

Introduction:
Leptomeningeal amyloidosis (LA) represents a rare subtype of familial transthyretin (TTR) amyloidosis, characterized by deposition of amyloid in cranial and spinal leptomeninges. Of >120 TTR mutations identified, few have been associated with LA.
Case Report:
A 27-year-old male presented with a 2-year history of progressive symptoms including cognitive decline and right-sided weakness and numbness. Cerebrospinal fluid (CSF) analyses demonstrated high protein level. Gadolinium-enhanced magnetic resonance imaging (MRI) revealed extensive leptomeningeal enhancement over the surface of the brain and spinal cord. Pathologic analyses revealed a TTR mutation c.113A>G (p.D38G).
Review Summary:
Fifteen mutations and genotype-phenotype correlation of 72 LA patients have been summarized to provide an overview of LA associated with transthyretin mutations. The mean age of clinical onset was 44.9 years and the neurological symptoms primarily included cognitive impairment, headache, ataxia seizures and hearing, visual loss. CSF analysis showed elevated high CSF protein level and MRI revealed extensive leptomeningeal enhancement.
Conclusion:
Clinicians should be aware of this rare form of familial transthyretin amyloidosis as well as its typical MRI enhancement and high CSF protein. The important role of biopsy, genetic testing and the potential early diagnosis value of contrast MRI were suggested. Early recognition of these characteristics is important to provide misdiagnosis and shorten the time before correct diagnosis. These findings expand the phenotypic spectrum of TTR gene and have implications for the diagnosis, treatment, and systematic study of LA.
Key Words: leptomeningeal amyloidosis, transthyretin, leptomeningeal enhancement, TTR mutations
Familial transthyretin amyloidosis is a life-threatening and gain-of-toxic-function autosomal dominant disorder commonly caused by mutation of the transthyretin gene (TTR; OMIM 176300) located at chromosome 18q12.1.1 So far, >120 mutations in the TTR gene have been found worldwide.2 The TTR gene product forms a plasma and cerebrospinal fluid (CSF) homotetramer, synthesized by the liver, choroid plexus and retina, and involved in the transport of retinol and thyroxine.3 TTR mutations render the gene product prone to dissociate into its constituent monomers leading to extracellular build-up of amyloid fibrils in major organs and neural tissues.4 The 3 main phenotypes of hereditary transthyretin amyloidosis are familial amyloid polyneuropathy (FAP), familial amyloid cardiomyopathy (FAC), and familial leptomeningeal amyloidosis (LA).5
Familial LA is a rare manifestation of TTR amyloidosis, characterized by amyloid deposition in the meninges of the brain and spinal cord, often with ocular involvement.6 Patients present with a wide spectrum of central nervous system (CNS) including cognitive difficulties, ataxia, epilepsy, headache, visual, and hearing loss.6–9 The wide variety of clinical phenotypes frequently leads to misdiagnosis, a considerable time before correct diagnosis and poorer patient outcomes.10 While many TTR mutations could cause FAP, familial LA was extremely rare. Since first described by Goren et al in 1980,11 only 15 TTR mutations causing LA have been identified. Until now, these mutations have been found exclusively in European, American, and Japanese patients.
Here we described leptomeningeal involvement secondary to a TTR c.113A>G (p.D38G, formerly labeled as “D18G”) mutation in a patient of Chinese origin, corroborated by clinical, radiologic, and histopathologic findings. As the LA syndrome remains extremely rare, for a clearer delineation of the clinical evaluation and genetic presentation, the relevant literature for LA have been reviewed. The aim of this article is to demonstrate TTR mutations and phenotypic spectrum in order to have potential value in the diagnosis, treatment, and elucidation of its pathogenic mechanisms.
METHODS
The spectrum of clinical symptoms and the genetic analysis of a LA with p.D38G mutation were identified and investigated. Genetic testing was performed on DNA obtained from a peripheral blood sample. DNA isolation and high throughput sequencing Genomic DNA was extracted from peripheral blood according to the manufacturer’s instructions (QIAGEN, Hilden, Germany). DNA sequencing libraries were then prepared according to Illumina standard protocol. Fragments in the exonic regions of targeted genes were captured by a specific Amyloidosis Disease GenePanel using biotinylatedoligo-probes (MyGenostics GenCap Enrichment Technologies, MyGenostics, Baltimore, MD). The capture experiment was conducted according to the manufacturer’s protocol. SIFT (http://sift.jcvi.org/) and POLYPHEN2 (http://genetics.bwh.harvard.edu/pph2/) were used to analyze the amino acid substitutions of p.D38G.
Due to the syndrome remains extremely rare, we searched PubMed and MEDLINE databases to find LA cases in reports published till August 2019 using the terms: “leptomeningeal amyloidosis” or “oculoleptomeningeal amyloidosis”; all identified articles published in English and articles referenced therein were reviewed. The patient inclusion criteria consisted of transthyretin mutations and presented LA symptoms.
CASE REPORT
A 27-year-old right-handed Chinese man was admitted in our department with rapidly progressive cognitive decline over a 2-year period. He presented initially in February 2017 with headache, nausea and vomit for 1 months. He also had a fever with highest temperature 39°C. At his local hospital, a noncontrast head magnetic resonance imaging (MRI) was normal. He was diagnosed with viral encephalitis and received antiviral therapy. Three months later, he developed progressive confusion, fatigue, and weakness in limbs. In June 2017, he suffered from recurrent episodes of memory loss, slurred speech, right-sided weakness and numbness, lasting 4 to 5 hours. The episodes were initially every 3 weeks but increased in frequency over time. In October 2017, the patient was found blurred consciousness and right limb weakness. Then he was referred to our department.
Cardiovascular, ophthalmic, audiometric, respiratory, and abdominal examination was unremarkable. The neurological examination revealed cognitive decline. Screening cognitive testing demonstrated short-term and long-term memory decline and impairment of attention and calculation [Mini-mental State Examination, (MMSE)=24/30; Montreal Cognitive Assessment (MoCA)=21/30]. Examination of the limbs revealed brisk reflexes in right lower limbs. There was no relevant past medical history. His father had suffered from coronary heart disease and renal dysfunction. He died of a myocardial infarction in his 40s. His mother died of car accident in her 50s.
The blood and urine tests were normal or negative including full blood count, electrolytes, liver function, renal function, thyroid function, cardiac enzymes, B12 and folate, serum ACE and ANA, ANCA, antineuronal antibodies, and immunofixation. Lumbar puncture demonstrated CSF protein was elevated at 339 mg/dL and IgA, IgG was increased at 4.64 and 39.20 mg/dL. CSF analyses were negative for encephalitis and paraneoplastic autoimmune encephalitis. No organisms were cultured (including Mycobacterium tuberculosis) and no cellular atypia was seen. The patient had normal levels of myelin basic protein and aquaporin-4 with no neuromyelitis optica and a normal cell count without oligoclonal bands.
Gadolinium-enhanced MRI of the brain and spine revealed extensive leptomeningeal enhancement over the surface of the brain and spinal cord (Figs. 1A–F). Cerebral and spinal angiography was normal. Nerve conduction studies and autonomic function testing were negative. ECG, echocardiogram, and cardiac MRI demonstrated normal cardiac function.
FIGURE 1.
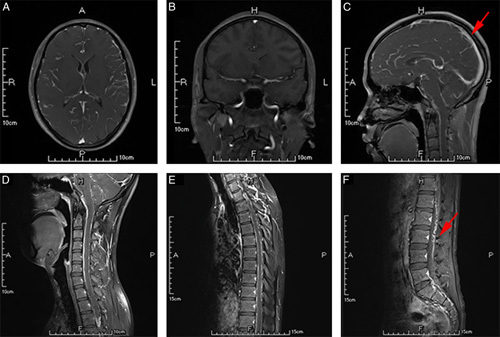
Leptomeningeal enhancement over brain and spinal cord. (A–C) Gadolinium-enhanced transverse, coronal and sagittal T1-weighted magnetic resonance imaging (MRI) brain demonstrating enhancement extending along the meninges. (D–F) Gadolinium-enhanced sagittal T1-weighted MRI of the cervical thoracic and lumbar vertebra spine demonstrating thick irregular leptomeningeal enhancement (red arrow) over the spinal cord surface.
The patient was ultimately referred to neurosurgery for consideration of leptomeningeal biopsy for the purpose of providing a tissue diagnosis. He underwent a L5 to S1 laminectomy for biopsy of the intradural lesion. Intraoperatively, the arachnoid mater was markedly thickened and of a grayish, discolored appearance. These rust-colored, thicken tissues were sampled as specimen for biopsy. Pathologic analysis revealed congo red positive. In this rare form of the disease, this pattern is diagnostic (Figs. 2A, B). Proteomic analysis by mass spectrometry of congo red positive material indicated amyloid deposition of the transthyretin type.
FIGURE 2.
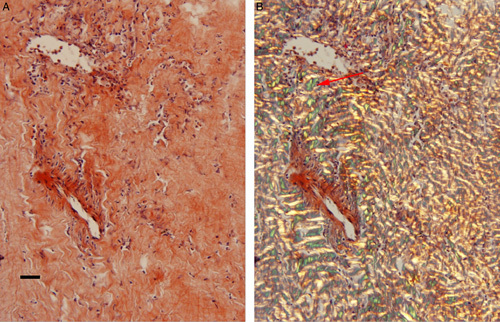
Brain biopsy of meningeal amyloid angiopathy. (A) Hematoxylin and eosin stain of the L5 to S1 intradural lesion. (B) Congo red stain shows apple green birefringence under polarized light (black scale bars=200 μm).
Genetic testing performed on DNA obtained from a peripheral blood sample revealed a heterozygous adenine to guanine transversion at nucleotide 113 of the TTR gene, resulting in an asparagicacid (Asp) to glycine (Gly) substitution (p.D38G). Both SIFT (http://sift.jcvi.org/) and POLYPHEN2 (http://genetics.bwh.harvard.edu/pph2/) predicted the mutation c.113A> G to be deleterious, which means they probably damage and affect protein functions.
REVIEW OF THE LITERATURE
LA was first described by Goren et al11 in 1980, since then 15 TTR mutations causing LA have been identified.3,6–9,12–40 Until now, these have been found exclusively in European, American, and Japanese patients. In addition to the present case, a total of 72 genetically confirmed cases (including our case) were identified and summarized to provide an overview of the clinical and genetic features of LA (Table 1).
TABLE 1.
Mutations Reported Associated With Leptomeningeal Amyloidosis
| Amino Acid Change (Mature Protein) Nucleotide Change | Cases Refs | Countries | Age of Onset (y) | Disease Duration (y) | CNS Neurological Symptoms | Autonomic Neuropathy | Peripheral Neuropathy | Hearing Loss | Visual Loss | Cardiac Dysfunction | CNS Protein | Neuroimaging |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p.D38G (D18G) c.113A>G | 13 our case8,12,15,18 | Hungary, Japan, USA, China | 35.9±8.5 | 16.7±10.6 | Cognitive impairment, headache, ataxia, stroke, SAH | + | − | + | + | − | High | Leptomeningeal enhancement superficial siderosis |
| p.L32P (L12P) c.95T>C | 428–31 | UK, Nigeria, Germany | 36.1±6.0 | 15 | Cognitive impairment, headache, ataxia, stroke, seizures, SAH | + | + | + | − | + | High | Leptomeningeal enhancement |
| p.A45T (A25T) c.133G>A | 316,21,32 | Japan, Spain | 46.0±4.4 | 7.5±4.9 | Cognitive impairment, ataxia, stroke, seizures, SAH | − | + | + | − | − | High | Leptomeningeal enhancement, superficial siderosis |
| p.V50G (V30G) c.148G>A | 77,33,34 | USA, Germany | 46.1±8.9 | 5.6±3.6 | Cognitive impairment, headache, ataxia, stroke, seizures | − | − | − | + | − | High | Leptomeningeal enhancement |
| p.V50M (V30M) C.148G>A | 1117,35 | Mexico, Japan | 56.5±4.3 | — | Headache, seizures, hydrocephalus | + | + | − | − | − | High | Leptomeningeal enhancement Hydrocephalus |
| p.A56P (A36P) c.166G>C | 336 | Italy | 38±1.7 | 17.5±20.5 | Cognitive impairment, ataxia, stroke | − | + | + | + | − | High | Leptomeningeal enhancement Hydrocephalus |
| p.F64S F44S c.191T>C | 137 | USA | 26 | — | Cognitive impairment, headache | + | + | + | − | + | — | Leptomeningeal enhancement |
| p.T69P T49P c.205A>C | 119 | Ireland | 53 | — | Cognitive impairment, headache, seizures | − | + | − | − | + | High | Leptomeningeal enhancement |
| p.G73R G53R c.217G>A | 238 | USA | 51.0±7.1 | 17 | Cognitive impairment, stroke, seizures, hydrocephalus | − | − | − | − | − | — | Leptomeningeal enhancement Hydrocephalus |
| p.G73E G53E c.218G>A | 314 | France | 38.3±3.2 | 4.5±3.5 | Cognitive impairment, ataxia, headache, SAH | + | − | − | − | + | High | Leptomeningeal enhancement |
| p.G73A G53A c.218G>C | 113 | UK | 40 | — | Cognitive impairment, ataxia, stroke | − | + | − | − | + | High | Leptomeningeal enhancement |
| p.F84S F64S c.215T>C | 339,40 | Canada | 28 | 12 | Cognitive impairment, headache, ataxia, stroke, seizures, hydrocephalus | + | + | + | + | + | — | Leptomeningeal enhancement |
| p.Y89H Y69H c.265T>C | 139,41–43 | USA, Sweden, Canada | 50.4±9.2 | 7.7±3.1 | Cognitive impairment, headache, ataxia, stroke, seizures, SAH | − | + | + | + | − | High | Leptomeningeal enhancement |
| p.I127M I107M c.381T>C | 144 | Canada | 51 | — | Ataxia | + | + | − | − | + | High | — |
| p.Y134C Y114C c.401A>G | 66 | Japan | — | — | Cognitive impairment | + | + | + | − | + | — | Leptomeningeal enhancement |
CNS indicates central nervous system; SAH, subarachnoid hemorrhage.
Epidemiology and Pathophysiology
All of the LA patients included in the present report had their diagnoses genetically confirmed. c.113A>G (16.7%), c.265T>C (16.7%), and C.148G>A (15.2%) are most common mutations associated with LA. Of all mutations, c.148G>A and c.217G>A only show the CNS symptoms without autonomic neuropathy, peripheral neuropathy, or cardiac involvement. These 2 genes only responded to LA but not FAP or FAC. These mutations exclusively founded in European (60%), American (33.3%), and Japanese (26.7%) patients. The only mutation founded in China is c.113A>G.
Recent studies on LA pathogenesis came from an electron microscopic study of sural nerve biopsy specimens from 49 Val30Met.45 This work has shown that the direct damage of amyloid fibers leads to the damage of Schwann cells and basal lamina. The loss of small fiber axons and vasculopathy may also contribute to the pathogenesis of neuropathy.
Clinical Phenotype
The average onset age of all LA patients is 44.9±9.1. Among these mutations, c.191T>C, c.215T>C, and c.113A>G are early-onset mutations. The prior studies suggest that the c.113A>G mutation caused LA with later onset age. However, in our study, we found that c.113A>G is an early-onset variant. The average disease duration is 9.3±7.1, which means LA is a life-threatening disease. All reported cases showed CNS symptoms. More specifically, of the 15 patients, 13 (86.7%) suffered from cognitive impairment, 10 (66.7%) exhibited ataxia, and 9 (60%) showed headache. Seizures and hydrocephalus were also evident in the patients. Stroke and subarachnoid hemorrhage (SAH) were also detected in many patients which suggest that amyloid angiopathy could also destroy dura mater, leptomeninges, and neocortical blood vessels. Autonomic neuropathy and peripheral neuropathy were also accompanied with LA in 8 (53.3%) and 11 (73.3%) patients. These cases usually presented predominant loss of superficial sensation including nociception and thermal sensation, and marked autonomic dysfunction, including orthostatic hypotension, sexual impotence, neurogenic bladder, and disturbed bowel movement. Eight patients (53.3%) showed hearing dysfunction. Just like prior Hungarian family reported, hearing loss is a frequent and early symptom.7 Five patients (33.3%) detected visual dysfunction. LA often accompanied with ocular involvement. Eight patients (53.3%) with cardiac involvement showed congestive heart failure, intractable arrhythmia, and conduction blocks and occasionally require implantation of a pacemaker and/or implantable cardioverter defibrillator.
Diagnostic Tools
Several approaches prove to be useful for early diagnosis. MRI of the brain and spine in all reported patients showed extensive leptomeningeal enhancement over the surface of the brain and spinal cord. Superficial siderosis was reveal in two patients with TTR mutations. 3 patients (20%) presented hydrocephalus. However, the frequent headache symptom in LA patients suggest hydrocephalus is probably more frequent than recorded. Besides, cerebrospinal fluid (CSF) analysis in 11 LA patients showed elevated high CSF protein level.
Treatment
Liver transplant was first-line therapeutic intervention for patients with TTR mutations and has been used for >30 years.46 Since most transthyretin is produced in the liver, it was initially assumed that WT TTR could be used to instead mutated TTR to prevent the amyloidosis progression.47 A 20-year retrospective analysis of the FAP World Transplant Registry revealed 20-year survival after liver transplant was 55.3%.48 However, after liver transplantation, neuropathy can progress. One study pointed out that regardless of liver transplantation, 31% of patients still had clinical manifestations of focal neurological episodes.49 Altogether, long-term outcomes of liver transplantation with LA are not usually reversed.
Considering the limitations of liver transplantation, pharmacological agents are needed. There are actually 3 innovative pharmacological approaches: TTR tetramer stabilizer, gene modifying therapy, and drugs targeting amyloid fibrils. Oral TTR stabilizers, such as Tafamidis and Diflunisal, were able to prevent amyloid deposition and slow disease progression. Tafamidis has been approved in the Japan and European countries.50 Tafamidis binds to the thyroxine-binding sites, thereby blocking TTR amyloidogenesis, preventing dissociation of the native TTR quaternary structure.51 In a randomized, double-blind trial, Tafamidis significantly reduced neurological decline in most variables examined. Diflunisal, a nonsteroidal anti-inflammatory drug (NSAID) developed in the 1970s, stabilizes TTR tetramers in healthy volunteers and FAP patients, at a dose of 250 mg twice daily.52 Besides TTR stabilizers, 2 types of gene silencing therapies have been evaluated in phase 3 clinical trials: antisense oligonucleotides53 and small interfering RNAs.54 By strongly reducing both mutant and wild-type TTR plasma levels, it is hoped that such approaches will stop disease progression. In addition to technologies mortifying genes, to clear amyloid deposits already in the tissue to reduce the amyloid load and ultimately to restore organ function. In this regard, anti-Serum Amyloid P agents or anti-TTR antibodies are the main approaches now being developed.55,56
In conclusion, thanks to a better understanding of the TTR amyloid formation, therapeutic developments which are less invasive than liver transplantation have been founded. TTR stabilizer drugs are safe and seem to delay the disease progression in some groups of patients. Indeed, gene modifying and amyloid fibrils targeting drugs open a new area in the field with the hope that we can safely bring about long-term stabilization of the disease.
DISCUSSION
In our study, 15 mutations and its genotype-phenotype correlation for LA have be summarized, while its underlying pathogenic mechanism is still somewhat unclear. A proposed mechanism which may explain CNS selectivity is that highly destabilized TTR proteins encoded by these variants were completely degraded through the endoplasmic reticulum-associated degradation mechanism in peripheral tissues. They could cause later CNS impairment, because these mutants were secreted with lower efficacy from choroid plexus due to a stabilizing effect of thyroxine chaperoning, which is highly concentrated in choroid plexus cells.
Based on previous studies, we summarized the genotype-phenotype correlations of LA. LA is a rare TTR mutation subtype, mainly causing stroke, SAH and/or hydrocephalus. It presents with CNS symptoms, including cognitive impairment, headaches, ataxia, seizures, and hearing loss. These CNS pathologic changes are caused by the deposition of mutant TTR protein secreted from choroid plexus.41 LA diagnosis hallmarks include extensive leptomeningeal enhancement on contrast MRI and increased CSF protein levels.
In this study, we identified a 27-year-old with c.113A>G (p.D38G, D18G) mutation which showed rapidly progressive cognitive decline over a 2-year period. Four previously discovered p.D38G Hungary cases were characterized by dementia, spasticity or hearing loss without ocular disease or polyneuropathy.17 Two Japanese brothers were also reported carried p.D38G mutations.12 These 2 patients had recurrent SAH, presumably secondary to amyloid laden, friable vessels.42 The third report due to D18G mutation was a US woman.20 She had developmental delay, headache, and vomiting. Last year, another Chinese group also identified the p.D38G variant in an early-onset LA family.21 The hydrocephalus-associated clinical symptoms and the absence of peripheral neuropathy, vitreous opacity, and cardiac impairment in that family further support the high CNS selectivity of the p.D38G variant.43 These four findings indicate that besides genetic factors, environment factors may also modify the manifestations of LA.
Medical management of LA involved liver transplantation and pharmacological agents. However, best medical management specific for LA is lacking. Some previous studies suggest LA cannot be blocked by liver transplantation as mutant TTR protein are mainly secreted from choroid plexus but not liver.44 Given the associated imaging findings, patients with LA may be referred for consideration of neurosurgical intervention. Awareness of this rare pathology, and its consideration among the differential diagnosis, may limit unnecessary surgery in cases for which diagnosis can be made by less invasive means or through genetic testing. Neurosurgical management, as in this case, is limited to biopsy for cases in which less invasive strategies fail to yield a diagnosis.
CONCLUSION
The present report summarizes 72 patients with LA to provide an overview of the clinical and genetic characteristics of this disorder. Fifteen mutations and genotype-phenotype correlation of these 72 patients have be summarized. The mean age of clinical onset was 44.9 years and the neurological symptoms primarily included cognitive impairment, headache, ataxia seizures and hearing, visual loss. In addition, strokes and SAH due to small blood vessels also occurred. Furthermore, several patients suffered from autonomic, peripheral neuropathy, and cardiac dysfunction. CSF analysis showed elevated high CSF protein level and MRI revealed extensive leptomeningeal enhancement. These findings suggest that clinicians should be aware of this rare form of familial transthyretin amyloidosis as well as its typical MRI enhancement and high CSF protein. The important role of biopsy, genetic testing and the potential early diagnosis value of contrast MRI were suggested. No curative treatment for LA is presently available, and thus early recognition of these characteristics is important to provide misdiagnosis and shorten the time before correct diagnosis. These findings expand the phenotypic spectrum of TTR gene and have implications for the diagnosis, treatment, and systematic study of LA.
Footnotes
The institutional review board of Xuan Wu Hospital affiliated to Capital Medical University approved the study. Written informed consent was obtained from the patient to participate.
Written informed consent is available for publication of this case report and accompanying brain MRI images and mutation results.
Q.Q.: drafted the manuscript. C.W. and Q.Q.: examined and acquired patient’s data. J.J. and C.W.: revised the article. F.L. and Y.S.P.: performed and analyzed the pathologic analysis. H.W.: did leptomeningeal biopsy. F.W., X.Z., and Y.H.: analyzed the mutation. X.Z., D.G., and J.L.: participated in the literature review and helped to draft the manuscript.
This study was supported by Capital Medical University (PYZ19137); Capital’s Funds for Health Improvement and Research (CFH2020-4-1033) and Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (No. ZYLX201837). Also funded by the National Key R&D Program of China (No. 2017YFC1310103).
The authors declare no conflict of interest.
Contributor Information
Qi Qin, Email: qinqibao@126.com.
Cuibai Wei, Email: chuibainews@126.com.
YueShan Piao, Email: yueshanpiao@126.com.
Fang Lian, Email: lianfang910@163.com.
Hao Wu, Email: 13901397527@139.com.
Aihong Zhou, Email: zahxwh@163.com.
Fen Wang, Email: wangfenwangfen@hotmail.com.
Xiumei Zuo, Email: zuoxm111@126.com.
Yue Han, Email: anitahy1978@163.com.
Jihui Lyu, Email: lvjihui@139.com.
Dongmei Guo, Email: gdmei0718@163.com.
Jianping Jia, Email: jjp@ccmu.edu.cn.
REFERENCES
- 1.Plante-Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018;265:976–983. [DOI] [PubMed] [Google Scholar]
- 2.Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–1097. [DOI] [PubMed] [Google Scholar]
- 3.Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86:1036–1043. [DOI] [PubMed] [Google Scholar]
- 4.Phull P, Sanchorawala V, Connors LH, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018;25:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finsterer J, Scorza FA, Fiorini AC, et al. The variable phenotype of familial transthyretin-related amyloidosis. Acta Neurol Belg. 2018;120:209–210. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura M, Yamashita T, Ueda M, et al. Neuroradiologic and clinicopathologic features of oculoleptomeningeal type amyloidosis. Neurology. 2005;65:1051–1056. [DOI] [PubMed] [Google Scholar]
- 7.Dowell JD, Fleck JD, Vakili ST, et al. Familial oculoleptomeningeal amyloidosis associated with primary angiitis of the CNS. Neurology. 2007;68:77–78. [DOI] [PubMed] [Google Scholar]
- 8.Garzuly F, Vidal R, Wisniewski T, et al. Familial meningocerebrovascular amyloidosis, Hungarian type, with mutant transthyretin (TTR Asp18Gly). Neurology. 1996;47:1562–1567. [DOI] [PubMed] [Google Scholar]
- 9.Schweitzer K, Ehmann D, Garcia R, et al. Oculoleptomeningeal amyloidosis in 3 individuals with the transthyretin variant Tyr69His. Can J Ophthalmol. 2009;44:317–319. [DOI] [PubMed] [Google Scholar]
- 10.Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016;29(suppl 1):S3–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goren H, Steinberg MC, Farboody GH. Familial oculoleptomeningeal amyloidosis. Brain. 1980;103:473–495. [DOI] [PubMed] [Google Scholar]
- 12.Bevers MB, McGuone D, Jerath NU, et al. Leptomeningeal transthyretin-type amyloidosis presenting as acute hydrocephalus and subarachnoid hemorrhage. J Clin Neurosci. 2016;29:203–205. [DOI] [PubMed] [Google Scholar]
- 13.Douglass C, Suvarna K, Reilly MM, et al. A novel amyloidogenic transthyretin variant, Gly53Ala, associated with intermittent headaches and ataxia. J Neurol Neurosurg Psychiatry. 2007;78:193–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellie E, Camou F, Vital A, et al. Recurrent subarachnoid hemorrhage associated with a new transthyretin variant (Gly53Glu). Neurology. 2001;57:135–137. [DOI] [PubMed] [Google Scholar]
- 15.Fan K, Zhu H, Xu H, et al. The identification of a transthyretin variant p.D38G in a Chinese family with early-onset leptomeningeal amyloidosis. J Neurol. 2019;266:232–241. [DOI] [PubMed] [Google Scholar]
- 16.Hagiwara K, Ochi H, Suzuki S, et al. Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology. 2009;72:1358–1360. [DOI] [PubMed] [Google Scholar]
- 17.Herrick MK, DeBruyne K, Horoupian DS, et al. Massive leptomeningeal amyloidosis associated with a Val30Met transthyretin gene. Neurology. 1996;47:988–992. [DOI] [PubMed] [Google Scholar]
- 18.Jin K, Sato S, Takahashi T, et al. Familial leptomeningeal amyloidosis with a transthyretin variant Asp18Gly representing repeated subarachnoid haemorrhages with superficial siderosis. J Neurol Neurosurg Psychiatry. 2004;75:1463–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakagawa K, Sheikh SI, Snuderl M, et al. A new Thr49Pro transthyretin gene mutation associated with leptomeningeal amyloidosis. J Neurol Sci. 2008;272:186–190. [DOI] [PubMed] [Google Scholar]
- 20.Salvi F, Pastorelli F, Plasmati R, et al. Genotypic and phenotypic correlation in an Italian population of hereditary amyloidosis TTR-related (HA-TTR): clinical and neurophysiological aids to diagnosis and some reflections on misdiagnosis. Amyloid. 2012;19(suppl 1):58–60. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu Y, Takeuchi M, Matsumura M, et al. A case of biopsy-proven leptomeningeal amyloidosis and intravenous Ig-responsive polyneuropathy associated with the Ala25Thr transthyretin gene mutation. Amyloid. 2006;13:37–41. [DOI] [PubMed] [Google Scholar]
- 22.Tawara S, Nakazato M, Kangawa K, et al. Identification of amyloid prealbumin variant in familial amyloidotic polyneuropathy (Japanese type). Biochem Biophys Res Commun. 1983;116:880–888. [DOI] [PubMed] [Google Scholar]
- 23.Maia LF, Magalhaes R, Freitas J, et al. CNS involvement in V30M transthyretin amyloidosis: clinical, neuropathological and biochemical findings. J Neurol Neurosurg Psychiatry. 2015;86:159–167. [DOI] [PubMed] [Google Scholar]
- 24.Obici L, Merlini G. An overview of drugs currently under investigation for the treatment of transthyretin-related hereditary amyloidosis. Expert Opin Investig Drugs. 2014;23:1239–1251. [DOI] [PubMed] [Google Scholar]
- 25.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–249. [DOI] [PubMed] [Google Scholar]
- 26.Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66:2451–2466. [DOI] [PubMed] [Google Scholar]
- 28.Barreiros AP, Post F, Hoppe-Lotichius M, et al. Liver transplantation and combined liver-heart transplantation in patients with familial amyloid polyneuropathy: a single-center experience. Liver Transpl. 2010;16:314–323. [DOI] [PubMed] [Google Scholar]
- 29.Brett M, Persey MR, Reilly MM, et al. Transthyretin Leu12Pro is associated with systemic, neuropathic and leptomeningeal amyloidosis. Brain. 1999;122(Pt 2):183–190. [DOI] [PubMed] [Google Scholar]
- 30.McColgan P, Viegas S, Gandhi S, et al. Oculoleptomeningeal amyloidosis associated with transthyretin Leu12Pro in an African patient. J Neurol. 2015;262:228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urban PP, Hertkorn C, Schattenberg JM, et al. Leptomeningeal familial amyloidosis: a rare differential diagnosis of leptomeningeal enhancement in MRI. J Neurol. 2006;253:1238–1240. [DOI] [PubMed] [Google Scholar]
- 32.Llull L, Berenguer J, Yague J, et al. Leptomeningeal amyloidosis due to A25T TTR mutation: a case report. Neurologia. 2014;29:382–384. [DOI] [PubMed] [Google Scholar]
- 33.Petersen RB, Goren H, Cohen M, et al. Transthyretin amyloidosis: a new mutation associated with dementia. Ann Neurol. 1997;41:307–313. [DOI] [PubMed] [Google Scholar]
- 34.Roe RH, Fisher Y, Eagle RC, Jr, et al. Oculoleptomeningeal amyloidosis in a patient with a TTR Val30Gly mutation in the transthyretin gene. Ophthalmology. 2007;114:e33–e37. [DOI] [PubMed] [Google Scholar]
- 35.Furuya H, Yoshioka K, Sasaki H, et al. Molecular analysis of a variant type of familial amyloidotic polyneuropathy showing cerebellar ataxia and pyramidal tract signs. J Clin Invest. 1987;80:1706–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mascalchi M, Salvi F, Pirini MG, et al. Transthyretin amyloidosis and superficial siderosis of the CNS. Neurology. 1999;53:1498–1503. [DOI] [PubMed] [Google Scholar]
- 37.Klein CJ, Nakumura M, Jacobson DR, et al. Transthyretin amyloidosis (serine 44) with headache, hearing loss, and peripheral neuropathy. Neurology. 1998;51:1462–1464. [DOI] [PubMed] [Google Scholar]
- 38.Liepnieks JJ, Dickson DW, Benson MD. A new transthyretin mutation associated with leptomeningeal amyloidosis. Amyloid. 2011;18(suppl 1):160–162. [DOI] [PubMed] [Google Scholar]
- 39.Uemichi T, Uitti RJ, Koeppen AH, et al. Oculoleptomeningeal amyloidosis associated with a new transthyretin variant Ser64. Arch Neurol. 1999;56:1152–1155. [DOI] [PubMed] [Google Scholar]
- 40.Uitti RJ, Donat JR, Rozdilsky B, et al. Familial oculoleptomeningeal amyloidosis. Report of a new family with unusual features. Arch Neurol. 1988;45:1118–1122. [DOI] [PubMed] [Google Scholar]
- 41.Purrucker JC, Hund E, Hinderhofer K, et al. Doxycycline in ATTRY69H (p.ATTRY89H) amyloidosis with predominant leptomeningeal manifestation. Amyloid. 2013;20:279–280. [DOI] [PubMed] [Google Scholar]
- 42.Ziskin JL, Greicius MD, Zhu W, et al. Neuropathologic analysis of Tyr69His TTR variant meningovascular amyloidosis with dementia. Acta Neuropathol Commun. 2015;3:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suhr OB, Andersen O, Aronsson T, et al. Report of five rare or previously unknown amyloidogenic transthyretin mutations disclosed in Sweden. Amyloid. 2009;16:208–214. [DOI] [PubMed] [Google Scholar]
- 44.Mathieu F, Morgan E, So J, et al. Oculoleptomeningeal amyloidosis secondary to the rare transthyretin c.381T>G (p.Ile127Met) mutation. World Neurosurg. 2018;111:190–193. [DOI] [PubMed] [Google Scholar]
- 45.Koike H, Ikeda S, Takahashi M, et al. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology. 2016;87:2220–2229. [DOI] [PubMed] [Google Scholar]
- 46.Grande-Trillo A, Baliellas C, Llado L, et al. Transthyretin amyloidosis with cardiomyopathy after domino liver transplantation: results of a cross-sectional study. Am J Transplant. 2020;21:372–381. [DOI] [PubMed] [Google Scholar]
- 47.Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015;21:282–292. [DOI] [PubMed] [Google Scholar]
- 48.Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99:1847–1854. [DOI] [PubMed] [Google Scholar]
- 49.Nelson LM, Penninga L, Villadsen GE, et al. Outcome in patients treated with isolated liver transplantation for familial transthyretin amyloidosis to prevent cardiomyopathy. Clin Transplant. 2015;29:1098–1104. [DOI] [PubMed] [Google Scholar]
- 50.Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, Xin Y, Song Z, et al. Tafamidis, a noninvasive therapy for delaying transthyretin familial amyloid polyneuropathy: systematic review and meta-analysis. J Clin Neurol. 2019;15:108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sekijima Y, Tojo K, Morita H, et al. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22:79–83. [DOI] [PubMed] [Google Scholar]
- 53.Benson MD, Kluve-Beckerman B, Zeldenrust SR, et al. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve. 2006;33:609–618. [DOI] [PubMed] [Google Scholar]
- 54.Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369:819–829. [DOI] [PubMed] [Google Scholar]
- 55.Bodin K, Ellmerich S, Kahan MC, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468:93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Higaki JN, Chakrabartty A, Galant NJ, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid. 2016;23:86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]