

The crystal structure of 4-chloro-1H-pyrazole has been determined at 170 K, showing a hydrogen-bonded trimeric molecular assembly that is isostructural to its bromo analogue, 4-bromo-1H-pyrazole.
Keywords: crystal structure, pyrazole, proton disorder, low temperature
Abstract
The structure of 4-chloro-1H-pyrazole, C3H3ClN2, at 170 K has orthorhombic (Pnma) symmetry and is isostructural to its bromo analogue. Data were collected at low temperature since 4-chloro-1H-pyrazole sublimes when subjected to the localized heat produced by X-rays. The structure displays intermolecular N—H⋯N hydrogen bonding and the packing features a trimeric molecular assembly bisected by a mirror plane (m normal to b) running through one chlorine atom, one carbon atom and one N—N bond. The asymmetric unit therefore consists of one and one-half 4-chloro-1H-pyrazole molecules. Thus, the N—H proton is crystallographically disordered over two positions of 50% occupancy each.
Chemical context
Pyrazoles are a family of five-membered, π-excess aromatic heterocycles with adjacent nitrogen atoms (Krishnakumar et al., 2011 ▸; Karrouchi et al., 2018 ▸). They serve as scaffolds for numerous pharmaceutically active compounds and agrochemicals as they are stable and amenable to substitution (Naim et al., 2016 ▸). They readily coordinate to metals, forming a large group of complexes where typically pyrazoles are monodentate or bridging bidentate pyrazolido ligands (Pettinari et al., 2010 ▸; Viciano-Chumillas et al., 2010 ▸). In the solid state, pyrazoles form hydrogen-bonded aggregates whose topology depends on the steric bulk of peripheral substituents and the acidity of the N1—H proton and basicity of the N2 atom. For example, 4-bromo-pyrazole and 3-methyl-pyrazole form approximately planar triangular assemblies (Foces-Foces et al., 1999 ▸; Goddard et al., 1999 ▸), 3,5-diphenyl-pyrazole and 3,5-bis(trifluoromethyl)pyrazole form tetranuclear assemblies (Raptis et al., 1993 ▸; Alkorta et al., 1999 ▸), while pyrazole forms a one-dimensional polymeric chain (Krebs Larsen et al., 1970 ▸).
Structural commentary
4-Chloro-1H-pyrazole crystallizes with one and one-half molecules in the asymmetric unit (Z′ = 1.5) as shown in Fig. 1 ▸. The second molecule is bisected by a mirror plane normal to b (x, 3/4, z), which runs through C5, Cl2, and the N3—N3i bond [symmetry code: (i) x, 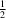 − y, z]. As a result of this mirror plane, the NH protons are crystallographically disordered over two positions. As with the bromo analogue (Foces-Foces et al., 1999 ▸), 4-chloro-1H-pyrazole crystallizes in the Pnma space group and forms trimeric units (Fig. 2 ▸).
− y, z]. As a result of this mirror plane, the NH protons are crystallographically disordered over two positions. As with the bromo analogue (Foces-Foces et al., 1999 ▸), 4-chloro-1H-pyrazole crystallizes in the Pnma space group and forms trimeric units (Fig. 2 ▸).
Figure 1.

Perspective view of the asymmetric unit of 4-chloro-1H-pyrazole. Displacement ellipsoids are shown at 50% probability and half the disordered protons are removed for clarity.
Figure 2.

Packing of 4-chloro-1H-pyrazole, viewed parallel to the c axis, showing the formation of trimeric units. Half the disordered protons have been removed for clarity.
Also disordered in this structure are the C and N atoms. Without disorder, pyrazoles have one ‘pyrrole-like’ side and one ‘pyridine-like’ side, as was discussed in the neutron diffraction study of 1H-pyrazole (Krebs Larsen et al., 1970 ▸). The C—N, C—C, and C—H bonds on either side of the molecule are crystallographically distinct and resemble either pyrrole or pyridine. However, due to the disorder of the N—H protons in the current structure, only average positions of the C and N atoms have been obtained. Therefore, the C—N, C—C, and C—H bonds on either side of the molecule are indistinguishable. This is most apparent in the C—N bonds. In the current structure, the C—N bonds are 1.335 (2), 1.334 (2), and 1.334 (2) Å for C1—N1, C3—N2, and C4 —N3, respectively. In the 1H-pyrazole structure, the C—N bond lengths are 1.356 and 1.350 Å for the ‘pyrrole-like’ side and the ‘pyridine-like’ side, respectively. The C—N bond lengths of the current structure are in agreement with those previously reported in the 4-bromo analogue with C—N bond lengths of 1.343 (10), 1.331 (10), and 1.327 (10) Å (Foces-Foces et al., 1999 ▸). Furthermore, these bond lengths are also in agreement with the 4-chloro-1H-pyrazol-2-ium chloride salt, which exhibits C—N bond lengths of 1.334 (2) and 1.331 (2) Å (Farmiloe et al., 2019 ▸). The N—N bonds of the present molecule are 1.346 (2) and 1.345 (3) Å, for N1—N2 and N3—N3i, respectively, and are similar to those of 4-methyl-1H-pyrazole (which is refined without proton disorder in Pca21) with N—N bond lengths of 1.343 (2), 1.344 (2), and 1.349 (2) Å (Goddard et al., 1999 ▸). However, the N—N bonds of the 4-bromo analogue are slightly shorter at 1.335 (9) Å each (Foces-Foces et al., 1999 ▸).
Supramolecular features
Pyrazoles are known to crystallize in a variety of hydrogen-bonded motifs – dimers, trimers, tetramers, and catemers (Infantes & Motherwell, 2004 ▸; Pérez & Riera, 2009 ▸). The current structure of 4-chloro-1H-pyrazole crystallizes in trimeric units, as do the 4-bromo and 4-methyl analogues. The intermolecular N1⋯N1i, N2⋯N3ii, and N3⋯N2ii [symmetry codes: (i) x, −y +  , z; (ii) −x + 1, −y + 1, −z] are 2.885 (3), 2.858 (2), and 2.858 (2), respectively, as shown in Table 1 ▸. These values are in agreement with other intermolecular hydrogen-bond interactions of pyrazoles. Just as with the 4-bromo derivative, 4-chloro-1H-pyrazole packs in a herringbone arrangement when viewed down the b axis (Fig. 3 ▸). The current structure exhibits no π-stacking interactions.
, z; (ii) −x + 1, −y + 1, −z] are 2.885 (3), 2.858 (2), and 2.858 (2), respectively, as shown in Table 1 ▸. These values are in agreement with other intermolecular hydrogen-bond interactions of pyrazoles. Just as with the 4-bromo derivative, 4-chloro-1H-pyrazole packs in a herringbone arrangement when viewed down the b axis (Fig. 3 ▸). The current structure exhibits no π-stacking interactions.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯N1i | 0.88 | 2.03 | 2.885 (3) | 165 |
| N2—H2⋯N3ii | 0.88 | 1.99 | 2.8582 (19) | 169 |
| N3—H3A⋯N2ii | 0.88 | 1.99 | 2.8582 (19) | 169 |
Symmetry codes: (i) x, -y+{\script{3\over 2}}, z; (ii) -x+1, -y+1, -z.
Figure 3.

Packing of 4-chloro-1H-pyrazole, viewed parallel to the b axis, showing the formation of a herringbone motif.
Synthesis and crystallization
4-Chloro-1H-pyrazole was purchased commercially and crystals were grown from the slow evaporation of a methylene chloride solution.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. The data were collected at 170 K on a Bruker D8 CMOS diffractometer equipped with a Photon II detector. CrysAlis PRO was used for scaling and absorption correction. All C—H protons were refined freely while the N—H protons were fixed using an AFIX command and constrained to half occupancy due to the proton disorder.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C3H3ClN2 |
| M r | 102.52 |
| Crystal system, space group | Orthorhombic, P n m a |
| Temperature (K) | 170 |
| a, b, c (Å) | 14.9122 (10), 17.6410 (9), 4.9878 (3) |
| V (Å3) | 1312.13 (14) |
| Z | 12 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.69 |
| Crystal size (mm) | 0.29 × 0.14 × 0.08 |
| Data collection | |
| Diffractometer | Bruker D8 CMOS |
| Absorption correction | Multi-scan (CrysAlis PRO; Rigaku OD, 2019 ▸) |
| Tmin, Tmax | 0.760, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 25891, 1798, 1481 |
| R int | 0.043 |
| (sin θ/λ)max (Å−1) | 0.682 |
| Refinement | |
| R[F2 > 2σ(F 2)], wR(F 2), S | 0.033, 0.092, 1.06 |
| No. of reflections | 1798 |
| No. of parameters | 97 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.27, −0.26 |
To remove the proton disorder, an attempt was made to refine the molecule in the non-centrosymmetric Pn21 a space group, which is also consistent with the systematic absences. In Pn21 a, hydrogen atoms were refined in both possible positions – on the odd-labeled N atoms or on the even-labeled N atoms. However, as Foces-Foces et al. (1999 ▸) found with the 4-bromo-analogue, refinement in the lower symmetry space group did not improve the refinement. For the trial Pn21 a structure with protons on the even-labeled N atoms, the R 1 and wR 2 values slightly increased to 3.67% and 9.67%, respectively, and the shifting of the carbon atoms could not be consolidated. For the trial Pn21 a structure with protons on the odd-labeled N atoms, the R 1 and wR 2 values increased even more to 3.71% and 9.69%, respectively, and the coordinates of the non-protonated N atoms could not converge. Therefore, the best refined model was chosen to be in the centrosymmetric Pnma space group with NH protons disordered over two positions.
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989021008604/zl5013sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989021008604/zl5013Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989021008604/zl5013Isup3.cml
CCDC reference: 2103822
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors would like to acknowledge Dr Indranil Chakraborty and Dr Horst Puschmann for their assistance and crystallographic expertise.
supplementary crystallographic information
Crystal data
| C3H3ClN2 | Dx = 1.557 Mg m−3 |
| Mr = 102.52 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, Pnma | Cell parameters from 6701 reflections |
| a = 14.9122 (10) Å | θ = 2.3–30.2° |
| b = 17.6410 (9) Å | µ = 0.69 mm−1 |
| c = 4.9878 (3) Å | T = 170 K |
| V = 1312.13 (14) Å3 | Rect. Prism, clear colourless |
| Z = 12 | 0.28 × 0.14 × 0.08 mm |
| F(000) = 624 |
Data collection
| Bruker D8 CMOS diffractometer | 1481 reflections with I > 2σ(I) |
| ω scans | Rint = 0.043 |
| Absorption correction: multi-scan (CrysAlisPro; Rigaku OD, 2019) | θmax = 29.0°, θmin = 2.3° |
| Tmin = 0.760, Tmax = 1.000 | h = −20→20 |
| 25891 measured reflections | k = −23→23 |
| 1798 independent reflections | l = −6→6 |
Refinement
| Refinement on F2 | Primary atom site location: dual |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.033 | Hydrogen site location: mixed |
| wR(F2) = 0.092 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0418P)2 + 0.5547P] where P = (Fo2 + 2Fc2)/3 |
| 1798 reflections | (Δ/σ)max = 0.001 |
| 97 parameters | Δρmax = 0.27 e Å−3 |
| 0 restraints | Δρmin = −0.26 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| Cl1 | 0.65912 (3) | 0.45830 (2) | 0.73672 (9) | 0.04510 (15) | |
| C1 | 0.66638 (11) | 0.61504 (9) | 0.6893 (4) | 0.0375 (3) | |
| C2 | 0.63595 (10) | 0.54554 (8) | 0.6008 (3) | 0.0330 (3) | |
| N1 | 0.63427 (9) | 0.66822 (7) | 0.5248 (3) | 0.0390 (3) | |
| H1A | 0.644141 | 0.717242 | 0.538844 | 0.047* | 0.5 |
| C3 | 0.58468 (11) | 0.56001 (10) | 0.3776 (3) | 0.0389 (4) | |
| N2 | 0.58435 (9) | 0.63462 (8) | 0.3340 (3) | 0.0405 (3) | |
| H2 | 0.556236 | 0.657939 | 0.202649 | 0.049* | 0.5 |
| Cl2 | 0.70256 (4) | 0.250000 | 0.59928 (11) | 0.03892 (16) | |
| C5 | 0.62343 (15) | 0.250000 | 0.3495 (4) | 0.0326 (4) | |
| N3 | 0.52730 (9) | 0.28813 (7) | 0.0432 (3) | 0.0395 (3) | |
| H3A | 0.494432 | 0.317237 | −0.061095 | 0.047* | 0.5 |
| C4 | 0.58543 (12) | 0.31226 (10) | 0.2276 (3) | 0.0396 (4) | |
| H3 | 0.5571 (14) | 0.5266 (11) | 0.253 (4) | 0.043 (5)* | |
| H1 | 0.7031 (12) | 0.6279 (11) | 0.839 (4) | 0.040 (5)* | |
| H4 | 0.5962 (12) | 0.3661 (12) | 0.261 (4) | 0.051 (6)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0525 (3) | 0.0303 (2) | 0.0525 (3) | 0.00392 (16) | −0.00952 (19) | 0.00342 (16) |
| C1 | 0.0379 (8) | 0.0335 (8) | 0.0412 (8) | −0.0017 (6) | −0.0027 (7) | −0.0020 (7) |
| C2 | 0.0340 (7) | 0.0281 (7) | 0.0371 (8) | 0.0020 (6) | 0.0008 (6) | −0.0006 (6) |
| N1 | 0.0408 (7) | 0.0330 (6) | 0.0431 (7) | −0.0007 (6) | 0.0017 (6) | −0.0012 (6) |
| C3 | 0.0429 (9) | 0.0345 (8) | 0.0391 (8) | 0.0028 (6) | −0.0053 (7) | −0.0032 (7) |
| N2 | 0.0450 (8) | 0.0383 (7) | 0.0384 (7) | 0.0053 (6) | −0.0022 (6) | 0.0028 (6) |
| Cl2 | 0.0416 (3) | 0.0395 (3) | 0.0356 (3) | 0.000 | −0.0053 (2) | 0.000 |
| C5 | 0.0359 (11) | 0.0311 (10) | 0.0309 (10) | 0.000 | −0.0011 (9) | 0.000 |
| N3 | 0.0432 (7) | 0.0370 (7) | 0.0384 (7) | 0.0068 (6) | −0.0036 (6) | −0.0001 (6) |
| C4 | 0.0483 (10) | 0.0306 (8) | 0.0401 (8) | 0.0052 (7) | −0.0035 (7) | −0.0040 (7) |
Geometric parameters (Å, º)
| Cl1—C2 | 1.7169 (15) | N2—H2 | 0.8800 |
| C1—C2 | 1.380 (2) | Cl2—C5 | 1.716 (2) |
| C1—N1 | 1.335 (2) | C5—C4 | 1.377 (2) |
| C1—H1 | 0.952 (19) | C5—C4i | 1.377 (2) |
| C2—C3 | 1.374 (2) | N3—N3i | 1.345 (3) |
| N1—H1A | 0.8800 | N3—H3A | 0.8800 |
| N1—N2 | 1.3457 (19) | N3—C4 | 1.334 (2) |
| C3—N2 | 1.334 (2) | C4—H4 | 0.98 (2) |
| C3—H3 | 0.951 (19) | ||
| C2—C1—H1 | 130.6 (11) | N1—N2—H2 | 125.7 |
| N1—C1—C2 | 108.04 (14) | C3—N2—N1 | 108.50 (14) |
| N1—C1—H1 | 121.4 (11) | C3—N2—H2 | 125.7 |
| C1—C2—Cl1 | 127.15 (13) | C4i—C5—Cl2 | 127.11 (10) |
| C3—C2—Cl1 | 126.74 (12) | C4—C5—Cl2 | 127.11 (10) |
| C3—C2—C1 | 106.09 (14) | C4—C5—C4i | 105.8 (2) |
| C1—N1—H1A | 125.6 | N3i—N3—H3A | 125.7 |
| C1—N1—N2 | 108.87 (13) | C4—N3—N3i | 108.61 (9) |
| N2—N1—H1A | 125.6 | C4—N3—H3A | 125.7 |
| C2—C3—H3 | 131.0 (12) | C5—C4—H4 | 129.2 (11) |
| N2—C3—C2 | 108.49 (15) | N3—C4—C5 | 108.50 (15) |
| N2—C3—H3 | 120.2 (12) | N3—C4—H4 | 122.3 (11) |
| Cl1—C2—C3—N2 | −178.73 (13) | N1—C1—C2—Cl1 | 178.76 (12) |
| C1—C2—C3—N2 | 0.03 (19) | N1—C1—C2—C3 | 0.01 (18) |
| C1—N1—N2—C3 | 0.07 (18) | Cl2—C5—C4—N3 | 179.74 (15) |
| C2—C1—N1—N2 | −0.05 (18) | N3i—N3—C4—C5 | −0.20 (16) |
| C2—C3—N2—N1 | −0.06 (19) | C4i—C5—C4—N3 | 0.3 (3) |
Symmetry code: (i) x, −y+1/2, z.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···N1ii | 0.88 | 2.03 | 2.885 (3) | 165 |
| N2—H2···N3iii | 0.88 | 1.99 | 2.8582 (19) | 169 |
| N3—H3A···N2iii | 0.88 | 1.99 | 2.8582 (19) | 169 |
Symmetry codes: (ii) x, −y+3/2, z; (iii) −x+1, −y+1, −z.
Funding Statement
This work was funded by U.S. Nuclear Regulatory Commission grant NRC-HQ-84-14-G-0040 to Kelly Rue.
References
- Alkorta, I., Elguero, J., Donnadieu, B., Etienne, M., Jaffart, J., Schagen, D. & Limbach, H.-H. (1999). New J. Chem. 23, 1231–1237.
- Bruker (2020). APEX3. Bruker AXS Inc., Madison, Wisconsin, USA.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Farmiloe, S. E., Berdiell, I. C. & Halcrow, M. E. (2019). CSD Communication (deposition No. 1944671). CCDC, Cambridge, England. https://doi.org/10.5517/ccdc.csd.cc238lbb
- Foces-Foces, C., Llamas-Saiz, A. L. & Elguero, J. (1999). Z. Kristallogr. 214, 237–241.
- Goddard, R., Claramunt, R. M., Escolástico, C. & Elguero, J. (1999). New J. Chem. 23, 237–240.
- Infantes, L. & Motherwell, S. (2004). Struct. Chem. 15, 173–184.
- Karrouchi, K., Radi, S., Ramli, Y., Taoufik, J., Mabkhot, Y., Al-aizari, F. A. & Ansar, M. (2018). Molecules, 23, 134. [DOI] [PMC free article] [PubMed]
- Krishnakumar, V., Jayamani, N. & Mathammal, R. (2011). Spectrochim. Acta A Mol. Biomol. Spectrosc. 79, 1959–1968. [DOI] [PubMed]
- Krebs Larsen, F., Lehmann, M. S., Søtofte, I. & Rasmussen, S. E. (1970). Acta Chem. Scand. 24, 3248–3258.
- Naim, M. J., Alam, O., Nawaz, F., Alam, J. & Alam, P. (2016). J. Pharm. Bioallied Sci. 8, 2–17. [DOI] [PMC free article] [PubMed]
- Pérez, J. & Riera, L. (2009). Eur. J. Inorg. Chem. pp. 4913–4925.
- Pettinari, C., Masciocchi, N., Pandolfo, L. & Pucci, D. (2010). Chem. Eur. J. 16, 1106–1123. [DOI] [PubMed]
- Raptis, R. G., Staples, R. J., King, C. & Fackler, J. P. (1993). Acta Cryst. C49, 1716–1719.
- Rigaku OD (2019). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, England.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Viciano-Chumillas, M., Tanase, S., de Jongh, L. J. & Reedijk, J. (2010). Eur. J. Inorg. Chem. pp. 3403–3418. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989021008604/zl5013sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989021008604/zl5013Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989021008604/zl5013Isup3.cml
CCDC reference: 2103822
Additional supporting information: crystallographic information; 3D view; checkCIF report