 months using quantitative real-time PCR and an ELISA, respectively (n = 9-10 mice/group). The bar graphs show means ± SD. Data were analyzed using one-way ANOVAs followed by pair-wise testing with two-tailed Student’s t-tests. *, P ≤ 0.004 versus air-exposed mice belonging to the same genotype control or the group indicated. In C, C57BL/6 WT mice were exposed to air or CS for 1 week, and alveolar macrophages (AMs) were isolated using bronchoalveolar lavage. Surface Adam15 protein levels were quantified on AMs using immunostaining techniques, as described in Methods. The boxes in the box-plots show the medians and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles; n = 5 mice/group with 200-300 cells analyzed/mouse. Data were analyzed using a Kruskal-Wallis One-Way ANOVA followed by pair-wise testing with Mann-Whitney U tests. *, P = 0.008 versus air group. In D-E, AMs were isolated from naïve WT mice and incubated for 24 h at 37°C with varying concentrations of CS extract (CSE; D) or for varying times with 2.5% CSE (E). Cells were then fixed and immunostained for surface Adam15, as described in Methods. Data are mean ± SEM; n = 150-200 cells/group. The experiments shown are representative of at least three independent experiments yielding similar results. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.001 compared to unstimulated cells. In F-G, CD8+ lymphocytes were isolated from the spleens of naïve WT mice and incubated at 37°C for 2 h with varying concentrations of CSE (F) or with or without 2.5% CSE for varying times up to 60 min (G). Cells were fixed and immunostained for surface Adam15. Data are mean ± SEM; n = 150-200 cells/group. The experiments shown are representative of at least three independent experiments yielding similar results. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.001 compared to unstimulated cells. In H, steady state ADAM15 mRNA levels were measured in normal human bronchial epithelial cells (HBECs) incubated at 37°C for varying times with or without 2.5% CSE using quantitative realtime PCR. Data are mean ± SD; n = 3 experiments. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.03 versus cells incubated without CSE. In I and J, human monocytes and CD8+ T cells were isolated from the blood of healthy donors by positive section for CD14 or CD8α using immunomagnetic beads. Monocytes and CD8+ T cells were incubated with or without CSE for varying times and/or with concentrations of CSE. Cells were fixed and non-permeabilized cells were immunostained for surface ADAM15 as described in Methods. Data are mean ± SEM; n = 150-200 cells/group. The experiments shown are representative of at least three independent experiments yielding similar results. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.001 compared to unstimulated cells.
Surface Adam15 staining levels were measured on AMs isolated from WT mice that were exposed to air or CS for 1 week. Acute CS exposure increased surface Adam15 levels on AMs ~3-fold (Fig. 1C). CS extract (CSE) induced concentration- and time-dependent increases in surface Adam15 protein levels on AMs isolated from unchallenged WT mice (Fig. 1D–1E). CD8+ T cells are increased in the lungs of CS-exposed mice and COPD patients and are required for CS-induced lung inflammation and airspace enlargement in mice15–17. We investigated whether CSE regulates surface Adam15 levels on CD8+ T cells. CSE induced concentration-dependent increases in surface Adam15 levels on CD8+ T cells isolated from unchallenged WT mice (Fig. 1F). Maximal increases in surface Adam15 levels occurred rapidly (within 30 min of adding CSE [(Fig. 1G]) as reported for release of perforin and granzyme B from the lytic granules of antigen-activated CD8+ T cells15. CSE also increased ADAM15 expression in human cells including ADAM15 transcript levels in human bronchial epithelial cells (HBECs), and surface ADAM levels on monocytes and CD8+ T cells in vitro (Fig. 1H–1J)
Adam15−/− mice develop exaggerated CS-induced COPD-like disease and pulmonary inflammation:
Adam15−/− mice have normal lung development and a normal phenotype in the unchallenged state18. When exposed to CS for 6 months, Adam15−/− mice had greater emphysema development (Fig. 2A–2B) and small airway fibrosis (Fig. 2C–2D), and more α-SMA-positive cells in their small airways (Fig. 2E–2F) than WT mice. WT and Adam15−/− mice had similar CS-induced airway mucus metaplasia (Fig. E2).
Figure 2: Adam15 deficiency in mice increases CS-induced emphysema development and small airway fibrosis.
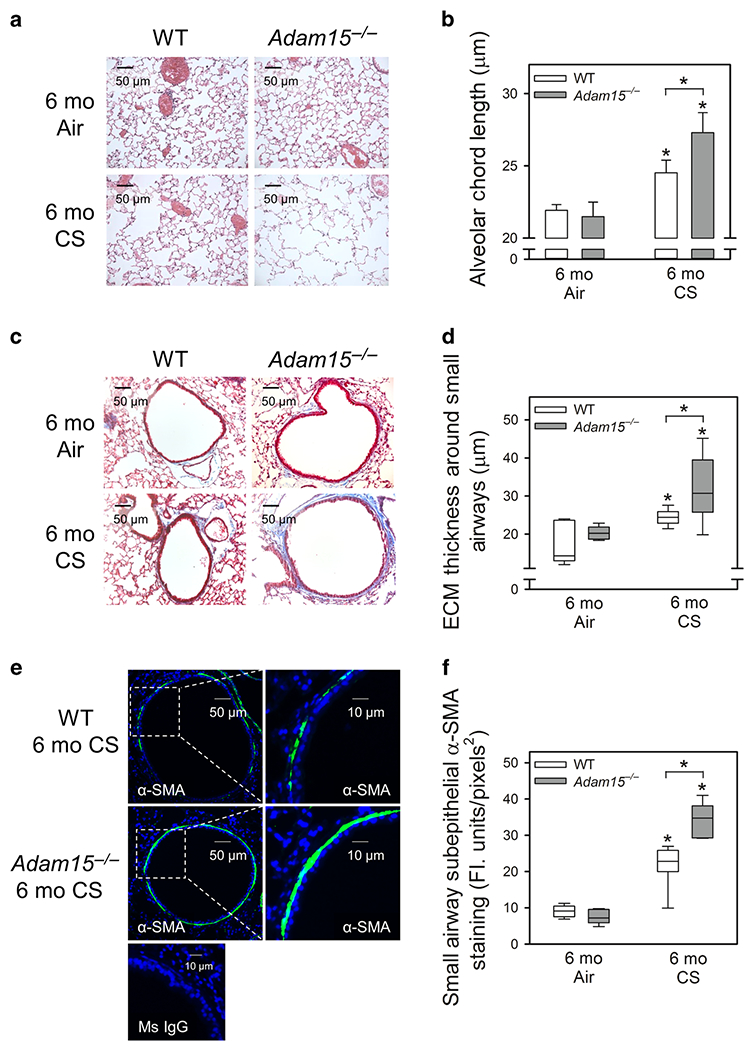
Adult Adam15−/− mice and WT mice were exposed to air or CS 6 days-a-week for 6 months (Mo). A: Images of hematoxylin and eosin-stained inflated lung sections from mice. B: Airspace size was quantified in Gills-stained lung sections using Scion image analysis software, as described in the Methods. The mean chord length is the average of the horizontal and vertical distances between the alveolar walls. Data are mean ± SD; n = 8-11 mice exposed to air and 11-14 mice exposed to CS. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P ≤ 0.035 versus the same genotype exposed to air or the group indicated. C shows representative images of extracellular matrix (ECM) deposition around small airways (300-699 μm in diameter) in Masson Trichrome-stained lung sections from WT and Adam15−/− mice exposed to air or CS for 6 months. D: The thickness of the ECM protein layer deposited around small airways was quantified using morphometry methods, as described in the Methods. The boxes in the box-plots show the medians and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles; n = 7-10 mice/group. Data were analyzed using a Kruskal-Wallis One-Way ANOVA followed by pair-wise testing with Mann-Whitney U tests. *, P ≤ 0.038 versus the same genotype exposed to air or the group indicated. In E, murine lung sections were immunostained for α-smooth muscle actin (α-SMA, a marker of myofibroblasts or smooth muscle cells) around small airways of WT and Adam15−/− mice that were exposed to air or CS for 6 months. Nuclei were counterstained blue with 4′,6-diamidino-2-phenylindole. As a control, lung sections were stained with a non-immune murine (Ms) IgG (bottom panel). The merged images shown in E are representative of 5-7 mice/group. F: Box-plots of α-SMA staining that was quantified per unit area of the walls of small airways having a luminal diameter of 300-699 μm using MetaMorph software (5 air-exposed mice/group and 6-7 CS-exposed mice/group). The boxes in the box-plots show the medians and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles. Data were analyzed using a Kruskal-Wallis One-Way ANOVA followed by pair-wise testing with Mann-Whitney U tests. *, P ≤ 0.005 versus air-exposed mice belonging to the same genotype or the group indicated.
Inhaling CS induces a chronic pulmonary inflammatory response in mice and humans which promotes airspace enlargement2,15,19. Thus, we evaluated whether the exaggerated airspace enlargement observed in CS-exposed Adam15−/− mice is associated with increased pulmonary inflammation. CS-exposed Adam15−/− mice had greater increases in bronchoalveolar lavage (BAL) total leukocyte, macrophage, and lymphocyte counts than CS-exposed WT mice (Fig. 3A–3C). Adam15−/− mice had lower increases in BAL PMN counts than WT mice after 6 months CS exposure (Fig. E3A). Mean total blood leukocyte and leukocyte subset counts were similar in air- and CS-exposed Adam15−/− and WT mice (Table E1).
Figure 3: Deficiency of Adam15 in mice increases CS-induced lung inflammation.
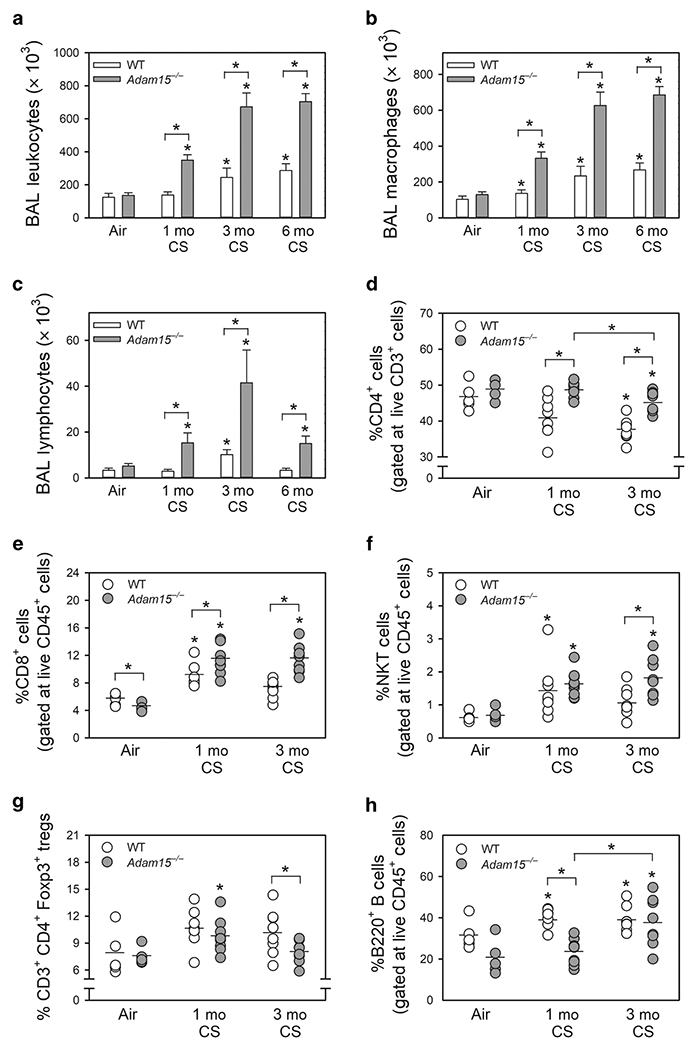
Wild-type (WT) and Adam15−/− mice were exposed to air or CS for 1 month (Mo), 3 months or 6 months. Absolute numbers of all leukocytes (A), macrophages (B), and lymphocytes (C) were counted in bronchoalveolar lavage (BAL) samples. Data are mean ± SEM; n = 12-14 mice exposed to air and 6-11 mice exposed to CS. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P ≤ 0.005 versus the same genotype exposed to air or the group indicated. In D-H, WT and Adam15−/− mice were exposed to air or CS for 1 or 3 months, lungs were enzymatically digested, and leukocytes were immunostained for markers of lymphocyte subsets including CD4+ lymphocytes (D), CD8+ lymphocytes (E), CD3-positive NKT cells (F), Tregs (G), and B cells (H). Lymphocyte subsets were quantified using flow cytometry. Data are mean ± SD; n = 5 mice exposed to air and 8-9 mice exposed to CS. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 air-exposed mice belonging to the same genotype or the group indicated.
When exposed to CS for 1 and/or 3 months, Adam15−/− mice had a higher percentage of CD4+ lymphocytes (Fig. 3D), CD8+ lymphocytes (Fig. 3E), and CD3-positive NKT cells (Fig. 3F) but a lower percentage of lung CD3+CD4+Foxp3+ T regulatory cells (Tregs; Fig. 3G) in enzymatic lung digest samples than WT mice. The percentage of B cells in lung digest samples was lower in Adam15−/− mice than WT mice after 1 month of CS exposure, but similar after 3 months of CS exposure (Fig. 3H). Adam15−/− mice had a lower percentage of CD3-negative natural killer (NK) cells in enzymatic lung digest samples when exposed to air or CS for up to 3 months than WT mice (Fig. E3B). Flow cytometry dot plots are shown in Fig. E4.
Lung Ccl-2, Cxcl-10, Gm-csf, and Mmp-12 levels were higher in CS-exposed Adam15−/− versus WT mice (Fig. 4A–4D). Lung Il-10, Il-6, and Il-1β levels were lower in CS-exposed Adam15−/− versus WT mice (Fig. 4E–4G). Lung Tgf-β, Tnf-α, Ccl-5, M-csf, and Ccl-3 levels were similar in CS-exposed WT and Adam15−/− mice (Fig. E5). CS exposure increased lung levels of granzyme A (a CD8+ T cell product which injures pulmonary epithelial cells16) in Adam15 but not WT mice (Fig. 4H).
Figure 4: Deficiency of Adam15 in mice increases lung levels of mediators that promote CS-induced lung inflammation and destruction.
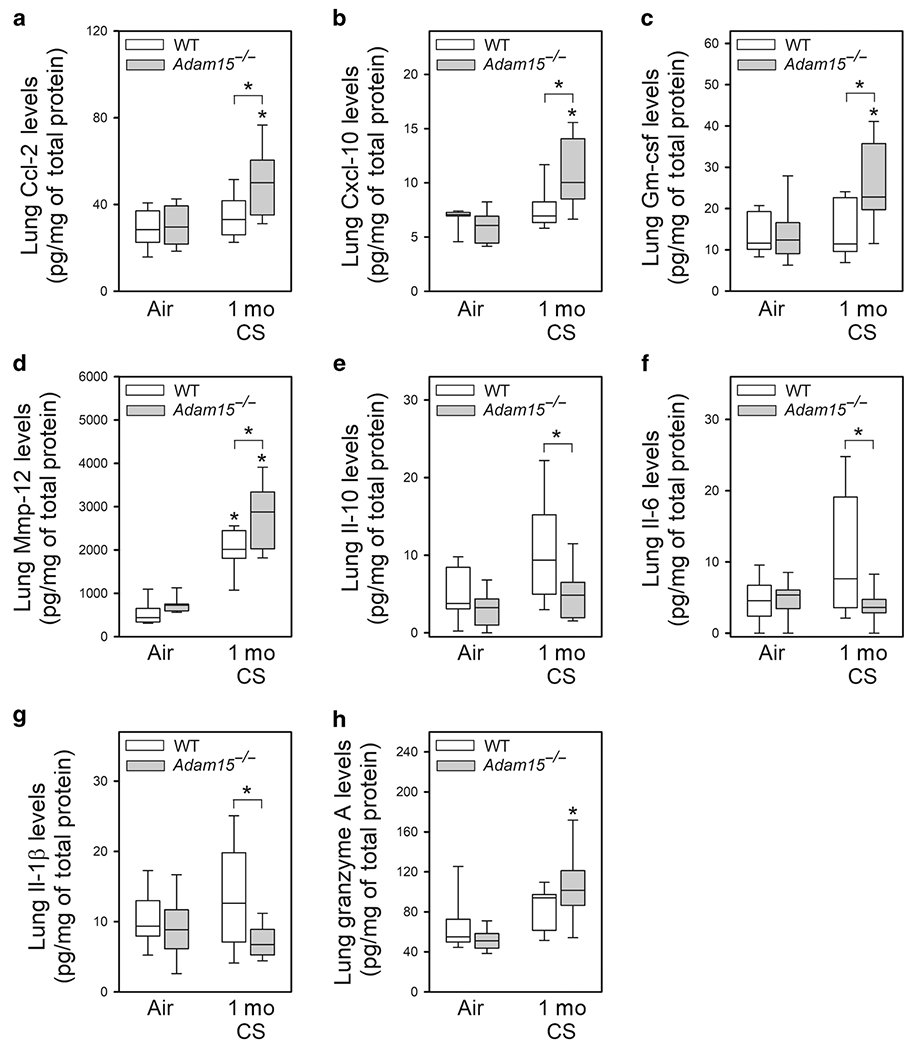
WT and Adam15−/− mice were exposed to air or CS for 1 month (Mo), lung levels of Ccl-2 (A; n = 10-12 mice/group), Cxcl-10 (B; n = 7 mice/group), Gm-csf (C; n = 10-12 mice/group), Mmp-12 (D; n = 8 mice/group), Il-10 (E; n = 10-12 mice/group), Il-6 (F; n = 14-22 mice/group), Il-1β (G; n = 13-22 mice/group) and granzyme A (H; n = 8 mice/group) were measured in homogenates of lung samples using commercial ELISA kits. Data were normalized to total protein levels measured in the same samples. The boxes in the box-plots show the medians and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles. Data were analyzed using a Kruskal-Wallis One-Way ANOVA followed by pair-wise testing with Mann-Whitney U tests. *, P ≤ 0.025 versus air-exposed mice belonging to the same genotype or the group indicated.
Adam15 deficiency in leukocytes leads to exaggerated pulmonary inflammation and COPD-like disease:
Adam15 is expressed by leukocytes and lung parenchymal cells. Thus, Adam15 bone marrow (BM) chimeras were generated and exposed to air or CS for 6 months to identify the Adam15-deficient cells that drive the greater CS-induced COPD-like disease. Adam15 deficiency in leukocytes (not lung parenchymal cells) increased CS-induced emphysema development (Fig. 5A), small airway fibrosis (Fig. 5B) and lung inflammation as: 1) WT recipients transplanted with Adam15−/− BM had greater CS-induced emphysema, small airway fibrosis, and lung macrophage counts (Fig. 5C) than WT recipients transplanted with WT BM; and 2) Adam15−/− recipients transplanted with WT BM had reduced CS-induced emphysema development, small airway fibrosis, and macrophage counts than Adam15−/− recipients transplanted with Adam15−/− BM (Fig. 5A–5C).
Figure 5: Deficiency of Adam15 in bone marrow (BM)-derived leukocytes drives the exaggerated COPD-like disease detected in CS-exposed Adam15−/− mice.
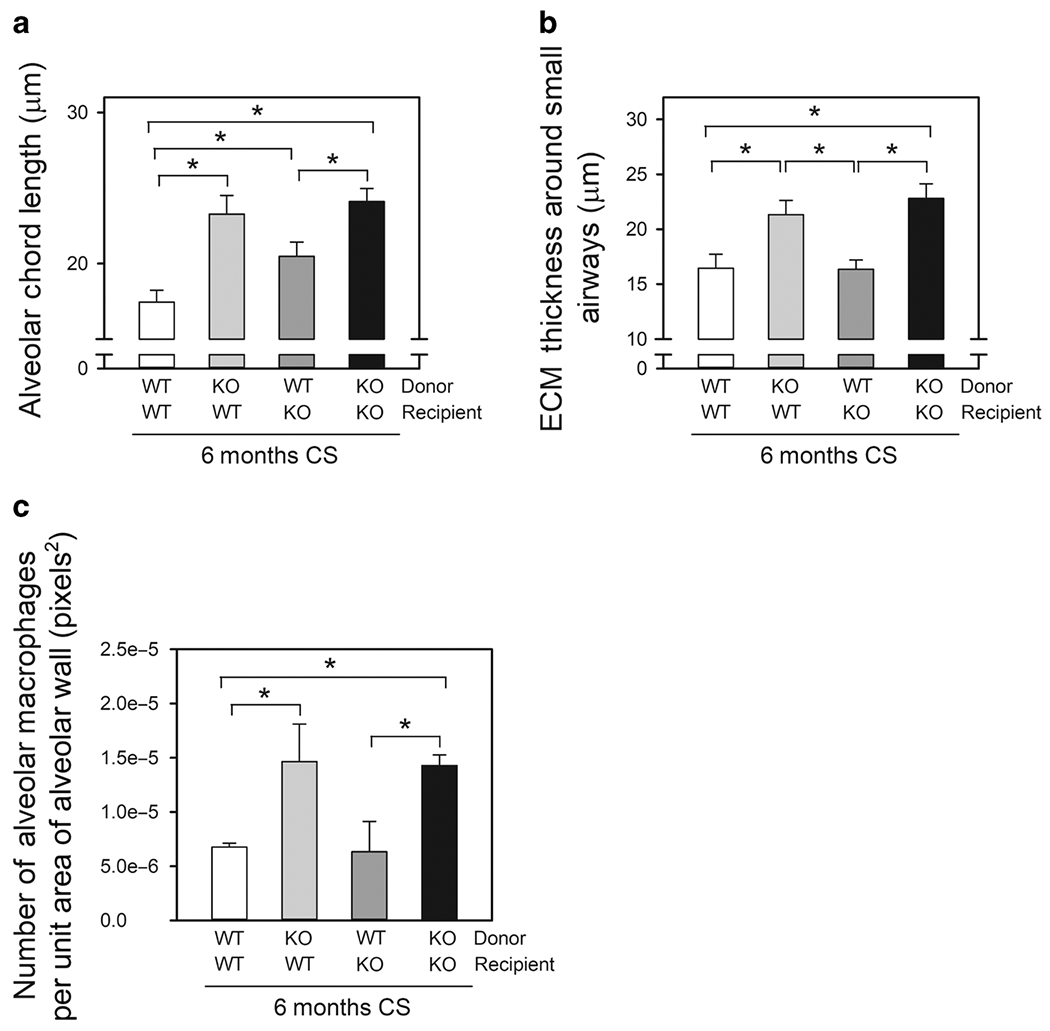
In A-C, 4 groups of Adam15 BM chimeric mice (WT BM into WT recipients [WT WT], Adam15−/− BM into WT recipients [KO WT], WT BM into Adam15−/− recipients [WT KO], and Adam15−/− BM into Adam15−/− recipients [KO KO]) were exposed to air or CS for 6 months. Panel A shows the mean alveolar chord lengths measured in the CS-exposed mice. The mean chord length in air-exposed mice was 14 to 19 μm in all groups (data not shown). Data are mean ± SEM; n = 4-6 mice/group. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 versus the group indicated. In B, the thickness of the ECM protein layer deposited around small airways was quantified in microns, as described in Methods. Data are mean ± SEM; n = 5-8 mice/group. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P ≤ 0.022 versus the group indicated. In C, the number of alveolar macrophages in lung sections normalized to alveolar wall area was measured, as described in the Methods. Data are mean ± SEM; n = 5-6 mice/group. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 versus the group indicated.
As Cd8−/− mice are protected from CS-induced accumulation of macrophages in the lung and emphysema development15, Adam15−/−xCd8−/− compound mutant mice were generated and exposed to air or CS for up to 6 months. Adam15 deficiency in CD8+ T cells was crucial for driving the enhanced pulmonary inflammation and emphysema development in Adam15−/− mice as CS-exposed Adam15−/−xCd8−/− mice had lower BAL total leukocyte, macrophage, and lymphocyte counts and reduced emphysema development than CS-exposed Adam15−/− or WT mice (Fig. 6).
Figure 6: Deficiency of Adam15 in CD8+ T cells is crucial for driving the exaggerated lung inflammation and destruction detected in CS-exposed Adam15−/− mice.
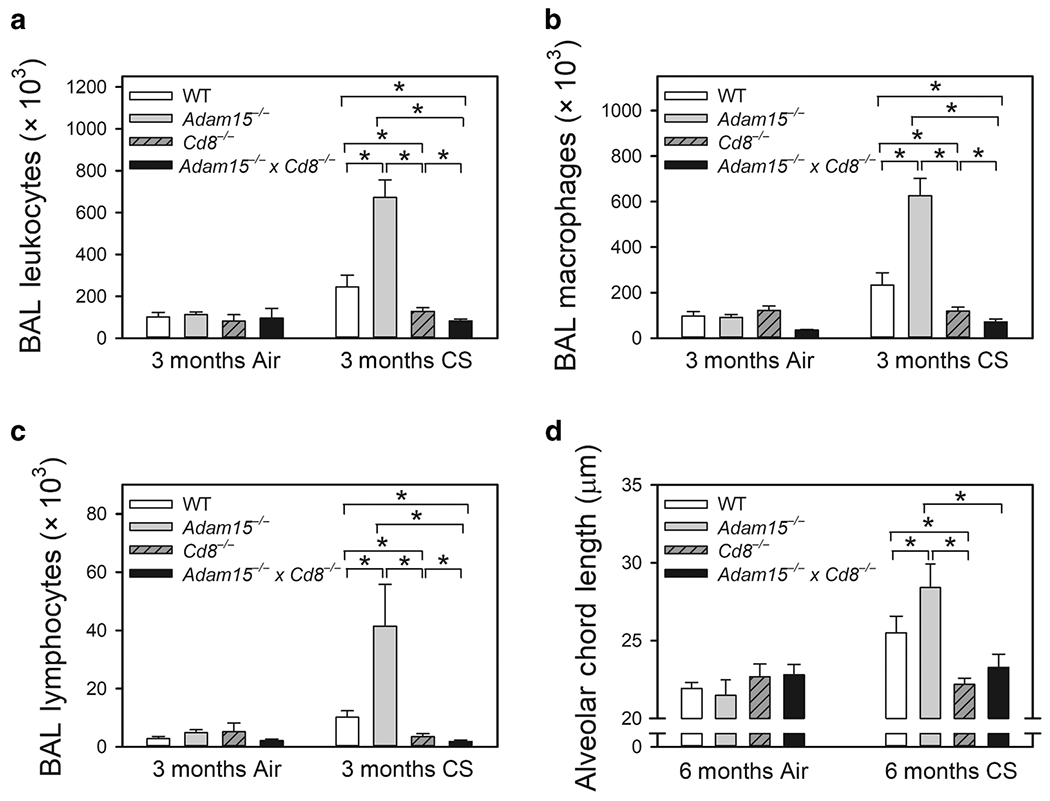
Adult WT mice, Adam15 mice, Cd8−/− mice and Adam15−/− x Cd8−/− compound mutant mice were exposed to air or CS 6 days-a-week for 3 months or 6 months. Absolute numbers of all leukocytes (A), macrophages (B), and lymphocytes (C) were counted in bronchoalveolar lavage (BAL) samples. Data are mean ± SEM; n = 7-17 mice exposed to air for 3 months air and 6-11 mice exposed to CS for 3 months. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 versus the group indicated. D: alveolar chord length was measured on lung sections as a measure of airspace enlargement. Data are mean ± SEM; n = 5-11 mice exposed to air for 6 months and 10-16 mice exposed to CS for 6 months. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 versus the group indicated.
Adam15 is not required for CD8+ T cell or macrophage adhesion or migration:
ADAM15 increases glomerular mesangial cell migration and cell-cell interactions of a fibroblast cell line12,14. Whether Adam15 regulates the adhesion of migration of leukocytes is unknown. WT and Adam15−/− macrophages and CD8+ T cells did not differ in their capacity to adhere to an ECM protein (Fig. E6) or migrate towards chemoattractants (Fig. E7) in vitro. Thus, it is unlikely that Adam15 deficiency increases CS-induced pulmonary inflammation by increasing the trans-endothelial migration of monocytes or CD8+ T cells into the lung.
Adam15 deficiency in CD8+ T cells and macrophages abrogates CS-induced activation of the mitochondrial apoptosis pathway to increase cell survival by reducing CS-induced inactivation of mTOR signaling:
CS-induced apoptosis of leukocytes regulates leukocyte accumulation in the lung2. Compared with CS-exposed WT mice, CS-exposed Adam15−/− mice had reduced rates of apoptosis in their alveolar macrophages (AMs) and CD8+ T cells (assessed as lower intracellular active caspase-3 levels; Fig. 7A and 7B, respectively) and reduced CS-induced activation of the mitochondrial apoptosis pathway in their AMs (as assessed by a lower percentage of cells that did not stain with the mitochondrial dye, JC-1 indicative of loss of mitochondrial membrane potential; Fig. 7C). CSE-treated macrophages and CD8+ T cells isolated from naive Adam15−/− mice exhibited reduced activation of the mitochondrial apoptosis pathway in vitro (Fig. 7D–7E).
Figure 7: Deficiency of Adam15 in CD8+ T cells and macrophages protects the cells from CS-induced activation of the mitochondrial apoptosis pathway in vivo and in vitro.
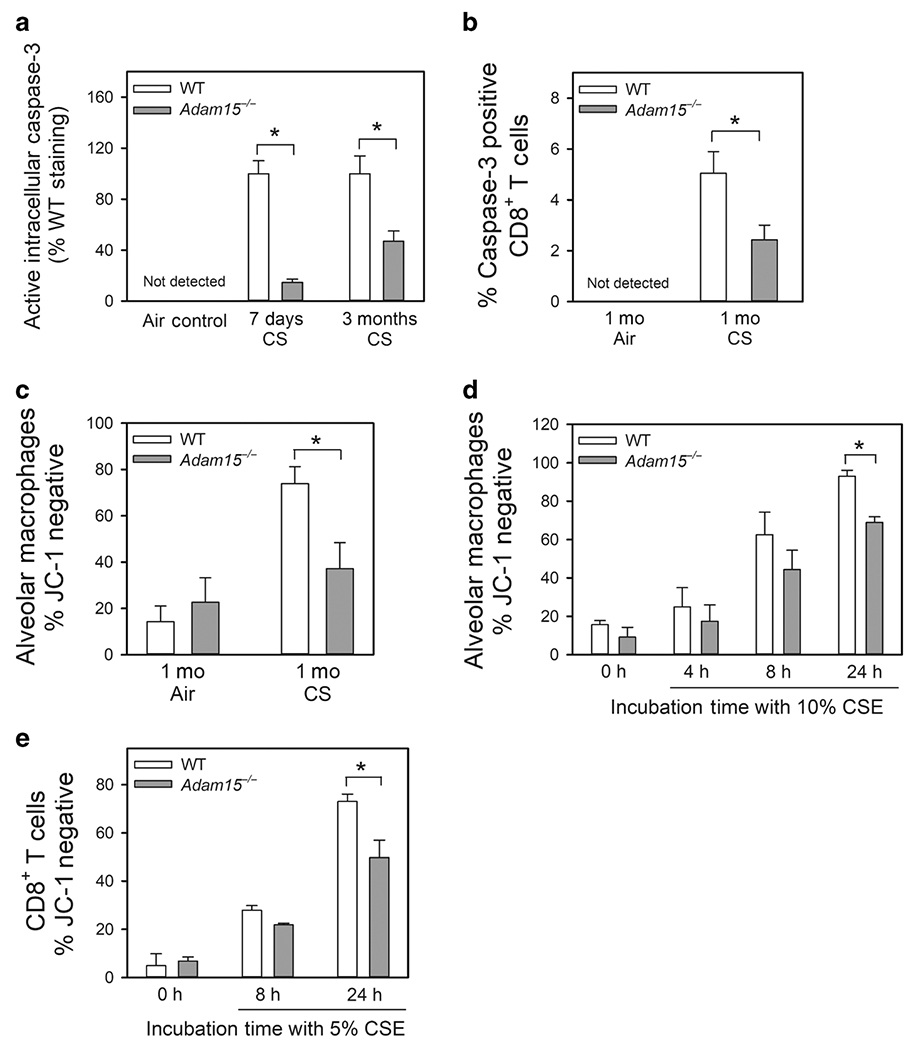
A: Alveolar macrophages (AMs) were isolated from the lungs of WT and Adam15−/− mice that had been exposed to air or CS for 1 week or 3 months (Mo) using BAL. B: CD8+ T cells were isolated from the lungs of WT and Adam15−/− mice exposed to air or CS for 1 month as described in Methods. In A and B, immediately after the AMs and CD8+ T cells were isolated they were fixed and then immunostained for intracellular active caspase-3. Staining was quantified, as described in the Methods. Active caspase-3 was not detected in macrophages or CD8+ T cells isolated from air-exposed WT or Adam15−/− mice. Data are mean ± SEM; n = 5 mice/group in A, and n = 4-5 mice/group in B. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 versus the group indicated. C: AMs were isolated from the lungs of WT and Adam15−/− mice exposed to air or CS for 1 month using BAL. Activation of the mitochondrial apoptosis pathway was measured as loss of mitochondrial membrane potential (lack of staining of the cells with the mitochondrial dye, JC-1 which stains only viable mitochondria). Data are mean ± SEM; n = 5 mice/group. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 versus the group indicated. D: AMs isolated from naïve WT and Adam15−/− mice were incubated at 37°C for up to 24 h with or without 10% CS extract (CSE). E: CD8+ T cells isolated from spleen of naïve WT Adam15−/− mice were incubated at 37°C for up to 24 h with or without 5% CSE. In D and E, activation of the mitochondrial apoptosis pathway was measured as loss of mitochondrial membrane potential (lack of staining of the cells with JC-1). Data are mean ± SEM; n = 3 separate experiments in D and E. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.05 versus the group indicated.
The PI3 kinase-Akt-mTOR pathway inhibits activation of the mitochondrial apoptosis pathway by increasing the expression of anti-apoptotic Bcl-2 family members, especially intracellular myeloid cell leukemia sequence-1 (Mcl-1) in macrophages and T cells20,21. Translocation of Bcl-2 proteins to the mitochondrial membrane prevents mitochondrial membrane depolarization and cytochrome C release22. The mTOR-Mcl-1 pathway pro-survival pathway was measured in CD8+ T cells isolated from the lungs mice that had been exposed to CS for 3 months, or in macrophages isolated from unchallenged mice and exposed to CSE in vitro. CD8+ T cells isolated from the lungs of Adam15−/− mice had greater intracellular levels of phosphorylated ribosomal protein S6 (P-S6; a marker of mTOR signaling; Fig. 8A–8B) and higher intracellular levels of Mcl-1 (Fig. 8C–8D). CSE exposure substantially reduced intracellular levels of P-S6 (Fig. 8E) and Mcl-1 in WT but not Adam15−/− macrophages in vitro (Fig. 8F–8G). Thus, Adam15 deficiency promotes the survival of CD8+ T cells and macrophages in the CS-exposed lungs by rendering these cells resistant to CS-triggered activation of the mitochondrial apoptosis pathway by preserving their mTOR-Mcl-1 signaling when they are exposed to CS.
Figure 8: Deficiency of Adam15 in macrophages and CD8+ T cells presences activation of the mTOR-Mcl-1 pro-survival pathway when cells are exposed to CS.
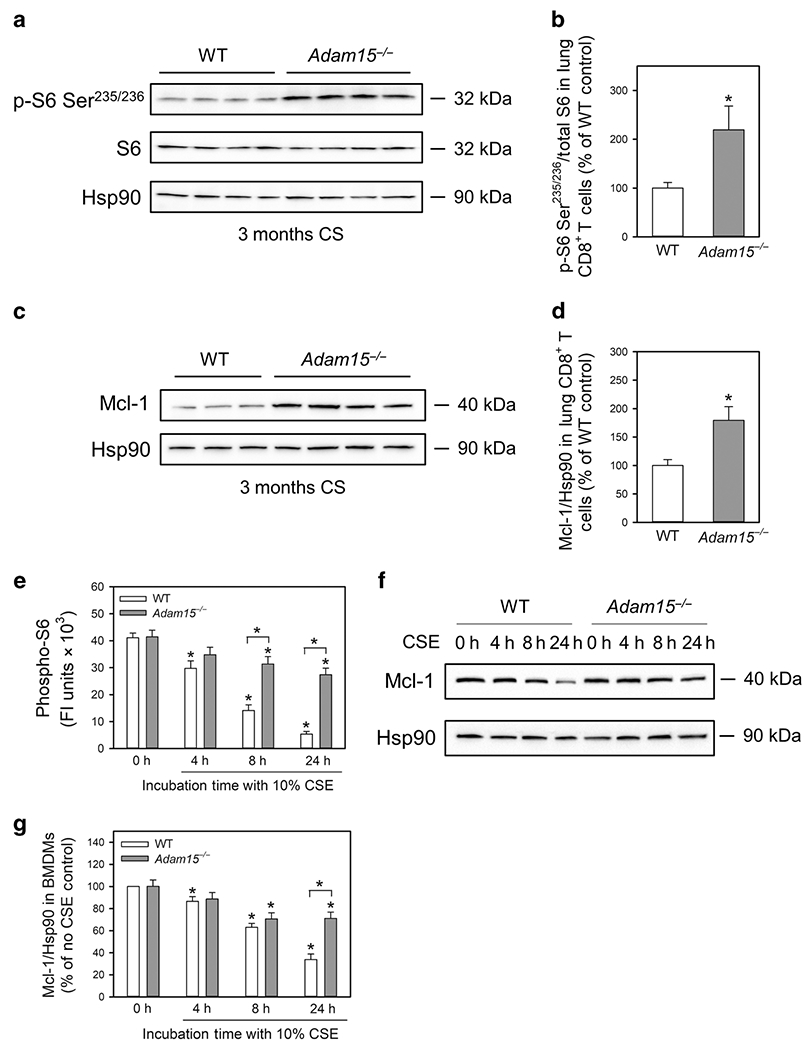
In A-D, CD8+ T cells were isolated from the lungs of WT and Adam15−/− mice that had been exposed to CS for 3 months. In A, phosphorylated S6 (p-S6-Ser235/236), total S6 (S6), and a housekeeping control (heat shock protein-90; Hsp90) were quantified in extracts of the pulmonary CD8+ T cells using Western blotting and densitometry. The p-S6-Ser235/236 signals were normalized to total S6 levels and expressed as a % of the values for WT mice. The images shown in A are representative of 4 mice per group. B: The bars show means ± SD (n = 4 mice/group). Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Students t-tests. *, P ≤ 0.01 versus the WT control. In C-D, intracellular protein levels of Mcl-1 and Hsp90 were quantified in pulmonary CD8+ T cells from CS-exposed mice using Western blotting and densitometry. The Mcl-1 signals were normalized to Hsp90 levels and expressed as a % of the values for WT mice. The images shown in C are representative of 3-4 mice per group. D: The bars show means ± SD (n = 3-4 mice/group). Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Students t-tests. *, P ≤ 0.004 versus the WT control. In E-G, bone marrow-derived macrophages (BMDMs) were isolated from unchallenged WT and Adam15−/− mice and exposed to 10% CSE for up to 24 h. In E, BMDMs were fixed, permeabilized, and immunostained with a green fluorophore for intracellular phosphorylated ribosomal protein S6 (p-S6), and the nuclei were counterstained blue with 4′,6-diamidino-2-phenylindole. Control cells were stained with a primary non-immune rabbit (Rb) IgG (right panels). P-S6 staining in BMDMs was quantified, as described in the Methods. Data are mean ± SD and data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Students t-tests. *, P ≤ 0.01 versus the control. In F-G, intracellular protein levels of Mcl-1 and Hsp90 were quantified in CSE-treated BMDMs using Western blotting and densitometry. The images shown in G are representative of 7 mice/group. In G, the Mcl-1 signals were normalized to Hsp90 levels and expressed as a % of the values for controls cells incubated without CSE. Data are mean ± SD; n = 7 mice/group. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P ≤ 0.004 versus no CSE control belonging to the same genotype or the group indicated.
ADAM15 over-expression reduces CSE-induced activation of the mitochondrial apoptosis pathway in monocyte/macrophage-like cells:
ADAM15 was over-expressed in human differentiated THP-1 (monocyte-macrophage-like) cells using a vector containing an ADAM15 complementary DNA (cDNA) and electroporation. Robust ADAM15 expression was detected using real-time quantitative PCR in THP-1 cells 24 h after electroporation but not in cells treated with the control vector (Fig. 9A). THP-1 cells over-expressing ADAM15 had increased CSE-induced activation of the mitochondrial apoptosis pathway (as assessed by a higher percentage of cells that did not stain with JC-1; Fig. 9B) and reduced intracellular MCL-1 protein levels compared with cells treated with the control vector (Fig. 9C–9D). Thus, ADAM15 overexpression promotes CSE-induced inactivation of mTOR-Mcl-1 signaling to increase the mitochondrial apoptosis pathway in monocyte/macrophage-like cells.
Figure 9: Overexpression of ADAM15 in human THP-1 cells promotes CSE-induced apoptosis and reductions in intracellular Mcl-1 protein levels.
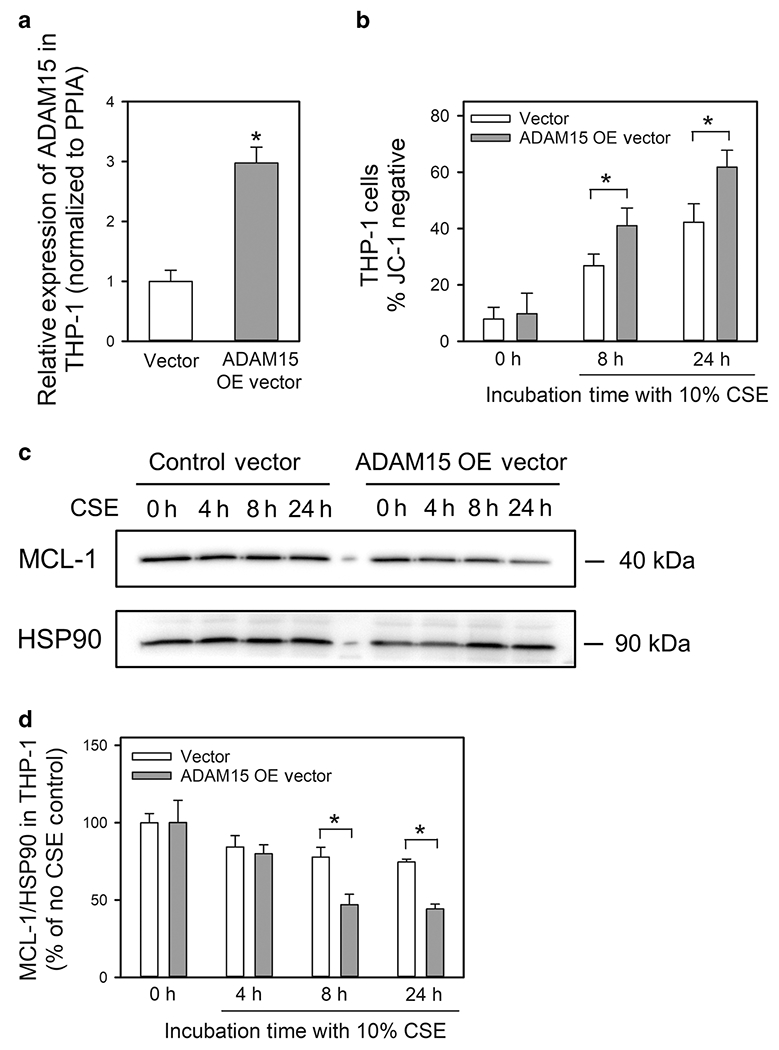
In A, ADAM15 steady state mRNA levels were measured in total cellular RNA samples from differentiated THP-1 24 h after electroporation of an ADAM15 over-expression [OE] vector (pcDNA3.1-ADAM15, a plasmid containing an ADAM15 complementary DNA) or a control vector (Vector) using quantitative real-time PCR. In B-D, differentiated THP-1 cells were electroporated with either the ADAM15 OE vector or the control vector and then incubated with 10% CSE for the time indicated. In B, loss of mitochondrial membrane potential was measured by staining the cells with JC-1 dye and quantifying the staining using MetaMorph software. In C-D, intracellular protein levels of myeloid cell leukemia-1 (MCL-1) and heat shock protein-90 (HSP90; a housekeeping control) were quantified in CSE-treated THP-1 cells using Western blotting and densitometry. The MCL-1 signals were normalized to HSP90 levels and expressed as a % of the values for controls cells incubated without CSE. Data are mean ± SD. Data were analyzed using a One-Way ANOVA followed by pair-wise testing with two-tailed Student’s t-tests. *, P < 0.03 versus the group indicated.
Discussion
This study links Adam15 to the pathogenesis of COPD-like disease, leukocyte function, and the mTOR pathway in CS-exposed mice for the first time. Acute CS exposure increased lung Adam15 transcript and protein levels in WT mice, and Adam15 was localized to pulmonary macrophages, CD8+ T cells, epithelial cells, and airway α-ASM-positive cells in CS-exposed WT mice. When exposed to CS for 6 months, Adam15−/− mice developed exaggerated pulmonary inflammation, emphysema development, and small airway fibrosis compared with WT mice. Adam15 deficiency in BM-derived leukocytes (especially CD8+ T cells) was crucial for driving the exaggerated COPD-like disease in CS-exposed Adam15−/− mice. Adam15 deficiency increased CD8+ T cell and macrophage counts in the CS-exposed murine lung by increasing the resistance of these cells to CS-induced activation of the mitochondrial apoptosis pathway thereby increasing the survival of these cells in the lung. Adam15 deficiency reduced activation of the mitochondrial apoptosis pathway in pulmonary CD8+ T cells and macrophages by abrogating CS-induced inactivation of mTOR signaling in CD8+ T cells and macrophages. The greater mTOR signaling in CS-exposed CD8+ T cells and macrophages leads to higher intracellular levels of Mcl-1 (a Bcl-2 pro-survival protein) which translocates to the mitochondrial membrane to reduce loss of mitochondrial membrane potential, downstream caspase-3 activation, and apoptosis [Fig. 10]. Conversely, ADAM15 over-expression in THP-1 cells increases CSE-induced inactivation of mTOR-MCL-1 signaling and promotes CSE-induced activation of the mitochondrial apoptosis pathway.
Figure 10: cartoon illustrating the mechanisms by which deficiency of Adam15 leads to exaggerated COPD in mice.
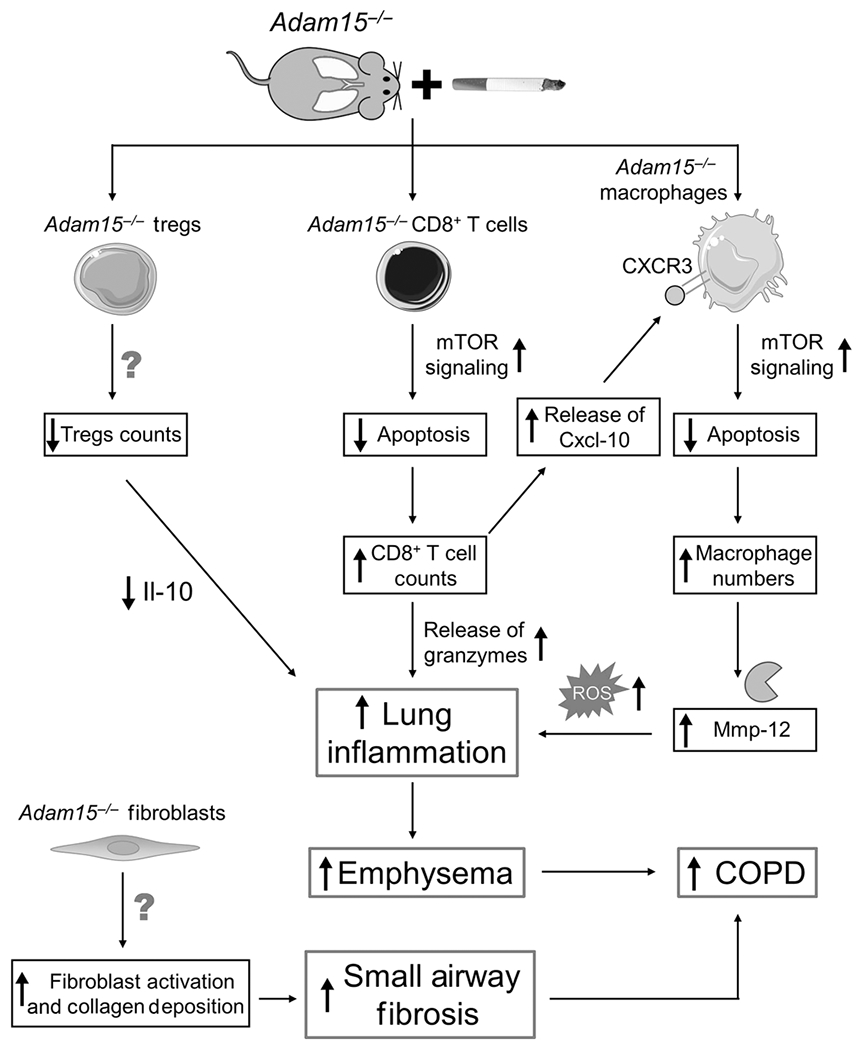
Exposing Adam15−/− mice to CS leads to a phlogistic pulmonary response characterized by increases in the number of innate and adaptive immune cells (alveolar macrophages [AMs], CD4+ lymphocytes, CD8+ lymphocytes and CD3-positive NKT cells) and reductions in leukocytes having anti-inflammatory activities (Foxp3+ T regulatory cells) in the lung, along with exaggerated emphysema development and small airway fibrosis. The results of studies of Adam15 bone marrow chimeras and Adam15−/− X Cd8−/− mice indicate that deficiency of Adam15 in leukocytes (especially CD8+ T cells) drives the increased emphysema development and small airway fibrosis observed in CS-exposed Adam15−/− mice. Likely, increased release of Mmp-12 and oxidants from the increased numbers of macrophages and the increased release of granzymes from the increased numbers of CD8+ T cells detected in the lungs of CS-exposed Adam15−/− mice contributed to their increased emphysema development. The exaggerated small airway fibrosis in CS-exposed Adam15−/− mice may be due to: 1) increased release of mediators from leukocytes and other cells that activate (myo)fibroblasts; and 2) Adam15 deficiency in (myo)fibroblasts increasing the survival and/or profibrotic activities of these cells in the small airways. Adam15 deficiency increases CS-induced pulmonary inflammation by several mechanisms. First, Adam15−/− CD8+ T cells (and macrophages) are protected from CS-induced activation of the mitochondrial apoptosis pathway and consequently accumulate in, and injure, the lung. Adam15 deficiency preserves mammalian target of rapamycin (mTOR) signaling in CD8+ T cells (and macrophages), leading to preserved expression of myeloid cell leukemia sequence-1 (Mcl-1) protein (an anti-apoptotic protein which binds to the mitochondrial membrane to prevent loss of mitochondrial membrane potential, mitochondrial release of cytochrome c and downstream caspase activation). The increased number of CD8+ T cells in lungs of Adam15−/− mice also promotes the survival of macrophages in the lung via Adam15−/− CD8+ T cells releasing increased quantities of Cxcl-10 (and possibly other ligands for Cxcr3) which bind to Cxcr3 on Adam15−/− macrophages to increase mTOR signaling in macrophages leading to increased macrophage survival and activation and release of Mmp-12, oxidants and other mediators that promote lung inflammation and destruction. Second, the accumulation of leukocytes leads to increased release of pro-inflammatory mediators that amplify pulmonary inflammation and destruction in Adam15−/− lungs. Third, lung levels of anti-inflammatory mediators, such as interleukin-10 (Il-10) are reduced in CS-exposed Adam15−/− lung, which may be due to the reduced numbers of Foxp3+ T regulatory cells (which are a source of Il-10) in their lungs. A reduced anti-inflammatory immune response likely contributes to the phlogistic environment that increases emphysema development and small airway fibrosis in CS-exposed Adam15−/− lungs.
ADAM15 has not been studied previously in COPD. ADAM15 gene expression is upregulated macrophages and fibroblasts when they are activated9. Although ADAM15 was reported not to be expressed by CD8+ T cells in healthy subjects23, we report increased Adam15 staining in CD8+ T cells in CS-exposed WT lungs and on the surface of CSE-activated murine CD8+ T cells in vitro.
The increased Adam15 gene expression in the lungs of WT mice that were exposed acutely to CS could be mediated by the increased lung levels of pro-inflammatory mediators present in their lungs. (Epi)genetic processes could also increase Adam15 expression in CS-exposed murine lungs. The murine Adam15 promoter contains three Sp1-binding sites24. CS increases Sp1 binding to the promoter regions of other genes to activate their transcription25. Whether Sp1 binding to the Adam15 promoter contributes to the increased Adam15 expression observed in CS-exposed WT murine lungs mice is unclear. Single-nucleotide polymorphisms in the ADAM15 locus are linked to rheumatoid arthritis26, but to our knowledge have not linked to human COPD. MicroRNAs (miRNAs) regulate gene transcription in human and murine COPD27. MiR-147b binds the 3′ untranslated region of the ADAM15 mRNA in human endothelial cells to reduce ADAM15 expression in these cells28. Whether other miRNAs increase Adam15 expression in CS-exposed murine lungs will be investigated in our future studies.
CSE increased ADAM15 surface levels on human monocytes and CD8+ T cells and ADAM15 transcript levels in HBECs in vitro indicating that the expression data obtained from murine cells are translatable to humans. In publicly-available Gene Expression Omnibus (GEO) microarray and RNA sequencing data-sets, ADAM15 transcript levels were higher in bronchial epithelial cells from COPD patients versus non-smokers and smokers but were not related to GOLD stage (GSE5058 and GSE 63127; data not shown). ADAM15 transcript levels in AMs were higher in smokers versus non-smokers, but not increased in AMs from COPD patients (GSE130928; data not shown).
CS-exposed Adam15−/− mice developed exaggerated COPD-like disease suggesting that Adam15 has protective activities in lungs exposed chronically to CS. However, WT mice exposed to CS for up to 2 months had increased lung levels of Adam15. Heme oxygenase-1 (HO-1) is a cyto-protective protein which is upregulated during various acute insults in humans and mice29,30 including hypoxia- and ischemia-reperfusion (IR)-induced tissue injury. However, HO-1−/− mice have exaggerated disease compared with WT mice in these injury models29,30. While 2 months of CS exposure increased lung Adam15 protein levels in WT mice 7-fold, 6 month CS exposures did not further increase Adam15 lung levels. Thus, increases in Adam15 lung levels induced by acute CS exposures may represent an acute cyto-protective stress response similar to that observed in HO-1−/− mice with acute hypoxia- or ischemia-reperfusion (IR)-induced tissue injury31,32. We hypothesize that the failure to upregulate Adam15 lung levels further during more chronic CS exposures contributes to inadequate protection of the lungs of WT mice from the deleterious effects of CS. The absence of Adam15 in the lungs of Adam15−/− mice likely sensitizes them to the development of CS-induced COPD-like disease.
Studies of Adam15 BM chimeras and Adam15−/−xCd8−/− mice revealed that Adam15 deficiency in leukocytes (especially CD8+ T cells) was responsible for the exaggerated pulmonary inflammation and COPD-like disease in CS-exposed Adam15−/− mice. Macrophages are required for CS-induced emphysema development in mice19,33 as their products (e.g. Mmp-12 and oxidants) injure the alveolar walls34. However, CD8+ T cells are required for macrophage accumulation and activation in the CS-exposed lung15 (Fig. 10). CD8+ T cells release ligands (e.g. Cxcl-10) for Cxcr3 expressed on macrophages which induce macrophage activation and Mmp-12 release15,16. CD8+ T cells also induce alveolar epithelial cell apoptosis by releasing perforin and granzymes15. It is noteworthy that CS-exposed Adam15−/− mice had elevated lung levels of Cxcl-10, granzyme A, and Mmp-12. CS-exposed Adam15−/− mice also had reduced Treg counts in their lungs which may have contributed to their increased pulmonary inflammatory response to CS as Treg release anti-inflammatory molecules such as Il-1035, and COPD patients have a blunted Treg response to CS36.
Adam15 deficiency promotes pulmonary inflammation and emphysema development, in part, by reducing CS-induced activation of the mitochondrial apoptosis pathway in CD8+ T cells and macrophages thereby increasing their survival in the lung by abrogating CSE-induced reductions in mTOR signaling and downstream synthesis of Mcl-1 protein. Mcl-1 binds to and stabilizes the mitochondrial membrane thereby preventing CS-induced mitochondrial depolarization, cytochrome c release, and cellular apoptosis. Adam15 deficiency in macrophages rendered these cells resistant to CSE-induced apoptosis in vitro. However, studies of CS-exposed Adam15−/−xCd8−/− mice demonstrated that Adam15 deficiency in CD8+ T cells was required for the increased lung inflammation (including the increased lung macrophage counts) in CS-exposed Adam15−/− mice. CD8+ T cells release Cxcl10 which activates mTOR signaling in other leukocytes by binding to Cxcr3 expressed on leukocytes and lung Cxcl-10 levels were increased in CS-exposed Adam15−/− mice37. We hypothesize that the increased numbers of apoptosis-resistant CD8+ T cells in the lungs of CS-exposed Adam15−/− mice release increased quantities of ligands for Cxcr3 expressed by macrophages which, in turn, drives increased mTOR signaling (and survival) of macrophages in the lungs of Adam15−/− mice (Fig. 10). The mechanism by which Adam15 deficiency maintains mTOR signaling in CS-exposed CD8+ T cells and macrophages is unclear, but Adam15 has a cytoplasmic tail which binds to intracellular proteins including Src kinases which regulate the PI3 kinase-Akt pathway upstream of mTOR signaling38.
Adam15 deficiency may also increase macrophage counts in CS-exposed lungs by increasing lung levels of mediators that promote macrophage recruitment (Ccl-2) or survival (Gm-csf), or by decreasing lung levels of anti-inflammatory mediators including Il-10. The reduced lung Il-10 levels in CS-exposed Adam15−/− mice may be due to their reduced pulmonary Treg counts as Il-10 is a Treg product35. To our knowledge this is the first report linking deficiency of an Adam family member to Treg counts in any organ.
Adam15 deficiency increased CS-induced small airway fibrosis and the numbers of α-SMA-positive cells (which could be myofibroblasts or smooth muscle cells) in the small airways of mice. Adam15 expression was increased in small airway α-SMA-positive cells in CS-exposed WT mice, and human ADAM15 increases human fibroblast migration and survival14,39. Thus, Adam15 deficiency in myofibroblasts may promote exaggerated small airway fibrosis in CS-exposed Adam15−/− mice by increasing myofibroblast activation, survival, and/or ECM protein deposition. Although Tgf-β promotes small airway fibrosis in mice34, CS-exposed WT and Adam15−/− mice had similar lung Tgf-β levels. The greater CS-induced small airway fibrosis in Adam15 mice was linked to their greater lung inflammation by BM transplant experiments. It is noteworthy that macrophage products promote small airway remodeling in CS-exposed rodents40.
A limitation of this study is that we employed only a loss-of-function approach in mice. Some of our findings could be due to compensatory changes in lung levels of other proteinases in Adam15−/− mice. Small molecules inhibitors which are selective for Adam15 do not exist. However, our ADAM15 over-expression results in THP-1 cells support our hypothesis that ADAM15 has protective activities in CS-exposed lungs by limiting the survival of leukocytes in the lung. Future studies will employ gain-of-function approaches in CS-exposed mice to test the hypothesis that Adam15 has protective activities in the CS-exposed lung. We did not fully elucidate the mechanisms by which Adam15 regulates small airway fibrosis or the contributions of PMN-derived Adam15 to CS-induced COPD-like disease which will be explored in future studies. Our in vitro studies evaluated the effects of CSE on leukocytes and epithelial cells using CSE concentrations that were optimized for each cell type and readout. These studies only provide information on the effects of aqueous-soluble CS components, and it is not possible to relate the CSE concentrations that were tested to concentrations of gaseous-phase CS which contains other noxious components.
Our results suggest that strategies that increase lung levels of ADAM15 have therapeutic potential in COPD. However, increased expression of ADAM15 has been detected in various tumors including non-small cell lung cancer and is linked to tumor cell migration and metastasis by increasing MMP-9 expression41,42. Additional studies are needed to evaluate ADAM15 as a potential therapeutic target for COPD.
In conclusion, our results strongly link Adam15 deficiency to the pathogenesis of COPD-like disease in mice and mTOR signaling in pulmonary CD8+ T cells and macrophages in CS-exposed mice for the first time. In CS-exposed mice, Adam15 deficiency leads to exaggerated COPD, in part, by increasing the pulmonary inflammatory response to CS by protecting CD8+ T cells and macrophages from CS-induced activation of the mitochondrial apoptosis pathway by preserving mTOR-Mcl-1 signaling in these leukocytes.
Methods
Ethics
Studies conducted on mice were approved by the Institutional Care and Use Committee at Brigham and Women’s Hospital (BWH). Studies conducted on human subjects were approved by the BWH Institutional Review Board.
Mice:
Adam15 mice were obtained from Dr. CP Blobel (Cornell University, New York, NY) and backcrossed into a pure C57BL/6 strain (to the F8 generation). Adam15−/− mice have normal lung development and no abnormal phenotype in the unchallenged state18. Pure strain C57BL/6 WT mice and Cd8−/− mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Adam15−/−xCd8−/− mice were generated by breeding Adam15−/− mice and Cd8−/− mice. Genotypes were confirmed using PCR methods performed on DNA extracted from tail biopsies. Mice were housed under specific pathogen-free conditions until they were 8-12 weeks of age before experiments were conducted. Male and female were studied and randomized to the experimental groups by an observer not involved in the conduct of the experiments.
Adam15 bone marrow (BM) chimeric mice:
Unchallenged C57BL/6 WT and Adam15−/− recipient mice aged 10-12 weeks were lethally-irradiated (using 450 centigray dose from a 137Cesium source8,43,44 twice 4 h apart) and then received complete BM (erythrocytes and leukocytes) containing two million leukocytes from non-irradiated donor mice via a tail vein injection. Four groups of Adam15 BM chimeras were generated by transplanting: 1) WT BM into WT recipients; 2) WT BM into Adam15−/− recipients; 3) Adam15−/− BM into WT recipients; and 4) Adam15−/− BM into Adam15−/− recipients. Mice were house for 10 weeks to permit engraftment of donor BM and then 6 month air and CS exposures were initiated and lungs were removed (at ~46 weeks of age)8.
Cigarette smoke (CS) exposures:
Mice were exposed to air or mixed mainstream and side-stream CS from 3R4F Kentucky Research cigarettes for 2 h/day 6 days-a-week in Teague TE 10z chambers for 1-6 months45.
CS extract (CSE):
Smoke from two 3R4F cigarettes was bubbled through 20 ml of DMEM medium yielding a 100% CSE solution which was filter sterilized (0.2 μm syringe filter), diluted, and used within 5 min of preparation7,8. Pilot experiments optimized the CSE concentrations needed to induce Adam15 expression or apoptosis for each cell type studied.
Adam15 protein and mRNA levels:
Adam15 protein levels were measured in homogenates of WT lungs using an ELISA (CUSABIO, College Park, MD) and normalized to total protein levels measured in the same samples using a kit (Thermo-Fisher Scientific, Rockford, IL). Adam15 mRNA levels were measured using a SYBR green-based real-time PCR assay with Ppia as the housekeeping gene (Table E2) and the comparative threshold method7.
Immunostaining of alveolar macrophages (AMs) and CD8+ T cells for surface Adam15:
AMs were isolated from murine lungs using BAL. Macrophages were isolated from the peritoneal cavities of mice 4 days after intraperitoneal injection of 40 mg of thioglycollate in 1 ml of DMEM medium using peritoneal lavage. Macrophages were cultured in DMEM containing 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin for 48 h in 8 well Lab-Tek® Chamber slides™ to render them quiescent. Macrophages were then incubated at 37°C for 8-48 h with 0-5% CSE.
CD8+ lymphocytes were isolated from the spleens of unchallenged mice using EasySep™ Mouse CD8+ T Cell Isolation Kits (STEMCELL Technologies, Cambridge, MA). CD8+ T cells were incubated at 37°C for up to 2 h with 0-2.5% CSE. Cells were fixed for 3 min at 4°C in 3% paraformaldehyde and 0.25% glutaraldehyde in PBS, and immunostained for surface Adam15 using rat anti-Adam15 IgG or non-immune rat IgG followed by Alexa-488-conjugated goat anti-rat F(ab)2. Cell surface immunofluorescence was quantified on images of the cells using MetaMorph software (Molecular Devices LLC, Sunnyvale, CA)7,46.
ADAM15 mRNA levels in HBECs:
Normal HBECs (Lonza, Allendale, NJ) were incubated for 4-24 h with or without 2.5% CSE. Adam15 mRNA levels were measured using quantitative real-time-PCR using PPIA as the housekeeping gene (Table E2).
Immunostaining of human leukocytes for surface ADAM15:
Monocytes and CD8+ T cells were isolated from the blood of healthy donors by positive section for CD14 or CD8α using immunomagnetic beads (STEMCELL Technologies). Monocytes and CD8+ T cells were incubated with and without CSE and then immunostained for surface ADAM15, as described in the online supplement.
COPD-like disease:
Mice were exposed to air or CS for 6 months, and serum samples were obtained post-euthanasia via right ventricular puncture. Lungs were inflated to 25 cm H2O pressure and fixed in a 10% buffered formalin. Distal airspace size or fibrosis around small airways having a mean diameter of 300-699 microns was quantified on Gill’s-stained or Masson’s Trichrome-stained lung sections, respectively, using morphometric methods7,8. Lung sections were immunostained for airway epithelial α-SMA (a myofibroblast or smooth muscle cell marker)7.
BAL leukocyte counts:
Total and differential leukocyte counts were performed on BAL samples47.
Lymphocyte subsets in enzymatic lung digests:
Mice were exposed to air or CS for 1-3 months. Lung tissue was digested with 1 mg/ml collagenase D (Sigma Aldrich, St. Louis, MO) in medium at 37°C for 45 min. The digests were passed through a 70 μm strainer and washed with cold PBS containing 0.5% bovine serum albumin. The cells were fixed and permeabilized using the Foxp3 Staining Buffer Set (eBioscience, San Diego, CA). Cells were incubated with Fc block solution (Biolegend, San Diego, CA), and incubated with antibodies (Table E3) or isotype-matched control antibodies (rat IgG1κ, IgG2aκ, or IgG2bκ) for 45 min at 4°C (Table E3) and washed. After dead cell and doublet exclusion, lymphocyte subsets were identified as follows: a) CD4+ T cells (CD45+CD3+CD4+), b) T regulatory cells (Tregs; CD45+CD3+CD4+ Foxp3+), c) CD8+ (CD45+CD3+CD8α+), c) B cells (CD45+B220+), f) CD3-negative natural killer cells (CD45+CD3−Nk1.1+), or g) CD3-positive NKT cells (CD45+CD3+NK1.1+). Cells were identified and quantified using flow cytometry with a BD LSR Fortessa Cell Analyzer (San Jose, CA) and counting beads (ThermoFisher, Waltham, MA). Data were analyzed with FlowJo software (Tree Star Inc., Palo Alto, CA).
Lung mediator levels:
Mediators were measured in lung homogenates using ELISA kits and normalized to lung total protein levels measured on the aliquots of the same samples7.
Leukocyte apoptosis:
AMs and splenic CD8+ T cells were isolated from unchallenged mice, as described above, and induced to undergo apoptosis by incubating them at 37°C with up to 10% CSE for up to 24 h at 37°C. Cells were fixed (as described above), permeabilized with 100% methanol at 4°C for 20 min, and immunostained for intracellular active caspase-3 using Alexa-488-conjugated rabbit anti-caspase-3 IgG or Alexa-488-conjugated rabbit IgG as a control (Cell Signaling, Beverly, MA). The mean integrated fluorescence of 200 cells/group was quantified using MetaMorph software. Data were corrected for non-specific staining in the presence of the non-immune antibody48. Other aliquots of cells were immediately stained for 30 min at 4°C with JC-1 dye using a Mitoprobe® kit (Thermo-Fisher Scientific, Rockford, IL). JC-1 staining in >500 cells/group was measured using MetaMorph software.
Ribosomal protein S6 phosphorylation in bone marrow-derived macrophage (BMDMs):
BMDMs were isolated from naïve mice7,49, cultured in 8-well chamber-slides in L929-conditioned medium for 7 days. The media were changed to complete DMEM medium (DMEM [Corning, Manassas, VA] supplemented with 10% heat-inactivated FBS [Sciencell Research Laboratories, Carlsbad, CA], 100 units/ml penicillin, and 100 μg/mL streptomycin [ThermoFisher Scientific, Waltham, MA]). Cells were incubated with or without 10% CSE and, at intervals, were fixed and incubated with rabbit antimurine phospho-S6 (Ser235/236) IgG (Cell Signaling Technology, Beverly, MA), or non-immune rabbit IgG (Dako, Santa Clara, CA) followed by Alexa-488-conjugated goat anti-rabbit F(ab)2 (Invitrogen, Charlestown, MA). MetaMorph software was used to measure intracellular staining for phospho-S6.
Myeloid cell leukemia sequence-1 (Mcl-1) protein levels in CSE-treated BMDMs:
BMDMs were isolated from naïve mice, and then cultured to render them quiescent7. Cells were incubated with or without 10% CSE and lyzed at intervals in radioimmunoprecipitation assay (RIPA) buffer containing proteinase inhibitors. Western Blot analysis for Mcl-1 and heat shock protein 90 (Hsp90; as a loading control) was performed and Mcl-1 signals were normalized to Hsp90 signals7.
Ribosomal protein S6 phosphorylation and Mcl-1 protein levels in pulmonary CD8+ T cells:
Mice were exposed to air or to CS for 3 months. CD8+ T cells were isolated from enzymatic digests of lungs, as outlined above. Cells were immediately lyzed in RIPA buffer containing proteinase inhibitors and PhosSTOP (Sigma, St. Louis, MO). Western Blot analysis for Mcl-1, phospho-S6 (Ser235/236), total S6, and heat shock protein 90 (Hsp90; as a loading control) was performed. Mcl-1 signals were normalized to Hsp90 signals, phospho-S6 (Ser235/236) signals were normalized to total S6 signals7.
Overexpression ADAM15 in human THP-1 cells:
Human ADAM15 cDNA (isoform 6 pre-proprotein as the longest transcript variant) amplified by RT-PCR was subcloned into the BamHI and XhoI sites of pcDNA3.1 vector to obtain pcDNA-ADAM15 (the primer sequences used for cloning were: forward: 5′ -CGCGGTACCATGCGGCTGGCGCTGCTCTGGG-3’; reverse: 5′ -CCGCTCGAGTCAGAGGTAGAGCGAGGACACT-3’). Human THP-1 monocytes (from ATCC) were maintained in RPMI-1640 containing 10% FBS and differentiated into macrophage-like cells by incubating them with 150 nM phorbol myristate acetate for 24 h. Cells were electroporated (Neon Transfection System; Thermo Fisher Scientific, Waltham, MA) with the ADAM15 or control (pcDNA3.1) vector50. ADAM15 expression was measured using qRT-PCR and the THP-1 cells were treated with 10% CSE for up to 24 h. JC-1 staining and intracellular levels of MCL-1 were measured as described above.
Statistics:
Data are presented as box-plots showing medians and 25th and 75th percentiles and whiskers showing 10th and 90th percentiles (for data that were not normally distributed) or mean ± SD (or SEM; for data that were normally distributed). Data were analyzed with Kruskal-Wallis One-Way ANOVA followed by pair-wise comparisons using Mann-Whitney U-tests (non-parametric data), or One-Way ANOVAs followed by pair-wise comparisons using two-tailed Student’s t-tests (parametric data). P values ≤ 0.05 were considered significant.
Supplementary Material
Acknowledgments:
This work was supported by the following grants: Public Health Service, National Heart, Lung, and Blood Institute Grants HL063137, HL086814, HL111835, and AI111475-01, Flight Attendants Medical Research Institute grants #CIA123046 and CIA052349, and The Department of Defense (CDMRP) grant # PR152060 (to CO). Public Health Service, National Heart, Lung, and Blood Institute Grants F32HL147437 (to XW).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Disclosure: CO is an employee of Vertex Pharmaceuticals Inc. and may own stock in the company. The other authors have no conflicts of interest.
References
- 1.Quaderi SA & Hurst JR The unmet global burden of COPD. Glob Health Epidemiol Genom 3, e4, doi: 10.1017/gheg.2018.1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owen CA Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 3, 253–268 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabe KF et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 176, 532–555, doi: 10.1164/rccm.200703-456SO (2007). [DOI] [PubMed] [Google Scholar]
- 4.Kasahara Y et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106, 1311–1319, doi: 10.1172/JCI10259 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro SD et al. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol 163, 2329–2335, doi: 10.1016/S0002-9440(10)63589-4 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molet S et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res 54, 31–36, doi: 10.1007/s00011-004-1319-4 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Wang X et al. A Disintegrin and A Metalloproteinase-9 (ADAM9): A Novel Proteinase Culprit with Multifarious Contributions to COPD. Am J Respir Crit Care Med, doi: 10.1164/rccm.201711-2300OC (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polverino F et al. A Disintegrin and Metalloproteinase Domain-8: A Novel Protective Proteinase in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 198, 1254–1267, doi: 10.1164/rccm.201707-1331OC (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Owen CA Leukocyte cell surface proteinases: regulation of expression, functions, and mechanisms of surface localization. Int J Biochem Cell Biol 40, 1246–1272, doi: 10.1016/j.biocel.2008.01.020 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dreymueller D, Uhlig S & Ludwig A ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Lung Cell Mol Physiol 308, L325–343, doi: 10.1152/ajplung.00294.2014 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Seals DF & Courtneidge SA The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev 17, 7–30, doi: 10.1101/gad.1039703 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Martin J, Eynstone LV, Davies M, Williams JD & Steadman R The role of ADAM 15 in glomerular mesangial cell migration. J Biol Chem 277, 33683–33689, doi: 10.1074/jbc.M200988200 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Maretzky T et al. Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem J 420, 105–113, doi: 10.1042/BJ20082127 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Herren B et al. ADAM15 overexpression in NIH3T3 cells enhances cell-cell interactions. Exp Cell Res 271, 152–160, doi: 10.1006/excr.2001.5353 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Maeno T et al. CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol 178, 8090–8096, doi: 10.4049/jimmunol.178.12.8090 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Grumelli S et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med 1, e8, doi: 10.1371/journal.pmed.0010008 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodge S, Hodge G, Nairn J, Holmes M & Reynolds PN Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD 3, 179–187 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Horiuchi K et al. Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol 23, 5614–5624, doi: 10.1128/mcb.23.16.5614-5624.2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiemstra PS Altered macrophage function in chronic obstructive pulmonary disease. Ann Am Thorac Soc 10Suppl, S180–185, doi: 10.1513/AnnalsATS.201305-123AW (2013). [DOI] [PubMed] [Google Scholar]
- 20.Marriott HM et al. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest 115, 359–368, doi: 10.1172/JCI21766 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bosque A, Marzo I, Naval J & Anel A Apoptosis by IL-2 deprivation in human CD8+ T cell blasts predominates over death receptor ligation, requires Bim expression and is associated with Mcl-1 loss. Mol Immunol 44, 1446–1453, doi: 10.1016/j.molimm.2006.04.029 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Fulda S Synthetic lethality by co-targeting mitochondrial apoptosis and PI3K/Akt/mTOR signaling. Mitochondrion 19Pt A, 85–87, doi: 10.1016/j.mito.2014.04.011 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Melgert BN et al. Short-term smoke exposure attenuates ovalbumin-induced airway inflammation in allergic mice. Am J Respir Cell Mol Biol 30, 880–885, doi: 10.1165/rcmb.2003-0178OC (2004). [DOI] [PubMed] [Google Scholar]
- 24.Khammas H et al. Characterisation of the human ADAM15 promoter. Nephron Exp Nephrol 118, e27–38, doi: 10.1159/000320698 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Mercer BA, Wallace AM, Brinckerhoff CE & D’Armiento JM Identification of a cigarette smoke-responsive region in the distal MMP-1 promoter. Am J Respir Cell Mol Biol 40, 4–12, doi: 10.1165/rcmb.2007-0310OC (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aporntewan C et al. Gene hunting of the Genetic Analysis Workshop 16 rheumatoid arthritis data using rough set theory. BMC Proc 3Suppl 7, S126, doi: 10.1186/1753-6561-3-s7-s126 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osei ET et al. Unravelling the complexity of COPD by microRNAs: it’s a small world after all. Eur Respir J 46, 807–818, doi: 10.1183/13993003.02139-2014 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee V et al. MicroRNA-147b regulates vascular endothelial barrier function by targeting ADAM15 expression. PLoS One 9, e110286, doi: 10.1371/journal.pone.0110286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryter SW, Alam J & Choi AM Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86, 583–650, doi: 10.1152/physrev.00011.2005 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Wei J, Peng DH, Layne MD & Yet SF Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes 54, 778–784, doi: 10.2337/diabetes.54.3.778 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Richards JA, Wigmore SJ & Devey LR Heme oxygenase system in hepatic ischemia-reperfusion injury. World J Gastroenterol 16, 6068–6078, doi: 10.3748/wjg.v16.i48.6068 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sunderram J et al. Heme oxygenase-1-dependent central cardiorespiratory adaptations to chronic intermittent hypoxia in mice. J Appl Physiol (1985) 121, 944–952, doi: 10.1152/japplphysiol.00036.2016 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Hautamaki RD, Kobayashi DK, Senior RM & Shapiro SD Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277, 2002–2004, doi: 10.1126/science.277.5334.2002 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Owen CA Proteinases and oxidants as targets in the treatment of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2, 373–385; discussion 394-375, doi: 10.1513/pats.200504-029SR (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmitt EG et al. IL-10 produced by induced regulatory T cells (iTregs) controls colitis and pathogenic ex-iTregs during immunotherapy. J Immunol 189, 5638–5648, doi: 10.4049/jimmunol.1200936 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barcelo B et al. Phenotypic characterisation of T-lymphocytes in COPD: abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J 31, 555–562, doi: 10.1183/09031936.00010407 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Schwarz JB et al. Novel role of the CXC chemokine receptor 3 in inflammatory response to arterial injury: involvement of mTORC1. Circ Res 104, 189–200, doi: 10.1161/CIRCRESAHA.108.182683 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Poghosyan Z et al. Phosphorylation-dependent interactions between ADAM15 cytoplasmic domain and Src family protein-tyrosine kinases. J Biol Chem 277, 4999–5007, doi: 10.1074/jbc.M107430200 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Bohm BB, Freund I, Krause K, Kinne RW & Burkhardt H ADAM15 adds to apoptosis resistance of synovial fibroblasts by modulating focal adhesion kinase signaling. Arthritis Rheum 65, 2826–2834, doi: 10.1002/art.38109 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Churg A et al. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 62, 706–713, doi: 10.1136/thx.2006.068353 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong DD, Zhou H & Li G ADAM15 targets MMP9 activity to promote lung cancer cell invasion. Oncol Rep 34, 2451–2460, doi: 10.3892/or.2015.4203 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Fehringer G et al. Cross-Cancer Genome-Wide Analysis of Lung, Ovary, Breast, Prostate, and Colorectal Cancer Reveals Novel Pleiotropic Associations. Cancer Res 76, 5103–5114, doi: 10.1158/0008-5472.CAN-15-2980 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Craig VJ et al. Profibrotic activities for matrix metalloproteinase-8 during bleomycin-mediated lung injury. J Immunol 190, 4283–4296, doi: 10.4049/jimmunol.1201043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knolle MD et al. Adam8 limits the development of allergic airway inflammation in mice. J Immunol 190, 6434–6449, doi: 10.4049/jimmunol.1202329 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laucho-Contreras ME, Taylor KL, Mahadeva R, Boukedes SS & Owen CA Automated measurement of pulmonary emphysema and small airway remodeling in cigarette smoke-exposed mice. J Vis Exp, 52236, doi: 10.3791/52236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X et al. Tissue Inhibitor of Metalloproteinase-1 Promotes Polymorphonuclear Neutrophil (PMN) Pericellular Proteolysis by Anchoring Matrix Metalloproteinase-8 and −9 to PMN Surfaces. J Immunol 202, 3267–3281, doi: 10.4049/jimmunol.1801466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quintero PA, Knolle MD, Cala LF, Zhuang Y & Owen CA Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol 184, 1575–1588, doi: 10.4049/jimmunol.0900290 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owen CA, Hu Z, Lopez-Otin C & Shapiro SD Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol 172, 7791–7803, doi: 10.4049/jimmunol.172.12.7791 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Lee H, Zhang D, Zhu Z, Dela Cruz CS & Jin Y Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci Rep 6, 35250, doi: 10.1038/srep35250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang D et al. A potential role of microvesicle-containing miR-223/142 in lung inflammation. Thorax 74, 865–874, doi: 10.1136/thoraxjnl-2018-212994 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.