

Abstract
ARID1B haploinsufficiency is a frequent cause of intellectual disability (ID) and autism spectrum disorder (ASD), and also leads to emotional disturbances. In this review, we examine past and present clinical and preclinical research into the neurobiological function of ARID1B. The presentation of ARID1B-related disorders (ARID1B-RD) is highly heterogeneous, including varying degrees of ID, ASD and physical features. Recent research includes the development of suitable clinical readiness assessments for the treatment of ARID1B-RD, as well as similar neurodevelopmental disorders. Recently developed mouse models of Arid1b haploinsufficiency successfully mirror many of the behavioral phenotypes of ASD and ID. These animal models have helped to solidify the molecular mechanisms by which ARID1B regulates brain development and function, including epigenetic regulation of the Pvalb gene and promotion of Wnt/β-catenin signaling in neural progenitors in the ventral telencephalon. Finally, preclinical studies have identified the use of a positive allosteric modulator of the GABAA receptor as an effective treatment for some Arid1b haploinsufficiency-related behavioral phenotypes, and there is potential for the refinement of this therapy in order to translate it into clinical use.
Keywords: ARID1B, intellectual disability, autism, BAF, neurodevelopment
Introduction
Advances in genetic sequencing and enhancements in the development of animal models provide avenues for discovering mutations underlying neurodevelopmental disorders and deciphering the roles of these mutated genes in brain development and behavior. In 2010 a patient that presented with agenesis of the corpus callosum, intellectual disability (ID) and features of autism spectrum disorder (ASD) was found to harbor a mutation resulting in haploinsufficiency of the ARID1B gene 1. Not long thereafter, a separate research group identified 8 additional patients with similar symptoms caused by ARID1B haploinsufficiency 2. Then, in 2012, three groups independently identified haploinsufficiency of ARID1B as a relatively frequent cause of syndromic and non-syndromic intellectual disability 3–5. Since that time, numerous studies have further validated and defined the important role of ARID1B mutations in neurodevelopmental disorders 6–16.
ARID1B encodes the AT-rich interactive domain-containing protein 1B (ARID1B), also known as BAF250B, a large subunit of the BRG1/BRM-associated factor (BAF) chromatin remodeling complex (mammalian SWI/SNF complex) 17, 18. Following the discovery of ARID1B mutations as a monogenic cause for ID and ASD, preclinical research examining the neurodevelopmental and neurobiological functions of ARID1B has accelerated. This work has begun to shed light on the potential for pharmacological interventions targeting the root causes of ID and ASD due to ARID1B haploinsufficiency, which ameliorate a host of symptoms in animal models 9, 19. Translating similar treatments for neurodevelopmental disorders into clinical use has proved challenging 20, 21 and controversial 22–24 up to this point.
In this review we discuss recent advances in preclinical and clinical research regarding ARID1B haploinsufficiency, with a lens toward developing targeted and effective treatments for affected individuals.
Disorders and neurobehavioral phenotypes associated with ARID1B mutations
ID is a developmental disorder affecting approximately 1–8% of the overall population 25–27. It is characterized by significant limitations in cognitive function and adaptive behaviors, imposing a considerable burden on affected individuals and their caregivers. Several studies report that the underlying cause of ID is of primarily genetic origin, with over two thousand associated genes identified. Mutational analysis in 887 patients with nonspecific ID revealed nine patients (0.9%) with de novo nonsense or frameshift mutations resulting in a truncated copy of ARID1B 3. Exome sequencing studies on large cohorts of children with undiagnosed developmental disorders from the Deciphering Developmental Disorders (DDD) study in the United Kingdom also identified ARID1B mutations as the most frequent cause of ID; out of the 271 DDD diagnoses examined, 11 patients exhibited ARID1B mutations, which represent 4.7% of total ID diagnoses 28. Two of these individuals also exhibited comorbid ASD (9.1% of total ASD diagnoses), and one of those also presented with seizures (2.6% of patients with seizures) 28.
While ID is a highly heterogenous disorder, there are multiple syndromic subtypes. Coffin-Siris syndrome (CSS) is characterized by mild to severe ID, speech impairment, coarse facial features, growth deficiencies, and hypoplastic or absent fifth fingernail or toenail 29. Additionally, agenesis of the corpus callosum is frequently seen in CSS patients 2, 30, 31. Utilizing whole-exome sequencing to identify the genetic origins of CSS in three diagnosed individuals, Santen et al. revealed heterozygous, de novo truncating mutations in the ARID1B gene in all cases. Additionally, copy-number variation analysis in 2,000 individuals with ID showed that three subjects with ARID1B gene deletions exhibited phenotypes overlapping with CSS 4. Exome sequencing done by Tsurusaki et al. also revealed that out of 23 individuals diagnosed with CSS, six patients had de novo heterozygous mutations in ARID1B, further suggesting haploinsufficiency of ARID1B as a cause of CSS 5. Further targeted sequencing studies also identified mutations in other BAF complex genes in individuals affected with CSS (SMARCA4, SMARCB1, SMARCA2, SMARCE1, ARID1A, DPF2, and ARID2) 5, 32–36 and the specific gene that is mutated appears to influence the presentation and severity of associated symptoms 37. Disorders caused by ARID1B are better described as being on a spectrum including non-syndromic ID and CSS cases, either with or without additional physical and/or neurological symptoms, and referred to under the umbrella term, ARID1B-related disorders (ARID1B-RD) 13, 29.
ASD is a neurodevelopmental disorder characterized by significant social and communication deficits as well as stereotyped behaviors, affecting approximately 1 in 54 individuals 38, 39. ID is also a prevalent phenotype in ASD patients, seen in 30–75% of affected individuals 40–43. Next generation sequencing and microarray analysis from eight ID patients showed de novo translocations or deletions that resulted in a truncated copy of the ARID1B gene in each case 2. Of these, five patients exhibited phenotypes consistent with ASD. Further, brain imaging indicates that four patients also had corpus callosum defects. An additional study revealed that ASD patients have a decreased transcript level of the ARID1B gene 44. The SFARI Gene initiative, a comprehensive database of genes and copy number variants associated with ASD, also classified ARID1B as one of 25 high confidence genes related to autism.
ASD often presents with other co-occurring conditions, including epilepsy and attention-deficit/hyperactivity disorder (ADHD) 45, 46. Approximately one third of individuals with ARID1B mutations experience epileptic seizures, particularly of the tonic-clonic type, characterized by full-body muscle stiffening followed by rhythmic jerking of the body 29. Epilepsy is a common comorbidity in pediatric patients with ID/global developmental delay (ID/GDD-EP), with an estimated prevalence of about 22.2% 47. In a study of 143 ID patients with ARID1B mutations, 27.5% individuals suffered from epileptic seizures 13. Additionally, a copy number variation analysis on seven pediatric patients with nonspecific ID/GDD-EP revealed one individual with an ARID1B gene deletion presenting with CSS phenotypes and epilepsy 48. While the overall prevalence is unknown, individuals with ARID1B-related disorders also appear to be at an increased risk of attention-deficit/hyperactivity disorder (ADHD) diagnoses 29.
A list of all the human studies discussed in this review can be found in Table 1. The results of these studies underscore the crucial role that ARID1B plays in normal brain development and behavior. Several mutations leading to ARID1B haploinsufficiency cause developmental disorders including ID, CSS, and ASD, as well as abnormalities of the corpus callosum. These findings further support the hypothesis that deficits in chromatin remodelers play a significant role in neurodevelopmental disorders 49–51.
Table 1.
Summary of human genetic studies examining ARID1B haploinsufficiency
| STUDY REFERENCE | TOTAL NUMBER OF INDIVIDUALS | INDIVIDUALS WITH ARID1B HAPLOINSIFFUCIENCY | KEY FINDINGS |
|---|---|---|---|
|
| |||
| PIROLA ET AL. 1998 | 1 | 1 | Agenesis of the corpus callosum in a patient with a deletion including ARID1B |
| NAGAMANI ET AL. 2009 | 4 | 4 | |
| BACKX ET AL. 2011 | 1 | 1 | |
| NORD ET AL. 2011 | 41 | 1 | |
| HALGREN ET AL. 2012 | 8 | 7 | |
| HOYER ET AL. 2012 | 887 | 2 | |
| SANTEN ET AL. 2012 | 2000 | 3 (6) | 6 patients with deletions including ARID1B; 3 individuals with only ARID1B haploinsufficiency. |
| TSURUSAKI ET AL. 2012 | 22 | 5 | |
| SANTEN ET AL. 2013 | 63 | 28 | Mutations affecting different BAF complex subunits lead to divergent phenotypes |
| TSURUSAKI ET AL. 2012 | 52 | 15 | |
| WIECZOREK ET AL. 2013 | 46 | 19 | ARID1B mutations account for 76% of the 21 identified mutations leading to Coffin-Siris syndrome and 43% of the 7 mutations detected in patients with Nicolaides-Baraitser syndrome |
| SIM ET AL. 2014 | 1 | 1 | Evidence of dysregulated cell-cycle in patient-derived cells |
| VENGOECHEA ET AL. 2014 | 1 | 1 | |
| WRIGHT ET AL. 2015 | 271 | 11 | |
| MIGNOT ET AL. 2016 | 99 | 10 | ARID1B haploinsufficiency is a chief cause of corpus callosum defects in individuals with ID |
| DEMILY ET AL. 2019 | 8 | 8 | Severity of corpus callosum defects may be used to predict other symptoms |
| GOROKHOVA ET AL. 2019 | 44 | 44 | |
| VAN DER SLUIJS ET AL. 2019 | 143 | 143 | ARID1B-related disorders exist on a spectrum and should be treated as such |
| KRUIZINGA ET AL. 2020 | 24 | 12 | Suggestions on tests and clinical endpoints to be used in treating patients with ARID1B-related disorders |
List of human studies including references to ARID1B discussed in this review. The total number of cases, individuals with ARID1B-RD, and key findings are given for each study, where applicable.
Behavioral phenotypes of Arid1b haploinsufficient mice
Three groups recently developed mouse models of ARID1B haploinsufficiency. Two groups generated heterozygous mice by removing exon 5 of the Arid1b gene 9, 19, while the other group removed exon 3 52. Celen et al. and Shibutani et al. used the CRISPR/Cas9 gene editing system to generate mutant mice, while Jung et al. used a more traditional knockout strategy. All methods resulted in frameshift mutations and loss of function of one copy of the gene. Each group performed a variety of assays to characterize the behavioral phenotypes of Arid1b haploinsufficient mice. A summary of each group’s results is described in Table 2.
Table 2.
Summary of behavioral phenotypes observed in Arid1b haploinsufficient mice
| BEHAVIOR | BEHAVIORAL ASSAY | CELEN ET AL. 2017 | JUNG ET AL. 2017 | SHIBUTANI ET AL. 2017 |
|---|---|---|---|---|
|
| ||||
| SPATIAL REFERENCE MEMORY | Morris Water Maze | − | ↓ | N/A |
| SPATIAL REFERENCE MEMORY | T Maze | N/A | ↓ | − |
| RECOGNITION MEMORY | Novel Object Recognition | N/A | ↓ | N/A |
| MOTOR LEARNING | Rotarod Test | N/A | ↓ | N/A |
| FEAR LEARNING | Fear Conditioning | − | N/A | ↑ |
| SOCIABILITY | Open Field Social Interaction | ↓ | ↓ | − |
| SOCIABILITY | Home-Cage Social Interaction | N/A | N/A | ↓ |
| SOCIABILITY | 3-Chamber Test | N/A | ↓ | − |
| SOCIAL NOVELTY | 3-Chamber Test | N/A | ↓ | − |
| COMMUNICATION | Ultrasonic Vocalizations | Altered Communication | N/A | N/A |
| REPETITIVE BEHAVIOR | Grooming | ↑ | ↑ | − |
| ANXIETY | Elevated Plus Maze | ↑ | ↑ | ↑ |
| ANXIETY | Open Field | ↑ | ↑ | Unclear |
| ANXIETY | Light-Dark Box | ↑ | N/A | − |
| DEPRESSION | Forced Swim | N/A | ↑ | Unclear |
| DEPRESSION | Tail Suspension | N/A | ↑ | N/A |
Comparison between the three published mouse models of global Arid1b heterozygous knockout.
As mentioned previously, ID is characterized by severe limitations in cognitive function 25, 53. Arid1b heterozygous mice present with significant deficits in learning and memory. Using the Morris water maze to assess spatial reference memory, Jung et al. showed that heterozygous mice have increased escape latencies during training and spend less time in the target quadrant during probe trials 9. Surprisingly, Celen et al. detected no cognitive deficits using Morris water maze test 19. The T-maze is another test used to assess spatial reference memory. Jung et al. reported that Arid1b heterozygous mice are less successful in the T-maze test 9, however Shibutani et al did not find any deficits in mutant mice 52. To assess recognition memory, Jung et al. also performed the novel object test. They found that heterozygous mice show no preference for a novel object over a familiar one, while control mice prefer the novel object 9.Additionally, Jung et al. assessed motor learning ability using the rotarod test and report that mutant mice exhibit a decreased latency to fall off the rotating rod and limited ability to learn throughout training days 9. Both other groups also assessed fear learning. While Shibutani et al. showed that Arid1b heterozygous mice have enhanced performance in fear conditioning tests 52, Celen et al. reported that mutant mice perform similarly to controls 19.
The core characteristics of ASD include deficits in social interaction and communication as well as repetitive, stereotyped behaviors 38, 39. All three groups assessed sociability with an array of tests. In the open field social interaction test, both Jung et al. and Celen et al. reported reduced interaction time when an Arid1b heterozygous mouse is introduced to an unfamiliar mouse in an open arena 9, 19. Using the 3-chamber test for sociability, Jung et al. also showed that mutant mice spend more time in the empty chamber while controls prefer the chamber with an unfamiliar mouse 9. The 3-chamber test for social novelty also indicated that mutant mice spend less time with a novel stranger than they do with a more familiar stranger 9. Interestingly, Shibutani et al. reported no change in sociability/social novelty in either the open field or 3-chamber tests; however, they did observe reduced interaction between Arid1b heterozygous mice in a home-cage environment 52. To assess changes in communication between mice, Celen et al. examined ultrasonic vocalizations (USVs) and reported that mutant mice emit USVs that are longer in duration and of abnormal pitch compared to controls 19. All three groups examined the incidence of repetitive behaviors by assessing grooming. Both Jung et al. and Celen et al. reported an increase in time spent self-grooming in Arid1b heterozygous mice 9, 19, while Shibutani et al. detected no change in grooming times compared to controls 52.
Anxiety and depression-like behaviors are common comorbidities seen with ASD 54, 55. All three groups utilized the elevated plus maze to assess anxiety-like behavior in Arid1b heterozygous mice. Mutant mice spend less time in, and exhibit fewer entries into, the open arms of the maze in all cases 9, 19, 52. In the open field test, Arid1b heterozygous mice also spend less time in, and have fewer entries into, the center of the arena 9, 19. Additionally, Celen et al. reported that mutant mice avoid exploring the brightly-lit section of the light-dark box test 19, while Shibutani et al. identified no changes in exploratory behavior compared to controls 52. In assessing depression-like behavior, Jung et al. reported that Arid1b heterozygous mice exhibit greater immobility time in the forced swim and tail suspension tests 9. Interestingly, Shibutani et al. reported contradictory results in the forced swim test 52.
Together, results from these three studies consistently show that Arid1b heterozygous mice recapitulate the majority of behavioral phenotypes seen in ASD and ID, thus providing a useful tool moving forward to better understand the pathology underlying these disorders. The few discrepancies among the results of individual behavioral assays can likely be explained by differences in specific protocols, mouse handling and stressors, and environmental stimuli.
Interneuron subtype-specific behavior
Many neurodevelopmental disorders are characterized by significant deficits in inhibitory interneuron development 56–58. In a recent study, Smith and colleagues highlighted distinct roles of two interneuron subtypes in mediating ASD- and ID-associated behaviors 59. To examine interneuron subtype-specific behavior in mice, they generated conditional knockout mice exhibiting Arid1b haploinsufficiency in either parvalbumin- (PV) or somatostatin-expressing (SST) interneurons. Smith et al. showed that haploinsufficiency in PV subtypes alters social and emotional behaviors, while haploinsufficiency in SST subtypes affects cognitive function and repetitive behavior 59, together recapitulating the phenotypes of global Arid1b haploinsufficiency 9. Table 3 summarizes the behavioral phenotypes observed with global Arid1b haploinsufficiency in comparison to haploinsufficiency specifically in PV and SST interneurons. These results provide further insight into how individual interneuron subtypes may contribute to neurodevelopmental disorders, paving the way for even more targeted therapeutic strategies.
Table 3.
Summary of behavioral deficits in global and conditional Arid1b heterozygous mice
| BEHAVIOR | ARID1B +/− | F/+;PV-CRE | F/+;SST-CRE |
|---|---|---|---|
|
| |||
| SOCIAL BEHAVIOR | ↓ | ↓ | − |
| REPETITIVE BEHAVIOR | ↑ | − | ↑ |
| ANXIETY | ↑ | ↑ | − |
| DEPRESSION-LIKE BEHAVIOR | ↑ | ↑ | − |
| RECOGNITION MEMORY | ↓ | − | ↓ |
| MOTOR LEARNING | ↓ | − | ↓ |
| SPATIAL REFERENCE MEMORY | ↓ | − | ↓ |
Comparison between global, PV-Cre conditional, and SST-Cre conditional heterozygous Arid1b knockout mice. ↓ = a decrease in a particular behavior; ↑ = an increase in a particular behavior; − = no change in behavior
The role of ARID1B in neural development
The BAF complex, including ARID1B, is essential for neurodevelopmental processes such as neural stem cell generation, proliferation, migration, and differentiation into neuronal subtypes. ARID1B haploinsufficiency causes abnormal regulation of cell cycle re-entry in developmentally arrested cells 60. Thus, ARID1B mutations impair developmental processes by abnormal initiation of progenitor cell proliferation 6. In addition, homozygous knockout of Arid1b in mice is embryonic-lethal, but Arid1b knockout embryonic stem cells demonstrate a reduced proliferation rate and perturbation of differentiation and the cell cycle 61. It is unclear, however, how ARID1B deletion is linked to neural proliferation and eventual functional neurodevelopmental deficits, but recent studies exploring these questions are addressed in greater detail below.
The BAF complex is important for neuronal morphogenesis during brain development. Arid1b is highly expressed in differentiated neurons in the developing and postnatal mouse brain, and knockdown of Arid1b influences the expression of several genes known to promote neuronal migration and neurite outgrowth, such as Gap43, Gprn1, and Stmn2 8. It was previously reported that another BAF subunit, BAF53b, also has a critical role in activity-dependent dendritic outgrowth via regulation of Gap43 and Ephexin1 transcription 62. Knockdown of ARID1B in cortical pyramidal neurons leads to abnormal dendrite arborization and dendritic spine formation through suppression of c-Fos and Arc 8, which play critical roles in dendritic and synaptic development 63, 64. Moreover, knockdown of ARID1B markedly decreases dendritic innervation into cortical layer I, with fewer apparent attachments of dendritic terminals at the pial surface 8. Apical dendritic attachments in layer I are crucial for feedback interactions in the cerebral cortex involved in associative learning and attention 65–67. Furthermore, targeting the let-526 gene in C. elegans, an ARID1B ortholog, also leads to aberrant dendritic arborization, and the severity of these effects are gene dose-dependent 68.
Consistently, many BAF complex subunits are involved in regulating neurite architecture during brain development. For example, the BAF complex subunit, BAF100a, is required for terminal maturation and morphogenesis of dorsal spinal neurons during spinal cord development 69. Moreover, the BAF complex mechanistically plays a role in the calcium-mediated transcription activation function of calcium-responsive trans-activator (CREST), which is a key factor for proper dendrite outgrowth, arborization, and refinement 70. Thus, ARID1B and the BAF complex play a crucial role in neuronal morphogenesis and dendrite formation. Importantly, dendritic impairments are found in neurodevelopmental disorders associated with ARID1B mutations 71, 72.
Role of ARID1B in inhibitory neural communication
Gamma-aminobutyric acid-ergic (GABAergic) inhibitory interneurons represent around 10–20% of the total cortical cell population 73. GABAergic inhibitory interneurons inhibit a complex network of excitatory and inhibitory neurons in multiple cortical circuits 74. GABAergic inhibitory interneurons, such as parvalbumin- (PV), somatostatin- (SST), calretinin, calbindin 1- and neuropeptide Y-expressing subtypes are the source of GABA in the nervous system and play an important role in neural function and activity 75–77. Cortical inhibitory interneurons are generated by progenitor cells originating from the ganglionic eminence 78. Alterations in GABAergic inhibitory interneuron density and number are involved in human neurodevelopmental disorders and associated mouse models 79, 80. For instance, GABA levels are lower in frontal, motor, somatosensory and auditory cortices in ASD patients 81–84. Haploinsufficiency of the gene encoding the voltage-gated sodium channel SCN1A, another monogenic cause of ASD, in mice decreases GABAergic inhibitory interneuron density 85, while SH3 And Multiple Ankyrin Repeat Domains 1 (Shank1) mutations lead to elevated PV-positive cell numbers in the mouse brain 86. In addition, knockout of the gene encoding Contactin-associated protein-like 2 (Cntnap2) leads to downregulation of PV expression levels in various brain regions 87.
Malfunctions in GABAergic inhibitory interneurons induce social deficits and repetitive behavior 88. Jung and colleagues showed that PV-positive and total GABAergic interneuron numbers are significantly decreased in Arid1b haploinsufficient mice 9. In addition, the number of total and PV-positive GABAergic interneurons are also decreased following heterozygous conditional deletion of Arid1b in interneuron progenitors 9. However, GABAergic interneuron migration routes and speeds remain normal in Arid1b haploinsufficient mice 9. Furthermore, the numbers of vesicular GABA transporter- (VGAT) and glutamic acid decarboxylase-positive (GAD) inhibitory synapses, which are responsible for GABA transport and synthesis in synaptic vesicles, are decreased in Arid1b haploinsufficient mice 9. However, the number of excitatory synapses expressing vesicular glutamate transporter 1 (VGLUT1), is unchanged in Arid1b haploinsufficient mice, compared to wild type littermates 9. Moreover, heterozygous knockout of Arid1b reduces the number of GABAergic inhibitory interneurons by inhibiting proliferation of ganglionic eminence progenitors and by accelerating apoptosis of developing interneurons 9. The altered number and density of GABAergic interneurons in Arid1b haploinsufficient brains likely leads to abnormal neuronal connectivity, thus breaking a systemic balance between excitation and inhibition (E/I imbalance) and influencing behavior.
Normal morphology and molecular composition of synapses are essential for proper synaptic function 89, 90. Deletion of the gene encoding ELKS2alpha/CAST limits the size of the readily-releasable pool of synaptic vesicles at the active zone of inhibitory synapses and engenders abnormal behavior 91. In addition to the reduced number of inhibitory interneurons, Jung et al. showed that Arid1b haploinsufficiency results in an expanded inhibitory synaptic cleft and shortened postsynaptic density length, potentially contributing to E/I imbalance 9. Miniature excitatory postsynaptic currents (mEPSCs) and miniature inhibitory postsynaptic currents (mIPSC) coincide with the spontaneous release of small quantities of excitatory or inhibitory chemical neurotransmitters from presynaptic terminals 92. Therefore, the mEPSC or mIPSC frequency and amplitude in postsynaptic neurons is assumed to relate to factors operating presynaptically 93. Using whole-cell patch-clamp recording on cortical slices, Jung et al. reported that the mIPSC frequency and amplitude in pyramidal neurons diminish with heterozygous deletion of Arid1b 9. In contrast, there are no significant changes in the frequency or amplitude of mEPSCs between control and Arid1b haploinsufficient neurons. These results suggest that Arid1b haploinsufficiency disrupts inhibitory presynaptic inputs via abnormal formation and transmission of inhibitory synapses, resulting in an E/I imbalance and influencing behavior (Figure 1).
Figure 1: Abnormal epigenetics and gene expression related to inhibitory neurons.

Arid1b haploinsufficiency leads to a reduction in body size and impaired inhibitory synaptic function, including decreased GAD, VGAT, and Gephyrin levels. Inhibitory synaptic cleft width is also increased. The resulting shift in E/I balance leads to ASD- and ID-like behaviors.
The E/I balance in neural circuits is important for normal development and function of the brain 94, 95. In ASD, the E/I balance in key cortical neuronal circuits is disrupted. For this reason, ASD patients have irregular brain rhythms due to abnormal connectivity and neural integration 96, 97. GABAergic interneurons are important for maintaining E/I balance and many ASD-associated genes are expressed in interneurons 80. Specifically, PV-positive GABAergic interneurons drive gamma rhythms and promote cortical circuit performance, and have been shown to be dysregulated in patients with ASD 98. Recent clinical reports indicated that ASD-related behaviors can be caused by altered development of PV-positive GABAergic interneurons 85, 99–101. In mice, Pvalb knockout results in impaired social interaction and communication, along with repetitive and stereotyped behavior patterns 102. Also, Mecp2 deletion restricted to PV-positive interneurons leads to social deficits 103. Furthermore, ASD animal models such as Fmr1 knockout mice 104, the prenatal valproate mouse model 105, and Cntnap2 knockout mice 87 all show abnormal PV-positive GABAergic interneuron function. These results collectively suggest a pattern of E/I imbalance caused by malfunctions in PV-expressing interneurons in ASD patients and mouse models. Importantly, Arid1b haploinsufficiency induces ASD-like neuroanatomical and electrophysiological phenotypes, similar to those observed in other ASD mouse models, especially in regard to PV-positive interneurons.
Molecular insights of ARID1B function
ARID1B participates in ATP-dependent chromatin remodeling as the largest subunit in certain BAF complexes 106. BAF complexes, sometimes referred to as mammalian SWI/SNF complexes, regulate chromatin organization by ejecting, sliding, and rearranging nucleosomes in order to regulate gene transcription 107, 108. Over 20% of human cancers are caused by a mutation to a BAF complex subunit 109–111. All BAF complexes are composed of multiple subunits, and the composition of each multi-subunit complex is determined in a cell type-specific manner 112. For instance, distinct BAF complexes are found in neuronal progenitors and post-mitotic neurons 112. ARID1B contains a helix-turn-helix AT-rich interactive domain (ARID) DNA-binding domain, which recognizes and binds to linear duplex DNA, and, despite the name, does not preferentially bind to AT-rich DNA regions in humans 113. ARID1B is more than 60% identical with another BAF complex subunit, ARID1A, and their incorporation in BAF complexes is mutually exclusive 17, 18. Recent structural analyses showed that ARID1A and ARID1B serves as the structural core of BAF complexes, and do not appear to bind to nucleosomal DNA when included in a BAF complex 114, 115, as had previously been suggested. In 2007, Nagl et al. reported that ARID1A and ARID1B display opposing roles in regulating cell-cycle progression, with ARID1A repressing the cell-cycle and ARID1B driving the expression of pro-proliferation genes 60. In addition, cell-cycle arrest is not disrupted in ARID1B-depleted cells, but cell-cycle re-entry is delayed in a parental pre-osteoblast cell line 60. These results suggest that ARID1B haploinsufficiency may be due to dysregulation of the cell-cycle, potentially leading to aberrant neuronal precursor proliferation.
Considering the role ARID1B plays in epigenetic regulation, it is no surprise that ARID1B haploinsufficiency leads to large-scale changes in gene expression. Shibutani et al. utilized RNA-sequencing to compare the Arid1b+/− brain transcriptome with microarray data from patients with ASD and Chd8 haploinsufficient mice 52. They reported widespread overlap in gene expression between Arid1b+/− mice and both ASD patients and Chd8 mutant mice, but they did not further explore any mechanistic explanations for these similarities, beyond mentioning that CHD8 and ARID1B are both chromatin remodeling factors 52. The authors did, however, perform a further RNA expression level comparison between Arid1b+/− brains and mouse fast-spiking neonatal neurons, presumably PV-expressing inhibitory interneurons, and reported that Arid1b haploinsufficiency generates a gene-expression profile more similar to immature fast-spiking cells 52, a hallmark of ASD brains 116. Celen et al. also performed RNA-sequencing and determined widespread gene expression differences in the hippocampus of Arid1b+/− mice compared to wild-type mice, including differential regulation of 14 autism risk genes 19. They also showed that plasma IGF1 level and liver Igf1 mRNA expression are lower in Arid1b+/− mice compared to control animals, and suggested that this IGF1 deficiency may explain the smaller stature of Arid1b+/− mice and some human patients with ARID1B haploinsufficiency 19. The authors failed to suggest any direct mechanistic link between IGF1 levels and ARID1B. However, they did report that conditional, brain-specific, heterozygous knockout of Arid1b leads to reduced mouse size and IGF1 deficiencies, while liver-specific Arid1b manipulations influence neither stature nor IGF1 levels 19. This implies that ARID1B deficits within the brain are, in large part, responsible for growth impairments and brain dysfunction in Arid1b haploinsufficient mice.
In addition to chromatin remodeling, ARID1B is involved in gene regulation via the posttranslational epigenetic modification of histones 7, 9, 117, though this too may be attributed to increased physical access for histone modifying enzymes to nucleosome-depleted DNA regions created by BAF complex activity 109, 111, 114. In their mouse model of Arid1b haploinsufficiency, Jung et al. showed that histone acetylation and methylation are decreased in Arid1b+/− mice, compared with wild type littermates 9. ARID1B modulates histone acetyltransferase (HAT) and histone deacetyltransferase (HDAC) activity in the periphery 60 and in cell lines 7, but Jung and colleagues did not detect any changes in the level or activity of HAT or HDAC in the brains of Arid1b haploinsufficient mice 9. However, they reported that heterozygous deletion of Arid1b leads to decreased acetylation of histone H3 at lysine 9 (H3K9ac), a marker of transcriptional activation, in the promoter region of the Pvalb gene, which encodes PV 9. Accordingly, Jung et al. also reported a decrease in phosphorylation of the (ser5)-carboxy-terminal domain of RNA polymerase II in the Pvalb promoter region, which indicates decreased transcriptional initiation of the gene (Figure 2) 9.
Figure 2: Abnormal epigenetics and gene expression related to inhibitory neurons.
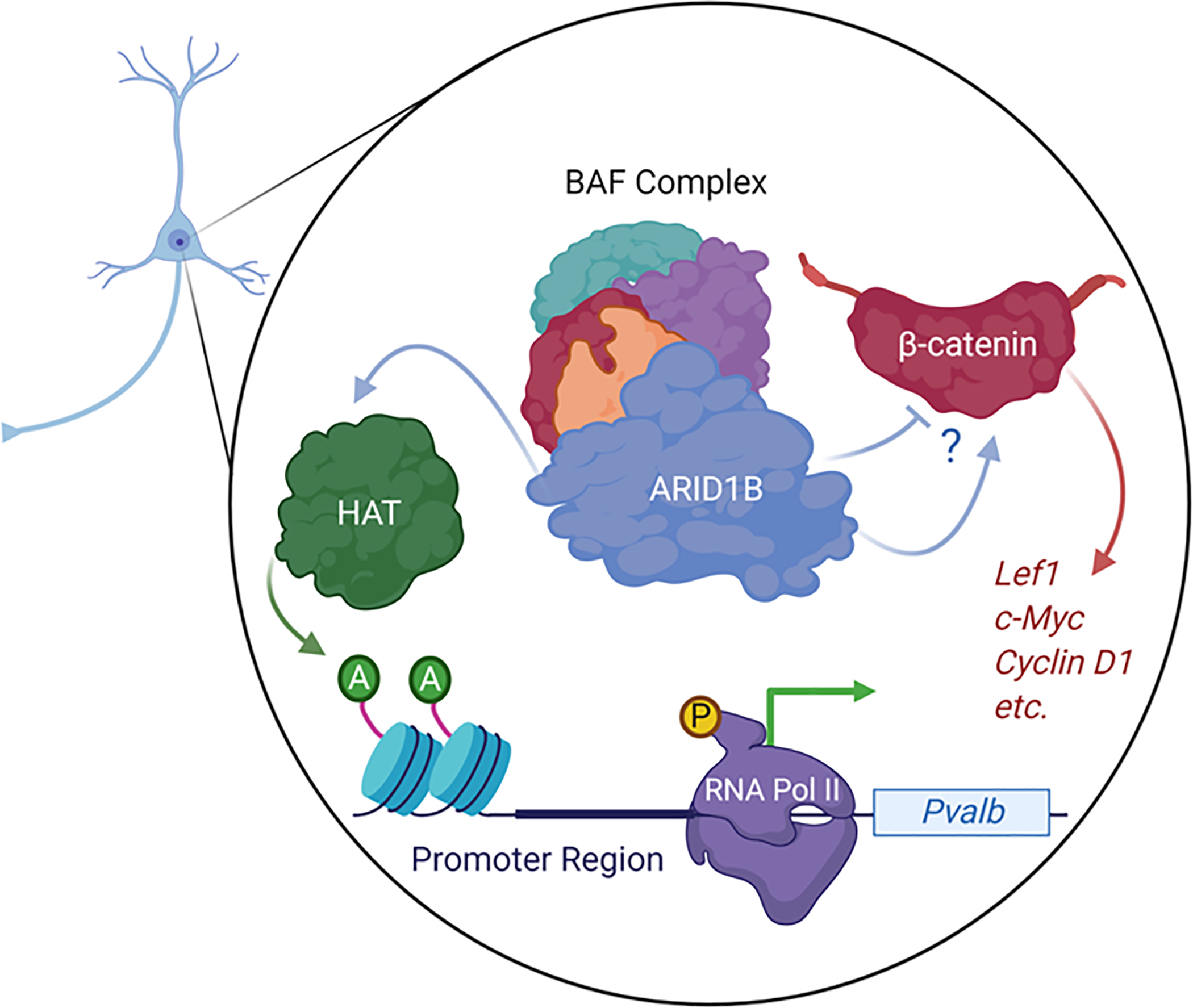
ARID1B, as a member of the BAF complex, maintains HAT activity in the Pvalb promoter region, leading to increased gene expression. ARID1B also interacts with β-catenin to up- or down-regulate β-catenin target gene expression in a cell-type specific manner.
In accordance with prior research demonstrating the role of ARID1B in cell-cycle regulation 60, Jung et al. showed that Arid1b haploinsufficiency-induced loss of PV-positive interneurons is due to impaired proliferation of interneuron precursors in the ganglionic eminences 9. They reported that Wnt/β-catenin signaling-related genes, including Lef1, c-Myc, Cyclin D1 and others, are downregulated in the ventral telencephalon of Arid1b+/− mice (Figure 2) 9. ARID1B was previously shown in vitro to enable access for the BAF chromatin remodeling complex to the c-Myc promoter and activating c-Myc gene expression, a well-established β-catenin target 60. The same report also showed that ARID1B associates with repressive transcription factors at the c-Myc promotor, but concluded that ARID1B is not essential for repressing cell-cycle activity via c-Myc 60. Another study reported, however, that ARID1B represses the expression of Wnt/β-catenin target genes in a BRG1-dependent manner 7. ARID1B does not directly interact with β-catenin, rather BRG1 complexes with β-catenin 118, and ARID1B influences β-catenin’s transcriptional regulation via its interaction with BRG1 7. Initially, the physical interaction between BRG1 and β-catenin was shown to promote the expression of β-catenin target genes 118, but the specific BAF complex subunits recruited to the BRG1-β-catenin complex appear to determine whether expression of a specific target gene is promoted or repressed 7, 49, 60, 119. Liu et al. recently confirmed that ARID1B regulates Wnt/β-catenin transcriptional regulation in HEK293T cells, and that this regulation is indeed dependent on BRG1-β-catenin interaction 16. They further showed that knockout of Arid1b in a mouse chondrogenic cell line impedes cell proliferation and reduces the expression level of downstream β-catenin target genes, however, Arid1b/ARID1B knockout in undifferentiated chondrogenic cells and in HEK293T cells upregulates β-catenin target gene expression 16. They concluded that the growth deficits they and others observe in human patients with ARID1B haploinsufficiency, as well as in their own zebrafish arid1b knockdown model, are likely due to impaired Wnt/β-catenin activity 16. These findings provide perhaps a more complete explanation for the causative role of ARID1B in regulating growth. Overall, due to the physical interaction between β-catenin and BRG1/ARID1B and ARID1B’s upstream regulation of Wnt/β-catenin gene expression, it is likely that the effects of ARID1B haploinsufficiency on Wnt/β-catenin signaling will largely come down to specific cell-types and developmental timepoints.
Research into the direct and indirect roles of ARID1B in brain development and neuronal function is still in its nascent stages. Future elucidation of the epigenetic and signaling regulatory roles of ARID1B will contribute to the development of more targeted therapies. Furthermore, a new human embryonic stem cell (hESC) heterozygous ARID1B line could allow for more investigation of the molecular function of ARID1B in human cells 14, though it is vital to remember that different cell-types utilize distinct BAF complex conformations 112, which can have an extreme influence on the apparent molecular function of ARID1B 60.
Development of clinical readiness and outcome assessment for ARID1B disorders
One hurdle to addressing the treatment of ARID1B-RD and other neurodevelopmental disorders, is the lack of reliable clinical endpoints for treatment, or targeted outcome measurements indicating therapeutic success 120. In a recent report, Kruizinga and colleagues grappled with the specific challenges in assessing the efficacy of therapeutic interventions to treat ARID1B-RD 15. They recruited 12 patients with pathogenic ARID1B mutations and performed a battery of non-invasive tests to determine appropriate clinical endpoints that could be incorporated into a clinical trial testing potential ARID1B-RD treatments 15. The authors argued that the tolerability, accuracy, stability, and significance of each test needs to be evaluated to determine its efficacy for use as a clinical endpoint. Specifically, they demonstrated that cognitive assessments (such as the animal fluency test 121), eye tracking measurements, executive function tests, and EEG analyses were all found to effectively differentiate between individuals with an ARID1B-RD and a control group 15. They also equipped subjects with smart watches that tracked their step count, heart rate and sleep patterns, and report that these may be an effective and well-tolerated tool for evaluating clinical outcomes for ARID1B-RD and similar rare neurodevelopmental disorders 15.
Overall, this study demonstrates an important first step toward the effective evaluation of pharmacological agents for treating ARID1B-RD. Due to the relatively small sample size, and the necessary exclusion of patients with severe ID 15, it is possible that additional tests may be more effective in evaluating outcomes in some cases.
Reversing E/I imbalance as a treatment tool for ARID1B haploinsufficiency-induced neurodevelopmental conditions
Developing pharmacological interventions for neurodevelopmental and neuropsychiatric disorders has proved challenging, but as preclinical and clinical tools for measuring the underlying causes of these disorders improve, the opportunity for targeted therapies is expanding 122. For instance, new research suggests that the gut/brain axis may play a role in the pathogenesis of ASD and other neurological disorders, which intimates that guided repopulation of the gut microbiome could be a viable treatment option in the future 123, 124. In 2012, Han et al. demonstrated that treatment with a low dose of the GABAA receptor positive allosteric modulator, clonazepam, is effective in reversing several ASD-like behaviors in the Scn1A haploinsufficiency mouse model of autism 85. The same group later showed that the low-dose clonazepam treatment is effective in another ASD mouse model, BTBR mice 125. As Jung et al. detected a marked decrease in the number of PV-expressing interneurons in Arid1b+/− mice, they examined the efficacy of clonazepam in reversing ID- and ASD-like behavior in this model 9. Acute clonazepam treatment produced a significant improvement in social behavior, anxiety-like behavior, and recognition memory, but was unable to rescue all aberrant behavioral phenotypes 9. It is likely that that some of these behaviors would benefit from interventions during a particular developmental window 122, but it is promising that positive GABA modulation is effective in treating some behavioral aspects in multiple ASD mouse models 9, 85, 125, 126.
A benzodiazepine, clonazepam is a potent sedative and patients who take clonazepam can develop dependence and undergo withdrawal following treatment cessation 127. In addition to its function as a positive allosteric modulator of the GABAA receptor, clonazepam also has serotonergic effects and is commonly prescribed to treat panic disorder and seizures 127. On a promising note, the doses commonly used in animal models for ASD, including Arid1b haploinsufficient mice, are much lower than those typically required to treat other neurological disorders 9, 85, 125. This implies that clonazepam may be effective in treating ARID1B-RD without the complications and side effects associated with higher doses. Nevertheless, there is still work to be done in developing more targeted treatments.
One potential avenue for future exploration is developing GABA modulators that specifically target interneuron subtypes. As discussed above, Jung et al. showed that Arid1b haploinsufficient mice display a significant reduction in PV-expressing interneurons 9 and, in a follow-up study, Smith et al. demonstrated that conditional deletion of Arid1b in PV- or SST-expressing interneurons yields divergent behavioral outcomes 59. Due to the heterogeneity of ARID1B-RD, and of ID and ASD in general, developing specific drugs targeting a small subset of affected cells would allow for a more personalized treatment regimen. Achieving this level of specificity, however, will require a concerted effort. Due to the rise of chemogenetic and optogenetic technologies in preclinical research, it is getting easier to manipulate specific neurons in real-time in rodents, but there is currently limited potential for translation into clinical use 128. On the other hand, cortical PV- and SST-positive GABAergic interneurons may have distinct surface receptors, as is the case with nicotinic acetylcholine receptors 129, that could be targeted individually to improve specific symptoms. Characterizing and utilizing the intrinsic diversity present in neuronal subtypes, will hopefully lead to effective and targeted therapies.
Concluding remarks
It has now been 10 years since the first reported case of ID caused by ARID1B haploinsufficiency was published 1. In the intervening decade, our collective understanding of the role of ARID1B in normal brain development and function has rapidly expanded. With the development of Arid1b haploinsufficiency mouse models 9, 19, 52 and, now, a heterozygous ARID1B knockout hESC line 14, the requisite tools are in place to develop novel therapies to treat ARID1B-RD. The discovery that Arid1b haploinsufficiency leads to E/I imbalance in the mouse brain due to a significant loss of PV-positive interneurons, and that treatment with a GABA positive allosteric modulator effectively rescues several ASD- and ID-like behaviors 9, could have a large impact on future drug development to treat ARID1B-RD and other neurological disorders with a convergent E/I imbalance root. Moreover, as we continue to disentangle the apparently complex interaction between ARID1B and the Wnt/β-catenin signaling cascade in different cell-types, we stand to receive fresh insight into epigenetic regulation of the cell-cycle, with potentially outsized roles in neurodevelopmental and emotional disorders and cancer. Finally, the initiative to test ARID1B-RD patients for suitable clinical endpoints 15, will help to ensure that clinical trials provide an accurate accounting of drug efficacy.
Looking forward, the future is bright in the sphere of ARID1B-RD research, and the next 10 years have great promise to produce new breakthroughs. As technologies and computational strategies are developed and improved, the potential to untangle the complicated molecular roles of ARID1B, on its own and within BAF complexes, will continue to grow, as will the prospect of translating these molecular insights into clinical treatments.
Acknowledgements
This work was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R01NS091220 to W.Y. Kim. Figures were created in part using Biorender.
Footnotes
Competing interests
None
References
- 1.Backx L, Seuntjens E, Devriendt K, Vermeesch J, Van Esch H. A balanced translocation t(6;14)(q25.3;q13.2) leading to reciprocal fusion transcripts in a patient with intellectual disability and agenesis of corpus callosum. Cytogenet Genome Res 2011; 132(3): 135–143. [DOI] [PubMed] [Google Scholar]
- 2.Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM et al. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet 2012; 82(3): 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 2012; 90(3): 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet 2012; 44(4): 379–380. [DOI] [PubMed] [Google Scholar]
- 5.Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 2012; 44(4): 376–378. [DOI] [PubMed] [Google Scholar]
- 6.Sim JC, White SM, Fitzpatrick E, Wilson GR, Gillies G, Pope K et al. Expanding the phenotypic spectrum of ARID1B-mediated disorders and identification of altered cell-cycle dynamics due to ARID1B haploinsufficiency. Orphanet J Rare Dis 2014; 9: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vasileiou G, Ekici AB, Uebe S, Zweier C, Hoyer J, Engels H et al. Chromatin-Remodeling-Factor ARID1B Represses Wnt/beta-Catenin Signaling. Am J Hum Genet 2015; 97(3): 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ka M, Chopra DA, Dravid SM, Kim WY. Essential Roles for ARID1B in Dendritic Arborization and Spine Morphology of Developing Pyramidal Neurons. J Neurosci 2016; 36(9): 2723–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung EM, Moffat JJ, Liu J, Dravid SM, Gurumurthy CB, Kim WY. Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat Neurosci 2017; 20(12): 1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu D, Jiao X, Cao T, Huang F. Serum miRNA expression profiling reveals miR-486–3p may play a significant role in the development of autism by targeting ARID1B. Neuroreport 2018; 29(17): 1431–1436. [DOI] [PubMed] [Google Scholar]
- 11.Demily C, Duwime C, Lopez C, Hemimou C, Poisson A, Plasse J et al. Corpus callosum metrics predict severity of visuospatial and neuromotor dysfunctions in ARID1B mutations with Coffin-Siris syndrome. Psychiatr Genet 2019; 29(6): 237–242. [DOI] [PubMed] [Google Scholar]
- 12.Gorokhova S, Mortreux J, Afenjar A, Attie-Bitach T, Blanluet M, Cormier-Daire V et al. Significant contribution of intragenic deletions to ARID1B mutation spectrum. Genet Med 2019; 21(11): 2654–2655. [DOI] [PubMed] [Google Scholar]
- 13.van der Sluijs PJ, Jansen S, Vergano SA, Adachi-Fukuda M, Alanay Y, AlKindy A et al. The ARID1B spectrum in 143 patients: from nonsyndromic intellectual disability to Coffin-Siris syndrome. Genet Med 2019; 21(6): 1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boerstler T, Wend H, Krumbiegel M, Kavyanifar A, Reis A, Lie DC et al. CRISPR/Cas9 mediated generation of human ARID1B heterozygous knockout hESC lines to model Coffin-Siris syndrome. Stem Cell Res 2020; 47: 101889. [DOI] [PubMed] [Google Scholar]
- 15.Kruizinga MD, Zuiker R, Sali E, de Kam ML, Doll RJ, Groeneveld GJ et al. Finding Suitable Clinical Endpoints for a Potential Treatment of a Rare Genetic Disease: the Case of ARID1B. Neurotherapeutics 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Hu G, Ye J, Ye B, Shen N, Tao Y et al. De Novo ARID1B mutations cause growth delay associated with aberrant Wnt/beta-catenin signaling. Hum Mutat 2020; 41(5): 1012–1024. [DOI] [PubMed] [Google Scholar]
- 17.Nie Z, Yan Z, Chen EH, Sechi S, Ling C, Zhou S et al. Novel SWI/SNF chromatin-remodeling complexes contain a mixed-lineage leukemia chromosomal translocation partner. Mol Cell Biol 2003; 23(8): 2942–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Nagl NG, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J 2004; 383(Pt 2): 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Celen C, Chuang JC, Luo X, Nijem N, Walker AK, Chen F et al. Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. Elife 2017; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee AW, Ventola P, Budimirovic D, Berry-Kravis E, Visootsak J. Clinical Development of Targeted Fragile X Syndrome Treatments: An Industry Perspective. Brain Sci 2018; 8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. J Neurodev Disord 2017; 9: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapp SK, Gillespie-Lynch K, Sherman LE, Hutman T. Deficit, difference, or both? Autism and neurodiversity. Dev Psychol 2013; 49(1): 59–71. [DOI] [PubMed] [Google Scholar]
- 23.Pantazakos T Treatment for whom? Towards a phenomenological resolution of controversy within autism treatment. Stud Hist Philos Biol Biomed Sci 2019; 77: 101176. [DOI] [PubMed] [Google Scholar]
- 24.Eady N, Courtenay K, Strydom A. Pharmacological management of behavioral and psychiatric symptoms in older adults with intellectual disability. Drugs Aging 2015; 32(2): 95–102. [DOI] [PubMed] [Google Scholar]
- 25.Ellison JW, Rosenfeld JA, Shaffer LG. Genetic basis of intellectual disability. Annu Rev Med 2013; 64(1): 441–450. [DOI] [PubMed] [Google Scholar]
- 26.Simonoff E, Pickles A, Chadwick O, Gringras P, Wood N, Higgins S et al. The Croydon Assessment of Learning Study: prevalence and educational identification of mild mental retardation. J Child Psychol Psychiatry 2006; 47(8): 828–839. [DOI] [PubMed] [Google Scholar]
- 27.Westerinen H, Kaski M, Virta L, Almqvist F, Iivanainen M. Prevalence of intellectual disability: a comprehensive study based on national registers. J Intellect Disabil Res 2007; 51(Pt 9): 715–725. [DOI] [PubMed] [Google Scholar]
- 28.Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 2015; 385(9975): 1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vergano SA, van der Sluijs PJ, Santen G. ARID1B-Related Disorder. BTI - GeneReviews(®). University of Washington, Seattle: 1993. [PubMed] [Google Scholar]
- 30.Santen GW, Clayton-Smith J, consortium ABC. The ARID1B phenotype: what we have learned so far. Am J Med Genet C Semin Med Genet 2014; 166C(3): 276–289. [DOI] [PubMed] [Google Scholar]
- 31.Mignot C, Moutard ML, Rastetter A, Boutaud L, Heide S, Billette T et al. ARID1B mutations are the major genetic cause of corpus callosum anomalies in patients with intellectual disability. Brain 2016; 139(11): e64. [DOI] [PubMed] [Google Scholar]
- 32.Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ et al. Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum Mutat 2013; 34(11): 1519–1528. [DOI] [PubMed] [Google Scholar]
- 33.Wieczorek D, Bogershausen N, Beleggia F, Steiner-Haldenstatt S, Pohl E, Li Y et al. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet 2013; 22(25): 5121–5135. [DOI] [PubMed] [Google Scholar]
- 34.Miyake N, Tsurusaki Y, Matsumoto N. Numerous BAF complex genes are mutated in Coffin-Siris syndrome. Am J Med Genet C Semin Med Genet 2014; 166C(3): 257–261. [DOI] [PubMed] [Google Scholar]
- 35.Bramswig NC, Caluseriu O, Ludecke HJ, Bolduc FV, Noel NC, Wieland T et al. Heterozygosity for ARID2 loss-of-function mutations in individuals with a Coffin-Siris syndrome-like phenotype. Hum Genet 2017; 136(3): 297–305. [DOI] [PubMed] [Google Scholar]
- 36.Vasileiou G, Vergarajauregui S, Endele S, Popp B, Buttner C, Ekici AB et al. Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome. Am J Hum Genet 2018; 102(3): 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosho T, Okamoto N, Coffin-Siris Syndrome International C. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet 2014; 166C(3): 262–275. [DOI] [PubMed] [Google Scholar]
- 38.Pasca SP, Veenstra-VanderWeele J, McPartland JC. Research and training in autism spectrum disorder to catalyze the next genomic and neuroscience revolutions. Mol Psychiatry 2020. [DOI] [PubMed] [Google Scholar]
- 39.Knopf A Autism prevalence increases from 1 in 60 to 1 in 54: CDC. The Brown University Child and Adolescent Behavior Letter 2020; 36(6): 4–4. [Google Scholar]
- 40.Fombonne E The epidemiology of autism: a review. Psychol Med 1999; 29(4): 769–786. [DOI] [PubMed] [Google Scholar]
- 41.O’Brien G, Pearson J. Autism and learning disability. Autism 2004; 8(2): 125–140. [DOI] [PubMed] [Google Scholar]
- 42.Perou R, Bitsko RH, Blumberg SJ, Pastor P, Ghandour RM, Gfroerer JC et al. Mental health surveillance among children--United States, 2005–2011. MMWR supplements 2013; 62(2): 1–35. [PubMed] [Google Scholar]
- 43.Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ 2018; 67(6): 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nord AS, Roeb W, Dickel DE, Walsh T, Kusenda M, O’Connor KL et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur J Hum Genet 2011; 19(6): 727–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wing L, Gould J. Severe impairments of social interaction and associated abnormalities in children: epidemiology and classification. J Autism Dev Disord 1979; 9(1): 11–29. [DOI] [PubMed] [Google Scholar]
- 46.Purpura G, Fulceri F, Puglisi V, Masoni P, Contaldo A. Motor coordination impairment in children with autism spectrum disorder: a pilot study using Movement Assessment Battery for Children-2 Checklist. Minerva Pediatr 2016; (1827–1715 (Electronic)). [DOI] [PubMed] [Google Scholar]
- 47.Robertson J, Hatton C, Emerson E, Baines S. Prevalence of epilepsy among people with intellectual disabilities: A systematic review. Seizure 2015; 29: 46–62. [DOI] [PubMed] [Google Scholar]
- 48.Kessi M, Xiong J, Wu L, Yang L, He F, Chen C et al. Rare Copy Number Variations and Predictors in Children With Intellectual Disability and Epilepsy. Front Neurol 2018; 9: 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 2013; 14(5): 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lopez AJ, Wood MA. Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci 2015; 9: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabriele M, Lopez Tobon A, D’Agostino G, Testa G. The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Prog Neuropsychopharmacol Biol Psychiatry 2018; 84(Pt B): 306–327. [DOI] [PubMed] [Google Scholar]
- 52.Shibutani M, Horii T, Shoji H, Morita S, Kimura M, Terawaki N et al. Arid1b Haploinsufficiency Causes Abnormal Brain Gene Expression and Autism-Related Behaviors in Mice. Int J Mol Sci 2017; 18(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daily DK, Ardinger HH, Holmes GE. Identification and evaluation of mental retardation. American family physician 2000; 61(4): 1059–1067, 1070. [PubMed] [Google Scholar]
- 54.Fakhoury M Autistic spectrum disorders: A review of clinical features, theories and diagnosis. International journal of developmental neuroscience : the official journal of the International Society for Developmental Neuroscience 2015; 43: 70–77. [DOI] [PubMed] [Google Scholar]
- 55.Reid KA, Smiley E, Cooper SA. Prevalence and associations of anxiety disorders in adults with intellectual disabilities. J Intellect Disabil Res 2011; 55(2): 172–181. [DOI] [PubMed] [Google Scholar]
- 56.Nelson SB, Valakh V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015; 87(4): 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramamoorthi K, Lin Y. The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends in molecular medicine 2011; 17(8): 452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marin O Interneuron dysfunction in psychiatric disorders. Nature reviews Neuroscience 2012; 13(2): 107–120. [DOI] [PubMed] [Google Scholar]
- 59.Smith AL, Jung EM, Jeon BT, Kim WY. Arid1b haploinsufficiency in parvalbumin- or somatostatin-expressing interneurons leads to distinct ASD-like and ID-like behavior. Sci Rep 2020; 10(1): 7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagl NG Jr., Wang X, Patsialou A, Van Scoy M, Moran E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J 2007; 26(3): 752–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan Z, Wang Z, Sharova L, Sharov AA, Ling C, Piao Y et al. BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells 2008; 26(5): 1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA et al. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 2007; 56(1): 94–108. [DOI] [PubMed] [Google Scholar]
- 63.Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 2002; 34(6): 999–1010. [DOI] [PubMed] [Google Scholar]
- 64.Kwon M, Fernandez JR, Zegarek GF, Lo SB, Firestein BL. BDNF-promoted increases in proximal dendrites occur via CREB-dependent transcriptional regulation of cypin. J Neurosci 2011; 31(26): 9735–9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rubio-Garrido P, Perez-de-Manzo F, Porrero C, Galazo MJ, Clasca F. Thalamic input to distal apical dendrites in neocortical layer 1 is massive and highly convergent. Cereb Cortex 2009; 19(10): 2380–2395. [DOI] [PubMed] [Google Scholar]
- 66.Gilbert CD, Sigman M. Brain states: top-down influences in sensory processing. Neuron 2007; 54(5): 677–696. [DOI] [PubMed] [Google Scholar]
- 67.Sjostrom PJ, Hausser M. A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron 2006; 51(2): 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aguirre-Chen C, Stec N, Ramos OM, Kim N, Kramer M, McCarthy S et al. A Caenorhabditis elegans Model for Integrating the Functions of Neuropsychiatric Risk Genes Identifies Components Required for Normal Dendritic Morphology. G3 (Bethesda) 2020; 10(5): 1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.John A, Brylka H, Wiegreffe C, Simon R, Liu P, Juttner R et al. Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development 2012; 139(10): 1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I et al. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 2004; 303(5655): 197–202. [DOI] [PubMed] [Google Scholar]
- 71.Martinez-Cerdeno V Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev Neurobiol 2017; 77(4): 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gilbert J, Man HY. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front Cell Neurosci 2017; 11: 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Overstreet-Wadiche L, McBain CJ. Neurogliaform cells in cortical circuits. Nature reviews Neuroscience 2015; 16(8): 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nature reviews Neuroscience 2004; 5(10): 793–807. [DOI] [PubMed] [Google Scholar]
- 75.Tremblay R, Lee S, Rudy B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016; 91(2): 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Urban-Ciecko J, Barth AL. Somatostatin-expressing neurons in cortical networks. Nature reviews Neuroscience 2016; 17(7): 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2001; 25(1): 1–27. [DOI] [PubMed] [Google Scholar]
- 78.Lim L, Mi D, Llorca A, Marin O. Development and Functional Diversification of Cortical Interneurons. Neuron 2018; 100(2): 294–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chattopadhyaya B, Cristo GD. GABAergic circuit dysfunctions in neurodevelopmental disorders. Frontiers in psychiatry 2012; 3: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee E, Lee J, Kim E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biological psychiatry 2017; 81(10): 838–847. [DOI] [PubMed] [Google Scholar]
- 81.Puts NAJ, Wodka EL, Harris AD, Crocetti D, Tommerdahl M, Mostofsky SH et al. Reduced GABA and altered somatosensory function in children with autism spectrum disorder. Autism research : official journal of the International Society for Autism Research 2017; 10(4): 608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robertson CE, Ratai EM, Kanwisher N. Reduced GABAergic Action in the Autistic Brain. Curr Biol 2016; 26(1): 80–85. [DOI] [PubMed] [Google Scholar]
- 83.Harada M, Taki MM, Nose A, Kubo H, Mori K, Nishitani H et al. Non-invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-editing proton MR spectroscopy using a clinical 3 tesla instrument. J Autism Dev Disord 2011; 41(4): 447–454. [DOI] [PubMed] [Google Scholar]
- 84.Gaetz W, Bloy L, Wang DJ, Port RG, Blaskey L, Levy SE et al. GABA estimation in the brains of children on the autism spectrum: measurement precision and regional cortical variation. NeuroImage 2014; 86: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012; 489(7416): 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mao W, Watanabe T, Cho S, Frost JL, Truong T, Zhao X et al. Shank1 regulates excitatory synaptic transmission in mouse hippocampal parvalbumin-expressing inhibitory interneurons. The European journal of neuroscience 2015; 41(8): 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lauber E, Filice F, Schwaller B. Dysregulation of Parvalbumin Expression in the Cntnap2−/− Mouse Model of Autism Spectrum Disorder. Front Mol Neurosci 2018; 11: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wiebe S, Nagpal A, Truong VT, Park J, Skalecka A, He AJ et al. Inhibitory interneurons mediate autism-associated behaviors via 4E-BP2. Proceedings of the National Academy of Sciences of the United States of America 2019; 116(36): 18060–18067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harris KM, Weinberg RJ. Ultrastructure of synapses in the mammalian brain. Cold Spring Harbor perspectives in biology 2012; 4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harbor perspectives in biology 2012; 4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaeser PS, Deng L, Chavez AE, Liu X, Castillo PE, Sudhof TC. ELKS2alpha/CAST deletion selectively increases neurotransmitter release at inhibitory synapses. Neuron 2009; 64(2): 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci 1998; 18(21): 8740–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wall MJ, Usowicz MM. Development of the quantal properties of evoked and spontaneous synaptic currents at a brain synapse. Nature neuroscience 1998; 1(8): 675–682. [DOI] [PubMed] [Google Scholar]
- 94.Rho HS, Ahn SM, Hwang JS. Inhibitory effect of N-adamantyl-3,4-dihydroxybenzamide on melanogenesis in melan-a cells and brown guinea pigs. Archives of dermatological research 2011; 303(3): 153–159. [DOI] [PubMed] [Google Scholar]
- 95.Selten M, van Bokhoven H, Nadif Kasri N. Inhibitory control of the excitatory/inhibitory balance in psychiatric disorders. F1000Res 2018; 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry 2019; 24(9): 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ajram LA, Horder J, Mendez MA, Galanopoulos A, Brennan LP, Wichers RH et al. Shifting brain inhibitory balance and connectivity of the prefrontal cortex of adults with autism spectrum disorder. Translational psychiatry 2017; 7(5): e1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lunden JW, Durens M, Phillips AW, Nestor MW. Cortical interneuron function in autism spectrum condition. Pediatric research 2019; 85(2): 146–154. [DOI] [PubMed] [Google Scholar]
- 99.Karayannis T, Au E, Patel JC, Kruglikov I, Markx S, Delorme R et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature 2014; 511(7508): 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gogolla N, Leblanc JJ, Quast KB, Sudhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. Journal of neurodevelopmental disorders 2009; 1(2): 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pizzarelli R, Cherubini E. Alterations of GABAergic signaling in autism spectrum disorders. Neural plasticity 2011; 2011: 297153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wohr M, Orduz D, Gregory P, Moreno H, Khan U, Vorckel KJ et al. Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Translational psychiatry 2015; 5: e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY. Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes. Neuron 2015; 88(4): 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Selby L, Zhang C, Sun QQ. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neuroscience letters 2007; 412(3): 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lauber E, Filice F, Schwaller B. Prenatal Valproate Exposure Differentially Affects Parvalbumin-Expressing Neurons and Related Circuits in the Cortex and Striatum of Mice. Front Mol Neurosci 2016; 9: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 2011; 21(3): 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol 2017; 18(7): 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou CY, Johnson SL, Gamarra NI, Narlikar GJ. Mechanisms of ATP-Dependent Chromatin Remodeling Motors. Annu Rev Biophys 2016; 45: 153–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 2015; 1(5): e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013; 45(6): 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Masliah-Planchon J, Bieche I, Guinebretiere JM, Bourdeaut F, Delattre O. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol 2015; 10: 145–171. [DOI] [PubMed] [Google Scholar]
- 112.Ho L, Crabtree GR. Chromatin remodelling during development. Nature 2010; 463(7280): 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dallas PB, Pacchione S, Wilsker D, Bowrin V, Kobayashi R, Moran E. The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol Cell Biol 2000; 20(9): 3137–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X et al. Structure of nucleosome-bound human BAF complex. Science 2020; 367(6480): 875–881. [DOI] [PubMed] [Google Scholar]
- 115.Han Y, Reyes AA, Malik S, He Y. Cryo-EM structure of SWI/SNF complex bound to a nucleosome. Nature 2020; 579(7799): 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gandal MJ, Nesbitt AM, McCurdy RM, Alter MD. Measuring the maturity of the fast-spiking interneuron transcriptional program in autism, schizophrenia, and bipolar disorder. PLoS One 2012; 7(8): e41215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li XS, Trojer P, Matsumura T, Treisman JE, Tanese N. Mammalian SWI/SNF--a subunit BAF250/ARID1 is an E3 ubiquitin ligase that targets histone H2B. Mol Cell Biol 2010; 30(7): 1673–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barker N, Hurlstone A, Musisi H, Miles A, Bienz M, Clevers H. The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J 2001; 20(17): 4935–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Son EY, Crabtree GR. The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am J Med Genet C Semin Med Genet 2014; 166C(3): 333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Budimirovic DB, Berry-Kravis E, Erickson CA, Hall SS, Hessl D, Reiss AL et al. Updated report on tools to measure outcomes of clinical trials in fragile X syndrome. J Neurodev Disord 2017; 9: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tombaugh TN, Kozak J, Rees L. Normative data stratified by age and education for two measures of verbal fluency: FAS and animal naming. Arch Clin Neuropsychol 1999; 14(2): 167–177. [PubMed] [Google Scholar]
- 122.Basilico B, Morandell J, Novarino G. Molecular mechanisms for targeted ASD treatments. Curr Opin Genet Dev 2020; 65: 126–137. [DOI] [PubMed] [Google Scholar]
- 123.Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA et al. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019; 101(2): 246–259 e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cryan JF, O’Riordan KJ, Sandhu K, Peterson V, Dinan TG. The gut microbiome in neurological disorders. Lancet Neurol 2020; 19(2): 179–194. [DOI] [PubMed] [Google Scholar]
- 125.Han S, Tai C, Jones CJ, Scheuer T, Catterall WA. Enhancement of inhibitory neurotransmission by GABAA receptors having alpha2,3-subunits ameliorates behavioral deficits in a mouse model of autism. Neuron 2014; 81(6): 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rhine MA, Parrott JM, Schultz MN, Kazdoba TM, Crawley JN. Hypothesis-driven investigations of diverse pharmacological targets in two mouse models of autism. Autism Res 2019; 12(3): 401–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nardi AE, Perna G. Clonazepam in the treatment of psychiatric disorders: an update. Int Clin Psychopharmacol 2006; 21(3): 131–142. [DOI] [PubMed] [Google Scholar]
- 128.Delbeke J, Hoffman L, Mols K, Braeken D, Prodanov D. And Then There Was Light: Perspectives of Optogenetics for Deep Brain Stimulation and Neuromodulation. Front Neurosci 2017; 11: 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Demars MP, Morishita H. Cortical parvalbumin and somatostatin GABA neurons express distinct endogenous modulators of nicotinic acetylcholine receptors. Mol Brain 2014; 7: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pirola B, Bortotto L, Giglio S, Piovan E, Janes A, Guerrini R et al. Agenesis of the corpus callosum with Probst bundlesowing to haploinsufficiency for a gene in an 8 cM region of 6q25. Journal of medical genetics 1998; 35(12): 1031–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nagamani SC, Erez A, Eng C, Ou Z, Chinault C, Workman L et al. Interstitial deletion of 6q25.2-q25.3: a novel microdeletionsyndrome associated with microcephaly, developmental delay, dysmorphic features and hearing loss. Eur J Hum Genet 2009;17(5): 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vengoechea J, Carpenter L, Zarate YA. Papillary thyroid cancer in a patient with interstitial 6q25 deletion including ARID1B. American journal of medical genetics Part A 2014; 164A(7): 1857–1859. [DOI] [PubMed] [Google Scholar]