

SUMMARY
Despite wide use of anti-vascular endothelial growth factor (VEGF) therapy for many solid cancers, most individuals become resistant to this therapy, leading to disease progression. Therefore, new biomarkers and strategies for blocking adaptive resistance of cancer to anti-VEGF therapy are needed. As described here, we demonstrate that cancer-derived small extracellular vesicles package increasing quantities of VEGF and other factors in response to anti-VEGF therapy. The packaging process of VEGF into small extracellular vesicles (EVs) is mediated by the tetraspanin CD63. Furthermore, small EV-VEGF (eVEGF) is not accessible to anti-VEGF antibodies and can trigger intracrine VEGF signaling in endothelial cells. eVEGF promotes angiogenesis and enhances tumor growth despite bevacizumab treatment. These data demonstrate a mechanism where VEGF is partitioned into small EVs and promotes tumor angiogenesis and progression. These findings have clinical implications for biomarkers and therapeutic strategies for ovarian cancer.
Graphical abstract
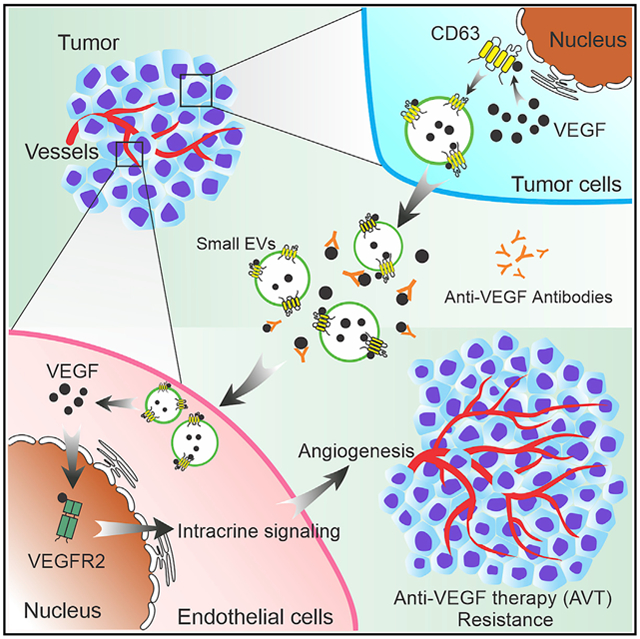
In brief
Ma et.al report that cancer-cell-derived small EVs contain increasing amounts of VEGF (eVEGF) and contribute to resistance to anti-VEGF therapy (AVT). CD63 is a potential mediator that regulates packaging of VEGF into small EVs. eVEGF can trigger intracrine VEGF signaling in endothelial cells and promote angiogenesis despite AVT.
INTRODUCTION
Angiogenesis is well recognized as a major factor in promoting tumor growth and progression (Carmeliet and Jain, 2011). Among the many angiogenic factors, vascular endothelial growth factor (VEGF; also known as VEGF-A) is arguably the most dominant (Apte et al., 2019). Therefore, pharmaceutical companies have developed multiple anti-VEGF therapies (AVTs) and anti-VEGF receptor (VEGFR) therapies (Jain et al., 2006). The U.S. Food and Drug Administration has approved bevacizumab, a humanized monoclonal anti-VEGF antibody, for treatment of many solid tumors, including recurrent ovarian cancer (Ma et al., 2018). Despite the initial efficacy of AVTs, adaptive resistance and progressive disease will develop in most individuals with cancer (Bergers and Hanahan, 2008; Jain et al., 2009). Several mechanisms, including hypoxia-induced alterations of vascularization, metabolic symbiosis, and cell-to-cell communication, contribute to this adaptive resistance (Ma et al., 2018). However, a broader understanding of these resistance mechanisms is needed to identify reliable biomarkers for drug response and develop new therapeutic strategies for cancer.
Small extracellular vesicles (EVs) play important roles in cell-to-cell communication and tumor progression (Simons and Raposo, 2009; Tkach and Théry, 2016). A plethora of biomolecular cargoes, such as proteins, lipids, and nucleic acids, can be packaged in small EVs and transferred to recipient cells (Choi et al., 2013; Thakur et al., 2014; Théry et al., 2009). Several studies have demonstrated that the contents of small EVs can shape the tumor microenvironment by modifying drug response or tumor angiogenesis (Li and Nabet, 2019; Todorova et al., 2017). Studies have shown that EVs, including exosomes, can carry angiogenic factors such as VEGF and promote tumor angiogenesis (Baruah and Wary, 2020). A recent study has shown that the VEGF189 isoform localizes to the EV surface and promotes angiogenesis regardless of cell uptake (Ko et al., 2019). However, the extent to which various mechanisms contribute to sorting of VEGF into small EVs is not well understood. Here, we found that after AVT increasing quantities of VEGF and other angiogenesis-related proteins in small EVs evaded recognition by therapeutic antibodies, promoting angiogenesis in an intracrine manner. These findings have implications for identification of biomarkers of drug response in small EVs and for development of effective therapies to block adaptive resistance to AVT.
RESULTS
VEGF121 and VEGF189 isoforms present in cancer-cell-derived small EVs
We first isolated small EVs from cancer cell culture supernatant via sucrose density gradient ultracentrifugation (SUC) and demonstrated the presence of VEGF in small EVs. We characterized the EV particles by transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA), and western blotting (Figures 1A, 1B, S1A, and S1B). We selected CD63, Alix, and TSG101 as small EV-positive markers and GRP94 as a small EV-negative marker (Figures 1B and S1B). We loaded recombinant human VEGF proteins (isoforms 121, 165, and 189) in parallel as indicators (Figures 1B and S1B). The results revealed that small EVs contain VEGF121 monomers and dimers and VEGF189 monomers and dimers while lacking the VEGF165 isoform (Figures 1B and S1B). Because AVTs can cause hypoxia in tumors, we further collected small EVs from cancer cells cultured under normal (21% O2) or hypoxic (1% O2) conditions and performed a human angiogenesis array (Figure 1C). Characterization of isolated EV particles was carried out using TEM and NTA (Figure 1D). We first confirmed the hypoxic status of these cells by detecting induction of hypoxia-inducible factor 1α (Figure 1E). We next measured the amount of secreted VEGF in the cell culture supernatant using a human VEGF enzyme-linked immunosorbent assay (ELISA) kit and found an increasing trend of secreted VEGF levels under hypoxia (Figure 1E). The angiogenesis array results revealed that several angiogenic factors, including VEGF, can be detected in small EV extracts (EVE) (Figure 1F). We further validated the expression of VEGF in small EVs using western blotting and observed increased VEGF expression under hypoxia after normalization with CD63 expression (Figure 1G). To determine whether hypoxia can affect the ability of cells to secrete small EVs, we compared the number of small EVs released from cancer cells at the same cell density under normal and hypoxic conditions. However, there was no significant difference in small EV secretion or the protein amount per 1 million small EVs (Figure S1C). In addition, we found that the VEGF level in small EVs was lower under hypoxia than normoxia in RF24 human immortalized umbilical endothelial cells, although the secreted VEGF level was higher under hypoxia (Figures S1D and S1E).
Figure 1. VEGF isoforms present in sEVs.

(A) Characterization of sEVs by transmission electron microscopy (TEM) and Nanoparticle tracking analysis (NTA). Scale bar, 100 nm.
(B) Western blotting of VEGF isoforms in sEV extract (EVE). A total of 200 ng of recombinant human VEGF121, VEGF165, and VEGF189 protein was loaded into SDS-PAGE gel in a reduced condition.
(C) Schematic of the experimental design for identifying angiogenic factors in small EVs (sEVs).
(D) Characterization of sEVs isolated from SKOV3 cells under normal and hypoxic conditions according to TEM and NTA. Scale bar, 100 nm.
(E) Western blotting showing induction of hypoxia-inducible factor-1α (HIF-1α). Secreted VEGF levels in OVCAR5 and SKOV3 cells culture supernatants were determined by human VEGF ELISA kit. **p < 0.01; n.s., not significant.The p value was determined by a Student’s t test for comparison between two groups. Data represent mean ± SD.
(F) Human angiogenesis array data for EVEs. Red rectangles show the location of VEGF. Array membranes were developed at the same time.
(G) Western blotting showing the levels of VEGF in sEVs. Arrowheads point to the location of VEGF isoforms.
WCE, whole-cell extract. See also Figure S1.
eVEGF expression is increased in mouse models of resistance to AVT and individuals receiving bevacizumab-containing therapy
Next we established an orthotopic tumor xenograft mouse model of resistance to AVT (Figure S2A). We examined the tumor burden in OVCAR5 and SKOV3 xenograft mouse models via in vivo imaging system (IVIS) in the same mice after 2–4 weeks of bevacizumab treatment. At the end of the experiment, we placed mice in bevacizumab-sensitive and -resistant groups based on bioluminescence imaging results. Representative bioluminescence images of sensitive mice (n = 2) are shown in Figures 2A and 2B and of resistant mice (n = 3) in Figures 2C and 2D. We isolated small EVs from the serum of mice in both groups and analyzed them using a human angiogenesis array. We characterized the EVs using a NTA assay (Figure S2B) and western blotting (Figure S2C). As described above, we used CD63, Alix, and TSG101 as small EV-positive markers and GRP94 as a small EV-negative marker. Then we performed an angiogenesis array and found that the small EV-VEGF (eVEGF) levels in small EVs were higher in the resistant OVCAR5 and SKOV3 xenograft mouse models than in the sensitive ones (Figures 2E and 2F). In addition to VEGF, expression of serpin E1 and urokinase-type plasminogen activator (uPA) was increased in small EVs in the resistant models (Figures 2E and 2F).
Figure 2. eVEGF levels are elevated in mice with ovarian cancer resistant to bevacizumab and serum from individuals receiving bevacizumab-containing therapy.

(A and B) In vivo imaging system (IVIS) images of tumor-bearing mice becoming sensitive to bevacizumab treatment in (A) OVCAR5 and (B) SKOV3 xenograft models. Sen, sensitive.
(C and D) IVIS images of tumor-bearing mice becoming resistant to bevacizumab treatment in (C) OVCAR5 and (D) SKOV3 xenograft models. Res, resistant.
(E and F) Angiogenic factors in sEVs isolated from the (E) OVCAR5 and (F) SKOV3 xenograft mouse models. The relative pixel densities of the data arrays were calculated using ImageJ software after background subtraction. For comparison purposes, we loaded the same amount of proteins and developed the arrays at the same time. Red rectangles show the location of VEGF. uPA, urokinase-type plasminogen activator; DPPIV, dipeptidyl peptidase IV.
(G) Translational study of eVEGF levels in serum samples from three individuals receiving bevacizumab-containing therapy. Pt. patient; C1D1, cycle 1 day 1; C3D1, cycle 3 day 1; C5D1, cycle 5 day 1; EOT, end of treatment.
(H) VEGF expression in sEVs isolated from serum samples using western blotting. CD63, TSG101, and HSP70 were used as sEV-positive markers, and GRP94 was used as a sEV-negative marker. Arrowheads point to the location of VEGF isoforms.
See also Figure S2.
We next wanted to find out whether eVEGF levels can be affected by bevacizumab treatment in human serum. We harvested serum samples from five individuals with cancer before each cycle of treatment from a clinical trial (ClinicalTrials.gov: NCT02923739) designed to test the efficacy of paclitaxel and bevacizumab with emactuzumab in individuals with platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer. We isolated small EVs from 2 mL of serum from affected individuals using ultracentrifugation and measured the eVEGF level using a human VEGF ELISA kit. For two individuals, we had available pre-and post-treatment samples and observed substantially lower secreted VEGF levels at the end of treatment (EOT) compared with initiation of treatment (cycle 1 day 1 [C1D1]) (Figure 2G). Notably, we observed a substantial increase in eVEGF levels in the relevant small EVs from these two individuals at EOT (Figure 2G). We found decreased secreted VEGF levels in serum and enriched eVEGF levels in serum in the third individual at cycle 3 day 1 and EOT (Figure 2G). We further validated these findings by determining VEGF expression in small EVs using western blotting (Figure 2H). We did not detect eVEGF expression in the two other individuals at C1D1 (Figure S2D). The size distribution and particle numbers for small EVs in all serum samples were determined by NTA (Figure S2E).
eVEGF evades recognition by anti-VEGF antibodies
We next wanted to find out whether eVEGF can be recognized by anti-VEGF antibodies. First we measured the ratio of small EVs from cancer cells that can bind to anti-VEGF antibodies to the total small EV population. To achieve this, we employed flow cytometry and gated the channels of interest using a flow cytometry sub-micron particle size reference kit with four sizes of green fluorescent beads (100, 200, 500, and 1000 nm) to exclude any particles larger than 200 nm (Figures S3A and S3B). We labeled the small EVs with CellMask Green plasma membrane stain and detected EVs using Amnis Imaging flow cytometers (Yokoi et al., 2019). After analysis using Image Data Exploration and Analysis Software (IDEAS), we observed that only 2.8% ± 1.6% and 2.82% ± 0.5% of the small EVs in OVCAR5 and SKOV3 cells, respectively, were recognized by the human VEGF phycoerythrin (PE)-conjugated antibodies (Figures 3A and 3B). The percentages of PE-conjugated CD63-positive small EVs were 23.7% ± 25.0% and 46.6% ± 19.0% in OVCAR5 and SKOV3 cells, respectively. We then incubated the same samples of small EVs with detergent (2% NP-40 lysis buffer) for 20 min and observed a substantial reduction in the number of particles, which suggested that most of the detected particles had lipid bilayer membranes (Figure S3C). The intracellular flow cytometry experiments revealed that VEGF PE-conjugated antibodies can bind to free VEGF protein maintained in the cells (Figure 3C). These results indicate that VEGF antibodies can bind to free VEGF protein but are unable to bind to eVEGF.
Figure 3. eVEGF is not recognized by anti-VEGF antibodies.

(A and B) Amnis flow cytometry results for (A) OVCAR5-derived and (B) SKOV3-derived sEVs. CD63 phycoerythrin (PE)-conjugated (PE-CD63) antibodies were used as positive controls. Data represent mean ± SD.
(C) Intracellular flow cytometry using VEGF PE-conjugated antibodies in OVCAR5 and SKOV3 cells. PE-A, PE area; Ctrl, control.
CM+, CellMask plasma membrane stain positive particles. See also Figure S3.
Last, we examined whether bevacizumab can recognize and neutralize eVEGF. We isolated small EVs from bevacizumab-resistant RF24 (RF24-Bev) endothelial cells, which we treated with 1 μg/μL bevacizumab to maintain their resistance, and then examined eVEGF levels. We first checked the efficacy of bevacizumab in neutralizing secreted VEGF using a human VEGF ELISA kit. The results demonstrated that VEGF levels in the supernatants were substantially reduced under normal and hypoxic conditions (<15.6 pg/mL; Figure S3D). Of note, we could still detect eVEGF in small EVs even though VEGF was absent from the supernatant (Figure S3E). These data demonstrate that exposure to bevacizumab cannot neutralize eVEGF.
CD63 regulates packaging of VEGF into small EVs
Next, we explored the mechanisms that orchestrate VEGF incorporation into small EVs. CD63, a tetraspanin protein, has been demonstrated to play a key role in EV secretion and regulation of protein packaging (Hurwitz et al., 2016, 2017). Therefore, we examined whether CD63 is a potential regulator for packaging VEGF into small EVs. Using confocal immunofluorescence, we demonstrated that VEGF does co-localize with CD63, suggesting a possible mechanism for packaging VEGF in small EVs (Figure 4A). We further explored their interactions using a co-immunoprecipitation (coIP) assay. Our results showed that VEGF is associated with CD63 in OVCAR5 and SKOV3 cells (Figure 4B). We next established CD63 overexpression and CD63 knockdown cell lines by transducing OVCAR5 and SKOV3 cells with pCT-CD63-GFP virus particles and CD63 shRNA lentiviral transduction particles, respectively. CD63-GFP-expressing cells were further selected via flow cytometry, whereas shCD63-expressing cells were established via puromycin (2 μg/mL) treatment. Our results showed that overexpression of CD63 can increase the eVEGF levels (mostly VEGF121 dimers, VEGF189 monomers, and VEGF189 dimers) in OVCAR5 and SKOV3 cells (Figure 4C). After knockdown of CD63, there was a decrease in eVEGF levels (VEGF121 dimers and VEGF189 dimers) in OVCAR5 cells and a decrease in VEGF189 dimers in SKOV3 cells (Figure 4D).
Figure 4. CD63 mediates the packaging of VEGF into sEVs.

(A) Confocal images of cellular co-localization of VEGF and CD63. Scale bar, 20 μm.
(B) CoIP experiments of VEGF and CD63 proteins. Five micrograms of WCE from OVCAR5 and SKOV3 cells were used as input control. Arrowheads point to the location of VEGF isoforms.
(C) Upregulation of eVEGF levels in cells transduced with pCT-CD63-GFP virus particles. Arrowheads point to the location of VEGF isoforms.
(D) Downregulation of eVEGF levels after the knockdown of CD63 expression using shRNA. Arrowheads point to the location of VEGF isoforms.
eVEGF triggers intracrine VEGF signaling
Given that VEGF/VEGFR2 signaling is among the most important pathways for regulating angiogenesis (Simons et al., 2016), we further determined the effects of eVEGF on activation of this pathway. We first wanted to find out whether endothelial cells take up cancer-cell-derived small EVs using immunofluorescence. After incubation with small EVs (labeled with CellMask Green plasma membrane stain in advance) for 3 h, we detected small EVs taken up by RF24 cells, and most of the small EVs were located in the perinuclear region (Figure 5A). To determine whether eVEGF can activate the VEGFR2/Akt/extracellular signal-regulated kinase (Erk) pathway, we treated RF24 cells with cancer-cell-derived small EVs along with bevacizumab. The western blotting results showed that OVCAR5-derived small EVs had a stronger effect in activating phosphorylated Akt and Erk expression than SKOV3-derived EVs regardless of bevacizumab treatment (Figures S4A and S4B). Next, we collected small EVs from VEGF knockout colorectal cancer (CRC) RKO (RKO-VEGF−/−) cells (Yamagishi et al., 2013). The depletion of secreted VEGF was validated using a human VEGF ELISA kit (Figure 5B). We characterized the small EVs from RKO parental (RKO-PAR) cells as well as RKO-VEGF−/− cells using TEM, NTA, and western blotting (Figures 5C and 5D). The western blotting results also revealed complete depletion of eVEGF expression (Figure 5D). Next, we performed subcellular protein fractionation on RF24 endothelial cells after treating them with VEGF-positive small EVs (VEGF+ sEVs) from RKO-PAR cells and VEGF-negative sEVs (VEGF− sEVs) from RKO-VEGF−/− cells. The results showed that neither VEGF+ sEVs nor VEGF− sEVs can activate VEGFR2 phosphorylation (p-VEGFR2) in membrane extract (ME) (Figure 5E). Importantly, we observed an increased level in p-VEGFR2 in nuclear extract (NE) from cells treated with VEGF+ sEVs (Figure 5E). This suggested that eVEGF could induce intracrine VEGF signaling. We then silenced the kinase insert domain receptor (KDR) gene in RF24 cells using small interfering RNA (siRNA) and evaluated the roles of VEGFR2 in eVEGF-induced intracrine signaling. To increase the knockdown efficacy by siRNAs, we pooled four individual siRNAs to achieve a substantial reduction in mRNA level (almost 70%) and nearly complete depletion of VEGFR2 protein (Figure 5F). We found that VEGF+ sEVs did not activate several intracellular kinases (e.g., Erk, c-Jun, p70S6, p38, and STAT1) after knockdown of VEGFR2 in RF24 cells, which means that induction of intracrine signaling is partially dependent on VEGFR2 expression (Figure 5G).
Figure 5. eVEGF triggers intracrine VEGF signaling in a VEGFR2-dependent manner.

(A) Representative confocal images showing the uptake of sEVs by RF24 cells after 3 h of incubation. 3D structures were constructed using Imaris software. CellMask Green staining, sEVs; CellMask Deep Red staining, cell membrane. Scale bar, 20 μm.
(B) Secreted VEGF levels in VEGF−/− and RKO-PAR cells as revealed by a human VEGF ELISA kit. Data represent mean ± SD.
(C) Characterizations of sEVs from RKO-PAR and RKO-VEGF−/− cells according to TEM and NTA. Scale bar, 100 nm.
(D) Knockout of VEGF in sEVs confirmed by western blotting. CD63, TSG101, and Alix were used as sEV-positive markers, and GRP94 was used as a sEV-negative marker. An arrowhead points to the location of the VEGF isoform.
(E) Subcellular protein fractionation assessment in RF24 cells after treatment with VEGF+ and VEGF− sEVs. ME, membrane extract; CE, cytoplasmic extract; NE, nuclear extract.
(F) KDR mRNA level in RF24 cells 48 h after transfection with individual siRNA and the pool of four siRNAs. Data were normalized to the control group (RF24 only) and represent mean ± SD. The protein level of VEGFR2 and p-VEGFR2 in RF24 cells of different treatment groups after silencing KDR using pooled siRNAs. **p < 0.01; ***p < 0.001. p values were determined by a Student’s t test for comparison between two groups.
(G) A human phospho-kinase array on RF24 cells after silencing KDR and treatment with sEVs. The pixel density was calculated using ImageJ software and the bar graph lists the most upregulated proteins in VEGF+ sEVs-treated RF24 cells. All array membranes were developed at the same time. Erk1/2, extracellular signal-regulated protein kinase; C-jun, cellular Jun; PDGFRβ, platelet-derived growth factor receptor beta; PLC-γ1, phospholipase C-γ1; STAT, signal transducer and activator of transcription;
See also Figure S4.
Cancer cell-derived sEVs promote angiogenesis and tumor growth
Next, we assessed tube formation by RF24 cells following treatment with cancer-cell-derived sEVs in the presence or absence of bevacizumab. We observed an increase in the number of tubes formed by RF24 cells treated with sEVs isolated from OVCAR5 and SKOV3 cells under normal and hypoxic conditions (N-sEVs and H-sEVs, respectively) (Figures 6A and 6B). Specifically, addition of bevacizumab to treatment with sEVs did not abolish the effect of sEVs on tube formation. These findings indicate that the pro-angiogenic functions of cancer cell-derived sEVs are not affected by exposure to bevacizumab.
Figure 6. eVEGF promotes angiogenesis and tumor growth in vivo.

(A and B) The numbers of tubes formed by RF24 cells after treatment with sEVs from (A) OVCAR5 and (B) SKOV3 cells in the presence or absence of bevacizumab. Bev, 1 μg/μL bevacizumab. N-sEVs, sEVs isolated under normal conditions; H-sEVs, sEVs isolated under hypoxic conditions; rhVEGF, recombinant human VEGF protein. **p < 0.01, ***p < 0.001, ****p < 0.0001. p values were determined by one-way ANOVA for comparison among multiple groups followed by Dunnett’s multiple comparisons test. Scale bar, 500 μm. Data represent mean ± SD.
(C) Quantification of mouse tumor volumes during the study. Tumor lengths and widths were recorded twice a week. *p < 0.05. The p value was determined by one-way ANOVA for comparison among multiple groups followed by Dunnett’s multiple comparisons test. Data represent mean ± SD.
(D) Micro-vessel densities in frozen tumor sections were assessed using immunochemical staining for CD31. The CD31 score was determined using ImageJ software. No significant difference was identified among groups by one-way ANOVA. Data represent mean ± SD.
(E) Representative images of in vivo angiogenesis Matrigel plugs and hemoglobin amounts in Matrigel plugs measured using a hemoglobin assay kit. rhVEGF was used as a positive control. VEGF+ sEVs, Matrigel plug with sEVs isolated from RKO-PAR cells; VEGF− sEVs, Matrigel plug with sEVs isolated from RKO-VEGF−/− cells. *p < 0.05, **p < 0.01, ***p < 0.001; p values were determined by one-way ANOVA for comparison among multiple groups followed by Dunnett’s multiple comparisons test. Data represent mean ± SD.
To determine the effects of cancer-cell-derived sEVs on tumor growth during AVT in vivo, we harvested sEVs from SKOV3 cells cultured under normal and hypoxic conditions. We then injected sEVs into subcutaneous ovarian tumors in mice (n = 8 per group). We administered bevacizumab (5 mg/kg) to mice intraperitoneally twice a week and then measured their tumor sizes twice a week until the endpoint of the study. The results revealed that sEVs isolated under hypoxia had markedly increased tumor volumes and that addition of bevacizumab to this treatment did not affect the rate of tumor growth (Figure 6C). We also observed greater tumor volumes in mice given treatment with N-sEVs and H-sEVs than in the control group, but the difference was not significant. To assess tumor vessel density, we sectioned the tumors and stained them with an anti-CD31 antibody. Microvessel density was higher in mice that received sEVs and bevacizumab than in those treated with bevacizumab only (Figure 6D). However, we did not observe a significant difference in microvessel density among the groups. These results are consistent with the increased tumor volumes in mice given treatment with sEVs, suggesting that sEVs can promote tumor growth during AVT.
Our in vitro results demonstrated that several angiogenesis factors apart from VEGF can be packaged into sEVs. Also, other factors in sEVs, such as microRNAs, long noncoding RNAs, and cytokines, may contribute to tumor growth. To further validate the role of eVEGF in tumor growth, we harvested sEVs from RKO-VEGF−/− cells for an in vivo Matrigel plug assay. The in vivo Matrigel plug assay showed that VEGF+ sEVs had the strongest effects in increasing angiogenesis in mice, as reflected by the highest hemoglobin level in comparison with other groups (Figure 6E). The hemoglobin level was decreased in a group of mice given treatment with VEGF− sEVs, which suggested that eVEGF has an important role in angiogenesis. However, the hemoglobin levels in groups of mice given VEGF− sEVs were higher than those in the control mice, suggesting that other factors may also contribute to angiogenesis.
DISCUSSION
VEGF is known to play an important role in angiogenesis during the growth and metastasis of ovarian and other tumors (Boocock et al., 1995; Mesiano et al., 1998). Thus, investigators have developed many different drug-based approaches that target VEGF or its receptors (Duda et al., 2007). Among these, bevacizumab is used widely in individuals with ovarian, colon, and other cancers (Aghajanian et al., 2012, 2015; Coleman et al., 2017). Unfortunately, most individuals will eventually experience resistance to this therapy and have progressive disease; the underlying mechanisms of this remain poorly understood. Several studies have shown that sEVs can deliver VEGF into endothelial cells and may cause resistance to AVTs (Ko et al., 2019; Todorova et al., 2017). In the present study, we identify a mechanism whereby CD63 can help package VEGF into sEVs and eVEGF can activate intracrine VEGF signaling to mediate AVT resistance. Studies have demonstrated that sEVs participate in reshaping the tumor microenvironment to promote tumor development (Kahlert and Kalluri, 2013; Li and Nabet, 2019). In addition, researchers have demonstrated that EVs derived from cancer, stromal, or immune cells confer drug resistance (Binenbaum et al., 2018; Qin et al., 2019; Qu et al., 2016). Also, several studies have demonstrated that sEVs can cause chemotherapy resistance by delivering RNAs, including microRNAs and long noncoding RNAs, to cancer cells (Liu et al., 2019; Qin et al., 2019; Qu et al., 2016). Similar effects can be achieved with transfer of certain proteins, such as Wnt, between stromal cells and cancer cells (Hu et al., 2019). Notably, sEVs released by endothelial cells also carry a decent amount of VEGF as well as other angiogenic factors. Therefore, the autocrine effects of these sEVs should be considered to be involved in AVT resistance.
In the present study, we also observed stimulation of VEGFR2 phosphorylation in the NE by sEVs. This phenomenon can be explained by the intracrine signaling of VEGF released by sEVs after their internalization (Bhattacharya et al., 2016; Ruan et al., 2011). Furthermore, we validated that induction of intracrine VEGF signaling is VEGFR2-dependent. Indeed, we observed that knockout of VEGF in sEVs can substantially reduce angiogenesis in vivo. Although our in vivo Matrigel plug assay showed a significant increase in hemoglobin levels in mice with VEGF-negative sEVs, this is not surprising because other angiogenesis factors, such as uPA and serpin E1, are packaged in sEVs. These factors together with VEGF may contribute to cancer resistance to AVTs, which should be explored in future studies.
We provided evidence of a sEV-mediated mode of cancer resistance to AVTs using serum samples from individuals with cancer in a recent clinical trial (ClinicalTrials.gov: NCT02923739). Although the number of participants was limited (n = 5), and not all samples were available during each treatment course, we can still assess the translational potential of our findings. We observed elevated eVEGF levels in serum, which could contribute to AVT resistance. However, further work on the response of individuals with cancer to AVT is needed to examine the relationship between eVEGF levels and drug response. To precisely define the contribution of eVEGF to AVT resistance, studies of large cohorts are warranted.
We successfully established a model of adaptive cancer resistance to bevacizumab. However, this in vivo study has some limitations. Although we used a human angiogenesis array kit to detect the EV proteins derived from human cancer cells, we cannot rule out the possibility that the kit can cross-react with mouse EV proteins. It is also challenging to distinguish tumor-derived sEVs from mouse cell-derived ones using this experiment. Nevertheless, this study provides a model of bevacizumab-resistant cancer, and additional translational studies are needed. Future experiments must also explore the cell sources responsible for these candidates and determine whether they can be used for cancer diagnosis. This model will be essential to identify the potential for predicting AVT resistance of cancer and developing therapeutic strategies targeting cancer cell-derived sEVs.
Overall, our findings have important implications regarding the potential mechanism of resistance of cancer to AVTs. Therapeutic strategies for overcoming AVT resistance will have to take into account the mechanism of eVEGF-mediated resistance in ovarian cancer. Cloaking VEGF in sEVs by CD63 and evading recognition by AVTs likely compromises the response to such therapies. Therefore, therapeutic interventions targeting the negative effects of sEVs could be synergistic when combined with AVTs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for all original resources and reagents presented in this manuscript should be directed to the lead contact, Anil K. Sood (asood@mdanderson.org)
Materials availability
There are no unique/stable reagents generated in this study.
Data and code availability
- All data reported in this paper will be shared by the lead contact upon request.
- This paper does not report original code.
- Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
In Vivo Studies
Mice.
Four to eight-week NCRNU-Female nude (NCr) mice were obtained from Taconic. All mice were housed and bred in specific pathogen-free conditions at MDACC animal facility. All animal euthanasia methods were approved on the animal protocol and performed at the end of the experiment or when any mice became moribund. The mice were euthanized by CO2 exposure followed by cervical dislocation. All animal protocols were approved by the Institutional Animal Care and Use Committee of the MD Anderson Cancer Center.
Bevacizumab-resistant mouse model.
Twenty NCRNU-Female nude mice were used to generate resistant models to bevacizumab therapy. Ten mice were inoculated intraperitoneally with 1 × 106 SKOV3-luciferase cells and the other 10 were inoculated with 1 × 106 OVCAR5-luciferase cells. Bevacizumab (5 mg/kg) was administered intraperitoneally to the mice 21 days after cell inoculation. IVIS imaging (PerkinElmer) of the mice was performed every week to monitor tumor burdens after treatment. The mice were placed in non-responding (resistant) and responding (sensitive) groups after 2-4 weeks of treatment based on the IVIS imaging findings. At the end of the study, tumors and blood samples were collected from the mice, and stored at −80°C.
Intratumoral injection of small EVs.
SKOV3 cells (1 × 106) were subcutaneously inoculated into NCRNU-Female nude mice (n = 8 per group). Treatment in the mice was started when tumors formed. Bevacizumab (5 mg/kg) was given intraperitoneally twice a week followed by small EVs via intratumoral injection (5 μg/mouse). Small EVs were isolated from SKOV3 cells under normal and hypoxic conditions and injected into the mice twice a week. Tumor lengths and widths were measured twice a week to calculate tumor volumes. Mice were sacrificed when those in any group became moribund, and their tumors were harvested, fixed, and sectioned for histochemical staining.
In vivo Matrigel plug assay.
Small EVs were isolated from the cell culture supernatants of RKO-PAR and RKO-VEGF−/− cells cultured with 2% exosome-depleted FBS medium for 48 hr. Five micrograms of small EVs were mixed with phenol red-free Matrigel (2:3 proportion; 500 μL in total; BD Biosciences). Next, the mixture was injected subcutaneously into NCRNU-Female nude mice in the small EV groups (three per group). PBS with Matrigel served as a negative control (n = 3), and recombinant human VEGF (0.25 nM) served as a positive control. After 8 days, the mice were sacrificed, and Matrigel plugs were extracted for a hemoglobin content assay (BioAssay Systems).
Patient samples
Patient (Female, median age 74 years, range 60 to 77 years) serum samples were obtained from a clinical trial (ClinicalTrials.gov identifier: NCT02923739) which was conducted at MDACC. Blood serum samples were taken pre-dose on day 1 of each treatment cycle. All the specimens were approved for processing and analysis by the MD Anderson IRB protocol.
Cell culture
SKOV3, HeyA8, HeyA8MDR, A2780, and A2780CP20 cells were cultured in RPMI 1640 supplemented with 10% FBS and 0.1% gentamycin. OVCAR5 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and 0.1% gentamycin. The human immortalized umbilical endothelial cell line RF24 and bevacizumab-resistant RF24 (RF24-Bev) cells were cultured in minimal essential medium supplemented with 10% FBS, sodium pyruvate, nonessential amino acids, minimal essential medium vitamins, and glutamine. HT29 cells were cultured using McCoy’s 5a medium supplemented with 10% FBS and 0.1% gentamycin. All above cells were purchased from the ATCC or provided by the MD Anderson Cytogenetics and Cell Authentication Core. The colon carcinoma cell lines RKO-PAR and RKO-VEGF−/− were kindly provided by Dr. Long H. Dang and cultured in Eagle’s Minimum Essential Medium supplemented with 10% FBS and 0.1% gentamycin. All cells were characterized using short tandem repeat DNA profiling, and mycoplasma testing of the cells was done with an ATCC universal mycoplasma detection kit. All cells were grown at 37°C in a 5% CO2 incubator.
METHOD DETAILS
Small EVs Isolation and Characterization
Isolating small EVs from cell culture supernatants
Cells passaged less than 20 times were cultured in a medium containing 2% exosome-depleted FBS for 48 hr before EV isolation. The harvested conditioned medium was pooled together (around 600 mL in total) and centrifuged at 300 × g for 10 min and 2000 × g for 20 min to remove cell debris and apoptotic bodies, respectively. The medium was subjected to centrifugation at 10,000 × g for 40 min to remove large vesicles. Next, we centrifuged the medium at 100,000 × g for 2 hr at 4°C using a Beckman 45Ti rotor. The supernatant was discarded, and the pellet (small EVs and protein) was re-suspended in PBS and centrifuged at 100,000 × g for 2 hr. The small EVs were finally re-suspended in 50 μL of PBS and stored at −80°C. The isolation procedures used in this study followed the standard methods of the International Society of Extracellular Vesicles (Théry et al., 2018; Witwer et al., 2013).
Isolation from serum
Serum samples were collected and stored at −80°C before use. Briefly, 0.5 to 2 mL of serum was used to isolate small EVs. After thawing, the serum samples were diluted with 50 mL of sterile PBS and spun down for 30 min at 2000 × g at 4°C. The supernatant was then transferred to a fresh tube and subjected to centrifugation at 10,000 × g and 4°C for 40 min. The supernatant was carefully transferred to an ultracentrifuge bottle, and the bottle was filled with 60 mL of PBS. The supernatant was centrifuged at 100,000 × g and 4°C for 2 hr, and the pellet was re-suspended in 3 mL of PBS. Next, the solution was transferred to a fresh polypropylene centrifuge tube (Beckman Coulter) and centrifuged for 2 hr at 100,000 × g and 4°C (repeated once). After this, the pellet was re-suspended in 20 μL of PBS and stored at −80°C.
Purification of small EVs by 30% Sucrose/Deuterium oxide (D2O) cushion
To demonstrate the presence of VEGF in small EVs, we performed a sucrose/D2O cushion ultracentrifugation (SUC) to deplete potential protein contaminations from cell culture supernatants as previously described (Ma et al., 2021).
Transmission electron microscopy (TEM)
TEM was performed by Kenneth Dunner Jr. with the High Resolution Electron Microscopy Facility of MDACC as described previously (Ma et al., 2021).
Small EVs quantification
The isolated small EVs were characterized independently using a Nano-sight tracking analysis (NTA), a Qubit protein assay kit (Thermo Fisher Scientific), and western blotting. Small EV fractions were diluted accordingly and analyzed using NTA to record their numbers and sizes. The Qubit protein assay kit was used to measure the protein concentrations in small EVs, and western blotting was performed to measure the expression of surface markers of small EVs.
Small EVs uptake assay
Ten micrograms of cancer cell-derived small EVs were isolated from OVCAR5 and SKOV3 cells and stained with CellMask Green plasma membrane stain (Thermo Fisher Scientific; Cat. #C37608, 1:200) for 30 min at 37°C. The small EV samples were then diluted in 3 mL of PBS and transferred to a fresh polypropylene centrifuge tube, which was followed by centrifugation for 2 hr at 100,000 × g and 4°C. The small EV samples were added into RF24 cells in a final concentration of 5 μg/mL. After 3 hr incubation in a cell culture incubator, cells were washed with ice-cold PBS twice and fixed in 4% formaldehyde for 15 min at room temperature. CellMask Deep Red plasma membrane stain (Thermo Fisher Scientific; Cat. #C10046, 1:1000) was used to stain the membranes of RF24 cells, and Hoechst 33342 (Invitrogen; Cat. #H3570, 1:10000) was used to stain the nuclei after fixation. The samples were stored at 4°C in the dark until they were analyzed using confocal microscopy. Images of the cells were acquired using an Andor Spinning Disk Confocal Microscope (Oxford Instruments). Analysis of confocal images was performed using ImarisViewer software (Oxford Instruments).
Co-Immunoprecipitation
For immunoprecipitation, we used a Universal Magnetic Co-IP kit (Active Motif; Cat. #54002) to extract the proteins according to the manufacturer’s procedures. Briefly, equivalent amounts (250 μg) of whole-cell extracts were incubated with primary anti-VEGF (Santa Cruz Biotechnology; Cat. #sc-7269, 1:100) or anti-CD63 (Santa Cruz Biotechnology; Cat. #sc-5275, 1:100) antibodies on a rolling shaker for 3 hr at 4°C. Control mouse IgG (Santa Cruz Biotechnology; Cat. #sc-2025, 1:100) was used as a negative control. Then, 25 μL protein G magnetic beads were added and then incubated for 1 hr at 4°C. After washing four times with complete Co-IP/wash buffer and eluting samples with 2X reducing loading buffer (130 mM Tris pH 6.8, 4% SDS, 0.02% Bromophenol blue, 20% glycerol, 100 mM DTT), we proceeded to detect the interaction of VEGF and CD63 by western blotting using rabbit antibodies against VEGF (Abcam; Cat. #ab46154, 1:1000) and CD63 (System biosciences; Cat. #EXOAB-CD63A-1, 1:1000). We used 5 μg of WCE as an input control.
Quantitative real-time reverse-transcriptase polymerase chain reaction (quantitative real-time RT-PCR)
Adherent cells (1 × 106) were washed twice with cold PBS and lysed with 350 μL of TRIzol (Thermo Fisher Scientific). Total RNA samples were further extracted using a Direct-zol RNA isolation kit (Zymo Research) and the sample quality was assessed using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). One microgram of RNA samples was subjected to a Verso cDNA synthesis kit (Thermo Fisher Scientific) and 2 μL of synthesized cDNA was used as a template for quantitative real-time RT-PCR. The primers for the human KDR gene were as follows: Forward 5′-GGCCCAATAATCAGAGTGGCA-3′, Reverse 5′-CCAGTGTCATTTCCGATCACTTT-3′. The 2−ΔΔCt method was used to calculate the relative gene expression and 18S was used as a housekeeping gene.
Western blotting
Small EVs isolated from cancer cell supernatants were quantified using a Qubit protein assay kit (Thermo Fisher Scientific) and cell protein samples from whole-cell lysates were quantified using a Pierce BCA protein assay kit (Thermo Fisher Scientific). Small EVs and cell proteins were extracted using RIPA buffer (25 mM Tris, pH 7.5, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1% Triton X-100). An equal amount of protein (5-10 μg for EVE and 20 μg for WCE) from each sample was loaded onto a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and transferred to a nitrocellulose membrane. After blocking with 5% nonfat milk, the membrane was blotted with primary antibodies (anti-VEGF [Santa Cruz Biotechnology; Cat. #sc-7269, 1:500], anti-HIF-1α [Cell Signaling Technology; Cat. #14179, 1:1000], anti-Alix [Santa Cruz Biotechnology; Cat. #sc-53538, 1:500], anti-CD63 [System Biosciences; Cat. #EXOAB-CD63A-1, 1:1000], anti-GRP94 [Santa Cruz Biotechnology; Cat. #sc-32249, 1:500], anti-TSG101 [Abcam; Cat. #ab30871, 1:1000], anti-HSP70 [Santa Cruz Biotechnology; Cat. #sc-24, 1:500], anti-p44/42 MAPK (Erk1/2) [Cell Signaling Technology; Cat. #4695, 1:1000], anti-p44/42 MAPK (phosphorylated Erk1/2) [Cell Signaling; Cat. #9101, 1:1000], anti-Akt (phospho ser473) [Cell Signaling Technology; Cat. #9271, 1:1000], anti-Akt [Cell Signaling Technology; Cat. #9272, 1:1000], anti-VEGFR2 [Cell Signaling Technology; Cat. #2479, 1:1000], anti-phosphoVEGFR2 [Abcam; Cat. #ab5473, 1:1000], anti-Lamin A/C [Santa Cruz Biotechnology; Cat. #sc-7292, 1:500], anti-Lamin B1 [Cell Signaling Technology; Cat. #12586 1:1000], anti-GAPDH [Sigma-Aldrich; Cat. #G8795, 1:3000], anti-vinculin [Sigma-Aldrich; Cat. #V9131, 1:3000], or anti-β-actin [Sigma-Aldrich; Cat. #A5441, 1:3000]) at 4°C overnight. The membrane was then washed three times with 0.1% Tris-buffered saline and Tween 20 (TBS-T) for 10 min each and then blotted with secondary antibodies (anti-rabbit or anti-mouse) for 1 hr at room temperature. Next, the membrane was washed three times with TBS-T, and the chemiluminescence signal was detected using X-ray film or Azure 400 Visible Fluorescent Western Blot Imaging System (Azure Biosystems, USA). After stripping with Restore Plus Western blot stripping buffer (Thermo Fisher Scientific), the membranes were then blocked with 5% non-fat milk and probed with another antibody. All the experiments were performed at least twice unless otherwise indicated.
Proteome profiler angiogenesis and phospho-kinases array
Total proteins of small EVs (50 μg) or whole-cell lysates (70 μg) were extracted and applied for the arrays following the manufacturer’s instructions. The measurement of the signal was performed according to the instructions of the manufacturer and the pixel density of each array spot was analyzed using ImageJ software program (National Institutes of Health).
Tube formation assay
Briefly, RF24 cells (8000 per well) were plated using a μ-Slide Angiogenesis (Ibidi) pre-coated with Matrigel (10 μL/well). Cells were cultured in a minimal essential medium for 4-6 hr in a 37°C incubator and treated with PBS only (control cells), normoxic small EVs (10 μg/mL) with or without bevacizumab (1 μg/ μL; N-sEVs), hypoxic small EVs (10 μg/mL) with or without bevacizumab (1 μg/μL; H-sEVs), or recombinant human VEGF (100 ng/mL) with or without bevacizumab (1 μg/μL). Images of tubes were acquired using a Leica camera, and tubes in three individual wells were counted using the ImageJ software program. The experiments were performed in triplicate independently.
Detection of Akt/Erk activation in RF24 cells by small EVs
RF24 cells (2 × 105 per well) were plated in a six-well plate and allowed to attach overnight. Next, cells were starved by depleting FBS from the medium for 6 hr and treated with PBS only (control cells), normoxic small EVs (10 μg/mL) with or without bevacizumab (1 μg/μL; N-sEVs), recombinant human VEGF (100 ng/mL) with or without bevacizumab (1 μg/μL), bevacizumab only (1 μg/μL). The cells were harvested after 6 hr, and protein was extracted for western blotting analysis.
Subcellular protein fractionation assay for cultured cells
RF24 cells (1 × 106 per 10-cm Petri dish) were plated in a complete cell culture medium. The next day, cells were washed twice with PBS and treated with VEGF positive and negative EVs (5 μg/mL) in 2% exosome-depleted FBS medium twice a day. Forty-eight hours after EV treatment, we harvested the cells and proceeded to the subcellular protein fractionation kit following the manufacturer’s instructions. Briefly, RF24 cells were harvested after trypsinization by spinning down at 500 × g and 4°C for 3 min. We then added ice-cold cytoplasmic extraction buffer (CEB) containing protease inhibitors to the pellet and incubated them at 4°C for 10 min with gently mixing. The mixture was then spun down for 5 min at 500 × g and 4°C and the supernatant (cytoplasmic extract) was transferred to a fresh ice-cold tube. The membrane extraction buffer (MEB) containing protease inhibitors was then added to the pellet and vortexed at the highest speed for 5 s. The suspension was incubated at 4°C for 10 min with gently mixing. We centrifuged the mixture for 5 min at 3000 × g and 4°C and collected the supernatant for membrane extract. The pellet was subjected to ice-cold nuclear extraction buffer (NEB) containing protease inhibitor and vortexed at the highest speed for 15 s. The resultant suspension was further incubated for 30 min at 4°C. Finally, the mixture was centrifuged for 5 min at 5000 × g and 4°C, and the supernatant was collected as a soluble nuclear extract. The protein concentration was measured by Pierce BCA kit and an equal amount of protein (20 μg) was loaded into SDS-PAGE gel for western blotting.
Intracellular VEGF staining via flow cytometry
First, we seeded cells in a 15-cm Petri dish at a density of 1-2 × 106 cells. After cell attachment, we replaced the culture medium with a complete medium containing a protein transport inhibitor cocktail (eBioscience, Cat. #00-4980-03). The next day, the cells were harvested using trypsin and washed twice with ice-cold PBS. We then spun down cells at 500 × g and 4°C for 5 minutes and re-suspended cell pellets with 4% paraformaldehyde (PFA, in PBS) for 10 min at room temperature. After fixation, the cells were washed twice with ice-cold PBS and pelleted down at 500 × g and 4°C for 5 minutes. We added 200 μL of 0.2% Triton X-100 (in PBS) to permeabilize cells for 10 min at room temperature. Afterward, cells were incubated with blocking buffer (10% FBS, 1% BSA, and human Fc-blocker (BD Biosciences; 1:10) in PBS, filtered through 0.22 μm filtration) for 20 min at room temperature. After blocking, cells were spun down at 500 × g and 4°C for 5 minutes, and a human PE-conjugated anti-VEGF-A antibody (R&D Systems; Cat. #IC2931P, diluted in blocking buffer, 1:10) was added. A mouse IgG2A PE-conjugated antibody (R&D Systems; Cat. #IC003P, 1:10) was used as a negative control. The cell suspension was further incubated for 30 min in the dark at room temperature. Then, we spun down cells and washed cells twice with ice-cold PBS. The final cell pellets were suspended in 500 μL of PBS and analyzed by BD FACS Celesta Flow Cytometer (BD Biosciences, US). Unstained cells were used as negative color control.
Amnis image flow cytometry
Small EVs (5 × 1010) were stained with CellMask Deep Red plasma membrane stain for 30 min at 37°C. The stained small EVs were placed in three groups of equal numbers and incubated with PBS only, a human PE-conjugated anti-CD63 antibody (R&D Systems; Cat. #IC5048P-025, 1:10), and a human PE-conjugated anti-VEGF-A antibody (R&D Systems; Cat. #IC2931P, 1:10), respectively. After 1 hr of incubation at room temperature, the samples were diluted with 3 mL of PBS and centrifuged for 3 hr at 100,000 × g and 4°C. The pellets were re-suspended in 50 μL of PBS and immediately analyzed using an Amnis Image StreamX MKII flow cytometer (Luminex). For single-color control samples, 20 μL of OneComp eBeads Compensation Beads (Thermo Fisher Scientific) was incubated with the PE-conjugated anti-CD63 antibody for 20 min at room temperature. The beads were then diluted with 1 mL of PBS and centrifuged for 5 min at 1500 rpm and 4°C. The PE-conjugated bead pellet was eventually re-suspended in 50 μL of PBS. The samples exposed to PBS only, PBS with CellMask Deep Red plasma membrane stain, or PBS with PE-conjugated anti-VEGF antibodies served as negative controls. Experiments were performed three times independently.
Immunofluorescence staining
Cultured cells were seeded onto a thin cover glass in six-well plates one day before staining. The next day, cells were fixed by 4% paraformaldehyde followed by permeabilization with 0.2% Triton X-100. The fixed cells were then incubated with a blocking buffer containing 3% FBS and 1% bovine serum albumin (BSA) for 1 hr at room temperature. After blocking, the cells were incubated with primary anti-VEGF (Abcam; Cat. #ab52917; 1:100) at 4°C overnight. After washing thrice with PBS, the cells were incubated with Alexa 488-labeled secondary antibodies (Jackson ImmunoResearch Laboratories; 1:250) for 1 hr at room temperature. Then the cells were washed three times with PBS and incubated with second primary anti-CD63 (Santa Cruz; Cat. #sc-5275, 1:250) antibodies overnight at 4°C. After incubation, cells were washed with PBS followed by incubation with Alexa 594-labeled secondary antibodies (Jackson ImmunoResearch Laboratories; 1:250) 1 hr at room temperature. The cells were then washed three times with PBS and incubated with Hoechst 33342 (Invitrogen; 1:10,000) for 20 min. Prolong Diamond Antifade Mountant media was used to mount the slides. The fluorescence signal was imaged under Andor Spinning Disk Confocal Microscope (Oxford Instrument) and analyzed using ImarisViewer software (Oxford Instruments). All experiments were performed in triplicates.
Immunohistochemistry
The frozen tumor samples from in vivo experiments were used to detect the expression of CD31 by immunohistochemistry. Tissue sections were fixed in cold acetone for 10 min and transferred to PBS for 3 times wash. The sections were blocked with 3% fish gelatin for 20 min at room temperature and then incubated with primary anti-CD31 antibodies (Abcam; 1:800) at 4°C overnight. After washing with PBS, the sections were incubated with horseradish peroxidase-conjugated goat anti-rat IgG (Jackson ImmunoResearch Laboratories; 1:500) for 1 hr at room temperature. Then, the slides were stained with DAB working solution, hematoxylin, and fresh PBS. The slides were examined under an Olympus microscope (Waltham, MA, USA), and images of each slide were captured using a Leica camera (Wetzlar, Germany). Five fields per slide and at least three slides per group were examined.
QUANTIFICATION AND STATISTICAL ANALYSIS
All results were presented as mean ± standard deviation (SD) values. All data were analyzed using the Mann-Whitney U test, Student’s t test or one-way analysis of variance (ANOVA) with the GraphPad Prism 8 (San Diego, CA, USA) unless otherwise indicated. The p values less than 0.05 were considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-VEGF (C-1) | Santa Cruz | Cat. #sc-7269; RRID: AB_628430 |
| Anti-VEGF | Abcam | Cat. #ab46154; RRID: AB_2212642 |
| Anti-VEGF | Abcam | Cat. #ab52917; RRID:AB_883427 |
| Anti-VEGFR2 | Cell Signaling Technology | Cat. #2479; RRID: AB_2212507 |
| Anti-VEGFR2 (phospho Y1054+Y1059) | Abcam | Cat. #ab5473; RRID: AB_304917 |
| Anti-Akt (phospho ser473) | Cell Signaling Technology | Cat. #9271; RRID: AB_329825 |
| Anti-Akt | Cell Signaling Technology | Cat. #9272; RRID: AB_329827 |
| Anti-p44/42 MAPK (phosphorylated Erk1/2) | Cell Signaling Technology | Cat. #9101; RRID: AB_331646 |
| Anti-p44/42 MAPK (Erk1/2) | Cell Signaling Technology | Cat. #4695; RRID: AB_390779 |
| Anti-GRP94 | Santa Cruz Biotechnology | Cat. #sc-32249, RRID: AB_627676 |
| Anti-CD63 | System Biosciences | Cat. #EXOAB-CD63A-1; RRID: AB_2561274 |
| Anti-CD63 | Santa Cruz | Cat. #sc-5275; RRID: AB_627877 |
| Anti-TSG101 | Abcam | Cat. #ab30871; RRID: AB_2208084 |
| Anti-Alix | Santa Cruz Biotechnology | Cat. #sc-53538; RRID: AB_673821 |
| Anti-HSP70 | Santa Cruz Biotechnology | Cat. #sc-24; RRID: AB_627760 |
| Anti-vinculin | Sigma-Aldrich | Cat. #V9131; RRID: AB_477629 |
| Anti-GAPDH | Sigma-Aldrich | Cat. #G8795; RRID: AB_1078991 |
| Anti-β-actin | Sigma-Aldrich | Cat. #A5441; RRID: AB_476744 |
| Anti-CD31 | Abcam | Cat. #ab56299; RRID: AB_940884 |
| Anti-HIF-1α | Cell Signaling Technology | Cat. #14179; RRID: AB_2622225 |
| Lamin A/C | Santa Cruz Biotechnology | Cat. #sc-7292; RRID: AB_627875 |
| Lamin B1 | Cell Signaling Technology | Cat. #12586; RRID: AB_2650517 |
| Human CD63 PE-conjugated antibody | R&D Systems | Cat. #IC5048P-025; RRID: N/A |
| Human VEGF PE-conjugated antibody | R&D Systems | Cat. #IC2931P; RRID: N/A |
| Mouse IgG2A PE-conjugated Antibody | R&D Systems | Cat. #IC003P; RRID: AB_357245 |
| Human BD Fc Block | BD Biosciences | Cat. #564219; RRID: AB_2728082 |
| Alexa Fluor 488 goat anti-rabbit IgG | Jackson ImmunoResearch | Cat. #111-546-047; RRID: AB_2338056 |
| Alexa Fluor 488 goat anti-rat IgG | Jackson ImmunoResearch | Cat. #112-546-072; RRID: AB_2338368 |
| Alexa Fluor 594 goat anti-mouse IgG | Jackson ImmunoResearch | Cat. #115-586-072; RRID: AB_2338897 |
| Peroxidase AffiniPure Goat Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat. #112-035-167; RRID: AB_2338139 |
| ECL anti-rabbit IgG, horseradish peroxidase | GE Healthcare | Cat. #GENA934; RRID: AB_2722659 |
| ECL anti-mouse IgG, horseradish peroxidase | GE Healthcare | Cat. #NA931; RRID: AB_772210 |
| Bacterial and virus strains | ||
| Exosome Cyto-Tracer, pCT-CD63-GFP | SYSTEM BIOSCIENCES | Cat. #CYTO120-VA-1 |
| pLKO MISSION® CD63 shRNA Lentiviral Transduction Particles | Sigma-Aldrich | Cat. #SHCLNV-NM_001780 |
| Firefly Luciferase Lentifect Purified Lentiviral Particles | Genecopoeia | Cat. #LPP-FLUC-Lv100c |
| Biological samples | ||
| Patients’ serum samples | MDACC | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human VEGF 165 protein | R&D Systems | Cat. #293-VE-010 |
| Recombinant Human VEGF 121 (aa 207-327) Protein | R&D Systems | Cat. #4644-VS-010 |
| Recombinant Human VEGF 189 (aa 27-215) Protein | R&D Systems | Cat. #8147-VE-025 |
| Bevacizumab | Genentech | NDC Code 50242-061-01 |
| Tris base | Thermo Fisher Scientific | Cat. #BP152-5 |
| NaCl | Thermo Fisher Scientific | Cat. #AC424290050 |
| Glycine | Thermo Fisher Scientific | Cat. #BP381-5 |
| NP-40 | Sigma-Aldrich | Cat. #74385 |
| Tissue Freezing Medium | Mercedes Medical | Cat. #MER 5000 |
| Halt Protease Inhibitor Cocktail (100X) | Thermo Fisher Scientific | Cat. #78438 |
| Permount | Thermo Fisher Scientific | Cat. #SP15-100 |
| Luciferin | Gold Biotechnology | Cat. #LUCK-1G |
| Hoechst 33342 | Thermo Fisher Scientific | Cat. #H3570 |
| CellMask Deep Red plasma membrane stain | Thermo Fisher Scientific | Cat. #C10046 |
| CellMask Green plasma membrane stain | Thermo Fisher Scientific | Cat. #C37608 |
| OneComp eBeads Compensation beads | Thermo Fisher Scientific | Cat. #01-1111-41 |
| Exosome-depleted FBS | System Biosciences | Cat. #EXO-FBS-250A-1 |
| Matrigel | BD Biosciences | Cat. #356231 |
| 16% paraformaldehyde | Electron Microscopy Sciences | Cat. #15710-S |
| Triton X-100 | Thermo Fisher Scientific | Cat. #BP151-500 |
| Hematoxylin Solution | Sigma-Aldrich | Cat. #GHS316-500ML |
| Stable DAB | Thermo Fisher Scientific | Cat. #750118 |
| ProLong Diamond Antifade Mountant | Thermo Fisher Scientific | Cat. #P36961 |
| Critical commercial assays | ||
| Proteome Profiler Human Angiogenesis Array Kit | R&D Systems | Cat. #ARY007 |
| Proteome Profiler Human Phospho-Kinase Array Kit | R&D Systems | Cat. #ARY003C |
| Subcellular Protein Fractionation Kit for Cultured Cells | Thermo Fisher Scientific | Cat. #78840 |
| Human VEGF Quantikine ELISA Kit | R&D Systems | Cat. #DVE00 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat. #23225 |
| Restore Plus Western Blot Stripping Buffer | Thermo Fisher Scientific | Cat. #46430 |
| Direct-zol RNA Kits | Zymo Research | Cat. #R2062 |
| Verso cDNA Synthesis Kit | Thermo Fisher Scientific | Cat. #AB1453B |
| Universal Mycoplasma Detection Kit | ATCC | Cat. #30-1012K |
| Qubit Protein Assay Kit | Thermo Fisher Scientific | Cat. #Q33212 |
| Universal Magnetic Co-IP Kit | Active Motif | Cat. #54002 |
| Flow Cytometry Sub-micron Particle Size Reference Kit | Thermo Fisher Scientific | Cat. #F13839 |
| Experimental models: Cell lines | ||
| RF24 | MDACC cell line bank | N/A |
| RF24-Bev | Anil Sood lab (MDACC) | N/A |
| SKOV3 | MDACC cell line bank | N/A |
| OVCAR5 | MDACC cell line bank | N/A |
| RKO-PAR | Kindly provided by Dr. Long H. Dang, Health First Cancer Institute | N/A |
| RKO-VEGF−/− | Kindly provided by Dr. Long H. Dang, Health First Cancer Institute | N/A |
| HeyA8 | MDACC cell line bank | N/A |
| HeyA8MDR | MDACC cell line bank | N/A |
| HT29 | Kindly provided by Lee Ellis lab (MDACC) | N/A |
| A2780 | MDACC cell line bank | N/A |
| A2780CP20 | MDACC cell line bank | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: NCRNU-F nude (NCr) | Taconic | Model #: NCRNU-F |
| Oligonucleotides | ||
| Primers for qRT-PCR, see Methods | This paper | N/A |
| Accell Human KDR siRNA, set of 4 | Horizon | Cat. #EQ-003148-00-0010 |
| Accell eGFP Control siRNA | Horizon | Cat. #D-001940-01-20 |
| Software and algorithms | ||
| Prism version 8.00 | GraphPad Software | https://www.graphpad.com/ |
| CorelDRAW Graphics Suite 2018 | CorelDRAW | http://www.coreldraw.com/e/ |
| Living Image software | PerkinElmer | https://www.perkinelmer.com/ |
| IDEAS 6.2 | Luminex | https://www.luminexcorp.com/imagestreamx-mk-ii/#software |
| Imaris Viewer x64 9.5.1 | Oxford Instruments | https://imaris.oxinst.com/ |
| ImageJ | NIH Image | https://imagej.nih.gov/ij/index.html |
Highlights.
Cancer cells package increasing amounts of VEGF in small EVs with anti-VEGF therapy
VEGF packaging into small EVs is mediated by the tetraspanin CD63
Anti-VEGF antibodies failed to recognize small EV-VEGF (eVEGF)
eVEGF triggers intracrine VEGF signaling and promotes angiogenesis
ACKNOWLEDGMENTS
This publication is part of the NIH Extracellular RNA Communication Consortium paper package and was supported by the NIH Common Fund’s ex-RNA Communication Program. This work was supported, in part, by NIH grants UH3TR000943, P50CA217685, R35CA209904, and P30CA016672; Interagency Agreement ACN15006-001 between the National Cancer Institute and the Pacific Northwest National Laboratory (PNNL); the Ovarian Cancer Research Alliance; the Blanton-Davis Ovarian Cancer Research Program; the American Cancer Society Research Professor Award; and the Frank McGraw Memorial Chair in Cancer Research. S.M. is supported by a Foundation for Women’s Cancer Research grant (Sponsor Award number FP00009883). S.L. is supported by a Sprint for Life Research Award and the MD Anderson Ovarian Cancer Moon Shot Program. O.D.L. was supported by an NIH institutional training grant (5T32CA009599). S.Y.W. was supported by the Cancer Prevention & Research Institute of Texas Research Training Program (grants RP101502, RP140106, and RP170067). A.V.P. was supported by the NIH Partnership for Excellence in Cancer Research (U54CA096300/U54CA096297). Short tandem repeat DNA fingerprinting was supported by the NIH/NCI under award number P30CA016672 and used the Cytogenetics and Cell Authentication Core. We thank Scientific Publications (Dr. Donald R. Norwood) Research Medical Library at The University of Texas MD Anderson Cancer Center for reviewing and editing this manuscript. PNNL is a multiprogram national laboratory operated by Battelle for the DOE under contract DE-AC05-76RL01830. We thank Kenneth Dunner Jr. with the High Resolution Electron Microscopy Facility (grant NIHP30CA016672) at The University of Texas MD Anderson Cancer Center for performing theTEM studies. Wethank Dr. Long H. Dang at Health First for providing RKO-PAR and RKO-VEGF−/− cells. We acknowledge the Flow Cytometry and Cellular Imaging Core Facility (FCCICF) (NCI grants P30CA16672) at The University of Texas MD Anderson Cancer Center for performing flow cytometry experiments.
Footnotes
DECLARATION OF INTERESTS
R.L.C. has received grant funding from Genentech, Merck, Janssen Pharmaceutical, Clovis Oncology, AstraZeneca, and AbbVie and serves as an investigator on the scientific steering committees for Tesaro, Clovis Oncology, AstraZeneca, and AbbVie. A.K.S. a consultant for KIYATEC, Merck, and AstraZeneca, has received research funding from M-Trap, and is a BioPath Holdings shareholder.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109549.
REFERENCES
- Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, Sovak MA, Yi J, and Nycum LR (2012). OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol 30, 2039–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanian C, Goff B, Nycum LR, Wang YV, Husain A, and Blank SV (2015). Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol 139, 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte RS, Chen DS, and Ferrara N (2019). VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 176, 1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruah J, and Wary KK (2020). Exosomes in the Regulation of Vascular Endothelial Cell Regeneration. Front. Cell Dev. Biol 7, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, and Hanahan D (2008). Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Ye XC, Wang R, Ling X, McManus M, Fan F, Boulbes D, and Ellis LM (2016). Intracrine VEGF Signaling Mediates the Activity of Prosurvival Pathways in Human Colorectal Cancer Cells. Cancer Res. 76, 3014–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binenbaum Y, Fridman E, Yaari Z, Milman N, Schroeder A, Ben David G, Shlomi T, and Gil Z (2018). Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res. 78, 5287–5299. [DOI] [PubMed] [Google Scholar]
- Boocock CA, Charnock-Jones DS, Sharkey AM, McLaren J, Barker PJ, Wright KA, Twentyman PR, and Smith SK (1995). Expression of vascular endothelial growth factor and its receptors flt and KDR in ovarian carcinoma. J. Natl. Cancer Inst 87, 506–516. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, and Jain RK (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DS, Kim DK, Kim YK, and Gho YS (2013). Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 13, 1554–1571. [DOI] [PubMed] [Google Scholar]
- Coleman RL, Brady MF, Herzog TJ, Sabbatini P, Armstrong DK, Walker JL, Kim BG, Fujiwara K, Tewari KS, O’Malley DM, et al. (2017). Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 18, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DG, Batchelor TT, Willett CG, and Jain RK (2007). VEGF-targeted cancer therapy strategies: current progress, hurdles and future prospects. Trends Mol. Med 13, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YB, Yan C, Mu L, Mi YL, Zhao H, Hu H, Li XL, Tao DD, Wu YQ, Gong JP, and Qin JC (2019). Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 38, 1951–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz SN, Conlon MM, Rider MA, Brownstein NC, and Meckes DG Jr. (2016). Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 5, 31295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz SN, Nkosi D, Conlon MM, York SB, Liu X, Tremblay DC, and Meckes DG Jr. (2017). CD63 Regulates Epstein-Barr Virus LMP1 Exosomal Packaging, Enhancement of Vesicle Production, and Noncanonical NF-κB Signaling. J. Virol 91, e02251–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK, Duda DG, Clark JW, and Loeffler JS (2006). Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 3, 24–30. [DOI] [PubMed] [Google Scholar]
- Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, Batchelor TT, and Sorensen AG (2009). Biomarkers of response and resistance to antiangiogenic therapy. Nat. Rev. Clin. Oncol 6, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlert C, and Kalluri R (2013). Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. (Berl.) 91, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko SY, Lee W, Kenny HA, Dang LH, Ellis LM, Jonasch E, Lengyel E, and Naora H (2019). Cancer-derived small extracellular vesicles promote angiogenesis by heparin-bound, bevacizumab-insensitive VEGF, independent of vesicle uptake. Commun. Biol 2, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li I, and Nabet BY (2019). Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 18, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Zhang X, Du L, Wang Y, Liu X, Tian H, Wang L, Li P, Zhao Y, Duan W, et al. (2019). Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol. Cancer 18, 43. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ma S, Pradeep S, Hu W, Zhang D, Coleman R, and Sood A (2018). The role of tumor microenvironment in resistance to anti-angiogenic therapy. F1000Res. 7, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, McGuire MH, Mangala LS, Lee S, Stur E, Hu W, Bayraktar E, Villar-Prados A, Ivan C, Wu SY, et al. (2021). Gain-of-function p53 protein transferred via small extracellular vesicles promotes conversion of fibroblasts to a cancer-associated phenotype. Cell Rep. 34, 108726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesiano S, Ferrara N, and Jaffe RB (1998). Role of vascular endothelial growth factor in ovarian cancer: inhibition of ascites formation by immunoneutralization. Am. J. Pathol 153, 1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Guo H, Wang X, Zhu X, Yan M, Wang X, Xu Q, Shi J, Lu E, Chen W, and Zhang J (2019). Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 20, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, et al. (2016). Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell 29, 653–668. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Han S, Jiang WG, Boulton ME, Chen ZJ, Law BK, and Cai J (2011). αB-crystallin, an effector of unfolded protein response, confers anti-VEGF resistance to breast cancer via maintenance of intracrine VEGF in endothelial cells. Mol. Cancer Res 9, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Simons M, and Raposo G (2009). Exosomes–vesicular carriers for intercellular communication. Curr. Opin. Cell Biol 21, 575–581. [DOI] [PubMed] [Google Scholar]
- Simons M, Gordon E, and Claesson-Welsh L (2016). Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol 17, 611–625. [DOI] [PubMed] [Google Scholar]
- Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, et al. (2014). Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 24, 766–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C, Ostrowski M, and Segura E (2009). Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol 9, 581–593. [DOI] [PubMed] [Google Scholar]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, et al. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, and Théry C (2016). Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 164, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Todorova D, Simoncini S, Lacroix R, Sabatier F, and Dignat-George F (2017). Extracellular Vesicles in Angiogenesis. Circ. Res. 120, 1658–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer KW, Buzás EI, Bemis LT, Bora A, Lässer C, Lötvall J, Nolte-’t Hoen EN, Piper MG, Sivaraman S, Skog J, et al. (2013). Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi N, Teshima-Kondo S, Masuda K, Nishida K, Kuwano Y, Dang DT, Dang LH, Nikawa T, and Rokutan K (2013). Chronic inhibition of tumor cell-derived VEGF enhances the malignant phenotype of colorectal cancer cells. BMC Cancer 13, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi A, Villar-Prados A, Oliphint PA, Zhang J, Song X, De Hoff P, Morey R, Liu J, Roszik J, Clise-Dwyer K, et al. (2019). Mechanisms of nuclear content loading to exosomes. Sci. Adv 5, eaax8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
- All data reported in this paper will be shared by the lead contact upon request.
- This paper does not report original code.
- Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.