

SUMMARY
Rapid removal of histone H2A.Z from neuronal chromatin is a key step in learning-induced gene expression and memory formation, but mechanisms underlying learning-induced H2A.Z removal are unclear. Anp32e was recently identified as an H2A.Z-specific histone chaperone that removes H2A.Z from nucleosomes in dividing cells, but its role in non-dividing neurons is unclear. Moreover, prior studies investigated Anp32e function under steady-state rather than stimulus-induced conditions. Here, we show that Anp32e regulates H2A.Z binding in neurons under steady-state conditions, with lesser impact on stimulus-induced H2A.Z removal. Functionally, Anp32e depletion leads to H2A.Z-dependent impairment in transcription and dendritic arborization in cultured hippocampal neurons, as well as impaired recall of contextual fear memory and transcriptional regulation. Together, these data indicate that Anp32e regulates behavioral and morphological outcomes by preventing H2A.Z accumulation in chromatin rather than by regulating activity-mediated H2A.Z dynamics.
Graphical abstract
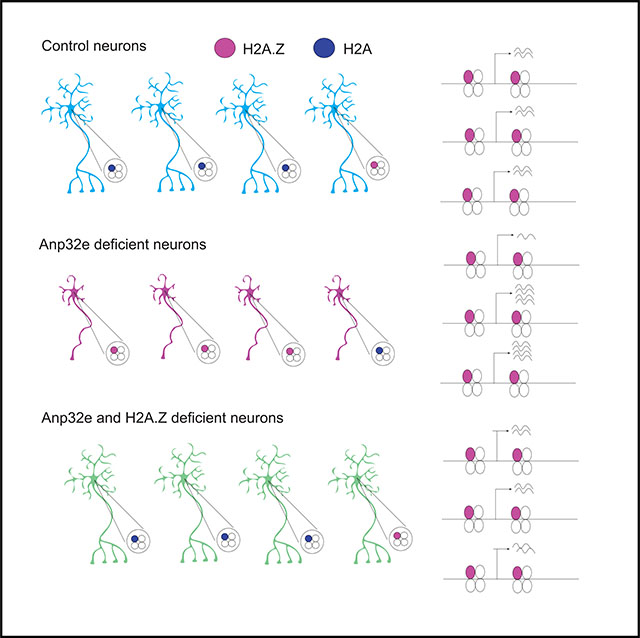
In brief
Histone variants are highly dynamic regulators of neural plasticity and memory formation. Stefanelli et al. show that histone H2A.Z in neurons is regulated by Anp32e, whereby Anp32e keeps H2A.Z binding in check. In cases of Anp32e deficiency, neurons accumulate H2A.Z, their development is impaired, and mice exhibit impaired memory formation.
INTRODUCTION
The dynamic turnover and exchange of histone variants in the brain were recently identified as epigenomic regulators of neural and behavioral plasticity, including memory formation (Zovkic et al., 2014; Stefanelli et al., 2018; Narkaj et al., 2018; Maze et al., 2015; Zovkic, 2021). Histone variants are structurally and functionally distinct counterparts of the canonical histones H2A, H2B, H3, and H4 (Weber and Henikoff, 2014) and their function is of special interest in the non-dividing neuronal cells of the brain. In contrast to replication-coupled canonical histones, histone variants are replication independent and, as such, variants become the primary source of histones as neurons mature (Stefanelli et al., 2018; Maze et al., 2015). Recent studies implicated histone variants as vital regulators of learning and memory, whereby the H3 variant H3.3 has a positive effect on memory and the H2A variant H2A.Z has a negative effect on memory (Zovkic et al., 2014; Maze et al., 2015; Ramzan et al., 2020; Stefanelli et al., 2018). The ability of each histone to influence memory is strongly tied to its dynamic regulation during learning, whereby H2A.Z and H3.3 binding is rapidly modified by a learning event and this dynamic regulation is vital for establishing long-term memories (Maze et al., 2015; Stefanelli et al., 2018; Narkaj et al., 2018). In particular, training on a memory task promotes H2A.Z removal from promoters of genes whose expression is upregulated with learning, and potentiation of this process with experimentally induced H2A.Z depletion improves memory (Stefanelli et al., 2018; Zovkic et al., 2014), suggesting that low levels of H2A.Z in chromatin promote memory formation.
Anp32e (acidic leucine-rich nuclear phosphoprotein 32 family member E) was recently identified as a highly specific H2A.Z chaperone that removes, but does not deposit, H2A.Z from nucleosomes (Mao et al., 2014; Obri et al., 2014), suggesting that Anp32e may also regulate H2A.Z binding in neuronal chromatin. Anp32e belongs to the functionally diverse acidic nuclear phosphoprotein 32 (Anp32) family, whose members are particularly abundant in the nucleus (Matilla and Radrizzani, 2005). The ability of Anp32e to regulate H2A.Z removal was identified in HeLa cells (Mao et al., 2014) and mouse embryonic fibroblasts (MEFs) (Obri et al., 2014), but whether this function extends to non-dividing neuronal cells is unknown. This is especially important because Anp32e was first identified as a regulator of synaptic function in the cerebellum (Costanzo et al., 2006), suggesting a potentially alternative function in the brain. Moreover, Anp32e has only been implicated in H2A.Z regulation under steady-state conditions (Mao et al., 2014; Obri et al., 2014; Gursoy-Yuzugullu et al., 2015), such that its role in regulating stimulus-induced H2A.Z removal (a key aspect of memory formation) is unknown.
Here, we addressed 3 questions regarding Anp32e function. (1) Does Anp32e regulate H2A.Z binding in the mouse hippocampus, a brain region vital for memory formation (Rempel-Clower et al., 1996)? (2) Does Anp32e regulate stimulus-induced H2A.Z removal? (3) Does neuronal Anp32e regulate transcription, neuronal morphology, and memory formation? Using a knockdown approach, we showed that Anp32e depletion results in H2A.Z accumulation under steady-state (i.e., baseline) conditions, indicating that Anp32e does regulate steady-state levels of H2A.Z. However, Anp32e depletion did not block activity-induced H2A.Z removal in cultured hippocampal cells, suggesting that the primary function of Anp32e is to regulate H2A.Z binding under basal conditions. Indeed, Anp32e depletion resulted in both up- and downregulation of gene expression in association with increased H2A.Z levels, and this transcriptional dysregulation was partly reversed by H2A.Z co-depletion, suggesting that Anp32e-mediated H2A.Z accumulation has bidirectional effects on transcription. Functionally, Anp32e depletion was associated with impaired dendritic arborization, whereas Anp32e overexpression had no effect, suggesting that H2A.Z accumulation induced by the absence of Anp32e is particularly detrimental to neuronal function. In line with these findings, Anp32e depletion in the mouse hippocampus led to an H2A.Z-dependent impairment in contextual fear memory, whereas Anp32e overexpression improved fear memory, indicating that Anp32e regulates memory and neuronal function by restricting H2A.Z binding in neuronal chromatin.
RESULTS
Anp32e and H2A.Z co-localize on neuronal chromatin
A key step in memory formation is the removal of H2A.Z from plasticity-related genes in the mouse hippocampus in response to a learning event (Zovkic et al., 2014; Stefanelli et al., 2018). Because prior studies with H2A.Z were conducted using mixed-cell populations in the mouse hippocampus, we first evaluated whether H2A.Z is regulated by neuronal activity in a pure neuron population using a primary hippocampal culture system. Neurons were depolarized with 55 mM KCl for 30 min and chromatin immunoprecipitation (ChIP) was used to compare H2A.Z binding in stimulated cells compared to unstimulated controls (Figure 1A). H2A.Z signal was normalized to H3 binding on the same loci to account for any possible changes in nucleosome occupancy. Consistent with findings in the mouse hippocampus, where H2A.Z is removed from Egr1 and Fos genes after contextual fear conditioning (Stefanelli et al., 2018; Zovkic et al., 2014), neuronal activity induced H2A.Z removal from the first nucleosome (+1 nucleosome) downstream of the transcription start site (TSS) of Fos (t27 = 3.54, p = 0.001) and Egr1 (t29 = 3.32, p = 0.002) genes, as well as the coding region of Fos (t28 = 3.21, p = 0.003) (Figure 1B). Notably, we did not observe reduction in H2A.Z binding at the +1 nucleosome of P2ry10b or Bdnf4 genes (Figure 1B), indicating that H2A.Z removal is gene specific.
Figure 1. H2A.Z and Anp32e binding is reduced with neural depolarization.
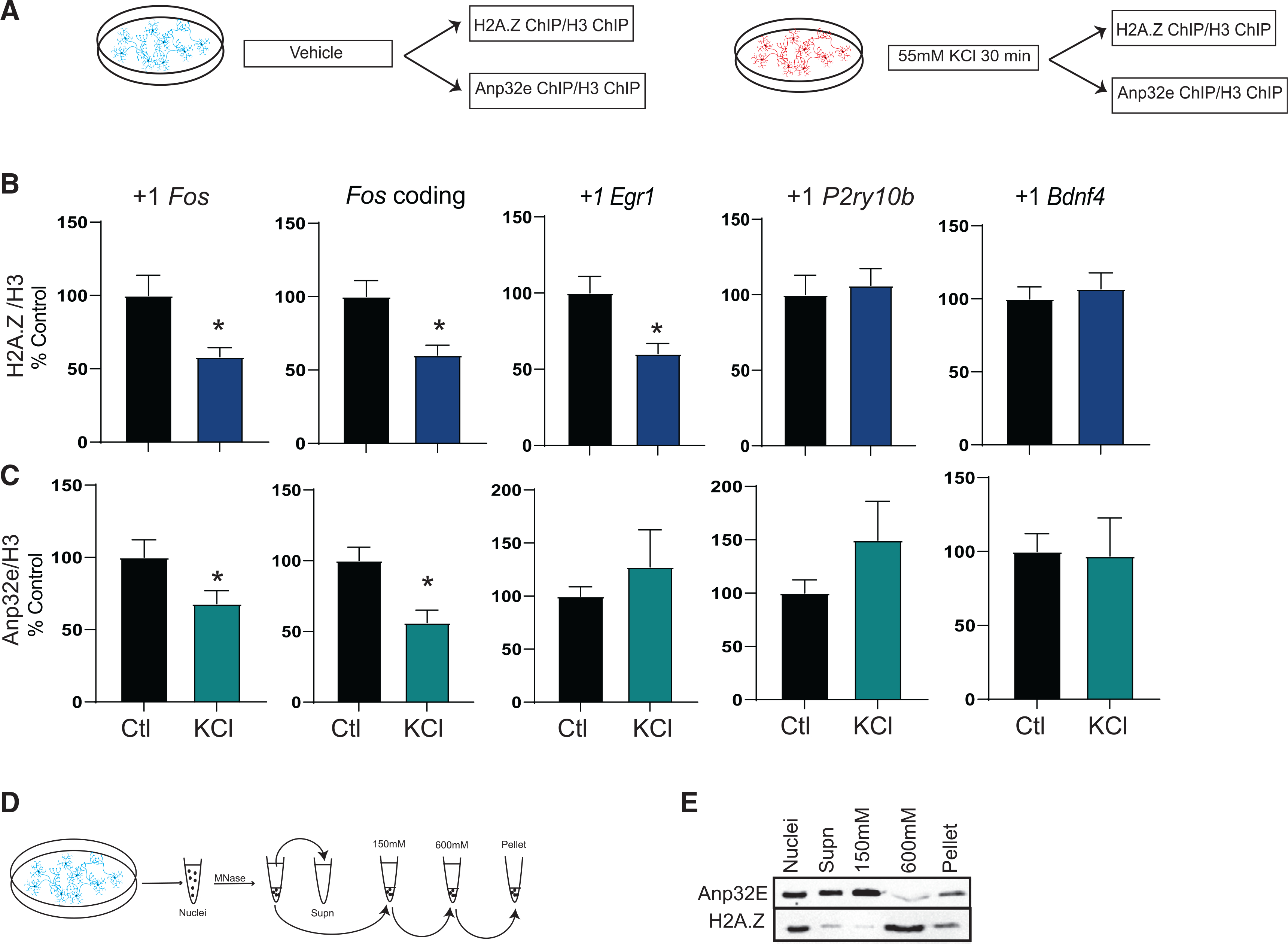
(A) Schematic representation of the experimental workflow in cultured hippocampal neurons.
(B and C) Chromatin immunoprecipitation (ChIP) was used to assess binding of (B) H2A.Z (n = 13–17/group) or (C) Anp32e (N = 8–10/group) in the first nucleosome downstream (+1 nucleosome) of the transcription start site (TSS) and in the coding region 30 min after KCl depolarization. H2A.Z binding is normalized to H3 and data are expressed as mean ± SEM. *p ≤ 0.05.
(D) Schematic representation of nucleosome salt fractionation.
(E) MNase fractionation and salt extraction of primary hippocampal neuronal nuclei were used to assess Anp32e binding to H2A.Z in distinct chromatin fractions.
Having demonstrated that H2A.Z is subject to activity-mediated removal in cultured hippocampal neurons, we next assessed whether Anp32e is involved in this process by conducting Anp32e ChIP in primary hippocampal neurons treated with KCl (Figure 1A). Anp32e has a similar removal pattern as H2A.Z (Figure 1C), whereby Anp32e was removed from the +1 nucleosome of the Fos gene (t15 = 2.15, p = 0.049) and the Fos gene body (t15 = 3.38, p = 0.004), although there was no change in Anp32e binding at the Egr1 gene. Regions that showed lack of H2A.Z removal in Figure 1B also showed no Anp32e removal (P2ry10b, Bdnf4) in response to depolarization (Figure 1C). Importantly, both Anp32e and H2A.Z are enriched compared to immunoglobulin G (IgG) on all the loci we examined (Figure S1). Overall, these data indicate that Anp32e and H2A.Z occupy similar genomic loci and are co-regulated in response to neuronal activity.
We further characterized the relative location of H2A.Z and Anp32e in chromatin using a nucleosome salt fractionation protocol (Teves and Henikoff, 2012) that digests nuclei with MNase, and then extracts nucleosomes at different salt concentrations to separate chromatin into low-salt-soluble (150 mM), high-salt-soluble (600 mM), and insoluble (pellet) fractions (Figure 1D). H2A.Z levels are highest in the 600 mM fraction that contains condensed chromatin (Figure 1E), followed by the insoluble pellet fractions that contain large, transcriptionally active multiprotein complexes (Teves and Henikoff, 2012), indicating that H2A.Z is present in multiple chromatin fractions with distinct physical properties. The majority of Anp32e is found in the 150 mM fraction that contains mostly mono-nucleosomes and corresponds to accessible chromatin (Teves and Henikoff, 2012) (Figure 1E), followed by non-chromatinized supernatant. Anp32e and H2A.Z are both found in the pellet, and a small amount of Anp32e is also found in the high-salt fraction that contains the majority of H2A.Z (Figure 1E). Overall, these data suggest that the interaction between Anp32e and H2A.Z may be most stable in highly multiplexed chromatin regions that are associated with transcriptional activation. However, the majority of Anp32e and H2A.Z is not found in overlapping fractions, indicating that Anp32e’s interaction with chromatin may be dynamic.
Anp32e depletion promotes steady-state H2A.Z accumulation in neuronal chromatin
Given that depolarization triggers co-removal of H2A.Z and Anp32e in cultured hippocampal neurons, we used this culture system to directly evaluate whether Anp32e regulates neuronal H2A.Z binding. Neurons were infected with adeno-associated virus (AAV) vectors containing short hairpin RNA (shRNA) against Anp32e (shAnp32e) or scramble control (shScr) at 1 DIV (day in vitro; for knockdown validation, see Figure S2A) and ChIP sequencing (ChIP-seq) for H2A.Z was performed at 8 DIV (Figure 2A). First, we assessed H2A.Z signal on sites corresponding to H2A.Z peaks in the mouse hippocampus (Stefanelli et al., 2018) and showed that Anp32e depletion produced an overall increase in H2A.Z signal in these regions (Figure 2B). Moreover, Anp32e depletion increased H2A.Z binding at CpG islands (Figure 2B, middle) and TSS-flanking regions (Figure 2B, right), suggesting that Anp32e depletion promotes H2A.Z accumulation at sites that were previously associated with H2A.Z binding in the mouse brain. We previously showed that H2A.Z binding is particularly high in CpG islands (Stefanelli et al., 2018), which tend to have low levels of DNA methylation (a modification whose occupancy is inversely related to H2A.Z binding in non-neuronal cells; Zilberman et al., 2008), indicating that Anp32e may maintain appropriate H2A.Z levels in these regions. Notably, Anp32e depletion did not increase total H2A.Z protein or mRNA levels (Figure S2B), indicating that the increase we observed is caused by increased levels of chromatinized histone. To verify that Anp32e depletion does indeed increase H2A.Z binding, we confirmed that Anp32e depletion with an alternate shRNA construct also increases H2A.Z binding on several genes that accumulate H2A.Z in the ChIP-seq, including Nr4a2 (t8 = 3.76, p = 0.006), Bdnf4 (t9 = 2.29, p = 0.048), and Mis18bp1 (t9 = 3.80, p = 0.004), and not on the negative control gene, P2ry10b (Figures S2C and S2D).
Figure 2. Anp32e depletion results in H2A.Z accumulation in neuronal chromatin.
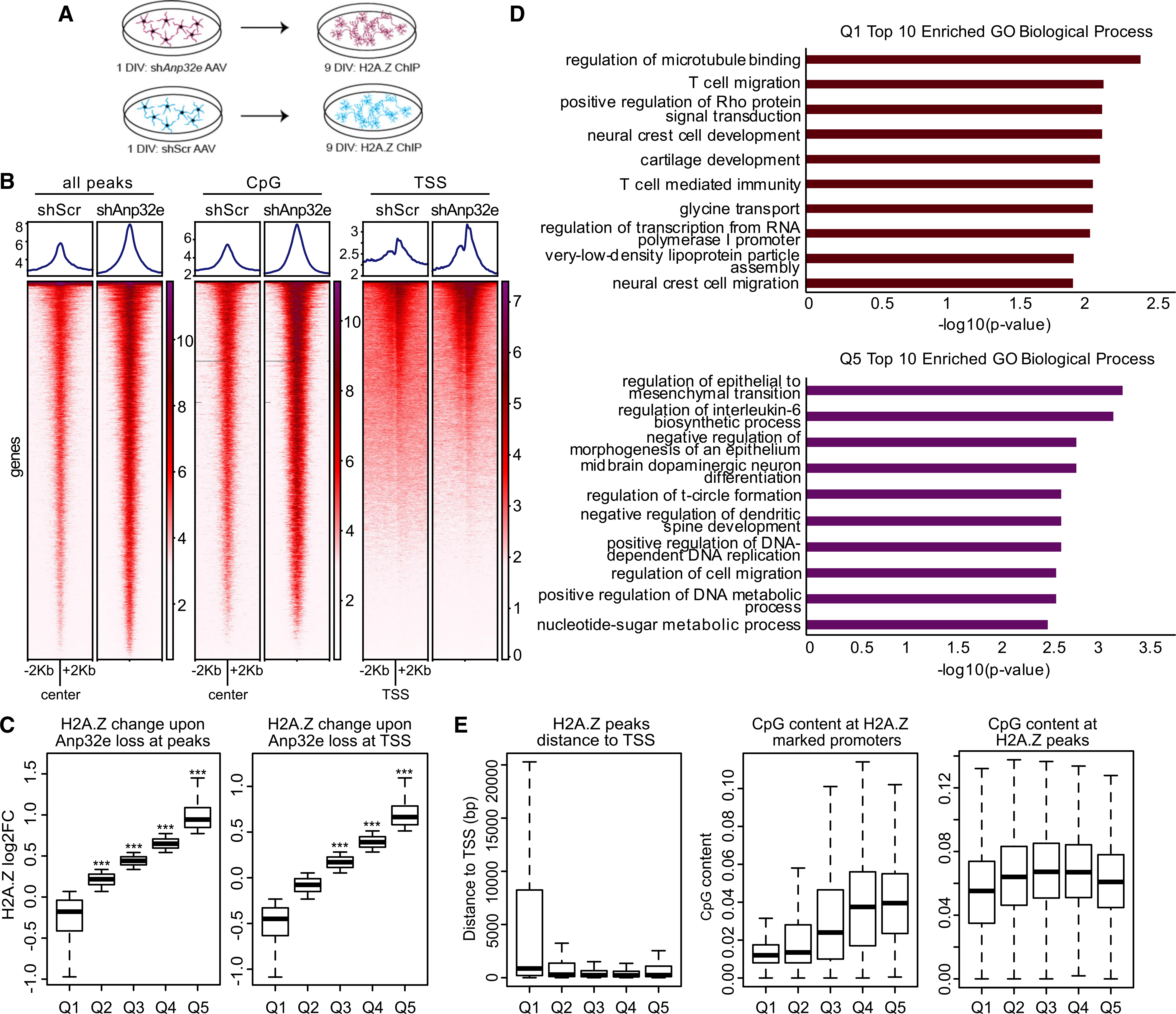
(A) Schematic representation of the experimental workflow in cultured hippocampal neurons.
(B) H2A.Z signal was assessed with ChIP sequencing (ChIP-seq) and analyses were conducted in several regions of interest, including sites of H2A.Z binding in the mouse hippocampus (left), CpG islands (middle), and TSS-flanking regions (right) in scramble control and Anp32e-depleted neurons.
(C) Boxplot representing change in H2A.Z enrichment at hippocampally determined H2A.Z peaks (left) or TSS-flanking loci (right). p values were generated from t tests comparing quantiles to a randomized normal distribution of log2 fold change (log2FC) change values with the mean of zero.
(D) Gene Ontology of biological processes for genes showing H2A.Z decrease, Q1 (top), or increase, Q5 (bottom), with Anp32e depletion at promoters identified in the mouse hippocampus.
(E) Boxplot representing the distance of H2A.Z peaks from the TSS (left), CpG content at H2A.Z promoters (middle), and CpG content at H2A.Z peaks (right).
To further validate the observation that Anp32e depletion promotes H2A.Z accumulation, differentially bound regions (shAnp32e versus shScr) were split into quintiles based on lowest (Q1) to highest (Q5) levels of H2A.Z binding (Figure 2C). Using this approach, we showed that quintiles 2–4 had higher H2A.Z signal at loci corresponding to hippocampal peaks (Figure 2C, left) and quintiles 3–5 had higher H2A.Z signal at TSS-flanking regions (Figure 2C, right) in Anp32e-depleted compared to control neurons. None of the quintiles exhibited significant overall reductions in H2A.Z binding with Anp32e depletion. Gene Ontology analyses showed that genes with no Anp32e-induced change in H2A.Z binding (Q1) and genes with the highest (Q5) H2A.Z enrichment in Anp32e-depleted neurons were associated with non-overlapping processes, whereby genes with the greatest level of H2A.Z enrichment (Q5) were associated with developmental regulation of plasticity-related factors, such as dopaminergic neuron differentiation and negative regulation of spine development. In contrast, H2A.Z signal in Q1 was associated with factors such as regulation of microtubule binding. Moreover, H2A.Z signal in the quintiles that showed H2A.Z accumulation upon Anp32e depletion (Q2–Q5) mapped in close proximity to the TSS (Figure 2E, left), and CpG density was highest at loci with the most H2A.Z accumulation (Figure 2E, middle). In fact, CpG levels were consistently high across all hippocampal H2A.Z peaks, as compared with unmarked promoters (Figure 2E, right). These data suggest that Anp32e is particularly relevant for maintaining H2A.Z levels at CpG-dense promoter regions.
Anp32e depletion affects mRNA expression at H2A.Z-bound genes
Having established that Anp32e depletion causes H2A.Z accumulation in neurons, we conducted RNA sequencing to evaluate the transcriptional consequences of Anp32e depletion. Primary hippocampal neurons were infected with shAnp32e or shScr AAV vectors at 1 DIV and RNA was extracted on 8 DIV (Figure 3A). Anp32e depletion resulted in up- (2,894) and down- (3,382) regulation of gene expression (Figure 3B; Table S1) and Gene Ontology analyses revealed distinct biological processes for up- and downregulated genes (Figure 3C).
Figure 3. Anp32e depletion regulates neuronal gene expression.
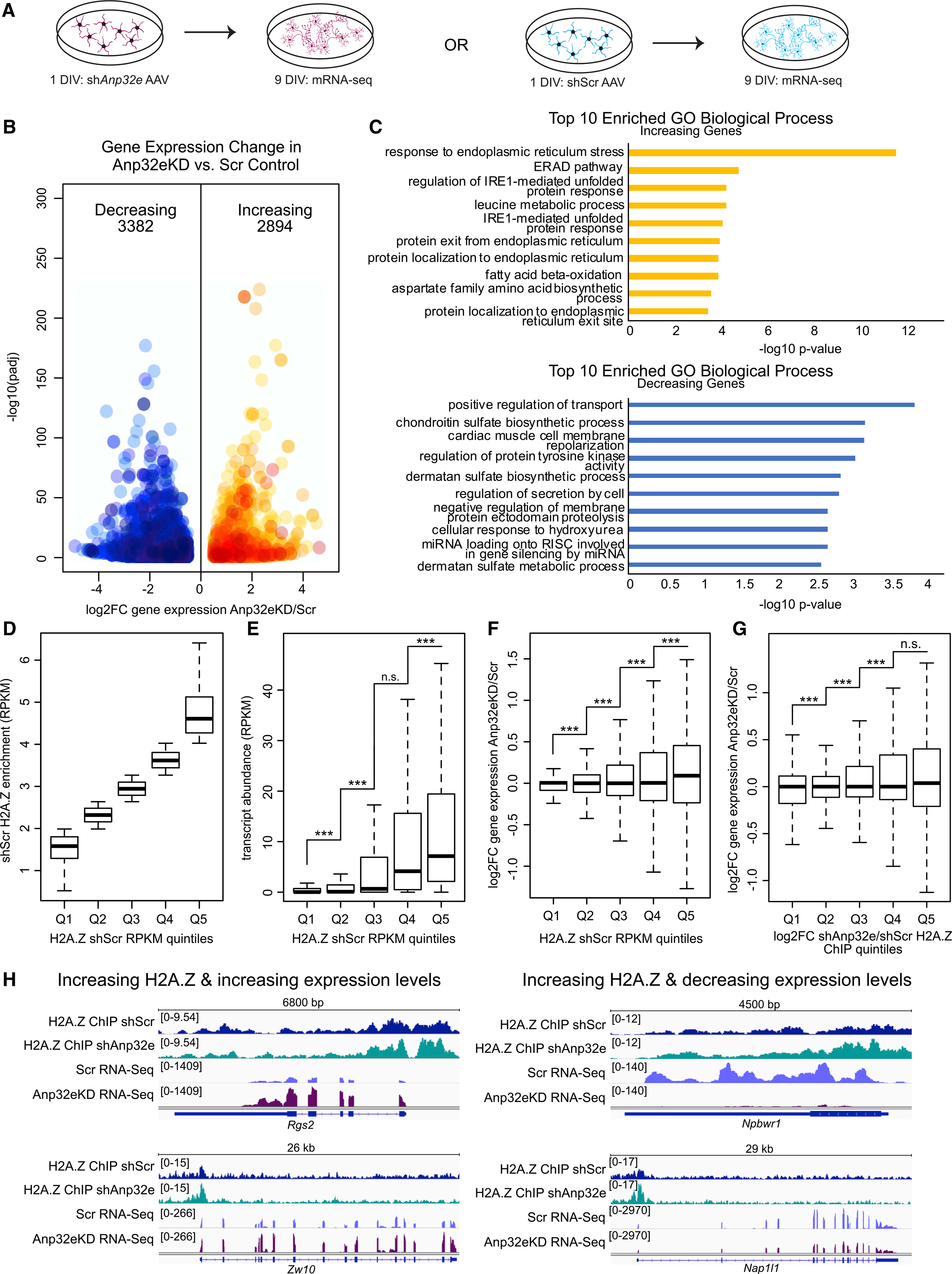
(A) Experimental strategy for measuring gene expression changes in Anp32e-depleted neurons (left) compared with control (right). 3 replicates per condition.
(B) Volcano plot depicting gene expression changes with decreases in blue and increases in orange. DESeq2 was used to determine significance (y axis) and log2FC (x axis) upon Anp32e loss.
(C) Bar charts depicting results from Gene Ontology analysis for increasing (top) and decreasing (bottom) genes with significance measurements on the x axis.
(D) Quintiles were assigned based on H2A.Z levels in control samples.
(E) RNA transcript abundance (RPKM) was measured at promoters based on quintiles defined in (D). p values are from two-tailed Student’s t tests.
(F) Changes in gene expression were measured for all gene promoters separated based on quintiles defined in (D). p values are from two-tailed Student’s t tests.
(G) Quintiles were assigned based on H2A.Z enrichment changes upon Anp32e depletion (Q1–Q5) and log2FC gene expression changes were measured at promoters. p values are from two-tailed Student’s t tests.
(H) Genome browser screen shots provide examples where changes in H2A.Z coincide with gene expression changes.
See also Table S1.
Given that H2A.Z is enriched at highly expressed genes in the mouse hippocampus (Stefanelli et al., 2018), we explored the relationship between H2A.Z and gene expression in cultured neurons. Genes in scramble control neurons were first sorted into quintiles according to their H2A.Z enrichment, from lowest (Q1) to highest (Q5) levels of H2A.Z (Figure 3D), and promoter expression levels (RPKM; reads per kilo base per million mapped reads) were calculated for each quintile of H2A.Z (Figure 3E). In accordance with previous reports, the highest levels of H2A.Z were associated with higher transcript abundance (Figure 3E), suggesting that H2A.Z is positively associated with steady-state gene expression. In support of H2A.Z functioning to regulate transcription, expression changes upon Anp32e depletion were most significant at promoters with the highest levels of H2A.Z (Figure 3F), and these promoters gained the most H2A.Z in the absence of Anp32e (Figure S3A). Moreover, Anp32e-mediated gene dysregulation occurred in Anp32e-deficient neurons at promoters where H2A.Z accumulated the most (Figures 3G and 3H), with significantly more genes increasing than decreasing expression (Figure S3B).
Co-depletion of Anp32e and H2A.Z partially restores gene transcription
Considering that Anp32e induces H2A.Z accumulation and alters transcription in relation to H2A.Z levels, we postulated that co-depleting Anp32e and H2A.Z would restore normal levels of H2A.Z and gene transcription. We first confirmed that co-depleting H2A.Z with Anp32e normalizes H2A.Z levels in chromatin, using soluble and chromatin-bound protein fractionation from neurons infected with shAnp32e alone or a combination of shAnp32e and shH2afz. When compared to scramble controls, Anp32e depletion caused an accumulation of H2A.Z, and H2A.Z depletion caused a reduction of H2A.Z in the chromatin fraction, whereas co-depletion of Anp32e and H2A.Z reinstated chromatin abundance of H2A.Z to control levels (Figure S4).
RNA sequencing revealed that gene expression changes induced by Anp32e depletion were often reversed upon co-depletion of H2A.Z (Figure 4A; Tables S2 and S3), suggesting that H2A.Z levels contribute to gene dysregulation. To further explore the capacity of H2A.Z co-depletion to rescue gene dysregulation, we parsed all genes into quintiles based on expression changes from Anp32e depletion alone, from lowest (Q1) to highest (Q5) (Figure 4B). Genes that were activated upon Anp32e depletion (Q5) tended to become less active upon co-depletion, and genes that were silenced (Q1) tended to become more active (Figure 4C). Of 3,382 downregulated differentially expressed genes (DEGs), 568 show strong rescue and 787 show weak rescue upon Anp32e and H2A.Z co-depletion (Figures 4D and 4E), and for upregulated (2,894) genes, 192 show strong rescue and 1,168 show weak rescue in double-knockdown conditions (Figures 4D and 4E). Together, these data suggest that a large proportion of Anp32e-mediated changes in gene expression involve a change in H2A.Z binding. Notably, just over half of up- and downregulated genes were not rescued by H2A.Z co-depletion, suggesting either incomplete H2A.Z knockdown or additional functions of Anp32e were involved. Finally, rescued genes that were up- versus downregulated by Anp32e depletion are involved in distinct biological processes, whereby top terms for downregulated genes include metabolic processes and developmental pattern formation, and top terms for upregulated genes include G protein receptor signaling and cation transport (Figure 4F). Together, these data indicate that Anp32e is a key factor in neuronal gene regulation, in large part through its downstream effects on H2A.Z abundance in chromatin.
Figure 4. Co-depletion of Anp32e and H2A.Z rescues gene expression.
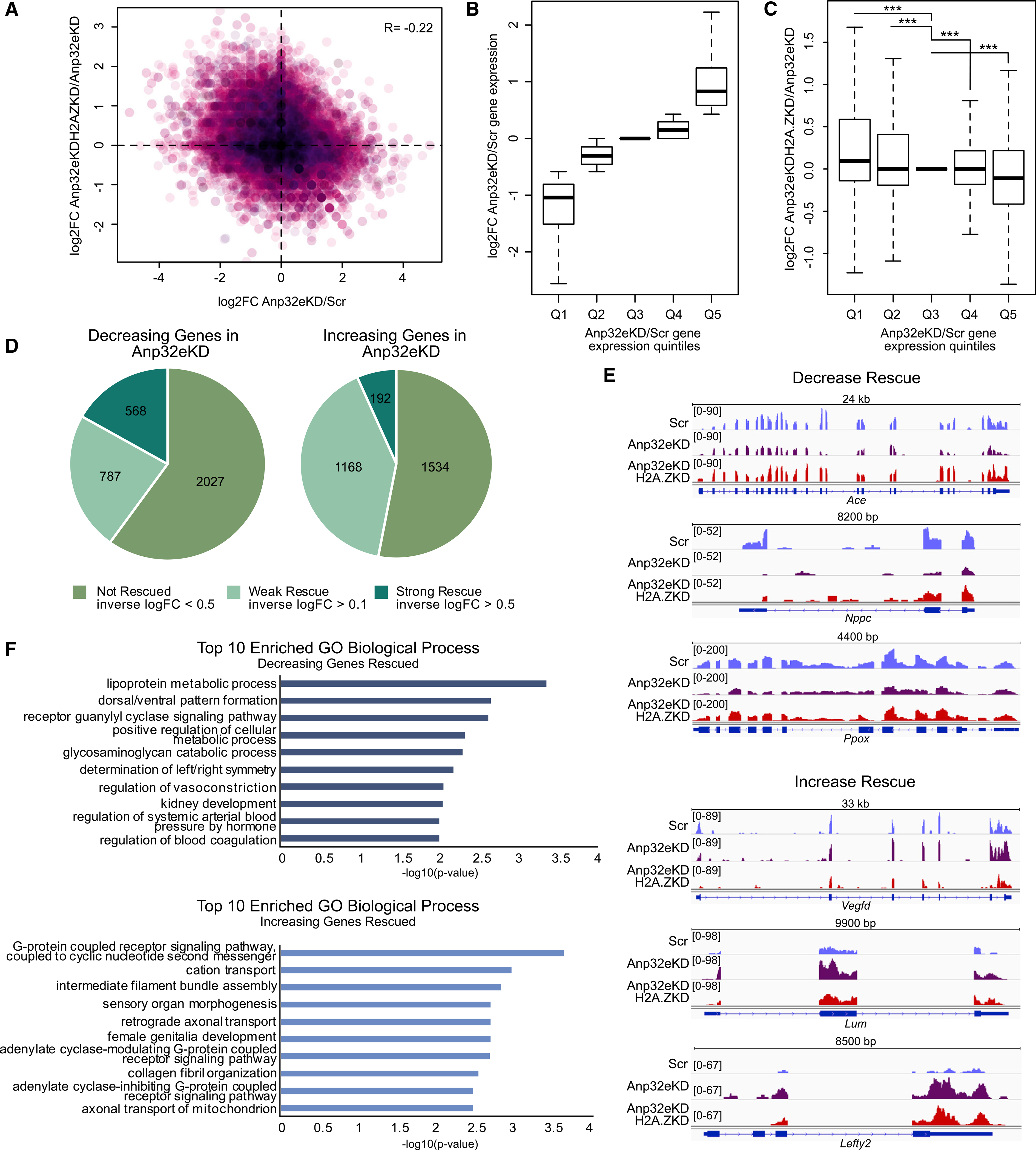
RNA sequencing was conducted on 8 day in vitro (DIV) neurons that were co-infected with shAnp32e and shH2afz at 1 DIV (3 replicates/condition).
(A) Scatterplot illustrating gene expression changes in shAnp32e neurons compared with shAnp32e + shH2afz. Pearson correlation R = −0.22.
(B) Genes were parsed by expression changes in Anp32e-depleted neurons (Q1–Q5).
(C) Boxplot showing gene expression changes in shAnp32e + shH2afz across quintiles in (B). ***p < 0.05 using one-tailed Student’s t tests.
(D) Pie charts indicating the numbers of decreasing (left) and increasing (right) genes displaying rescue upon shAnp32e + shH2afz.
(E) Representative tracks of genes showing a decrease (top) or increase (bottom) in shAnp32e and normal expression in shAnp32e + shH2afz.
(F) Gene Ontology of biological processes of decreasing (top) or increasing (bottom) genes in shAnp32e showing rescue in shAnp32e + shH2afz.
Anp32e regulates dendritic branching through effects on H2A.Z
Analyses of Anp32e-mediated changes in H2A.Z binding revealed an accumulation of H2A.Z at genes that regulate dendritic development (Figure 2D), generating the hypothesis that Anp32e-mediated H2A.Z accumulation may impact neuronal morphology. To test this hypothesis, primary hippocampal neurons were plated on coverslips and infected 1 DIV with AAV vectors to either deplete or overexpress Anp32e (Figure 5A). GFP expression levels for infected neurons were used to visualize and quantify neuronal morphology using Sholl analysis. The efficiency of Anp32e knockdown and overexpression was validated at the RNA (shAnp32e: t28 = 15.09, p < 0.0001; mAnp32e (mouse Anp32e overexpression): t28 = 6.062, p < 0.0001) and protein level (shAnp32e: t6 = 3.924, p = 0.007; mAnp32e: t6 = 10.74, p < 0.0001) (Figures 5A and S2A).
Figure 5. Anp32e depletion interferes with dendritic arborization in an H2A.Z-dependent way.
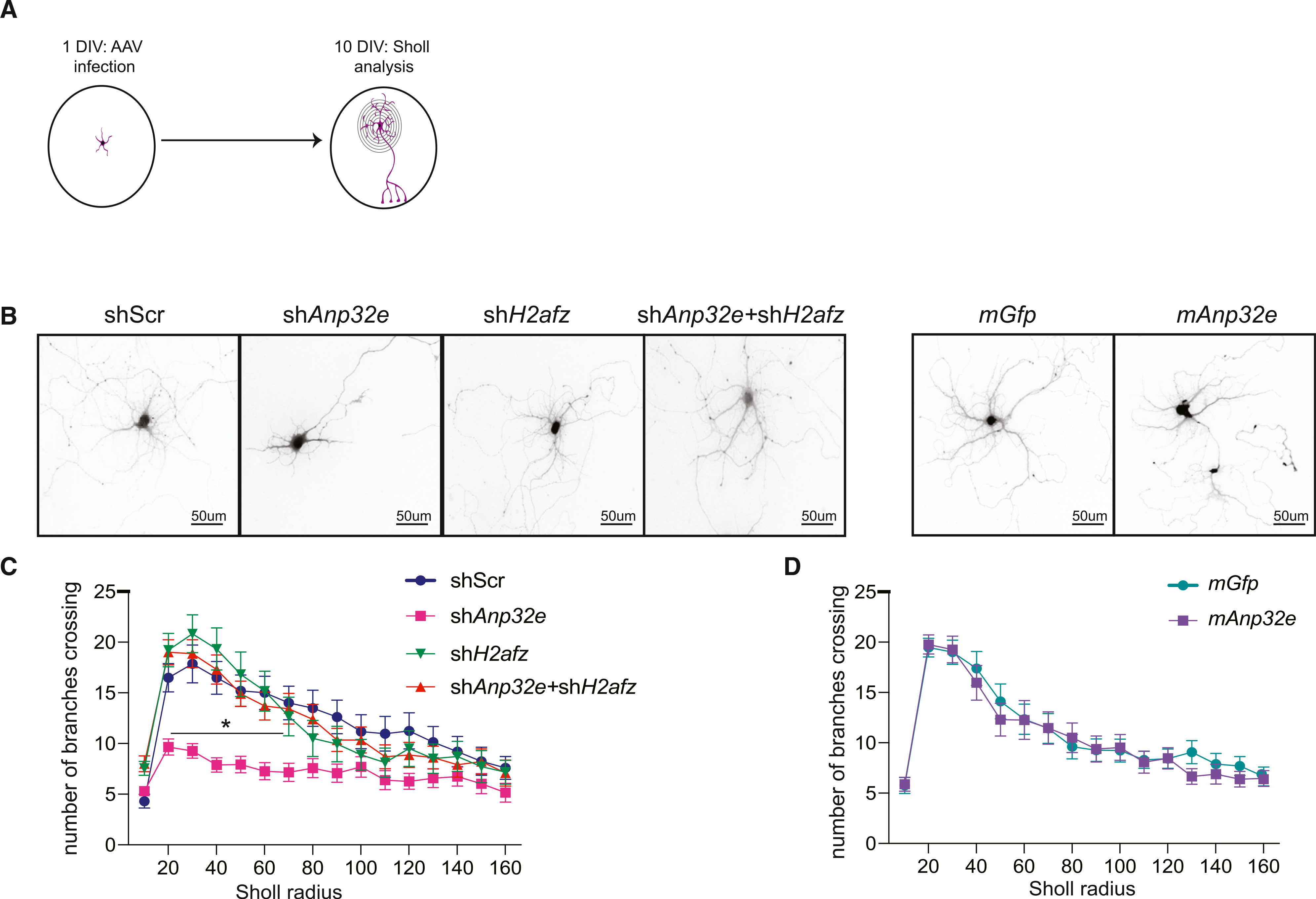
(A) Schematic representation of the experimental workflow.
(B) Representative images of neurons infected with each of the AAVs used.
(C and D) Quantification of dendritic branching by Sholl analysis (D; shScr: n = 29 neurons; shAnp32e: n = 26 neurons; shH2afz: n = 18 neurons; shAnp32e + shH2afz: n = 23 neurons) (E; mGfp: n = 26 neurons; mAnp32e: n = 26 neurons) (across 7 or 8 wells/virus; 2 or 3 neurons per coverslip). Data are expressed as mean ± SEM. *p ≤ 0.05.
Anp32e depletion produced profound morphological deficits, characterized by fewer primary dendritic branches compared to scramble control neurons (AAV × Sholl radius interaction: F19, 1,160 = 2.2773, p = 0.01; Tukey’s LSD [least significant difference]: shAnp32e, p < 0.05 at a radius of 20–90 μm) (Figures 5B and 5C). Given that Anp32e depletion increased H2A.Z binding, and that co-depletion of Anp32e and H2A.Z reinstates normal H2A.Z chromatin levels and rescues gene expression of genes involved in neurodevelopment and signaling (Figure 4F), we tested whether effects of Anp32e depletion on dendritic branching were mediated by H2A.Z accumulation. To test this hypothesis, we simultaneously depleted H2A.Z (shH2afz) and Anp32e in the same neurons and found that their co-depletion completely rescued the morphological deficits associated with Anp32e knockdown and restored a physiologically normal number of dendrites (Figures 5B and 5C). Importantly, H2A.Z depletion alone did not influence dendritic branching (Figure 5C) (AAV × Sholl radius interaction: F57, 1,810 = 1.696, p = 0.001; Tukey’s LSD: shAnp32e, p < 0.05 at a radius of 20–70 μm from all other viruses), indicating that the ability of H2A.Z co-depletion to rescue dendritic arborization deficits is not caused by an independent effect of H2A.Z depletion on dendritic branching and instead reflects a rescue of abnormal H2A.Z accumulation caused by Anp32e depletion (for shH2afz validation, see Figure S5B). Consistent with the observation that H2A.Z depletion alone had no effect on dendritic arborization (Figure 5C), artificially increasing Anp32e expression to promote H2A.Z removal also had no impact on dendritic arborization (main effect of Sholl radius: F19, 1,000 = 30.963, p < 0.001) (Figures 5B and 5D).
Anp32e depletion modifies, but does not prevent, activity-mediated H2A.Z removal
Our findings thus far indicated that Anp32e was vital for regulating basal levels of H2A.Z in neuronal chromatin, but it remained unknown whether Anp32e also regulated stimulus-induced H2A.Z removal. To address this question, we conducted H2A.Z ChIP-seq on Anp32e-depleted neurons after 30 min of depolarization with KCl, a treatment intended to mimic neuronal activity (Figure 6A) and that triggers H2A.Z removal in neurons (Figure 1B). Despite promoting steady-state H2A.Z accumulation, Anp32e depletion did not block depolarization-induced reduction in H2A.Z signal at TSS-flanking regions, or at regions defined by hippocampal H2A.Z peaks (Figure 6B), indicating that Anp32e depletion does not block dynamic, signal-induced loss of H2A.Z. Notably, H2A.Z accumulation resulting from Anp32e loss was not anti-correlated with activity-induced H2A.Z removal in scramble control neurons (Figure 6C, top), as we initially hypothesized, thus suggesting that basal H2A.Z accumulation occurs at regions that are not activity regulated. In support of this finding, sites of activity-induced H2A.Z removal in Anp32e-deficient and scramble control neurons were positively correlated (Figures 6C, bottom and S6A), suggesting that activity-mediated H2A.Z removal can occur in part through an Anp32e-independent mechanism.
Figure 6. Anp32e depletion partially interferes with activity-induced H2A.Z removal and alters activity-induced transcription in neurons.
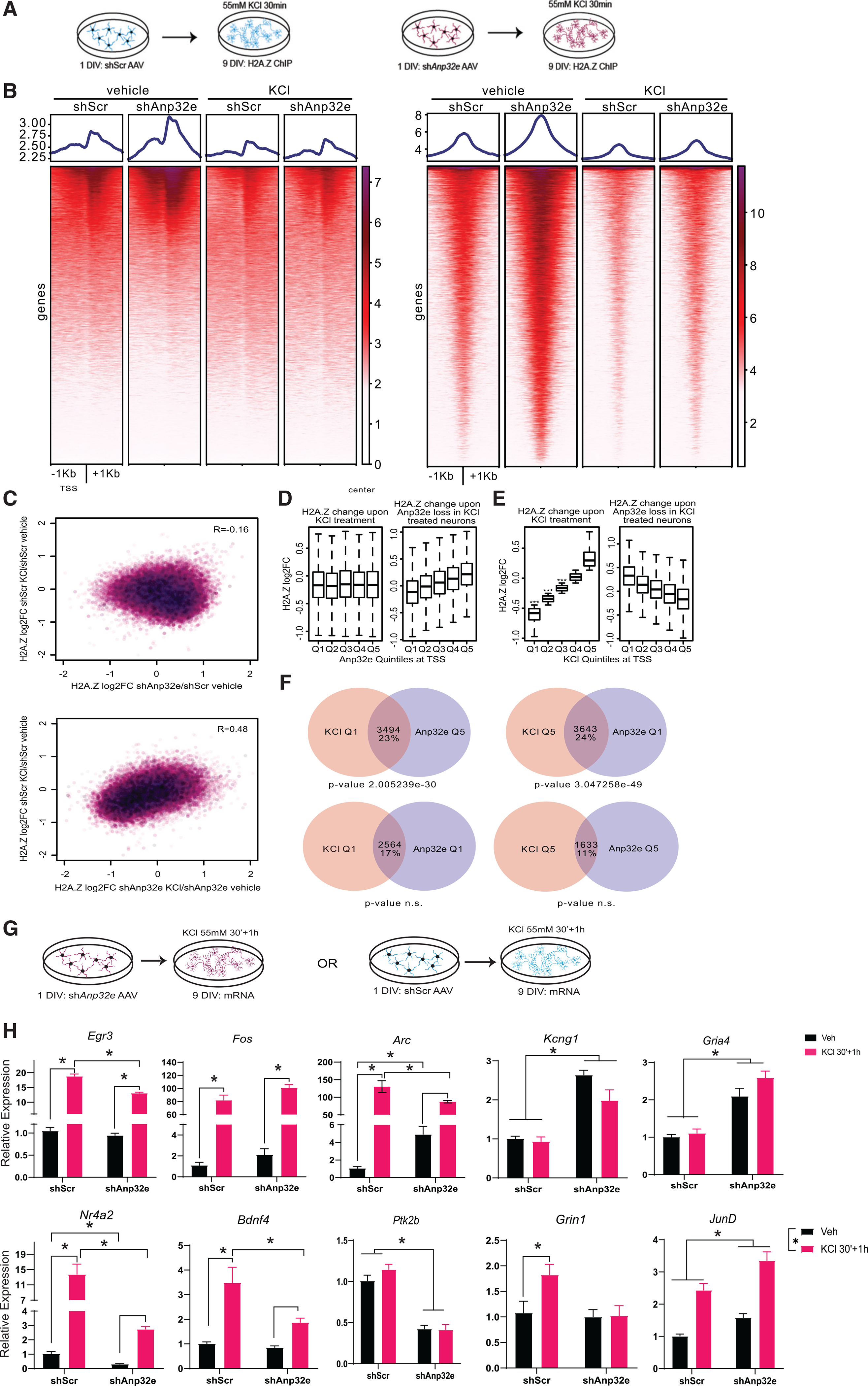
(A) Schematic representation of the experimental workflow in cultured hippocampal neurons (n = 3 replicates/condition). H2A.Z ChIP-seq was used to assess the effect of Anp32e depletion on activity-induced H2A.Z binding in neurons treated with vehicle (left) or KCl (right).
(B) Heatmaps illustrating H2A.Z enrichment at TSSs (left) and hippocampal H2A.Z peaks (right) in scramble control and Anp32e-depleted neurons with and without KCl treatment.
(C) Top: scatterplot illustrating H2A.Z changes at promoters comparing KCl treatment in controls with Anp32e-depletion-mediated H2A.Z accumulation at baseline. Bottom: scatterplot illustrating H2A.Z changes at promoters comparing KCl treatment in control versus Anp32e-depleted neurons. R values are from Pearson correlation tests.
(D) Promoters were parsed by H2A.Z changes in Anp32e-depleted neurons (Q1–Q5) and measurements were made comparing H2A.Z change upon KCl treatment (left) or upon Anp32e depletion in KCl-treated neurons (right).
(E) Promoters were parsed by largest to smallest H2A.Z changes in KCl-treated neurons (Q1–Q5) and the magnitude of H2A.Z change was measured upon KCl treatment (left) or upon Anp32e depletion in KCl-treated neurons (right). Significance tests are from paired t tests comparing quantiles to a randomized normal distribution with a mean of zero.
(F) Overlap between promoters parsed by H2A.Z changes upon KCl treatment (pink) or upon Anp32e depletion (purple). Promoters were then intersected based on whether changes in H2A.Z binding occurred in the opposite manner (top) or in the same manner (bottom).
(G) Schematic representation of the experimental workflow. Neurons were infected with shAnp32e AAV DJ to deplete Anp32e at 1 DIV and depolarized with KCl for 30 min (30′) at 8 DIV. Effects on activity-induced gene expression were assessed 1 h after washout of KCl.
(H) AAV-mediated gene depletion of Anp32e alters activity-induced gene expression (N = 5/group).Data are expressed as mean ± SEM. *p ≤ 0.05.
One possibility for these findings is that activity-regulated H2A.Z changes occur mostly at regions that are not regulated by Anp32e, although there may still be some degree of Anp32e influence on KCl-induced H2A.Z removal at a subset of important loci, most notably at promoters. In fact, we did find evidence for H2A.Z accumulation in Anp32e-deficient cells at several promoters displaying weak activity-mediated H2A.Z removal (Figure S6B). To investigate this further, we conducted additional analyses restricted only to gene promoters. Promoters were arranged into the same quintiles as in Figure 2, from lowest (Q1) to highest (Q5) levels of steady-state H2A.Z binding in Anp32e-depleted neurons. In scramble controls, all quintiles showed reduced H2A.Z binding upon KCl treatment (Figure 6D, left), but Anp32e-depleted neurons displayed notable H2A.Z loss only in Q1 (Figure 6D, right). Alternatively, when promoters were parsed into quintiles based on H2A.Z enrichment changes based on depolarization (Q1, greatest reduction; Q5, greatest increase with KCl), 3 of the 5 quintiles showed significant loss of H2A.Z in scramble control (Figure 6E, left), but in Anp32e-depleted neurons there was an opposite pattern (Figure 6E, right). This indicates that promoters that lost H2A.Z upon depolarization (Q1 and Q2) gained H2A.Z in Anp32e-deficient neurons. Thus, even though Anp32e depletion does not prevent global H2A.Z removal (Figure 6B), H2A.Z levels do appear to be oppositely influenced by depolarization and Anp32e loss at a similar subset of genomic loci.
To test whether activity-induced H2A.Z changes were statistically more likely to be regulated by Anp32e, we quantified the number of promoters exhibiting opposite changes in H2A.Z levels. Specifically, we compared gene promoters with the highest KCl-mediated H2A.Z reduction in scramble control (Q1) to genes with the highest H2A.Z accumulation upon Anp32e loss (Q5). This revealed that 23% of promoters were regulated by Anp32e and are subject to activity-mediated H2A.Z removal (p < 2.01e-30) (Figures 6F and S6C), suggesting that Anp32e may indeed operate during or upstream of activity-mediated H2A.Z removal. In fact, we observed a similar level of overlap (24%) when we compared promoters that displayed the opposite patterns, H2A.Z increases upon KCl treatment and reduction upon Anp32e depletion (p < 3.05e-49). This overlap of seemingly indirect H2A.Z changes from each independent experiment further supports mechanistic interaction between Anp32e function and activity-mediated H2A.Z removal. We observed no significant overlap in promoters where H2A.Z changes occurred in parallel directions (e.g., Q1 with Q1, and Q5 with Q5) (Figure 6F). Thus, even though Anp32e depletion does not prevent H2A.Z removal from chromatin, there is imperfect overlap between the sites that exhibit activity-induced H2A.Z removal and baseline Anp32e-dependent H2A.Z regulation.
Anp32e loss alters the transcription of activity-induced genes
To test whether Anp32e regulates activity-induced gene expression, neurons were infected with shAnp32e on 1 DIV and treated with 55 mM KCl for 30 min, followed by 1 h of fresh media on 8 DIV (Figure 6G). Gene expression changes followed several patterns, including decreased or impaired induction in Anp32e-depleted neurons despite similar basal expression levels (interaction: Egr3: F1, 16 = 9.94, p = 0.006; Gadd45b: F1, 16 = 11.50, p = 0.004; Bdnf4: F1, 16 = 4.98, p = 0.04), decreased basal and KCl-induced expression with Anp32e depletion (interaction: Arc: F1, 16 = 7.65, p = 0.01; Nr4a2: F1, 16 = 14.92, p = 0.001), and reduced overall expression with Anp32e depletion (main effect of virus: Ptk2b: F1, 16 = 121.69, p < 0.0001; Grin1: F1, 16 = 5.09, p = 0.038). Additionally, some genes had higher overall expression with Anp32e depletion (main effect of virus: Kcng1: F1, 16 = 69.08, p < 0.0001; Gria4: F1, 16 = 70.13, p < 0.0001; Tufm: F1, 16 = 34.58, p < 0.0001; JunD: F1, 16 = 16.07, p = 0.001; Kcnf1: F1, 16 = 40.38, p < 0.0001), or higher overall and KCl-induced expression (interaction: Cited2: F1, 16 = 9.13, p = 0.008; Megf10: F1, 16 = 10.82, p = 0.005). Only Fos was not affected by Anp32e depletion (Figure 6H). Together, these data indicate that Anp32e depletion has a complex effect on activity-mediated gene induction, such that some genes show decreased and others show increased inducibility when Anp32e is absent.
Anp32e and H2A.Z co-localize in mouse hippocampal chromatin and are co-evicted during memory consolidation
Having demonstrated that Anp32e regulates H2A.Z binding and neuronal function in cultured hippocampal neurons, we investigated whether the same relationship holds true in vivo. First, we confirmed that H2A.Z and Anp32e bind to similar loci in the mouse hippocampus using ChIP experiments with IgG as control (Figures S7A–S7C). Next, we performed sequential ChIP, in which we first immunoprecipitated H2A.Z-bound chromatin and subsequently reprobed the isolated material with an antibody against Anp32e to assess their co-occupancy in hippocampal chromatin (Figure 7A). IgG was used as a control for non-specific binding in the second ChIP. Using candidate regions with known H2A.Z binding sites in the hippocampus (based on our previous studies; Stefanelli et al., 2018; Zovkic et al., 2014), we found that Anp32e co-localized with H2A.Z compared to IgG on most of the sites we examined (Figure 7B), including the +1 nucleosome of genes encoding Fos (t10 = 2.59, p = 0.027) and Pmvk (t10 = 2.62, p = 0.026), as well as the coding regions of Fos (t10 = 4.20, p = 0.002) and Egr2 (t10 = 3.20, p = 0.01) (Figure 7B), suggesting that Anp32e also regulates H2A.Z binding in vivo.
Figure 7. Anp32e affects learning-induced transcription and memory formation.
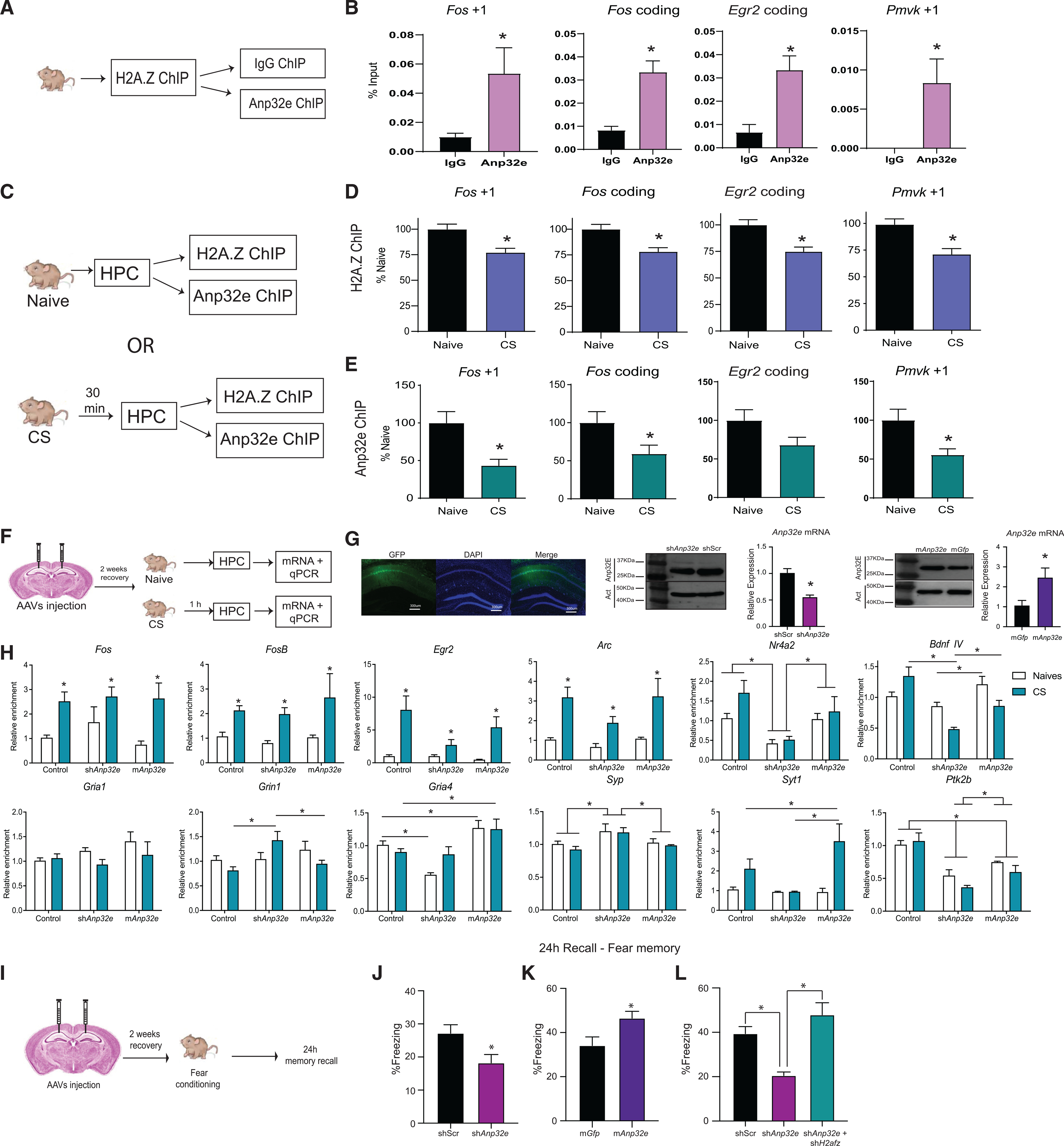
(A) Schematic representation of the ChIP-re-ChIP workflow. Mouse hippocampus was extracted and processed first for H2A.Z ChIP, followed by a second ChIP against Anp32e, using IgG as a control for non-specific binding.
(B) ChIP-re-ChIP showing Anp32e (pink) enrichment on H2A.Z-bound genomic sites (n = 5 or 6/group).
(C) Schematic representation of the experimental workflow.
(D and E) Separate ChIP analyses for (D) H2A.Z and (E) Anp32E were conducted for samples from the same mice under baseline conditions and 30 min after contextual fear conditioning. Both proteins had reduced binding at the first nucleosome downstream (+1 nucleosome) of the TSS and in the coding region of several H2A.Z-associated genes 30 min after fear conditioning (n = 15–25/group). Data are expressed as mean ± SEM. p values are from two-tailed unpaired t tests. *p ≤ 0.05.
(F) Schematic representation of the experimental workflow. Mice received stereotaxic injections of AAV vectors to either deplete or overexpress Anp32e and effects on learning-induced gene expression were assessed 1 h after contextual fear conditioning.
(G) Representative image of virus spread in the hippocampus (left) and mRNA levels and western blots showing Anp32e depletion and overexpression in areaCA1.
(H) AAV-mediated gene depletion or overexpression of Anp32e alters baseline and learning-induced gene expression of neuronal genes 1 h after fear conditioning(n = 3–6/group). *p ≤ 0.05.
(I) Schematic representation of the surgeries and behavioral testing timeline.
(J) Anp32e depletion impairs fear memory (n = 16/group).
(K) Anp32e overexpression enhances fear memory (n = 16/group).
(L) Simultaneous knockdown of Anp32e and H2afz (green bar) restores fear memory (n = 8 or 9/group). Data are represented as mean ± SEM. *p ≤ 0.05.
To determine whether Anp32e is removed along with H2A.Z during learning, we conducted standard ChIP protocols against H2A.Z or Anp32e from the same mouse sample 30 min after fear conditioning (Figure 7C). Fear conditioning resulted in H2A.Z removal from nucleosomes directly downstream (+1 nucleosome) of the TSS on genes encoding Fos (t16 = 3.005, p = 0.008) and Pmvk (t17 = 2.96, p = 0.009), and from coding regions of Fos (t16 = 2.27, p = 0.037) and Egr2 (t17 = 2.71, p = 0.015) (Figure 7D). Notably, Anp32e was also removed from H2A.Z binding sites after fear conditioning at +1 nucleosomes of Fos (t19 = 3.02, p = 0.007) and Pmvk (t19 = 2.50, p = 0.02), as well as the coding region of Fos (t19 = 2.09, p = 0.050) (Figure 7E). These data indicate that, like in cultured neurons, Anp32e co-occupies H2A.Z binding sites and is removed along with H2A.Z in response to fear conditioning.
Anp32e impacts learning-induced transcription in the mouse hippocampus
Learning-induced H2A.Z removal in the mouse hippocampus is associated with increased transcription 30 min and 1 h after learning (Stefanelli et al., 2018). Given that Anp32e promotes H2A.Z accumulation, we tested whether altering Anp32e levels impacts baseline and learning-induced transcription of known H2A.Z targets in vivo. We induced AAV-mediated depletion or overexpression of Anp32e in area CA1 of the hippocampus via stereotaxic injection. Efficient targeting of area CA1 and knockdown or overexpression of Anp32e mRNA (shAnp32e: t8 = 5.130, p = 0.0009; mAnp32e: t7 = 2.401, p = 0.04) and protein were validated (Figures 7F, 7G, S7D, and S7E). After 2 weeks of recovery, mice were separated into 2 groups: naive mice that did not undergo any behavioral testing, or fear-conditioned mice (context and shock; CS) that were trained to associate a novel context with a foot shock (3 foot shocks, 0.5 mA, 2 s in duration) 1 h before tissue collection (Figure 7F). An assessment of gene expression in the infected region showed that the immediate-early genes Egr2 (main effect of training: F1, 31 = 11.73, p = 0.002), Arc (main effect of training: F1, 31 = 25.54, p < 0.0001), Fos (main effect of training: F1, 30 = 18.73, p < 0.001), and FosB (main effect of training: F1, 31 = 31.80, p < 0.0001) showed no change at baseline, and fear conditioning resulted in increased transcription irrespective of Anp32e manipulation (Figure 7H).
In contrast, Anp32e impacted the expression of the activity-dependent genes Nr4a2 and Bdnf4, whereby Anp32e depletion reduced Nr4a2 levels compared to controls (p < 0.0001) and Anp32e-overexpressing mice (p = 0.03), whereas Anp32e overexpression had no effect (Figure 7H). At baseline, Anp32e overexpression increased Bdnf4 levels compared to Anp32e depletion (treatment × training interaction: F2, 31 = 6.33, p = 0.005), but neither group differed from controls. After fear conditioning, Anp32e depletion reduced Bdnf4 compared to controls (p = 0.001), whereas Anp32e overexpression showed a trend for reduced Bdnf4 expression compared to controls (p = 0.058) (Figure 7H). Among genes encoding glutamate receptor subunits, there were no differences in Grin1 at baseline (treatment × training interaction: F2, 31 = 4.13, p = 0.03), but Anp32e depletion increased Grin1 after fear conditioning (F2, 15 = 8.31, p = 0.004) compared to controls (p = 0.001) and Anp32e-overexpressing mice (p = 0.03), which did not differ from each other (Figure 7H). Gria4 expression was reduced by Anp32e depletion (treatment × training interaction: F2, 31 = 3.91, p = 0.03) at baseline (F2, 16 = 19.72, p < 0.0001) compared to controls (p < 0.0001) and enhanced by Anp32e overexpression (p < 0.0001), but only Anp32e overexpression increased Gria4 expression compared to controls after fear conditioning (F2, 15 = 3.91, p = 0.04). Gria1 did not display any group differences (Figure 7H).
The synaptic marker synaptophysin (Syp) (main effect of virus: F2, 31 = 7.24, p = 0.003) was higher with Anp32e depletion compared to controls (p = 0.001) and Anp32e-overexpressing mice (p = 0.02) (Figure 7H). Anp32e overexpression only increased synaptotagmin (Syt1) expression (interaction: F2, 31 = 3.93, p = 0.03) after fear conditioning (F2, 15 = 3.59, p = 0.05) compared to Anp32e depletion (p = 0.02) (Figure 7H). Lastly, Ptk2b, a protein involved in neuronal signaling (main effect of virus: F2, 31 = 20.25, p < 0.0001), was reduced by Anp32e depletion (p < 0.0001) and overexpression (p = 0.002) compared to controls (Figure 7H), whereas levels were increased with Anp32e overexpression compared to Anp32e depletion (p = 0.05). Taken together, these results suggest that Anp32e influences transcription of a subset of H2A.Z target genes, and many transcriptional changes occur in the opposite direction when Anp32e is increased versus decreased, suggesting that both Anp32e manipulations may be functionally relevant for memory formation.
Manipulation of Anp32e regulates memory formation in an H2A.Z-dependent fashion
To assess the functional role of Anp32e in memory, mice were injected with AAV vectors into area CA1 of the hippocampus to overexpress or deplete Anp32e. After 2 weeks of recovery, mice were trained on contextual fear conditioning (single foot shock, 0.5 mA, 2 s in duration), where they learned to associate a mild foot shock with a novel context. Contextual fear memory was assessed based on the amount of freezing observed 24 h later in the absence of shock (Figure 7I). Consistent with a negative effect of H2A.Z on fear memory (Stefanelli et al., 2018; Zovkic et al., 2014), Anp32e depletion impaired (t31 = 2.485, p = 0.01) (Figure 7J) and Anp32e overexpression enhanced (t30 = 2.48, p = 0.02) (Figure 7K) fear memory compared to control mice. To directly evaluate the hypothesis that impaired memory is a result of H2A.Z accumulation, we co-depleted Anp32e and H2A.Z in area CA1 and observed a complete rescue of fear memory, whereas Anp32e again impaired memory compared to scramble controls (one-way ANOVA: F2, 23 = 15.30, p < 0.0001; Tukey’s multiple comparison: shScr versus shAnp32e, p = 0.002; shAnp32e versus shAnp32e + shH2afz, p < 0.0001) (Figure 7L). These data provide independent validation of our previous assertion that H2A.Z accumulation in chromatin has a negative impact on memory strength (Zovkic et al., 2014; Zovkic and Walters, 2015; Stefanelli et al., 2018) and suggest that Anp32e is a target for regulating memory formation through its actions on the histone variant H2A.Z.
DISCUSSION
Our data reveal a function of Anp32e in behavioral regulation and provide evidence for a dissociable role of Anp32e in steady-state versus activity-mediated regulation of H2A.Z binding. Specifically, our data support prior observations that Anp32e regulates H2A.Z removal from chromatin of dividing cells (Mao et al., 2014; Obri et al., 2014) and demonstrate that Anp32e also regulates basal chromatin levels of H2A.Z in post-mitotic neurons. We show that Anp32e is responsible for keeping H2A.Z levels in check, whereby the loss of Anp32e leads to H2A.Z accumulation. However, Anp32e depletion did not prevent H2A.Z removal in response to neuronal activity, indicating that the primary role of Anp32e may be to regulate basal H2A.Z turnover, whereas alternative mechanisms are employed to achieve activity-induced H2A.Z removal. Several studies indicate that H2A.Z in the +1 nucleosome is repressive to transcriptional elongation (Cole et al., 2021; Mylonas et al., 2021), whereas active transcription antagonizes excessive H2A.Z incorporation at transcribed regions in human U2OS cells (Hardy et al., 2009). In addition, global inhibition of transcription with alpha-amanitin increases H2A.Z incorporation at gene bodies in HCT116 cells (Lashgari et al., 2017), suggesting that some aspect of active transcription is involved in H2A.Z removal. In yeast, H2A.Z removal from promoters is regulated by RNA polymerase II (RNA Pol II) during transcription initiation (Ranjan et al., 2020), suggesting that transcription by RNA Pol II may also regulate H2A.Z occupancy and activity-mediated removal from neuronal chromatin. Thus, we speculate that activity-mediated H2A.Z removal is regulated by an interplay of chaperones and active transcription, similar to reports that H3.3 binding is regulated both by its chaperone HIRA and proteasome-mediated degradation (Maze et al., 2015). Together, these data indicate that histone variant dynamics in the brain are tightly regulated through multiple interacting factors.
Consistent with prior evidence for bidirectional effects of H2A.Z on transcription in the mouse hippocampus (Zovkic et al., 2014), our data show that Anp32e depletion also has a bidirectional impact on gene expression. Notably, H2A.Z binding was positively associated with basal transcription, as previously reported (Stefanelli et al., 2018), and DEGs that accumulated H2A.Z with Anp32e depletion were more strongly linked with increased than with decreased gene expression, reinforcing a tendency toward a positive link between H2A.Z and basal transcription. Indeed, co-depletion of H2A.Z with Anp32e partially reversed transcriptional consequences of H2A.Z depletion, indicating that Anp32e mediates transcription in large part through effects on H2A.Z.
We also describe an H2A.Z-dependent function of Anp32e in regulating dendritic arborization and memory formation. Anp32e was first identified in the cerebellum as a protein belonging to the Anp32 family of protein phosphatase 2 (PPP2A) inhibitors (Matilla and Radrizzani, 2005), where it regulates cerebellar synaptogenesis by inhibiting PPP2 activity at synapses (Costanzo et al., 2006). However, studies in cell lines and primary cells show that Anp32e is vital for regulating H2A.Z functions (Mao et al., 2014; Obri et al., 2014; Gursoy-Yuzugullu et al., 2015; Dunn et al., 2017) and our data extend these findings to the hippocampus. Using multiple strategies in cultured neurons and the mouse hippocampus, we show that Anp32e has extensive effects on dendritic morphology, transcription, and memory that are mediated by its effects on H2A.Z binding in neuronal chromatin. Specifically, we show that Anp32e and H2A.Z co-depletion partially rescues the effects of Anp32e loss on transcription and completely abolishes the detrimental effect of Anp32e depletion on dendritic arborization and memory formation, suggesting that Anp32e exerts functional effects in the hippocampus primarily by regulating H2A.Z. In contrast, Anp32e overexpression, which reduces chromatin levels of H2A.Z in HeLa cells (Mao et al., 2014), improved memory without influencing dendritic branching. Indeed, H2A.Z depletion alone also did not impact dendritic branching, indicating that H2A.Z accumulation has greater influence than its depletion. These data are consistent with evidence that knocking down H2A.Z enhances fear memory (Stefanelli et al., 2018; Zovkic et al., 2014; Ramzan et al., 2020) and suggest that Anp32e may be a useful therapeutic target for improving memory impairment. Indeed, basal H2A.Z binding increases in the aging mouse hippocampus (Stefanelli et al., 2018), and future experiments will determine whether age-related loss of Anp32e plays an active role in this process.
Notably, our finding that H2A.Z depletion does not impact dendritic morphology in cultured neurons contrasts with prior reports of impaired cortical neurogenesis with H2A.Z deletion (Shen et al., 2018). This difference is likely associated with methodological differences, whereby previous work utilized neural progenitor cells (Shen et al., 2018) derived from conditional knockout mice and we used primary neurons infected with shRNA against H2A.Z. In our preparation, neurons have already committed to their cell fate (in contrast to progenitor cells) before H2A.Z depletion and spend ~2 days in culture before infection begins. As such, the timing and modality of H2A.Z depletion likely account for distinct outcomes, indicating a precise developmental role for H2A.Z in regulating neuronal morphology.
Results of this study and others (Mao et al., 2014; Obri et al., 2014) are consistent with Anp32e functioning as a chaperone that regulates steady-state H2A.Z levels in chromatin through H2A.Z removal. Our data suggest that Anp32e primarily maintains homeostatic levels of H2A.Z to avoid improper accumulation of the histone, while playing a restricted role in activity-dependent removal. This conclusion is consistent with evidence that H2A.Z accumulates upon double-strand breaks, followed by recruitment of Anp32e to remove excess H2A.Z and resolve the break (Gursoy-Yuzugullu et al., 2015). Notably, we show that Anp32e is present in chromatin with H2A.Z and is simultaneously removed when neurons are depolarized. Although the reason for this co-occupancy is unclear, the fact that H2A.Z accumulates when Anp32e is knocked down suggests that steady-state binding of Anp32e to chromatin may be important for keeping H2A.Z levels in check. The observation that H2A.Z accumulation at double-strand breaks precedes Anp32e binding (Gursoy-Yuzugullu et al., 2015) indicates that H2A.Z levels may regulate Anp32e recruitment, such that their co-removal with KCl treatment may be a consequence of reduced H2A.Z levels in chromatin. Although H2A.Z is subject to activity-mediated removal even when Anp32e is depleted, this removal occurs on distinct loci, indicating that Anp32e may nevertheless have some capacity to regulate activity-mediated H2A.Z removal. Indeed, it is likely that Anp32e contributes to H2A.Z removal in 2 ways, including direct removal that primarily regulates to steady-state H2A.Z levels, and a less predominant pathway in which Anp32e, directly or indirectly, contributes to activity-dependent H2A.Z removal.
Conclusions
Overall, our findings elucidate the function of Anp32e in the brain, demonstrating that Anp32e is a key regulator of H2A.Z binding at TSS-proximal sites and CpG islands, but that alternative mechanisms are also involved in activity-induced H2A.Z removal. Notably, we showed that effects of H2A.Z on memory can be reproduced by indirect manipulation through a chaperone rather than direct knockdown of the histone itself. Combined with the overwhelmingly negative impact of H2A.Z accumulation on several functional aspects in the mouse hippocampus, these data suggest that Anp32e may be a strong therapeutic candidate for H2A.Z regulation in age-related cognitive decline, neurodegenerative conditions, as well as cancers with altered H2A.Z binding.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| H2A.Z | Millipore | ABE1348 |
| H3 | Millipore | Millipore Cat# 05–499, RRID:AB_309763 |
| Anp32e | Abcam | Ab5993 |
| GFP | Thermo Fisher | Cat# PA1–9533, RRID:AB_1074893 |
| Alexa Fluor 488 | Thermo Fisher | Cat# A-11039, RRID:AB_2534096 |
| Beta actin | Cell Signaling | Cat# 4967, RRID:AB_330288 |
| Anti rabbit HRP | Cell Signaling | Cat# 7076, RRID:AB_330924 |
| Anti mouse HRP | Cell Signaling | Cat# 7074, RRID:AB_2099233 |
| Protein G magnetic beads | Millipore | 16–662 |
|
Bacterial and virus strains | ||
| AAV/DJ | Cell Biolabs | VPK-400-DJ |
|
Biological samples | ||
| Mouse hippocampi | C57BL/6j mice | N/A |
|
Critical commercial assays | ||
| Bradford reagent | Bioshop | BRA222.250 |
| EZ-10 spin column total RNA extraction kit | Biobasic | BS82322 |
| cDNA Reverse Transcription | Applied biosystems | 4368814 |
| Direct-zol RNA mini prep Kit | Zymo research | R2050 |
| PCR purification kit | Biobasic | BS664 |
| QIAquick PCR purification Kit | QIAGEN | 28104 |
|
Deposited data | ||
| ChIP-seq data | This paper | GEO: GSE157006 |
| RNA-seq data | This paper | GEO: GSE157006 |
|
Experimental models: Cell lines | ||
| Primary hippocampal neurons | E16 mice brain | N/A |
|
Experimental models: Organisms/strains | ||
| C57BL/6J mice | Jackson laboratories | 00664 |
|
Oligonucleotides | ||
| cDNA primers | Sigma Aldrich and Life Technologies. See Table S5 for sequences | N/A |
| gDNA Primers | Sigma Aldrich and Life Technologies. See Table S4 for sequences | N/A |
|
Software and algorithms | ||
| ImageJ/Fiji | NIH | https://imagej.net/software/fiji/ |
| GraphPad Prism 9 | GraphPad Software Inc, USA | https://www.graphpad.com/scientific-software/prism/ |
| SPSS | IBM | https://www.ibm.com/products/spss-statistics? |
| Trimmomatic | Bolger et al., 2014 | The Usadella lab |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| SAMtools suite (v1.9) | Li et al., 2009 | https://sourceforge.net/projects/samtools/files/samtools/ |
| MACS2 (v 2.1.4) | ||
| deepTools (v 3.1.3) | https://deeptools.readthedocs.io/en/develop/ | |
| Bedtools (v 2.29.2) | https://bedtools.readthedocs.io/en/latest/ | |
| HOMER (v 4.10) | http://homer.ucsd.edu/homer/ | |
| EnrichR | Chen et al., 2013 | https://maayanlab.cloud/Enrichr/ |
| GREAT | McLean et al., 2010 | http://great.stanford.edu/public/html/ |
| RNA-STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| DESEQ2 | Love et al., 2014 | https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8 |
|
Other | ||
| ProLong Gold anti-fade reagent | Cell Signaling | 9071 |
| Protease inhibitors | Cell Signaling | 5871S |
| LiCl immune complex | Millipore | 20–156 |
| Proteinase K | Roche | 3115828001 |
| DAPI | Cell Signaling | 4083S |
| HBSS | GIBCO | 14175095 |
| Trypsin | Life Technologies | 15050065 |
| Poly-L-Lysine | Sigma | P2636 |
| Neurobasal | GIBCO | 21103049 |
| B-27 | Life Technologies | 17504044 |
| L-glutamine | GIBCO | 25030081 |
| DNase | QIAGEN | 79254 |
| PVDF | Millipore | IPVH00010 |
| MNase | Cell Signaling | 10011S |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Iva Zovkic (iva.zovkic@utoronto.ca).
Materials availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Data and code availability
ChIP-seq and RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Male C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) at 10 weeks of age. Mice were housed in groups on 12h light cycle and had ad libitum access to food and water. All procedures were approved by the University of Toronto animal care committee and complied with institutional guidelines and the Canadian Council on Animal Care.
Primary hippocampal neurons and treatments
Hippocampi were isolated from E17 pups, washed in HBSS (GIBCO, 14175095), digested with Trypsin 0.25% (Life Technologies, 15050065), and mechanically dissociated by pipetting. Cells were plated on poly-L-lysine (Sigma; P2636, 0.1 mg/mL) at a density of 250.000/well in a 6-multiwell plate for qPCR experiments and 266.000/well for ChIP seq. Neurons were grown in a culture medium containing Neurobasal (GIBCO, 21103049), B-27 (Life Technologies, 17504044) and L-glutamine (GIBCO, 25030081). Neurons were infected at 1 day in vitro (DIV) using 30.000 vg/cell. 8 days after infection, KCl was added to a final concentration of 55mM directly into culture media for 30 minutes. Vehicle groups were left untreated.
METHOD DETAILS
Fear conditioning
Mice were habituated to handling for 3 days before fear conditioning by transferring them from the colony to the testing room for 30 s of handling daily. For gene expression and ChIP studies, mice were assigned to one of 2 groups: Home cage Naive (N) controls or Context and Shock (CS) fear conditioned mice. On the test day, CS mice were placed in the fear conditioning chamber and given 2 minutes to explore, followed by 3 electric foot-shocks (0.5 mA; 2 s in duration) administered 1 min apart, with an additional minute of exploration before removal from the chamber. Mice were euthanized 30 min after fear conditioning for ChIP and 1h after fear conditioning for gene expression. Naive mice remained in their home cage until tissue collection.
For tests of memory retention, training consisted of 3 min of exploration and a single foot-shock (0.5 mA, 2 s duration), with an additional 30 s for exploration. This group was returned to the shock context 24 h after CS and allowed to explore for 3 minutes. Freezing behavior was scored by unbiased automated software (FreezeFrame, Coulbourn Instruments).
Stereotaxic surgery for viral delivery
Mice were anaesthetized with isoflurane and secured in a Kopf stereotaxic apparatus. Viral particles were bilaterally delivered into the hippocampus (anterior/posterior −2; medial/lateral 1.5; dorsal/ventral −1.6; 1.0 μL per hemisphere) at a rate of 225 nl min −1. Mice were allowed 2 weeks to recover before testing.
AAV production
Viral knockdown of Anp32e was achieved using a bicistronic vector encoding an Anp32e short hairpin RNA (shRNA) driven by the U6 promoter and CMV driving GFP expression. Viral overexpression of Anp32e was achieved using a vector encoding Anp32e ORF under CMV promoter followed by p2A autocleaving sequence and GFP ORF. All vectors were packaged in AAV serotype DJ at St. Jude Children’s Research Hospital. Briefly, AAV vector production was initiated by PEI- (polyethyleneimine “max,” Polysciences cat no 24765) mediated plasmid transfection. After three days, cell pellets were harvested and lysed by repeated freeze-thawing, while supernatants were pegylated with 40% polyethylene glycol. Both were treated with benzonase and then subjected to cesium chloride step gradient separation. The lower, virus-containing fraction was collected and dialyzed against PBS and concentrated by Amicon filter (100kDa) before titering by qPCR. AAV vectors were tittered by qPCR using serial dilutions of virus compared to linearized plasmid standards (25 ul reactions with primers to CMV promoter: Forward CMV primer: ATATGCCAAGTACGCCCCCTATTGAC and Reverse CMV primer: ACTGCCAAGTAGGAAAGTCCCATAAGGTC, performed on Applied Biosystems 7500 machine). The following shRNA sequence for Anp32e was employed: TTGGAACTTAGTGACAATAT. A scrambled shRNA sequence expressing eGFP was used a control. Anp32e overexpression construct is made from the coding sequence of Anp32e with the stop codon removed for P2A and GFP incorporation.
Morphological analyses
Neurons were plated on glass coverslips in 24 well plates at a density of 10.000 cells/well. One day after plating, neurons were infected using 1000 vg/cell. 7 days after infection, neurons were fixed in 4% PFA for 20 min at RT, permeabilized and blocked for 1 h with 5% Horse serum, 0.2% Triton X-100 in TBS. An anti GFP antibody (Thermo Fisher, PA19533, 1:1000) was added overnight (4 °C). Slides were incubated with the secondary antibody (Thermo Fisher, Alexa Fluor 488, A11039, 1:5000) for 1 h at RT and washed. DNA was counter stained with DAPI (Cell Signaling, 4083S). Slides were mounted with ProLong Gold Antifade Reagent (Cell Signaling, 9071). Images were acquired using Cytation5 (BioTek). Sholl analysis was performed using the dedicated plugin of Fiji (ImageJ, NIH).
mRNA extraction for RT-PCR and sequencing
A fluorescent lamp was used to dissect the area of the hippocampus expressing eGFP and RNA was extracted using the EZ-10 spin column total RNA extraction kit (BioBasic, BS82322) with an added DNase step (QIAGEN, 79254). For primary neurons, media was removed and cells were scraped in lysis buffer provided in the EZ-10 total RNA extraction kit. Complementary DNA was synthesized using high-capacity cDNA Reverse Transcription Kit (Applied BioSystems, 4368814). Primers were designed in the lab and used to detect levels of the indicated transcripts, and data were normalized to the geometric mean of β-actin and GAPDH. The list of primer sequences is provided in Table S4.
For RNA sequencing, neurons from 3 biological replicates per condition were lysed in trizol and mRNA was extracted using Direct-zol RNA mini prep kit (Zymo Research R2050). mRNA was purified using standard Illumina polyA selection and then sequenced using paired-end 150bp Illumina sequencing.
Protein extraction and western blotting
A fluorescent lamp was used to dissect the area of the hippocampus expressing eGFP. Tissues were homogenized using a dounce homognizer in RIPA buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 10% NP-40, 0.5% sodium deoxycolate, 0.1% SDS) supplemented with Protease Inhibitor Cocktail (Cell Signaling, 5871S). Homogenates were incubated for 20 min on ice, centrifuged at maximum speed at 4 °C for 15 minutes, and the supernatant was collected and quantified using the Bradford assay (BioShop, BRA222.250). For primary hippocampal neurons, media was removed, and wells were washed with HBSS (GIBCO, 14175095). 200ul of 6X sample buffer was added to each well, cells were scraped, and lysates collected. Equal volumes of each sample were loaded on the gel. All protein lysates were boiled at 95°C for 5 min before loading. Proteins were separated on a 15% gel using SDS-PAGE, and then transferred to a PVDF membrane (Millipore, IPVH00010). Membranes were blocked in TBS-T containing 5% milk for 1hr at RT, and then incubated with primary antibodies (Anp32e 1:1000, Abcam ab5993; β-Actin 1:10.000, Cell Signaling 4967S, H3 1:1000, Millipore 05–499) overnight at 4 °C. After washing, membranes were incubated with the appropriate secondary antibody (1:10.000 Cell Signaling, 7076 or 7074) for 1 hr at RT. Detection was performed by enhanced chemiluminescence (Pierce, 32106). For quantitative measures, autoradiographs were acquired using ImageQuant LAS 500 (GE). Exposure times were determined experimentally and varied according to antibody and sample, and images were saved as tiff files. Images were not manipulated from acquisition and image analysis and quantification were performed using Fiji (ImageJ, NIH).
Chromatin salt fractionation
Protocol was adapted from the Henikoff lab (Teves and Henikoff, 2012). Briefly, approximately 1.5 million neurons were resuspended in 1 mL of TM2 buffer (10mM Tris-HCl, pH7.4, 2mM MgCl2, 0.5mM PMSF). Cells were lysed by addition of NP-40 to a final concentration of 0.5% and incubation on ice for 3 min. Nuclei were collected by centrifugation (100 × g 10 min 4°C) and resuspended in 400ul of TM2 buffer. Nuclei were pre warmed at 37°C for 5 min and MNase (Cell Signaling Technologies 10011S) was added to a final concentration of 1.25 U/ml for 10 min at 37°C. EGTA to a final concentration of 2mM was added to stop the reaction. 50ul was taken as nuclei, remaining 350ul was spun down (100 × g 10 min 4°C). Supernatant was collected and represents soluble nuclear fraction. At this point nuclei were resuspended in 300ul of 150mM salt buffer (140mM NaCl, 10mM Tris-HCl pH7.4, 2mM MgCl2, 2mM EGTA, 0.1% Triton X-100, 0.5mM PMSF) 1h at 4°C. Nuclei were centrifuged (100 × g 10 min 4°C) and supernatant collected as 150mM fraction. 300ul of 600mM salt buffer (585mM NaCl, 10mM Tris-HCl pH7.4, 2mM MgCl2, 2mM EGTA, 0.1% Triton X-100, 0.5mM PMSF) was added 1h 4°C. Nuclei were centrifuged and supernatant collected as 600mM fraction. Nuclei were then resuspended in 300ul of TNE buffer (10mM Tris-HCl pH7.4, 200mM NaCl, 1mM EDTA) and labeled as Pellet fraction. Equal volume of each fraction was loaded on a 15% SDS-PAGE for protein detection.
Chromatin immunoprecipitation (ChIP)
ChIP from hippocampi was conducted in accordance with previously reported methods (Stefanelli et al., 2018). Briefly, hippocampi were incubated in PBS 1% formaldehyde for 10 min at 37°C, after which 1.25M glycine was added to quench the reaction. Next, samples were washed with PBS, and SDS lysis buffer (50 mM Tris pH7.4, 10mM EDTA, 1% SDS) was added to all samples prior to sonication [40% power, 6X for 10 s with 50 s rest (Fisher Scientific)]. Samples were centrifuged at 17,000x g for 10 minutes, aliquoted, and diluted with ChIP dilution buffer (16mM Tris pH 7.4, 0.01% SDS, 1% Triton X-100, 1.2mM EDTA, 170 mM NaCl)
For primary hippocampal neurons, cultures were fixed on the plates and then scraped and resuspended in buffer B (50mM NaCl, 10mM Pipes pH6.8, 5mM MgCl2, 1mM CaCl2), incubated for 5 min at 37°C and digested using 100U of MNase (Cell Signaling, 1011S) for 15 min at 37°C. MNase digestion was halted by adding EDTA to a final concentration of 5mM. SDS was added to a final concentration of 1% and samples were aliquoted and diluted with ChIP dilution buffer.
Both hippocampal and neuronal samples were treated with 20ul of Protein G magnetic beads (Millipore, 16–662) and 1ul of H2A.Z (Millipore, ABE1348) or 1 ul of Anp32e (Abcam, Ab5993) antibody overnight at 4°C. The next day, samples were washed sequentially with low-salt (20mM Tris pH7.4, 0.1% SDS, 1% Triton X-100, 2mM EDTA, 150mM NaCl), high-salt (20mM Tris pH7.4, 0.1%SDS, 1% Triton X-100, 1mM EDTA, 500mM NaCl), LiCl (Millipore, 20–156) and TE (10mM Tris pH7.4, 1mM EDTA) buffers, and incubated while rotating for 5 min between washes. Immune complexes were extracted using TE Buffer and proteinase K (Roche, 3115828001) for both ChIP and Input samples, and heated at 65°C for 2 h, followed by 95°C for 10 min before purification with PCR Purification Kit (Biobasic, BS664) or QIAquick PCR Purification Kit (QIAGEN, 28104) for sequencing samples. Primers were designed to detect specific sequences (Table S5) and ChIP data were calculated as % input, then normalized to the respective control samples.
For the ChIP-re-ChIP assay, an additional step was added after the overnight incubation with H2A.Z Ab. Specifically, immune complexes were washed and released from beads using TE 1%SDS with the addition of 50mM DTT 15 min at 37°C. H2A.Z bound material was diluted again in ChIP dilution buffer and 1ul of Anp32e antibody or IgG was added overnight with 20ul of beads. The remining steps were carried out as outlined above.
Bioinformatics
DNA libraries were sequenced from 3 biological replicates for each condition (~25 million reads, 100 bp pair end) on an Illumina sequencing platform (HiSeq 4000) by Genome Quebec in Montreal, Canada. Trimmomatic (Bolger et al., 2014) was used to trim paired and adaptors and filter out low quality reads using standard settings. Reads were aligned to the mouse NCBI genome version 38 (mm10) using Bowtie2 (v2.3.4.3) in local mode (Langmead and Salzberg, 2012). The resulting Sequence Alignment Map (SAM) files were converted to Binary Alignment Map (BAM) files and indexed using the SAMtools suite (v1.9) (Li et al., 2009). The data were analyzed using relative enrichment measurements at genomic landmarks, including established H2A.Z peaks in the mouse hippocampus (called by MACS2, version 2.1.4), CpG islands, and transcription start sites. Read counts were normalized using deepTools (version 3.1.3) and replicates were assessed for similarities and merged based upon spearman correlations. Data was parsed and log fold change was calculated using standard methods in R, based on values acquired from deepTools multiBigWigSummary. Intersection of regions was conducted with bedtools (2.29.2.) HOMER (version 4.10) was used for regional annotation, which allowed for the scoring of CpG density and identification of nearest proximal genes. Gene ontology was performed using Enrichr (Chen et al., 2013; Kuleshov et al., 2016) and GREAT (McLean et al., 2010) top ten GO Terms were reported. For RNA-Seq, sequencing data was aligned to the mm10 genome using RNA-STAR (Dobin et al., 2013). BAM files were input to DESEQ2 (Love et al., 2014) in order to identify differentially expressed genes. DeepTools was used to collect/compared normalized ChIP-Seq enrichment values and RNA levels (RPKM) at promoters.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analyses were conducted using IBM SPSS Statistics version 26 and consisted of indepdent-samples t tests when only a single variable was manipulated or two-way ANOVA when multiple variables were manipulated. When the omnibus test was significant, post hoc analyses were conducted with LSD post hoc or independent samples t test when appropriate. Number of animals per group is stated in figure legends, as is data representation. Significance was set at p < 0.05. Significant outliers were identified using the Grubbs’s Test for Outliers in GraphPad Prism (https://www.graphpad.com/quickcalcs/Grubbs1.cfm).
Supplementary Material
Highlights.
Anp32e depletion leads to accumulation of H2A.Z in neuronal chromatin
Anp32e depletion impairs dendritic branching via H2A.Z accumulation
Anp32e has a limited role in activity-mediated H2A.Z removal
Anp32e impairs memory formation through H2A.Z accumulation
ACKNOWLEDGMENTS
This research was supported by an NSERC Discovery Grant, NSERC Discovery Accelerator Supplement, and CIHR PJT-156414 (to I.B.Z.). Funding to P.J.M. was from NIGMS MIRA R35 GM137833-01.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109551.
REFERENCES
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole L, Kurscheid S, Nekrasov M, Domaschenz R, Vera DL, Dennis JH, and Tremethick DJ (2021). Multiple roles of H2A.Z in regulating promoter chromatin architecture in human cells. Nat. Commun. 12, 2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo RV, Vilá-Ortíz GJ, Perandones C, Carminatti H, Matilla A, and Radrizzani M (2006). Anp32e/Cpd1 regulates protein phosphatase 2A activity at synapses during synaptogenesis. Eur. J. Neurosci. 23, 309–324. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CJ, Sarkar P, Bailey ER, Farris S, Zhao M, Ward JM, Dudek SM, and Saha RN (2017). Histone hypervariants H2A.Z.1 and H2A.Z.2 play independent and context-specific roles in neuronal activity-induced transcription of Arc/Arg3.1 and other immediate early genes. eNeuro 4, ENEURO.0040–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursoy-Yuzugullu O, Ayrapetov MK, and Price BD (2015). Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl. Acad. Sci. USA 112, 7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy S, Jacques PE, Gévry N, Forest A, Fortin ME, Laflamme L, Gaudreau L, and Robert F (2009). The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet 5, e1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashgari A, Millau JF, Jacques PE, and Gaudreau L (2017). Global inhibition of transcription causes an increase in histone H2A.Z incorporation within gene bodies. Nucleic Acids Res 45, 12715–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Pan L, Wang W, Sun J, Shan S, Dong Q, Liang X, Dai L, Ding X, Chen S, et al. (2014). Anp32e, a higher eukaryotic histone chaperone directs preferential recognition for H2A.Z. Cell Res 24, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla A, and Radrizzani M (2005). The Anp32 family of proteins containing leucine-rich repeats. Cerebellum 4, 7–18. [DOI] [PubMed] [Google Scholar]
- Maze I, Wenderski W, Noh KM, Bagot RC, Tzavaras N, Purushothaman I, Elsässer SJ, Guo Y, Ionete C, Hurd YL, et al. (2015). Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87, 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylonas C, Lee C, Auld AL, Cisse II, and Boyer LA (2021). A dual role for H2A.Z.1 in modulating the dynamics of RNA polymerase II initiation and elongation. Nat. Struct. Mol. Biol 28, 435–442. [DOI] [PubMed] [Google Scholar]
- Narkaj K, Stefanelli G, Wahdan M, Azam AB, Ramzan F, Steininger CFD Jr., Walters BJ, and Zovkic IB (2018). Blocking H2A.Z incorporation via Tip60 inhibition promotes systems consolidation of fear memory in mice. eNeuro 5, ENEURO.0378–18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obri A, Ouararhni K, Papin C, Diebold ML, Padmanabhan K, Marek M, Stoll I, Roy L, Reilly PT, Mak TW, et al. (2014). ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 505, 648–653. [DOI] [PubMed] [Google Scholar]
- Ramzan F, Creighton SD, Hall M, Baumbach J, Wahdan M, Poulson SJ, Michailidis V, Stefanelli G, Narkaj K, Tao CS, et al. (2020). Sex-specific effects of the histone variant H2A.Z on fear memory, stress-enhanced fear learning and hypersensitivity to pain. Sci. Rep 10, 14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Nguyen VQ, Liu S, Wisniewski J, Kim JM, Tang X, Mizuguchi G, Elalaoui E, Nickels TJ, Jou V, et al. (2020). Live-cell single particle imaging reveals the role of RNA polymerase II in histone H2A.Z eviction. eLife 9, e55667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel-Clower NL, Zola SM, Squire LR, and Amaral DG (1996). Three cases of enduring memory impairment after bilateral damage limited to the hippocampal formation. J. Neurosci 16, 5233–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Ji F, Wang Y, Lei X, Zhang D, and Jiao J (2018). Brain-specific deletion of histone variant H2A.z results in cortical neurogenesis defects and neurodevelopmental disorder. Nucleic Acids Res 46, 2290–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanelli G, Azam AB, Walters BJ, Brimble MA, Gettens CP, Bouchard-Cannon P, Cheng HM, Davidoff AM, Narkaj K, Day JJ, et al. (2018). Learning and age-related changes in genome-wide H2A.Z binding in the mouse hippocampus. Cell Rep. 22, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, and Henikoff S (2012). Salt fractionation of nucleosomes for genome-wide profiling. Methods Mol. Biol. 833, 421–432. [DOI] [PubMed] [Google Scholar]
- Weber CM, and Henikoff S (2014). Histone variants: dynamic punctuation in transcription. Genes Dev 28, 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Coleman-Derr D, Ballinger T, and Henikoff S (2008). Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB (2021). Epigenetics and memory: an expanded role for chromatin dynamics. Curr. Opin. Neurobiol 67, 58–65. [DOI] [PubMed] [Google Scholar]
- Zovkic IB, and Walters BJ (2015). H2A.Z helps genes remember their history so we can remember ours. BioEssays 37, 596–601. [DOI] [PubMed] [Google Scholar]
- Zovkic IB, Paulukaitis BS, Day JJ, Etikala DM, and Sweatt JD (2014). Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature 515, 582–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq and RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.