

Autologous chimeric antigen receptor (CAR) T cell therapy targeting CD19 is effective in B-cell malignancies such as acute lymphoblastic leukemia (ALL)1–3. Prior to infusion, T cells must be collected, activated, transduced, expanded, and validated4. This creates a “bridging period,” lasting from the decision to treat until T cells are infused, during which patients may experience disease related complications that delay or prevent CAR T cell infusion. Clinical trials of CD19 CAR T therapy reported high rates of patient dropout after enrollment due to progressive disease (POD) or treatment-related complications2,5–8, which highlights the challenges of clinical management during the bridging period.
Chemotherapy is commonly given during bridging to prevent rapid disease progression prior to CAR T cell infusion in patients with ALL and lymphoma6,9–13. Furthermore, we and others have shown that a lower disease burden at time of CAR T cell infusion is correlated with improved long-term survival and decreased severe cytokine release syndrome (CRS) and neurotoxicity (NT)2,3,14. Thus, there may be a benefit to cytoreduction with bridging therapy prior to CAR T cell treatment. However, only a small number of studies have evaluated the optimal use of bridging chemotherapy, primarily in relapsed lymphoma patients15–17, and practice varies among providers and clinical trial protocols.
We therefore reviewed bridging strategies and outcomes for all patients enrolled on a single-center, phase 1 trial of CD19-specific CAR T cells for R/R adult ALL (ClinicalTrials.gov NCT01044069)2, for which we previously reported the results of 53 treated patients. The median time from study screening to infusion was 63 days (17–262 days). Of 94 patients screened, 8 patients had no evidence of disease at screening and either did not relapse or were lost to follow up, and therefore were never eligible for infusion; and 2 patients received treatment off-protocol. These ten patients were deemed not eligible for treatment and are not included in subsequent analyses (Supplementary Figure 1).
For the entire population of 84 enrolled patients, median overall survival (mOS) from enrollment was 11.5 months (95% CI 7.9–17.9) (Supplementary Figure 2A). Fifty-six patients received CAR T cells on protocol, with a mOS of 15.5 months (11.5–24.4) (Figure 1A). Ten patients received an alternate therapy, most commonly allogeneic hematopoietic stem cell transplantation (HSCT; n=6) or donor lymphocyte infusion (n=2) after attaining a complete remission (CR) to bridging chemotherapy, with a mOS of 27.2 months (8.9-NR) (Supplementary Figure 2B). The remaining 18 patients were not treated with CAR T cells or alternate therapy due to POD or disease-related complications and had a decreased mOS of 3.0 months (1.4–4.5) (Figure 1B). Characteristics of patients in each group are in Supplementary Table 1.
Figure 1:
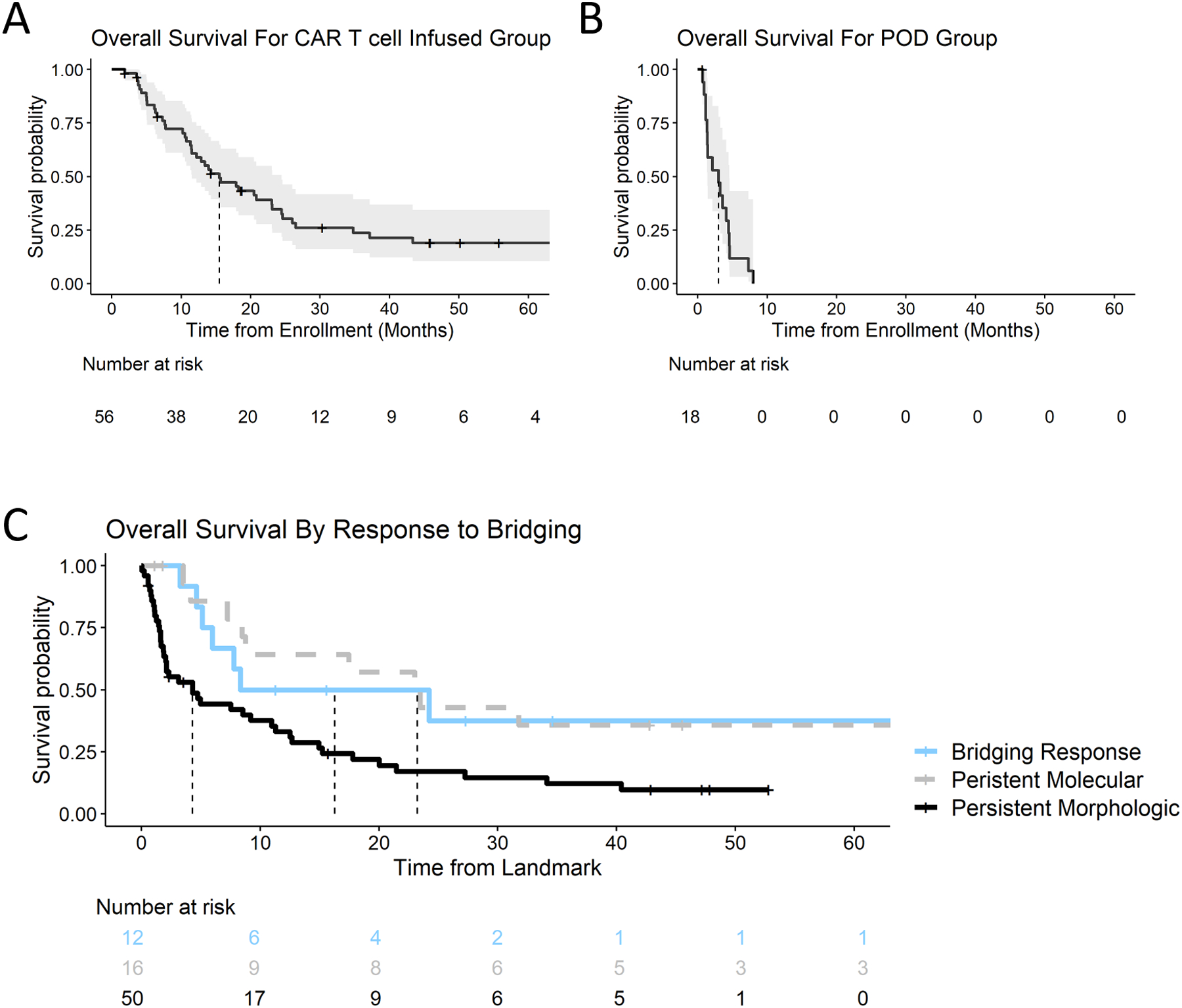
Overall survival from enrollment (start of bridging) for the (A) CAR T cell infused patients and (B) patients who were not infused due to POD. Dashed line – median overall survival. Due to immortal time arising from POD patient death during the bridging period, survival curves are not compared directly. POD: progression of disease. (C) Response to bridging at 3-month post-enrollment landmark as stratified by treatment group. Response categories are defined in text. Axis displayed as percent of patients in group, while numbers in box are absolute numbers of patients in group. p < 0.001 by Fisher’s exact test.
We sought to identify patient and disease factors at enrollment that predicted treatment with CAR T cells or alternate therapy, as opposed to progression of disease. Among factors examined by univariate logistic regression, only low bone marrow (BM) blast percentage at screening was significantly associated with receiving intended therapy (p<0.01; Supplementary Table 2).
We have previously shown that low disease burden at time of CAR T cell infusion correlates with improved post-CAR T cell treatment outcomes2. We therefore hypothesized that disease burden at time of infusion is a critical parameter and that patients who experienced cytoreduction during the bridging period would have improved clinical outcomes. The effect of changes in disease status during bridging was assessed by a landmark analysis at 3 months post-enrollment, as most patients were re-staged at 2–3 months. We characterized patients as 1) “responding to bridging”, 2) “persistent molecular disease,” 3) “persistent morphologic disease”, or 4) “progression during bridging” (see Supplementary Methods for full definitions).
Twelve of 62 patients (19%) with morphologic disease at enrollment had a response to bridging therapy by 3 months (Supplementary Figure 2C). Disease status at the 3-month landmark was significantly associated with OS (p=0.007) (Figure 1C). Median OS for the bridging response group (n=12) was 16.3 months (CI 6.0-NA), compared to 4.3 months (CI 2.0–11, p=0.04 for pairwise comparison) in the persistent morphologic disease group (n=42). Patients with persistent molecular disease (n=16) had a median OS of 23.2 months (8.8-NA), comparable to patients in the bridging response group. Thus, both patients who maintained low disease burden and patients who responded to bridging chemotherapy had favorable outcomes.
A theoretical drawback of cytoreduction with bridging therapy is reduced CAR T cell expansion, since expansion and persistence require CD19 expressing target cells. However, we found no difference in peak expansion or median duration of detectable CAR T cells between patients with morphologic disease, measurable residual disease, and no evidence of disease (Supplementary Figure 3).
The favorable association between bridging response and survival led us to search for bridging strategies that increased response rates and improved outcomes. We classified bridging chemotherapy regimens into either low- or high-intensity based on their myelosuppressive potential (Supplementary Table 3; Supplementary Figure 4). Of the 84 eligible patients, 3 received bridging at outside institutions and had insufficient records to accurately stratify therapy; 2 patients (2%) received no bridging therapy; 46 patients (57%) received low intensity therapy including blinatumomab (n=2) and inotuzumab (n=4); and 33 patients (41%) received high intensity therapy. The high and low intensity groups were well-matched for demographic and disease characteristics, and although there was a greater number of MRD patients in the low compared to high intensity groups, this difference was not statistically significant (27% vs. 15%; p=0.2) (Supplementary Table 4).
High intensity treatment was not associated with response to bridging at the 3-month landmark regardless of prior lines of treatments (Supplementary Figure 5A–D), nor with higher rates of CAR T cell infusion or alternate therapy (Supplementary Table 5). In multivariate logistic regression, BM blast percentage at enrollment but not bridging intensity was significantly associated with a higher likelihood of infusion (Supplementary Table 5). Furthermore, there was no significant difference in OS from start of bridging as stratified by intensity of bridging therapy (p=0.1 by log-rank) (Figure 2A). Median OS was 7.3 months (4.6–21) in the high intensity group compared to 14.0 months (11.5–25) in the low intensity group.
Figure 2:

Comparison of high- and low-intensity bridging strategies. (A) Overall survival from start of bridging stratified by bridging treatment intensity for entire cohort (A, left, p=0.1 by log-rank) or only patients with morphologic disease (bone marrow blasts > 5% or EMD) (B, right, p=0.8 by log-rank). (C) Rates of grade 3–4 infectious toxicity during the bridging period for all patients with available data (n=81). Y-axis reported as frequency of total population, with labels showing total number of patients.
Since the decision between high and low intensity bridging therapy may have been impacted by disease burden, we looked at outcomes only for patients with morphologic disease at time of enrollment (defined as BM blasts >5% and/or EMD; see Supplementary Figure 6). Even in this high disease burden sub-group, there was no difference in survival between high and low intensity treatment (p=0.5 by log-rank), with median OS of 7.3 months (4.5–21) and 11.5 months (8.0–24), respectively (Figure 2B).
The use of high intensity bridging chemotherapy regimens, however, was associated with therapy-related toxicity, with 23/33 (70%) patients in the high intensity group compared to 15/48 (31%) in the low intensity group experiencing grade 3–4 infectious complications during the bridging period (Figure 2C, p <0.001 by Fishers Exact Test). Infectious complications included bacteremia, pneumonia, pulmonary aspergillosis, and septic shock requiring ICU admission. In contrast, we did not observe a difference in rate of grade 3–4 CRS (38% vs. 22% for high and low intensity, p=0.11 by Fishers Exact test) or grade 3–4 NT (44% vs. 36% for high vs. low intensity, p>0.99 by Fishers Exact test) in patients who received CAR T cell infusion (Supplementary Figure 5E–F). Thus, high intensity bridging therapy was associated with an increased rate of infections during the bridging period, but not with post-CAR T cell infusion toxicities.
Targeted monoclonal antibodies can lead to deep responses in certain patients without significant myelosuppression18,19. Two and four patients received blinatumomab and inotuzumab as bridging therapy, respectively. Three of the six patients had morphologic disease prior to bridging. All six patients were successfully treated with either CAR T cells or alternate therapy, and none experienced a grade 3–4 infectious complication. Of these six patients, one had persistent morphologic disease during bridging, two experienced a response to bridging, and three had persistent molecular disease. Three of six patients were in remission at last follow up.
In summary, we demonstrate that low disease burden prior to the bridging period is predictive of successful CAR T cell infusion in ALL. We and others previously demonstrated that low disease burden immediately prior to CAR T cell infusion correlates with decreased toxicity and increased survival in ALL, and we now demonstrate that reductions in disease burden during the bridging period are associated with favorable outcomes2,3,14. This suggests that chemosensitivity during bridging either selects a favorable sub-group of patients or that effective cytoreduction may augment the efficacy of subsequent CAR T therapy, leading us to search for optimal strategies to reduce disease burden during bridging.
However, increased treatment intensity with highly myelosuppressive therapy was not associated with enhanced response to bridging, rates of CAR T cell infusion, or overall survival, but was associated with increased rates of severe infections during the bridging period. Our finding is likely driven by the low rates of response to bridging chemotherapy in this heavily pre-treated patient cohort. If CAR T therapy is explored as an earlier line of treatment, bridging chemotherapy will likely generate a higher response rate and more effective cytoreduction, and there may be a greater role for high-intensity chemotherapy.
Therefore, optimal bridging strategies are likely to be patient and disease specific. We propose a schema for selection of appropriate bridging strategies in Supplementary Figure 7. We emphasize that patients with low disease burden are likely to proceed to CAR therapy with low intensity bridging, and that patients with late relapses after responding to prior chemotherapy may be potential candidates for bridging intensification. Further studies will be needed to prospectively evaluate the role of bridging in CAR T cell therapies, including incorporation of targeted monoclonal antibodies.
Supplementary Material
Footnotes
Conflict of Interest Statement
KP discloses patent or royalties (Neximmune, Inc.) and acknowledge funding from NCI (t32-CA009207). KJC discloses consulting or advisory role (Novartis) and research funding (Juno Therapeutics). CD discloses travel accommodations and expenses from Juno Therapeutics. MG discloses consulting or advisory role with Tesaro. IR discloses stock or ownership interest, consulting or advisory role, research funding, and patents and royalties, all from Juno Therapeutics. MS discloses consulting or advisory role (Juno Therapeutics), research funding (Atara Biotherapeutics; Fate Therapeutics; Juno Therapeutics; Takeda; Takeda; Takeda; Takeda), and patients or royalties (Juno Therapeutics). RB discloses stock and ownership interests, consulting or advisory role, research funding, and patents or royalties, all from Juno Therapeutics. JHP discloses consulting or advisory roles (Adaptive Biotechnologies; Amgen; Kite Pharma; Novartis; Pfizer) and research funding (Genentech/Roche; Juno Therapeutics).
References
- 1.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med 2018; 379: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 2018; 378: 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016; 126: 2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther — Oncolytics 2016; 3: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017; : NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGurik JP et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. NEJM 2018; : 1–12. [DOI] [PubMed] [Google Scholar]
- 7.Turtle CJ, Hanafi L, Berger C, Hudecek M, Pender B, Robinson E et al. Immunotherapy of non-Hodgkin ‘ s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor – modified T cells. Sci Transl Med 2016; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017; 129: 3322–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain M, Chavez JC, Shah BD, Khimani F, Davila ML, Kim S et al. Radiation Therapy As a Bridging Strategy for Refractory Diffuse Large B Cell Lymphoma Patients Awaiting CAR T Manufacturing of Axicabtagene Ciloleucel. 2018.https://ash.confex.com/ash/2018/webprogram/Paper117133.html (accessed 5 Jan2019).
- 10.Gupta S, Alexander S, Zupanec S, Maude SL, Krueger J. High Vs. Low-Intensity Bridging Chemotherapy in Children with Acute Lymphoblastic Leukemia Awaiting Chimeric Antigen Receptor T-Cell Therapy: A Population-Based Study from Ontario, Canada 2018.https://ash.confex.com/ash/2018/webprogram/Paper115593.html (accessed 3 Jan2019).
- 11.DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK et al. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol Ther 2018; 26: 2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nastoupil L, Jain MD, Spiegel D, Ghobadi A, Dahiya S, Lunning MA et al. Axicabtagene Ciloleucel (Axi-cel) CD19 Chimeric Antigen Receptor (CAR) T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma: Real World Experience. 2018.https://ash.confex.com/ash/2018/webprogram/Paper114152.html (accessed 31 Dec2018).
- 13.Kuhnl A, Roddie C, Martinez-Cibrian N, Menne TF, Linton K, Lugthart S et al. Real-World Data of High-Grade Lymphoma Patients Treated with CD19 CAR-T in England. Blood 2019; 134: 767–767. [Google Scholar]
- 14.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (London, England) 2015; 385: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sim AJ, Jain MD, Figura NB, Chavez JC, Shah BD, Khimani F et al. Radiation Therapy as a Bridging Strategy for CAR T Cell Therapy With Axicabtagene Ciloleucel in Diffuse Large B-Cell Lymphoma. Int J Radiat Oncol Biol Phys 2019; 105: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain M, Jacobs MT, Nastoupil LJ, Y SJ, Ghobadi A, Locke FL. Characteristics and Outcomes of Patients Receiving Bridging Therapy While Awaiting Manufacture of Standard of Care Axicabtagene Ciloleucel CD19 Chimeric Antigen Receptor (CAR) T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma: Results from the. In: ASH Annual Meeting. ASH, 2019. [Google Scholar]
- 17.Imber BS, Sadelain M, DeSelm C, Batlevi C, Brentjens RJ, Dahi PB et al. Early experience using salvage radiotherapy for relapsed/refractory non‐Hodgkin lymphomas after CD19 chimeric antigen receptor (CAR) T cell therapy. Br J Haematol 2020; : bjh.16541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med 2016; 375: 740–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fielding AK, Ph D, Schuh AC, Dombret H, Foà R, Bassan R et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med 2017. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.