

Abstract
Isolated central nervous system lymphomatoid granulomatosis (CNS‐LYG) can mimic aggressive glioblastomas. We describe a complex presentation of CNS‐LYG coexisting with immune thrombocytopenia successfully managed with rituximab and ultra‐low‐dose radiation therapy.
Keywords: EBV‐associated B‐cell lymphoproliferative disorder, Epstein‐Barr virus, immune thrombocytopenia, lymphomatoid granulomatosis, pulmonary embolism, thrombosis
Isolated central nervous system lymphomatoid granulomatosis (CNS‐LYG) can mimic aggressive glioblastomas. We describe a complex presentation of CNS‐LYG coexisting with immune thrombocytopenia successfully managed with rituximab and ultra‐low‐dose radiation therapy.
1. CASE PRESENTATION
Epstein‐Barr virus‐associated B‐cell lymphoproliferative disorders (EBV‐LPDs) are a heterogeneous spectrum of hematologic disorders. Defects in immune surveillance are linked to pathogenesis of EBV‐LPD. Lymphomatoid granulomatosis (LYG) is a unique, extra‐nodal EBV‐LPD that can involve organs like the lungs, central nervous system (CNS), skin, kidneys and liver. Primary CNS‐LYG is extremely rare and can masquerade as primary brain tumors like glioblastomas. We describe a challenging case of a 59‐year‐old gentleman with isolated primary CNS‐LYG and a discordant low‐grade B‐cell lymphoma involving the bone marrow causing chronic immune thrombocytopenia. Clinical course was complicated with disseminated zoster and sub‐massive pulmonary embolism. There are no established standards‐of‐care in managing LYG. Treatment strategies that have been used for LYG include expectant observation, surgical resection, corticosteroids, radiation, chemo and immunotherapies. Our patient was successfully managed with a combination radioimmunotherapy strategy with rituximab and ultra‐low‐dose radiation therapy (ULD‐RT) of 2 Gy × 2. The biologic rationale for synergy with this combination, published literature and future directions in managing EBV‐LPDs and LYG are discussed.
A 59‐year‐old gentleman, with a past medical history of hypertension, hyperlipidemia, chronic immune thrombocytopenia (ITP) not requiring therapy (diagnosed 7 years prior), and coronary artery disease status post percutaneous intervention/drug‐eluting stent 2 months prior, presented to the emergency department after a fall.
Physical examination was unremarkable with the exception of a left facial droop. Laboratory evaluation showed mild thrombocytopenia (94 × 109/L).
Magnetic resonance imaging (MRI) revealed a 4.5 × 5.4 × 3.3 cm heterogeneous, peripherally T1 post‐contrast enhancing mass lesion involving the right frontal lobe with significant vasogenic edema (Figure 1A). This was concerning for primary CNS neoplasm like glioblastoma multiforme.
FIGURE 1.
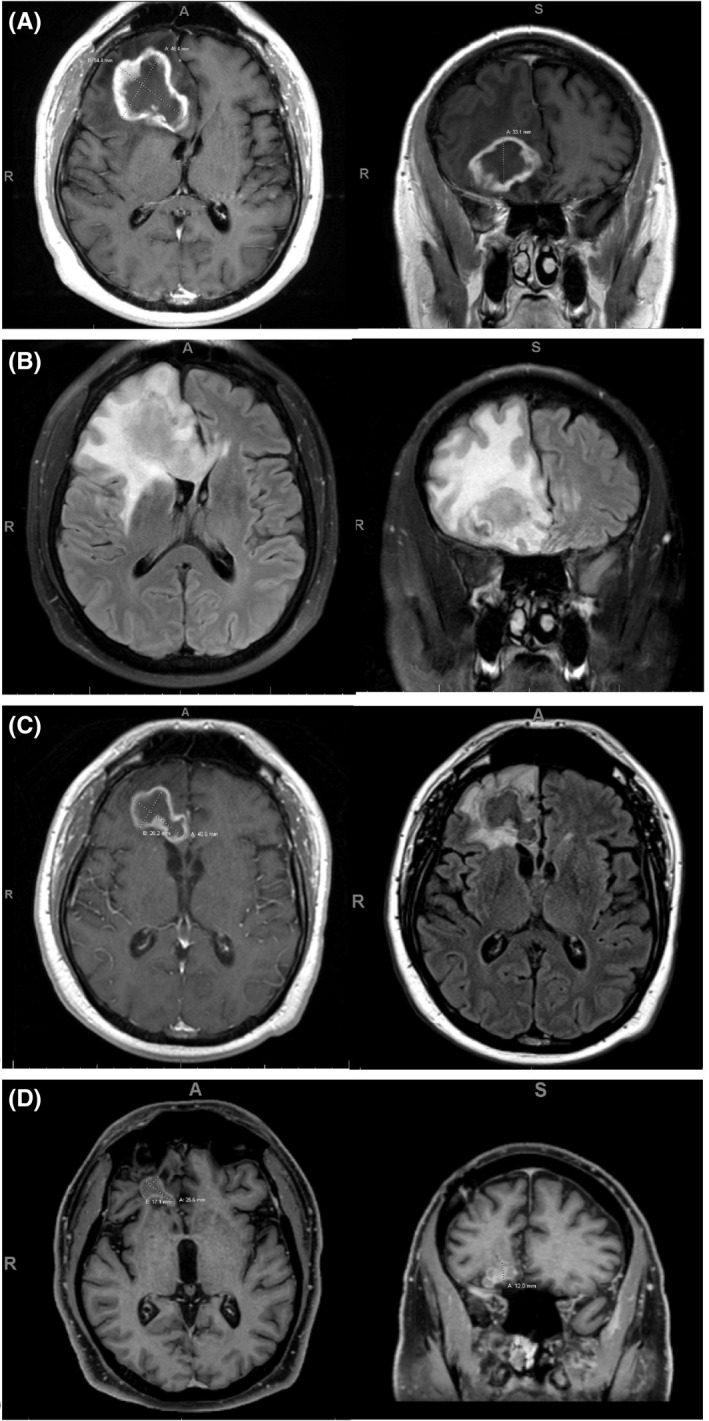
(A) MRI brain at time of diagnosis, T1 post‐contrast axial and coronal images. (B) MRI brain at time of diagnosis, T2 FLAIR axial and coronal images. (C) MRI brain at first follow‐up 3 months after radioimmunotherapy, axial T1 post‐contrast (left) and T2 FLAIR (right) images. (D) MRI brain at last follow‐up 16 months after radioimmunotherapy, T1 post‐contrast axial (left) and coronal (right) images
Pathologic analysis after stereotactic biopsy of the right frontal mass showed an angiocentric polymorphous lymphoid infiltrate composed of numerous small T cells admixed with histiocytes and scattered intermediate‐to‐large, atypical, EBV (+) B cells. The remainder of the lesion showed areas of lymphocytic vasculitis, angiodestruction, hemorrhage, and large areas of central necrosis (Figure 2A–F). The number of EBV (+) B cells was variable, but overall, less than 5 per high‐power field (hpf). After multi‐disciplinary discussion, the lesion was classified as an EBV‐LPD with features suggestive of low‐grade (grade I) lymphomatoid granulomatosis (LYG).
FIGURE 2.
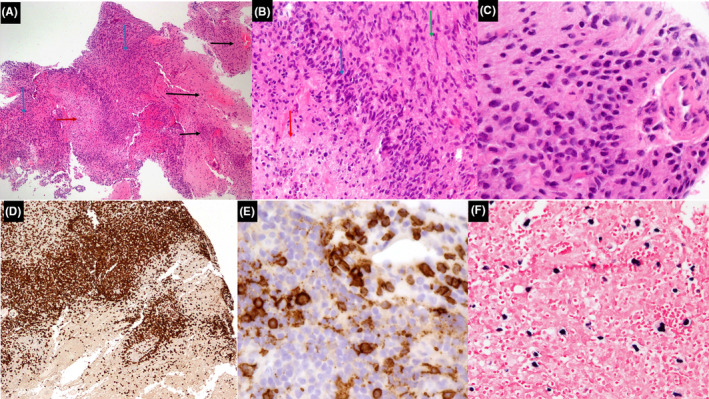
(A) Histologic sections show central nervous system tissue with an extensive, polymorphous lymphoid infiltrate (blue arrows) showing numerous areas of angioinvasion and destruction (black arrows), and a focal area of necrosis (red arrow) (hematoxylin and eosin, 4×). (B) Higher power shows area of central necrosis (red arrow) surrounded by atypical lymphocytes (blue arrow) admixed with numerous histiocytes (green arrow) (hematoxylin and eosin, 20×). (C) High power shows numerous medium to large‐sized atypical lymphocytes (hematoxylin and eosin, 40×). (D) Immunohistochemistry shows that the majority of lymphocytes are mature T cells (CD3, 20×). (E) Immunohistochemistry highlights scattered large atypical B cells with focal involvement of vessels (CD20, 40×). (F) In situ hybridization for EBV‐encoded small RNA (EBER) is positive in rare large atypical lymphocytes (EBER, 20×)
His platelet count dropped after the brain biopsy to 30 × 109/L. There was evidence of significant platelet dysfunction on whole blood impedance platelet aggregometry consistent with ITP. He received intravenous immunoglobulin (IVIG) and romiplostim along with a steroid taper. Clinical course was further complicated with vesicular rash over right arm and multiple dermatomes suggestive of disseminated zoster necessitating antiviral therapy.
Staging CT scans did not show any pulmonary or nodal involvement of LYG, but incidentally detected sub‐massive pulmonary embolism for which therapeutic anticoagulation was started. Bone marrow aspiration/biopsy showed 30%–40% involvement by low‐grade CD5(−)/CD10(−) B‐cell lymphoma. In situ hybridization for EBV‐encoded small RNA (EBER) was negative. Immunologic studies showed borderline low total IgG (681 mg/dl) and slightly decreased amounts of CD3(+)/CD8(+) T cells. Serum protein electrophoresis did not show evidence of monoclonal gammopathy. EBV PCR was detectable but non quantifiable. HIV, hepatitis C and hepatitis B serologies were negative. Anti‐nuclear and platelet antibodies were negative. Genetic testing for primary immunodeficiency and next‐generation sequencing of the brain biopsy did not reveal any pathologic mutations.
The patient was started on rituximab 375 mg/m2 weekly for 4 weeks. This was immediately followed by ultra‐low‐dose radiation therapy (ULD‐RT) to the right frontal brain mass at a total dose of 4 Gray in 2 fractions (2 Gray/fraction). The patient had an excellent clinical and radiologic response to therapy with serial imaging showing significant decrease in size of the brain mass (Figure 1B). Platelet count normalized with rituximab, allowing discontinuation of romiplostim.
The patient remains on maintenance Rituximab every 2 months for a planned 2 years of therapy. He has tolerated the therapy well with continued response noted on imaging, 20 months after treatment initiation (Figure 1C).
2. DISCUSSION
Since its discovery in 1964, Epstein‐Barr virus (EBV) has been causally linked to a variety of B‐cell lymphoproliferative disorders (EBV‐LPD). EBV‐LPD range from indolent and/or self‐limited inflammatory disorders to aggressive lymphomas. These include infectious mononucleosis, Burkitt lymphoma, Hodgkin lymphoma, post‐transplant‐ and other iatrogenic immunodeficiency associated‐ lymphoproliferative disorders (PTLD and Oii‐LPD), EBV positive diffuse large B‐cell lymphoma (not‐otherwise‐specified and chronic inflammation associated), primary CNS lymphoma, lymphomatoid granulomatosis (LYG), EBV positive mucocutaneous ulcer, germinotropic B‐LPD, primary effusion and plasmablastic lymphomas.1
First described in 1972 by Liebow et al., lymphomatoid granulomatosis (LYG) is a rare form of EBV‐LPD. LYG is characterized by an angiocentric and angiodestructive, polymorphous infiltrate of numerous small T‐lymphocytes, histiocytes, and variable numbers of large, atypical EBV (+) B cells. Despite its name, granuloma formation is rarely observed.
Lymphomatoid granulomatosis is a complex, extra‐nodal EBV‐LPD which usually affects middle‐aged adults in the fourth to sixth decades of life with a 2:1 male predominance. LYG is most commonly encountered in pulmonary tissue but can rarely show primary central nervous system (CNS) involvement.2, 3, 4, 5 Vascular damage and tissue necrosis in LYG is postulated to be cytokine‐mediated by CXCL9 and CXCL10. The cellular infiltrate is predominantly composed of CD3(+)/CD4(+) helper T cells and variable amounts of histiocytes. EBV (+) B cells range from rare, scattered cells to numerous clusters of cells.3, 5, 6
Lymphomatoid granulomatosis is histologically divided into low‐grade (Grade I/II) and high‐grade (Grade III) disease. Grade I lesions contain rare large atypical B cells and minimal necrosis whereas Grade II and III lesions contain greater number of large atypical B cells and extensive necrosis. There is a paucity of EBER+B cells (<5/high‐power field [hpf]) in Grade I lesions, but higher numbers are observed in Grade II (5–50/hpf) and Grade III (>50–100/hpf) lesions. Lesions with higher numbers of EBV (+) large B cells behave aggressively, usually necessitating chemotherapy. Low‐grade lesions, in contrast, are often treated aiming to improve immune surveillance of EBV.3, 5
The lung is the most common site of involvement in LYG, followed by the CNS (40%), skin (34%), kidney (19%), and liver (17%). Lymph node and bone marrow involvement are rare. Immunologic studies have shown a decrease in CD3+ T cells, specifically CD8+ cells in LYG.5 Though initial reports had noted LYG in setting of immunodeficiency states like Wiskott‐Aldrich syndrome and HIV, no underlying immunodeficiency state has been demonstrated in most patients.3, 5, 7
Central nervous system involvement in LYG is seen in approximately one‐third of patients and clinical presentation may include variable neurologic symptoms such as hearing loss, diplopia, dysarthria, hemiparesis, ataxia, peripheral neuropathy and atonic bladder. In a series of LYG from the National Cancer Institute (NCI), CNS involvement was reported in 28 (40%) patients.5 Isolated CNS involvement (primary CNS‐LYG) was reported in 19 patients in another series.8
As in our case, MRI commonly reveals focal intraparenchymal brain lesions with multiple foci of increased signal intensity on fluid attenuation inversion recovery (FLAIR) and T2‐weighted imaging. Lesions may evolve over time into ring‐enhancing lesions with a non‐enhancing core. Similar rates of CNS involvement by low‐ and high‐grade LYG have been reported.5, 8, 9, 10
There are no established consensus treatments for LYG due to its rarity. Treatment modalities that have been employed include observation, surgical resection, radiation therapy, corticosteroids, interferon (IFN), intravenous immunoglobulin (IVIG), chemotherapy and rituximab, with more intense strategies used for high‐grade disease.2, 3, 5, 6
Historically, 14%–27% of patients, achieved durable remissions without therapy. However, early series showed overall mortality of over 60%, with most deaths from progressive lung damage and infections. A prospective treatment strategy was formulated at the NCI in 1994 with low‐grade LYG managed with dose‐escalated IFN‐alfa2b, while high‐grade disease was treated with chemo‐immunotherapy with dose‐adjusted R‐EPOCH (rituximab, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin). Crossover therapy was allowed if alternate grade disease developed after initial therapy. With this approach, in their cohort of 70 patients, after 13 years of median follow‐up, more than half of treated patients are alive for at least 10 years.5
Published experience with isolated CNS‐LYG has been limited. The anti‐CD20 monoclonal antibody, rituximab as a single agent has shown efficacy in treating CNS‐LPD. We used the combination strategy of rituximab and ULD‐RT (2 Gy × 2). ULD‐RT has shown immune modulating effects and long‐lasting remissions in treating indolent lymphomas. Though the mechanism of action of ULD‐RT is not entirely clear, induction of the pro‐apoptotic pathways including p53, and immunogenic tumor cell death is postulated for its beneficial effects.11 Rituximab may also enhance the radio‐sensitivity of lymphoma cells, providing a synergistic rationale for this radioimmunotherapy combination.12 In the prospective MIR trial, the combination of involved field RT and rituximab was shown to be successful with preventing out‐of‐field relapses in early‐stage follicular lymphoma and a 2‐year PFS of 85%.13 In our experience with ULD‐RT in indolent lymphomas, the progression‐free and overall survival at 1‐year was 58% and 82%, respectively. In the seven patients who received combined ULD‐RT and single‐agent rituximab, the response rate was 100% (7/7), of which 5 had complete response at the time of latest follow‐up.14 ULD‐RT is being explored in combination with various immunotherapies in clinical trials for lymphomas.
Our case is unique due to the co‐existence of a low‐grade B‐cell lymphoma in the bone marrow along with CNS‐LYG, which has not yet been described before. While the clonal relatedness of these two processes is unknown, the low‐grade B‐cell lymphoma was likely responsible for ITP in our patient and both disorders responded well to rituximab therapy. The underlying immunodeficiency state with LYG combined with steroid use led to disseminated zoster. Acute illness and ITP likely resulted in hypercoagulability and pulmonary embolism.
Treatment of relapsed/refractory LYG remains a clinical challenge. Autologous and allogeneic stem cell transplantation has been utilized in this setting with some success. Novel therapies being evaluated for LYG include immunotherapies like anti‐PD1 (programmed death‐1) agents, anti‐CD30 monoclonal antibodies like brentuximab vedotin, targeted agents like histone deacetylase inhibitors (HDACis) in combination with antiviral agents and cellular therapies like EBV‐specific cytotoxic T‐lymphocytes and chimeric antigen receptor T‐cell (CAR T‐cell) therapies.5, 15
In summary, we present a unique and challenging case of CNS‐LYG, presenting as a large ring‐enhancing mass, mimicking glioblastoma, but with clinicopathologic features of low‐grade LYG. The disease occurred concurrently with chronic immune thrombocytopenia (ITP), disseminated zoster, sub‐massive pulmonary embolism, and bone marrow involvement by low‐grade B‐cell lymphoma. Despite the complexity of the presentation, our patient was successfully managed with a combination of immunotherapy with rituximab and ultra‐low‐dose radiation therapy (ULD‐RT). This approach has shown promise in low‐grade B‐cell lymphomas but requires further study and more comprehensive investigation. This highlights the complex spectrum of EBV‐LPD and supports the need for increased awareness and further study of this unique form of disease.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Authors KK and PRG were clinically responsible for the care of the patient and conceptualized the manuscript. All other authors contributed equally to the design, editing and submission of the manuscript.
CONSENT
Appropriate informed consent was obtained from the patient prior to manuscript preparation and submission to the journal.
ACKNOWLEDGMENT
The authors would like to acknowledge Rasmi Nair, PhD for her contribution in editing the final figures for the manuscript.
Soleja M, Jaso JM, Chen W, et al. Lymphomatoid granulomatosis of the central nervous system (CNS‐LYG) posing a management challenge. Clin Case Rep. 2021;9:e04808. 10.1002/ccr3.4808
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1.Crombie JL, LaCasce AS. Epstein barr virus associated B‐cell lymphomas and iatrogenic lymphoproliferative disorders. Front Oncol. 2019;9:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fauci AS, Haynes BF, Costa J, Katz P, Wolff SM. Lymphomatoid granulomatosis. Prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68‐74. [DOI] [PubMed] [Google Scholar]
- 3.Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:e35‐e48. [DOI] [PubMed] [Google Scholar]
- 4.Liebow AA, Carrington CR, Friedman PJ. Lymphomatoid granulomatosis. Hum Pathol. 1972;3:457‐558. [DOI] [PubMed] [Google Scholar]
- 5.Melani C, Jaffe ES, Wilson WH. Pathobiology and treatment of lymphomatoid granulomatosis, a rare EBV‐driven disorder. Blood. 2020;135:1344‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson WH, Kingma DW, Raffeld M, Wittes RE, Jaffe ES. Association of lymphomatoid granulomatosis with Epstein‐Barr viral infection of B lymphocytes and response to interferon‐alpha 2b. Blood. 1996;87:4531‐4537. [PubMed] [Google Scholar]
- 7.Sebire NJ, Haselden S, Malone M, Davies EG, Ramsay AD. Isolated EBV lymphoproliferative disease in a child with Wiskott‐Aldrich syndrome manifesting as cutaneous lymphomatoid granulomatosis and responsive to anti‐CD20 immunotherapy. J Clin Pathol. 2003;56:555‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benouaich‐Amiel AY‐KS, Fichman S, Siegal T. Lymphomatoid granulomatosis of the central nervous system successfully treated with corticosteroid alone: a case report and review of the literature. Neurosurg Cases Rev. 2018;1:9. [Google Scholar]
- 9.Lucantoni C, De Bonis P, Doglietto F, et al. Primary cerebral lymphomatoid granulomatosis: report of four cases and literature review. J Neurooncol. 2009;94:235‐242. [DOI] [PubMed] [Google Scholar]
- 10.Zaidi A, Kampalath B, Peltier WL, Vesole DH. Successful treatment of systemic and central nervous system lymphomatoid granulomatosis with rituximab. Leuk Lymphoma. 2004;45:777‐780. [DOI] [PubMed] [Google Scholar]
- 11.Knoops L, Haas R, de Kemp S, et al. In vivo p53 response and immune reaction underlie highly effective low‐dose radiotherapy in follicular lymphoma. Blood. 2007;110:1116‐1122. [DOI] [PubMed] [Google Scholar]
- 12.Skvortsova I, Skvortsov S, Popper BA, et al. Rituximab enhances radiation‐triggered apoptosis in non‐Hodgkin's lymphoma cells via caspase‐dependent and ‐ independent mechanisms. J Radiat Res. 2006;47:183‐196. [DOI] [PubMed] [Google Scholar]
- 13.Witzens‐Harig M, Hensel M, Unterhalt M, Herfarth K. Treatment of limited stage follicular lymphoma with Rituximab immunotherapy and involved field radiotherapy in a prospective multicenter Phase II trial‐MIR trial. BMC Cancer. 2011;11:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brocklehurst SDN, Shah JL, Rizvi S, Ramakrishnan Geethakumari P, Kumar K. Combined modality low‐dose radiation therapy (2 Gy x 2) and single‐agent rituximab immunotherapy in the treatment of indolent non‐hodgkin lymphoma. Blood. 2019;134:5262. [Google Scholar]
- 15.Bollard CM, Gottschalk S, Torrano V, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein‐Barr virus latent membrane proteins. J Clin Oncol. 2014;32:798‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.