

Abstract
Background and Objectives:
Niemann-Pick disease type C (NPC) is a rare autosomal recessive lysosomal storage disease that is also associated with progressive neurodegeneration. NPC shares many pathological features with Alzheimer's disease including neurofibrillary tangles, axonal spheroids, β-amyloid deposition, and dystrophic neurites. Here, we examined if these pathological features could be detected in induced pluripotent stem cell (iPSC)-derived neurons from NPC patients.
Methods:
Brain tissues from 8 NPC cases and 5 controls were analyzed for histopathological and biochemical markers of pathology. To model disease in culture, iPSCs from NPC patients and controls were differentiated into cortical neurons.
Results:
We found hyperphosphorylated tau, altered processing of amyloid precursor protein and increased Aβ42 in NPC postmortem brains, and in iPSC-derived cortical neurons from NPC patients.
Conclusion:
Our findings demonstrated that the main pathogenic phenotypes typically found in NPC brains were also observed in patient-derived neurons, providing a useful model for further mechanistic and therapeutic studies of NPC.
Keywords: Niemann-Pick disease, iPSC, disease modeling, brain pathology
Introduction
Niemann-Pick disease type C (NPC) is a rare autosomal-recessive storage disorder characterized by progressive neurological deterioration and premature death1. It is caused by mutations that lead to partial deficiency or complete loss of function of NPC1 (95% of NPC cases) or NPC2 (5%) that is believed to interact with NPC12. Both NPC1 and NPC2 are lysosomal proteins that facilitate intracellular cholesterol trafficking3. Thus, one of the hallmarks of the disease is the intracellular accumulation of unesterified cholesterol and glycosphingolipids in many tissues including the brain. This is accompanied by gliosis and loss of neurons in selected brain regions triggering widespread neurological deficits such as ataxia, dystonia, seizures and dementia, with onset typically in childhood, but may also have late onset in adulthood4. Interestingly, although NPC differs in many aspects from Alzheimer's disease (AD), both diseases also share pathologic features5. NPC is one of the very few disorders in which hallmark pathologies of AD including neurofibrillary tangles (NFTs) develop in the absence of tau mutations. NFTs contain aberrantly hyperphosphorylated tau as paired helical filaments that contain a similar composition with equal ratio of isoforms found in AD (3R/4R)6. In addition, AD patients exhibit abnormal amyloid precursor protein (APP) metabolism, and have amyloid plaques composed of the APP metabolite, the amyloid β-protein (Aβ)7. These pathological hallmarks of AD are also typical of NPC, especially in cases with a prolonged course of disease8.
Although much of our knowledge about NPC has been acquired using pharmacologically and genetically modified models, these systems do not accurately reproduce human pathology.9-12 The analysis of disease-specific human neurons could significantly advance our understanding of how the pathologies develop and lead to neuronal dysfunction and death. Cell culture neuronal models of NPC patients have been previously developed,13-16 but the hallmark pathologies found in NPC brains have not been reported in patient-derived neuronal models.
In this study, we investigated NPC cortical neurons generated through iPSCs from patients’ somatic cells. We found pathological phenotypes, including tau and Aβ pathology as well as altered APP processing, resembling patient brain pathology. We therefore propose that NPC patient-derived cortical neurons may serve a valuable model for studying mechanisms of NPC pathogenesis and may prove beneficial for development of novel therapies.
Results
Tau pathology, abnormal APP processing and Aβ42 accumulation in NPC patient brains
Cortical and cerebellar tissue samples from 8 NPC patients and 5 healthy controls were analyzed for histopathological and biochemical abnormalities using thioflavin-S, Hematoxylin and eosin (H and E) staining and tau immunostaining. NFTs’ main constituent is hyperphosphorylated tau protein that can be visualized immunohistochemically using highly specific monoclonal antibodies such as RD3 (for 3R tau isoform), ET3 (for 4R tau isoform), and AT8 (for phospho-tau). Whereas in patients with prolonged disease course (4 subjects, age range 17–32) major pathological abnormalities, including NFTs (thioflavin-S staining), tau pathology, ballooned neurons (H and E staining), and dystrophic neurites (Fig. 1A), were detected, those features were absent in cortices from younger subjects (4 subjects, age range 2–11) (Supplementary Table S1).
Figure 1: Biochemical characterization of NPC (Niemann-Pick disease type C) pathology from human brain tissue.

(A) Immunohistochemistry was conducted on sections of mid-frontal cortex of case 4237 (see Supplementary Table 1). Hematoxylin and eosin staining shows numerous ballooned neurons, magnification 200x. Thioflavin-S staining shows thioflavin-S positive tangles, magnification 400x. Tau pathology including dystrophic neurites was detected using anti-tau antibodies ET3 and RD3 as well as phospho-tau antibody AT8, magnification 200x. Arrows indicate histopathological abnormalities. (B) Representative Western blot showing Triton-soluble total tau in cortical brain tissue lysates from NPC patients (n = 3) and controls (n = 3). Statistical analysis represent n = 5 controls and n = 8 NPC patients. (C) Representative Western blots showing formic acid-soluble total tau (red) in cortical brain tissue lysates from controls (n = 5) and NPC patients (n = 8) (left: 3 controls, 4 NPC patients; right: 2 controls, 4 NPC patients) and corresponding phospho-tau (green). Arrows point to the most severe patients, 4237 (left) and M40002M (right). Asterisks mark the youngest patients, 4770 (left) and M4004M (right) (compare Supplementary Table S1). Equal volumes/tissue wet weight of each sample were loaded. Statistical analysis represent n = 5 controls and n = 8 NPC patients. (D) Representative Western blot showing Triton-soluble amyloid precursor protein (APP) in lysates of cerebellar tissue. APP-full length (APP-FL) protein and C-terminal fragments of APP (APP-CTF) protein are indicated. Statistical analysis represent n = 5 controls and n = 6 NPC patients. (E) Aβ42 and Aβ40 levels were quantified using enzyme-linked immunosorbent assay (ELISA) in Triton-soluble fractions from control (n=3) and NPC (n=5) cortical samples, and the Aβ42/40 ratio was determined. Error bars, mean ± standard error of the mean. *P < 0.05, Student's t test; n.s., not significant.
Reduced amounts of soluble tau were detected in NPC patient cortical tissue (Fig. 1B), but increased insoluble total tau (red signal, Fig. 1C) and phosphorylated tau (green signal, Fig. 1C) in 2 NPC cases with prolonged course of disease (arrows). Tau aggregation was not observed in the youngest NPC cases (asterisks, Fig. 1C). This heterogeneity, in particular when comparing older and younger NPC cases contributed to non-significant differences in ratios of phosphorylated and total tau. Among the fragments generated from APP processing, CTFs are of particular interest since they are direct precursors of Aβ peptides17, a major constituent of senile plaques seen in AD and NPC. We detected increased ratios of CTFs over APP full-length protein in NPC cases compared to controls (Fig. 1D). CTFs are further cleaved generating various forms of Aβ with different C-terminal lengths whereas the Aβ42 species is the most aggregation-prone and pathogenic species deposited in AD brains18. We therefore quantitatively analyzed Aβ40 and Aβ42 levels in soluble fractions of cortical tissue by ELISA and observed higher Aβ42/Aβ40 ratios in NPC compared to control samples (Fig. 1E).
Modeling NPC brain pathology using iPSC-derived cortical neurons from NPC patients
iPSCs were generated from monozygotic twins with NPC (T1, T2), their maternal NPC carrier (P), and from unrelated control (Ctrl) (Supplementary Table 2). Skin fibroblasts, iPSC-derived hepatic and neural cells from these NPC patients, were characterized for reduced NPC1 protein, cholesterol accumulation, and autophagy defects elsewhere.16,19,20. Here, we differentiated iPSCs into cortical glutamatergic neurons21 (Supplementary Figure 1) and grew them in culture for 55 days. We found that neurons from NPC patients demonstrated lower levels of soluble tau protein (Fig. 2A) that inversely correlated with higher levels of insoluble phosphorylated tau (Fig. 2B). We further detected increased APP-CTF fragments in NPC neurons but not in neurons from the unaffected carrier or control (Fig. 2C). Using ELISA, we also found increased Aβ42/Aβ40 ratios in neurons from NPC patients (Fig. 2D). These results demonstrated that iPSC-derived cortical neurons from NPC patients faithfully recapitulate a majority of features of NPC pathology observed in patient brains (compare Fig. 1).
Figure 2: Cortical neurons from NPC (Niemann-Pick disease type C) patients recapitulate NPC brain pathology.
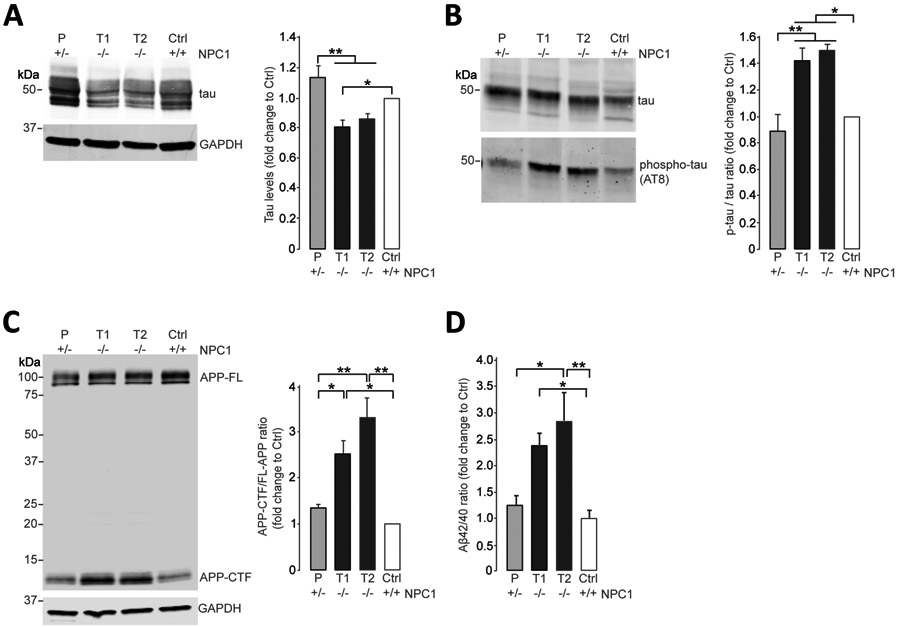
(A) Immunoblot analysis of total tau in Triton-soluble lysates of iPSC-derived cortical neurons from NPC monozygotic twins (T1, T2), an unaffected parent (P) and a healthy control (Ctrl) at day 55 of differentiation. GAPDH was used as a loading control. Results are shown relative to the healthy control individual (N = 8 independent experiments). (B) Immunoblot analysis of total tau and phospho-tau in Triton-insoluble lysates of iPSC-derived cortical neurons from NPC monozygotic twins (T1, T2), an unaffected parent (P), and a healthy control (Ctrl) at day 55 of differentiation. Results are shown relative to the healthy control (N = 3 independent experiments). (C) Immunoblot analysis of APP (amyloid precursor protein)-full length (APP-FL) and C-terminal fragments (APP-CTF) in Triton-soluble lysates of iPSC-derived cortical neurons from NPC monozygotic twins (T1, T2), an unaffected parent (P), and a healthy control (Ctrl) at day 55 of differentiation. GAPDH was used as a loading control. Results are shown relative to the healthy control (N = 3 independent experiments). (D) Aβ42 and Aβ40 levels were quantified by enzyme-linked immunosorbent assay in Triton-soluble lysates of iPSC-derived cortical neurons from NPC monozygotic twins (T1, T2), an unaffected parent (P), and a healthy control (Ctrl) at day 55 of differentiation (N = 4 independent experiments). Error bars, mean ± standard error of the mean. *P < 0.05 and **P < 0.01, one-way analysis of variance with Tukey's post hoc test.
Discussion
The ability to use iPSCs to model brain diseases is a powerful tool for unraveling mechanistic alterations22. Rodent models have advanced our understanding of brain pathology, but they do not always recapitulate the full spectrum of human neuropathology. For example, disease modeling using iPSCs has been most helpful in Parkinson's disease (PD) research, as currently available animal models do not fully recapitulate human pathology,23 and recent work has highlighted some fundamental differences between human and rodent midbrain neurons that at least in part explain the preferential vulnerability of human dopaminergic neurons in PD.24 Consistent with this notion and despite continued efforts, currently available NPC1 mouse models do not reflect the hallmark features of the disease in their entirety.25-29 The recent emergence of iPSC technology therefore offers an opportunity to study pathogenic phenotypes in a patient-specific manner with the endogenous expression of disease-linked mutations.
The clinical spectrum of NPC is heterogeneous and can vary depending on the age of onset and rate of clinical progression. For example, NFTs composed of aggregates of hyperphosphorylated tau have been most consistently reported in late-onset patients with a progressive chronic disease course but inconsistently in younger patients.8,30-32 In line with these reports are our histopathological and immunocytochemical findings confirming NFTs and abnormal, phosphorylated tau only in older NPC cases (Figure 1C; Supplementary Table 1). However, a reduction of soluble tau protein was observed for all patients compared to controls (Figure 1B).
In this study, we generated a patient-derived neuronal model of NPC to examine whether iPSC-derived neurons from patients recapitulate typical features of NPC brain pathology. Previous studies reported lysosomal cholesterol accumulation, alterations in autophagic pathways and reduced viability in NPC neuronal models.13-16 There is growing evidence that cholesterol accumulation contributes to the pathogenesis of AD by affecting APP processing and leading to increased amyloid plaque formation.33,34 The link between cholesterol, altered APP processing and Aβ has also been suggested in NPC,9-12 yet the functional significance of Aβ peptides in NPC pathology remains unclear.35 However, these in vitro studies were mainly based on pharmacologically and genetically induced NPC cell models and may therefore not recapitulate the whole spectrum of disease mechanisms. Our results demonstrate that cortical neurons from NPC patients recapitulated a majority of typical features of patient brain pathology, i.e. hyperphosphorylated tau, altered processing of APP, and increased aggregation-prone Aβ42 peptide.
Whereas animals are indispensable to study pharmacokinetics and pharmacodynamics in drug trials, human neuronal cultures may serve as more appropriate models for testing neuroprotective compounds, in particular during the early development of therapeutic interventions. This has also been shown in a study that used iPSC-derived midbrain dopamine neurons to test the efficacy of small-molecule modulators as a potential therapeutic approach for treating multiple forms of PD.36
Although current treatment can alleviate some symptoms of NPC, there is an immediate need to identify neuroprotective therapies for this progressively debilitating neurodegenerative disorder. Patient-derived neuronal models therefore provide a useful platform for understanding the disease mechanisms underlying NPC pathophysiology and may be used for testing of novel therapies.
MATERIALS AND METHODS
Human post-mortem brain collection
Human tissue samples from 8 patients with clinical diagnosis of NPC (age range 2–32 years) and 5 controls (age range 6–80 years) were obtained from National Institute of Child Health and Human Development (NICHD).
Human iPSC cultures and differentiation into cortical neurons
Skin fibroblasts from monozygotic twins (T1, T2) carrying a 1 bp deletion in exon 12 of the NPC1 gene (1920delG) and a missense mutation (IVS9-1009G>A), and one unaffected parent (P, heterozygous for 1920delG) were obtained from the NINDS Repository/Coriell Institute for Medical Research (T1, # GM22870; T2, #GM22871; P, #GM23151). Fibroblasts and a blood sample from an unrelated control were reprogrammed into iPSCs using Sendai Virus vectors and characterized for expression of pluripotency markers and genomic integrity as described previously.24
iPSCs were differentiated into cortical glutamatergic neurons using the NGN2 overexpression protocol21 as previously described.37
Sequential biochemical extraction
Sequential biochemical extraction of proteins from iPSC-derived neurons has been previously described.24 Human brain extracts were prepared as described in Mc Donald et al.38 to yield three biochemical fractions (TBS (soluble), TBS-TX (Triton-soluble) and FA (formic-acid soluble)).
Antibodies
Primary antibodies used: tau (K9J8; Dako, Burlington, ON, Canada; cat. no. A002401-2, 1:1000), phospho-tau (AT8; Thermo Fisher Scientific, Cleveland, OH, USA; cat no. MN1020, 1:1000), APP (clone 22C11; Millipore, Burlington, MA, USA; cat. no. MAB348, 1:1000), vGlut1 (Synaptic Systems, Göttingen, Germany; cat. no. 135 303), β-III-tubulin (BioLegend, San Diego, CA, USA; cat. no. 801202, 1:5000) and Glyceraldehyde 3-phosphate dehydrogenase (Millipore, Burlington, MA, USA; cat. no. MAB374, 1:5000).
ELISA
Brain tissue was homogenized in 1% Triton X-100 lysis buffer according to weight. Aβ40 ELISA (Invitrogen, Carlsbad, CA, USA; cat. no. KHB3481) and Aβ42 ELISA (Invitrogen, Carlsbad, CA, USA; cat. no. KHB3544) were done according to manufacturer’s instructions.
Immunohistochemistry procedure for paraffin-embedded brain tissue sections
Immunohistochemical analysis and microscopy was performed in standard fashion as previously reported.39,40 Primary antibodies: RD3 antibody to 3R tau (Millipore, clone 8E6/C11, 05-803), ET3 antibody to 4R tau (from Dr. Peter Davies) and phosphorylated tau (AT8, Thermo Scientific, MN1020).
Statistical Analysis
Student’s t-test analysis and one-way analysis of variance followed by Tukey’s post-hoc test were performed. P-values less than 0.05 were considered significant. All errors bars shown in the figures are standard error of the mean (SEM).
Supplementary Material
Supplementary Figure 1: Glutamatergic and neuronal markers in iPSC-derived cortical glutamatergic neurons from NPC cases and controls. Western blot analysis of vGlut1 and β-III-tubulin levels in T-soluble neuronal lysates from NPC mutant (T1, T2), NPC carrier (P) and unrelated control (Ctrl) cortical glutamatergic neurons at d55. GAPDH was used as a loading control.
Supplementary Table 1: Histopathological findings in subjects with a clinical diagnosis of NPC
PMI = post mortem interval, NFTs = neurofibrillary tangles, NBs = ballooned cells, DNs = dystrophic neurites
Supplementary Table 2: Clinical information of subjects that donated somatic cells for reprogramming into iPSCs
P = Parent; T1 = Twin 1; T2 = Twin 2; Ctrl = unrelated control
Acknowledgments
We are grateful to Niccolo E. Mencacci for helpful comments, Callen L. Spencer for staining of brain tissue sections, Northwestern Stem Cell Core Facility for generation of iPSC lines, NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore for brain tissues.
Funding:
This work was supported by NINDS grants R01 NS076054 and R37 NS096241 (to D.K.), NIA grant AG 13854 (to E.H.B.), and in part by Lysosomal Therapeutics, Inc. (to J.M.M.).
Footnotes
Full financial disclosures for the previous 12 months: D.K. is Founder of Lysosomal Therapeutics, Inc.; Vanqua Bio and serves on the scientific advisory boards of The Silverstein Foundation, Intellia Therapeutics, Prevail Therapeutics and is a Venture Partner at OrbiMed. J.M.M. is a shareholder in Lysosomal Therapeutics, Inc. All other authors have nothing to disclose.
Competing interests: The authors declare no competing interests.
REFERENCES
- 1.Vanier MT Niemann-Pick disease type C. Orphanet J Rare Dis 5, 16, doi: 10.1186/1750-1172-5-16 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sleat DE et al. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci U S A 101, 5886–5891, doi: 10.1073/pnas.0308456101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deffieu MS & Pfeffer SR Niemann-Pick type C 1 function requires lumenal domain residues that mediate cholesterol-dependent NPC2 binding. Proc Natl Acad Sci U S A 108, 18932–18936, doi: 10.1073/pnas.1110439108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patterson MC et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab 106, 330–344, doi: 10.1016/j.ymgme.2012.03.012 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Malnar M, Hecimovic S, Mattsson N & Zetterberg H Bidirectional links between Alzheimer's disease and Niemann-Pick type C disease. Neurobiol Dis 72 Pt A, 37–47, doi: 10.1016/j.nbd.2014.05.033 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Auer IA et al. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta neuropathologica 90, 547–551 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Haass C & Selkoe DJ Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 8, 101–112, doi: 10.1038/nrm2101 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Suzuki K et al. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol 89, 227–238, doi: 10.1007/BF00309338 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Kosicek M, Malnar M, Goate A & Hecimovic S Cholesterol accumulation in Niemann Pick type C (NPC) model cells causes a shift in APP localization to lipid rafts. Biochem Biophys Res Commun 393, 404–409, doi: 10.1016/j.bbrc.2010.02.007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malnar M et al. Niemann-Pick type C cells show cholesterol dependent decrease of APP expression at the cell surface and its increased processing through the beta-secretase pathway. Biochim Biophys Acta 1802, 682–691, doi: 10.1016/j.bbadis.2010.05.006 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamazaki T, Chang TY, Haass C & Ihara Y Accumulation and aggregation of amyloid beta-protein in late endosomes of Niemann-pick type C cells. J Biol Chem 276, 4454–4460, doi: 10.1074/jbc.M009598200 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Mattsson N et al. Amyloid-beta metabolism in Niemann-Pick C disease models and patients. Metab Brain Dis 27, 573–585, doi: 10.1007/s11011-012-9332-8 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Bergamin N et al. A human neuronal model of Niemann Pick C disease developed from stem cells isolated from patient's skin. Orphanet J Rare Dis 8, 34, doi: 10.1186/1750-1172-8-34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu D et al. Niemann-Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J Biomol Screen 19, 1164–1173, doi: 10.1177/1087057114537378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sung EA et al. Generation of patient specific human neural stem cells from Niemann-Pick disease type C patient-derived fibroblasts. Oncotarget 8, 85428–85441, doi: 10.18632/oncotarget.19976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maetzel D et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Reports 2, 866–880, doi: 10.1016/j.stemcr.2014.03.014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinha S et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540, doi: 10.1038/990114 (1999). [DOI] [PubMed] [Google Scholar]
- 18.De Strooper B & Annaert W Novel research horizons for presenilins and gamma-secretases in cell biology and disease. Annu Rev Cell Dev Biol 26, 235–260, doi: 10.1146/annurev-cellbio-100109-104117 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Takamura A et al. The useful preliminary diagnosis of Niemann-Pick disease type C by filipin test in blood smear. Mol Genet Metab 110, 401–404, doi: 10.1016/j.ymgme.2013.08.006 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Hoglinger D et al. NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat Commun 10, 4276, doi: 10.1038/s41467-019-12152-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798, doi: 10.1016/j.neuron.2013.05.029 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Unternaehrer JJ & Daley GQ Induced pluripotent stem cells for modelling human diseases. Philos Trans R Soc Lond B Biol Sci 366, 2274–2285, doi: 10.1098/rstb.2011.0017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawson TM, Golde TE & Lagier-Tourenne C Animal models of neurodegenerative diseases. Nat Neurosci 21, 1370–1379, doi: 10.1038/s41593-018-0236-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burbulla LF et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 357, 1255–1261, doi: 10.1126/science.aam9080 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karten B, Vance DE, Campenot RB & Vance JE Trafficking of cholesterol from cell bodies to distal axons in Niemann Pick C1-deficient neurons. J Biol Chem 278, 4168–4175, doi: 10.1074/jbc.M205406200 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Pentchev PG et al. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J Biol Chem 259, 5784–5791 (1984). [PubMed] [Google Scholar]
- 27.Miyawaki S, Yoshida H, Mitsuoka S, Enomoto H & Ikehara S A mouse model for Niemann-Pick disease. Influence of genetic background on disease expression in spm/spm mice. J Hered 77, 379–384, doi: 10.1093/oxfordjournals.jhered.a110265 (1986). [DOI] [PubMed] [Google Scholar]
- 28.Morris MD, Bhuvaneswaran C, Shio H & Fowler S Lysosome lipid storage disorder in NCTR-BALB/c mice. I. Description of the disease and genetics. Am J Pathol 108, 140–149 (1982). [PMC free article] [PubMed] [Google Scholar]
- 29.Maue RA et al. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum Mol Genet 21, 730–750, doi: 10.1093/hmg/ddr505 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Distl R et al. Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain. J Pathol 200, 104–111, doi: 10.1002/path.1320 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Sherriff FE, Bridges LR & De Souza DS Non-Alzheimer neurofibrillary tangles show beta-amyloid-like immunoreactivity. Neuroreport 5, 1897–1900, doi: 10.1097/00001756-199410000-00014 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Love S, Bridges LR & Case CP Neurofibrillary tangles in Niemann-Pick disease type C. Brain 118 ( Pt 1), 119–129, doi: 10.1093/brain/118.1.119 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Maulik M, Westaway D, Jhamandas JH & Kar S Role of cholesterol in APP metabolism and its significance in Alzheimer's disease pathogenesis. Mol Neurobiol 47, 37–63, doi: 10.1007/s12035-012-8337-y (2013). [DOI] [PubMed] [Google Scholar]
- 34.Hartmann T Cholesterol, A beta and Alzheimer's disease. Trends Neurosci 24, S45–48, doi: 10.1016/s0166-2236(00)01990-1 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Martins IJ et al. Cholesterol metabolism and transport in the pathogenesis of Alzheimer's disease. J Neurochem 111, 1275–1308, doi: 10.1111/j.1471-4159.2009.06408.x (2009). [DOI] [PubMed] [Google Scholar]
- 36.Burbulla LF et al. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson's disease. Sci Transl Med 11, doi: 10.1126/scitranslmed.aau6870 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valdez C, Ysselstein D, Young TJ, Zheng J & Krainc D Progranulin mutations result in impaired processing of prosaposin and reduced glucocerebrosidase activity. Hum Mol Genet 29, 716–726, doi: 10.1093/hmg/ddz229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mc Donald JM et al. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 133, 1328–1341, doi: 10.1093/brain/awq065 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao Q et al. Disease and Region Specificity of Granulin Immunopositivities in Alzheimer Disease and Frontotemporal Lobar Degeneration. J Neuropathol Exp Neurol 76, 957–968, doi: 10.1093/jnen/nlx085 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng HX et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67, 739–748, doi: 10.1002/ana.22051 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Glutamatergic and neuronal markers in iPSC-derived cortical glutamatergic neurons from NPC cases and controls. Western blot analysis of vGlut1 and β-III-tubulin levels in T-soluble neuronal lysates from NPC mutant (T1, T2), NPC carrier (P) and unrelated control (Ctrl) cortical glutamatergic neurons at d55. GAPDH was used as a loading control.
Supplementary Table 1: Histopathological findings in subjects with a clinical diagnosis of NPC
PMI = post mortem interval, NFTs = neurofibrillary tangles, NBs = ballooned cells, DNs = dystrophic neurites
Supplementary Table 2: Clinical information of subjects that donated somatic cells for reprogramming into iPSCs
P = Parent; T1 = Twin 1; T2 = Twin 2; Ctrl = unrelated control