

Abstract
Although cardiovascular toxicity from traditional chemotherapies has been well recognized for decades, the recent explosion of effective novel targeted cancer therapies with cardiovascular sequelae has driven the emergence of cardio-oncology as a new clinical and research field. Cardiovascular toxicity associated with cancer therapy can manifest as a broad range of potentially life-threatening complications, including heart failure, arrhythmia, myocarditis, and vascular events. Beyond toxicology, the intersection of cancer and heart disease has blossomed to include discovery of genetic and environmental risk factors that predispose to both. There is a pressing need to understand the underlying molecular mechanisms of cardiovascular toxicity to improve outcomes in patients with cancer. Preclinical cardiovascular models, ranging from cellular assays to large animals, serve as the foundation for mechanistic studies, with the ultimate goal of identifying biologically sound biomarkers and cardioprotective therapies that allow the optimal use of cancer treatments while minimizing toxicities. Given that novel cancer therapies target specific pathways integral to normal cardiovascular homeostasis, a better mechanistic understanding of toxicity may provide insights into fundamental pathways that lead to cardiovascular disease when dysregulated. The goal of this scientific statement is to summarize the strengths and weaknesses of preclinical models of cancer therapy–associated cardiovascular toxicity, to highlight overlapping mechanisms driving cancer and cardiovascular disease, and to discuss opportunities to leverage cardio-oncology models to address important mechanistic questions relevant to all patients with cardiovascular disease, including those with and without cancer.
Keywords: AHA Scientific Statements; cardiotoxicity; heart failure; medical oncology; models, cardiovascular; myocarditis; neoplasms
Cardiovascular toxicity resulting from traditional cytotoxic cancer therapies has been well recognized for decades. However, the recent explosion of effective novel targeted cancer therapies with cardiovascular and metabolic sequelae has driven the emergence of cardio-oncology as a new field. Whereas cardiovascular toxicity manifests primarily as cardiomyopathy with traditional agents such as the anthracyclines, newer targeted agents and immunotherapies have been associated with a range of potentially life-threatening complications, including arrhythmia, myocarditis, and vascular events. Beyond toxicology, the intersection of cancer and heart disease has prompted the discovery of genetic and environmental risk factors that predispose to both. With increasing recognition of the short- and long-term consequences of cardiovascular toxicity, there is a pressing need to understand the underlying molecular mechanisms to improve cardiovascular outcomes in patients with cancer.
Preclinical cardiovascular models, ranging from cellular assays to large animals, serve as the foundation for mechanistic studies that ultimately aim to identify biologically sound biomarkers and cardioprotective therapies. Given that novel cancer therapies target specific pathways integral to normal cardiovascular homeostasis, a better mechanistic understanding of toxicity may provide insights into fundamental pathways that lead to cardiovascular disease when dysregulated. For instance, the role of HER2 (human epidermal growth factor receptor 2) in cardiomyocyte signaling was elucidated as a direct result of observations made in patients with breast cancer who developed cardiomyopathy while treated with inhibitors of this pathway.1
The goal of this scientific statement is to highlight strengths and weaknesses of preclinical models used to study cancer therapy–associated cardiovascular toxicity. We also discuss emerging platforms with great potential for use in cardio-oncology, as well as the increasing recognition that the biological mechanisms driving cancer and cardiovascular disease may overlap. Finally, we discuss opportunities for basic and translational investigators to harness preclinical models to address unmet needs in cardiovascular pathophysiology, with the goal of improving cardiovascular care in patients with and without cancer.
PRECLINICAL MODELS TO STUDY CARDIOVASCULAR TOXICITY
Cultured Cell Models
Both traditional and novel cancer therapies can adversely affect a broad range of cell types in the cardiovascular system, from cardiomyocytes to nonmyocytes, including fibroblasts, pericytes, and endothelial cells. However, clinical observations of cardiomyopathy in patients treated with anthracyclines (eg, doxorubicin) motivated an early focus on recapitulating the drug effects in cultured cardiomyocytes, which are uniquely sensitive to perturbations in bioenergetic pathways. Rodent-derived cardiomyocytes, especially neonatal rat ventricular myocytes, are widely used cell models of cardiovascular disease (Table 1) and have been used extensively to decipher mechanisms driving doxorubicin-induced cardiotoxicity, including oxidative stress, impaired energy metabolism, and abnormal activation of cell death pathways.
Table 1.
Preclinical Models Available to Study Cancer Therapy-Associated Cardiovascular Toxicity
| Model system | Strengths | Weaknesses | Examples |
|---|---|---|---|
| Cultured cell and organoid models | |||
| Rodent-derived CMs | Primary cells Neonatal cardiomyocytes: easily harvested with well-established protocols; easy to transfect |
Neonatal cardiomyocytes: immature; express fetal gene program Adult cardiomyocytes: isolation and transfection are more challenging; contraction requires pacing; shorter time frame for performing assays |
Anthracyclines2 |
| Human cardiovascular cell lines (eg, hiPSC-CMs) | Can be expanded endlessly; derived from multiple easily accessible cell types; ability to study pathways specific to humans | Immature cells; experimental and genetic variability; optimal media conditions that replicate in vivo cardiotoxicity have not been clearly defined | Anthracyclines3 Trastuzumab4 Proteasome inhibitors5 |
| Microtissues and organoid systems | More physiologically relevant than isolated cell culture; potential to study multiple cell types simultaneously | Need for manual cell injection, resulting in nonstandardized organoids; absence of in vivo cell-to-cell communication and environment | Sunitinib6 |
| In vivo models | |||
| Zebratish | Vertebrate model with conservation of most disease-causing human genes; small size enables high-throughput screening; optical transparence facilitates imaging | Often used in embryonic/larval stage; 2-chamber heart without pulmonary circulation; zebrafish-specific reagents (eg, antibodies) may not be readily available | Anthracyclines7 Tyrosine kinase inhibitors8 Amyloid light-chain cardiotoxicity9 |
| Rodents (mice, rats) | Most well-established in vivo model for studying toxicity; transgenic models and antibodies often already exist | Most models do not accurately reflect human comorbidities (hypertension, diabetes, aging) or concomitant cardiotoxic therapies | Anthracyclines10–12 HER2 inhibition1 Sunitinib13 |
| Large animals (rabbits, swine, dogs) | Facilitate translation of diagnostic modalities and cardioprotective strategies | Costly and time-consuming; potential for gap in translation to humans | Anthracyclines14 |
HER2 indicates human epidermal growth factor receptor 2; HER2, human epidermal growth factor receptor 2; and hiPSC-CMs, human induced pluripotent stem cell-derived cardiomyocytes.
More recently, human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) have been harnessed to understand cardiovascular biology and specifically mechanisms of cancer therapy–associated toxicity, as summarized in a recent American Heart Association scientific statement15 and in Table 1. hiPSC-CMs enable high-throughput screening of toxicities from many cancer therapeutics in parallel, from conventional cytotoxic chemotherapies to newer targeted agents, and can offer insight into mechanisms of cardioprotection. Compared with rodent-derived cardiomyocytes, hiPSC-CMs display phenotypic characteristics that appear more representative of human cardiac physiology, including more typical ion channel expression, heart rate, contractility, and myofilament composition.16 Patient-specific hiPSC-CMs may recapitulate individual susceptibility to cardiovascular toxicity. For example, ex vivo treatment with doxorubicin identified those at risk of anthracycline cardiomyopathy3; however, the relationship between disease characteristics in patients and the in vitro phenotype is often unpredictable. hiPSC-CMs have an additional advantage in the modeling of cancer therapies that target human proteins but not their homologs in other species such as trastu-zumab, which specifically inhibits human HER2.4 Considerable experimental variability with hiPSC-CMs has been reported and highlights the need for more rigorous phenotyping, cross-validation between laboratories, and continued studies correlating phenotypic changes (eg, transcriptional changes, impaired contractility) with human disease end points in these models. Advances in gene therapy and engineered tissues, as discussed below, are expected to further improve the induced pluripotent stem cell methodologies.17
Organoid Systems
Microphysiological systems integrating engineered tissue have emerged as novel approaches to tackle the limitations of cell culture models. These “organs on a chip” typically incorporate human cells plated onto a polymeric membrane to enable evaluation of toxicity mechanisms that necessitate cross talk between different cell types such as cardiomyocytes, fibroblasts, and endothelial cells. Recent progress in the engineering of these organoid chips has allowed parallelized creation of multiple physiologically relevant tissues, including the heart.18 One limitation to this approach is the inability to account for the unique mechanical forces critical to the cardiovascular system such as contractile forces, tissue stiffness, and the presence of pulsatile versus laminar versus oscillatory flow. These limitations may be partially overcome by the integration of microfluidics into organoid models.
The ability to mimic fully functional organs remains a challenge of organoid systems. Although physiological measurements can be performed on ex vivo organs isolated from animal models (eg, Langendorff heart preparations), these approaches have not generally been translated to human organs because of the difficulty in obtaining human tissues. Rejected donor hearts or isolated cardiac muscle segments from discarded surgical tissue could represent a unique means to define the impact of anticancer therapy on cardiac muscle, although downstream effects on other tissues may not be captured. Similarly, the use of pressurized isolated vessel preparations alongside indirect measurements of vascular reactivity has recently been tested in human coronary vessels incubated with doxorubicin.19 With the use of a biomimetic culture system, human heart slices can be preserved and remain functional for 6 days with regard to calcium homeostasis, mitochondrial structure, and contraction and relaxation kinetics.20 However, the physiological or clinical relevance of these techniques has not yet been validated, prompting the National Toxicology Program to develop a new initiative, the National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods. This program focuses on the development and evaluation of alternatives to animal use for chemical safety testing and could contribute to the understanding of cancer therapy–associated cardiovascular toxicity.
In Vivo Models
Comprehensive assessment of cardiovascular toxicity often necessitates the use of an appropriate in vivo model that ideally incorporates all the distinctive pathological features of cardiovascular toxicity observed in patients. These models range from high-throughput experimental models (eg, zebratish) to large animal models (eg, swine), which have their own strengths and weaknesses (Table 1). For instance, larval zebratish are small and therefore well suited to chemical screening, an approach that has been used to identify small molecules that protect against doxorubicin cardiomyopathy.21 On the other hand, large animal models may phenocopy human cardiovascular toxicity more closely than small animal models, allowing detailed phenotyping of early cardiotoxicity by advanced imaging techniques such as cardiac magnetic resonance,14 but these studies are often limited by the high cost of maintaining and treating large animals. As with other models, it is important to consider time course, route of administration, and use of doses comparable to human therapeutic doses (see the Anthracyclines and Kinase Inhibitors sections). Animal models can be particularly useful in elucidating mechanisms of action contributing to specific cardiovascular phenotypes observed in patients. For instance, rodent and rabbit models have been harnessed to dissect the roles of vascular smooth muscle cell–mediated vasoconstriction and direct endothelial toxicity in 5-fluorouracil–mediated coronary vasospasm. Finally, rodent models have been applied extensively to elucidate mechanisms of cardioprotection associated with carvedilol, dexrazoxane, and other established cardiovascular therapies.
Once a relevant in vivo model has been established, a key element to consider is the end point for assessing cardiovascular toxicity, which should be robust and easily reproducible. Echocardiographic monitoring of left ventricular ejection fraction represents the most widely used approach, although the time course of echocardiographic changes does not always correlate with microstructural changes observed by histopathologic analyses. The incorporation of myocardial strain and strain rate, as detected with Doppler echocardiography, and cardiac magnetic resonance imaging can serve as more sensitive tools to detect early cardiac injury in rodents and large animals.22 Cardiac magnetic resonance imaging in particular can provide more objective and precise quantification of left ventricular function over time. Given the possible effects of sedatives on cardiac function (eg, heart rate changes, depression of cardiac function), many laboratories perform echocardiography in nonsedated rodents when possible. Additional markers of cardiomyocyte injury such as cardiac troponins and natriuretic peptides also can be helpful in assessing the severity of cardiac injury.
Despite the attempt to recapitulate key features of human disease, currently available in vivo models of cardiotoxicity display important shortcomings that should be taken into consideration for future studies (Table 1). Most experiments are carried out in healthy young rodents, whereas cancer therapy–associated cardiovascular toxicity in humans is a complex process that can be exacerbated by preexisting comorbidities. Although studies in mice have shown how previous exposure to doxorubicin increases the susceptibility to myocardial infarction or impairs adaptation to hypertension later in life, the opposite approach of modeling cardiotoxicity in animals with preexisting heart disease or cardiovascular comorbidities has not been attempted to date. Efforts have been made to establish models of juvenile doxorubicin exposure, demonstrating that hearts from young mice, unlike adult hearts, express the molecular machinery that primes them for apoptosis.23 Although similar cardiovascular toxicity has not been fully explored in aged mice, models of telomere dysfunction have been used to study the cardiotoxic effects of anthracyclines.24
Furthermore, to faithfully model cancer therapy–associated cardiovascular toxicity, it is important to take into consideration multimodal cancer therapies that can interact to potentiate cardiac dysfunction. For instance, anthracycline cardiotoxicity can be exacerbated by concurrent use of other cancer therapies, including traditional chemotherapy (cyclophosphamide, paclitaxel), targeted therapies (trastuzumab), and mediastinal radiotherapy. In addition, the potential interactions between cancer therapies and other medications intended for the treatment of cardiovascular comorbidities such as hypertension and diabetes should be considered and included in preclinical models.
The variability of intraspecies and interspecies phenotypes should also be considered in preclinical models. Cardiovascular phenotypes might differ in severity according to the genetic background of the animal strain used,25 with the potential for differences even between the N and J substrains of C57BL/6 mouse lines attributable to genetic variations. Sex is an additional variable that has been inadequately studied in preclinical models of cardiovascular toxicity. Despite the use of rigorous experimental techniques, there may still be a translational gap between animal models and clinical observations in human patients; for instance, imatinib causes cardiomyopathy in rodents but does not seem to have the same effect in patients.26 Integration of data from genomic and proteomic studies in patients can be helpful in determining the relevance of molecular mechanisms identified in preclinical models.
BALANCING CARDIOPROTECTION WITH ANTITUMOR EFFICACY
Many cardiotoxicity preclinical platforms fail to incorporate simultaneous modeling of the cancer. From a therapeutic perspective, it is critical to assess the impact of novel candidate cardioprotectants on the antitumor activity of cancer therapy, typically with the use of xenograft models in zebrafish or rodents.7,12 Syngeneic models, in which tumor cells are derived from the same genetic background as a given mouse strain, are straightforward to establish, but the aggressiveness of the tumor cells may require euthanasia of the animal before the development of cardiotoxicity. As an alternative strategy, genetically engineered models promote spontaneous and less aggressive tumor growth that more closely recapitulates human malignancies.
Any preclinical model should also consider interactions of the immune system with the tumor and the cardiovascular system. Immunocompetent preclinical models will be necessary to study emerging toxicities associated with immunotherapies. Both syngeneic and genetically engineered models provide effective platforms to study cardiovascular toxicity in the presence of a functional immune system. Humanized tumor models, particularly patient-derived xenografts, may recapitulate genetic and phenotypic heterogeneity of tumors more closely, but they typically require immunocompromised hosts such as athymic nude or severe combined immunodeficiency mice.
BASIC MECHANISMS OF CARDIOVASCULAR TOXICITY ASSOCIATED WITH COMMON CANCER THERAPIES
Anthracyclines
Anthracycline cardiotoxicity was first recognized in patients in the 1970s and has been studied extensively in preclinical models. Although several mechanisms have been implicated in the pathogenesis of anthracycline cardiomyopathy (Table 2 and the Figure), the interplay between these mechanisms has not yet been fully defined. Future studies should aim to develop an integrated mechanistic understanding of the complex networks involved in the pathogenesis of cardiotoxicity. For instance, it will be essential to understand the effects of perturbation of 1 pathway such as intracellular iron overload on other proposed pathways such as autophagy or modulation of PI3Kγ (phosphoinositide 3-kinase-γ) in order to identify new therapeutic approaches that can translate clinically. Despite the variety of established models to study anthracycline cardiotoxicity, few have demonstrated the ability to reproduce the complex pathophysiology of the disease as it manifests in humans.
Table 2.
Conventional Cancer Therapies and Proposed Mechanisms of Cardiovascular Toxicity
| Cancer therapy | Proposed pathways | Reported toxicity | Preclinical models | Unexplored questions |
|---|---|---|---|---|
| Anthracyclines Doxorubicin Daunorubicin Epirubicin Idarubicin Valrubicin Mitoxantrone |
Multiple proposed models: DNA damage (p53) Reactive oxygen species formation DNA damage caused by disruption of Top2β10 Iron metabolism (ABCB8)11 Autophagy/mitophagy (Beclin, PI3Kγ, BNIP3, p53)12,27 Calcium signaling Mitochondrial biogenesis HIF signaling AhR/Cyp121 Apoptosis and necrosis (BAX) Induction of inflammatory cytokines |
Cardiomyopathy (acute and delayed onset) Arrhythmia Ultrastructural changes on endomyocardial biopsy (myofibrillar loss and disarray, dilation of the sarcoplasmic reticulum, mitochondrial swelling, cytoplasmic vacuolization) |
hiPSC-CMs3 Zebrafish7 Rodents12,27 Swine14 |
How do proposed mechanistic pathways interact to cause cardiotoxicity? What is the role of vascular dysfunction in anthracycline cardiotoxicity? How can preclinical studies be harnessed to identify novel biomarkers of anthracycline cardiotoxicity in patients? Which molecular pathways are druggable for prevention or treatment of anthracycline cardiotoxicity? What is the genetic basis for interindividual susceptibility to anthracycline cardiotoxicity? |
| Fluoropyrimidines 5-Flurouracil Capecitabine |
DNA via thymidylate synthase Protein kinase C eNOS Metabolism to fluorocitrate, resulting in inhibition of TCA cycle Apoptosis and autophagy |
Coronary vasospasm Acute coronary syndrome Cardiomyopathy Arrhythmia Sudden cardiac death Pericarditis |
Rodents Rabbits |
Which risk factors predispose to endothelial dysfunction and vasospasm? |
| Alkylating agents Cyclophosphamide Ifosfamide Bendamustine Chlorambucil Cisplatin |
Heart fatty acid binding proteins Cardiomyocyte apoptosis Inflammation Endothelial dysfunction Calcium dysregulation Mitochondrial and ER damage Oxidative stress Direct DNA damage |
Cardiomyopathy Capillary microthrombosis Arrhythmia Hypotension QT prolongation Pericarditis Supraventricular arrhythmias Diastolic dysfunction |
Rodents | What are the genetic or metabolic factors that predispose certain patients to accumulation of toxic metabolites? |
| Antimicrotubule agents Paclitaxel Docetaxel |
DNA damage Histamine release |
Cardiomyopathy Hypertension Myocardial infarction Conduction abnormalities (QT prolongation, bradycardia, atrial fibrillation) |
Rodents | Which cell types and subcellular organelles are affected by these agents? |
| Hormonal therapies Androgen deprivation therapy (GnRH agonists, adrenal androgen receptor inhibitors, direct androgen receptor inhibitors) |
Modulation of sodium and potassium currents28 Testosterone deficiency29 |
QT prolongation Arrhythmia Metabolic syndrome Hypertension Vascular events |
hiPSC-CMs Rodents Rabbits |
Is there a central pathway affected by androgen deprivation that results in hypercholesterolemia, hyperinsulinemia, and obesity? What is the best preclinical model to recapitulate baseline cardiovascular risk factors that predispose to metabolic syndrome? What are the differences between classes of ADT and the risk of cardiovascular toxicity? |
| Chest radiation | Nuclear and mitochondrial DNA Upregulation of NF-κB Oxidative stress Inhibition of angiogenesis |
Accelerated atherosclerosis Dilated or restrictive cardiomyopathy Constrictive pericarditis Microvascular disease Valvular heart disease (stenosis or regurgitation) Conduction system disease Autonomic dysfunction |
Rodents30 Rabbits Pigs Dogs |
Are there genetic factors that predispose to radiation-induced cardiovascular toxicity? Which specific profibrotic and inflammatory pathways are induced by radiation? What is the best model of long-term cardiac complications of chest radiation? What are new therapeutic approaches to mitigate the cardiovascular toxicity of chest radiation? |
ABCB8 indicates ATP-binding cassette subfamily B member 8; ADT, androgen deprivation therapy; AhR, aryl hydrocarbon receptor; BAX, Bcl-2–associated X protein; BNIP3, BCL2/adenovirus E1B 19 kd-interacting protein 3; Cyp1, cytochrome P450 family 1 enzymes; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; GnRH, gonadotropin-releasing hormone; HIF, hypoxia-inducible factor; hiPSC-CMs, human induced pluripotent stem cell–derived cardiomyocytes; NF-κB, nuclear factor-κB; PI3Kγ, phosphoinositide 3-kinase-γ; TCA, tricarboxylic acid cycle; and Top2β, topoisomerase 2β.
Figure. Mechanisms of anthracycline cardiotoxicity.
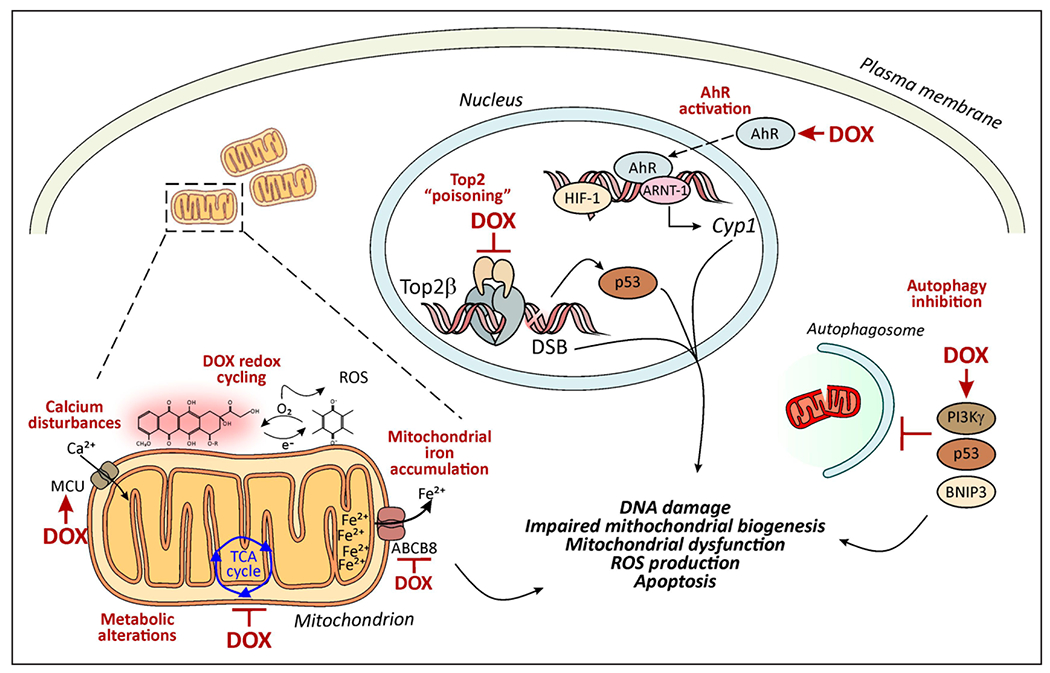
Several mechanisms have been proposed to contribute to the cardiotoxic effects of anthracyclines such as doxorubicin (DOX). These include topoisomerase “poisoning,” alterations of cell survival and metabolic pathways, and excessive generation of reactive oxygen species (ROS). In the nucleus, DOX binds Top2β (topoisomerase 2β), thereby promoting p53-dependent DNA damage responses and altering the transcription of genes involved in mitochondrial biogenesis, for example, PGC-1α (peroxisome proliferator-activated receptor-γ coactivator 1-α). As a polyaromatic hydrocarbon, DOX activates the AhR (aryl hydrocarbon receptor), a ligand-activated transcription factor that translocates to the nucleus to induce the expression of Cyp1 (cytochrome P450 family 1 enzymes). DOX can inhibit the binding of the HIF (hypoxia-inducible factor) heterodimer (HIF-1 α/ARNT [aryl hydrocarbon receptor nuclear translocator]) to the hypoxia response element, leading to a decrease in the HIF transcriptional response. DOX induces defects in mechanisms of organelle quality control such as autophagy and mitophagy, which are under the control of PI3Kγ (phosphoinositide 3-kinase-γ), p53, and BNIP3 (BCL2/adenovirus E1B 19 kd-interacting protein 3) and exacerbate DOX-associated mitochondrial dysfunction and ROS production. DOX can trigger ROS production directly or after being reduced to semiquinone radicals that undergo redox cycling, generating superoxide and hydrogen peroxide. DOX can bind to iron, resulting in iron cycling and subsequent production of cytotoxic hydroxyl radicals via the Fenton reaction. These events occur in the mitochondria where DOX preferentially accumulates on its binding to the mitochondrial phospholipid cardiolipin. ROS production also can be secondary to accumulation of calcium or iron within mitochondria, resulting from activation of the MCU (mitochondrial calcium uniporter) and inhibition of the iron transporter ABCB8 (ATP-binding cassette subfamily B member 8), respectively. DSB indicates double-strand break; and TCA, tricarboxylic acid cycle.
Careful consideration of drug dosing, formulation, and delivery is paramount to ensure reproducibility of cardiac phenotypes with anthracyclines and for other cardiotoxic cancer therapies. For instance, many anthracycline cardiotoxicity models in mice have used either very high doses or intraperitoneal injection of doxorubicin, which is more practical than intravenous delivery for regimens that require repeated administration or concomitant delivery of other agents such as cardioprotective small molecules. However, when possible, the intravenous route should be favored because doxorubicin and other cancer therapies may elicit local injury of the peritoneum and fibrosis, with consequent malaise, anorexia, weight loss, and reduced drug absorption in subsequent injections. For example, acute models of anthracycline cardiotoxicity rely on a single administration of high-dose doxorubicin in mice (eg, 20 mg/kg), whereby cardiac phenotypes are typically analyzed within 1 week. Although these models can provide insight into the acute side effects of doxorubicin, primarily arrhythmias and electrocardiographic abnormalities in humans, they are usually characterized by a high mortality rate that is unrelated to cardiac toxicity.
A major challenge of modeling anthracycline cardiotoxicity in preclinical models relates to the difficulties of recapitulating the chronic and typically delayed nature of the cardiomyopathy observed in patients. In adults, cardiotoxicity is dose dependent and typically manifests within the first year after completion of treatment, although cardiotoxicity may manifest years after anthracycline exposure, as observed in the pediatric patient population. To better recapitulate typical dosing regimens in humans, protocols featuring repeated injection of low-dose doxorubicin (eg, 2–5 mg/kg weekly for multiple weeks) have been developed that result in chronic cardiotoxicity, with the total cumulative dose varying slightly, depending on the differing sensitivity of specific mouse strains to doxorubicin.27 These models are characterized by low mortality and the development of cardiomyopathy, which is similar to that observed clinically, which typically manifests as a decrease in fractional shortening of >10% at 6 to 8 weeks after therapy completion. Models that recapitulate late cardiotoxicity occurring years after anthracycline exposure have not yet been developed and represent an important unmet need.
Kinase Inhibitors
Aberrant activation of kinases is implicated in many tumor types and has led to the development of kinase inhibitors (KIs) for cancer therapy. KIs include monoclonal antibodies that bind the receptor kinase or its ligand, as well as small-molecule inhibitors that interfere with the binding of the kinase to ATP or its substrates.31 Kinases also play critical roles in cardiovascular homeostasis, with KIs potentially leading to diverse cardiovascular sequelae, as summarized in Table 3. These may be “on target,” whereby the kinase inhibited for the treatment of cancer also has a critical role in cardiovascular biology, or “off target,” which can occur when the inhibitor binds to multiple kinase receptors simultaneously or other targets. When preclinical studies of cardiovascular toxicity associated with KIs are designed, it is critical to select an appropriate dose that reflects the kinase binding profile observed in patients and recapitulates the pharmacokinetics seen in humans.8
Table 3.
New Cancer Therapies and Proposed Mechanisms of Cardiovascular Toxicity
| Oncological target(s) | Agents | Reported toxicity | Preclinical models | Proposed pathways |
|---|---|---|---|---|
| KIs | ||||
| HER2 (ErbB2) | Trastuzumab Trastuzumab-emtansine Pertuzumab Lapatinib Neratinib |
Cardiomyopathy | hiPSC-CMs4 Primary cardiomyocytes Mice (genetic deletion of ERBB2)32 |
Alterations in cellular and metabolic pathways (eg, AMP kinase4) |
| VEGF, PDGF | Sunitinib Sorafenib Pazopanib Vandetanib Axitinib Regorafenib Cabozantinib Bevacizumab |
Cardiomyopathy Vascular disease Hypertension Thrombotic microangiopathy QT prolongation |
hiPSC-CMs Primary cardiomyocytes Mice |
Inhibition of VEGF/PDGF Capillary rarefaction Induction of HIFs in the myocardium33 Suppression of PI3K signaling34 Decreased NO production |
| BCR-ABL* | Nilotinib Dasatinib Imatinib Ponatinib Bosutinib |
Hyperglycemia (nilotinib) Pulmonary hypertension, pericardial and pleural effusion (dasatinib) Vascular events (nilotinib, ponatinib) |
VWF-mediated platelet adhesion leading to thrombotic microangiopathy (ponatinib)35 Suppression of PI3K signaling34 Decreased NO production |
|
| EGFR | Dacomitinib Erlotinib Gefitinib Osimertinib Cetuximab Panitumumab |
Osimertinib: QT prolongation Atrial fibrillation Heart failure Cardiomyopathy Pericardial effusion None with other agents |
hiPSC-CMs | Not defined |
| BTK | Ibrutinib Acalabrutinib |
Arrhythmia (supraventricular, particularly atrial fibrillation/flutter; ventricular) | Mice | Abnormal Ca2+ handling in atrial myocytes and reduced sarcoplasmic Ca2+ capacity Atrial fibrosis |
| Raf/MEK | Trametinib Cobimetinib Selumetinib |
Cardiomyopathy | Mice (Erk1−/− and Erk2+/−) |
Not defined |
| CDK4/6 | Ribociclib | QT prolongation | Rodent | Not defined |
| PI3K/mTOR | Temsirolimus Everolimus |
Metabolic Hyperlipidemia Hyperglycemia Peripheral edema |
Rodent | Decreased insulin secretion |
| MCL-1 | Investigational | Cardiomyopathy | Rodent (Mcl-1 conditional KO mice) | Not defined |
| ALK | Crizotinib Ceritinib Alectinib Brigatinib Lorlatinib |
Bradycardia | Rodent | Not defined |
| Drugs affecting protein degradation | ||||
| Proteasome | Bortezomib Carfilzomib Ixazomib |
Vascular events Hypertension Carfilzomib: Cardiomyopathy |
Rodent Primary cardiomyocytes |
Myocardial protein turnover |
| E3 ubiquitin ligase | IMiDs: Thalidomide Lenalidomide Pomalidomide |
Venous and arterial thrombotic events | Rodents Human embryonic stem cells |
Upregulation of prothrombotic factors (VWF, platelet activation) |
| Immune therapies | ||||
| Immune checkpoints Cancer-specific antigens T-cell engagers High-dose cytokines (eg, IL-2) CD52 CD20 |
ICIs: Anti–CTLA-4: Ipilimumab Tremelimumab Anti–PD-1 Nivolumab Pembrolizumab Cemiplimab Anti–PDL1: Atezolizumab Avelumab Durvalumab CAR T-cell therapy: Axicabtagene ciloleucel Tisagenlecleucel Brexucabtagene autoleucel Bispecific T-cell engager antibodies: Blinatumomab |
Myocarditis Cardiomyopathy Pericarditis Pleural effusion Vasculitis Vascular leak Thrombosis Tachyarrhythmia Bradyarrhythmia/heart block Cytokine release syndrome Atherosclerosis |
Mice (CTLA4−/−, PD-1−/−) | IgG autoantibodies36 T-cell activation T-cell and macrophage infiltration into myocardium37 |
ALK indicates anaplastic lymphoma kinase; BCR-ABL, breakpoint cluster region-abelson protooncogene; BTK, Bruton tyrosine kinase; CAR, chimeric antigen receptor; CDK, cyclin-dependent kinase; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; EGFR, epidermal growth factor receptor; ErbB2, erythroblastic oncogene B; HER2, human epidermal growth factor receptor 2; HIF, hypoxia-inducible factor; hiPSC-CM, human induced pluripotent stem cell—derived cardiomyocyte; ICI, immune checkpoint inhibitor; IgG, immunoglobulin G; IL-2, interleukin-2; IMiD, immunomodulatory imide drug; KI, kinase inhibitor; KO, knockout; MCL-1, myeloid-cell leukemia 1; MEK, mitogen-activated protein kinase; mTOR, mechanistic target of rapamycin; NO, nitric oxide; PD-1, programmed cell death protein-1; PDGF, platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; RAF, rapidly accelerated fibrosarcoma; VEGF, vascular endothelial growth factor; and VWF, von Willebrand factor.
Agents that target BCR-ABL may also inhibit the VEGF pathway (eg, ponatinib).
The cardiomyopathy observed with trastuzumab provided an initial model of on-target cardiotoxicity. Although pharmacological mouse models of trastuzumab cardiotoxicity have been challenging because trastuzumab does not bind mouse HER2, genetic deletion of HER2 in a mouse model resulted in ventricular dilatation and increased susceptibility of isolated cardiomyocytes to myocardial stress, including anthracycline exposure.1,32 Recently, the advent of hiPSC-CMs has allowed the implementation of a pharmacological model of cardiomyocyte toxicity in which trastuzumab exposure impaired contractile function, calcium handling, and cardiac energy metabolism.4
Another class of KIs (eg, sunitinib, sorafenib) inhibit VEGF (vascular endothelial growth factor) or PDGF (platelet-derived growth factor) and can lead to various cardiovascular adverse effects, including hypertension, vascular disease, and cardiomyopathy.31 Mouse models of genetic or pharmacological inhibition of VEGFor PDGF suggest that inhibition of these pathways leads to capillary rarefaction with subsequent myocyte hypoxia and induction of hypoxia-inducible factors, which are sufficient to cause a reversible cardiomyopathy.13,33 Consistent with these models, the cardiomyopathy observed in patients treated with sunitinib and sorafenib is often reversible.38 Although mouse models have provided insights into VEGF inhibitor–associated hypertension, human translational studies will be critical to elucidate underlying mechanisms.
Given the explosion of KIs in oncology and their associated cardiovascular sequelae, adoption of appropriate preclinical models of cardiotoxicity not only may dissect mechanisms of toxicity but also may lead to recognition of new kinase pathways in cardiovascular biology. For example, ibrutinib, a highly specific KI against Bruton tyrosine kinase used for the treatment of B-cell malignancies, is associated with arrhythmias, including atrial fibrillation. It is unclear whether this arrhythmogenic effect is an on-target (resulting from inhibition of Bruton tyrosine kinase) or off-target (caused by inhibition of another kinase) toxicity. Preclinical studies may illuminate novel kinase pathways that are critical to the development of arrhythmia, which could inform the development of new Bruton tyrosine kinase inhibitors. This concept has already been demonstrated with the observation that several multitargeted tyrosine KIs (eg, dasatinib, sunitinib, nilotinib) cause QT prolongation. This clinical finding prompted preclinical experiments suggesting a long-term, PI3K–dependent, QT-prolonging effect that altered multiple ion currents, especially the late sodium current (INa-L),34 in contrast to other medications that cause QT prolongation immediately and almost invariably target repolarizing potassium channels (eg, IKr).
Immune-Based Therapies
Immune-based therapies have revolutionized treatment for patients with cancer by harnessing the immune system to treat cancer. These therapies may be very specific; for example, CAR T-cell (chimeric antigen receptor T-cell) therapy repurposes genetically modified immune cells to target specific tumor antigens. On the other hand, immune checkpoint inhibitors (ICIs) are monoclonal antibodies against immune checkpoints such as CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), PD-1 (programmed cell death protein-1), and PD-L1 (programmed cell death protein-ligand 1) that release the brakes on T-cell activation in a more generic way compared with adoptive T-cell therapies. These act as inhibitory regulators of T cells during the adaptive immune response, preventing excessive immune responses and allowing self-tolerance. However, by activating the immune system, these therapies can cause immune-mediated side effects. A broad range of cardiovascular toxicities, including myocarditis, pericarditis, vasculitis, accelerated atherosclerosis, arrhythmia, and potentially thrombosis, have emerged as infrequent but severe and life-threatening complications associated with ICIs.39 ICI-associated myocarditis is characterized pathologically by myocardial infiltration of macrophages and T lymphocytes, resulting in myocyte death, consistent with the pathological definition for myocarditis.40
The emergence of immune-mediated cardiovascular diseases in cardio-oncology adds a new level of complexity for preclinical models, with animal models potentially best suited to recapitulate the resulting cellular interactions. Early data suggested that an on-target effect attributable to perturbation of the Ctla4 and Pdcd1 genes individually led to myocardial pathology.36 However, these models are background dependent and do not fully recreate the clinical and pathophysiological features of ICI-associated myocarditis. Monoallelic loss of Ctla4 in the context of complete genetic absence of PD-1 leads to premature death in mice as a result of myocardial infiltration by T cells and macrophages and causes severe electrocardiographic abnormalities, closely recapitulating the clinical and pathological hallmarks of ICI-associated myocarditis observed in patients.40a In another example of discordance between mouse models and clinical observations, CAR T-cells expressing a receptor with high affinity for the tumor antigen MAGE-A3 (melanoma-associated antigen 3) were found to be safe in mice but led to fatal cardiogenic shock in patients.37 Subsequent studies in hiPSC-CMs determined that these T cells cross-reacted with a peptide from the cardiac sarcomeric protein titin in humans but not the equivalent peptide in mice.41 The ongoing development of preclinical models that align with clinical phenotypes will be critical to understand the pathogenesis of cardiovascular toxicities, especially as the use of immunotherapies continues to expand. Emerging modalities such as CAR T-cell therapy and bispecific T-cell engager antibodies can also cause cardiomyopathy in the setting of cytokine release syndrome through mechanisms that have not yet been defined.
Chest Radiation
The adverse effects of chest radiation therapy have been studied in a variety of species, including rabbits, rats, and mice. Historically, γ- or x-ray radiation at doses up to 30 Gy has been deployed as a 1-time treatment in preclinical models, with few studies using clinically relevant fractionation protocols or proton therapy. Although modern image-guided radiation delivery platforms that recapitulate treatment in patients have become available for small animals, most prior studies have used traditional methods such as lead shielding. Apolipoprotein E–deficient (ApoE−/−) mice on normal chow develop atherosclerotic coronary disease ≥20 weeks after receiving radiation doses of ≥8 Gy.30 Systolic dysfunction in mice treated with chest radiation appears modest and occurs late after therapy. Histopathological analysis of the myocardium may show loss of microvascular density and fibrosis, a hallmark of radiation-induced organ dysfunction. Pericardial thickening and inflammatory cell infiltration have also been reported in several rodent studies. Despite some limitations, preclinical models have been used to identify genetic modulators of radiation-induced cardiovascular toxicity42 and to assess additive effects of chemotherapeutic agents (eg, tyrosine kinase inhibitors)43 and ICIs.44 Of note, models of radiation-induced valvular disease have not yet been established.
FUTURE DIRECTIONS
Shared Risk Factors for Cancer and Cardiovascular Disease
Preclinical models must also capture novel clinical frontiers in cardio-oncology. The tumor itself may affect cardiovascular function and exacerbate the toxicities of cancer therapy. On the other hand, shared risk factors could predispose to both cancer and cardiovascular disease, a concept that has enormous public health implications. For example, in clonal hematopoiesis of indeterminate potential, somatic mutations in specific genes in hematopoietic cells are associated not only with an increased risk for hematologic malignancies but also with early mortality driven by cardiovascular events.45,46 In parallel with human “omics” studies,47 it will be necessary to establish mouse models in which the direct causal effects of specific mutations on the cardiovascular system can be tested. Even more intriguing are recent data suggesting that heart failure itself is a risk factor for tumor growth.48 Conversely, excess levels of the oncometabolite D-2-hydroxyglutarate have been associated with the development of dilated cardiomyopathy.49 Perturbations in systemic inflammation, oxidative stress, AMP-activated protein kinase, plasminogen activator inhibitor-1, and Wnt signaling have similarly been proposed as shared mediators of cancer and cardiovascular disease, and cardioprotective therapies such as statins could improve cancer-related outcomes. Appropriate preclinical models will be critical to better define the mechanisms of these complex interactions between the cardiovascular system and tumor biology.
Translation to the Clinic
Establishment of preclinical models that recapitulate key features of human cardiovascular toxicity (eg, aging, comorbidities, tumor biology) will be an essential step toward translation of potential biomarkers or therapeutics. Given the strengths and weaknesses of each preclinical model discussed in this statement, multiple complementary models may be necessary to fully understand the complex and interrelated pathways involved in the pathogenesis of toxicity and their implications for patient care. High-throughput technologies such as metabolomics, proteomics, and genomics are increasingly being used to illuminate candidate pathways in humans that can be interrogated in preclinical models to understand underlying biological mechanisms. Computational modeling could accelerate the generation and interpretation of vast amounts of data, for instance, by predicting potential cardiovascular toxicities on the basis of existing drug/cell interaction databases. In the age of precision medicine, identification of genetic risk factors that predispose certain patients to severe cardiovascular toxicity, or conversely that offer cardioprotection, will be of the utmost importance. To maximize applications of these techniques to cardio-oncology, there is a need for large, multicenter biorepositories that include blood samples, endomyocardial biopsy samples, and autopsy tissues when relevant. Proper recording and adjudication of cardiovascular end points in cancer trials and, conversely, cancer end points in cardiovascular registries and trials will be equally important. Advances in molecular imaging techniques can also provide insights into mechanisms of toxicity in real time during treatment, both in patients and in preclinical models. Finally, new artificial intelligence and machine learning approaches may be valuable in integrating genetic risk with environmental and individual risk factors.
Cardio-Oncology as an Emerging Platform for Physician-Scientists
Cardio-oncology has come to the forefront of academic medicine as a field ripe with excellent opportunities to make impactful advances in science and patient care. The diverse cardiovascular sequelae of targeted cancer therapies represent an opportunity for closer collaboration between basic scientists and clinicians to interrogate the cardiovascular and cardiometabolic changes caused by the modulation of critical signaling pathways in patients with cancer. In this regard, cardio-oncology represents a novel and growing platform for basic and translational cardiovascular investigation located at the intersection of molecular biology, cardiovascular research, and drug discovery.50 Cancer therapies targeting specific molecular pathways can serve as tools for understanding the role of these pathways in other noncancer disease states; for instance, a detailed understanding of the sequelae of VEGF inhibition has contributed to the development of novel biomarkers for peripartum cardiomyopathy. In addition to cardiovascular toxicities associated with cancer therapies, malignancies and related conditions such as amyloidosis exert direct and indirect effects on the heart through mechanisms that remain underexplored. The National Heart, Lung, and Blood Institute and the National Cancer Institute have recognized cardio-oncology as a research area of importance.51 The public health impact of cancer therapy–associated cardiovascular toxicity will continue to expand with the rapid development of new cancer treatment paradigms; several new targeted agents and immunotherapies are approved by the US Food and Drug Administration each year. Cardiovascular injury mechanisms arising from cancer and cancer therapy present a unique opportunity not only to improve care for a growing patient population but also, more broadly, to understand fundamental signaling pathways in the heart that are modulated by specific cancer therapies.
Writing Group Disclosures
| Writing group member | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board | Other |
|---|---|---|---|---|---|---|---|---|
| Aarti Asnaniǂ | Beth Israel Deaconess Medical Center and Harvard Medical School Cardiovascular Institute | NIH/NHLBI K08HL145019†; R21HL148748† | None | None | None | None | Cytokinetics* | None |
| Javid J. Moslehi | Vanderbilt University Medical Center | NIH/NHLBI R01HL141466†; R01HL155990†; R01HL156021 † | None | None | None | None | Amgen*; AstraZeneca*; Audentes Pharmaceuticals*; Boehringer*; Boston Biomedical*; Bristol-Myers Squibb†; Cytokinetics*; Deciphera*; GlaxoSmithKline*; GSK*; ImmunoCore*; Janssen*; Myovant*; Nektar*; Pfizer* Pharmacyclics*; Regeneron*; Takeda* | None |
| Bishow B. Adhikari | National Institutes of Health, NHLBI | None | None | None | None | None | None | None |
| Alan H. Baik | University of California, San Francisco | None | None | None | None | None | None | None |
| Andreas M. Beyer | Medical College of Wisconsin | NIH (R01 level funding to support ongoing research laboratory)†; Bayer Pharmaceutical (contract to test efficacy of new drug in human source material, no personal gain or salary support)*; other academic investigators (reagents, tools, and compounds for research use)* | None | None | None | None | None | None |
| Rudolf A. de Boer | University Medical Center Groningen (The Netherlands) | DELIVER study (dapagliflozin in HFpEF) (executive board member [unpaid])*; MONITOR HF study (PA pressure monitoring in severe heart failure) (co-PI [unpaid])*; European Research Council, CoG 818715 (unpaid)* | None | None | None | None | None | None |
| Alessandra Ghigo | University of Torino (Italy) | None | None | None | None | None | None | None |
| Isabella M. Grumbach | University of Iowa | NIH (research grant to study vascular toxicity)† | None | None | None | None | None | None |
| Salvia Jain | Beth Israel Deaconess Medical Center | Abcuro, Inc. (research funding was obtained from company)†; NCI (K08 Career Development Grant was obtained)† | None | None | None | None | None | Abcuro, Inc. (0.2% salary support is being provided by Abcuro)*; NCI (K02 Career Development Grant)† |
| Han Zhu | Stanford University | None | None | None | None | None | None | None |
This table represents the relationships of writing group members that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all members of the writing group are required to complete and submit. A relationship is considered to be “significant” if (a) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (b) the person owns 5% or more of the voting stock or share of the entity or owns $10 000 or more of the fair market value of the entity A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Significant.
Dr. Asnani took on a Consulting position at Cytokinetics after acceptance of this manuscript and with the approval of the AHA. Since then, she has not made any edits to the manuscript other than responses to author queries.
Reviewer Disclosures
| Reviewer | Employment | Research grant | Other research support | Speakers’ bureau/honoraria | Expert witness | Ownership interest | Consultant/advisory board | Other |
|---|---|---|---|---|---|---|---|---|
| Charlotte Lee | Massachusetts General Hospital, Harvard Medical School | None | None | None | None | None | None | None |
| Christopher J. O’Donnell | VA Boston Healthcare System | None | None | None | None | None | None | None |
| Rupal O’Quinn | University of Pennsylvania | None | None | None | None | None | None | None |
| Heinrich Taegtmeyer | University of Texas Health Science Center at Houston | None | None | None | None | None | None | None |
| Sean M. Wu | Stanford University School of Medicine | Sanofi (research project on immune checkpoint inhibitor myocarditis)† | None | None | None | None | None | None |
This table represents the relationships of reviewers that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all reviewers are required to complete and submit. A relationship is considered to be “significant” if (a) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (b) the person owns 5% or more of the voting stock or share of the entity, or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Significant.
Footnotes
The American Heart Association makes every effort to avoid any actual or potential conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the writing panel. Specifically, all members of the writing group are required to complete and submit a Disclosure Questionnaire showing all such relationships that might be perceived as real or potential conflicts of interest.
This statement was approved by the American Heart Association Science Advisory and Coordinating Committee on January 6, 2021, and the American Heart Association Executive Committee on February 23, 2021. A copy of the document is available at https://professional.heart.org/statements by using either “Search for Guidelines & Statements” or the “Browse by Topic” area. To purchase additional reprints, call 215-356-2721 or email Meredith.Edelman@wolterskluwer.com.
The American Heart Association requests that this document be cited as follows: Asnani A, Moslehi JJ, Adhikari BB, Baik AH, Beyer AM, de Boer RA, Ghigo A, Grumbach IM, Jain S, Zhu H; on behalf of the American Heart Association Council on Basic Cardiovascular Sciences; Cardio-Oncology Science Subcommittee of Council on Genomic and Precision Medicine and Council on Clinical Cardiology; Council on Peripheral Vascular Disease; and Council on Arteriosclerosis, Thrombosis and Vascular Biology. Preclinical models of cancer therapy–associated cardiovascular toxicity: a scientific statement from the American Heart Association. Circ Res. 2021;128:e•••–e•••. doi: 10.1161/RES.0000000000000473
The expert peer review of AHA-commissioned documents (eg, scientific statements, clinical practice guidelines, systematic reviews) is conducted by the AHA Office of Science Operations. For more on AHA statements and guidelines development, visit https://professional.heart.org/statements. Select the “Guidelines & Statements” drop-down menu, then click “Publication Development.”
REFERENCES
- 1.Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy Nat Med. 2002;8:459–465. doi: 10.1038/nm0502-459 [DOI] [PubMed] [Google Scholar]
- 2.Dorr RT, Bozak KA, Shipp NG, Hendrix M, Alberts DS, Ahmann F. In vitro rat myocyte cardiotoxicity model for antitumor antibiotics using adenosine triphosphate/protein ratios. Cancer Res. 1988;48:5222–5227. [PubMed] [Google Scholar]
- 3.Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmström A, Chang AC, Coronado MJ, Ebert AD, et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. 2016;22:547–556. doi: 10.1038/nm.4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitani T, Ong SG, Lam CK, Rhee JW, Zhang JZ, Oikonomopoulos A, Ma N, Tian L, Lee J, Telli ML, et al. Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation. 2019;139:2451–2465. doi: 10.1161/CIRCULATIONAHA.118.037357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Judge LM, Perez-Bermejo JA, Truong A, Ribeiro AJ, Yoo JC, Jensen CL, Mandegar MA, Huebsch N, Kaake RM, So PL, et al. A BAG3 chaperone complex maintains cardiomyocyte function during proteotoxic stress. JCI Insight. 2017;2:e94623. doi: 10.1172/jci.insight.94623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Truitt R, Mu A, Corbin EA, Vite A, Brandimarto J, Ky B, Margulies KB. Increased afterload augments sunitinib-induced cardiotoxicity in an engineered cardiac microtissue model. JACC Basic Transi Sci. 2018;3:265–276. doi: 10.1016/j.jacbts.201712.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Asnani A, Zou L, Bentley VL, Yu M, Wang Y, Dellaire G, Sarkar KS, Dai M, Chen HH, et al. Visnagin protects against doxorubicin-induced cardiomyopathy through modulation of mitochondrial malate dehydrogenase. Sci Transl Med. 2014;6:266ra170. doi: 10.1126/scitranslmed.3010189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng H, Kari G, Dicker AP, Rodeck U, Koch WJ, Force T. A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ Res. 2011;109:1401–1409. doi: 10.1161/CIRCRESAHA.111.255695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra S, Joshi S, Ward JE, Buys EP Mishra D, Mishra D, Morgado I, Fisch S, Lavatelli F, Merlini G, et al. Zebrafish model of amyloid light chain cardiotoxicity: regeneration versus degeneration. Am J Physiol Heart Circ Physiol. 2019;316:H1158–H1166. doi: 10.1152/ajpheart.00788.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–1642. doi: 10.1038/nm.2919 [DOI] [PubMed] [Google Scholar]
- 11.Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124:617–630. doi: 10.1172/JCI72931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M, Sala V, De Santis MC, Cimino J, Cappello P Pianca N, Di Bona A, Margaria JP Martini M, Lazzarini E, et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation. 2018;138:696–711. doi: 10.1161/CIRCULATIONAHA.117.030352 [DOI] [PubMed] [Google Scholar]
- 13.Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, Dunner K Jr, Pati S, Bankson JA, Pasqualini R, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med. 2013;5:187ra69. doi: 10.1126/scitranslmed.3005066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galán-Arriola C, Lobo M, Vílchez-Tschischke JP López GJ, de Molina-Iracheta A, Pérez-Martínez C, Agüero J, Fernández-Jiménez R, Martín-García A, Oliver E, et al. Serial magnetic resonance imaging to identify early stages of anthracycline-induced cardiotoxicity. J Am Coll Cardiol. 2019;73:779–791. doi: 10.1016/j.jacc.2018.11.046 [DOI] [PubMed] [Google Scholar]
- 15.Gintant G, Burridge P Gepstein L, Harding S, Herron T, Hong C, Jalife J, Wu JC; on behalf of the American Heart Association Council on Basic Cardiovascular Sciences. Use of human induced pluripotent stem cell-derived cardiomyocytes in preclinical cancer drug cardiotoxicity testing: a scientific statement from the American Heart Association. Circ Res. 2019;125:e75–e92. doi: 10.1161/RES.0000000000000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117:80–88. doi: 10.1161/CIRCRESAHA.117.305365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheng CC, Amiri-Kordestani L, Palmby T, Force T, Hong CC, Wu JC, Croce K, Kim G, Moslehi J. 21st Century cardio-oncology: identifying cardiac safety signals in the era of personalized medicine. JACC Basic Transl Sci. 2016;1:386–398. doi: 10.1016/j.jacbts.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards DJ, Li Y, Kerr CM, Yao J, Beeson GC, Coyle RC, Chen X, Jia J, Damon B, Wilson R, et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat Biomed Eng. 2020;4:446–462. doi: 10.1038/s41551-020-0539-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hader SN, Zinkevich N, Norwood Toro LE, Kriegel AJ, Kong A, Freed JK, Gutterman DD, Beyer AM. Detrimental effects of chemotherapy on human coronary microvascular function. Am J Physiol Heart Circ Physiol. 2019;317:H705–H710. doi: 10.1152/ajpheart.00370.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ou Q, Jacobson Z, Abouleisa RRE, Tang XL, Hindi SM, Kumar A, Ivey KN, Giridharan G, El-Baz A, Brittian K, et al. Physiological biomimetic culture system for pig and human heart slices. Circ Res. 2019;125:628–642. doi: 10.1161/CIRCRESAHA.119.314996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asnani A, Zheng B, Liu Y, Wang Y, Chen HH, Vohra A, Chi A, Cornella-Taracido I, Wang H, Johns DG, et al. Highly potent visnagin derivatives inhibit Cyp1 and prevent doxorubicin cardiotoxicity. JCI Insight. 2018;3:e96753. doi: 10.1172/jci.insight.96753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauer M, Cheng S, Jain M, Ngoy S, Theodoropoulos C, Trujillo A, Lin FC, Liao R. Echocardiographic speckle-tracking based strain imaging for rapid cardiovascular phenotyping in mice. Circ Res. 2011;108:908–916. doi: 10.1161/CIRCRESAHA.110.239574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, Cantlon A, Fisch S, Golomb-Mello G, Ryan JA, et al. Developmental regulation of mitochondrial apoptosis by c-Myc governs age-and tissue-specific sensitivity to cancer therapeutics. Cancer Cell. 2017;31:142–156. doi: 10.1016/j.ccell.2016.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahin E, Colla S, Liesa M, Moslehi J, MQller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeiss CJ, Gatti DM, Toro-Salazar O, Davis C, Lutz CM, Spinale F, Stearns T, Furtado MB, Churchill GA. Doxorubicin-induced cardiotoxicity in collaborative cross (CC) mice recapitulates individual cardiotoxicity in humans. G3 (Bethesda). 2019;9:2637–2646. doi: 10.1534/g3.119.400232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12:908–916. doi: 10.1038/nm1446 [DOI] [PubMed] [Google Scholar]
- 27.Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–1687. doi: 10.1161/CIRCULATIONAHA.115.017443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salem JE, Yang T, Moslehi JJ, Waintraub X, Gandjbakhch E, Bachelot A, Hidden-Lucet F, Hulot JS, Knollmann BC, Lebrun-Vignes B, et al. Androgenic effects on ventricular repolarization: a translational study from the International Pharmacovigilance Database to iPSC-Cardiomyocytes. Circulation. 2019;140:1070–1080. doi: 10.1161/CIRCULATIONAHA.119.040162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herring MJ, Oskui PM, Hale SL, Kloner RA. Testosterone and the cardiovascular system: a comprehensive review of the basic science literature. J Am Heart Assoc. 2013;2:e000271. doi: 10.1161/JAHA.113.000271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gabriels K, Hoving S, Seemann I, Visser NL, Gijbels MJ, Pol JF, Daemen MJ, Stewart FA, Heeneman S. Local heart irradiation of ApoE(−/−) mice induces microvascular and endocardial damage and accelerates coronary atherosclerosis. Radiother Oncol. 2012;105:358–364. doi: 10.1016/j.radonc.2012.08.002 [DOI] [PubMed] [Google Scholar]
- 31.Moslehi JJ. Cardiovascular toxic effects of targeted cancer therapies. N Engl J Med. 2016;375:1457–1467. doi: 10.1056/NEJMra1100265 [DOI] [PubMed] [Google Scholar]
- 32.Ozcelik C, Erdmann B, Pilz B, Wettschureck N, Britsch S, Hübner N, Chien KR, Birchmeier C, Garratt AN. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci USA. 2002;99:8880–8885. doi: 10.1073/pnas.122249299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.May D, Gilon D, Djonov V, Itin A, Lazarus A, Gordon O, Rosenberger C, Keshet E. Transgenic system for conditional induction and rescue of chronic myocardial hibernation provides insights into genomic programs of hibernation. Proc Natl Acad Sci USA. 2008;105:282–287 doi: 10.1073/pnas.0707778105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Z, Wu CY, Jiang YP Ballou LM, Clausen C, Cohen IS, Lin RZ. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med. 2012;4:131ra50. doi: 10.1126/scitranslmed.3003623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Latifi Y, Moccetti F, Wu M, Xie A, Packwood W, Qi Y, Ozawa K, Shentu W, Brown E, Shirai T, et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood. 2019;133:1597–1606. doi: 10.1182/blood-2018-10-881557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319 [DOI] [PubMed] [Google Scholar]
- 37.Linette GP Stadtmauer EA, Maus MV, Rapoport AP Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uraizee I, Cheng S, Moslehi J. Reversible cardiomyopathy associated with sunitinib and sorafenib. N Engl J Med. 2011. ;365:1649–1650. doi: 10.1056/NEJMc1108849 [DOI] [PubMed] [Google Scholar]
- 39.Salem JE, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, Gobert A, Spano JP Balko JM, Bonaca MP et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018;19:1579–1589. doi: 10.1016/S1470-2045(18)30608-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–1755. doi: 10.1056/NEJMoa1609214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40a.Wei SC, Meijers WC, Axelrod ML, Anang NAS, Screever EM, Wescott EC, Johnson DB, Whitley E, Lehmann L, Courand PY, et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 2021;11:614–625. doi: 10.1158/2159-8290.CD-20-0856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlaak RA, Frei A, Schottstaedt AM, Tsaih SW, Fish BL, Harmann L, Liu Q, Gasperetti T, Medhora M, North PE, et al. Mapping genetic modifiers of radiation-induced cardiotoxicity to rat chromosome 3. Am J Physiol Heart Circ Physiol. 2019;316:H1267–H1280. doi: 10.1152/ajpheart.00482.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sridharan V, Thomas CJ, Cao M, Melnyk SB, Pavliv O, Joseph J, Singh SP Sharma S, Moros EG, Boerma M. Effects of local irradiation combined with sunitinib on early remodeling, mitochondria, and oxidative stress in the rat heart. Radiother Oncol. 2016;119:259–264. doi: 10.1016/j.radonc.2016.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du S, Zhou L, Alexander GS, Park K, Yang L, Wang N, Zaorsky NG, Ma X, Wang Y, Dicker AP et al. PD-1 modulates radiation-induced cardiac toxicity through cytotoxic T lymphocytes. J Thorac Oncol. 2018;13:510–520. doi: 10.1016/j.jtho.201712.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaiswal S, Natarajan P Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847 doi: 10.1126/science.aag1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bick AG, Pirruccello JP Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S, Natarajan P. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. 2020;141:124–131. doi: 10.1161/CIRCULATIONAHA.119.044362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meijers WC, Maglione M, Bakker SJL, Oberhuber R, Kieneker LM, de Jong S, Haubner BJ, Nagengast WB, Lyon AR, van der Vegt B, et al. Heart failure stimulates tumor growth by circulating factors. Circulation. 2018;138:678–691. doi: 10.1161/CIRCULATI0NAHA.117.030816 [DOI] [PubMed] [Google Scholar]
- 49.Karlstaedt A, Zhang X, Vitrac H, Harmancey R, Vasquez H, Wang JH, Goodell MA, Taegtmeyer H. Oncometabolite d-2-hydroxyglutarate impairs α-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci US A. 2016;113:10436–10441. doi: 10.1073/pnas.1601650113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bellinger AM, Arteaga CL, Force T, Humphreys BD, Demetri GD, Druker BJ, Moslehi JJ. Cardio-oncology: how new targeted cancer therapies and precision medicine can inform cardiovascular discovery. Circulation. 2015;132:2248–2258. doi: 10.1161/CIRCULATIONAHA.115.010484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shelburne N, Simonds NI, Adhikari B, Alley M, Desvigne-Nickens P, Dimond E, Filipski K, Gallicchio L, Minasian L. Changing hearts and minds: improving outcomes in cancer treatment-related cardiotoxicity. Curr Oncol Rep. 2019;21:9. doi: 10.1007/s11912-019-0751-0 [DOI] [PubMed] [Google Scholar]