

Abstract
Phospholipids are key components of cellular membranes and are emerging as important functional regulators of different membrane proteins, including pentameric ligand-gated ion channels (pLGICs). Here, we take advantage of the prokaryote channel ELIC (Erwinia ligand-gated ion channel) as a model to understand the determinants of phospholipid interactions in this family of receptors. A high-resolution structure of ELIC in a lipid-bound state reveals a phospholipid site at the lower half of pore-forming transmembrane helices M1 and M4 and at a nearby site for neurosteroids, cholesterol or general anesthetics. This site is shaped by an M4-helix kink and a Trp–Arg–Pro triad that is highly conserved in eukaryote GABAA/C and glycine receptors. A combined approach reveals that M4 is intrinsically flexible and that M4 deletions or disruptions of the lipid-binding site accelerate desensitization in ELIC, suggesting that lipid interactions shape the agonist response. Our data offer a structural context for understanding lipid modulation in pLGICs.
Increasing knowledge of membrane protein structures is shedding light on the molecular interplay between lipids and proteins that regulates both membrane protein and lipid bilayer function1–6. For pLGICs, this interplay has been the subject of intense investigation since the earliest attempts at defining the structural domains responsible for ligand binding and channel gating in the nicotinic acetylcholine receptor (nAChR)7. Although these studies suggest that lipids regulate nAChR function by stabilizing different proportions of activatable versus nonactivatable conformations, and by controlling the transitions between these states, the molecular interactions that underlie these mechanisms remain poorly defined8–11.
All members of the pLGIC family share a common topology, composed of an extracellular ligand-binding domain, a pore-forming transmembrane domain and an intracellular regulatory domain, which is absent in prokaryote receptors. The ligand-binding domain of each subunit is largely composed of β-sheets, β1–10, which harbor the residues on ‘loops’ A–C of the principal (+) subunit and ‘loops’ D–F of the complementary (−) subunit that together form the neurotransmitter binding site. The transmembrane pore domain (TMD) of each subunit is α-helical, M1–M4, with the M2 helix facing the central ion pore and the M4 helix facing the lipid membrane.
A wide range of experimental evidence obtained from different pLGICs has highlighted a role for the outermost transmembrane α-helix, M4, of each subunit in both channel function and lipid sensing12–16. M4 is conformationally dynamic during channel gating17. M4 is implicated in lipid-dependent desensitization18,19. Mutations along the lipid–protein interface influence channel gating, demonstrating that altered M4–lipid interactions influence channel function20,21. M4 may act as a lipid sensor, influencing channel function via interactions with the adjacent transmembrane α-helices, M1 and M3 (refs. 22–24).
An important role for M4 in mediating lipid–protein interactions is further suggested by recent structures, which have identified lipid recognition sites in pLGICs. One crystal structure of the bacterial homolog, GLIC, reveals three phospholipids bound at the interface between M4 and M1/M3 in each of the five pLGIC subunits25, although the bound lipid in the cytoplasmic leaflet extends to interact with M3 from the adjacent subunit. Interestingly, the binding of one of these lipids to the M4 and M1/M3 interface is displaced by the inhibitory anesthetic, propofol26. A cytoplasmic leaflet site at the interface between M4 and M1/M3 has also been shown to bind neurosteroids in various α1- or α5-based GABAA receptor chimeras27–29. The M3/M4 interface also serves as a binding site for cholesterol or PIP2 in structures of the heteromeric human GABAA receptor30,31.
Here, we take advantage of the prokaryotic channel ELIC as a model to understand the structural determinants of lipid action at this family of receptors. Using nanobodies to facilitate structural studies, we determined new ELIC structures, revealing bound lipids as well as distinct conformational states of the M4 helix. A phospholipid site is formed in the lower half of the pore-forming transmembrane domain near a previously identified binding site for neurosteroids, cholesterol and general anesthetics. A major part of this site is shaped by the M4 α-helix, which contains a characteristic kink at a conserved proline residue and is also found in eukaryote anion-selective channels. Using mutagenesis studies, we relate structure to function and demonstrate that perturbations of the lipid-binding site accelerate desensitization in ELIC, a phenomenon that is mimicked by reconstitution of ELIC into membranes with different lipid compositions. Together with mutant ELIC structures, crosslinking studies, molecular dynamics (MD) simulations and voltage clamp fluorometry (VCF), we further demonstrate the intrinsic flexibility of M4, thus providing novel mechanistic detail into the role of M4 in lipid sensing and offering a structural context for understanding desensitization in the family of pLGICs.
Results
High-resolution ELIC structure in a lipid-bound state.
In this study, we took advantage of nanobodies, which are antibody fragments derived from llamas that facilitate structural studies by promoting crystallogenesis and by stabilizing conformationally transient states32. This is especially useful for the prokaryote channel ELIC, for which almost 30 structures have been determined, all in the same nonconducting conformation and at relatively moderate resolution33. We screened a range of ELIC constructs, in combination with different nanobodies, to identify previously unresolved conformational states and crystals with improved diffraction quality. We focus here on the structure of one of these constructs, ELIC with a cysteine mutation at the 7′ position of the pore-lining M2 helix bound to nanobody 72 (Nb72). This structure was solved at a resolution of 2.5 Å (Supplementary Table 1), which is the highest resolution yet for an ELIC structure (Fig. 1a,b). At this resolution, densities that were absent from previous lower resolution structures are now revealed, enabling a more detailed view with bound ions, lipids and detergent molecules (Supplementary Figs. 1–3). ELIC 7′C is functionally almost identical to wild-type ELIC (half-maximum effective concentration (EC50) value of GABA = 7.6 ± 0.3 mM, n = 9), but gave better electron density for bound lipids.
Fig. 1 |. High-resolution ELiC structure in a lipid-bound state.
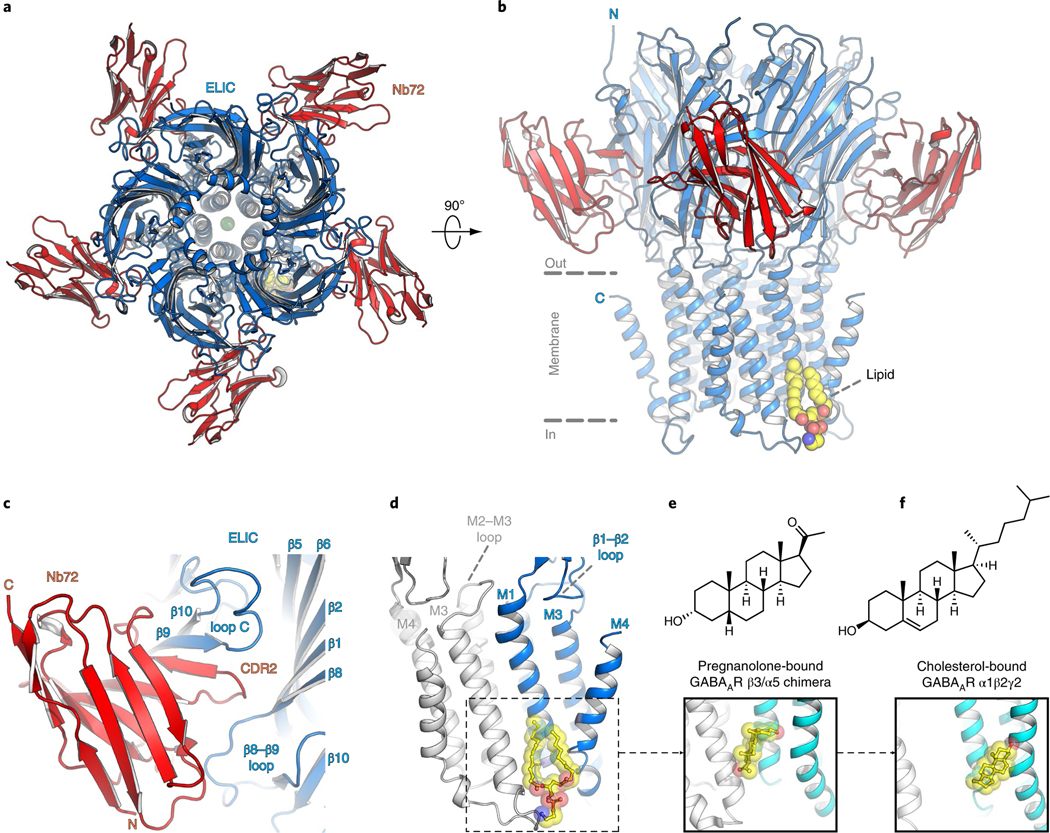
a,b, Top and side views of the crystal structure of ELIC 7′C pore mutant (blue) in complex with nanobody 72 (Nb72, red) in a lipid-bound state. Protein is shown in cartoon representation, lipid in sphere representation. Yellow corresponds to carbon, blue to nitrogen and red to oxygen. c, Detailed view of the interaction between Nb72 and orthosteric binding site in ELIC, including loop C. d, Detailed view of the lipid interaction at a transmembrane site. One subunit is shown in blue and its neighboring subunit in white. PE is shown in transparent sphere and sticks. The lipid site is formed at the M1–M4 interface of one subunit and M3 of the neighboring subunit. This site overlaps with a known neurosteroid binding site in the β3/α5 chimeric GABAA-R29 (e) and a cholesterol-binding site in the α1β2γ2 GABAA-R30 (f).
The ELIC pentamer is bound by five Nb72 molecules, one to each monomer. Nb72 interacts with the extracellular ligand-binding domain forming extensive contacts on each subunit with the β9 and β10 strands of the C-loop, which forms a flexible part of the orthosteric binding site, as well as with N-terminal residues of the β6–β7 loop. The β6–β7 loop extends down toward the TMD and plays a role in coupling agonist binding to channel gating. The nanobody also extends across the subunit interface to form contacts with the β8–β9 loop of the adjacent subunit. These intersubunit contacts could prevent intersubunit movements to stabilize the receptor in a single conformational state (Fig. 1c and Supplementary Figs. 1 and 2). Indeed, functional data show that Nb72 reduces the agonist-induced response (Supplementary Fig. 4).
The Nb72-stabilized conformation of ELIC is similar to previously published structures, although we observe that the 7′C pore is slightly more closed compared to wild-type ELIC (Supplementary Fig. 5). At the 6′ pore position, the T237 side chain points toward the center of the pore and interacts with a sodium ion (Supplementary Fig. 3). Significantly, we identify a lipid molecule at a site in the lower half of the transmembrane domain located between the M1 and M4 helices of one subunit and the M3 helix of a neighboring subunit (Fig. 1b,d). We identify the lipid as phosphatidylethanolamine (PE) based on electron density features (Supplementary Fig. 3) and in line with biochemical conditions. Indeed, PE is the most abundant lipid component of the Escherichia coli lipid extract (58% PE, 15% phosphatidyl glycerol (PG), and 10% cardiolipin), which is added as an essential part of the crystallization mix. The lipid site lies close to a previously identified binding site for neurosteroids in α1- or α5-based GABAA receptor chimeras27–29 as well as a cholesterol-binding site in the α1β2γ2 GABAA receptor structure30 (Fig. 1d–f). This conservation of a lipid-binding site between prokaryote and eukaryote channels suggests an important functional role. In addition to lipids and ions, we are able to assign a previously unexplained region of density in lower resolution ELIC structures, near W206 at the top of the M1 helix, as the head group of an undecylmaltoside detergent molecule (Supplementary Fig. 3). The lipid tail of the undecylmaltoside molecule points down and in between the upper half of the M3 and M4 helices. Together, these results offer a detailed view of the ELIC channel and interacting ions, lipids and detergents.
Lipid site conservation in ELIC and eukaryote channels.
A detailed view of amino acids composing the lipid-binding site in ELIC (Fig. 2a,b) reveals that this site is shaped in part by a characteristic kink, induced by P305, in the M4 helix. This orients R301 one helical turn down along M4 to interact with W224 in the M1 helix via a cation–π interaction (Fig. 2b). Consequently, W224 is oriented nearly perpendicular to the membrane and creates a wedge between the two lipid tails of the PE lipid molecule (Fig. 2b). This triad of interactions W–R–P is strictly conserved between ELIC and anion-selective eukaryote pLGICs (shown in the sequence alignment, Supplementary Fig. 6) but not in cation-selective eukaryote pLGICs, and suggests an important structural and functional role. In addition to the triad residues, we find other aromatic residues in M4, F308, and M1, W220, which are also strongly conserved. These residues on the complementary (−) subunit form the majority of the lipid-binding site, with additional residues, F274 (strongly conserved), L278 (semiconserved) and I281 (Ala in most anionic pLGICs), on the M3 helix of the principal (+) subunit also contributing to lipid binding (Fig. 2b). Consequently, the sequence alignment reveals characteristic motifs in anion-selective pLGICs, namely SWxxFW (M1), FxALx (M3) and SRxxFPxxFx (M4), with residues of the triad interaction underlined. From a structural perspective (Fig. 2d), the W–R–P triad creates a characteristic bend in the M4 helix, which is observed in all anion-selective pLGICs determined to date, α1 and α3 GlyR34,35, human β3 GABAAR36, human α1β(1–3)γ2 GABAAR30,31,37,38, glutamate-gated chloride channel from Caenorhabditis elegans GluCl39,40, but not in cation-selective pLGICs, for example human α4β2 nAChR41,42. In this last example, the triad amino acids structurally align with Y–I–L (Fig. 2d). As a consequence of M4 helix bending in anion-selective pLGICs the top of M4 moves back toward the M1 and M3 helices and the conserved Cys-loop, forming an interaction interface that plays a crucial role in the coupling of ligand binding to the channel opening. The absence of the M4 helix bend in cation-selective pLGICs suggests that the two branches of the family have evolved different signaling mechanisms via M4.
Fig. 2 |. Lipid interactions at a highly conserved Pro kink in the M4 helix.

a,b, Surface/sticks and cartoon/sticks representation of the lipid-binding site in ELIC. One subunit is shown in blue, the neighboring subunit in white, and lipid in yellow (carbon). The dashed line indicates a cation–π interaction between R301 and W224. c, Electrophysiological recordings of purified ELIC reconstituted in vesicles with known lipid content. All recordings were repeated in 4 to 26 independent experiments. The time constant for desensitization (τ) is mentioned as the mean ± s.e.m. d, Architectural conservation of the M4 helix Pro kink and cation–π (R–W) interaction in anion-selective eukaryotic receptors, but not in cation-selective receptors, for example, α4β2 nAChR41,42.
Lipids influence rate of agonist-induced desensitization.
Preliminary studies suggested that in defined membranes composed exclusively of phosphatidylcholine (PC), ELIC is locked in a nonconducting state, while in membranes composed of soybean asolectin, ELIC responds to agonist, albeit with enhanced rates of desensitization23. Using electrophysiological recordings from giant unilamellar vesicles, we confirm here that ELIC in membranes composed of soybean asolectin, a complex mixture of phospholipids, responds to agonist, but with relatively rapid desensitization kinetics (Fig. 2c and Supplementary Table 2). Desensitization is slowed in membranes composed of asolectin with 25% cholesterol, an intriguing observation given that the PE binding site in ELIC overlaps with the cholesterol-binding site in the α1β2γ2 GABAA receptor30. Strikingly, the rapid decay of the agonist-induced response in asolectin is eliminated when ELIC is reconstituted into either PC/ PG or PC/PG/PE membranes (Fig. 2c and Supplementary Table 2). These functional data show that lipids have a profound effect on agonist-induced desensitization and thus have the capacity to shape the agonist-induced response.
Lipid site mutagenesis accelerates ELIC desensitization.
We used single point mutations with channels expressed in Xenopus oocytes to test whether the composition of the lipid-binding site plays a role in ELIC function. Single Ala mutations of implicated residues (Fig. 2b and Supplementary Fig. 7) lead to relatively small changes in the EC50 for the agonist cysteamine relative to the EC50 of wild-type ELIC16 (Supplementary Table 3), suggesting that the lipid-binding site does not strongly affect receptor activation. The F274A, I281A, W220A and P305A mutations, however, each lead to a more rapid desensitizing phenotype (Fig. 3a,b and Supplementary Fig. 7), as did the W224R mutation, which should disrupt the lipid-binding site through electrostatic repulsion with the adjacent R301 on M4. Significantly, P305A, which is expected to disrupt the M4-helix kink that shapes the lipid-binding site, exhibits a particularly rapid rate of desensitization (Fig. 3b). To reposition the kink in the M4 helix we inserted new Pro residues onto the P305A background, with each Pro insertion occurring either one turn higher (P305A+L309P) or one turn lower (P305A+R301P) or two turns lower (P305A+I297P) on the M4 α-helix (Fig. 3c–e). Each of these mutants exhibits more rapid desensitization rates than wild-type ELIC, but less rapid than ELIC with an unkinked M4 (P305A). To quantify the differences in desensitization kinetics for all mutants, we measured the time constant for the rate of decay of the whole-cell agonist-induced response over multiple agonist concentrations. We then plotted the reciprocal of the decay constant versus the agonist concentration (Supplementary Fig. 8) and summarize slope values in Supplementary Table 4. Note that desensitization of the wild-type ELIC is too slow to accurately determine a slope value (slope value < 0.01 in Supplementary Table 4).
Fig. 3 |. Site-directed mutagenesis of the ELiC M4 helix accelerates desensitization.

a–h, Whole-cell two-electrode voltage clamp electrophysiological traces recorded from oocytes expressing wild-type ELIC (a), P305A mutant (b), M4 proline-shifted (c–e) and M4 deletion (f–h) mutants of ELIC. Ligand (cysteamine) concentration jumps (mM) are indicated by the horizontal bars. Representative traces are shown from four to eight independent experiments for each construct. Cartoon representations of the M4 helix indicate the proline-induced kink and deletion sites.
The P305A mutation and additional Pro insertions on the P305A background probably not only distort the lipid-binding pocket or affect lipid binding, but more generally alter how the M4 helix interacts with the remainder of the TMD. To further probe the role of M4–M1/M3 interactions in channel function, we deleted progressive parts of M4. Functional recordings revealed that the deletion of up to seven residues in ELIC has little effect on either expression or function, with all the C-terminal deletion mutants exhibiting EC50 values for the agonist cysteamine that are within 0.5 log units of the wild-type EC50 value (Fig. 3f). Notably, these deletions are not tolerated in the related prokaryote channel GLIC, where deletion of the final three or four residues at the M4 C terminus leads to a strong reduction or a complete loss of expression and/or function, respectively15. Additionally, deletions of the entire M4 are also not tolerated in 5-HT3Rs or GlyRs13. Remarkably, even ELIC mutants with up to eight or more residues deleted from the M4 C terminus (Δ7 and Δ8, Fig. 3f,g), including a mutant that lacked the entire M4 α-helix (ΔM4, Fig. 3h), still gave rise to robust agonist-induced currents. In each case, however, the agonist-induced currents exhibit very rapid desensitization compared to both wild-type ELIC and the ELIC deletion mutants with up to seven residues removed from the C terminus (Δ7, Fig. 3f). The rapid desensitization of the ELIC deletion mutants is similar to that observed for the therapeutically important α7 nAChR (Supplementary Fig. 8 and Supplementary Table 4), which is one of the fastest desensitizing pLGICs in this channel family. We determined that the M4 deletion mutants have rates of agonist-dependent desensitization similar to that of P305A (Supplementary Fig. 8 and Supplementary Table 4). These results demonstrate that perturbations of the M4 helix and specifically the residues that compose the lipid-binding site profoundly affect desensitization, consistent with the above observation that the lipid composition of the environment surrounding ELIC influences desensitization.
Mutant structure and MD simulation reveal M4 flexibility.
To obtain a structural context for understanding M4 dynamics we used X-ray crystal structures and MD simulations of M4 deletion mutants and P305A single mutants. We solved the crystal structure of the Nb72-bound ELIC Δ8 at 2.78 Å resolution (Fig. 4a), which is the construct displaying the fastest desensitization in Xenopus oocytes. Remarkably, proper density is lacking for the whole M4 helix, the M3–M4 linker and the bound lipid. The absence of density, extending far beyond the eight C-terminal residues, indicates that the remainder of M4 and the connecting loop are intrinsically disordered in ELIC Δ8.
Fig. 4 |. M4-helix truncation increases the dynamics of the lower half of the channel pore region.

a, Cartoon ‘putty’ representation of the ELIC 7′C structure and ELIC Δ8 deletion mutant both in complex with Nb72 (not shown). Residues are colored according to the average temperature B-factor, ranging from 25 Å2 (blue) to 250 Å2 (red) and for intermediate values according to the color shading indicated in the spectrum bar. The M4 helix and M3–M4 linker are structurally flexible in the ELIC Δ8 structure and are indicated with a gray dashed line. b, Plot of the average B-factor per residue as a function of residue number for ELIC 7’C and ELIC Δ8 structures. c, MD simulations of ELIC 7’C (blue) and P305A (red) reveal that the M4 undergoes a conformational change in P305A, which unkinks the helix and causes a conformational tilt toward the complementary subunit. The solid line represents the average of five subunits and the shading represents the s.e.m. The dashed line indicates the distance measured between the Cα of residue V316 in both conformations.
Additionally, we also observe that the entire lower half of the M1–M3 transmembrane pore domain becomes less resolved in the electron density map, resulting in a marked increase of the average B-factor per residue, which is used as an indicator of vibrational movement in different parts of the structure. A visual representation of the B-factor distribution is given by the familiar sausage or ‘putty’ representation in Fig. 4a with yellow-red residues having the highest B-factors. The plot of average B-factors per residue (Fig. 4b) indicates that the B-factor distribution in the extracellular domain is overall similar between ELIC and ELIC Δ8, with the most pronounced increase seen for the tip of loop C (residues 175–185). The biggest difference for ELIC Δ8 is seen in the lower half of the transmembrane pore domain, with B-factors sharply rising for the lower half of M1 (residues 214–224), the M1–M2 linker (225–230), the lower half of M2 (231–241) and the lower half of M3 (274–285) (Fig. 4b).
MD simulations also indicate a particularly dynamic Δ8 M4 helix: the r.m.s. displacement of the ELIC M4 backbone is 4.3 ± 0.5 Å (n = 5) in the ELIC Δ8 structure, compared to 3.6 ± 0.2 Å (n = 5) in the reference ELIC structure. The largest displacement observed in a single subunit is 6.3 Å for ELIC Δ8 and 4.1 Å for wild type. This result confirms the enhanced flexibility of the M4 helix in the Δ8 mutant.
Together, the ELIC Δ8 structure and dynamics suggest that the M4 is intrinsically flexible and probably dissociates from the bound lipid. In addition, the Δ8 mutation leads to destabilization of the lower half of the pore domain. Previous deletion and complementation studies have shown that constructs of the human GlyR and mouse 5-HT3AR lacking M4 do not form pentamers and are not functional13. These truncated constructs can be rescued by co-expression of M4 (ref. 13). In contrast, ELIC still forms functional pentamers when part of, or the entire, M4 is removed. The ELIC Δ8 structure, with its destabilized lower membrane domain, provides a mechanistic hint on how the absence of M4 could lead, in more fragile receptors, to the loss of receptor integrity. In particular, recent studies have suggested that the activation and desensitization gates of pLGICs are distinct, with the desensitization gate located near the lower half (the intracellular side) of the M2 helices43. The increased dynamics of the lower half of the pore domain with the Δ8 mutation may directly influence the dynamics of the desensitization gate leading to the fast desensitizing phenotype.
To further substantiate our understanding of M4 dynamics we conducted MD simulations of ELIC P305A, which produces fast desensitizing channels. In the MD simulations, we observe that M4 changes its conformation and undergoes a straightening at the Pro kink, effectively forming a continuous transmembrane helix rather than two helices separated by the Pro kink in the reference structure. In addition, the simulations indicate that the orientation of M4 is highly dependent on lipid exposure to the M1 and M3 helices. The analysis indicates a continuum of conformational shifts between two principal directions. In the first direction, the conformational change of the M4 helix results in a tilt toward the complementary (−) subunit and amounts to a distance of 5.0–7.5 Å when measured between the Cα atoms of the carboxyterminal residue V316 in both conformations, after 450 ns of simulation (Fig. 4c). By 450 ns, M4 is continuing to tilt, and this distance will likely increase further as the structure reaches equilibrium. In the second principal direction, the simulations suggest that the M4 helix could tilt away from the pore domain and into the membrane, similar to an uncoupled conformation9. These MD simulations are consistent with the observations from the crystal structures presented in this and the subsequent section that the M4 helix is intrinsically flexible and possibly adopts different conformations depending on the lipid environment.
ELIC structure with an alternate conformation of M4.
In over more than a decade of structural studies on ELIC, we have obtained three different datasets in a resolution range of 3.45–3.92 Å that each have several subunits with M4 in an alternate conformation compared to previously published structures of pLGICs (Supplementary Fig. 9). These structures were obtained serendipitously and might be the result of subtle variations in the solubilization and/or relipidation of ELIC before crystallization. Additionally, a detailed survey of the Protein Data Bank (PDB) revealed several ELIC structures solved by other groups and that contain densities indicating occupancy of M4 in this same alternate conformation. Examples are ELIC F16′A (PDB code 2yks)44 and ELIC F16′L (PDB code 3uq4)45 (Supplementary Fig. 10), suggesting that a 16′ pore mutation might stabilize the alternate M4 conformation in ELIC.
Figure 5a shows the 3.45 Å resolution structure of ELIC with pore mutation F16′S, which illustrates this alternate M4 conformation. In this structure, the M4 helix is not kinked post R301 but instead continues along the helical trajectory of the pre-301 residues. Consequently, the M4 helix of one subunit extends over to form intersubunit interactions with the M3 helix of its neighboring subunit (Supplementary Videos 1 and 2). This amounts to a movement of 19.4 Å when measured between the Cα atoms of I317 in the two conformations. In its new conformation, M4 is pinched between W220 and W224 in M1, which we identified as key components of the lipid-binding site. Consequently, access to the lipid-binding site is sterically hindered. This alternate conformation could represent a mechanism for modulating lipid binding to the ELIC channel, or vice versa. Among the newly formed interactions are residues F304, F308 and I311 on M4 and residues F274, L278, I281 on M3 of the neighboring subunit. In the kinked conformation, these residues are 15–20 Å apart, for example, F308 and L278 or I311 and F274, whereas in the alternate conformation these residues come within less than 4 Å from each other. MD simulations further confirm that the alternate conformation is stable over a time span of 300 ns, with an r.m.s. deviation of 2.8 Å.
Fig. 5 |. Alternative conformation of an unkinked M4 helix forming novel intersubunit interactions.

a,b, Cartoon representation of the ELIC structure with the M4 helix in an alternative unkinked conformation (red) compared to the original kinked conformation (blue). In the unkinked conformation the M4 helix is pinched between W220 and W224 and sterically hinders lipid interactions. In this conformation residues of the M4 helix, including F308 and I311, come within close range of residues of the M3 helix (white) of its neighboring subunit, including L278 and F274, respectively. In the kinked M4 conformation these residues do not interact (15–20 Å apart). c, VCF of post-M4 ELIC mutants G319C, I320C, T321C and L322C labeled with methanethiosulfonate-rhodamine. Black traces represent cysteamine-induced current responses, red traces represent corresponding changes in fluorescence. Representative traces are shown from five to seven independent experiments for each construct. Panels on the right show normalized current and fluorescence responses as the mean ± s.e.m.
To investigate whether this conformational state is sampled in the M4 dynamic landscape, we designed two pairs of cysteine mutations, which are within crosslinking distance in the alternate conformation, but not in the kinked conformation. These double mutants are L278C/F308C or F274C/I311C engineered in the background of a cysteine-free ELIC. Using mutant protein expressed in E. coli and purified in detergent solution we investigated band shifts with SDS–PAGE analysis (Supplementary Fig. 11). Under reducing conditions, we observe that the ELIC band migrates exclusively as monomers with an apparent molecular size around 30 kDa. However, under oxidizing conditions, we identified a characteristic redistribution of bands, which we identified as 2-mer, 3-mer, 4-mer and 5-mer, consistent with the formation of intersubunit crosslinks as predicted by the ELIC crystal structure with the alternate M4 conformation (Supplementary Fig. 11). Similar observations were made for the F274C/I311C double mutant (Supplementary Fig. 11). This crosslinking does not occur with the cysteine-free ELIC (Supplementary Fig. 11), indicating specific disulfide bond formation between introduced cysteines in L278C/F308C and F274C/I311C. These experiments suggest that, at least under detergent-solubilized conditions, the ELIC M4 helix may transiently adopt a conformation that is similar to the alternate conformation observed in the crystal structure.
Finally, we used VCF to investigate the M4 dynamics in the context of ELIC expressed in a lipid bilayer environment. VCF simultaneously monitors current changes and fluorescence changes, obtained from an environmentally sensitive and thiol-reactive fluorophore attached to single cysteine mutants in the M4 domain. We focused on the post-M4 residues, which are unresolved in the crystal structure, namely G319C, I320C, T321C and L322C, labeled with MTS-rhodamine (Fig. 5c). In each of these mutants, we observe an upward deflection of the fluorescence signal, which correlates with channel opening induced by increasing concentrations of the agonist cysteamine. These results suggest that the local environment around these residues changes during channel opening and is indicative of a conformational change in the post-M4 domain. Second, we probed residues of M4 buried further in the lipid bilayer with the unnatural fluorescent amino acid ANAP46. We focused on residues around P305, namely F304, P305, L306 and G307. At each of these positions we observe that incorporation of ANAP results in a robust agonist-induced response, but none of these positions reported a fluorescence change (Supplementary Table 5). We do observe an acceleration of the desensitization kinetics, in agreement with the mutagenesis data in Fig. 3b. This does not exclude a possible conformational change of M4 in a lipid bilayer, but it indicates that the net hydrophobicity of the fluorophore environment is unchanged as the channel pore opens and closes.
Together, these results confirm the conformational dynamics of the M4 domain in ELIC and its sensitivity to lipid binding in this region. We have now obtained an alternate conformational snapshot of M4 as an example of a possible transient state of M4, which most likely regulates lipid interaction and may modulate channel desensitization.
Discussion
With this study, we have provided new insights into the conformational dynamics and lipid interactions of the M4 transmembrane domain of a pLGIC (Fig. 6). Alternate conformations of M4 have so far proved elusive from structural studies. By using ELIC as a model ion channel and applying a set of comprehensive techniques we have revealed an alternate conformational state of the M4 helix. In this alternate state, M4 adopts an unkinked conformation and undergoes a tilting motion to interact with the M3 helix of its neighboring subunit. The entry of the M4 helix into this state precludes interaction with a phospholipid molecule, which we identified near a highly conserved Pro kink in the ELIC M4, also found in eukaryote anion-selective pLGICs. Using mutagenesis studies we found that perturbations of the conserved Pro residue drastically accelerate receptor desensitization, suggesting that the lipid-binding site allosterically couples to channel gating. The phenomenon of accelerated desensitization in ELIC can be mimicked by altering the lipid environment, adding further evidence to a regulatory role of lipids. Together, these results provide a structural context for understanding how lipids influence channel function. Given the strong conservation of the kinked architectural fold of M4 in anion-selective channels, we suggest that the mechanistic insights derived from this study extend across the anion-selective subfamily of pLGICs. The phospholipid-binding site closely overlaps with known sites for neurosteroids, cholesterol and general anesthetics, thus offering strategies for the design of new therapeutics that can modulate channel function in human disease.
Fig. 6 |. Model for lipid modulation of channel desensitization.

A conserved architectural feature in ELIC and anion-selective pLGICs is a kinked M4 helix, which interacts with lipids. This M4 helix is conformationally dynamic and based on functional recordings from mutant channels, such as ELIC ΔM4, we establish that perturbations of the M4 Pro kink drastically accelerate receptor desensitization.
online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41589-019-0369-4.
Methods
ELIC expression and purification.
ELIC mutants were expressed and purified as previously described for wild-type ELIC47 (UniProt P0C7B7), but with minor modifications. ELIC cDNA was cloned as a synthetic gene with optimized codon usage (Genscript) into the pET-11a plasmid (Novagen) and expressed as an N-terminal fusion to maltose-binding protein (MBP) and a 3C protease cleavage site for removal of the fusion tag. Site-directed mutants were engineered using a QuikChange strategy and verified by sequencing. The MBP-ELIC fusion protein was expressed in the C43 E. coli strain, which was grown to an absorbance A600 ~ 1.8 and induced with 200 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 20 °C overnight. The membrane fraction was collected by ultracentrifugation at 125,000g and solubilized with 2% (w/v) anagrade n-undecyl-β-d-maltoside (Anatrace) at 4 °C overnight. The cleared supernatant was batch purified by affinity chromatography on amylose resin (New England Biolabs). Column-bound ELIC was cleaved off by 3CV protease in the presence of 1 mM EDTA + 1 mM DTT at 4 °C overnight and further purified on a Superdex 200 10/300 GL (GE Healthcare) column equilibrated with buffer containing 10 mM Na-phosphate (pH 8.0), 150 mM NaCl, and 0.15% n-undecyl-β-d-maltoside (UDM). Concentrated protein (10 mg ml−1) was supplemented with 0.5 mg ml−1 E. coli lipids (Avanti Polar Lipids). Cysteine pore mutants of ELIC were engineered in the background of a previously described cysteine-free channel48, which was obtained by substituting two nonconserved cysteines in the M4 domain (C300S/C313S) and functionally behaves as wild-type ELIC. For nanobody production and selection a series of ELIC pore mutants were produced, including 6′C (T237C), 7′C (L238C) and 8′C (M239C) as candidates to identify previously unidentified conformational states of ELIC. For the structural characterization of M4 domain mutants, we produced ELIC Δ8 (deletion L315VIRGITL322), ELIC ΔM4 (deletion entire M4 helix starting D294 until end) and P305A, which were identified as ELIC variants with fast desensitization kinetics using TEVC recordings (see below). For the crosslinking experiments, we engineered two constructs in the background of cysteine-free ELIC, namely L278C/F308C and F274C/I311C.
Nanobody selection, expression and purification.
A llama was immunized with 2 mg, in total, of purified wild-type ELIC over a period of 6 weeks using a previously established protocol49. Briefly, from the anticoagulated blood of the immunized llama, lymphocytes were used to prepare cDNA. This cDNA served as a template to amplify the open reading frames coding for the variable domains of the heavy-chain only antibodies, also called nanobodies. The PCR fragments were ligated into the pMESy4 phage display vector and transformed in E. coli TG1 cells. A nanobody library of 1.1 × 108 transformants was obtained. For the discovery of ELIC specific nanobodies, we selected four different targets, all fused to the MBP: wild-type ELIC-MBP, ELIC 6′C-MBP, ELIC 7′C-MBP and ELIC 8′C-MBP.
Phage particles displaying the nanobody repertoire of the library were added to each of the ELIC-MBP fusion proteins trapped on amylose resin (New England Biolabs). Selections were done in 10 mM Na-phosphate (pH 8.0), 150 mM NaCl, 0.15% UDM, 0.05% BSA in the presence of 10 mM GABA and 10 mM DDT. Either after 15 min or 2 h of incubation aspecific phage were washed off. Specific phages were eluted by incubating the differently coated amylose resin with 100 μl of trypsin (250 μg ml−1) for 30 min. Although a high background was seen on the MBP-only-coated amylose resin, 184 clones in total were sent for sequence analysis. Sequence alignment revealed eight nanobody families that were enriched. All clones were produced and purified as previously described49.
A series of 16 nanobodies were individually expressed in the periplasm of E. coli strain WK6, which was grown in TB media supplemented with 1 mM carbenicillin, 0.1% glucose and 2 mM MgCl2 to an absorbance A600 ~ 0.7 at 37 °C. The culture was induced with 1 mM IPTG and incubated overnight at 28 °C. Cells were harvested by centrifugation, resuspended in ice-cold TES buffer (200 mM TRIS, pH 8.0; 0.5 mM EDTA and 500 mM sucrose) and washed four times in diluted TES buffer. Next, this fraction was centrifuged at 10,000g to remove cell debris. The supernatant was applied on nickel sepharose resin (GE Healthcare) and incubated for 1 h at room temperature. The column was then consecutively washed with phosphate buffer 1 (50 mM Na-phosphate, pH 7.0 and 1 M NaCl) and phosphate buffer 2 (50 mM Na-phosphate, pH 6.0 and 1 M NaCl). Nanobody was eluted from the column with 50 mM Na-acetate (pH 4.5), 1 M NaCl and neutralized by 1 M TRIS (pH 7.5). The protein was concentrated to less than 1 ml on a 3 kDa cutoff Vivaspin concentrating column (Sartorius) and further purified on a Superdex 75 10/300 GL column (GE Healthcare) equilibrated with 10 mM Na-phosphate (pH 8.0) and 150 mM NaCl. Peak fractions corresponding to nanobody were pooled and spin-concentrated to ~50 mg ml−1. The formation of stable ELIC + nanobody complexes in solution was evaluated by size exclusion chromatography on a Superdex 200 10/300 GL (GE Healthcare) column based on a shift of the peak fraction containing pentameric ELIC and evaluation of co-eluting protein bands on a Coomassie-stained SDS–PA gel.
Protein crystallization.
A large combinatorial crystallization screen was set up using ELIC 6′C, 7′C or 8′C in combination with the selection of 16 nanobodies. ELIC mutant and nanobody were mixed in a molar ratio 1:1.2 calculated as monomers and incubated at room temperature 2 h before setting up crystallization trials at 20 °C by vapor diffusion of sitting drops. Automated dispensing was carried out using a nanoliter Mosquito crystallization robot and MRC plates (Molecular Dimensions). Crystals for ELIC 7′C + Nb72 grew under conditions containing 100 mM MES (pH 6.5) and 30% PEG400. Crystals for ELIC Δ8 + Nb72 grew under conditions containing 300 mM ammonium formate, 50 mM TRIS pH 9.0 and 33% PEG monomethylether 550. Cryo-protection was achieved by adding 10% glycerol to the mother liquor. Crystals for ELIC F16′S with the alternate M4 straight conformation grew under conditions containing 200 mM ammonium sulfate, 50 mM ADA (N-(2-acetamido)iminodiacetic acid) (pH 6.5), and 9–12% (v/v) PEG4000. Cryo-protection was achieved by adding 30% (v/v) glycerol to the mother liquor. Crystals were cryo-cooled by immersion in liquid nitrogen and transported to synchrotron radiation sources in a dry shipper.
X-ray crystallography.
The structure of ELIC 7′C in complex with the Nb72 was solved with X-ray diffraction data at a resolution of 2.50 Å, which were collected at the X06A beamline of the Swiss Light Source using 0.91958 Å wavelength light. Diffraction data were integrated with XDS50 and scaled in Aimless as part of the CCP4 suite51. For this and subsequent datasets, the high-resolution limit was set using a CC1/2 -value of 30% as a cutoff criterium. Initial phases were obtained using molecular replacement with Phaser51 using the ELIC pentamer coordinates of PDB deposition 2yoe as one ensemble and the nanobody coordinates of PDB deposition 2p0g as the second ensemble. All nonprotein atoms were removed from the coordinates before running the search procedure. As previously published47, all our ELIC structures contain a Gly residue at position 164, which was missing in the initially published ELIC structure with PDB deposition code 2vl0, and this changes the residue numbering +1 from position 164 onward. Additionally, we previously also indicated a change in the sequence register of the β8 strand compared to 2vl0 and these observations are now confirmed in the higher resolution structure. The ELIC + Nb72 structure was rebuilt and refined through iterative cycles of manual building in Coot52 and automated refinement in Phenix53. The final refinement steps were done in Buster54 using basic TLS (Translation/Libration/Screw) and automated NCS (non-crystallographic symmetry) restraints. The electron density of the lipid-bound to ELIC subunit A was improved after extending the protein mask by 6 Å. Mask extension was removed for the final refinement steps. The density was interpreted as phosphatidylethanolamine since this is the major lipid component in the E. coli lipid extract added before crystallization. The density was sufficiently clear to build a 12-carbon and 9-carbon lipid tail, respectively. The head group in this subunit benefits from a crystal interaction between the phosphate group and K49 in a neighboring pentamer. The lipid densities in other subunits are present but less clear than subunit A. Structure validation was done in MolProbity55. The MolProbity score of this structure is 1.09 and is in the 100th percentile for this resolution range. Ramachandran statistics indicate 96.04% of residues are in the favored region and 0.05% are outliers. All composite omit maps reported in this study were calculated in Phenix53 using Cartesian simulated annealing at start and final temperatures of 5,000 and 300 K, respectively.
The structure of ELIC Δ8 + Nb72 was solved with X-ray diffraction data at a resolution of 2.78 Å, which were collected at the ID30B beamline of the European Synchrotron Radiation Facility using 0.9754 Å wavelength light. Diffraction data were integrated with XDS50 and scaled in Aimless51. Initial phases were determined using molecular replacement in Molrep51 and the coordinates of ELIC + Nb72 from which the nonprotein atoms, as well as the last eight carboxyterminal residues in ELIC, were removed. The resulting electron density map revealed missing density not only for the last eight amino acids but the entire M4 helix and M3–M4 linker, suggesting this domain is flexible in the Δ8 structure. Consequently, the ELIC Δ8 structure was truncated at the end of M3 (H285) and further improved with iterative cycles of manual rebuilding in Coot52 and refinement in Refmac51. The final refinement steps were done in Buster54 using basic TLS and automated NCS restraints. The MolProbity score of this structure is 1.38 and is in the 100th percentile for this resolution range. Ramachandran statistics indicate 97.03% of residues are in the favored region and 0.4% are outliers.
Over years of structural studies with ELIC, thousands of crystals were screened and we obtained three datasets in which M4 adopts the alternate straight conformation, which we assume has occurred as a result of unexpected variations in the solubilization, lipidation or crystallization conditions for these crystals. The highest resolution set was for an ELIC F16′S mutant at 3.45 Å, which was collected at the X06DA beamline of the Swiss Light Source using 1.000 Å wavelength light. The same alternate M4 conformation could be observed in two other datasets for wild-type ELIC at 3.92 Å and 3.87 Å, so we conclude that the alternate M4 can occur independently of the F16′S mutation. Diffraction data were integrated with XDS50 and scaled in Scala51. The initial phases were obtained using molecular replacement with Phaser using the coordinates of PDB deposition 4twd from which the nonprotein atoms were removed. The structure was improved with iterative cycles of manual rebuilding in Coot52 and refinement in Refmac51. The MolProbity score of this structure is 1.82 and is in the 100th percentile for this resolution range. Ramachandran statistics indicate 95.56% of residues are in the favored region and 0.03% are outliers. All figures were prepared using PyMOL (Schrödinger).
Lipid vesicle recordings.
The ELIC channel was reconstituted at a 1:200 protein to lipid ratio (weight to weight) in liposomes of different lipid compositions, such as soy polar extract (as a control condition) or soy polar extract supplemented with 25% Cholesterol, 50% POPC (1-palmitoyl-2-oleoyl-glycero-3phosphocholine):25% POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-racglycerol)):25% POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) or 75 % POPC:25% POPG, following a published protocol56. The desired phospholipid composition dissolved in chloroform (25 mg ml−1) was dried for 4 h in a roto-evaporator. The dried phospholipid film was resuspended in resuspension buffer (5 mM MOPS pH 7.0, 200 mM KCl) and sonicated in a bath-sonicator until the liposome suspension was translucent. The ELIC channel freshly eluted from a size exclusion chromatography column, Enrich SEC 650 (Bio-Rad), equilibrated in (buffer 50 mM Tris-Cl, pH 8.0, 150 mM KCl, 1 mM n-dodecylβ-d-maltoside) was mixed with the liposome suspension for 30 min at room temperature. The ELIC containing liposomes suspension was incubated for 1 h with 30 mg of Bio-Beads SM-2 (Bio-Rad) at room temperature. Followed by two additional cycles of 30 mg of Bio-Beads additions + 1-h incubation. Finally, the ELIC containing proteoliposomes suspension was incubated at 4 °C overnight with an extra 100 mg of Bio-Beads. Next day, the ELIC containing proteoliposomes were harvested by pelleting them at 150,000g for 2 h. The resultant pellet was resuspended by pipetting in precisely 40 μl of resuspension buffer until the solution was homogeneous. Normally, three drops (10 μl each) were laid over a glass microscope slide and dehydrated, in the dark at 4 °C overnight. The next day, the ELIC containing proteoliposomes were rehydrated by adding 20 μl of resuspension buffer and incubated in the dark at 4 °C for 24 h, which normally produced giant liposomes, before performing patch clamp experiments. ELIC electrophysiological recordings in different phospholipid composition were performed in symmetrical 5 mM MOPS at pH 7.0 in the presence of 200 mM KCl. ELIC macroscopic currents were elicited by the application of 30 mM GABA with a BioLogic fast perfusion system RSC-200 and recorded from several independent experiments (n = 4 to 26 repeats) with a patch clamp amplifier Axopatch 200 B. ELIC currents were sampled at 40 kHz with an analog filter set to 10 kHz. Borosilicate patch pipettes, after fire-polishing, displayed a resistance of 2.0 MΩ. Patch pipettes were filled with 200 mM KCl, 5 mM MOPS buffer at pH 7.0 and 5 mM MgSO4. The time constant for desensitization (τ) of the ELIC channel reconstituted in different lipid compositions after an application of 30 mM GABA was calculated by fitting a single exponential curve to the experimental data. The reported values are the mean ± s.e.m. of 4 to 26 independent experiments.
Electrophysiological recordings.
Whole-cell currents were recorded from injected oocytes (0.2–12 ng cRNA) immersed in HEPES buffer (150 mM NaCl, 0.5 mM BaCl2 and 10 mM HEPES, pH 7.0) using a two-electrode voltage clamp apparatus (OC-725C oocyte clamp). In most cases, currents through the plasma membrane in response to cysteamine concentration jumps (from 0 mM up to the indicated values) were measured with the transmembrane voltage clamped at −40 mV. For α7 nAChR, whole-cell currents were recorded from injected oocytes (20–60 ng cRNA) immersed in HEPES buffer (96 mM NaCl, 2 mM KCl, 1.8 mM BaCl2, 1 mM MgCl2, 10 mM HEPES, 1 μM Atropine, pH 7.3). Currents in response to acetylcholine concentration jumps were measured with the transmembrane voltage clamped at −60 mV.
Cysteine crosslinking.
For cysteine crosslinking of ELIC, we designed two cysteine pair mutants, L278C/F308C and F274C/I311C. These cysteine mutants were engineered in the background of cysteine-free ELIC and cloned into the pET-11a plasmid with a native ELIC signal sequence, an aminoterminal hexahistidine tag and a 3CV protease cleavage site followed by the mature ELIC sequence. The protein was expressed and purified as described above, but here membranes were solubilized in buffer containing 2% (w/v) anagrade n-dodecyl-β-d-maltoside (Anatrace) at 4 °C overnight. The cleared supernatant was loaded on a 1 ml HisTrap Fast Flow column (GE Healthcare) with buffer containing 50 mM Na-phosphate (pH 8.0), 150 mM NaCl and 0.05% n-dodecyl-β-d-maltoside plus 40 mM imidazole. The column was washed until the UV absorbance at 280 nm reached a baseline value and then a second wash step with buffer plus 80 mM imidazole was used. The protein was eluted using buffer plus 250 mM imidazole. The peak fraction was spin-concentrated to a volume <1 ml and loaded on a Superdex 200 10/300 GL (GE Healthcare) column equilibrated with buffer containing 10 mM Na-phosphate (pH 8.0), 150 mM NaCl, and 0.05% n-dodecyl-β-d-maltoside. The peak fractions corresponding to pentameric ELIC were pooled and spin-concentrated to a concentration of 1–2 mg ml−1. Each cysteine mutant was reduced in the presence of β-mercaptoethanol or DTT and crosslinking was initiated by addition of a 1:2 molar ratio of Cu2+ and 1,10-phenanthroline at a final concentration of 100 μM. Reactions were quenched at given time points with 100 mM N-ethyl maleimide and sample buffer without reducing agent was added. Samples were then loaded onto Mini-PROTEAN TGX precast gradient (4–15%) gels for SDS–polyacrylamide gel electrophoresis analysis.
VCF using MTS-rhodamine or ANAP.
Adult female Xenopus laevis frogs (Xenopus Express) were anesthetized (1.3 mg ml−1 MS-222), and oocytes were surgically removed via a small incision to the abdomen. Oocytes were then defolliculated with 1.5 mg ml−1 collagenase (Sigma) for 2 h. VCF with rhodamine-labeled ELIC was used to characterize post-M4 residues in ELIC G319C, I320C, T321C and L322C engineered in the background of a cysteine-free channel. cDNA encoding ELIC was inserted into the pGEM-HE expression vector and mRNA was transcribed in vitro using the mMESSAGE mMACHINE T7 transcription kit (Ambion) as previously described47. Mutants were constructed using a QuikChange strategy (Stratagene) and confirmed by sequencing. Mature stage V or VI oocytes were isolated and rinsed with calcium-free OR-2 solution containing 82.5 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES (pH 7.4). Oocytes were injected with 10 ng ELIC cRNA using a Nanoliter 2000 microinjector (WPI Inc.) and incubated at 16 °C in ND-96 storage solution, containing 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, 50 μg ml−1 gentamicin, 2.5 mM sodium pyruvate, and 0.5 mM theophylline (pH 7.4). Fluorescent labeling of oocytes was conducted 3–5 days after injection by a 1 min incubation in a solution containing 10 mM methanethiosulfonate-rhodamine (MTSR, Toronto Research Chemicals) in ND-96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES, pH 7.5). The oocytes were then washed three times in ND-96 and stored at 18 °C in the dark until the start of recordings. A Lambda LS 175 W xenon arc lamp was used as a light source for an inverted Nikon Eclipse TE300 microscope (Nikon Instruments), equipped with a high-Q TRITC filter set (Chroma Technology). A Lambda 10–2 unit (Sutter Instruments) was used for selection of excitation and emission wavelengths. Light was focused onto the dark pole of the oocyte using a Plan Fluor ×40 objective lens (Nikon Instruments), and fluorescence detection was achieved with an H7360–03 photomultiplier (Hamamatsu Photonics) coupled to a PMT400R photomultiplier subsystem (Ionoptix).
ANAP-labeled ELIC was produced with the pANAP plasmid from Addgene (plasmid No. 48696). ANAP was a kind gift from Dr. S. Pless (University of Copenhagen). Amber stop-codon (TAG) mutants, ELIC F304TAG, P305TAG, L306TAG and G307TAG, were constructed using QuikChange mutagenesis. One day after oocyte isolation, 2 mM ANAP and 10–20 ng of ELIC mRNA were injected separately into the pANAP-expressed oocytes. Recordings were obtained 2–3 d after injection. ANAP fluorescence was detected using an inverted microscope (Ti-S, Nikon Instruments) equipped with a DAPI filter set (49000, Chroma Technology) and a CFI Fluor 40X water immersion objective (N.A. 0.80, MRF07420, Nikon Instruments). A Lambda LS 175 W xenon arc lamp served as a light source and was coupled to the microscope via a liquid light guide (Sutter Instruments). Fluorescence was detected using an H7360–03 photomultiplier (Hamamatsu Photonics) coupled to a PMT400R photomultiplier subsystem (Ionoptix). In both MTS-rhodamine and ANAP experiments, oocytes were placed securely in the bath and were continually perfused with ND-96 solution. Agonists were dissolved in the same solution and were applied to the oocytes via a gravity-fed perfusion system. Currents were recorded via two-electrode voltage clamp using 0.2–2 MΩ resistance glass pipettes filled with 3 M KCl. Oocytes were voltage-clamped at −40 mV, and currents were recorded using a Gene Clamp 500B amplifier (Molecular Devices). ΔI (change in current) and ΔF (change in fluorescence) signals were digitized at 2 kHz via a Digidata 1322A interface and pCLAMP 9.2 software (Molecular Devices).
MD simulation.
Simulation systems were prepared by embedding the ELIC 7′C pore mutant with bound lipid (mapped to 1,2-dilauroyl-sn-glycero-3-phosphoethanolamine, DLPE) in a lipid bilayer composed of 1-palmitoyl-2oleoyl-sn-glycero-3-phosphocholine (POPC). Nanobodies and detergent were removed from the simulated system, and the ELIC-DLPE complex was embedded in the membrane, solvated and ionized with 0.15 M of neutralizing ions using CHARMM-GUI Membrane builder57. In addition to the ELIC 7′C pore mutant, a second system was run with an additional P305A substitution in all five M4 helices, while a third terminated all M4 helices at residue 314 to form the Δ8 mutant. Both in silico modifications were also made using CHARMM-GUI Membrane builder. All simulated systems contained about 250 phospholipid molecules and about 160,000 total atoms.
All simulations used the NAMD2.12 MD package58 and the CHARMM36 forcefield59,60 for proteins, phospholipids, ions and water. A Langevin thermostat and barostat maintained a constant temperature of 310 K and a constant pressure of 1 atm, respectively. A cutoff of 1.2 nm was used for nonbonded interactions, with a switching function starting at 1.0 nm. The smooth particle mesh Ewald method with a grid spacing of 1 Å handled long-range electrostatics. Periodic boundary conditions were employed in a rectilinear box. All bonds to the hydrogen atoms were constrained using the SHAKE/RATTLE algorithm, which permitted a high-frequency time step of 2 fs within a multiple time-step rRESPA method. After 10,000 steps of energy minimization, the simulations followed the default equilibration protocol established in the files output by CHARMM-GUI, followed by 450 ns of production simulation. An r.m.s. displacement analysis was conducted by aligning the structure in each frame to minimize the r.m.s. displacement for all M1, M2 and M3 helices, and then calculating Cα r.m.s. displacement for each M4 helix separately.
Ethics statement.
llama immunization was performed in accordance with institutional guidelines, following experimental protocol reviewed and approved by the Vrije Universiteit Brussel Ethical Committee for Animals experiments (Project number 16–601-3). All Xenopus frog handling procedures were approved by the University of Queensland Animal Ethics Committee (approval number: QBI/059/13/ARC/NHMRC).
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files. Atomic coordinates and structure factors have been deposited with the Protein Data Bank under accession numbers 6HJX, 6HJY and 6HK0 for ELIC 7′C+Nb72, ELIC Δ8+Nb72 and ELIC F16′S, respectively.
Code availability
Any code or mathematical algorithm used in this study are available from the corresponding authors upon request.
Supplementary Material
Acknowledgements
We thank INSTRUCT-ERIC, part of the European Strategy Forum on Research Infrastructures and the Research Foundation-Flanders (FWO) for funding nanobody discovery (J.S.). We thank N. Buys and K. Willibal for technical assistance during nanobody discovery. SBO/IWT-project 1200261 and FWO-project G.0762.13 were awarded to J.S. and C.U. Additional support was from KU Leuven OT/13/095, C32/16/035 and C14/17/093 to C.U. We thank beamline staff at the Swiss Light Source and ESRF. J.E.B. was supported by a grant (113312) from the Natural Sciences and Engineering Research Council of Canada. G.B. was supported by research grant NIH P01GM55876–14A1. L.G.C. was supported by research grant NIH 2R01GM097159. Computational resources were provided by the National Science Foundation XSEDE program through allocation NSF-MCB110149 as well as the Rutgers Discovery Informatics Institute (G.B.).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-019-0369-4.
References
- 1.Norimatsu Y, Hasegawa K, Shimizu N & Toyoshima C Protein-phospholipid interplay revealed with crystals of a calcium pump. Nature 545, 193–198 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Dawaliby R et al. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol. 12, 35–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martens C et al. Lipids modulate the conformational dynamics of a secondary multidrug transporter. Nat. Struct. Mol. Biol. 23, 744–751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.She J et al. Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 556, 130–134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laganowsky A et al. Membrane proteins bind lipids selectively to modulate their structure and function. Nature 510, 172–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whorton MR & Mackinnon R Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heidmann T, Sobel A, Popot JL & Changeux JP Reconstitution of a functional acetylcholine receptor. Conservation of the conformational and allosteric transitions and recovery of the permeability response; role of lipids. Eur. J. Biochem. 110, 35–55 (1980). [DOI] [PubMed] [Google Scholar]
- 8.daCosta CJB et al. Anionic lipids allosterically modulate multiple nicotinic acetylcholine receptor conformational equilibria. J. Biol. Chem. 284, 33841–33849 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.daCosta CJB, Dey L, Therien JPD & Baenziger JE A distinct mechanism for activating uncoupled nicotinic acetylcholine receptors. Nat. Chem. Biol. 9, 701–707 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Barrantes FJ Phylogenetic conservation of protein-lipid motifs in pentameric ligand-gated ion channels. Biochim Biophys. Acta 1848, 1796–1805 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Baenziger JE, Hénault CM, Therien JPD & Sun J Nicotinic acetylcholine receptor-lipid interactions: mechanistic insight and biological function. Biochim. Biophys. Acta 1848, 1806–1817 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Cory-Wright J et al. Aromatic residues in the fourth transmembrane-spanning helix M4 are important for GABAρ receptor function. ACS Chem. Neurosci. 9, 284–290 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Haeger S et al. An intramembrane aromatic network determines pentameric assembly of Cys-loop receptors. Nat. Struct. Mol. Biol. 17, 90–98 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Hénault CM et al. The role of the M4 lipid-sensor in the folding, trafficking, and allosteric modulation of nicotinic acetylcholine receptors. Neuropharmacology 96, 157–168 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Carswell CL et al. Role of the fourth transmembrane α helix in the allosteric modulation of pentameric ligand-gated ion channels. Structure 23, 1655–1664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hénault CM, Juranka PF & Baenziger JE The M4 transmembrane α-helix contributes differently to both the maturation and function of two prokaryotic pentameric ligand-gated ion channels. J. Biol. Chem. 290, 25118–25128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitra A, Bailey TD & Auerbach AL Structural dynamics of the M4 transmembrane segment during acetylcholine receptor gating. Structure 12, 1909–1918 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Velisetty P, Chalamalasetti SV & Chakrapani S Structural basis for allosteric coupling at the membrane-protein interface in Gloeobacter violaceus ligand-gated ion channel (GLIC). J. Biol. Chem. 289, 3013–3025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basak S, Schmandt N, Gicheru Y & Chakrapani S Crystal structure and dynamics of a lipid-induced potential desensitized-state of a pentameric ligand-gated channel. eLife 6, e23886 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lasalde JA et al. Tryptophan substitutions at the lipid-exposed transmembrane segment M4 of Torpedo californica acetylcholine receptor govern channel gating. Biochemistry 35, 14139–14148 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Bouzat C, Roccamo AM, Garbus I & Barrantes FJ Mutations at lipid-exposed residues of the acetylcholine receptor affect its gating kinetics. Mol. Pharm. 54, 146–153 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Domville JA & Baenziger JE An allosteric link connecting the lipid-protein interface to the gating of the nicotinic acetylcholine receptor. Sci. Rep. 8, 3898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carswell CL, Sun J & Baenziger JE Intramembrane aromatic interactions influence the lipid sensitivities of pentameric ligand-gated ion channels. J. Biol. Chem. 290, 2496–2507 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Labriola JM et al. Structural sensitivity of a prokaryotic pentameric ligand-gated ion channel to its membrane environment. J. Biol. Chem. 288, 11294–11303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bocquet N et al. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Nury H et al. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Laverty D et al. Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nat. Struct. Mol. Biol. 24, 977–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q et al. Structural basis of neurosteroid anesthetic action on GABAA receptors. Nat. Commun. 9, 3972 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller PS et al. Structural basis for GABAA receptor potentiation by neurosteroids. Nat. Struct. Mol. Biol. 24, 986–992 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu S et al. Structure of a human synaptic GABAA receptor. Nature 559, 67–72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laverty D et al. Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer. Nature 565, 516–520 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manglik A, Kobilka BK & Steyaert J Nanobodies to study G protein-coupled receptor structure and function. Annu. Rev. Pharmacol. Toxicol. 57, 19–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilf RJC & Dutzler R X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Du J, Lü W, Wu S, Cheng Y & Gouaux E Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 526, 224–229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang X et al. Crystal structures of human glycine receptor α3 bound to a novel class of analgesic potentiators. Nat. Struct. Mol. Biol. 24, 108–113 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Miller PS & Aricescu AR Crystal structure of a human GABAA receptor. Nature 512, 270–275 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phulera S et al. Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABAA receptor in complex with GABA. eLife 7, 531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masiulis S et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 565, 454–459 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hibbs RE & Gouaux E Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Althoff T, Hibbs RE, Banerjee S & Gouaux E X-ray structures of GluCl in apo states reveal a gating mechanism of Cys-loop receptors. Nature 512, 333–337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morales-Perez CL, Noviello CM & Hibbs RE X-ray structure of the human α4β2 nicotinic receptor. Nature 538, 411–415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh RM et al. Structural principles of distinct assemblies of the human α4β2 nicotinic receptor. Nature 557, 261–265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gielen M & Corringer P-J The dual-gate model for pentameric ligand-gated ion channels activation and desensitization. J. Physiol. 596, 1873–1902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimmermann I & Dutzler R Ligand activation of the prokaryotic pentameric ligand-gated ion channel ELIC. PLoS Biol. 9, e1001101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez-Gutierrez G et al. Mutations that stabilize the open state of the Erwinia chrisanthemi ligand-gated ion channel fail to change the conformation of the pore domain in crystals. Proc. Natl Acad. Sci. USA 109, 6331–6336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chatterjee A, Guo J, Lee HS & Schultz PG A genetically encoded fluorescent probe in mammalian cells. J. Am. Chem. Soc. 135, 12540–12543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spurny R et al. Pentameric ligand-gated ion channel ELIC is activated by GABA and modulated by benzodiazepines. Proc. Natl Acad. Sci. USA 109, E3028–E3034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nys M et al. Allosteric binding site in a Cys-loop receptor ligand-binding domain unveiled in the crystal structure of ELIC in complex with chlorpromazine. Proc. Natl Acad. Sci. USA 113, E6696–E6703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pardon E et al. A general protocol for the generation of nanobodies for structural biology. Nat. Protoc. 9, 674–693 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabsch W XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smart OS et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cuello LG, Cortes DM & Perozo E The gating cycle of a K+ channel at atomic resolution. eLife 6, 213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jo S, Kim T, Iyer VG & Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Phillips JC et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks BR et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files. Atomic coordinates and structure factors have been deposited with the Protein Data Bank under accession numbers 6HJX, 6HJY and 6HK0 for ELIC 7′C+Nb72, ELIC Δ8+Nb72 and ELIC F16′S, respectively.