

Abstract
Background.
Genetic defects of innate immunity impairing intestinal bacterial sensing are linked to the development of Inflammatory Bowel Disease (IBD). Although much evidence supports a role of the intestinal virome in gut homeostasis, most studies focus on intestinal viral composition rather than on host intestinal viral sensitivity.
Aim.
To demonstrate the association between the development of Very Early Onset IBD (VEOIBD) and variants in the IFIH1 gene which encodes MDA5, a key cytosolic sensor for viral nucleic acids.
Methods.
Whole exome sequencing (WES) was performed in two independent cohorts of children with VEOIBD enrolled in Italy (n=18) and USA (n=24). Luciferase reporter assays were employed to assess MDA5 activity. An enrichment analysis was performed on IFIH1 comparing 42 VEOIBD probands with 1527 unrelated individuals without gastrointestinal or immunological issues.
Results.
We identified rare, likely loss-of-function (LoF), IFIH1 variants in eight patients with VEOIBD from a combined cohort of 42 children. One subject, carrying a homozygous truncating variant resulting in complete LoF, experienced neonatal-onset, pan-gastrointestinal, IBD-like enteropathy plus multiple infectious episodes. The remaining seven subjects, affected by VEOIBD without immunodeficiency, were carriers of one LoF variant in IFIH1. Among these, two patients also carried a second hypomorphic variant, with partial function apparent when MDA5 was weakly stimulated. Furthermore, IFIH1 variants were significantly enriched in children with VEOIBD as compared to controls (p=0.007).
Conclusions.
Complete and partial MDA5 deficiency is associated with VEOIBD with variable penetrance and expressivity, suggesting a role for impaired intestinal viral sensing in IBD pathogenesis.
Keywords: Inflammatory Bowel Disease (IBD), innate immunity, melanoma differentiation associated protein 5 (MDA5), pattern-recognition receptors (PRRs), RIG-I-like receptors (RLRs)
INTRODUCTION
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is characterized by spontaneous, chronic or recurring inflammation of the gastrointestinal tract and is hypothesized to result from a dysregulated mucosal immune response to commensal or pathogenic microbes of the intestinal microbiota in a genetically susceptible host1.
IBD has a peak incidence during the 2nd and 3rd decade of life but onset can occur at any age2. Children with very early onset IBD (i.e., diagnosed ≤ 6 years of age; VEOIBD) represent a unique group of IBD patients presenting with distinct clinical characteristics3−6. The observation that IBD displays age-dependent features has led to the hypothesis that the diverse IBD phenotypes occurring across the age spectrum may reflect different disease etiologies. While adult-onset IBD is a common complex disease integrating the effects of host polygenic susceptibility factors and environmental triggers, increasing evidence suggests that VEOIBD more frequently reflects rare pathogenic variants or monogenic defects in a proportion of patients. To date, variants in over 60 causative genes affecting intestinal immune homeostasis have been associated with monogenic IBD via different mechanisms. These include disruption of the intestinal epithelial barrier (e.g., TTC7A deficiency), impairment of bacterial killing (e.g., NOX1 deficiency), hyper- or auto-inflammatory responses (e.g., mevalonate kinase deficiency), dysfunction of T- and B- cells (e.g., Wiskott-Aldrich syndrome), and deficiency of immune inhibitory mechanisms (e.g., IL-10 receptor deficiency)4,7,8.
Impaired bacterial sensing by both cytoplasmic and membrane-bound pattern recognition receptors (i.e., NOD-like and Toll-like receptors, respectively) has also been increasingly linked to the development of IBD7,9. NOD2 variants are one of the main known genetic susceptibility factors for CD10, and monogenic defects deregulating NOD2 downstream signaling (i.e., XIAP, TRIM22, and NPC1 deficiencies) lead to precocious chronic intestinal inflammation11−13. Also, sensing of commensal bacteria by Toll-like receptors (TLRs) is critical to maintain intestinal epithelial homeostasis and to protect against injury. In humans, variants of TLR4 are associated with increased susceptibility to Crohn’s disease14, and biallelic loss-of-function variants in ALPI abrogate LPS-detoxification leading to diminished control of TLR4-dependent inflammatory signals induced by microbiota-derived endotoxins causing severe IBD15.
We herein report the identification, through an unbiased phenotype-driven exome-wide approach, of IFIH1 loss-of-function (LoF) variants in eight unrelated children with VEOIBD. The IFIH1 gene encodes for the MDA5 pattern recognition receptor, i.e., the interferon-induced helicase C domain-containing protein 1, also known as the melanoma differentiation associated protein 5 (MDA5), that acts as a key sensor of pathogen-associated molecular patterns (PAMPs)-containing viral nucleic acids. MDA5 participates in the innate immune response against a large number of viruses including common intestinal pathogens16−18. This observation expands the spectrum of disorders of microbial sensing underlying bowel inflammation.
MATERIALS and METHODS
Patients
Two cohorts of children with VEOIBD were included in this study: one cohort comprising patients referred to the Unit of Pediatric Gastroenterology of the University Hospital of Padova (n=18; patients 1–4) and the other consisting of children submitted to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) from the Johns Hopkins Hospital Pediatric Gastroenterology Clinic (n=23; patients 5, 7, 8) and from the Baylor College of Medicine and Texas Children’s Hospital Gastroenterology Clinic (n=1; patient 6). While patient 7 technically missed the cut-off for a diagnosis of VEOIBD by 1 month, he had severe Eosinophilic Gastrointestinal disease beginning at age 3 years old that did not respond to dietary therapy. The severity and progression of his inflammation toward a pattern consistent with CD, as well as his comorbid autoimmune conditions argued for his inclusion in this cohort.
The medical records of all patients were reviewed to confirm the diagnosis of VEOIBD in agreement with the Porto criteria19. For each patient the following data were gathered: sex, birthdate, growth parameters, date of diagnosis, disease type, extent and behavior based on the Paris classification of pediatric IBD3, eventual relapses, treatment, relevant concomitant medical conditions, and family history.
Informed consent was obtained from the adult participants, and parent or guardian of the children. This study was approved by the Johns Hopkins and Baylor College of Medicine Institutional Review Boards. Patient 1 was also studied under an NIAID IRB-approved protocol after obtaining written informed consent.
Whole Exome Sequencing
To screen for genetic causes of IBD, genomic DNA from children affected by VEOIBD (n=42) were submitted to whole exome sequencing (WES). Each site performed WES utilizing their own in-house pipelines. Uniform filtering and quality parameters were employed across both cohorts to guarantee that the data was harmonized
At University Hospital of Padova, genomic DNA from patients 1, 2, 3, and 4 were sequenced employing the Agilent All Exon V.6 kit (Agilent Technologies, Inc., Santa Clara CA, USA) and HiSeq2500 Illumina sequencer (125-bp paired end sequence mode). Alignment to the reference genome (GRCh37) was performed using the Burrows-Wheeler Alignment (BWA) tool and Genome Analysis Toolkit (GATK). WES data and read alignment analysis were checked for coverage depth and alignment quality employing the Bedtools software package.
At the Johns Hopkins BHCMG, libraries from genomic DNA for patients 5, 7, and 8 were created using the Agilent SureSelect XT kit to capture the total ~52 Mb CCDS exonic regions and flanking intronic regions. Sequencing was performed using the HiSeq2500 Illumina sequencer. Each read was aligned to the 1000 Genomes phase 2 (GRCh37) human genome reference with the BWA version 0.5.10-tpx20 and GATK21 version 2.3–9-ge5ebf34. Variants were then filtered using the Variant Quality Score Recalibration (VQSR) method22. SNVs were annotated by the MQRankSum, HaplotypeScore, QD, FS, MQ, ReadPosRankSum adaptive error model.
WES for patient 6 was performed in the Baylor College of Medicine Human Genome Sequencing Center as previously described23,24. Exome capture utilized the Human Genome Sequencing Center core design (52 Mb, Roche NimbleGen, RRID: nif-0000–31466), and sequencing was performed on the Illumina HiSeq platform. The in-house Mercury pipeline was used for alignment and mapping the human genome reference sequence (GRCh37). Variant calling was performed with ATLAS, with annotation using an in-house pipeline, Cassandra, which incorporates Annotation of Genetic Variants (ANNOVAR)25.
Variant analysis and validation
In the Italian patients, WES sequencing results were first checked for pathogenic variants known to be associated with very early onset inflammatory bowel disease (VEOIBD) (see Supplementary Table 1 for gene list)26. As no known disease-causing variants were identified, all WES “protein changing” variants were next filtered for allele frequency (gnomAD minor allele frequency <1%), considering all potential modes of inheritance, and prioritized according to phenotype overlap, gene function, conservation (phyloP) and in silico prediction scores (CADD, SIFT, PolyPhen2, MutationTaster, and MutationAssessor). Classification was conducted in accordance with the guidelines from the American College of Medical Genetics and Genomics25. In brief, variants were classified as follows: i) pathogenic variants; ii) likely pathogenic variants; iii) variants of uncertain significance; iv) benign; or v) likely benign variants. Sanger sequencing was performed to validate the presence of significant variants and for family segregation.
The Johns Hopkins BHCMG used the PhenoDB Variant Analysis Tool27 to prioritize rare (minor allele frequency (MAF) <1%) functional (missense, nonsense, stop loss, splice site variants, and indels) heterozygous and homozygous variants in each proband. We excluded variants with a MAF > 0.01 in the Exome Variant Server (release ESP6500SI-V2), 1000 Genomes Project, ExAC, gnomAD, and in our in-house BHCMG samples. Next, we selected the genes that were mutated in four or more probands in a homozygous, compound heterozygous, or heterozygous state. Finally, among the genes mutated in four or more probands, we prioritized the genes that had been associated with IBD, Crohn disease and ulcerative colitis by GWAS studies (825 genes as of April 2019; https://www.ebi.ac.uk/gwas/ Accessed 04/25/19). Mutation Significance Cutoff (MSC) were applied to CADD score to predict mutation impact of identified IFIH1 single nucleotide variants using 95% of interval of confidence28,33. Computational 3D-simulations of missense variants were obtained with the HOPE and the Pymol softwares29,30. An IFIH1 gene-specific connectome was generated employing the Human Gene Connectome server (https://lab.rockefeller.edu/casanova/HGC) to set up the distance and degrees of separation between our gene of interest (i.e., IFIH1) and a core of genes associated to very early IBD onset (gene list available in Supplementary Table 4). Gene ranking was established according to HCG-predicted biological distance and connectivity32.
All candidate variants were confirmed by Sanger Sequencing. Primer pairs designed for genomic DNA amplification and sequencing are reported in the Supplementary Material (Supplementary Table 2).
Functional validation of the IFIH1 variant alleles
Dual luciferase reporter assays were performed as previously described, with modifications33. In brief, site-directed mutagenesis was used to generate pcDNA3.1 mammalian expression plasmids encoding the MDA5 mutants that corresponded to the patients’ likely damaging variants, which were confirmed by Sanger dideoxy sequencing. 293T cells were plated at 50,000 cells per well in 48-well plates, one day prior to transfection. Empty vector (EV), wild-type (WT) or mutant MDA5 constructs (20 ng), along with firefly luciferase reporter plasmid driven by human IFN-β1 promoter (100 ng), and a constitutively expressed Renilla luciferase reporter plasmid (10 ng), were transiently transfected using Lipofectamine 2000 (Invitrogen). Cells were simultaneously transfected with or without high molecular weight poly(I:C) (50 ng) at a final concentration of 200 ng/mL. Twenty hours after transfection, cells were lysed for measurement of luciferase activity. Firefly luciferase activity, normalized to Renilla Luciferase activity, was reported as fold increase relative to unstimulated cells. Statistical comparisons were performed using one-way ANOVA with Dunnett’s múltiple comparisons in the Prism 8.2.0 statistical software package (GraphPad).
Western blots to assess expression of the MDA5 variants, normalized to HSP90 as a loading control, were performed in parallel after transient transfections of the constructs into 293T cells, as previously described33. Anti-MDA5 antibody was obtained from Cell Signaling Technology cat #5321. The untagged truncated protein Gln230Ter, which could not be detected by the anti-MDA5 antibody, was subcloned into the N-terminal FLAG-tagged pclNEO backbone using the In-Fusion® HD Cloning Kit from Takarobio. The expression of the truncated protein was assessed by blotting with an anti-DYKDDDDK (FLAG) Tag antibody (L5 clone from Biolegend).
Enrichment Analysis
An enrichment analysis was performed on IFIH1 using 42 probands with VEOIBD and 1527 unrelated individuals recruited by the Baylor-Hopkins Center for Mendelian Genomics. The phenotype of the controls was assessed and no known gastrointestinal or immunologic were reported. A contingency 2×2 table containing the number of individuals presenting rare (MAF<0.5%) single nucleotide variants (missense, nonsense, stop loss and splicing) was built. Fisher’s exact test was determined and p<0.05 was considered as statistically significant34. A Principal Component Analysis was performed to assess the ethnicity of cases and controls in the US cohort.
All data are incorporated into the article and its online supplementary material.
RESULTS
BIALLELIC IFIH1 COMPLETE LOSS-OF-FUNCTION VARIANTS IN THE INDEX PATIENT WITH VEOIBD AND IMMUNODEFICIENCY
Clinical presentation of the index patient
Patient 1 is a female born to first degree cousins originating from Morocco (Figure 1a, Table 1). Starting from 20 days of life, she presented with persistent vomiting and diarrhea that progressively worsened. When admitted to the emergency department at 1 month, the patient manifested a high output inflammatory diarrhea (stool output > 100 gr/kg/d, calprotectin > 2100 ug/g) with severe dehydration and transient acute kidney injury. Gastrointestinal endoscopy, performed at 2 months, showed absence of duodenal folds and a nonspecific colitis (Figure 1b–c). Duodenal, ileal and colonic histological specimens were consistent with an immune-mediated enteropathy (Figure 1d–f), while electron microscopy did not reveal ultrastructural enterocyte anomalies (data not shown). Anti-enterocyte and anti-harmonin antibodies were not detected. Therefore, at 3 months of life, methylprednisolone and sirolimus were started with rapid and complete resolution of gastrointestinal symptoms and growth recovery. At 5 months, while on steroid tapering, the patient experienced a relapse of enteritis which responded to transient corticosteroid re-induction. At the last evaluation, the patient was 3 years old and asymptomatic on sirolimus mono-therapy. Upper and lower GI endoscopy demonstrated a mild reduction of the villous height/crypt depth ratio in the duodenum and few apoptotic bodies with increased mitotic activity and Paneth cell metaplasia in colonic specimens (data not shown).
Figure 1. Phenotypic and genetic characterization of patient 1 with rare homozygous IFIH1 LoF variants.
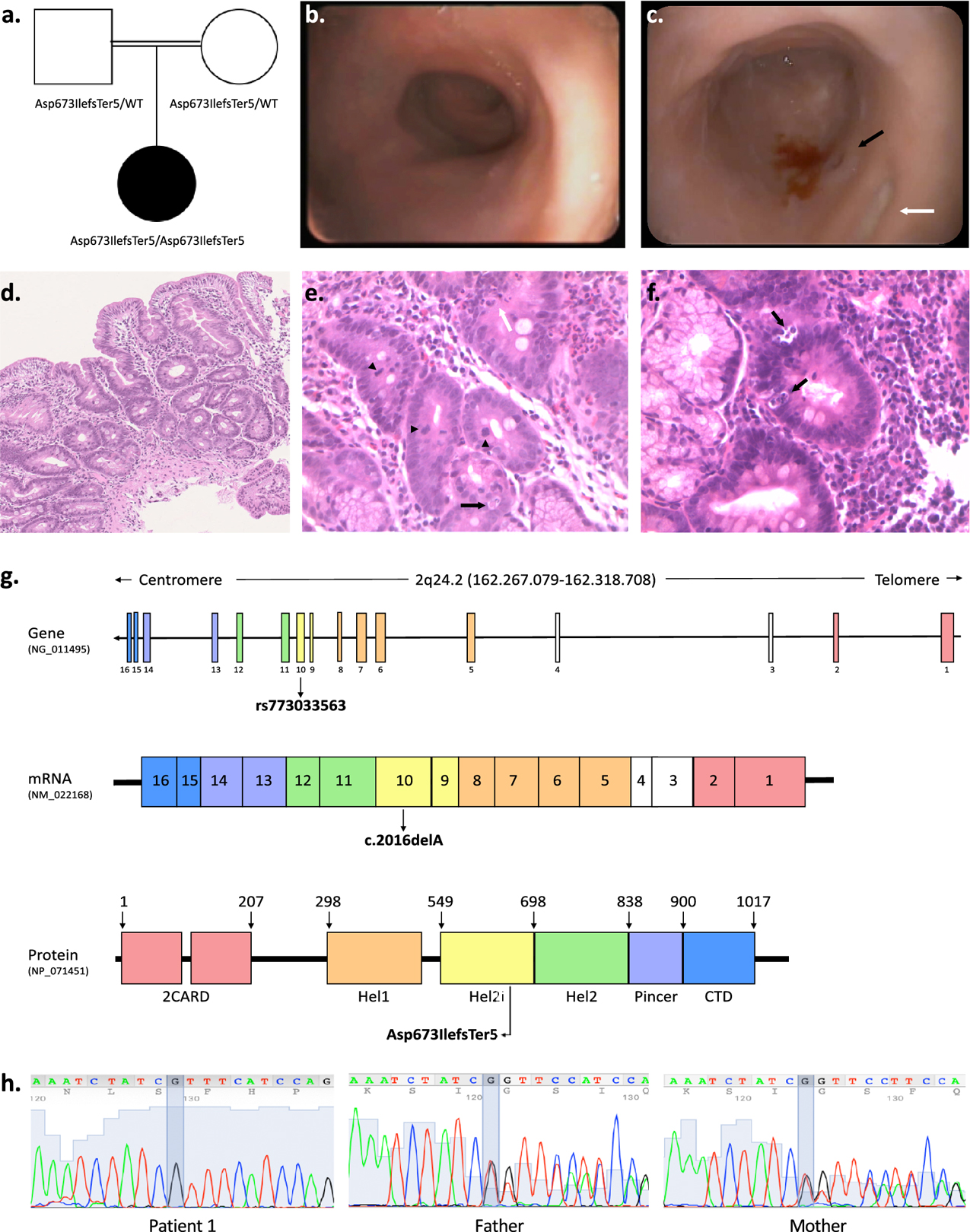
a. Pedigree of patient 1 indicating parental consanguinity and IFIH1 genotypes within the family. b. Upper gastrointestinal endoscopy showing absent duodenal folds with mucosal atrophy. c. Representative colonoscopy image of the left colon showing reduced haustral folds, a bleeding ulcer surrounded by mucosal inflammation (black arrow) and a fibrin-coated ulcer (white arrow). d-f. Duodenal mucosal histology (H&E; d 10x, e-f 20x) showing villous atrophy, crypt distortion, focal cryptitis (white arrow), mucin depletion, apoptotic bodies (black arrows), increased mitotic activity (arrow heads). g. Schematic of the human IFIH1 gene (NG_011495), mRNA (NM_022168) and protein (MDA5; NP_071451) showing the location of the homozygous frameshift deletion identified in patient 1 (rs773033563, c.2016delA, p. Asp673IlefsTer5). MDA5 is a 1025-residue protein (domain boundaries are marked with amino acid coordinates). The N-terminal of the protein contains two tandem caspase activation recruitment domains (2CARD) that activate MAVS. The central helicase domain is responsible for RNA-dependent ATP hydrolysis; it is made of two RecA-like domains (Hel1 and Hel2) and of the intervening Hel2i domain which facilitates the recognition of dsRNA. The Pincer domain is required for structural support and activation of the ATPase core of the protein. The C-terminal domain (CTD) is involved in dsRNA binding. h. Confirmatory Sanger sequencing demonstrating variant segregation within the family.
Table 1.
Synthesis of the clinical and genetic features of the nine patients carrying IFIH1 variants.
| PATIENT 1 | PATIENT 2 | PATIENT 3 | PATIENT 4 | PATIENT 5 | PATIENT 6 | PATIENT 7 | PATIENT 8 | |
|---|---|---|---|---|---|---|---|---|
| CLINICAL FEATURES | ||||||||
| Sex | F | M | F | M | F | F | M | M |
| Origin | Moroccan | Romanian | Hungarian | Albanese | Caucasian/Peruvian | Colombian/El Salvadorian | Caucasian | Caucasian |
| Intestinal phenotype * | IME | CD (A1a,L3,B1,G1)* | CD (A1a,L3,B1,G1)* | IBDU, colonic NLH | UC (E4,S1)* | UC (E4,S1)* | CD (A1bL3B1G 1)* | CD (A1a,L2,B 1,G1)* |
| Age at onset | 20 days | 6 months | 2 years | 3 months | 3 years | 1.5 years | 6 years | 2 years |
| Age at diagnosis | 2 months | 1 year | 2 years | 8 months | 4 years | 2 years | 6 years | 4 years |
| Infection(s) prior to IBD onset | + (CMV) | + (CMV) | - | - | - | - | - | - |
| Recurrent/severe infections | + | - | - | - | - | - | - | - |
| Other immune-mediated diseases | - | - | - | - | PSC | - | AIH, JIA, Psoriasis | - |
| Family history of IBD | - | - | - | - | + | - | - | + |
| IFIH1 COMPLETE LoF VARIANTS | ||||||||
| Zygosity | Homo | Het | Het | Het | Het | Het | Het | Het |
| Inheritance | P,M-both unaffected | P-unaffected | M-unaffected | M-unaffected | P-affected | P-unaffected | P-unaffected | P-unaffected |
| Nucleotide change | c.2016delA | c.2035_2036delTT | c.2807+1G>A | c.688C>T | c.1879G>T | c.1879G>T | c.2807+1G>A | c.2035_2036delTT |
| Amino acid change | p.Asp673IlefsTer5 | p.Leu679IlefsTer3 | Skipping of exon 14 | p.Gln230Ter | p.Glu627Ter | p.Glu627Ter | Skipping of exon 11 | p.Leu679IlefsTe r3 |
| gnomAD frequency § | 2.99e-4 | 1.06e-4 | 6.34e-3 | 2.79e-5 | 3.19e-3 | 3.19e-3 | 6.34e-3 | 1.06e-4 |
| CADD; MSC-CADD Impact Pred | NA | NA | 24; high | 37; high | 40; high | 40; high | 24; high | NA |
| HYPOMORPHIC AND NORMALLY FUNCTIONING IFIH1 VARIANTS | ||||||||
| Zygosity | - | Het | Het | - | - | Het | Het | Het |
| Inheritance | - | M-unaffected | 'P-unaffected | - | - | M-unaffected | P-unaffected | P-unaffected |
| Nucleotide change | - | c.1694G>T | c.2105C>T | - | - | c.C373A | c.C2105T | c.A1909G |
| Amino acid change | - | p.Cys565Phe | p.Thr702Ile | - | - | p.Leu125Met | p.Thr702Ile | p.Lys637Glu |
| gnomAD frequency § | - | 5.45e-5 | 2.34e-3 | - | - | 1.99e-5 | 2.34e-3 | 1.2e-5 |
| Protein function | - | Hypomorphic | Normal functioning | - | - | Hypomorphic | Normal functioning | Hypo morphic |
| CADD; MSC-CADD Impact Pred | - | 19; high | 10; low | - | - | 20; high | 10; low | 22; high |
* IBD phenotype according to the Paris Classification of Pediatric IBDs (Levine et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis 2011; 17(6):1314–21).
The gnomAD frequency is taken from gnomAD v2.1.1.
AIH: autoimmune hepatitis. CD: Crohn’s disease. CMV: Cytomegalovirus. IBDU: Undetermined Inflammatory Bowel Disease. IME: immune-mediated enteropathy. JIA: juvenile idiopathic arthritis. NLH: nodular lymphatic hyperplasia PSC: primary sclerosing cholangitis. UC: ulcerative colitis. URI: Upper Respiratory Infection.
LoF: loss-of-function. Hom: Homozygous. Het: Heterozygous. P: paternal. M: maternal. NA: not available.
Patient 1 also presented with multiple infections during the first year of life. Soon after birth she had an episode of early-onset neonatal sepsis. During the first three months of life, before the starting of any immunosuppressive medication, the patient experienced a primary post-natal CMV infection responsive to ganciclovir treatment (albeit with no amelioration of ongoing gastrointestinal symptoms), and two urinary tract infections due to P. aeruginosa. At four months, while on treatment with sirolimus and prednisone, patient 1 suffered from moderate-severe interstitial pneumonia due to Parainfluenza virus type 3, which required prolonged hospitalization and oxygen therapy. Moreover, at six months she experienced a central venous catheter-related sepsis due to Enterococcus faecium and Staphylococcus epidermidis.
Immunological workup at 30 days of life revealed: normal full blood count and immunoglobulin levels, increased memory CD4+ T cells (CD45RO+) and activated T cells (CD3+DR+), most probably due to active CMV infection. Subsequently, while on sirolimus monotherapy, persistent elevations in monocytes (range: 1.05–2.27 × 109/L) and lymphocytes (range: 8.47–11.77 × 109/L) were observed (see Supplementary Table 3).
No history of IBD or immune deficiency was reported in the family and both parents were healthy.
Whole exome sequencing and functional characterization of the IFIH1 variant identified
WES was performed and 84 genes known to be associated with IBD were analyzed (gene list available in Supplementary Table 1). No pathogenic or likely pathogenic variants in these genes were identified. Because of parental consanguinity, neonatal onset IBD, and recurrent infections, a monogenic immunological disorder with autosomal recessive inheritance was principally suspected. Further WES analysis (see Materials and Methods) highlighted the presence of a likely pathogenic homozygous variant (rs773033563, c.2016delA) in IFIH1, a gene reported to be associated with severe viral respiratory infections in children35. The c.2016delA frameshift deletion results in the insertion of a premature stop codon in exon 10 (p.Asp673IlefsTer5). This alteration is predicted by NMDEscPredictor36 to undergo nonsense-mediated decay. Any residual truncated MDA5 protein would lack domains required for RNA-dependent ATP hydrolysis, dsRNA binding, and MDA5 oligomerization (i.e., Hel2, Pincer, and CTD) important for protein function (Figure 1g, Table 1). To date, rs773033563 has been reported only in heterozygosity, mainly in European individuals (GnomAD allele frequency of 0.0002992), and is not described in the Inflammatory Bowel Disease Exomes Browser. Sanger sequencing confirmed the presence of this variant in both IFIH1 alleles of the proband and showed that both unaffected parents were heterozygous carriers (Figure 1h, Table 1).
To further explore the hypothesis that the IFIH1 predicted LoF variant had functional consequences, a standard dual luciferase reporter assay was used to test for activity of the allele driven by the IFNB1 promoter. As expected, only a small signal was seen at baseline (data not shown), but following stimulation by transfecting in poly(I:C), a synthetic ligand for MDA5 which mimics double-stranded RNA, robust activity was induced by wild-type MDA5. In contrast, the p.Asp673IlefsTer5 mutant MDA5 failed to activate the IFNB1 promoter (Figure 2a), a result suggestive of impairment of MDA5-mediated IFNβ1 production and increased cell vulnerability to viral infections16,33,37. Western blot performed in parallel showed that the p.Asp673IlefsTer5 mutant encoded a truncated MDA5 protein migrating at the predicted mass of ~ 81 KDa (Figure 2b).
Figure 2. Functional characterization of the IFIH1 variants identified in children with VEOIBD.
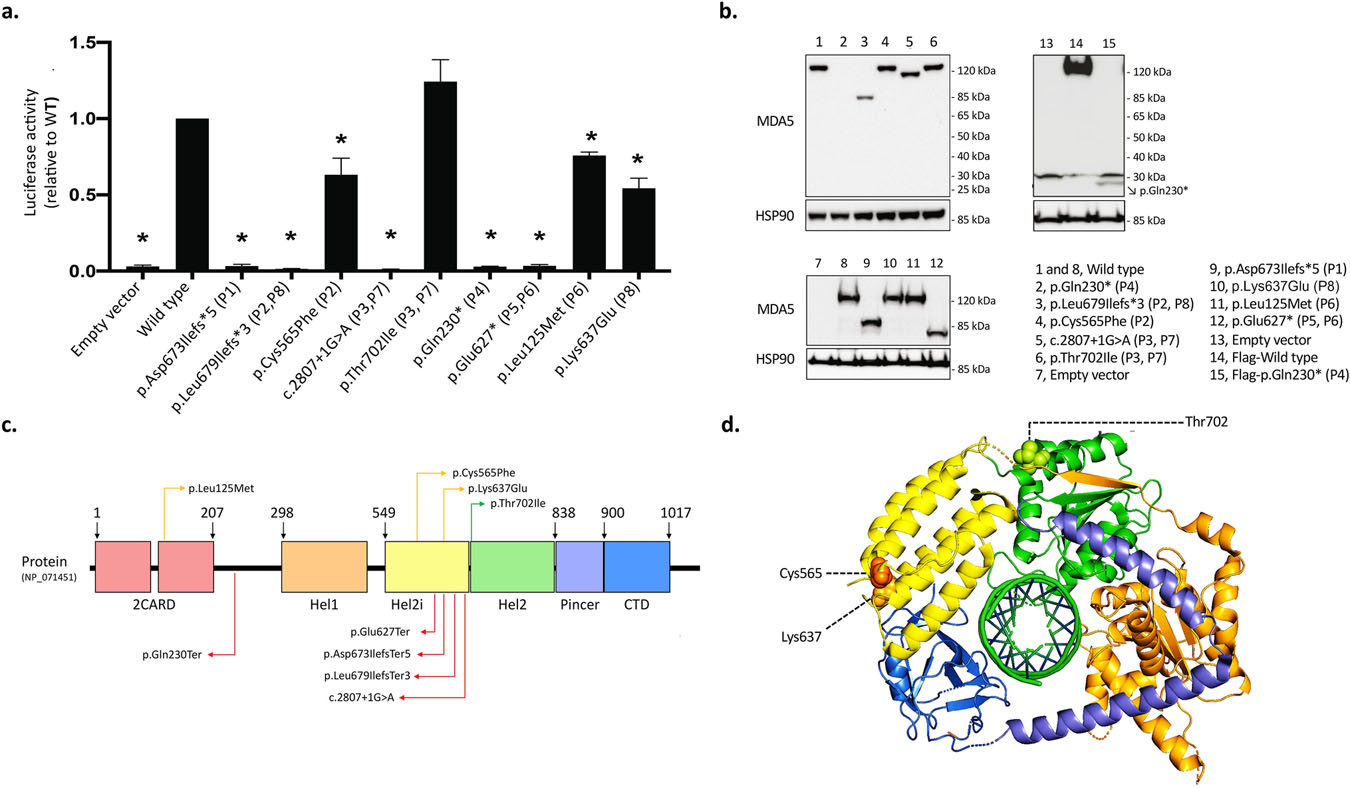
a-b. Functional activity of IFIH1 variants. Patient(s) carrying the tested variant are indicated in parentheses. a. Relative change in normalized luciferase activity driven by the IFNB1 promoter reporter construct. Cells were co-transfected with 20 ng WT or mutant MDA5 and with 200 ng/mL poly(I:C). Data show means ± SD from three independent experiments. *p <0.05, by one-way ANOVA. Unstimulated cells showed little activity at baseline (data not shown). b. Representative western blots for MDA5 protein expression and HSP90 loading control, of the mutant MDA5 constructs used in (a) as well as controls (wild type and empty vector). The N-terminally tagged p.Gln230* MDA5 mutant protein was detected using an anti-Flag antibody (lane 15). c. Schematic representation of MDA5 along with the variants identified in our cohort of children with VEOIBD. Complete LoF variants, partial functioning variants, and variants with no functional impact are indicated with red, orange and green arrows, respectively. d. Top view of the cryo-EM structure of the MDA5-dsRNA complex in cartoon representation (PDB ID: 7JL0)30,74. Individual domains are colored as in (c). No modelling template is available for the N-terminal of the protein containing the 2CARD domain. The amino acid residues affected by missense variants (with the exception of Leu125) are represented with spherical molecules. A 3D-animation is available in the supplementary material (Supplementary Video 1 with legend in the Supplementary Material).
BIALLELIC AND MONOALLELIC IFIH1 VARIANTS IN A COHORT OF CHILDREN WITH VEOIBD
Identification and clinical presentation of IFIH1 variant carriers
In parallel, we analyzed WES data in a cohort of patients (n= 41) with genetically unsolved VEOIBD, i.e., without pathogenic variants in genes known to be associated with monogenic IBD (see gene list in Supplementary Table 1). Of these, 17 children were from the University Hospital of Padova, and the remaining (n = 24) were recruited independently as part of the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) project. Collectively, we identified rare, likely LoF, IFIH1 variants in seven additional patients with genetically unsolved VEOIBD. In all cases IFIH1 variants were inherited from a heterozygous parent.
Of the cases identified (Table 1), three children belonged to the Padova cohort (patients 2, 3 and 4). As established by family segregation analysis, two of these patients carried biallelic variants in IFIH1 (patients 2: p.Leu679IlefsTer3 and p.Cys565Phe; patient 3: c.2807+1G>A and p.Thr702Ile), and one a monoallelic variant (patient 4: p.Gln230Ter). The remaining four children belonged to the cohort within the Baylor-Hopkins Center (patients 5, 6, 7 and 8). Three of these patients carried two IFIH1 variants either in trans (patient 6: p.Glu627Ter and p.Leu125Met) or in cis (patient 7: c.2807+1G>A and p.Thr702Ile; patient 8: p.Leu679IlefsTer3 and p.Lys637Glu), and one carried a single IFIH1 variant in heterozygosity (patient 5: p.Glu627Ter).
We predicted the biological distance and the connection route between the IFIH1 gene and a set of core genes correlated with VEO-IBD. Among the 84 genes known to be associated with VEOIBD we identified 37 genes with 2 degrees of separation with IFIH1 and 19 out of 37 genes with a significant BRP (best reciprocal p-value <0.05) (Supplementary Table 4), indicating indicates a close biological distance and connectivity between IFIH1 and other genes associated with VEO-IBD.
Clinically, among the seven children identified (four males and three females), CD was diagnosed in four subjects, UC in two, and IBDU with diffuse colonic nodular lymphatic hyperplasia in one (Table 1). Despite the different IBD phenotypes, all children shared the following clinical features: IBD onset within the first 6 years of age (range 3 months-6 years, median 24 months), prevalent and extensive colonic localization of inflammation, non-stricturing non-penetrating CD behavior, and absence of perianal disease. Two children experienced concomitant extra-intestinal immune-mediated disorders (primary sclerosing cholangitis in patient 5; autoimmune hepatitis, juvenile idiopathic arthritis, and psoriasis in patient 7). Two subjects (patients 5 and 8) had a positive family history of IBD (Table 1). The father of patient 5, carrying the same IFIH1 LoF variant, was diagnosed with UC during adulthood, and her deceased paternal grandmother, with an unknown genetic profile, had CD. Patient 8’s paternal great grandfather (unknown genetic profile) had CD. Patients were treated with different pharmacological treatments (aminosalicylates, corticosteroids, azathioprine, 6-mercaptopurine, methotrexate, infliximab, adalimumab) with variable responses and heterogeneous clinical courses. Four out of seven subjects (patients 2, 3, 4 and 8) reached IBD remission with first line pharmacological treatments (mesalamine, prednisone, azathioprine, 6-mercaptopurine, methotrexate). Three out of eight patients required a biological treatment to gain IBD control (infliximab and adalimumab in patients 5, infliximab in patient 6 and 7). One out of seven patients (patient 6), having failed multiple treatment regimens, required total colectomy at 4 years of age. One out of seven patients (patient 7) failed multiple treatments but therapy was hampered by concomitant immune-mediated disorders as well as by infectious complications occurring during immunosuppression. Clinical characteristics are detailed in Supplementary Material.
Functional characterization of the identified IFIH1 variants
The rare IFIH1 variants identified in patients 2 to 8 were all tested with the luciferase reporter assays driven by the IFNB1 promoter. Upon functional testing, all seven subjects (patients 2 to 8) were found to be heterozygous carriers of one monoallelic, complete LoF variant in IFIH1. Moreover, among these, patients 2 and 6 were carriers of a second hypomorphic variant, with partial function apparent when MDA5 was weakly stimulated. In patients 3, 7 and 8, the second variant either did not affect protein function or was on the same allele (Figure 2a and 2c, Figure 3, Table 1).
Figure 3. Visual summary of the IFIH1 variants identified in the seven children with partial MDA5 deficiency and VEOIBD.
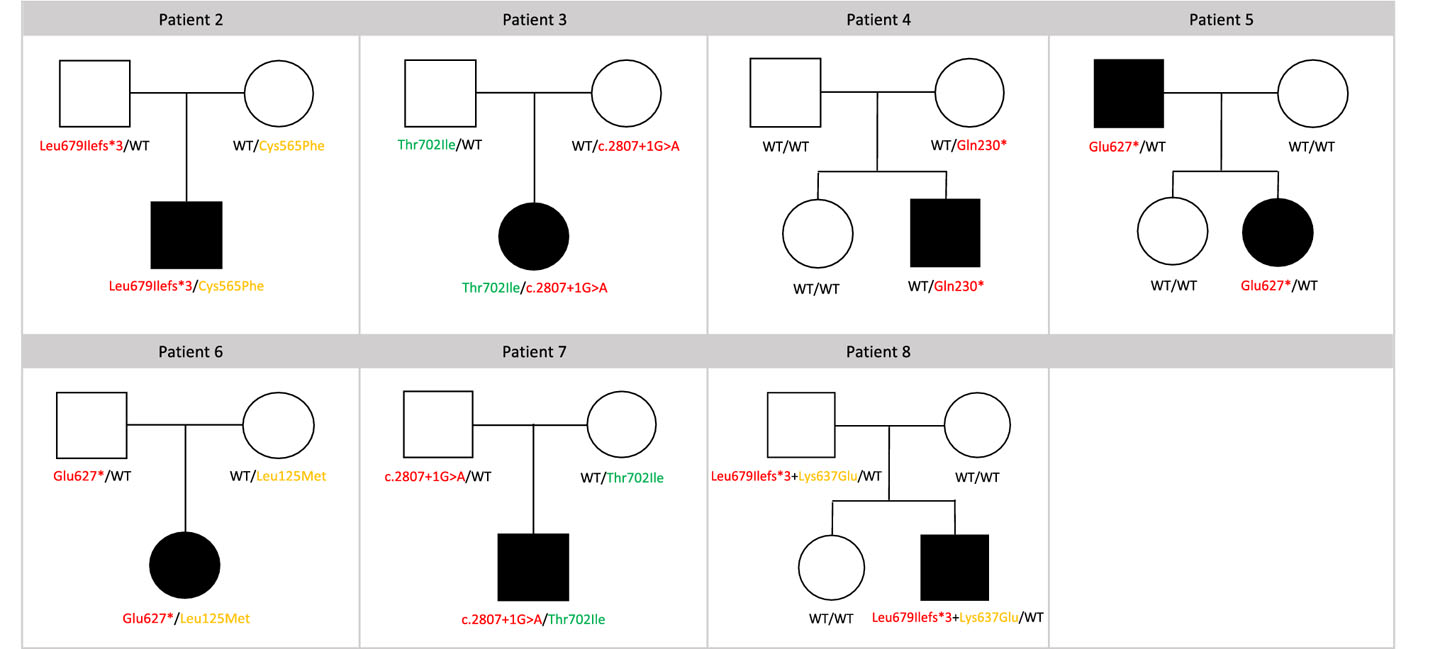
Pedigree indicating genotypes and variant segregation within the family are reported for each patient with partial MDA5 deficiency (patients 2 to 8). Complete LoF variants, partial functioning variants, and variants with no functional impact are indicated with red, orange and green arrows, respectively. WT, wild-type.
Among the above variants, four completely abolished MDA5 functional activity: p.Leu679IlefsTer3 (patients 2 and 8), c.2807+1G>A (patients 3 and 7), p.Gln230Ter (patient 4), and p.Glu627Ter (patient 5 and 6) (Figure 2a and 2c, Table 1). The frameshift variant p.Leu679IlefsTer3 and the two nonsense variants p.Gln230Ter and p.Glu627Ter are predicted to either undergo nonsense-mediated decay and/or to generate truncated proteins of ~75 kDa, ~30 kDa, and 69 kDa, respectively (Figure 2b). The p.Gln230Ter protein was expressed at the expected size but a much lower level than WT MDA5, suggesting protein instability; however, the other mutant proteins were overexpressed at the expected sizes and were missing domains required for RNA-dependent ATP hydrolysis, dsRNA binding, and MDA5 oligomerization (Figure 2b). The c.2807+1G>A variant is a rare donor splice-site variant with a predicted deleterious effect on splicing (NNSplice −0.98938, GeneSplicer −2.51739, MaxEntScan −8.18240) and protein function (CADD-PHRED 24/20), resulting in skipping of exon 14 and production of a non-functional protein lacking part of the Hel2 domain.
Three missense variants (p.Cys565Phe in patient 2, p.Leu125Met in patient 6, and p.Lys637Glu in patient 8) resulted in MDA5 partial functional impairment (Table 1, Figure 2a, 2c and 2d, Supplementary Video 1 with legend in the Supplementary Material) that normalized when poly(I:C) was increased to 1 μg/mL (data not shown). The p.Cys565Phe variant disrupts the cysteine bridge within the Hel2i domain, destabilizing MDA5 stability (Figure 2d, Supplementary Videos 2 and 3 with legends in the Supplementary Material)29,30. The p.Leu125Met variant substitutes a non-polar aliphatic amino acid with a bigger sulphur-containing amino in a highly conserved position of the CARD2 domain29. The p.Lys637Glu introduces a negative charged amino acid instead of a positive charged one, disrupting the ionic interactions and the hydrogen bonds driven by the original wild-type residue (Figure 2d, Supplementary Video 4 with legend in the Supplementary Material)29,30.
Despite different amino acid size and hydrophobicity, the variant p.Thr702Ile (patients 3 and 7), located in a non-conserved position of the Hel2 domain, did not result in any significant functional impairment of MDA5 (Table 1, Figure 2a, 2c and 2d).
ENRICHMENT ANALYSIS
Our enrichment analysis identified 97 rare single nucleotide variants in IFIH1 among 1527 individuals in whom exome sequencing was performed by the Baylor-Hopkins Center for Mendelian Genomics for indications other than gastrointestinal or immunological disorders. A statistically significant difference was observed in the burden for IFIH1 variants when comparing the patient group to the control group (p = 0.0079, Fisher’s exact test) (Table 2). Principal Component Analysis supports that this difference was not due to different ethnic background of cases and controls (Supplementary Figure 2).
Table 2.
A contingency 2x2 table displaying the statistical significance of enrichment of IFIH1 single nucleotide variants (missense, nonsense, stop loss and/or splicing) in cases with VEOIBD (n=42) compared to unaffected controls (n=1527). p-value of Fisher’s Exact test (FET) and Odds Ratio (OR) with a 95% confidence interval. p-value < 0.05 and OR > 1 have been considered statistically significant.
| Group | VEOIBD Samples | Control Samples | p(FET) | OR (95% CI) |
|---|---|---|---|---|
| with IFIH1 rare variants | 8 | 97 | 0.0079 | 3.47 (1.56 – 7.70) |
| without IFIH1 rare variants | 34 | 1430 |
DISCUSSION
Employing WES, we found mono- or biallelic variants in IFIH1 in 8 patients with VEOIBD. The nine variants discovered were overexpressed and tested for ability to drive luciferase expression from an IFNB1 promoter after stimulation with intracellular poly(I:C). Five abolished signaling and three were hypomorphic. Some of these findings were supported by previous work (Supplementary Table S3 in reference33). Our results were obtained in parallel and independently in two cohorts of patients with VEOIDB, one enrolled in Northern Italy and the second in North America. We also show that the population of children affected by VEOIBD is enriched in IFIH1 variants, as compared to the overall general population. These results indicate a role for MDA5 impairment as a risk factor for VEOIBD.
MDA5 is a ubiquitously-expressed pattern recognition receptor (PRR) that acts as a key sensor of pathogen-associated molecular patterns (PAMPs)-containing nucleic acids. Together with RIG-I, MDA5 belongs to the class of RIG-I-like receptors (RLRs), a group of cytoplasmic RNA helicases with caspase activation recruiting domains (CARDs) that are critical for host antiviral responses. MDA5 and RIG-I mainly recognize long and short double-stranded viral RNAs, respectively, but are also involved in the detection of viruses with single-stranded RNA and, in some studies, even double-stranded DNA16,41−45. Upon recognition of viral nucleic acids, MDA5 and RIG-I individually interact with the mitochondrial antiviral-signalling protein (MAVS), which serves as the essential adaptor for RLR signal transduction (i.e., RLR/MAVS pathway). Once stimulated, MAVS activates the transcription factors NF-kB and IRF3/IRF7 which induce the transcription of type I and III IFN genes41,46,47. Autocrine and paracrine IFN stimulation subsequently promotes transcription of hundreds of IFN-stimulated genes (ISGs), several of which encode proteins with direct antimicrobial functions16,17,48.
In humans, rare variants in IFIH1 have been implicated in monogenic autoinflammatory disorders as well as in extreme susceptibility to common viral infections. Aicardi-Goutieres Syndrome type 7 (AGS7, OMIM #615846)49, Singleton-Merten Syndrome 1 (SGMRT1, OMIM #182250)50 and early onset systemic lupus erythematosus with IgA deficiency51 are autoinflammatory conditions caused by IFIH1 heterozygous gain-of-function variants resulting in an enhanced or constitutively activated MAVS pathway. Vulnerability to viral infections has been associated with IFIH1 mono- and bi-allelic LoF variants leading to severely impaired response to polyI:C and recurrent life-threatening respiratory tract infections during infancy (for a summary of the variants and clinical phenotypes reported, see Supplementary Table 5 and Supplementary Figure 1)33,37,52. Also, polymorphic IFIH1 variants have been implicated in a number of common autoimmune and immune-mediated conditions (e.g., type 1 diabetes, systemic lupus erythematosus, multiple sclerosis, and sclerosing cholangitis)53−56. How these risk alleles relate to the development of these diseases is not clear, but may involve chronic induction of type I IFNs, which then initiate or enhance autoinflammatory and autoimmune responses16.
IFIH1 has been previously associated with IBD (both CD and UC) by GWAS signals56 (Supplementary Figure 1), but it has not been described as having a pathogenic role in IBD or VEOIBD. Support for a such role derives from different lines of evidence highlighting the role of the RLR/MAVS pathway in the maintenance of gut homeostasis. First, MDA5 and/or RIG-I signalling through MAVS have a functionally important role in limiting gastrointestinal infections by rotavirus and norovirus, both of which are leading global causes of morbidity and mortality in young children and immunocompromised individuals18,57, supporting the importance of this pathway for anti-viral responses in the intestine. Second, animal studies show that mice lacking either RIG-I or MAVS have increased susceptibility to dextran sulphate sodium (DSS)-induced colitis58−60 and to total body irradiation- or chemotherapy-induced intestinal damage61. A possible explanation for this susceptibility is that a defect in RLR/MAVS signalling results in failure of an intestinal insult to elicit the production of cytokines, antimicrobial peptides, and intracellular signals that are important for immune surveillance of the intestinal microbiota and for maintaining the integrity of the intestinal barrier58,59,61−63. This possibility is supported by data showing that the RLR/MAVS pathway is activated by RNA species derived from intestinal commensal bacteria and thus could be relevant in regulating mucosal immune responses towards different kinds of microorganisms59. Third, MDA5 is also critical for the production of type III interferon (IFN-λ) in response to viral dsRNA33,64. Recently, it has been shown that IFN-λ response to commensal enteric viruses in mice protects against intestinal inflammation by modifying transcriptional and non-translational neutrophil responses65. Fourth, a pronounced downregulation of RIG-I has been observed within the epithelial layer of the ileum in patients with CD66,67 and a rare variant in IFIH1 (p.Ile923Val) has been significantly associated with UC by fine mapping68 (Supplementary Figure 1). Lastly, by connectome analysis we show that IFIH1 and, consequently, the RLR/MAVS pathway is closely biologically related to other pathways implicated in the pathogenesis of VEO-IBD.
Patient 1 carries a homozygous IFIH1 variant resulting in a complete MDA5 deficiency and experienced a neonatal onset, life-threatening, pan-gastrointestinal immune-mediated enteropathy, in addition to multiple viral and bacterial infectious episodes throughout the first year of life. Viral infections were caused by known viral targets of the MDA5 sensor33,43,45 and bacterial superinfections have also been described in other homozygous MDA5-deficient patients33,37. Moreover, a role of MDA5 in the induction of immune responses during bacterial septicaemia has been recently hypothesized69. Notwithstanding the continuation of the immunosuppressive treatment, patient 1 did not present with any other infectious episode beyond the first year of age. Similarly, the homozygous MDA5-deficient patients reported by Lamborn et al. (2017)33 and by Asgari et al. (2017)37 experienced a spontaneous decrease in the incidence of infections beyond infancy. This phenomenon, as in other defects in innate immunity, might be explained by the development of adaptive antigen-specific lymphocyte responses that can mitigate the impairment of innate immunity over time 70−72. Another possible explanation for the decreased incidence of infections with age include the partial redundancy among intracellular viral sensors and cytosolic DNA receptors16,17,48,73.
In comparison to patient 1, the children with a partial MDA5 deficiency, here described, displayed a milder gastrointestinal VEOIBD phenotype with clinical manifestations presenting beyond the neonatal period and did not experience recurrent or severe infections. These differences may reflect variations in allelic dosage and are consistent with previous findings in which patients with complete MDA5 deficiency presented with life-threatening bacterial and viral infections (n=3), while patients with partial MDA5 deficiency (due to monoallelic LoF variants) suffered mostly from common viral infections (n=7) (Supplementary Table 5 and Supplementary Figure 1)33,37,52. The absence of an intestinal phenotype among these previously reported patients could be due to the fact that eight out of the ten patients were identified in a cohort of children with respiratory failure caused by a common viral respiratory infection, resulting in a marked selection bias.
Among our patients carrying a monoallelic complete LoF variant (patients 2–8), the presence of other genetic or environmental factors influencing the phenotype needs to be postulated to account for the incomplete penetrance of the variants. Indeed, in the majority of these patients, the variants were inherited by heterozygous unaffected parents and, in some cases, the predicted LoF IFIH1 variants occurred at a relatively high frequency (Table 1). Intriguingly, in two out of seven patients (patient 2 and 6), we identified a second rare IFIH1 variant in trans with both variants contributing to functional impairment of MDA5. Moreover, a dominant negative effect of IFIH1 LoF variants has been previously reported for the c.2807+1G>A (patients 3 and 7) and the c.1879G>T (patients 5 and 6) variants as mutant MDA5 isoforms might interfere with wild-type MDA5 enzymatic activity and protein stability37. Finally, the role of IFIH1 LoF variants as contributing factors with incomplete penetrance in the pathogenesis of VEOIBD is supported by our enrichment analysis that demonstrated a higher prevalence of rare IFIH1 variants in our cohort, as compared to a large control group.
Although the study has several limitations including the absence of in vivo studies exploring the causal relationship between MDA5 deficiency and bowel inflammation, our data support the possibility that MDA5 deficiency contributes to VEOIBD with incomplete penetrance. We propose that the compromised microbial sensing impairs MAVS activation and interferon production leading to impairment of innate immune defences and chronic intestinal inflammation. The incomplete penetrance may be related to several factors such as allele dosage (i.e., monoallelic vs. biallelic IFIH1 variants), age (i.e., major relevance of innate immunity during the first years of life33,71) and environmental factors (i.e., occurrence of infectious triggers). Additional genetic contributing factors (e.g., variants in genes involved in innate immune responses) could also modify the risk of developing IBD but further studies are needed to test this hypothesis.
In conclusion, the results herein reported support that MDA5 deficiency causes a novel primary defect of innate immunity with autosomal inheritance and variable penetrance characterized by increased susceptibility to infections and VEOIBD, and suggest a role for impaired viral sensing in IBD pathogenesis.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge the assistance of Ms. Helen Matthews. We thank the patients and their families for participating in the research study.
Grant support
The work was funded by a grant from Fondazione Città della Speranza ONLUS, the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, and the Baylor-Hopkins Center for Mendelian Genomics (NHGRI UM1 HG006542).
Abbreviations
- CD
Crohn’s Disease
- IBD
Inflammatory Bowel Disease
- IFN
Interferon
- LoF
Loss-of-function
- SNVs
Single Nucleotide Variants
- UC
Ulcerative Colitis
- IBDU
Undetermined Inflammatory Bowel Disease
- VEOIBD
Very Early Onset Inflammatory Bowel Disease
- WES
Whole Exome Sequencing
Footnotes
Competing interests
Baylor College of Medicine and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), formerly the Baylor Miraca Genetics Laboratories, which performs CMA and clinical ES. J.R.L. serves on the Scientific Advisory Board of the BG. J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. A.L. has a share in ownership of Research & Innovation (R&I Genetics) Srl. The other authors declare that they have no conflict of interest.
WEB RESOURCES
The URLs for data presented herein are as follows:
Combined Annotation Dependent Depletion (CADD), http://cadd.gs.washington.edu/
gnomAD Browser, https://gnomad.broadinstitute.org/
HGMD, http://www.hgmd.cf.ac.uk/ac/index.php
Hope, https://www3.cmbi.umcn.nl/hope/
IBD Exomes Browser Portal, Cambridge, MA; http://ibd.broadinstitute.org
OMIM, http://www.omim.org/
Picard Project, http://broadinstitute.github.io/picard/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
PDB Protein Data Bank, www.wwpdb.org
RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq
SIFT, http://sift.jcvi.org/
SnpEff, http://snpeff.sourceforge.net
The Human Gene Connectome (HGC), https://lab.rockefeller.edu/casanova/HGC
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version
REFERENCES
- 1.Ruel J, Ruane D, Mehandru S, et al. IBD across the age spectrum - Is it the same disease? Nat Rev Gastroenterol Hepatol 2014;11:88–98. [DOI] [PubMed] [Google Scholar]
- 2.Uhlig HH. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013;62:1795–1805. [DOI] [PubMed] [Google Scholar]
- 3.Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis 2011;17:1314–21. [DOI] [PubMed] [Google Scholar]
- 4.Kelsen JR, Baldassano RN, Artis D, et al. Maintaining Intestinal Health: The Genetics and Immunology of Very Early Onset Inflammatory Bowel Disease. Cmgh 2015;1:462–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uhlig HH, Schwerd T. From Genes to Mechanisms. Inflamm Bowel Dis 2016;22:202–212. [DOI] [PubMed] [Google Scholar]
- 6.Kerur B, Benchimol EI, Fiedler K, et al. Natural History of Very Early Onset Inflammatory Bowel Disease in North America: A Retrospective Cohort Study. Inflamm Bowel Dis 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pazmandi J, Kalinichenko A, Ardy RC, et al. Early-onset inflammatory bowel disease as a model disease to identify key regulators of immune homeostasis mechanisms. Immunol Rev 2019;287:162–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwerd T, Bryant RV., Pandey S, et al. NOX1 loss-of-function genetic variants in patients with inflammatory bowel disease. Mucosal Immunol 2018;11:562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corridoni D, Chapman T, Ambrose T, et al. Emerging Mechanisms of Innate Immunity and Their Translational Potential in Inflammatory Bowel Disease. Front Med 2018;5:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hugot J, Chamaillard M, Zouali H. Association of NOD2 leucine rich repeat variants with susceptibility to Crohn’s disease. Nature 2001;411:599–603 [DOI] [PubMed] [Google Scholar]
- 11.Zeissig Y, Petersen B-S, Milutinovic S, et al. XIAP variants in male Crohn’s disease. Gut 2015;64:66–76. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Lee CH, Peters LA, et al. Variants in TRIM22 That Affect NOD2 Signaling Are Associated With Very-Early-Onset Inflammatory Bowel Disease. Gastroenterology 2016;150:1196–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwerd T, Pandey S, Yang H-T, et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut 2017;66:1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Y, Li X, Liu S, et al. Toll-like receptors and inflammatory bowel disease. Front Immunol 2018;9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parlato M, Charbit-Henrion F, Pan J, et al. Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol Med 2018;10:e8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dias Junior AG, Sampaio NG, Rehwinkel J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol 2019;27:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ori D, Murase M, Kawai T. Cytosolic nucleic acid sensors and innate immune regulation. Int Rev Immunol 2017;36:74–88. [DOI] [PubMed] [Google Scholar]
- 18.Broquet AH, Hirata Y, McAllister CS, et al. RIG-I/MDA5/MAVS Are Required To Signal a Protective IFN Response in Rotavirus-Infected Intestinal Epithelium. J Immunol 2010;186:1618–1626. [DOI] [PubMed] [Google Scholar]
- 19.Levine A, Koletzko S, Turner D, et al. The ESPGHAN Revised Porto Criteria for the Diagnosis of Inflammatory Bowel Disease in Children and Adolescents. J Pediatr Gastroenterol Nutr 2013;58:1. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Depristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lupski JR, Gonzaga-Jauregui C, Yang Y, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med 2013;5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bainbridge MN, Wang M, Wu Y, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol 2011;12:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid JG, Carroll A, Veeraraghavan N, et al. Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics 2014;15:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smedley D, Robinson PN. Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med 2015;7:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobreira N, Schiettecatte F, Boehm C, et al. New tools for mendelian disease gene identification: PhenoDB variant analysis module; and genematcher, a web-based tool for linking investigators with an interest in the same gene. Hum Mutat 2015;36:425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itan Y, Shang L, Boisson B, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods 2016;13:109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venselaar H, Beek TAH te, Kuipers RKP, et al. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010;11:548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrodinger L The PyMOL Molecular Graphics System,. 2010.
- 31.Itan Y, Mazel M, Mazel B, et al. HGCS: An online tool for prioritizing disease-causing gene variants by biological distance. BMC Genomics 2014;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itan Y, Shang L, Boisson B, et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci U S A 2015;112:13615–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamborn IT, Jing H, Zhang Y, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med 2017;214:1949–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derkach A, Lawless JF, Sun L. Robust and Powerful Tests for Rare Variants Using Fisher’s Method to Combine Evidence of Association From Two or More Complementary Tests. Genet Epidemiol 2013;37:110–121. [DOI] [PubMed] [Google Scholar]
- 35.Asgari S et al. Loss-of-function mutations in IFIH1 predispose to severe viral respiratory infections in children, The Biology of Genomes Meeting In: ; 2016. [Google Scholar]
- 36.Coban-Akdemir Z, White JJ, Song X, et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am J Hum Genet 2018;103:171–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asgari S, Schlapbach LJ, Anchisi S, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci U S A 2017;114:8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reese MG, Eeckman FH, Kulp D, et al. Improved splice site detection in Genie. J Comput Biol 1997;4:311–23. [DOI] [PubMed] [Google Scholar]
- 39.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res 2001;29:1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeo G, Burge CB. Maximum Entropy Modeling of Short Sequence Motifs with Applications to RNA Splicing Signals. J Comput Biol 2004;11:377–394. [DOI] [PubMed] [Google Scholar]
- 41.Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brisse M, Ly H. Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front Immunol 2019;10:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y, Karijolich J. Know Thyself: RIG-I-Like Receptor Sensing of DNA Virus Infection. J Virol 2019;93:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coch C, Stümpel JP, Lilien-Waldau V, et al. RIG-I Activation Protects and Rescues from Lethal Influenza Virus Infection and Bacterial Superinfection. Mol Ther 2017;25:2093–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melchjorsen J, Rintahaka J, Søby S, et al. Early Innate Recognition of Herpes Simplex Virus in Human Primary Macrophages Is Mediated via the MDA5/MAVS-Dependent and MDA5/MAVS/RNA Polymerase III-Independent Pathways. J Virol 2010;84:11350–11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee HC, Chathuranga K, Lee JS. Intracellular sensing of viral genomes and viral evasion. Exp Mol Med 2019;51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu B, Peisley A, Richards C, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013;152:276–289. [DOI] [PubMed] [Google Scholar]
- 48.Kasumba DM, Grandvaux N. Therapeutic Targeting of RIG-I and MDA5 Might Not Lead to the Same Rome. Trends Pharmacol Sci 2019;40:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webb K, Desguerre I, Ariaudo G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 2014;46:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rutsch F, MacDougall M, Lu C, et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet 2015;96:275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eyck L Van, Somer L De, Pombal D, et al. Brief report: IFIH1 mutation causes systemic lupus erythematosus with selective IgA deficiency. Arthritis Rheumatol 2015;67:1592–1597. [DOI] [PubMed] [Google Scholar]
- 52.Zaki M, Thoenes M, Kawalia A, et al. Recurrent and prolonged infections in a child with a homozygous IFIH1 nonsense mutation. Front Genet 2017;8:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorman JA, Hundhausen C, Errett JS, et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat Immunol 2017;18:744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smyth DJ, Cooper JD, Bailey R, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 2006;38:617–9. [DOI] [PubMed] [Google Scholar]
- 55.Enevold C, Oturai AB, Sørensen PS, et al. Multiple sclerosis and polymorphisms of innate pattern recognition receptors TLR1–10, NOD1–2, DDX58, and IFIH1. J Neuroimmunol 2009;212:125–131. [DOI] [PubMed] [Google Scholar]
- 56.Ellinghaus D, Jostins L, Spain SL, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet 2016;48:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacDuff DA, Baldridge MT, Qaqish AM, et al. HOIL1 Is Essential for the Induction of Type I and III Interferons by MDA5 and Regulates Persistent Murine Norovirus Infection. Pfeiffer JK, ed. J Virol 2018;92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dai W, Liu Z-X, Li X-H, et al. Rig-I −/− mice develop colitis associated with downregulation of Gαi2. Cell Res 2007;17:858–868. [DOI] [PubMed] [Google Scholar]
- 59.Li X-D, Chiu Y-H, Ismail AS, et al. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proc Natl Acad Sci 2011;108:17390–17395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Solis M, Goubau D, Hiscott J. RIG-I has guts: Identification of a role for RIG-I in colitis development. Cell Res 2007;17:974–975. [DOI] [PubMed] [Google Scholar]
- 61.Fischer JC, Bscheider M, Eisenkolb G, et al. RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Transl Med 2017;9:eaag2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He L, Chen Y, Wu Y, et al. Nucleic acid sensing pattern recognition receptors in the development of colorectal cancer and colitis. Cell Mol Life Sci 2017;74:2395–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu H, Xu WY, Hu Z, et al. RNA virus receptor Rig-I monitors gut microbiota and inhibits colitis-associated colorectal cancer. J Exp Clin Cancer Res 2017;36:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee S, Baldridge MT. Interferon-lambda: A potent regulator of intestinal viral infections. Front Immunol 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Broggi A, Tan Y, Granucci F, et al. IFN-λ suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol 2017;18:1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawaguchi S, Ishiguro Y, Imaizumi T, et al. Retinoic acid-inducible gene-I is constitutively expressed and involved in IFN-gamma-stimulated CXCL9–11 production in intestinal epithelial cells. Immunol Lett 2009;123:9–13. [DOI] [PubMed] [Google Scholar]
- 67.Funke B, Lasitschka F, Roth W, et al. Selective downregulation of retinoic acid-inducible gene I within the intestinal epithelial compartment in Crohn’s disease. Inflamm Bowel Dis 2011;17:1943–54. [DOI] [PubMed] [Google Scholar]
- 68.Huang H, Fang M, Jostins L, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017;547:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asadpour-Behzadi A, Kariminik A. RIG-1 and MDA5 are the important intracellular sensors against bacteria in septicemia suffering patients. J Appl Biomed 2018;16:358–361. [Google Scholar]
- 70.Ciancanelli MJ, Huang SXL, Luthra P, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015;348:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Israel L, Wang Y, Bulek K, et al. Human Adaptive Immunity Rescues an Inborn Error of Innate Immunity. Cell 2017;168:789–800.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Picard C, Bernuth H von, Ghandil P, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 2010;89:403–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic Sensing of Viruses. Immunity 2013;38:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato K, Ahmad S, Zhu Z, et al. Structural analysis of RIG-I-like receptors reveals ancient rules of engagement between diverse RNA helicases and TRIM ubiquitin ligases. Mol Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.