

Abstract
Adipose tissue macrophages (ATM) are a major source of low-grade inflammation in obesity, and yet reasons driving ATM accumulation in white adipose tissue (WAT) are not fully understood. Emerging evidence suggested that ATM underwent extensive remodeling in obesity. In addition to abundance, ATM in obesity were lipid-laden and metabolically reprogrammed, which in turn was tightly related to their functional alterations and persistence in obesity. Herein, we aimed to discuss that activation of lipid sensing signaling associated with metabolic reprogramming in ATM was indispensible for their migration, retention, or proliferation in obesity. Likewise, lipolysis also induced similar but transient ATM remodeling. Therefore, we assumed that obesity might share overlapping mechanisms with lipolysis in remodeling ATM. Formation of crown-like structures (CLS) in WAT was presumably a common event initiating ATM remodeling, with a spectrum of lipid metabolites released from adipocytes being potential signaling molecules. Moreover, adipose interlerkin-6 (IL-6) exhibited homologous alterations by obesity and lipolysis. Thus, we postulated a positive feedback loop between ATM and adipocytes via IL-6 signaling backing ATM persistence by comparison of ATM remodeling under obesity and lipolysis. An elucidation of ATM persistence could help to provide novel therapeutic targets for obesity-associated inflammation.
1. Introduction
The prevalence of Western dietary pattern and sedentary lifestyle boosted the pandemic of obesity worldwide, which was closely associated with increased risks of various metabolic disorders [1]. To tackle this problem, the development of new therapeutics for obesity is urgent. Strongly involved in the pathogenesis of obesity was white adipose tissue (WAT) [2]. Expansion of fat in obesity was associated with adipose tissue macrophages (ATM) accumulation, which caused a state of chronic, low-grade inflammation, and insulin resistance [3]. To date, reasons for the persistence of excessive ATM in obesity have not been fully understood. Canonically, it was known that inflamed adipose tissue could overexpress chemokines such as monocyte chemoattractant protein-1 (MCP-1) which would lead to increased infiltration of macrophages [4]. Despite the role in inflammation, ATM were also key players for systemic energy homeostasis as lipid scavengers. ATM would increase the uptake of lipid fluxes from adipocytes in mice in response to fasting or pharmacologically induced lipolysis, while intra-abdominal ATM depletion resulted in elevated fasting serum free fatty acid (FFA) levels in mice [5]. The fact that most of ATM were localized within crown-like structures (CLS) in obesity [6] indicated CLS might be a vital initiator for obesity-related ATM recruitment. Indeed, adipocyte death per se can trigger ATM infiltration [7] because ATM were important in digesting dead adipocytes within CLS via secretion of cytokines or lysosomal lipases which activated lipolysis and breakdown of triglyceride (TG) in adipocytes [8, 9]. The group of Becker identified a “metabolically activated” (MMe) phenotype of ATM in obese mice, which were beneficial by clearing dead adipocytes through lysosomal exocytosis at the late onset of obesity [10].
Obesity influenced not only ATM abundance but also their metabolic process, indicated by accumulated cellular lipid droplets (LD) in ATM [11, 12]. Mechanistically, ATM in obese mice and humans overexpressed various cell surface receptors to uptake lipids derived from adipocytes in CLS, many of which were indispensable for ATM accumulation in obesity [13, 14]. Thus, in this regard, it was postulated that adipocytes could crosstalk with ATM via secretion of diverse lipid metabolites and then activated lipid sensing signaling in ATM, which could be another key mechanism underlining obesity-linked ATM accumulation.
Compared to obesity, the process of adipocyte lipolysis was also accompanied with CLS formation, lipid liberation in adipocytes, and inflammation in WAT [6, 15, 16]. The accumulation of ATM occurred in response to fat mobilization activated by fasting or catecholamine signaling activation in healthy lean mice, which would be restored later possibly due to cease of FFAs fluxes from adipocytes [5]. Moreover, ATM also became lipid-laden during lipolysis [5, 17, 18]. Therefore, lipolysis might share overlapping mechanisms with obesity to recruit and remodel ATM. Thus, we postulated that comparison of ATM remodeling in these two conditions would lead to discoveries of related mechanisms for excessive ATM in obesity.
Adipose interlerkin-6 (IL-6) appeared to be a potential shared molecule exhibiting similar upregulation by both obesity and adipocyte lipolysis [16, 19, 20]. In obese mice, IL-6/signal transducer and activator of transcription 3 (STAT3) signaling played an important role in liberation of FFAs from adipocytes [21]. Recently, Yu et al. demonstrated that ATM in obesity could also crosstalk with adipocytes through secretion of IL-6 [22]. Therefore, in this review, supported with current literature, we proposed the existence of a positive feedback loop between adipocytes and ATM via IL-6 signaling in obesity and WAT lipolysis concerning ATM persistence, in hoping to provide new insights for studies of obesity-related inflammation.
2. Remodeling of ATM in Obesity
2.1. Metabolic Reprogramming of ATM by Obesity
The initial event leading to infiltration, retention, or proliferation of ATM under obesity was of limited understanding. The phenomenon that up to 90% of ATM was localized within CLS in adipose tissue in obese db/db mice and obese humans [6] implied CLS might have a pivotal role in obesity-associated ATM accumulation. By means of immunofluorescence, Haase et al. demonstrated that the microenvironment of CLS predominantly contributed to a time-dependent increase in ATM proliferation in mice fed with high fat diet (HFD) [23]. At the late state of obesity, expansion of fat was dominated with CLS clustering and adipocyte death in obese mice [10, 24]. Meanwhile, the number of ATM could rise up to 10-folds in adipose tissue in diet-induced obesity (DIO) in mice [3]. Thus, obesity-linked CLS formation was possibly one of the events orchestrating ATM accumulation. Then, what were the signaling molecules within the loci of CLS that gave rise to obesity-associated ATM abundance? It was well acknowledged that obesity was featured by inefficiency of lipid storage and increased lipid liberation from adipocytes in WAT [25], creating a lipid-rich microenvironment where cells including ATM were metabolically challenged. Would this metabolic challenge to ATM relate to their persistence in obesity? (Figure 1).
Figure 1.
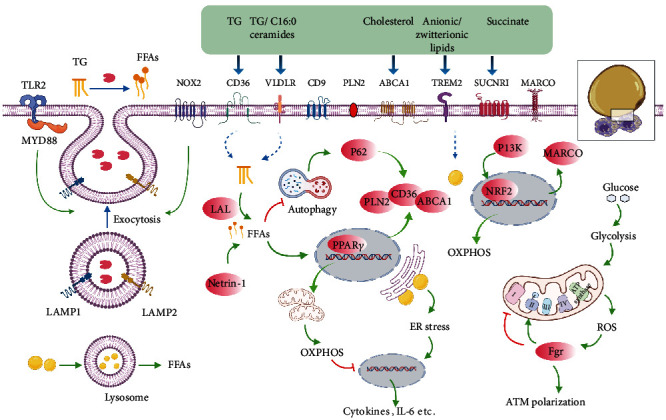
Integrative model of ATM remodeling in obesity. Representation of ATM surrounding apoptotic adipocytes in CLS in obesity, with the small black square enlarging alterations of ATM in connection with dysfunctional adipocytes. The signaling pathways that link these components by activation (green arrows) or inhibition (red inhibitory lines) are also indicated. Briefly, in obesity, ATM are dynamically recruited to apoptotic adipocytes to form CLS and becoming lipid lading associated with increased synthesis of lysosome responsible for catabolism of LD. This alteration might involve these following processes. Firstly, to digest dead adipocytes and facilitate FFA liberations, exocytosis of lysosomal hydrolases from ATM is activated by NOX2 and TLR2/MYD88 signaling pathways with the detection of LAMP1 and LAMP2 on cell surface of ATM. Secondly, expressions of specific cell surface markers including CD36, VLDLR, CD9, PLN2, ABCA1, TEM2, SUCNR1, and MARCO are boosted in ATM in response to stimulation of metabolites like TG, ceramides, cholesterol, anionic/zwitterionic lipids, or succinate, contributing to increased lipid uptake and LD formation in ATM, which seems to be essential for ATM migration and accumulation in adipose tissue by obesity. Mechanistically, metabolism of cellular TG to FFAs mediated by LIPA activates PPARγ responsible for increased expressions of PLN2, CD36, and ABCA1, while activation of PI3K-NRF2 signaling leads to increased MARCO expression. Finally, lipid accumulation in ATM might lead to metabolic reprogramming associated with ER stress as well as shifting to aerobic glycolysis in which building up of ROS results in Fgr activation, mitochondrial complex II activation, and complex I inhibition, all of which induce preferential production of inflammatory cytokines and ATM polarization. By contrast, OXPHOS promoted by FFA metabolism and p62 activation due to reduced autophagy or PI3K signaling activation exhibits anti-inflammatory functions.
The concentration of lipids in ATM in obesity was higher than that of the lean counterpart in mice [11, 12, 18]. In fact, obesity-associated ATM were metabolically remodeled to be preferential of lipid handling. Firstly, stimulated by the lipid-rich microenvironment, ATM boosted cellular lipid contents by upregulation of lipid uptake in response to obesity in mice. This process was mediated by increased expressions of a variety of cell surface markers, including the ATP-binding cassette transporters A1 (ABCA1), very low-density lipoprotein receptor (VLDLR), perilipin (PLIN) 2, and cluster of differentiation (CD)36 [13, 14] as well as the scavenger receptor of macrophage receptor with collagenous structure (MARCO) in mouse models of obesity [18]. Moreover, the large quantity of FFAs released from apoptotic adipocytes activated proteins like nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2), myeloid differentiation primary response 88 (MYD88) and toll-like receptor 2 (TLR2) in ATM, and modeled ATM to increase the uptake of lipids through lysosomal exocytosis indicated by detection of lysosomal associated membrane protein (LAMP)1 and LAMP2 on cell surface of ATM in obese mice [10]. In the process of lysosomal exocytosis, ATM formed close synapse with dead adipocytes and digested them with lysosomal enzymes reported by Haka et al. using mouse models of obesity, human adipose tissue, and ex vivo cell model [26]. In addition, around three percent of ATM in WAT possibly conjugated to form the multinucleated giant cells (MGCs) in order to phagocytize oversized dead adipocytes in CLS in obese patients and mice [27]. Furthermore, biogenesis of lysosome was driven in ATM to enable high rate of lipid catabolism, because inhibition of lysosome function by chloroquine treatment resulted in accumulation of LD in ATM in obese mice [12, 18]. Lipid metabolism in ATM was also altered by obesity. Petkevicius et al. demonstrated that ATM isolated from obese mice and humans exhibited increased rate of de novo phosphatidylcholine (PC) biosynthesis [28]. Consistently, the cholesterol efflux capacity (CEC) was enhanced in ATM in obesity associated with increased expressions of the transporters like ABCA1 compared to that of lean mice [29].
Taken together, the available data suggested that metabolic reprogramming of ATM featuring lipid handling and LD deposition occurred in obesity, which was likely related to CLS formation. We further set out to discuss in detail the implications of metabolic reprogramming in ATM accumulation and inflammation in obesity.
2.2. ATM Accumulation Driven by Lipid Sensing Signaling
The finding that the rate of adipocyte death in WAT coincided with ATM accumulation in obese mice suggested a role of ATM in adipose tissue remodeling [8]. Metabolically, the lipid-laden macrophages in insulin-resistant adipose tissue were also regarded as a protective measure. Because selective silencing of lipid handling genes including lipoprotein lipase (LPL) or CD36 in ATM resulted in less LD formation in ATM as well as elevated circulating serum FFAs levels in mice fed with HFD [30]. On the contrary, enhancing the lipid storage capability of ATM by depletion of lysosome with chloroquine treatment decreased basal lipolysis and serum FFAs in obese mice [12]. In addition, HFD feeding to mice led to generation of a subset of metabolically beneficial ATM defined by markers of F4/80, CD11b, CD11c, and CD206 (termed as FBC ATM), featuring increased lipid uptake and catabolism mediated by the activation of phosphoinositide 3-kinase- (PI3K-) nuclear factor erythroid 2-related factor 2- (NRF2-) MARCO signaling pathway. Similarly, this lipid-buffering subset of ATM antagonized obesity-associated metabolic disorders to some extent in mouse models of obesity [18].
More importantly, activation of lipid handling signaling was vital for macrophages to accumulate in WAT. In a study of Sharma et al., myeloid-specific depletion of the neuroimmune guidance cue molecule netrin-1 in mice attenuated ATM accumulation in visceral WAT as well as obesity-associated metabolic dysfunctions, mainly due to improved lipid handling and metabolic function in residual ATM [31]. By genetic depletion of netrin-1 coding gene Ntn 1 in a mouse model of obesity, adipose tissue inflammation and insulin resistance were reduced, due to restored migratory capacity and facilitated emigration of ATM [32]. Likewise, the group of Amit identified a subset of ATM with a special transcriptional signature of triggering receptor expressed on myeloid cell 2 (TREM2), lysosomal acid lipase (LIPA), LPL, Cathepsin B (CTSB), Cathepsin L (CTSL), fatty acid-binding protein (FABP)4, FABP5, lectin, galactoside-binding, soluble (LGALS)1, LGALS3, CD9, and CD36 and characteristic of lipid metabolism and phagocytosis, which they named as “lipid-associated macrophages” (LAM) in human and mouse adipose tissue [33]. Deletion of TREM2 abrogated the lipid uptake and storage function of LAM, as well as their recruitment to enlarged adipocytes for the assembly of CLS in obese mice, thereby enforcing weight gain, hypercholesterolemia, and glucose intolerance [33, 34]. TREM2 could sense a broad array of anionic and zwitterionic lipids to sustain the microglial response in Alzheimer's disease in mice [35]. However, the specific species and source of lipids sensed by ATM via TREM2 in obesity remained largely unexplored. Besides TG, unesterified free cholesterol was also deposited in adipocytes [36]. Cellular cholesterol efflux was initiated and propagated by ABCA1 and the ATP-binding cassette transporters G1 (ABCG1) whose expressions and functions might be rate-limiting for cholesterol efflux [37]. During obesity in mice, expression of ABCG1 was reported to be essential for M2-like macrophages to migrate towards CLS for handling the unesterified cholesterol released from dead adipocytes [38]. Additionally, succinate, a tricarboxylic acid (TCA) cycle intermediate which could serve as an activation signal in macrophages stimulated by lipopolysaccharide (LPS) [39], was overproduced from adipose tissue in mice under stressed conditions like hypoxia and hyperglycemia and induced upregulated chemotaxis in ATM towards CLS through activation of succinate receptor 1 (SUCNR1) [40].
In sum, we postulated that dysfunctional adipocytes would release a distinguished spectrum of lipid species or metabolites to signal ATM migration, retention, or proliferation in obesity, which might be another important mechanism underlining obesity-associated ATM accumulation. The effects of these lipid species or metabolites on ATM were possibly mediated by expressions of specific lipid sensors on cell surface of ATM. If so, blockage of these lipid sensors might be new therapeutic targets for obesity-related inflammation. Nevertheless, except results from genetically engineered mouse models, by far, there was no pharmacological component targeting these sensors in ATM. Therefore, further investigation is needed to characterize the spectrum of lipids or metabolites released from adipocytes during obesity, as well as to elucidate relevant signaling pathways in ATM.
2.3. Involvement of ATM-LD in Inflammation
Previous studies had demonstrated that inflammation of ATM compromised metabolic homeostasis in obese mice [3, 4, 21, 41]. ATM comprised proinflammatory M1 and anti-inflammatory M2 subsets. The M1-like ATM induced by T helper (Th) 1 mediators such as LPS were responsible for increased production of proinflammatory cytokines. However, the M2-like phenotype driven by Th2 mediators including IL-4 and IL-13 exhibited an anti-inflammation role with activation of the transcriptional factor peroxisome proliferator-activated receptor γ (PPARγ) [41]. Obesity was associated with an increased ratio of M1/M2-like ATM in WAT, favoring generation of cytokines such as IL-6 and promoting adipocyte lipolysis, ectopic lipid accumulation, and insulin resistance [21].
A report employing transmission electron microscopy (TEM) showed that M1-like macrophages contained more LD than M2 phenotype [14], indicating lipid accumulation correlated with inflammation in ATM. In addition, within CLS, a novel subset of ATM defined by cell surface marker CD9 promoted inflammation after becoming lipid-laden in mice fed a HFD [42, 43]. In fact, inflammatory macrophages would increase the uptake of lipids [44]. In a study of Castoldi et al., cellular TG was demonstrated to be indispensible for macrophages' inflammatory role [45]. Moreover, elevation of VLDLR expression in ATM in obese mice increased the uptake of TG and C16:0 ceramides and rendered the polarization of ATM to M1-like phenotype, thus promoting adipose tissue inflammation and glucose intolerance [46]. These data suggested that cellular LD could be an important contributor to inflammation in ATM.
Then, how is ATM-LD linked with inflammation? Based on the current literature, the answer might be partly related to the metabolic reprogramming accompanied with the formation of LD in ATM. The result of Na et al. suggested that increased glycolytic capacity was required for the inflammatory phenotype of macrophages [47]. Specifically, the reliance of aerobic glycolysis by M1-like macrophages led to production of mitochondrial reactive oxidative species (ROS) and oxidative stress, which contributed to secretion of cytokines such as IL-6 and tumor necrotic factor-α (TNF-α) and adipose tissue inflammation [48–50]. Indeed, in patients with morbid obesity, disturbances in the antioxidant barrier and enhanced oxidative damage to proteins and lipids were observed. Although bariatric surgery improved redox homeostasis in obese patients without metabolic syndrome, those with metabolic syndrome showed a continuous decrease in the antioxidant status [51]. ROS accumulation would also lead to activation of Fgr kinase (a member from the Src family), which induced phosphorylation and activation of mitochondrial complex II and degradation of mitochondrial complex I, contributing to ATM polarization and obesity-associated insulin resistance in mice [52].
Moreover, LD formation was often associated with endoplasmic reticulum (ER) stress. Exposure of RWA264.7 to saturated fatty acid palmitic acid led to cytoplasmic accumulation of lipids, ER stress, and M1 polarization in vitro [53], while attenuation of ER stress reduced M1 polarization of ATM and subsequent metabolic stress in mice fed by HFD [53, 54]. On the contrary, in M2-like macrophages which were less lipid-laden, PPARγ activation resulted in mitochondrial biogenesis and oxidative metabolism due to IL-4 stimulation and suppressed production of cytokines in mice [41, 55]. Interestingly, M2-like ATM were not using the LD-dependent TG hydrolysis pathway to generate FFAs for fatty acid oxidation. Instead, uptake of TG substrates via CD36 and subsequent lipolysis by LIPA was used to support elevated oxidative phosphorylation (OXPHOS), prolonged survival, and expressions of gene characteristic of M2 phenotype in mice [14]. What was more, promoting lipid metabolism via activation of PPARγ and p62/Sqstm1 by palmitate partly limited inflammation in macrophages in adipose tissue, explaining why there was a low-grade sterile inflammation in obese mice [13]. However, van Dierendonck et al. reported lipid contents in ATM were significantly reduced in mice with myeloid-specific deficiency of hypoxia-inducible lipid droplet-associated (HILPDA). But in DIO in mice, this decreased lipid storage in HILPDA-deficient ATM did not alter inflammatory and metabolic parameters. Thus, the data presented by van Dierendonck et al. questioned the contribution of LD accumulation in ATM to obesity-associated inflammation and metabolic complications [56]. Xu et al. also reported that by depletion of lysosome, lipid contents increased in ATM in mouse perigonadal fat in vitro. But this lipid accumulation was not associated with a clear inflammatory induction in ATM [12].
Taken together, the role of cellular LD in ATM inflammation merits further validation. Possibly, the cellular LD per se do not lead to polarization of ATM, but the metabolic reprogramming shifting to aerobic oxidation under LD formation is crucial to ATM inflammation. A case in point was HILPDA deficiency which led to LD removal associated with increased oxidative metabolism in ATM [56]. It was also demonstrated that upregulation of oxidative metabolism in ATM by stimulation with recombinant growth differentiation factor 15 (GDF15) led to M2-like polarization, reversing adipose inflammation as well as insulin resistance in mice [57].
In light of the finding that adoptive transfer of bone marrow cells from VLDLR knockout mice into wild-type mice relieved adipose tissue inflammation and improved insulin resistance in DIO in mice due to reduced TG and ceramides uptake into ATM [46], certain subsets of ATM could be new pharmacological targets in obesity-related inflammation and metabolic complications, which warrants further investigation.
3. Reversal Remodeling of ATM during Lipolysis
3.1. ATM Accumulating and Becoming Lipid-Laden during Lipolysis
Lipolysis induced by fasting resulted in mobilization of TG in adipocytes to maintain metabolism of other organs in the body [5]. Canonically, activation of β3-adrenergic receptor (ADRB3) in adipocytes by epinephrine or norepinephrine stimulated hydrolysis of TG to FFAs and glycerol mediated by cyclic adenosine monophosphate- (cAMP-) dependent protein kinase A (PKA) activation and subsequent phosphorylation of hormone sensitive lipase (HSL) [58]. In the process of lipolysis, ATM also underwent dramatic but transient remodeling, which showed certain similarities to that in the state of obesity (Figure 2).
Figure 2.
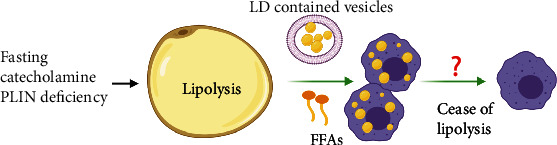
Scheme of transient ATM remodeling during lipolysis. Briefly, adipocyte lipolysis induced by fasting and catecholamine stimulation or PLIN deficiency leads to ATM accumulation and cellular LD abundance in ATM, possibly stimulated by LD contained vesicles and FFAs released from adipocytes. After lipolysis, remodeled ATM would return to normalization by unknown mechanisms.
On the one hand, accumulation of ATM in WAT was detected during lipolysis in normal lean mice. So what is the mechanism behind this alteration? Similar to obesity, lipolysis also resulted in the death of adipocytes and formation of CLS in normal mice [6, 15, 16]. Moreover, ATM accumulation was colocalized within CLS during lipolysis. Kim et al. reported increased formation of F4/80+ macrophage crown structure in gonadal WAT associated with augmented expression of CD11B, following activation of ADRB3 with isoproterenol treatment in mice [7]. While formation of CLS in obesity was known to persist for weeks or months, CLS induced by ADRB3 activation would quickly resolve and be updated by newly differentiated adipocytes during lipolysis in vivo in mice [15]. Thus, these data suggested that lipolysis-linked CLS formation might be one of the reasons responsible for the temporary recruitment of ATM in WAT. In a study by Lee et al., the result of triple staining of macrophage galactose-C type lectin 1 (MGL1, a marker of M2-like macrophages), LD enclosing protein PLIN1, and neutral lipid in whole-mount gonadal WAT showed that M2-like macrophages accumulated and surrounded LD in adipocytes devoid of PLIN1 in response to CL-316,243 treatment in mice, in order to function as a scavenger of dead adipocytes [15]. Thus, adipocyte death in CLS following ADRB3 activation could be a trigger for lipolysis-related ATM accumulation. Moreover, except the canonical manner, adipocytes could get rid of stored lipids directly by exocytosis of LD-contained vesicles during lipolysis in mice. These lipid-laden vesicles, after being internalized, would then induce migration, differentiation, and accumulation of ATM [59]. Unfortunately, the molecules and pathways involved were not well known by far.
On the other hand, metabolic reprogramming of ATM was also indicated by the results that lipolysis-derived FFAs or LD-containing vesicles eliciting neutral LD formation in ATM [5, 17, 59]. Functionally, some subsets of ATM were hypothesized to act as lipid buffer to prevent plasma TG from soaring up too much during fasting in mice [59]. In parallel to DIO, ADRB3 signaling could also lead to activation of the PI3K-NRF2-MARCO signaling pathway in myeloid, which contributed to accumulation of FBC ATM characteristic of facilitated lipid uptake and catabolism in mice [18]. Whether lipolysis could lead to activation of the lipid sensing signaling pathway in ATM similar to that in obesity as discussed above is worthy of further investigation. Of note, unlike in obesity where LD persisted in ATM, cellular lipid levels were restored in ATM post-lipolysis in mice, possibly due to ceasing of FFA flux. However, the specific mechanism is far from understanding. Since CLS were postulated to be an event initiating ATM remodeling in obesity, whether lipolysis-related temporary LD accumulation also associates CLS formation needs further validation. Meanwhile, it would be intriguing to answer the question how ATM were oriented to apoptotic adipocytes to form CLS in obesity or lipolysis. The discovery that the composition of FFAs generated by lipolysis in mice was similar to that released from dead adipocytes in cultured adipose tissue explant ex vivo [7] indicated the possibility that lipolysis might share with obesity a similar spectrum of adipocyte-secreted lipid metabolites in signaling ATM recruitment.
In sum, the available data suggested that lipolysis was associated with ATM remodeling characteristic of accumulation and metabolic reprogramming, possibly related to the role of ATM in clearance of lipid metabolites from adipocytes. Besides, the results also indicated the existence of mechanisms responsible for ATM renormalization postlipolysis which is not yet well investigated. In the following, we will address the alterations and implications of ATM during lipolysis in detail.
3.2. ATM Composition Altered by Lipolysis
Subpopulations of ATM were dynamically changed in lipolysis. As discussed above, deficiency in LD storage in adipocytes might trigger a signal for macrophages accumulation in lipolysis. Irregular and persistent lipolysis due to deficiency of PLIN1 in adipocytes initiated migration and M1-like polarization of monocytes into WAT in mice, mediated by lipid metabolites such as prostaglandin (PG) secreted from adipocytes. In this condition, macrophage clearance by clodronate alleviated inflammation in WAT in mice with PLIN1 deficiency [60]. Likewise, lipolysis in mice stimulated by CL-316,243 also led to PLIN1 deficiency in LD in adipocytes [15]. It was assumable that ADRB3 signaling resulted in ATM accumulation associated with M1 polarization. Intriguingly, during fasting in lean mice, Hu et al. reported that ATM accumulated but did not polarize, thus without altering the M1/M2 ratio of ATM in WAT. Nevertheless, it was demonstrated that adipocyte-derived prostaglandin E downstream of phospholipase A2 α (PLA2α) and cyclooxygenase 2 (COX2) activation played an important role in mediating ATM infiltration during adipocyte lipolysis ex vivo [61]. Allison et al. also demonstrated that recruitment of macrophages into fat depot was triggered by possible lipid metabolites derived from adipocytes in lipolysis both ex vivo and in vivo, followed by activation of the c-Jun N-terminal kinase (JNK)/nuclear factor kappa light chain enhancer of activated B cells (NFκB)/COX2 axis [62]. In contrast, intermittent fasting which was associated with metabolic benefits was found to increase the population of M2 macrophages without affecting M1 population in mice fed with HFD [63]. Additionally, Asterholm et al. showed that 24 h fasting in healthy mice increased pan macrophages indicated by F4/80 expression in WAT, with M1-like macrophage decrease indicated by CD11c and nitric oxide synthase 2 (NOS2) expressions and M2-like macrophage increase indicated by CD206, CD301, and CD163 expressions. However, this dynamic switch to M2 phenotype during nutrition deprivation was impaired in obese mice [64]. Camell et al. also reported that the young fasted mice showed greater reduction in CD11c+CD206− ATM (M1 population) with corresponding increase in CD11c+CD206+ ATM (M2 population) in comparison to fed ones [65]. Likewise, the team of James showed that chronic treatment of CL-316,243 for one week in mice increased M2 macrophages in gonadal WAT without affecting M1 macrophages [15]. The reason leading to these inconsistent data could be the composition of ATM in WAT was more complex than the M1/M2 diagram and distinct types of ATM dynamically adapted to different metabolic environments. Silva et al. found that a subset of ATM, termed as vasculature associated macrophages (VAM), was tightly associated with blood vessels in epididymal WAT. Remarkably, both fasting and ADRB3 activation could cause rapid depletion of VAM which would be restored by refeeding in healthy mice, confirming ATM subpopulations dynamically adapted to lipolysis-induced metabolic stress [66].
It is now clear that ATM composition in fat depots dynamically and transiently changes during acute metabolic stress such as fasting. But how exactly ATM subpopulations respond to lipolysis requires further characterization. Profound understanding of the mechanism responsible for ATM renormalization after lipolysis is of vital importance to prevent ATM accumulation in pathophysiological state-like obesity.
3.3. Role of ATM in Lipid Storage Efficiency of Adipocytes
The transient recruitment of ATM by lipolysis gives rise to a question concerning the significance of ATM in adipocyte lipolysis. According to the current literature, ATM could both positively and negatively influence lipid storage efficiency of adipocytes.
On the one hand, certain subsets of ATM were indispensible for maintaining lipid storage in adipocytes, as the demonstration that overall depletion of ATM by clodronate treatment promoted lipolysis and reduced fat weight in mice [5]. Thus, there might be a paracrine effect of ATM on lipid mobilization in adipocytes. Bu et al. revealed that CD11c+ ATM-derived growth differentiation factor 3 (GDF3), functioning as a ligand for activin receptor-like kinase 7 (ALK7) receptor on the surface of adipocytes, antagonized lipolysis via inhibition of lipase in mice [67]. In line with this result, age-associated impairment in fasting-induced lipolysis was partly accounted by activation of the Nod-like receptor (NLR) pathway and subsequent induction of GDF3 in ATM in visceral WAT in aged mice [65]. Moreover, M2-like ATM also had an inhibitory effect on lipolysis, with the impairment in their differentiation leading to lipolysis, diminished fat mass, and subsequent development of hypertriglyceridaemia as well as insulin resistance in mice, which could be restored by supplementation of M2-like ATM [68]. Nevertheless, the underlying mechanism responsible for actions of M2-like ATM on lipolysis remains elusive. Furthermore, certain subsets of ATM in CLS could induce quick differentiation of preadipocytes during lipolysis, since it was revealed that osteopontin (OPN) expressing M2-like ATM in CLS in mice attracted and initiated the differentiation of platelet-derived growth factor receptor alpha- (PDGFRα-) positive adipocyte progenitor that had a receptor CD44 for OPN [15]. Finally, the subpopulation of ATM expressing estrogen receptor β (ERβ) improved lipid storage capability of adipocytes and had a suppressive effect on CLS formation via the axis of ERβ/OPN/hypoxia inducible factor-1α (HIF-1α), with depletion of ERβ in ATM increasing the number of CLS in obese mice [69].
On the other hand, some subsets of ATM could also accelerate lipid depletion from adipocytes. It was well acknowledged that M1-like phenotype promoted lipolysis through secretion of cytokines like IL-6 and TNF-α [70]. Cytokines aside, ATM were able to secrete other molecules positively regulating lipolysis. Iwamura et al. reported that macrophage-derived soluble protein apoptosis inhibitor of macrophage (AIM) would induce ablation of PPARγ activity in adipocytes in mice, leading to declined expressions of genes indispensable for LD coating and TG storage including fat-specific protein 27 (FSP27) and PLIN [71]. Moreover, activation of the chemokine (C-C motif) receptor 2-positive (CCR2+) macrophages was required for adipocyte death-triggered lipolysis in mice, associated with increased serum levels of catecholamine [7].
It is likely that ATM of different subsets can secrete an array of molecules that have bilateral roles in the regulation of lipid storage efficiency of adipocytes. Therefore, further interrogation into the secretome of diverse subsets of ATM might be helpful for in-depth understanding of the crosstalk between adipocytes and ATM (Figure 3).
Figure 3.
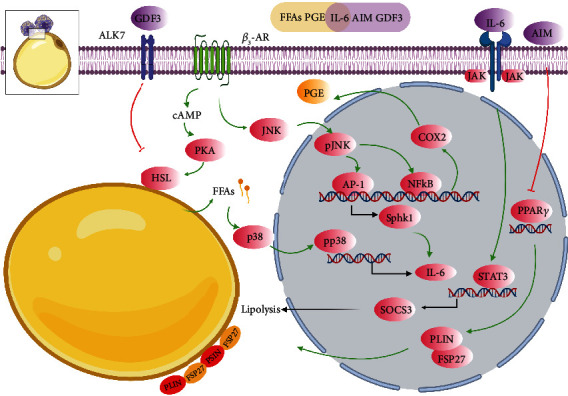
Scheme of relevant adipocyte alterations interacted with ATM in response to lipolysis. Representation of apoptotic adipocytes surrounded by ATM in CLS in lipolysis, with the small black square enlarging alterations of adipocytes. The signaling pathways that link these components by activation (green arrows) or inhibition (red inhibitory lines) are also indicated. Light yellow bar indicates secretome of adipocytes, while light purple one indicates secretome of ATM. Briefly, ADRB3 activation mediated by the canonical cAMP/PKA/HSL axis as well as the JNK/NFκB/COX2 axis liberates various lipid metabolites like FFAs and PGE from adipocytes, which would trigger infiltration of lipolysis-associated ATM. In addition, ADRB3 signaling also leads to IL-6 production from adipocytes, through p38 activation stimulated by FFAs building up or downstream of the JNK/AP-1/SPHK1 signaling pathway. IL-6 could induce adipocyte remodeling by activation of the JAK/STAT3/SOCS3 signaling, further enhancing catabolism and liberation of lipids from adipocytes. Also, IL-6 is an important signal node for ATM accumulation. Bilaterally, ATM participate in affecting adipocyte lipolysis via secretion of different molecules. On the one hand, ATM-derived GDF3 can inhibit lipolysis via activation of ALK on cell surface of adipocytes and subsequent lipase inhibition. Additionally, AIM secreted from ATM leads to irregular and persistent lipolysis due to inhibition of PPARγ and reduction of FSP27 and PLIN levels. On the other hand, M2-like ATM inhibit adipocyte lipolysis by undefined mechanisms.
Compared to obesity, lipolysis is also associated with ATM remodeling in terms of composition and metabolism. Future experiments on the comparison and characterization of ATM remodeling in these two conditions will help to comprehend the persistence of ATM in obesity. Next, we decipher a potential molecule (IL-6), which showed homologous alterations in WAT in both obesity and lipolysis and might have a role in ATM remodeling.
4. Role of IL-6 in the Crosstalk between ATM and Adipocytes
As summarized above, dysfunctional adipocytes in obesity or lipolysis might drive chemotactic response of ATM via a secretome of lipids/metabolites which induced activation of lipid sensing pathways in ATM. If this assumption was true, upstream molecules involved in regulation of this process must exist. We focused on the role of adipose IL-6 in ATM remodeling in the context of obesity and lipolysis for the reasons as follows. First of all, both obesity and lipolysis induced a surge in IL-6 in adipose tissue in mice [16, 19, 20]. Secondly, both adipocytes and ATM can secrete IL-6 [72, 73]. The level of IL-6 in adipocytes and macrophages would be enhanced when coculturing these two cells ex vivo, which would become more prominent in the presence of TLR4 ligand LPS [74], suggesting a crosstalk between adipocytes and macrophages in IL-6 production [75]. Thirdly, emerging evidence supported that IL-6 was associated with ATM accumulation in obesity as well as lipolysis [76–78]. Finally, IL-6 performed an important role in regulating lipid homeostasis in adipocytes [79].
4.1. Effects of Adipose IL-6 on ATM in Obesity
IL-6 expression in adipose tissue was persistently elevated in obese mice [19]. In fact, obesity was demonstrated to be a positive regulator of IL-6 and IL-6 receptor levels in subcutaneous WAT, which contributed to induction of inflammation and obesity-associated metabolic dysfunctions in mice [80]. In addition, IL-6 trans-signaling through the soluble IL-6 receptor (sIL-6R) had a major proinflammatory effect, with circulating sIL-6R more closely associated with insulin resistance status than waist-to-hip ratio or body mass index (BMI) in morbidly obese Taiwanese adults [81]. This inflammatory role of IL-6 possibly involved ATM, as it was demonstrated that adipocyte IL-6 promoted local inflammation by increasing ATM accumulation as well as M1 polarization in WAT, with conditional depletion of IL-6 in adipocytes ameliorating inflammation as well as glucose intolerance in mice fed a HFD [73]. On the contrary, blocking IL-6 trans-signaling with its inhibitor sgp 130Fc prevented HFD-induced ATM accumulation in mice [82]. Braune et al. reported that IL-6 was also produced by M2-like ATM in obese mice fed a HFD [83]. Paradoxically, IL-6/STAT3 signaling was required for M2-like ATM proliferation in obesity and could thus retard obesity-related insulin resistance in mice [84]. The augmentation and proliferation of M2 polarization by IL-6 were presumably due to upregulation of the IL-4 receptor α (IL-4 Rα) expression via STAT3 activation. IL-6 signaling could act as a Th2 cytokine and antagonize inflammation via favoring proliferation of M2-penothype macrophages and consequent clearance of dead adipocytes in obese mice [83, 84]. Taken together, adipose IL-6 signaling would exert diverse functions depending on where it stemmed from, with adipocyte-derived IL-6 inducing ATM accumulation and inflammation in WAT, while IL-6 from M2-like ATM ameliorating inflammation by clearance of dead adipocytes.
So what is the ultimate effect of adipose IL-6 on inflammation in obesity? It might depend on the onset of obesity. At the early stage of obesity, IL-6 probably induces ATM polarization and inflammation, whereas it antagonizes inflammation at the late onset of obesity when CLS formations are dominant. However, the mechanisms responsible for elevated IL-6 in adipocytes or ATM under obesity remain largely unexplored and merit further investigation.
4.2. Effects of Adipocyte IL-6 on ATM in Fasting
Lipolytic activation also triggered a proinflammatory response in WAT, with acute activation of ADRB3 eliciting expressions of proinflammatory genes including IL-6 in WAT in mice [16, 20]. Moreover, ADRB3 was demonstrated to be necessary and sufficient for acute stress-induced IL-6 production from brown adipocytes in mice [85]. In white adipocytes, building up of FFAs was proved to be a trigger for lipolytic signaling-induced IL-6 production through phosphorylation and activation of p38 downstream of HSL, with blockage of FFAs release minimizing the increased proinflammatory cytokines both in vivo and in vitro [86]. Furthermore, it was reported that expression of sphingosine kinase 1 (SPHK1) induced by activation of the JNK/activating protein-1 (AP-1) signaling pathway was indispensible for IL-6 production by ADRB3 in mice. Functionally, this IL-6 production was correlated with macrophage infiltration in epididymal fat during lipolysis in mice [76–78], which suggested a possible role of adipocyte IL-6 in ATM remodeling during lipolysis. Moreover, it was reported that lipolysis-derived FFAs per se only elicited formation of neutral LD but not inflammation in macrophages in vitro [17], supporting the possibility that other molecules like IL-6 released from adipocytes during lipolysis might trigger ATM recruitment and inflammation.
Therefore, lipolysis by ADRB3 activation could lead to IL-6 production in adipocytes partly mediated by the JNK/AP-1/SPHK1 signaling pathway (Figure 3). It would be fascinating to clarify whether obesity shares with lipolysis this mechanism to regulate IL-6 production in adipocytes.
4.3. Crosstalk between ATM and Adipocytes via IL-6
Except for its role in immunity, adipose IL-6 played a role in lipid homeostasis in both ATM and adipocytes. For one thing, IL-6 accelerated lipid removal from macrophages. IL-6 facilitated ABCA1-mediated cholesterol efflux from human macrophages to apolipoprotein AI (apoAI) via activation of the Janus kinase (JAK)-2/STAT3 signaling pathway in vitro [87]. The class A macrophage scavenger receptor (MSR) contributed to increased uptake of modified low density lipoproteins (LDL) and transformation of macrophages into lipid-laden foam cells. IL-6 dose-dependently inhibited the expression of MSR and reduced the uptake of acetylated LDL in THP-1 macrophages in vitro [88]. For another, IL-6 was also implicated in regulation of lipid metabolism in adipocytes [79]. In adipose tissue, IL-6 elevation could initiate a metabolic reprogramming. For example, in fasting state, metabolism was transiently switched from carbohydrate to lipid utilization in humans [89]. In this context, circulating IL-6 level was increased, contributing to TG mobilization and increased oxidation of FFAs in adipose tissue in mice [90]. In fact, exercise-induced IL-6 accelerated lipolysis and reduced visceral fat mass in humans [91]. Furthermore, IL-6 accounted for increased lipolysis in mesenteric adipose tissue in mice fed a HFD [21, 92]. Additionally, IL-6 was associated with cachexia-related lipolysis and WAT waste, with anti-IL-6 receptor antibody inhibiting WAT lipolysis in mice [93]. Mechanistically, IL-6 could induce the expressions of STAT3 and suppressor of cytokine signaling 3 (SOCS3) which inhibited insulin signaling in adipocytes [94]. Likewise, vesicle-contained IL-6 released from tumor would lead to activation of the JAK/STAT3 pathway and resulted in lipolysis in mice [95]. Hence, IL-6/STAT3 signaling had an important role in liberating lipids from adipocytes in various conditions. It was well known that ATM were major sources for increased systemic IL-6 levels in obesity in mice [49]. In fact, exposure to lipids or lipid metabolites such as PGE2 stimulated production of IL-6 in macrophages in vitro [96, 97]. As discussed above, a specific spectrum of lipid species or metabolites from adipocytes possibly functions as signals for remodeling and recruiting ATM during obesity or lipolysis. But the exact molecule signaling governing this process is scarcely known. Given the result that ATM surrounding dead adipocytes in CLS in obese mice expressed IL-6 [8], it is tempting to assume that ATM-derived IL-6 is one of the upstream molecules manipulating the secretion of those lipids/metabolites from adipocytes.
It is likely that there is a positive feedback loop between adipocytes and ATM via IL-6 signaling. Specifically, IL-6 stimulates lipid/metabolites release from adipocytes, which consequently recruits and remodels ATM into lipid-laden status. Then, ATM respond by releasing more IL-6 to get rid of their own lipids and further facilitate lipid liberation from adipocytes, thus forming a vicious circuit to enable ATM persistence. Rigorous investigation is needed to test this assumption.
5. Conclusions
ATM exhibited similar remodeling in terms of abundance and cellular LD contents in the contexts of obesity and lipolysis. Thus, comprehensive comparisons of ATM remodeling in these two situations might give rise to novel insight into obesity-associated persistence of ATM. Metabolic reprogramming associated with activation of lipid sensing signaling in ATM might be an important mechanism for ATM accumulation in obesity. Based on the comparison of ATM remodeling under obesity and WAT lipolysis, CLS formation seemed to be an overlapping event that initiated ATM remodeling, with a spectrum of lipids or metabolites released from dysfunctional adipocytes being potential signaling molecules. Thus, it is assumable that blockage of this lipid sensing signaling in ATM could antagonize obesity-related inflammation. ATM composition dynamically changed in response to metabolic stress signals from adipocytes, and characterization of distinct subpopulations of ATM is necessary for pharmacological targeting. How the transient abundance of ATM and their cellular LD contents are renormalized postlipolysis is poorly understood. There could be a positive feedback loop between ATM and adipocytes via IL-6 signaling which generates liberation of lipid metabolites from adipocytes to induce ATM remodeling, and ATM then in turn release more IL-6 enhancing its action on adipocytes. It should be noted that the majority of the reported information came from animal models. To verify these findings and develop new drugs, more data from human studies should be collected. Hopefully, investigation of adipose IL-6 signaling might be helpful in the discovery of the clearance mechanism of ATM from adipose tissue.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. U19A2009 and 32060169), Department of Human Resources and Social Security of Anhui Province (No. 2019LCX028), Education Department of Anhui Province (No. KJ2020A0385), and Hainan Provincial Natural Science Foundation of China (No. 320RC503). Figures in this article were created with http://Biorender.com.
Abbreviations
- ABCA1:
ATP-binding cassette transporter A1
- ABCG1:
ATP-binding cassette transporter G1
- ADRB3:
β3-adrenergic receptor
- AIM:
Apoptosis inhibitor of macrophage
- ALK7:
Activin receptor-like kinase 7
- AP-1:
Activating protein-1
- apoAI:
Apolipoprotein AI
- ATM:
Adipose tissue macrophages
- BMI:
Body mass index
- cAMP:
Cyclic adenosine monophosphate
- CD36:
Cluster of differentiation 36
- CEC:
Cholesterol efflux capacity
- CLS:
Crown-like structures
- COX2:
Cyclooxygenase 2
- CTSB:
Cathepsin B
- CTSL:
Cathepsin L
- DIO:
Diet-induced obesity
- ER:
Endoplasmic reticulum
- ERβ:
Estrogen receptor β
- FABP:
Fatty acid-binding protein
- FFAs:
Free fatty acids
- FSP27:
Fat-specific protein 27
- GDF3:
Growth differentiation factor 3
- GDF15:
Growth differentiation factor 15
- HFD:
High fat diet
- HIF-1α:
Hypoxia inducible factor-1α
- HILPDA:
Hypoxia-inducible lipid droplet-associated
- HSL:
Hormone sensitive lipase
- IL-6:
Interlerkin-6
- JAK:
Janus kinase
- JNK:
c-Jun N-terminal kinase
- LAM:
Lipid-associated macrophages
- LAMP:
Lysosomal associated membrane protein
- LD:
Lipid droplets
- LDL:
Low-density lipoproteins
- LGALS:
Lectin, galactoside-binding, soluble
- LIPA:
Lysosomal acid lipase
- LPL:
Lipoprotein lipase
- LPS:
Lipopolysaccharide
- MARCO:
Macrophage receptor with collagenous structure
- MCP-1:
Monocyte chemoattractant protein-1
- MGCs:
Multinucleated giant cells
- MGL1:
Macrophage galactose-C type lectin 1
- MSR:
Macrophage scavenger receptor
- MYD88:
Myeloid differentiation primary response 88
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NFκB:
Nuclear factor kappa light chain enhancer of activated B cells
- NLR:
Nod-like receptor
- NOX2:
NADPH oxidase 2
- NOS2:
Nitric oxide synthase 2
- NRF2:
Nuclear factor erythroid 2-related factor 2
- OPN:
Osteopontin
- OXPHOS:
Oxidative phosphorylation
- PC:
Phosphatidylcholine
- PDGFRα:
Platelet-derived growth factor receptor alpha
- PG:
Prostaglandin
- PI3K:
Phosphoinositide 3-kinase
- PKA:
Protein kinase A
- PLA2α:
Phospholipase A 2α
- PLIN2:
Perilipin 2
- PPARγ:
Peroxisome proliferator-activated receptor γ
- ROS:
Reactive oxidative species
- SOCS3:
Suppressor of cytokine signaling 3
- SPHK1:
Sphingosine kinase 1
- STAT3:
Signal transducer and activator of transcription 3
- SUCNR1:
Succinate receptor 1
- TCA:
Tricarboxylic acid
- TEM:
Transmission electron microscopy
- Th:
T helper
- TG:
Triglyceride
- TLR2:
Toll-like receptor 2
- TNF-α:
Tumor necrotic factor-α
- TREM2:
Triggering receptor expressed on myeloid cell 2
- WAT:
White adipose tissue
- VAM:
Vasculature-associated macrophages
- VLDLR:
Very low-density lipoprotein receptor.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Swinburn B. A., Sacks G., Hall K. D., et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378(9793):804–814. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- 2.Longo M., Zatterale F., Naderi J., et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. International Journal of Molecular Sciences. 2019;20(9) doi: 10.3390/ijms20092358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., Ferrante A. W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation. 2003;112(12):1796–1808. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanda H., Tateya S., Tamori Y., et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of Clinical Investigation. 2006;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosteli A., Sugaru E., Haemmerle G., et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. The Journal of Clinical Investigation. 2010;120(10):3466–3479. doi: 10.1172/JCI42845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cinti S., Mitchell G., Barbatelli G., et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of Lipid Research. 2005;46(11):2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Kim S. J., Feng D., Guillot A., et al. Adipocyte death preferentially induces liver injury and inflammation through the activation of chemokine (C-C motif) receptor 2-positive macrophages and lipolysis. Hepatology. 2019;69(5):1965–1982. doi: 10.1002/hep.30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strissel K. J., Stancheva Z., Miyoshi H., et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56(12):2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 9.Singh R. K., Barbosa-Lorenzi V. C., Lund F. W., Grosheva I., Maxfield F. R., Haka A. S. Degradation of aggregated LDL occurs in complex extracellular sub-compartments of the lysosomal synapse. Journal of Cell Science. 2016;129(5):1072–1082. doi: 10.1242/jcs.181743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coats B. R., Schoenfelt K. Q., Barbosa-Lorenzi V. C., et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Reports. 2017;20(13):3149–3161. doi: 10.1016/j.celrep.2017.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grijalva A., Xu X., Ferrante A. W., Jr. Autophagy is dispensable for macrophage-mediated lipid homeostasis in adipose tissue. Diabetes. 2016;65(4):967–980. doi: 10.2337/db15-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu X., Grijalva A., Skowronski A., van Eijk M., Serlie M. J., Ferrante A. W., Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metabolism. 2013;18(6):816–830. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kratz M., Coats B. R., Hisert K. B., et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metabolism. 2014;20(4):614–625. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang S. C., Everts B., Ivanova Y., et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature Immunology. 2014;15(9):846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee Y. H., Petkova A. P., Granneman J. G. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metabolism. 2013;18(3):355–367. doi: 10.1016/j.cmet.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varghese M., Griffin C., McKernan K., et al. Sex differences in inflammatory responses to adipose tissue lipolysis in diet-induced obesity. Endocrinology. 2019;160(2):293–312. doi: 10.1210/en.2018-00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caspar-Bauguil S., Kolditz C. I., Lefort C., et al. Fatty acids from fat cell lipolysis do not activate an inflammatory response but are stored as triacylglycerols in adipose tissue macrophages. Diabetologia. 2015;58(11):2627–2636. doi: 10.1007/s00125-015-3719-0. [DOI] [PubMed] [Google Scholar]
- 18.Brunner J. S., Vogel A., Lercher A., et al. The PI3K pathway preserves metabolic health through MARCO-dependent lipid uptake by adipose tissue macrophages. Nature Metabolism. 2020;2(12):1427–1442. doi: 10.1038/s42255-020-00311-5. [DOI] [PubMed] [Google Scholar]
- 19.Jonas M. I., Kurylowicz A., Bartoszewicz Z., et al. Interleukins 6 and 15 levels are higher in subcutaneous adipose tissue, but obesity is associated with their increased content in visceral fat depots. International Journal of Molecular Sciences. 2015;16(10):25817–25830. doi: 10.3390/ijms161025817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buzelle S. L., MacPherson R. E. K., Peppler W. T., Castellani L., Wright D. C. The contribution of IL-6 to beta 3 adrenergic receptor mediated adipose tissue remodeling. Physiological Reports. 2015;3(2, article e12312) doi: 10.14814/phy2.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perry R. J., Camporez J. P. G., Kursawe R., et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160(4):745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H., Dilbaz S., Coßmann J., et al. Breast milk alkylglycerols sustain beige adipocytes through adipose tissue macrophages. The Journal of Clinical Investigation. 2019;129(6):2485–2499. doi: 10.1172/JCI125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haase J., Weyer U., Immig K., et al. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia. 2014;57(3):562–571. doi: 10.1007/s00125-013-3139-y. [DOI] [PubMed] [Google Scholar]
- 24.Geng J., Zhang X., Prabhu S., et al. 3D microscopy and deep learning reveal the heterogeneity of crown-like structure microenvironments in intact adipose tissue. Science Advances. 2021;7(8) doi: 10.1126/sciadv.abe2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arner P., Langin D. Lipolysis in lipid turnover, cancer cachexia, and obesity-induced insulin resistance. Trends in Endocrinology and Metabolism. 2014;25(5):255–262. doi: 10.1016/j.tem.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Haka A. S., Barbosa-Lorenzi V. C., Lee H. J., et al. Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. Journal of Lipid Research. 2016;57(6):980–992. doi: 10.1194/jlr.M064089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braune J., Lindhorst A., Fröba J., et al. Multinucleated giant cells in adipose tissue are specialized in adipocyte degradation. Diabetes. 2021;70(2):538–548. doi: 10.2337/db20-0293. [DOI] [PubMed] [Google Scholar]
- 28.Petkevicius K., Virtue S., Bidault G., et al. Accelerated phosphatidylcholine turnover in macrophages promotes adipose tissue inflammation in obesity. eLife. 2019;8 doi: 10.7554/eLife.47990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Reilly M. E., Kajani S., Ralston J. C., Lenighan Y. M., Roche H. M., McGillicuddy F. C. Nutritionally derived metabolic cues typical of the obese microenvironment increase cholesterol efflux capacity of adipose tissue macrophages. Molecular Nutrition & Food Research. 2019;63(2):p. 1800713. doi: 10.1002/mnfr.201800713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aouadi M., Vangala P., Yawe J. C., et al. Lipid storage by adipose tissue macrophages regulates systemic glucose tolerance. American Journal of Physiology. Endocrinology and Metabolism. 2014;307(4):E374–E383. doi: 10.1152/ajpendo.00187.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma M., Schlegel M., Schlegel M., et al. Netrin-1 alters adipose tissue macrophage fate and function in obesity. Immunometabolism. 2019;1(2) doi: 10.20900/immunometab20190010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramkhelawon B., Hennessy E. J., Ménager M., et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nature Medicine. 2014;20(4):377–384. doi: 10.1038/nm.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaitin D. A., Adlung L., Thaiss C. A., et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. 2019;178(3):686–698.e14. doi: 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu C., Li P., Li H., et al. TREM2 regulates obesity-induced insulin resistance via adipose tissue remodeling in mice of high-fat feeding. Journal of Translational Medicine. 2019;17(1):p. 300. doi: 10.1186/s12967-019-2050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., Cella M., Mallinson K., et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160(6):1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Y., Chen C. Y., Li J., Cheng J. X., Jang M., Kim K. H. In vitro exploration of ACAT contributions to lipid droplet formation during adipogenesis. Journal of Lipid Research. 2018;59(5):820–829. doi: 10.1194/jlr.M081745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frambach S., de Haas R., Smeitink J. A. M., Rongen G. A., Russel F. G. M., Schirris T. J. J. Brothers in arms: ABCA1- and ABCG1-mediated cholesterol efflux as promising targets in cardiovascular disease treatment. Pharmacological Reviews. 2020;72(1):152–190. doi: 10.1124/pr.119.017897. [DOI] [PubMed] [Google Scholar]
- 38.Wei H., Tarling E. J., McMillen T. S., Tang C., LeBoeuf R. C. ABCG1 regulates mouse adipose tissue macrophage cholesterol levels and ratio of M1 to M2 cells in obesity and caloric restriction. Journal of Lipid Research. 2015;56(12):2337–2347. doi: 10.1194/jlr.M063354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tannahill G. M., Curtis A. M., Adamik J., et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Diepen J. A., Robben J. H., Hooiveld G. J., et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia. 2017;60(7):1304–1313. doi: 10.1007/s00125-017-4261-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill D. A., Lim H. W., Kim Y. H., et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(22):E5096–E5105. doi: 10.1073/pnas.1802611115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C., Menoret A., Farragher C., et al. Single cell transcriptomics based-MacSpectrum reveals novel macrophage activation signatures in diseases. JCI Insight. 2019;5 doi: 10.1172/jci.insight.126453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng J., Li A., Deng J., et al. miR-21 attenuates lipopolysaccharide-induced lipid accumulation and inflammatory response: potential role in cerebrovascular disease. Lipids in Health and Disease. 2014;13(1) doi: 10.1186/1476-511X-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castoldi A., Monteiro L. B., van Teijlingen Bakker N., et al. Triacylglycerol synthesis enhances macrophage inflammatory function. Nature Communications. 2020;11(1) doi: 10.1038/s41467-020-17881-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin K. C., Hwang I., Choe S. S., et al. Macrophage VLDLR mediates obesity-induced insulin resistance with adipose tissue inflammation. Nature Communications. 2017;8(1) doi: 10.1038/s41467-017-01232-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Na Y. R., Gu G. J., Jung D., et al. GM-CSF induces inflammatory macrophages by regulating glycolysis and lipid metabolism. Journal of Immunology. 2016;197(10):4101–4109. doi: 10.4049/jimmunol.1600745. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe R., Hilhorst M., Zhang H., et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight. 2018;3(20) doi: 10.1172/jci.insight.123047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boutens L., Hooiveld G. J., Dhingra S., Cramer R. A., Netea M. G., Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. 2018;61(4):942–953. doi: 10.1007/s00125-017-4526-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chimin P., Andrade M. L., Belchior T., et al. Adipocyte mTORC1 deficiency promotes adipose tissue inflammation and NLRP3 inflammasome activation via oxidative stress and de novo ceramide synthesis. Journal of Lipid Research. 2017;58(9):1797–1807. doi: 10.1194/jlr.M074518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choromańska B., Myśliwiec P., Łuba M., et al. A longitudinal study of the antioxidant barrier and oxidative stress in morbidly obese patients after bariatric surgery. Does the metabolic syndrome affect the redox homeostasis of obese people? Journal of Clinical Medicine. 2020;9(4) doi: 10.3390/jcm9040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Acín-Pérez R., Iborra S., Martí-Mateos Y., et al. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nature Metabolism. 2020;2(9):974–988. doi: 10.1038/s42255-020-00273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang F., Liu Y., Ren H., Zhou G., Yuan X., Shi X. ER-stress regulates macrophage polarization through pancreatic EIF-2alpha kinase. Cellular Immunology. 2019;336:40–47. doi: 10.1016/j.cellimm.2018.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Shan B., Wang X., Wu Y., et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nature Immunology. 2017;18(5):519–529. doi: 10.1038/ni.3709. [DOI] [PubMed] [Google Scholar]
- 55.Kang S., Nakanishi Y., Kioi Y., et al. Semaphorin 6D reverse signaling controls macrophage lipid metabolism and anti-inflammatory polarization. Nature Immunology. 2018;19(6):561–570. doi: 10.1038/s41590-018-0108-0. [DOI] [PubMed] [Google Scholar]
- 56.van Dierendonck X., de la Rosa Rodriguez M. A., Georgiadi A., et al. HILPDA uncouples lipid droplet accumulation in adipose tissue macrophages from inflammation and metabolic dysregulation. Cell Reports. 2020;30(6):1811–1822.e6. doi: 10.1016/j.celrep.2020.01.046. [DOI] [PubMed] [Google Scholar]
- 57.Jung S. B., Choi M. J., Ryu D., et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nature Communications. 2018;9(1) doi: 10.1038/s41467-018-03998-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang A., Mottillo E. P. Adipocyte lipolysis: from molecular mechanisms of regulation to disease and therapeutics. The Biochemical Journal. 2020;477(5):985–1008. doi: 10.1042/BCJ20190468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flaherty S. E., 3rd, Grijalva A., Xu X., Ables E., Nomani A., Ferrante A. W., Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science. 2019;363(6430):989–993. doi: 10.1126/science.aaw2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sohn J. H., Lee Y. K., Han J. S., et al. Perilipin 1 (Plin1) deficiency promotes inflammatory responses in lean adipose tissue through lipid dysregulation. The Journal of Biological Chemistry. 2018;293(36):13974–13988. doi: 10.1074/jbc.RA118.003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu X., Cifarelli V., Sun S., Kuda O., Abumrad N. A., Su X. Major role of adipocyte prostaglandin E2 in lipolysis- induced macrophage recruitment. Journal of Lipid Research. 2016;57(4):663–673. doi: 10.1194/jlr.M066530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gartung A., Zhao J., Chen S., et al. Characterization of Eicosanoids Produced by Adipocyte Lipolysis. The Journal of Biological Chemistry. 2016;291(31):16001–16010. doi: 10.1074/jbc.M116.725937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim K. H., Kim Y. H., Son J. E., et al. Intermittent fasting promotes adipose thermogenesis and metabolic homeostasis via VEGF-mediated alternative activation of macrophage. Cell Research. 2017;27(11):1309–1326. doi: 10.1038/cr.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Asterholm I. W., McDonald J., Blanchard P. G., et al. Lack of "immunological fitness" during fasting in metabolically challenged animals. Journal of Lipid Research. 2012;53(7):1254–1267. doi: 10.1194/jlr.M021725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Camell C. D., Sander J., Spadaro O., et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature. 2017;550(7674):119–123. doi: 10.1038/nature24022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silva H. M., Báfica A., Rodrigues-Luiz G. F., et al. Vasculature-associated fat macrophages readily adapt to inflammatory and metabolic challenges. The Journal of Experimental Medicine. 2019;216(4):786–806. doi: 10.1084/jem.20181049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bu Y., Okunishi K., Yogosawa S., et al. Insulin regulates lipolysis and fat mass by upregulating growth/differentiation factor 3 in adipose tissue macrophages. Diabetes. 2018;67(9):1761–1772. doi: 10.2337/db17-1201. [DOI] [PubMed] [Google Scholar]
- 68.Satoh T., Kidoya H., Naito H., et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature. 2013;495(7442):524–528. doi: 10.1038/nature11930. [DOI] [PubMed] [Google Scholar]
- 69.Wang L., Zhao R. P., Song X. Y., Wu W. F. Targeting ERβ in Macrophage Reduces Crown-like Structures in Adipose Tissue by Inhibiting Osteopontin and HIF-1α. Scientific Reports. 2019;9(1) doi: 10.1038/s41598-019-52265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frühbeck G., Méndez-Giménez L., Fernández-Formoso J. A., Fernández S., Rodríguez A. Regulation of adipocyte lipolysis. Nutrition Research Reviews. 2014;27(1):63–93. doi: 10.1017/S095442241400002X. [DOI] [PubMed] [Google Scholar]
- 71.Iwamura Y., Mori M., Nakashima K., et al. Apoptosis inhibitor of macrophage (AIM) diminishes lipid droplet-coating proteins leading to lipolysis in adipocytes. Biochemical and Biophysical Research Communications. 2012;422(3):476–481. doi: 10.1016/j.bbrc.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 72.Zoico E., di Francesco V., Olioso D., et al. In vitro aging of 3T3-L1 mouse adipocytes leads to altered metabolism and response to inflammation. Biogerontology. 2010;11(1):111–122. doi: 10.1007/s10522-009-9236-0. [DOI] [PubMed] [Google Scholar]
- 73.Han M. S., White A., Perry R. J., et al. Regulation of adipose tissue inflammation by interleukin 6. Proceedings of the National Academy of Sciences of the United States of America. 2020;117(6):2751–2760. doi: 10.1073/pnas.1920004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dowal L., Parameswaran P., Phat S., et al. Intrinsic properties of brown and white adipocytes have differential effects on macrophage inflammatory responses. Mediators of Inflammation. 2017;2017:11. doi: 10.1155/2017/9067049.9067049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamashita A., Soga Y., Iwamoto Y., et al. Macrophage-adipocyte interaction: marked interleukin-6 production by lipopolysaccharide. Obesity. 2007;15(11):2549–2552. doi: 10.1038/oby.2007.305. [DOI] [PubMed] [Google Scholar]
- 76.Zhang W., Mottillo E. P., Zhao J., et al. Adipocyte Lipolysis-stimulated Interleukin-6 Production Requires Sphingosine Kinase 1 Activity. The Journal of Biological Chemistry. 2014;289(46):32178–32185. doi: 10.1074/jbc.M114.601096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mohamed-Ali V., Flower L., Sethi J., et al. β-adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies. The Journal of Clinical Endocrinology and Metabolism. 2001;86(12):5864–5869. doi: 10.1210/jcem.86.12.8104. [DOI] [PubMed] [Google Scholar]
- 78.Fujimoto Y., Hashimoto O., Shindo D., et al. Metabolic changes in adipose tissues in response to β3‐adrenergic receptor activation in mice. Journal of Cellular Biochemistry. 2019;120(1):821–835. doi: 10.1002/jcb.27443. [DOI] [PubMed] [Google Scholar]
- 79.Ghanemi A., St-Amand J. Interleukin-6 as a "metabolic hormone". Cytokine. 2018;112:132–136. doi: 10.1016/j.cyto.2018.06.034. [DOI] [PubMed] [Google Scholar]
- 80.Sindhu S., Thomas R., Shihab P., Sriraman D., Behbehani K., Ahmad R. Obesity is a positive modulator of IL-6R and IL-6 expression in the subcutaneous adipose tissue: significance for metabolic inflammation. PLoS One. 2015;10(7, article e0133494) doi: 10.1371/journal.pone.0133494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuo F. C., Huang Y. H., Lin F. H., et al. Circulating soluble IL-6 receptor concentration and visceral adipocyte size are related to insulin resistance in Taiwanese adults with morbid obesity. Metabolic Syndrome and Related Disorders. 2017;15(4):187–193. doi: 10.1089/met.2016.0135. [DOI] [PubMed] [Google Scholar]
- 82.Kraakman M. J., Kammoun H. L., Allen T. L., et al. Blocking IL-6 trans -Signaling Prevents High-Fat Diet-Induced Adipose Tissue Macrophage Recruitment but Does Not Improve Insulin Resistance. Cell Metabolism. 2015;21(3):403–416. doi: 10.1016/j.cmet.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 83.Braune J., Weyer U., Hobusch C., et al. IL-6 regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. Journal of Immunology. 2017;198(7):2927–2934. doi: 10.4049/jimmunol.1600476. [DOI] [PubMed] [Google Scholar]
- 84.Mauer J., Chaurasia B., Goldau J., et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nature Immunology. 2014;15(5):423–430. doi: 10.1038/ni.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qing H., Desrouleaux R., Israni-Winger K., et al. Origin and function of stress-induced IL-6 in murine models. Cell. 2020;182(6):p. 1660. doi: 10.1016/j.cell.2020.08.044. [DOI] [PubMed] [Google Scholar]
- 86.Mottillo E. P., Shen X. J., Granneman J. G. β3-adrenergic receptor induction of adipocyte inflammation requires lipolytic activation of stress kinases p38 and JNK. Biochimica et Biophysica Acta. 2010;1801(9):1048–1055. doi: 10.1016/j.bbalip.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Frisdal E., Lesnik P., Olivier M., et al. Interleukin-6 Protects Human Macrophages from Cellular Cholesterol Accumulation and Attenuates the Proinflammatory Response. The Journal of Biological Chemistry. 2011;286(35):30926–30936. doi: 10.1074/jbc.M111.264325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liao H. S., Matsumoto A., Itakura H., et al. Transcriptional inhibition by interleukin-6 of the class A macrophage scavenger receptor in macrophages derived from human peripheral monocytes and the THP-1 monocytic cell line. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(8):1872–1880. doi: 10.1161/01.ATV.19.8.1872. [DOI] [PubMed] [Google Scholar]
- 89.Steinhauser M. L., Olenchock B. A., O’Keefe J., et al. The circulating metabolome of human starvation. JCI Insight. 2018;3(16) doi: 10.1172/jci.insight.121434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wueest S., Item F., Boyle C. N., et al. Interleukin-6 contributes to early fasting-induced free fatty acid mobilization in mice. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2014;306(11):R861–R867. doi: 10.1152/ajpregu.00533.2013. [DOI] [PubMed] [Google Scholar]
- 91.Wedell-Neergaard A. S., Lang Lehrskov L., Christensen R. H., et al. Exercise-induced changes in visceral adipose tissue mass are regulated by IL-6 signaling: a randomized controlled trial. Cell Metabolism. 2019;29(4):844–855.e3. doi: 10.1016/j.cmet.2018.12.007. [DOI] [PubMed] [Google Scholar]
- 92.Wueest S., Item F., Lucchini F. C., et al. Mesenteric fat lipolysis mediates obesity-associated hepatic steatosis and insulin resistance. Diabetes. 2016;65(1):140–148. doi: 10.2337/db15-0941. [DOI] [PubMed] [Google Scholar]
- 93.Han J., Meng Q., Shen L., Wu G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids in Health and Disease. 2018;17(1):p. 14. doi: 10.1186/s12944-018-0657-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Serrano-Marco L., Rodriguez-Calvo R., el Kochairi I., et al. Activation of peroxisome proliferator-activated Receptor- /- (PPAR- /- ) Ameliorates Insulin Signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6-stimulated adipocytes. Diabetes. 2011;60(7):1990–1999. doi: 10.2337/db10-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hu W., Ru Z., Zhou Y., et al. Lung cancer-derived extracellular vesicles induced myotube atrophy and adipocyte lipolysis via the extracellular IL-6-mediated STAT3 pathway. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids. 2019;1864(8):1091–1102. doi: 10.1016/j.bbalip.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 96.Williams J. A., Pontzer C. H., Shacter E. Regulation of macrophage interleukin-6 (IL-6) and IL-10 expression by prostaglandin E2: the role of p38 mitogen-activated protein kinase. Journal of Interferon & Cytokine Research. 2000;20(3):291–298. doi: 10.1089/107999000312423. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y. C., Hu Y. W., Sha Y. H., et al. Ox-LDL upregulates IL-6 expression by enhancing NF-κB in an IGF2-dependent manner in THP-1 macrophages. Inflammation. 2015;38(6):2116–2123. doi: 10.1007/s10753-015-0194-1. [DOI] [PubMed] [Google Scholar]