

Abstract
Ovarian cancer (OC) remains the leading cause of mortality due to gynecological malignancies. Epidemiological studies have demonstrated that steroid hormones released from the hypothalamic-pituitary-ovarian axis can play a role in stimulating or inhibiting OC progression, with gonadotropins, estrogens and androgens promoting OC progression, while gonadotropin-releasing hormone (GnRH) and progesterone may be protective factors in OC. Experimental studies have indicated that hormone receptors are expressed in OC cells and mediate the growth stimulatory or growth inhibitory effects of hormones on these cells. Hormone therapy agents have been evaluated in a number of clinical trials. The majority of these trials were conducted in patients with relapsed or refractory OC with average efficacy and limited side-effects. A better understanding of the mechanisms through which hormones affect cell growth may improve the efficacy of hormone therapy. In the present review article, the role of hormones (GnRH, gonadotropins, androgens, estrogens and progestins) and their receptors in OC tumorigenesis, and hormonal therapy in OC treatment is discussed and summarized.
Keywords: ovarian cancer, hormone hypothesis, hormone receptors, gonadotropin-releasing hormone, hormone replacement therapy
1. Introduction
Ovarian cancer (OC) is a highly common malignancy of the female reproductive system that ranks 4th among all causes of cancer-related mortality among women (1,2). The early symptoms of OC are atypical, and reliable methods in terms of early detection are insufficient, with ~70% of cases already presenting with International Federation of Gynecology and Obstetrics (FIGO) stage III/IV disease at clinical diagnosis (3). OC is currently treated with full-stage surgery or tumor cell reduction combined with first-line chemotherapy drugs (paclitaxel or platinum) as the standard of care. Chemotherapy results in first remission in ~80% of patients, whereas the majority of cases experience tumor recurrence and progressive chemoresistance within 5 years, achieving a 5-year survival rate of <47.6% (3). Thus, further investigations are required in order to gain insight into the pathogenesis OC and design novel therapeutic strategies.
OC is divided into various histopathological subtypes, including epithelial, germ cell and sex cord-stromal tumors. Almost 90% of malignant ovarian tumors are epithelial, originating in the ovarian surface epithelium (OSE). However, over the past few years, the fimbriae of the fallopian tubes were considered as the probable site of origin of such malignancies, particularly high-grade serous OC (HGSOC) (4,5). It is noteworthy that simple original OSE exhibiting mesenchyme-related characteristics is characterized by the Müllerian epithelium since its development towards malignancy. Based on the morphology, function and antigenic similarity to the Müllerian duct epithelium, epithelial OC (EOC) is divided into five major subtypes that are histologically defined as follows: Mucinous (3%), clear cell (10%), endometrioid (10%), low-grade serous OC (LGSOC; <5%) and HGSOC (70%) (6–8). The primary origin of HGSOC is the fimbriated end of the fallopian tube, and its characteristics include common TP53 mutations and aberrations in genes involved in cell cycle control [e.g., neurofibromin 1 (NF1), retinoblastoma 1 (RB1) and cyclin E1 (CCNE1)], or the inactivation of genes involved in homologous recombination DNA repair (e.g., BRCA1 and BRCA2) (9,10). LGSOC may be implicated in the activation of the MAPK pathway via NRAS, KRAS or BRAF mutations, and is associated with high levels of estrogen receptor (ER) and progesterone receptor (PR) expression (11). Endometrioid and clear cell OC may be associated with endometriosis, whereas certain lifestyle factors (e.g., smoking) noticeably increase the risk of developing mucinous OC (12,13). Moreover, the pathogenesis of OC is also associated with mutations in the AT-rich interaction domain 1A (ARID1A), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and phosphatase and tensin homolog (PTEN) genes (14–16). Ovarian germ cell tumors develop from primordial germ cells of the embryonic gonads, and the most common germ cell malignancies include yolk sac tumor, teratoma and dysgerminoma. Sex cord-stromal neoplasms may originate from a wide range of cell types within the sex cords and gonadal stroma. Granulosa cell tumors (GCTs) are the most common in this group, accounting for ~5% of all OC cases.
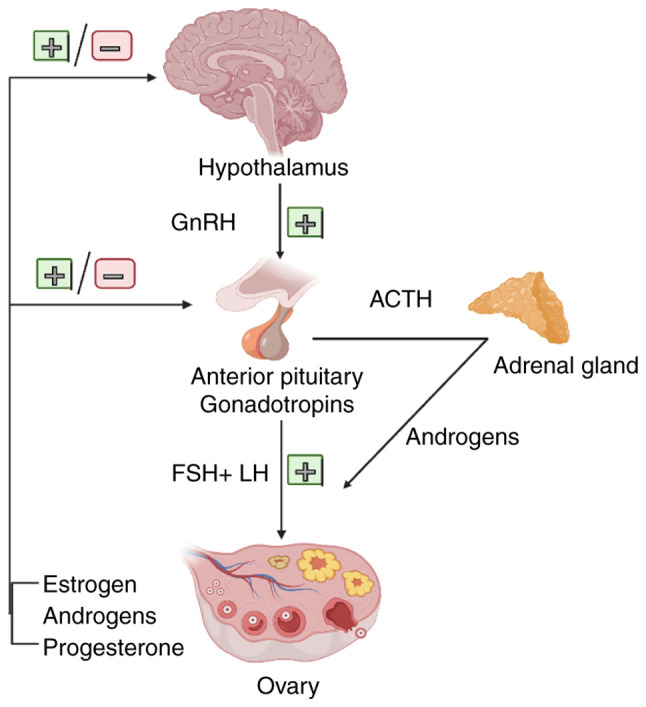
Despite its major impact on public health, the factors regulating the development and progression of OC have yet to be fully elucidated (17,18). In general, women who inherit mutations in either one of the two breast cancer susceptibility genes (BRCA1 or BRCA2) are at an increased risk of developing breast cancer and OC compared with women without a BRCA gene mutation. The overall lifetime risk for developing OC is 20–40% for women who have a BRCA1 mutation and 10–20% for women who have a BRCA2 mutation. Women in the general population have a <2% risk of developing OC (19). The National Comprehensive Cancer Network (NCCN) OC guidelines (2021) (20) recommend poly(ADP-ribose) polymerase (PARP) inhibitors for maintenance therapy in patients with platinum-sensitive recurrent OC, regardless of the biomarker status. In addition, the widespread application of hormone therapy in the clinical treatment of breast and endometrial cancers has made hormone receptors the primary research direction for a targeted cure for OC (21,22). It has been clearly stated in the NCCN OC guidelines (2021) (20) and in the European Society of Medical Oncology/European Society of Gynecological Oncology guidelines (23) that hormonal therapy may be used to treat patients with platinum-resistant and recurrent OC. Epidemiological studies have demonstrated that steroid hormones released through the hypothalamic/pituitary/ovarian axis can stimulate or suppress OC progression: Gonadotropins, estrogens and androgens promote OC progression, while gonadotropin-releasing hormone (GnRH) and progesterone may serve as protective factors against OC (3,24). The endocrine regulation of the ovary primarily relies on the neuroendocrine actions of the hypothalamic-pituitary-ovary axis (Fig. 1). Experimental studies and clinicopathological findings have demonstrated that hormone receptors are expressed in the normal ovarian surface epithelium, as well as in ovarian cancer cells and mediate the stimulatory or inhibitory effects of various hormones on the development of these cells. Moreover, hormonal therapeutic agents have been clinically evaluated in some patients with recurrent or refractory ovarian tumors, mainly exhibiting average efficiency and limited side-effects. For example, in the study by Sieh et al (25), data from almost 3,000 women with invasive EOC were analyzed u sing hormone receptor assay and evidence of the prognostic role of ER and PR and the potential hormonal sensitivity of EOC was provided. In addition, Paleari and DeCensi (26) conducted a meta-analysis of 53 clinical trials, including 2,490 patients and revealed an overall clinical benefit rate (CBR) of 41% [95% confidence interval (CI), 0.34–0.48] for any endocrine therapy. These results suggest that a greater understanding of the mechanisms through which hormones affect OC cell development may improve the effectiveness of hormone therapy for patients with this type of cancer.
Figure 1.
Hormonal environment of the ovaries. The endocrine regulation of ovary is primarily directed by the neuroendocrine actions of the HPO axis. Proper development and organization of the HPO axis are indispensable for normal female fertility. The basic molecule regulating the function of the HPO axis is GnRH. The episodic and timely secretion of GnRH from the hypothalamus and the activation of the pituitary GnRH-R are essential for the synthesis and secretion of gonadotropins (FSH and LH). The ovarian tissue secretes mainly estrogen, progesterone and androgens, which together with a small amount of androgens from the adrenal cortex regulate the development of ovarian tissue and the maturation of follicles for release (the figure was created using biorender.com). HPO, hypothalamic-pituitary-ovary; GnRH, gonadotropin-releasing hormone; GnRH-R, GnRH receptor; FSH, follicle-stimulating hormone; LH, luteinizing hormone; ACTH, adrenocorticotrophic hormone.
The present review conducted a literature search on PubMed, Web of Science and Clinical Trials for relevant articles published from inception to May, 2021 with no restrictions. The search terms included ‘hormone therapy’ (or ‘hormone replacement therapy’) and ‘ovarian cancer’ (or ‘ovarian carcinoma’ or ‘ovary cancer’); ‘peptide hormones (gonadotropin-releasing hormone/GnRH, gonadotropins, androgens, estrogens, and progestins) and ‘ovarian cancer’ (or ‘ovarian carcinoma’ or ‘ovary cancer’). The reference lists of the included studies were also reviewed for potential available studies. The mechanisms involved in the hormonal influences on the progression of OC (mainly EOC) are summarized, mentioning peptide hormones (GnRH, gonadotropins, androgens, estrogens and progestins), and the clinical efficacy and safety of various hormonal therapies for OC are discussed.
2. Hormone hypothesis of ovarian cancer pathogenesis
Epidemiological studies have implicated hormonal and reproductive factors in the pathogenesis of OC. Several hormonal hypotheses have been suggested thus far in an attempt to elucidate the etiology of OC, including gonadotropin signaling, direct influences exerted by progesterone and androgen, and incessant ovulation (Fig. 2). The first hypothesis of sex hormones as a potential mechanism underlying ovarian carcinogenesis is the ‘gonadotropin hypothesis’ (27–29). According to this hypothesis, OC advances due to excessive ovarian tissue excitation by pituitary gonadotropins [follicle-stimulating hormone (FSH) and luteinizing hormone (LH)] (27). Exposure to excess gonadotropins, which is associated with menopause, ovulation or infertility treatment, has been identified as an important risk factor for the development of OC. Moreover, such a theory would also explain the decreased risk of developing OC associated with the use of oral contraceptives and pregnancy, which results in the decreased exposure to gonadotropins owing to the negative feedback regulation of steroid hormones onto the pituitary gland (30,31). In contrast to the above, post-menopausal women with increased gonadotropin levels and women suffering from polycystic ovary syndrome (PCOS), who have upregulated circulating LH levels, are at an increased risk of developing EOC (32). Evidence from several epidemiological studies supports the gonadotropin theory. First, the rise in circulating gonadotropin levels exhibits a strong temporal association with the increased incidence of EOC (33,34). Menopause occurs at the age of ~51 years and is accompanied by variations in gonadotropin levels due to the cessation of the menstrual cycle and the deterioration of ovarian function. When ovarian function ceases completely, the negative feedback of ovarian steroids on gonadotropins is lost. Within 2–3 years following menopause, the gonadotropin levels can be particularly high. Accordingly, the LH and FSH concentrations may peak 3–4-fold (20–50 mIU/ml) and 10–20-fold (50–100 mIU/ml) compared with the values during the proliferation stage of the menstrual cycle, respectively. Subsequently, the levels of both gonadotropins slightly decline in a gradual manner. The incidence of EOC markedly increases at the age range in which the majority of women experience menopause, a phenomenon consistent with the gonadotropin theory. The mean age at onset of EOC is 57–59 years in the USA, and half of the cases are aged >65 years at the time of diagnosis, with 85–90% of the cases recorded in peri- or post-menopausal women, whereas EOC occurs in only 10–15% of pre-menopausal women (35,36). According to early information, multiple pregnancies and the use of oral contraceptives are established protective factors in terms of the incidence of EOC; each additional pregnancy is associated with a 10–16% reduction in the risk of developing EOC (37,38), and the protective effect of oral contraceptives increases by 7% with each year of use, reaching a 80% decrease among long-term users (over a decade) (39). These data regarding multiple pregnancies and the long-term use of oral contraceptives support the gonadotropic theory, since these factors are associated with low gonadotropin levels and the suppression of incessant ovulation. Furthermore, late menopause and early menarche are associated with a higher number of ovulations and exposure to high gonadotropin levels, thereby increasing the risk of developing EOC. The gonadotropin hypothesis is further supported by the elevated gonadotropin levels found in the cysts and peritoneal fluid of patients with EOC (40,41). In particular, high concentrations of FSH in ascitic fluid have been suggested to be inversely associated with survival (42), and EOC has been found to be associated with significantly higher levels of FSH in serum and capsular fluid compared with non-neoplastic ovarian lesions (43).
Figure 2.

Proposed hypotheses for the development of OC. There are several proposed hypotheses regarding the development of OC, although there is not sufficient evidence to support their association (the figure was created using biorender.com). FSH, follicle-stimulating hormone; LH, luteinizing hormone; ACTH, adrenocorticotrophic hormone.
Another major hypothesis in the development of EOC, namely incessant ovulation, was proposed in 1971 by Fathalla (44). The causal mechanism through which ovulation facilitates ovarian carcinogenesis remains unclear; however, some possible theories have been suggested. During the ovulatory process, the OSE is damaged and, subsequently, the repair process occurs through extensive cell proliferation, increasing the likelihood of spontaneous error and genome instability during DNA replication (45). Later studies under various disciplines proved the protective effect exerted by oral contraceptives, provided further insight into the biological mechanisms of ovulation, examined the possible pharmacological production of luteinized unruptured follicles and presented epidemiology-related evidence for this hypothesis (30,46). The lifetime number of ovulatory cycles or years exhibits a positive association with the risk of developing EOC, which supports the hypothesis of incessant ovulation (47,48). The levels of several inflammatory mediators (e.g., prostaglandins and cytokines) are increased during ovulation and may enhance mutagenesis (45). Moreover, the ‘androgen/progestin hypothesis’ is suggested to be a fundamental mechanism underlying the development of EOC. According to this hypothesis, the higher androgen levels observed in menopausal or obese women, and in patients with PCOS, are associated with an increased risk of developing EOC, whereas progesterone reduces the risk of developing EOC (49,50). Although several in vitro and animal studies have suggested a role for androgens in the development of EOC (50–52), the epidemiological evidence is not convincing.
3. Gonadotropins and GnRH
Role of FSH, LH and their receptors in OC and the normal ovarian surface epithelium
The two gonadotropins, FSH and LH, which share similar chemical and structural characteristics, are synthesized in the anterior pituitary, regulate gametogenesis and steroidogenesis in the testes and the ovary in an endocrine manner, and they are critical regulators of ovarian cell function (53). Specific receptors for gonadotropins have been reported in EOC, as well as in normal OSE cells and in fallopian tube cells (54,55). The LH receptor (LHR) and FSH receptor (FSHR) are G-protein coupled seven-transmembrane domain receptors (Fig. 3) that are highly expressed in theca and granulosa cells, and play essential roles in reproductive physiology when activated by their respective hormones (56). Over the past few years, FSHR expression has been identified in the tumor vasculature of a number of epithelial tumors, indicating a wider role for FSHR in carcinogenesis (57,58). The expression status of various gonadotropin receptors likely affects ovarian tumor progression through various mechanisms. According to Lenhard et al (59), patients with EOC expressing higher levels of FSHR than LHR had a worse prognosis, whereas cases expressing higher levels of LHR than FSHR exhibited an improved overall survival. Two previous studies investigated the effects of FSHR overexpression on immortalized OSE cells or benign ovarian epithelial tumor cells and the advanced EOC cell line, OVCAR-3 (60,61). According to these studies, FSHR overexpression may be associated with an increased oncogenic potential and the increased proliferation of pre-neoplastic OSE cells. In addition, EOC cells that expressed FSHR exhibited a superior invasive ability (61). These data indicate that FSHR may activate oncogenic pathways that promote cell proliferation and invasive phenotypes, even in the absence of FSH. Data from another study demonstrated that there was a positive association between FSHR and aryl hydrocarbon receptor levels, with their simultaneous expression observed in patients with the least favorable EOC outcomes (62). Cheung et al (63) investigated the functional roles of gonadotropin receptor expression in the progression of EOC and demonstrated that the knockdown of FSHR and LHR expression was associated with a more aggressive EOC phenotype and promoted pro-metastatic behavior. The effects exerted by gonadotropins on EOC have not yet been thoroughly investigated. Some studies have demonstrated that gonadotropins (FSH and LH) promote cell migration and invasion by inducing cyclooxygenase (COX)2 expression in EOC cells, and that specific COX2 inhibitors significantly prevent these effects (53,64). A recent study reported that sphingosine kinase (SphK) served as a key mediator of FSH-induced EOC cell proliferation, suggesting a novel strategy of using two isozymes of SphK as drug targets for the treatment of EOC (65). By contrast, another study demonstrated that in SKOV-3 cells, the overexpression of LHR upregulated the expression of ERBB2, and the addition of LH further increased ERBB2 expression, whereas it reduced cell proliferation and motility, indicating that the upregulation of ERBB2 independently failed to effectively decrease the anti-proliferative effect exerted by LH on these cells (66). LH has also been implicated in the development of EOC by inducing the secretion of VEGF via the PI3K/AKT/mTOR pathway (67) and upregulating survivin expression, leading to the inhibition of apoptosis (68).
Figure 3.

Mode of action of hormones binding to the corresponding receptors. The ER is a cytoplasmic monomer when not bound to a ligand. However, when bound to estrogen (E2), it is translocated to the nucleus and dimerizes. The ER then binds to the ERE in the upstream regulatory region of the estrogen response gene and recruits the cofactor complex [coenzyme A (CoA)] to induce or regulate gene transcription. The PR is a specific cellular protein with a high affinity and specificity for the progesterone (P) hormone. Progesterone binds to the progesterone receptor and enters the nucleus to form phosphorylated dimers. Subsequently, progesterone acts in concert with estrogen to promote proliferative and pro-survival gene programs. Androgen binding to AR is subsequently transferred to the nucleus where it binds to AREs in chromosomes and regulates the transcription of its target genes together with various transcription factors and co-regulators. GnRH I and the gonadotropins (FSH and LH) bind to the GPCRs and perform their respective functions (the figure was created using biorender.com). ER, estrogen receptor; ERE, estrogen response element; CoA, coenzyme A; PR, progesterone receptor; AREs, androgen response elements; GnRH I, gonadotropin-releasing hormone I; FSH, follicle-stimulating hormone; LH, luteinizing hormone; ACTH, adrenocorticotrophic hormone; GPCRs, G protein-coupled receptors.
GnRHs and their receptors in OC
GnRH is a decapeptide hormone synthesized by the hypothalamus and comprises 10 different amino acid residues. As a key neuromodulator of the reproductive system, GnRH is secreted in pulses and enters the anterior lobe of the pituitary gland via the hypothalamic/pituitary portal circulation, acts on gonadotropin-secreting cells in the anterior pituitary, and regulates gamete formation and sex hormone production in the gonads through the regulation of pituitary synthesis and the secretion of FSH and LH. At present, there are three forms of GnRH found in most vertebrates, namely GnRH-I, GnRH-II and GnRH-III, of which two subtypes, GnRH-I and GnRH-II, are expressed in humans (69,70). GnRH-I, also known as LH-releasing hormone, plays a role in regulating ovarian proliferative activity. GnRH-I and its receptor are expressed in ~80% of human ovarian epithelial tumors, EOC cell lines and OSE cells (71), and their activation by exogenous factors has been found to be associated with the notable (time- and dose-dependent) and specific inhibition of cell proliferation (72). Currently, the protein expression of the GnRH-I receptor is considered as a favorable prognostic factor in primary ovarian tumors (73,74). GnRH-II has been identified in the hypothalamus of chickens and has been reported to exhibit a conserved structure in vertebrates. Similar to GnRH-I, GnRH-II is expressed within a wide range of human ovarian cells, including OSE cells, granulosa luteinizing cells, as well as in EOC cells (75–77). It has been suggested that the anti-proliferative effects of GnRH-II on tumor cells are mediated via the GnRH-I receptor, as a functional GnRH-II receptor has not yet been identified (78). Further research on GnRH has demonstrated that, apart from the pituitary gland and hypothalamus, GnRH and the relevant receptors are also expressed in peripheral tissues (e.g., the ovaries, placenta, endometrium and smooth muscle) and in certain malignant tumors (e.g., OC, endometrial, breast and prostate cancers). GnRH must bind to the high-affinity GnRH receptor (GnRH-R) to exert its effects, i.e., the regulation of pituitary hormone release and extra-pituitary products.
Possible mechanisms of GnRH/GnRH analog (GnRH-a) therapy for OC
GnRH-as are produced by replacing or removing the 6th and 10th amino acids from naturally occurring GnRH, and their biological properties are 50–100-fold stronger compared with those of natural GnRH. The ultimate effects and efficacy of GnRH agonists and antagonists are similar, differing in that antagonists have a more rapid onset of action and do not have the initial surge in sex hormone release. For example, the GnRH-a, degarelix, which is an antagonist of gonadotropin receptors in the pituitary gland, directly inhibits the synthesis and release of LH and FSH, and does not have the initial surge characteristic of GnRH agonists (79). In addition to protecting ovarian function and improving the quality of life of patients, GnRH-as may also act as antitumor agents through various mechanisms. GnRH agonists and antagonists act by binding to GnRH-Rs and are used in the treatmetn of steroid-dependent conditions, including hormone-dependent tumors (80). The pharmacological application of GnRH or the relevant synthetic analogs (agonists and antagonists) may represent a valuable tool for stimulating or blocking gonadotropin secretion, regulating the fertility of women suffering from reproductive disorders and assisting with reproductive technologies (81). GnRH-as can inhibit the hypothalamic/pituitary axis and inhibit tumor growth by downregulating FSH and LH secretion and by binding to GnRH-I and GnRH-II receptors on the surface of cancer cells, thereby exerting direct inhibitory effects on cancer cells through the regulation of local autocrine and paracrine secretion (Table I). Following the administration of GnRH agonists, FSH and LH levels are increased for a short period of time; following long-term administration, the number of GnRH-Rs decreases, inhibiting FSH and LH release and thus, the secretion of estrogen and progesterone by the ovaries. GnRH antagonists exert opposite effects. By directly binding to the GnRH-R, they inhibit the release of FSH and LH without stimulating the pituitary gland, thereby reducing the level of progesterone and estrogen in the blood, and inhibiting sex hormone-dependent tumor development (82). GnRH does not only play a key role in mammalian reproductive regulation; however, it has also been widely reported to exert significant antitumor (anti-proliferative and anti-metastatic) effects by regulating the activation of local GnRH-R (83,84). The antitumor mechanisms of action of GnRH-I in OC are considered to involve the desensitization or downregulation of GnRH-I receptors within the pituitary gland, thereby reducing gonadotropin secretion, which in turn leads to a decrease in gonadotropin steroids acting as tumor growth-promoting factors (85,86). In addition to regulating gonadotropin and relevant receptors in vitro, GnRH-I and the relevant analogs have also been found to exert a direct inhibitory effect on normal epithelial cells and EOC cell lines in vitro and in vivo, leading to cell cycle arrest based on the increase in the protein levels of p53 and p21 (87).
Table I.
Hormonal therapy options for ovarian cancer.
| Hormone therapy | Mechanisms | Options |
|---|---|---|
| GnRH analogs | Competitively binds GnRH-R and reduces the secretion of FSH and LH | GnRH I agonists |
| Triptorelin | ||
| Goserelin | ||
| Histrelin | ||
| Leuprolide acetate | ||
| GnRH II antagonists | ||
| Cetrorelix | ||
| Degarelix acetate | ||
| Estrogen | Estrogen receptor blockade | Antiestrogens: |
| Tamoxifen | ||
| Toremifene | ||
| Estrogen synthesis suppression | Aromatase inhibitors: | |
| Anastrozole | ||
| Exemestane | ||
| Letrozole | ||
| Estrogen receptor downregulation | ER antagonist: | |
| Fulvestrant | ||
| Hormonal ablation | Surgery | |
| Radiation (infrequently used) | ||
| Androgen | Androgen receptor blockade | Anti-androgens: |
| Flutamide | ||
| Bicalutamide | ||
| Enzalutamide | ||
| Progesterone | Progesterone receptor blockade | PR antagonists: |
| Mifepristone | ||
| Medroxyprogesterone | ||
| Megestrol acetate | ||
| Increasing progesterone levels | Oral contraceptive pills | |
| Pregnancy | ||
| Breastfeeding |
GnRH-R, gonadotropin-releasing hormone receptor; FSH, follicle-stimulating hormone; LH, luteinizing hormone; ER, estrogen receptor; PR, progesterone receptor.
After binding to G-protein α(i), activated GnRH-R induces a phosphotyrosine phosphatase (PTP) that is subject to EGF-triggered tyrosine autophosphorylation of the EGF receptor, which leads to a reduction in cell proliferation and mitogenic signal transduction (88). This process prevents growth factor-driven mitogenic signaling, leading to the EGFR-induced expression of MAPK (72) and c-fos (89), as well as in the inhibition of cell proliferation (90). In addition, GnRH-I has been reported to have the ability to trigger JunD-DNA binding, leading to an increased cell number and a decreased DNA synthesis at the G0/G1 phase of the cell cycle, thereby reducing cell proliferation (91). The GnRH-I analog, leuprorelin, may also induce the apoptosis of tumor cells through the Fas-ligand/Fas mechanism. Data from two research teams suggested that elevated levels of Fas ligand in tumors expressing GnRH-R were likely to promote apoptotic cell death by targeting intratumoral Fas-positive cells, thereby exerting growth inhibitory effects on GnRH-sensitive tumors (92,93). However, the GnRH-I agonist, triptorelin (D-Trp-6-LH-RH), has been reported to reduce apoptosis induced by NF-κB activation triggered by the cytotoxic agent, doxorubicin (94–96). GnRH-II has been reported to exert a more prominent anti-proliferative effect on EOC cells compared with equimolar concentrations of GnRH-I agonists. The inhibitory effect exerted by GnRH-II on the proliferation of EOC cells in humans notably exceeds that exerted by the highly active GnRH-I agonist, triptorelin (97). GnRH-I and GnRH-II agonists inhibit the mitogenic signaling of growth factor receptors based on the activation of PTP, thus leading to decreased proliferation of cancer cells (88). Unlike GnRH-I and GnRH-II agonists, GnRH-II antagonists mainly promote the apoptosis of EOC cells (97). Apoptosis triggered by GnRH-II antagonists occurs through intrinsic apoptotic pathways: This is mediated via the activity of the pro-apoptotic protein, Bax, induced by MAPKs p38 and JNK, followed by caspase-3 activation, the release cytoplasmic cytochrome c, and the probable loss of mitochondrial membrane potential (97,98). The aforementioned antitumor effects were confirmed in nude mice, as a previous study demonstrated that antagonistic analogs of GnRH-II significantly reduced the growth of mouse EOC xenograft tumors without notable side-effects (97). Since the existence of a GnRH-II receptor in humans is controversial, there is a tendency to infer that the anti-proliferative effects of both GnRH-I and GnRH-II are mediated through the GnRH-I receptor (99,100). Of note, the GnRH antagonist, AEZ-115, has exhibited substantial antitumor activity in endometrial and EOC cells; however, this antitumor effect is not mediated by tumor GnRH-Rs (90). Based on the GnRH-R tumor-specific signaling in gynecological cancers, such as OC, and the particular distribution of GnRH-R, gene therapy by employing GnRH-a as inducers of therapeutically related gene transcription has been successfully developed and tested in vitro, as well as in athymic mice with EOC cell xenografts (101).
Clinical application of GnRH-as in OC
The inhibitory effect of gonadotropins and the anti-proliferative effect exerted by GnRH-a binding to the receptor form the rationale for their use in various hormone-dependent tumors. In a limited number of clinical studies, GnRH-I agonists have been assessed for their potential as third-line therapy in women with recurrent (mostly platinum-resistant) and refractory OC, and with the failure of at least one regimen of chemotherapy. A summary of 18 clinical trials (102–119), dating back to 1988, that have employed a wide range of GnRH-as (triptorelin, goserelin and leuprolide) for the treatment of patients with relapsed/platinum-resistant OC is presented in Table II. Initially, Parmar et al (102,120) reported on patients with advanced EOC who relapsed following conventional treatment and were treated with slow-release triptorelin microcapsules once per month. During treatment, they observed stable disease (SD) in 5 patients and evidence of clinical and/or radiological partial remission (PR) in 6 patients, where the tumor size decreased by >50%. This result suggests that the clinical benefit of therapy with GnRH agonists for recurrent OC is highly encouraging. Subsequently, based on the inhibition of LH/FSH secretion by GnRH agonists, several clinical trials using triptorelin and leuprolide acetate in advanced recurrent EOC demonstrated associated objective remission and/or disease stabilization in 10–50% of patients (103,112,113,117). In addition, in 2014, 42 women suffering from platinum-refractory or -resistant GnRH-R-positive EOC were evaluated for zoptarelin efficacy and toxicity; of these 42 patients, 6 (14.3%) achieved a partial response and 16 (38%) had SD (121). However, regardless of the aforementioned encouraging results, the benefits of employing GnRH-a as a treatment for advanced OC remain controversial.
Table II.
Overview of trials using GnRH analogs in ovarian cancer.
| Drug | Condition | Intervention | Histology (no. of samples) | CR | PR | SD | Phase | (Refs.) |
|---|---|---|---|---|---|---|---|---|
| Triptorelin | Advanced EOC | Long-acting depot/month | 41; | 0 | 6 | 5 | II | (102) |
| Serous, 28; | ||||||||
| endometrioid, 7; | ||||||||
| Mucinous, 1; | ||||||||
| Clear cell, 2; | ||||||||
| Mixed, 3 | ||||||||
| Triptorelin | Advanced EOC | 0.1 mg/daily or | 19; | 0 | 11 | 0 | II | (103) |
| 3.2 mg/month depot | Serous:19 | |||||||
| Triptorelin | Progressive ovarian | 3.75 mg for 4 weeks | 15; | 0 | 0 | 14 | II | (104) |
| cancer | Serous, 12; | |||||||
| Mucinous, 2; | ||||||||
| Brenner, 1 | ||||||||
| Triptorelin | Relapsed ovarian | 3.2 mg for 4 weeks | 14; | 0 | 0 | 8 | II | (105) |
| cancer | Serous, 8; | |||||||
| Endometrioid, 3; | ||||||||
| Mucinous, 1; | ||||||||
| Anaplastic, 1; | ||||||||
| Mixed, 1 | ||||||||
| Triptorelin | Relapsed EOC | 3.75 mg/on days 1, 8 and | 69; | 0 | 0 | 11 | II | (106) |
| 28 followed by 4-weekly | Serous, 19; | |||||||
| Endometrioid, 5; | ||||||||
| Mucinous, 4; | ||||||||
| Clear cell, 2; | ||||||||
| Mixed, 1; | ||||||||
| Undifferentiated, 5; | ||||||||
| Papillary, 6; | ||||||||
| Unspecified, 27; | ||||||||
| Triptorelin | Advanced EOC | 3.75 mg/month depot | 69; | 0 | 0 | 0 | II | (107) |
| Serous, 59; | ||||||||
| Endometrioid, 2; | ||||||||
| Mucinous, 4; | ||||||||
| Clear cell, 1; | ||||||||
| Undifferentiated, 3 | ||||||||
| Triptorelin and | Progressive ovarian | – | 40 | 0 | 0 | 1 | – | (108) |
| tamoxifen | cancer | – | ||||||
| Goserelin | Refractory EOC | 3.6 mg/month | 23; | 0 | 4 | 7 | II | (109) |
| Serous, 15; | ||||||||
| Endometrioid, 2; | ||||||||
| Mucinous, 1; | ||||||||
| Solid, 4; | ||||||||
| Clear cell, 1 | ||||||||
| Goserelin | Relapsed EOC | 3.6 mg/month | 30; | 0 | 2 | 5 | II | (110) |
| Serous, 14; | ||||||||
| Endometrioid, 1; | ||||||||
| Mucinous, 4; | ||||||||
| Clear cell, 1; | ||||||||
| Mixed, 1; | ||||||||
| Undifferentiated, 7; | ||||||||
| Borderline, 2 | ||||||||
| Goserelin and | Recurrent EOC | Goserelin, 3.6 mg once | 26 | 1 | 2 | 10 | II | (111) |
| tamoxifen | a month; Tamoxifen, 40 mg/daily | – | ||||||
| Leuprolide | Relapsed EOC | 3.75 mg intramuscularly | 32 | 0 | 4 | 5 | – | (112) |
| acetate | once a month | – | ||||||
| Leuprolide | Platinum-refractory | 3.75 mg intramuscularly | 32; | 1 | 2 | 4 | II | (113) |
| acetate | ovarian cancer | once a month | Serous, 25; | |||||
| Endometrioid, 4; | ||||||||
| Mucinous, 2; | ||||||||
| Unspecified, 1 | ||||||||
| Leuprolide | Platinum- and | 3.75 mg intramuscularly | 37; | 0 | 0 | 4 | – | (114) |
| acetate | paclitaxel-refractory | every 4 weeks | Serous, 19; | |||||
| ovarian cancer | Endometrioid, 6; | |||||||
| Mucinous, 11; | ||||||||
| Unspecified, 1 | ||||||||
| Leuprolide | Advanced EOC | 3.75 mg on days 1, 8, | 12; | 0 | 1 | 3 | II | (115) |
| acetate | 28 followed by 28-day | Serous, 9; | ||||||
| intervals | Endometrioid, 1; | |||||||
| Mucinous, 2 | ||||||||
| Leuprolide | Refractory or | 1 mg dose for a minimum | 18; | 0 | 4 | 2 | II | (116) |
| acetate | persistent EOC | of 8 weeks | Serous, 9; | |||||
| Endometrioid, 2; | ||||||||
| Mucinous, 3; | ||||||||
| Unspecified, 4 | ||||||||
| Leuprolide | Advanced or | 1 mg subcutaneously, daily | 5; | 1 | 4 | 0 | II | (117) |
| acetate | recurrent ovarian | Serous, 2; | ||||||
| cancer | Mucinous, 1; | |||||||
| Papillary, 1; | ||||||||
| Brenner, 1 | ||||||||
| Leuprolide | Advanced or | 1 mg subcutaneously daily | 25; | 0 | 1 | 15 | II | (118) |
| acetate | recurrent EOC | Serous, 19; | ||||||
| Endometrioid, 2; | ||||||||
| Mucinous, 1; | ||||||||
| Clear cell, 1; | ||||||||
| Mixed, 2 | ||||||||
| Cetrorelix | Platinum-resistant | 10 mg subcutaneously daily | 17; | 0 | 3 | 6 | II | (119) |
| ovarian cancer | Serous, 10; | |||||||
| Endometrioid, 5; | ||||||||
| Mixed, 2 |
EOC, epithelial ovarian cancer; CR, complete response; PR, partial response; SD, stable disease.
A prospective, double-blind, randomized clinical research was carried out to assess whether the addition of the GnRH agonist, triptorelin, to the course of common platinum-based chemotherapy prolongs the survival of patients with stage III or IV EOC who have previously undergone surgery; however, that study reported that the use of triptorelin to inhibit the release of gonadotropins did not inhibit disease progression or prolong the overall survival of the patients (107). In 2001, the European Organization for Research and Treatment of Cancer completed the largest trial of a GnRH agonist to date. In that study, 74 women with progressive OC who were treated with platinum-based chemotherapy received intramuscular injections of the GnRH agonist triptorelin. However, according to the results presented, triptorelin treatment was only moderately effective in patients already treated with platinum-based chemotherapy (106). In a recent phase II clinical trial, tamoxifen and goserelin were combined to treat patients with advanced EOC who had developed recurrence following chemotherapy. Although ‘endocrine responses’ were observed in ~50% of the patients, including SD (38.5%), partial response (7.7%) and complete response (3.8%), there was no consistent association observe4d between LH/FSH suppression and tumor progression (111). Similarly, in two other pilot studies in which patients with advanced OC were treated independently with platinum-containing chemotherapy or chemotherapy plus triptorelin, no significant differences were observed between the two groups in terms of drug response, survival and time to progression (122,123). The ineffectiveness of GnRH agonists when combined with chemotherapeutic agents may be due to their direct anti-proliferative effect being neutralized by chemotherapy-related anti-apoptotic activity, as confirmed by in vitro cell experiments. By contrast, Rzepka-Górska et al (124) reported that the combination of chemotherapy and the GnRH-a, goserelin, yielded favorable results in advanced OC and, in that study, patients in the combination group exhibited higher overall and 5-year survival rates compared with the chemotherapy group; in addition, serum LH levels were significantly lower in the combination group. Based on this result, it was concluded that combination therapy for advanced OC was effective and that GnRH-a may be an effective adjuvant therapy for OC (124).
As GnRH agonists have failed to meet the expectations in the clinical setting, the application of high doses of GnRH antagonists warrants further in-depth investigations. Since GnRH-I antagonists do not possess intrinsic gonadotropin activity, the initial ‘burst’ phenomenon observed with agonist therapy can be avoided, which renders antagonists more tolerable and blocks gonadotropin secretion within a short period of time. High-dose cetrorelix, a GnRH-I antagonist, was used in a phase II trial for the treatment of OC or Müllerian tumors refractory to platinum chemotherapy: All cases who relapsed following standard chemotherapy received 10 mg cetrorelix daily. A total of 3 patients (18%) experienced partial remission following cetrorelix treatment, lasting for 9, 16 and 17 weeks, respectively, and 6 patients (35%) had SD for 1–12 months (119). These results suggest that GnRH antagonists may be considered for the palliative treatment of patients with platinum-resistant OC.
4. Androgens
Androgens and their receptors in OC
The biological effects of androgens [testosterone and dihydrotestosterone (DHT), as well as certain androgens produced by the adrenal glands and ovaries] are typically mediated via the androgen receptor (AR), a steroid hormone receptor that is a member of the nuclear receptor superfamily (125–127). Under basal conditions, AR exhibits inactivity and can bind towards heat shock proteins and certain cellular partners. When activated by androgens, it induces a cascade of events (e.g., ligand binding, dissociation from heat shock protein, phosphorylation and dimerization) and is involved in nuclear translocation. Specific to the nucleus, AR binds to specific DNA sequences known as androgen response elements in the nucleus and binds to various AR cofactors to form complexes. The AR complex does not only alter the expression of genes involved in multiple physiological and pathological functions; however, under certain pathological conditions, AR may also become activated in the absence of androgens. For example, the activation of AR by IL-6 in human prostate carcinoma cells does not require the presence of androgens (128). Androgen/AR signals have been reported to promote metastasis and tumorigenesis in various malignancies, including prostate carcinoma, for which androgen deprivation therapy remains the primary treatment option (129), but also in other types of cancer, such as breast (130) and bladder cancer (131).
In addition, there is accumulating evidence to indicate that AR and related signaling pathways are involved in the development and progression of OC. The expression of AR in OC was first demonstrated by Hamilton et al (132) with the use of ligand binding tests. According to in-depth research, AR expression has been detected in ~90% of EOCs using biochemical receptor tests (133) and in 43.5–86% of EOCs using immunohistochemistry (134,135). Some investigators have attempted to determine whether AR is differentially expressed in various histological subtypes of OC. According to Cardillo et al (136) the expression of AR varies widely across various histological subtypes of OC. The aforementioned result was verified by Elattar et al (137), who reported that AR was expressed in 43.7% of EOC samples, with the highest expression levels observed in serous carcinomas (47.5%). Similarly, according to de Toledo et al (138), AR-positive expression tended to exhibit a higher prevalence in serous tumors compared with that in non-serous tumors. The association between AR expression and other clinicopathological characteristics of OC, such as tumor stage and grade, was also assessed. According to Jönsson et al (139), a negative AR expression was associated with high-grade cancer. By contrast, four other studies on malignant ovarian tumors reported that AR expression exhibited no association with tumor FIGO stage (135,136,138,140). Furthermore, according to a previous study, matched primary and metastatic serous EOC samples exhibited non-significant differences in the levels of nuclear AR (140). In summary, AR expression is more frequently detected in serous compared with non-serous ovarian tumors; however, whether AR expression levels are crucial for OC progression requires further in-depth investigation.
Androgen-mediated AR signaling in ovarian tumorigenesis
Currently, a number of factors are known to be involved in regulating the transcriptional activity of AR, including not only various androgenic and androgen-derived compounds, but also the expression levels and variants of AR per se. Edmondson et al (141) first demonstrated that OSE cells were androgen-responsive, and the addition of androgen to eight primary human OSE cell lines cultured in vitro was able to promote the proliferation of these cells and reduce cell apoptosis. In other in vitro experiments, DHT and testosterone were shown to notably stimulate the proliferation of ovarian tumor and normal ovarian cell lines/cultures (142). Co-treating these cells with the anti-androgen 4-hydroxyflutamide also demonstrated that this androgen-stimulating effect was reversible (142). In another study, DHT stimulation was reported to increase cell division in six of 11 primary cultured EOC cell lines. The proportion of cells in the S phase also increased from 4.4% in serum-free medium to 8.3% in 100 nM DHT-stimulated cells (137). In that study, it was also found that AR nuclear expression was positively associated with an increased fraction in the S phase in response to androgen stimulation, while the expression of AR in the immunohistochemical nuclear and cytoplasm was significantly decreased following chemotherapy (137). This may explain the low response rates observed in clinical trials among patients who have received aggressive prior chemotherapy. Other androgens, such as androstenedione (143) and methylosome (144), have also been shown to increase the proliferation and induce cell motility and invasion in EOC lines. Animal models have also been used to evaluate the effect of androgens on EOC progression. According to Silva et al (145), testosterone therapy in guinea pigs promoted ovarian epithelial cell proliferation, leading to the formation of papillomas on the ovarian surface, small adenomas in the ovarian parenchyma and benign cysts. Similarly, in a mouse xenotransplantation model, DHT treatment significantly induced tumor growth (146), and the AR inhibitor enzalutamide was shown to antagonize the effects of DHT (147). These observations in animal models and cell lines suggest that androgen/AR signaling can critically stimulate EOC growth and promote EOC progression. The activation of AR signaling is likely to be associated with the sensitivity of malignant tumors to conventional chemotherapeutic agents, as suggested by research on prostate cancer (148) and bladder urothelial carcinoma (125). AR expression has also been found to be considerably upregulated in the paclitaxel-resistant SKOV3 subline (149). Correspondingly, as previously demonstrated, in paclitaxel-resistant SKOV3 cells, the silencing of AR via RNA interference increased cell sensitivity to chemotherapy and enhanced paclitaxel-mediated apoptosis (149,150). Conversely, the activation of AR with the agonist DHT has been shown to upregulate the expression of paclitaxel resistance target genes (150).
Clinical use of androgen deprivation therapy in patients with OC
As aforementioned, preclinical evidence suggests that androgen/AR signaling is associated with the progression of OC, and targeting AR is a promising treatment strategy. However, to date, only a small number of clinical trials have been conducted to evaluate the efficacy of anti-androgen therapy in OC, with or without GnRH agonists (Table III) (151–155). Notably, androgen deprivation therapy (e.g., bicalutamide and flutamide), with or without GnRH agonists, is extensively applied in patients with prostate cancer without severe side-effects, and functions by inhibiting pituitary LH release and ultimately reducing androgen production (49,156). In a previous study, Tumolo et al (151) assessed the use of flutamide, a non-steroidal AR antagonist, in patients with EOC who relapsed and progressed following platinum-based chemotherapy. First, 68 eligible patients were included, 32 of whom had completed oral flutamide therapy (750 mg/day) for at least 2 months. Of the 32 patients in the study, only 2 responded to the treatment and the disease was stable over a mean period of 24 weeks in 9 patients. Based on the results of that trial, it was concluded that flutamide was ineffective as a treatment for patients who had previously received extensive chemotherapy, and that it is associated with certain side-effects, such as nausea and vomiting. In another phase II study, Vassilomanolakis et al (152) assessed the response of patients with stage III or IV EOC to flutamide (300 mg/day) following chemotherapy. The outcome of that clinical trial was not satisfactory, with only 1 (4.3%) of the 23 evaluated patients exhibiting a partial response and 2 patients (8.7%) having SD, with the remaining 20 patients experiencing disease progression within 3 months. In addition, bicalutamide, a non-steroidal drug exhibiting anti-androgen characteristics, was tested in patients with grade II or higher EOC in remission (154). However, the results of that study demonstrated that the oral bicalutamide administration (30 mg/day) combined with subcutaneous goserelin injection (3.6 mg/4 weeks) did not prolong progression-free survival (PFS) in 35 patients. In another later study, Gruessner et al (153) investigated the effects of preoperative oral flutamide therapy (125 mg/day for 6 weeks) on biomarker levels in blood and tissue samples from 12 patients at a high risk of developing OC and 47 controls, 47% of whom had BRCA mutations. The results demonstrated that flutamide significantly reduced the expression of colony stimulating factor (CSF)-1 and Erb-B2 receptor tyrosine kinase 4 (ERBB4) in the ovarian stroma and was well-tolerated by the patients. Enzalutamide is an oral AR signaling inhibitor capable of blocking the binding of androgens to AR and preventing AR nuclear translocation, DNA binding and coactivator recruitment (157). In 2012, enzalutamide was approved by the US Food and Drug Administration for the treatment of castration-resistant prostate carcinoma. A phase II study is currently underway to evaluate enzalutamide in women with AR-positive OC (NCT 01974765) (155).
Table III.
Overview of trials using androgen deprivation therapy in ovarian cancer.
| Drug | Condition | Intervention | Histology (no. of samples) | CR | PR | SD | Phase | (Refs.) |
|---|---|---|---|---|---|---|---|---|
| Flutamide | Relapsed EOC | 750 mg daily for at | 62; | 1 | 1 | 9 | II | (151) |
| least 2 months | Serous, 33; | |||||||
| Endometrioid, 1; | ||||||||
| Mucinous, 4; | ||||||||
| Clear cell, 1; | ||||||||
| Undifferentiated,1; | ||||||||
| Unspecified, 22 | ||||||||
| Flutamide | Advanced EOC | 100 mg three | 23; | 0 | 1 | 2 | II | (152) |
| times daily | Serous, 17; | |||||||
| Endometrioid, 2; | ||||||||
| Clear cell, 1; | ||||||||
| Mixed, 3 | ||||||||
| Flutamide | Pre-operative | 125 mg daily | 12 | – | – | – | II | (153) |
| treatment EOC | for 6 weeks | – | ||||||
| Bicalutamide + | Consolidation EOC | Bicalutamide, | 35; | – | – | – | II | (154) |
| goserelin | 50 mg daily; goserelin, | Serous, 30; | ||||||
| 3.6 mg every 4 weeks | Endometrioid, 3; | |||||||
| Clear cell, 2 | ||||||||
| Enzalutamide | Androgen | 160 mg daily | 58 | – | – | – | II | (155) |
| receptor-positive EOC | – |
EOC, epithelial ovarian cancer; CR, complete response; PR, partial response; SD, stable disease.
Evidence from limited clinical trials indicates that only a small proportion of patients with EOC benefit from androgen deprivation therapy consisting of non-steroidal anti-androgen drugs with generally tolerable side-effects. Therefore, further research is required to determine the actual benefit of androgen deprivation therapy and the optimal regimen, and to select appropriate drug candidates by evaluating AR expression, AR polymorphism/fragment variation and downstream targets of AR activity.
5. Estrogen
Estrogen and ER in OC
Estrogens belong to the family of steroidal organic compounds and are mainly synthesized from androgens through the aromatase activity of granulosa cells in the ovaries. In fact, the term ‘estrogen’ refers to a group of female hormones, the most common of which are estrone, estradiol and estriol. Serum estrogen concentrations increase from the metaphase, consistent with the formation of dominant follicles. In the follicular fluid chambers, estrogen levels are 1,000-fold higher compared with those in peripheral blood. All these estrogens can bind to nuclear and membrane ERs with varying affinities and response strengths (158). Estrogen signal transduction is mediated by several estrogen isomers, the most important of which are the classical ERs, namely ERα and ERβ. Estrogen, as a steroid hormone, can cross the plasma membrane and interact with ERα and ERβ in the cells, acting directly by binding to DNA sequences. ER complexes bind directly or indirectly to DNA; thus, estrogen-mediated signaling events can be genomic as well as non-genomic. Genomic effects involve the migration of ER complexes to the nucleus and direct interaction with chromatin on specific DNA sequences, known as estrogen response elements (158,159). On the other hand, non-genomic effects involve the indirect regulation of gene expression through a variety of intracellular signaling events. The G-protein coupled receptor (GPR)30, also known as G-protein coupled ER1, has been extensively investigated as a membrane ER that activates non-genomic estrogen signaling pathways in a variety of tissues (160).
ERα is closely associated with endocrine sensitivity in breast cancer and is a significant mediator of the estrogen response in OC (161,162). Multiple studies have investigated the expression of ERα in EOC (163–166); however, the largest study was conducted by the Ovarian Tumor Tissue Analysis consortium in 2013 (25). That study investigated 2,933 women and found that HGSOC, LGSOC and endometrioid OC exhibited the highest levels of ER positivity at 81, 88 and 77%, respectively; furthermore, these tumor types have exhibited responses to endocrine therapy (tamoxifen and aromatase inhibitors) in multiple clinical studies. By contrast, ERα expression was detected in only 20 and 21% of clear cells in mucinous and clear cell carcinomas, respectively. ERβ is known to be expressed as five different isoforms, namely ERβ1-5; however, only the full-length form of ERβ1 is able to bind to agonist or antagonist ligands and has a more complex function (167). Rutherford et al (168) found higher levels of ERβ expression in normal OSE cells, lower levels of ERβ expression in primary EOC, and the absence of ERβ expression in metastatic tumors. These results indicate that there may be fundamental differences in ERβ expression from the normal ovarian tissue to primary OC to metastatic tumor (168).
Estrogen signaling in OC
ER is highly expressed in several EOCs and is a potential target for endocrine therapy. Studies using in vitro experiments and in vivo animal models of EOC support the hypothesis that ER expression levels are a crucial determinant of the response to treatment with selective ER modulators. In addition, epidemiological studies have demonstrated that the long-term use of estrogen-only therapy increases the risk of developing OC in women, supporting the hypothesis that estrogen signaling contributes to the etiology of the disease.
In vitro cultured EOC cells exhibiting a high ERα expression can be stimulated by estrogen, while treatment with selective ER modulators (tamoxifen and fulvestrant) can inhibit the growth of EOC cell xenografts with a high ERα expression in vivo (161,163,169,170). By contrast, estrogen has been shown to exert no effect on ERα-negative cell lines (170). The silencing of ERα expression in SKOV3 EOC cells has been shown to inhibit estrogen-stimulated proliferation and reverse the inhibitory effects of 4-hydroxytamoxifen (an active metabolite of tamoxifen) and fulvestrant, indicating that the effects of these selective ER modulator were ERα-mediated (171). In another study, the treatment of patient-derived xenografts (PDXs) from patients with ERα-positive/ERβ-negative HGSOC with 4-hydroxytamoxifen or fulvestrant also provided support for the ERα-mediated action of these drugs (165). Of the four PDXs, two xenografts with higher levels of ERα expression exhibited reduced diffusion during drug therapy, while the two xenografts with lower levels of ERα expression were not affected, meeting the requirements for a higher level of ERα response. Fulvestrant decreased the protein expression of ERα in the reaction cells, which was consistent with its action pattern (165). In addition to regulating proliferation, estrogen promotes cell migration and epithelial-to-mesenchymal transition through ERα, and these changes are mediated by the Snail- and Slug-mediated downregulation of E-cadherin (172). The extracellular molecule, fibulin, which binds to fibronectin and laminin, is also regulated by estrogen through ERα and is considered to play a key role in cell attachment and motility (173).
The previous evaluation of the role of ERβ suggested that this receptor isoform functions as a tumor suppressor in EOC by reducing tumor growth and deterring metastasis. These actions rely on the fact that ERβ inhibits ERα expression and decreases estradiol-induced cell proliferation (174,175). In addition, the restoration of ERβ expression in EOC cells has been shown to lead to a decrease in proliferation and invasion, while apoptosis is enhanced (176). Similarly, the overexpression of ERβ1 induced by transfection with an overexpression vector in the SKOV3 EOC cell line (177) or the ES-2 cell line (178) has been shown to result in reduced proliferation and motility, and increased apoptosis. The gene ERβ, is localized on chromosome 14q, and this region is frequently deleted in OC (176). Consistent with its tumor-suppressive effect, the high expression of ERβ1 in the cytoplasm of EOC cells has been found to be strongly associated with a longer disease-free and overall survival of patients (179). In contrast to the inhibitory role of ERβ1, both ERβ2 and ERβ5 have been associated with pro-migratory and invasive activities. ERβ2 overexpression has been shown to increase cell migration and invasion, but not the proliferation of EOC cells (179). The mitochondrial activity of cytoplasmic ERβ2 signaling in serous carcinomas has been found to be associated with BAD binding, leading to reduced apoptosis, thereby serving a pro-survival role (174). ERβ5 also plays an important role in tumor progression and has been shown to promote cell proliferation, and enhance cell migration and invasion. High levels of ERβ5 expression have been found in the nuclei of tumor cells from patients with clinically advanced EOC (particularly serous and clear cell carcinomas), which has also been associated with poor patient survival, although some researchers have reported that cytoplasmic ERβ5 expression may be associated with more favorable patient outcomes (179). The cell cycle is affected by the presence of ERβ by reducing the proportion of cells in the S phase, increasing the number of cells in the G2/M phase, and inducing apoptosis (167,175). A suppressive effect of ERβ on cyclin D1 expression has been reported, and its expression appears to be enhanced in response to estrogen antagonists (180). Indirect evidence that ERβ is implicated in ovarian carcinogenesis is the fact that its expression has been found to be significantly associated with survival in human and animal models (181,182). A recent study revealed that ERβ-positive nuclear staining was associated with a decreased PFS (183). On the other hand, patients with hormone receptor-negative OC have been shown to have a favorable prognosis (184).
Targeting ER with selective ER modulators and aromatase inhibitors in OC
Several clinical trials have been conducted using selective ER modulators (mainly tamoxifen) and aromatase inhibitors (letrozole and anastrozole), which have exhibited activity against OC (low response rate, yet apparently SD) and these are discussed below (Tables IV and V) (108,185–213).
Table IV.
Overview of trials using tamoxifen in ovarian cancer.
| Drug | Condition | Intervention | Histology (no. of samples) | CR | PR | SD | Phase | (Refs.) |
|---|---|---|---|---|---|---|---|---|
| Tamoxifen | Advanced EOC | 40 mg daily | 6 | 0 | 1 | 1 | II | (185) |
| Serous, 3; | ||||||||
| Endometrioid, 1; | ||||||||
| Mucinous, 1; | ||||||||
| Clear cell, 1 | ||||||||
| Tamoxifen | Recurrent EOC | 40 mg daily | 47- | 1 | 2 | 22 | – | (186) |
| Tamoxifen | Recurrent ovarian | 30 or 40 mg daily | 65; | 2 | 2 | 50 | – | (187) |
| cancer | Endometrioid, 10 | |||||||
| Non-endometrioid, 55 | ||||||||
| Tamoxifen | Advanced EOC | 40 mg daily | 30; | 2 | 0 | 10 | II | (188) |
| Serous, 25; | ||||||||
| Endometrioid, 1; | ||||||||
| Mucinous, 1; | ||||||||
| Clear cell, 2; | ||||||||
| Adenosquamous, 1 | ||||||||
| Tamoxifen | Progressive ovarian cancer | 30 mg daily | 33- | 0 | 0 | 2 | II | (108) |
| Tamoxifen | Refractory EOC | 80 then 40 mg | 29; | 2 | 3 | 18 | II | (189) |
| daily | Serous, 5; | |||||||
| Endometrioid, 2; | ||||||||
| Mucinous, 1; | ||||||||
| Clear cell, 1; | ||||||||
| Papillary, 17; | ||||||||
| Undifferentiated, 3 | ||||||||
| Tamoxifen | Recurrent EOC | 30 mg daily | 26; | 0 | 0 | 7 | II | (190) |
| Serous,10; | ||||||||
| Endometrioid, 4; | ||||||||
| Mucinous, 5; | ||||||||
| Clear cell, 3; | ||||||||
| Undifferentiated, 4 | ||||||||
| Tamoxifen | Recurrent EOC | 20 mg daily | 21 | 1 | 7 | 12 | II | (191) |
| – | ||||||||
| Tamoxifen | Refractory ovarian | 100 then 40 mg | 51 | 0 | 1 | 0 | II | (192) |
| cancer | daily | – | ||||||
| Tamoxifen | Relapsed EOC | 40 mg daily | 22 | 0 | 0 | 1 | II | (193) |
| – | ||||||||
| Tamoxifen | Recurrent EOC | 20–40 mg daily | 23 | 0 | 0 | 19 | II | (194) |
| – | ||||||||
| Tamoxifen | Advanced ovarian | 40 mg daily | 105; | 10 | 8 | 40 | II | (195) |
| cancer | Serous, 57; | |||||||
| Endometrioid, 15; | ||||||||
| Mucinous, 6; | ||||||||
| Clear cell, 4; | ||||||||
| Mixed, 8; | ||||||||
| Adenosquamous, 1; | ||||||||
| Unspecified, 12; | ||||||||
| Undifferentiated, 2 | ||||||||
| Tamoxifen | Recurrent EOC | 20 mg daily | 29; | 1 | 2 | 6 | II | (196) |
| Serous, 22; | ||||||||
| Endometrioid, 3; | ||||||||
| Mucinous, 1; | ||||||||
| Mixed, 1; | ||||||||
| Undifferentiated, 2 | ||||||||
| Tamoxifen | Advanced ovarian | 20 mg daily | 13 | 0 | 1 | 4 | – | (197) |
| cancer | – | |||||||
| Tamoxifen | Relapsed EOC | 20 or 40 mg daily | 43; | 0 | 6 | 22 | – | (198) |
| Serous, 22; | ||||||||
| Endometrioid, 13; | ||||||||
| Clear cell, 6; | ||||||||
| Undifferentiated, 2 | ||||||||
| Tamoxifen | Recurrent EOC | 40 mg daily | 68; | 0 | 0 | 5 | III | (199) |
| Serous, 56 | ||||||||
| Endometrioid, 2; | ||||||||
| Mixed, 7; | ||||||||
| Unspecified, 3 | ||||||||
| Tamoxifen | Advanced EOC | Tamoxifen, 10 mg twice daily; | 49; | 0 | 0 | 0 | II | (200) |
| and cytotoxic | cis-diamminedichloroplatinum | Serous, 29; | ||||||
| chemotherapy | (50 mg/m2) and doxorubicin | Endometrioid, 10; | ||||||
| (50 mg/m2 every 4 weeks | Mucinous, 2; | |||||||
| Clear cell, 2; | ||||||||
| Mixed, 1; | ||||||||
| Undifferentiated, 5 | ||||||||
| Tamoxifen | Recurrent or | Tamoxifen, 80 mg daily for | 50; | 15 | 10 | 12 | II | (201) |
| and platinum | progressive EOC | 1 month then 40 mg daily; | Serous, 37; | |||||
| (P)-based | cisplatin, 100 mg/m2 or | Endometrioid, 1; | ||||||
| chemotherapy | carboplatin, 400 mg/m2, every | Clear cell, 1; | ||||||
| 21 days | Undifferentiated, 1; | |||||||
| Unspecified, 10 | ||||||||
| Tamoxifen | Platinum-resistant | Tamoxifen: 80 mg daily for | 14 | 0 | 0 | 0 | II | (202) |
| and carboplatin | EOC | 1 month then 40 mg daily; carboplatin: every 21 days | – | |||||
| Tamoxifen | Refractory or | Tamoxifen: 40 mg daily; | 56- | 0 | 0 | 16 | II | (203) |
| and gefitinib | resistant EOC | gefitinib: 500–250 mg daily |
EOC, epithelial ovarian cancer; CR, complete response; PR, partial response; SD, stable disease.
Table V.
Overview of trials using aromatase inhibitors in ovarian cancer.
| Drug | Condition | Intervention | Histology (no. of samples) | CR | PR | SD | Phase | (Refs.) |
|---|---|---|---|---|---|---|---|---|
| Letrozole | Relapsed EOC | 2.5 mg daily | 60; | 0 | 5 | 14 | II | (204) |
| Serous, 43; | ||||||||
| Endometrioid, 11; | ||||||||
| Undifferentiated, 6 | ||||||||
| Letrozole | Relapsed or recurrent | 2.5 mg daily | 27; | 1 | 3 | 5 | II | (205) |
| EOC | Serous, 15; | |||||||
| Endometrioid, 2; | ||||||||
| Clear cell, 1; | ||||||||
| Unspecified, 9 | ||||||||
| Letrozole | Estrogen receptor-positive | 2.5 mg daily | 44; | 0 | 7 | 11 | II | (206) |
| ovarian cancer | Serous, 23; | |||||||
| Endometrioid, 4; | ||||||||
| Mixed, 17 | ||||||||
| Letrozole | Recurrent and advanced | 2.5 mg daily | 13 | 2 | 2 | 5 | – | (207) |
| EOC | – | |||||||
| Letrozole | Recurrent platinum- | 2.5 mg daily | 33; | 0 | 1 | 7 | II | (208) |
| and taxane-resistant | Serous, 29; | |||||||
| ovarian cancer | Mixed, 3; | |||||||
| Adenocarcinoma, 1 | ||||||||
| Letrozole | Estrogen receptor-positive | 2.5 mg daily | 14 | 5 | 0 | 5 | – | (209) |
| ovarian cancer | – | |||||||
| Letrozole | Relapsed EOC | 2.5 mg daily | 54; | 0 | 8 | 22 | – | (198) |
| Serous, 25; | ||||||||
| Endometrioid, 15; | ||||||||
| Clear cell, 10; | ||||||||
| Unspecified, 4 | ||||||||
| Letrozole + | Relapsed estrogen | Letrozole: 2.5 mg daily; | 19; | 0 | 0 | 9 | II | (210) |
| everolimus | receptor-positive ovarian | Everolimus: 10 mg daily | Serous, 17; | |||||
| cancer | Carcinosarcoma, 1; | |||||||
| Transitional cell | ||||||||
| carcinoma, 1 | ||||||||
| Anastrazole | Recurrent/persistent | 1 mg daily | 53; | 0 | 1 | 22 | II | (211) |
| ovarian cancer | Serous, 38; | |||||||
| Endometrioid, 3; | ||||||||
| Clear cell, 1; | ||||||||
| Unspecified, 11 | ||||||||
| Anastrazole + | Recurrent EOC | Anastrazole, 1 mg daily; | 23 | 1 | 0 | 14 | II | (212) |
| gefitinib | Gefitinib, 250 mg daily | – | ||||||
| Exemestane | Refractory ovarian cancer | 25 mg daily | 22 | 0 | 0 | 8 | II | (213) |
| – |
EOC, epithelial ovarian cancer; CR, complete response; PR, partial response; SD, stable disease.
Tamoxifen was the first selective ER modulator to be evaluated in clinical trials for OC and is still in use today; it is considered to function as a selective ER modulator that competes with estrogen for binding to the ERα and, thus, functions as an ER antagonist. The majority of studies were designed as single-arm studies to assess whether tamoxifen can induce a response in EOC; however, they did not evaluate the effects of tamoxifen on symptom control, the quality of life or the survival time of patients. It has been reported that the overall mean response rate of this treatment is 10–15%, and the disease stability rate is 30–40% (214–216). In clinical trials in which tamoxifen was applied, the majority of patients had undergone major pre-treatment procedures, such as chemotherapy, and several studies did not differentiate between ER-positive patients. Perez-Gracia and Carrasco (215) reported an overall response rate of 26% and a complete response rate of 9% in the analysis of trials using tamoxifen in at least 50% of cases not having received multiple prior treatments, compared with a clinical study treating patients with severe disease with an effectiveness rate of only 4% (215). For 19 years, non-steroidal aromatase inhibitors have been studied for their antitumor activity, which appears to be comparable to that of tamoxifen, by inhibiting the conversion of androgens to estrogens, thereby reducing circulating estrogen levels. Paleari et al (216) reviewed 53 endocrine therapy trials including a total of 2,490 patients in their meta-analysis. The clinical benefit rate (CBR; percentage of total patients exhibiting complete response, partial response or SD in all endocrine therapy evaluations) reached 41%; the CBR for tamoxifen was 43% (23 trials) and the CBR for aromatase inhibitors was 39% (10 trials), demonstrating that the effectiveness of the two types of drugs was comparable for patients with advanced EOC (216). According to a recently conducted trial (PARAGON), the application of anastrozole in a phase II study on asymptomatic cases suffering from ER- and PR-positive recurrent EOC with CA125 progression was assessed (217). The response rate reached 4%, and the CBR reached 35%, which was disappointing, given that these cases only had limited disease and had previously received single chemotherapy treatment.
Two recent reports described studies evaluating the use of endocrine therapy at their respective centers and provided insight into settings outside of HGSOC trials. An analysis of 97 patients treated at the Royal Marsden Hospital (London, UK) investigated the use of tamoxifen and letrozole for advanced EOC (91% HGSOC) (198). More than a quarter of the patients had previously received five or more types of chemotherapy, and half of these had an unknown ER status, whereas the CBR reached 60% (tamoxifen, 65%; and letrozole, 56%). Cases responding to letrozole had a significantly longer response time (198). A 25-year analysis of 269 cases with HGSOC in Edinburgh revealed a comparable overall response rate for letrozole and tamoxifen (8 and 11%, respectively) and CBR (41 and 33%, respectively). Cases with a high ER expression and a longer treatment-free interval were most likely to benefit from these treatments (218). The conclusions of these two analyses are consistent with the findings of clinical trials, demonstrating that tamoxifen or letrozole constitute reasonable treatment options for patients with ER-positive HGSOC, with a comparable overall response rate, CBR and disease stability. Endocrine therapy may be a promising alternative therapy for LGSOC, which is less sensitive to chemotherapy. Gershenson et al (219) identified a 9% response rate and 61% disease stabilization rate in a retrospective analysis of 64 LGSOC cases who had received a total of 89 hormonal regimens. The PFS for cases receiving hormonal maintenance therapy (primarily letrozole or tamoxifen) was 65 months, compared with 26 months for cases under observation only (P<0.001) (219). That study was followed-up by Fader et al (220), who also retrospectively explored the use of adjuvant hormonal therapy following surgery without chemotherapy, with promising results. A stage III trial initiated in 2019 (NRG-GY019) is also currently ongoing. Specifically, the comparison is between paclitaxel/carboplatin + letrozole vs. letrozole independently for stage II–IV LGSOC (221). To date, there is limited information available on the sensitivity of endometrioid OC to hormone therapy. In the Royal Marsden High Grade Ovarian Cancer Study, a total of 5 patients with high-grade endometrioid OC were treated with endocrine therapy and, encouragingly, 3 patients exhibited partial remission, while the remaining 2 patients had SD (198). In a study on letrozole reported by Bowman et al (204), 4/11 endometrioid OC cases had SD compared with 4/43 serous carcinoma cases. Moreover, estrogen-targeting therapies have exhibited considerable promise in the treatment of GCTs. In a review article summarizing aromatase inhibitors as single agents, 25 cases with known outcomes were described. The response rate to aromatase inhibitors in these patients was 48% (12/25) and the clinical benefit rate was 76% (19/25) (222). According to a previous analysis, 9 out of 9 patients responded to aromatase inhibitors. Although the number of patients in those studies was limited, they supported aromatase inhibitors as a potential alternative to chemotherapy (223).
6. Progesterone and progestins
Progesterone (P4) and PR in OC
Progesterone (P4) is a steroid hormone mainly generated from the corpus luteum in the ovaries during the luteal phase or the second half of the menstrual cycle. The adrenal glands and the placenta during pregnancy also produce small amounts of progesterone (224,225). Thus, from the beginning of menstruation to the end of menopause, there is a monthly cycle of hormone exposure, and the regulation of the growth and differentiation of the female reproductive tract system and breast tissue (225). Pregnancy interrupts this cyclic process, as high progesterone levels are continuously required for fetal growth, the maintenance of uterine/placental integrity and breast development during lactation (226). Progesterone-dependent effects and related biological actions in different tissues and tumors are mediated by two PR subtypes, namely PR-A and PR-B. PR-B is the major subtype required for mammary gland development and expansion, while PR-A is required for normal uterine development and reproductive activity (227). PR is a polypeptide expressed in the cytoplasm, and once bound to progesterone, it translates into the nucleus and regulates the expression of a specific set of genes. Limited studies have reported information on the relative expression of PR in tissue samples from human OC subtypes. Diep et al (228) assessed the percentage of PR-positive tumors among primary histological subtypes of OSE-derived OC in 504 tissue samples. According to their results, 35% of ovarian tumors were PR-positive, with the maximal total PR expression found in the endometrioid (67%) and serous (35%; LGSOC, 64%) subtypes (228). Subsequently, the International Consortium for Ovarian Tissue Analysis analyzed the proportion of tumors positive for PR staining in ~3,000 invasive epithelial ovarian tumors and reported the highest expression in endometrioid (67.4%) and LGSOC (57.4%); intermediate expression in HGSOC (31.1%); and the lowest expression in the mucinous (16.4%) and clear cell subtypes (8.0%) (25). Additionally, that study investigated the prognostic implications of PR expression in ovarian tumors highly expressing PR (staining of 50% tumor cell nuclei). PR expression was associated with a significantly favorable survival for HGSOC, and significantly improved disease-specific survival independent of patient age, tumor grade, site and stage for endometrioid OC (25). To the best of our knowledge, only three studies to date have reported the differential expression of PR isoforms in ovarian tumors (229–231). These studies reported the dominance of PR-B expression in all subtypes of ovarian tumors; furthermore, PR-B is usually expressed in serous subtypes, whereas PR-A is weakly expressed in mucinous and serous OC, and its expression is low or absent in tumors compared with that of PR-B in comparison with normal and malignant ovarian tissues.
Progesterone signaling actions in OC
The first indication that progesterone may be involved in the regulation of OC is derived from the observation that the use of progestin-containing oral contraceptives appears to prevent the occurrence of OC (232). Moreover, the incidence of OC is increased among women with progesterone deficiency, while the high levels of serum progesterone during pregnancy are associated with a lower risk of developing OC (233,234). The protective effect of progesterone against OC development is lost in post-menopausal women, as the serum levels of progesterone are negligible following menopause (235). In addition, women with a history of twin pregnancies exhibit a lower risk of developing OC, which may be associated with the higher serum levels of progesterone in the maternal circulation in twin compared with singleton pregnancies (236,237). The molecular mechanisms through which progesterone exerts its protective effects against OC are not yet well understood. Both proliferative and inhibitory effects of progesterone on OC cells have been reported in cell line assays, which may be attributed, at least in part, to the differential effects of progesterone on the two receptors, PR-A and PR-B, and their relative expression in target cells. Several independent in vitro studies have demonstrated the anti-proliferative effects of progesterone at higher concentrations in EOC cells, primarily through the induction of apoptosis (238–240). The activation of progesterone signaling can inhibit ovulation, antagonize the growth-promoting effects of estrogen, and regulate EOC cell proliferation and apoptosis (241). High concentrations of estrogen combined with progesterone may induce the apoptosis of EOC cells by promoting the expression of let-7a and microRNA-34b, and decreasing the expression of Bcl-2 (241). In another study, however, it was demonstrated that the combination of tamoxifen and progesterone treatment induced apoptosis similar to that induced by treatment with progesterone independently, with no additional anticancer effect on EOC cells (242).
The precursor form of the steroid hormone pregnenolone reduces ovarian cell proliferation and viability by downregulating the expression of PR (243). Recently, Pedernera et al (244) observed that progesterone treatment significantly reduced cell survival in endometrioid OC. That study demonstrated the protective effects of progesterone against OC and indicated that the presence of PRs may suppress the progression of endometrioid OC. Of note, as demonstrated in a previous study, the progesterone metabolite, allopregnanolone, increased the proliferation of and Ki67 expression in EOC IGROV-1 cells, whereas the expression of cleaved caspase-3 was unaltered (239). Furthermore, progesterone and allopregnanolone increased the migratory capacity of IGROV-1 cells in a concentration-dependent manner (239). Moreover, it has been demonstrated that PR membrane component-1 (PGRMC1) plays an important role in promoting EOC cell viability by binding to progesterone, and that the attenuation of PGRMC1 function by small interfering RNA increases the sensitivity of EOC cells to cisplatin (237,245). The prolonged presence of the anti-progestin, mifepristone, has also been shown to block the repopulation of EOC cells that escaped platinum or platinum/paclitaxel treatment, providing evidence of the long-term use of anti-progestins as anti-repopulation therapy for cells that escape other effective chemotherapies (246–248). However, as progesterone/PR signaling is complex, further research on the intricate details of its role in tumor progression is required before it can be considered as a potential clinical target in OC treatment.
Clinical trials of progesterone in recurrent OC
Despite the high expression levels of endocrine response receptors, hormonal therapy plays merely a secondary role in EOC treatment. Epidemiological evidence coupled with the findings of in vivo and in vitro studies suggests the modulation of PR levels or activity as a form of endocrine therapy for EOC (Table VI) (189,249–262). In a phase II clinical trial conducted in 2000, 34 patients with recurrent EOC who no longer responded to cisplatin/paclitaxel chemotherapy were administered 200 mg oral mifepristone daily for 28 days. Of these patients, 9 responded to mifepristone, exhibiting a decrease in tumor size by at least 50% or a 50% decrease in the levels of CA125, which is used to assess disease recurrence (261). However, the results from another phase II clinical trial including 24 patients with advanced EOC who received standard chemotherapy and relapsed within 6 months, demonstrated that only 1 patient had an objective response to a 28-day regimen of 200 mg mifepristone administered daily (262). This clinical evidence appears to be less convincing in terms of the number of patients included, the lack of biomarkers to predict the response, and the fact that the studies did not report hormone receptor expression levels in OC tissue. Niwa et al (263) investigated the effects of the combination of medroxyprogesterone acetate (MPA) with primary adjuvant chemotherapy for advanced EOC in 2008. Both PFS and overall survival were significantly longer in the cases treated with the combination of MPA and platinum-based chemotherapy compared with those in the control group. Zheng et al (264) reviewed the value of PR ligands in OC treatment by examining 13 clinically related trials that included 432 cases with recurrent or refractory OC treated with megestrol acetate or MPA. A total of 10 cases (2.3%) had a complete response, 21 (4.9%) had a partial response and 47 (10.9%) had SD. The authors of that study concluded that the effectiveness achieved by progesterone preparations in recurrent EOC was not confirmed by existing research. In addition, the anti-progestin, mifepristone, is employed in the treatment of platinum-resistant EOC. PR can be induced by estrogen (and by tamoxifen, if serving as an agonist), and several trials have investigated the effectiveness achieved by combination hormonal therapy that targets the mentioned crosstalk (206,265,266). One noticeable clinically related response was observed in a trial of 65 patients with refractory EOC who were administered medroxyprogesterone and sequential ethinyl estradiol, achieving a response rate of 14% and SD in 20% of the patients (267). Given the recent preclinical data and basic research findings, it is expected that drugs targeting different PR subtypes (PR-A vs. PR-B), or drugs targeting other progesterone-binding receptors, will be developed and used in the near future.
Table VI.
Overview of trials using progestins (MA and MPA) and anti-progestins in persistent or recurrent EOC.
| Drug | Condition | Intervention | Histology (no. of samples) | CR | PR | SD | Phase | (Refs.) |
|---|---|---|---|---|---|---|---|---|
| Medroxyprogesterone | Advanced EOC | 800 mg daily | 30; | 0 | 0 | 2 | II | (249) |
| Serous, 16; | ||||||||
| Endometrioid, 7; | ||||||||
| Mucinous, 1; | ||||||||
| Clear cell, 4; | ||||||||
| Undifferentiated, 2 | ||||||||
| Medroxyprogesterone | Advanced EOC | 500 mg daily for | 33; | 0 | 5 | 2 | II | (249) |
| 4 weeks, then 500 mg | Serous, 20; | |||||||
| twice weekly for | Endometrioid, 8; | |||||||
| 4 weeks, then 500 mg | Mucinous, 2; | |||||||
| once weekly | Undifferentiated, 3 | |||||||
| Medroxyprogesterone | Advanced EOC | 1,000 mg weekly | 19 | 0 | 0 | 1 | II | (250) |
| by intramuscular injection | – | |||||||
| Medroxyprogesterone | Advanced | – | 27 | 0 | 1 | 0 | II | (251) |
| chemotherapy-resistant EOC | – | |||||||
| Medroxyprogesterone | Advanced ovarian | 500 mg daily | 25 | 0 | 1 | 9 | II | (252) |
| cancer | – | |||||||
| Medroxyprogesterone | Advanced EOC | 500 mg daily, then | 41; | 0 | 1 | 7 | II | (253) |
| 1,000 mg weekly | Serous, 18; | |||||||
| Endometrioid, 2; | ||||||||
| Mucinous, 3; | ||||||||
| Brenner, 3; | ||||||||
| Adenocarcinoma,12 | ||||||||
| Undifferentiated, 3 | ||||||||
| Medroxyprogesterone | Advanced or | 150 mg daily | 24; | 0 | 1 | 9 | II | (254) |
| recurrent EOC | Serous, 17; | |||||||
| Endometrioid, 4; | ||||||||
| Mucinous, 2; | ||||||||
| Undifferentiated, 1 | ||||||||
| Megestrol acetate | Ovarian | 800 mg daily for | 23; | 7 | 4 | 0 | I–II | (255) |
| adenocarcinoma | 4 weeks, then 400 mg daily | Adenocarcinoma, 23 | ||||||
| Megestrol acetate | Resistant EOC | 800 mg daily for | 45; | 1 | 3 | 5 | II | (256) |
| 4 weeks, then 400 mg | Serous, 37; | |||||||
| daily | Endometrioid, 6; | |||||||
| Clear cell, 2 | ||||||||
| Megestrol acetate | Progressive or | 160 mg daily | 33; | 0 | 0 | 12 | II | (257) |
| recurrent EOC | Serous, 28; | |||||||
| Endometrioid, 2; | ||||||||
| Clear cell, 1; | ||||||||
| Brenner, 1; | ||||||||
| Undifferentiated, 1 | ||||||||
| Megestrol acetate | Resistant ovarian | 800 mg daily for | 32; | 0 | 0 | 13 | II | (189) |
| cancer | 30 days, then 400 mg | Serous, 3; | ||||||
| daily | Endometrioid, 4; | |||||||
| Mucinous, 1; | ||||||||
| Clear cell, 2; | ||||||||
| Papillary, 18; | ||||||||
| Undifferentiated, 4 | ||||||||
| Megestrol acetate | Advanced ovarian | 800 mg daily for | 67; | 0 | 1 | 9 | II | (258) |
| cancer | 30 days, then 400 mg | Serous, 33; | ||||||
| daily | Endometrioid, 3; | |||||||
| Mucinous, 2; | ||||||||
| Clear cell, 2; | ||||||||
| Adenocarcinoma, 23; | ||||||||
| Undifferentiated, 4 | ||||||||
| Megestrol acetate | Resistant EOC | 200 mg four times | 30; | 0 | 0 | 0 | II | (259) |
| daily | Serous, 9; | |||||||
| Endometrioid, 4; | ||||||||
| Mucinous, 2; | ||||||||
| Adenocarcinoma, 11; | ||||||||
| Papillary, 4 | ||||||||
| Megestrol acetate | Refractory EOC | 800 mg daily for | 36; | 3 | 4 | NA | II | (260) |
| 4 weeks, then 400 mg | Serous, 7; | |||||||
| daily | Endometrioid, 9; | |||||||
| Mucinous, 8; | ||||||||
| Clear cell, 10; | ||||||||
| Adenocarcinoma, 2 | ||||||||
| Mifepristone | Refractory EOC | 200 mg daily | 34; | 3 | 6 | NA | II | (261) |
| Serous, 25; | ||||||||
| Endometrioid, 2; | ||||||||
| Clear cell, 1; | ||||||||
| Undifferentiated, 6 | ||||||||
| Mifepristone | Recurrent or EOC | 200 mg daily | 22; | 0 | 1 | 3 | II | (262) |
| Persistent | Serous, 19; | |||||||
| Endometrioid, 1; | ||||||||
| Adenocarcinoma, 2 |
EOC, epithelial ovarian cancer; CR, complete response; PR, partial response; SD, stable disease.
7. Hormone replacement therapy and the risk of ovarian cancer
Findings by epidemiology researches on menopausal hormone replacement therapy (HRT) and the risk of developing OC are inconsistent. HRT exhibits an association with an increased risk of developing OC. Nevertheless, some researchers have reported that they found no such association, whereas other researchers have reported a positive association within the single histology subdivided type. As concluded from previous a review and meta-analysis of information (1966 and 2006), examining the application of post-menopausal hormone therapy (HT), an upregulated risk of developing OC by 30% was observed in contrast to no HT; it was thus indicated that the risk of developing OC with estrogen therapy (ET) was independently higher than that related to estrogen plus progestin therapy (EPT) (268). Nevertheless, the Million Women Study examined 2,273 incident cases of OC and with 948,576 females, reported an increased risk of developing OC with hormone replacement therapy, whereas an insignificant differential influence exerted by ET vs. EPT was achieved (269); similarly, another nationwide study found no evidence of risk associated with vaginal ET or transdermal vs. oral EPT (270). Furthermore, in other studies, cyclic treating process were reported to increase the risk of OC, and no statistics-related significant differences were found from the cyclic combined regimen (28,271). A national prospective cohort study covering overall Danish females aged 50–79 years from 1995–2005 suggested an increased risk of developing OC with EPT and estrogen therapy, exhibiting an insignificant effect exerted by various doses, the length of use, routes of administration, progestin types and regimens (22). Based on the investigation of the identical cohort of cases recruited, as was reported, the risk of developing OC was altered in accordance with the histology of the tumor: In contrast to never users, females undergoing unopposed oral estrogen therapy exhibited an increased risk of developing ovarian serous and endometrial-like tumors, whereas they exhibited a decreased risk of developing mucinous tumors (272). According to a previous study analyzing the risk of developing OC in post-menopausal females treated with estradiol-progestin, the elevated risk of developing OC associated with EPT use over a period of 5 years was only observed in serous and mixed cancers, while the risk of mucinous cancers was reduced (270). A recently conducted meta-analysis reported by Liu et al (273) concluded that EPT during menopause may increase the risk of developing OC, particularly in terms of endometrioid and serous tumors. In a collaborative re-analysis of 52 epidemiological studies in the OC Epidemiology Research Collaborative (274), the relative risk (RR) of HRT for a duration of ≥5 years was more notable than that observed with past users who had terminated the therapy <5 years earlier. It was thus concluded that this increased risk was likely to be primarily or entirely involving a cause; if causal, female users who had been on hormone therapy for 5 years from the age of ~50 years would have one additional OC for every 1,000 users and, if their prognosis was characteristic, one additional OC-related death for every 1,700 users.
The effects of post-operative HRT on non-progression and total survival within cases of EOC is also controversial. The present review article can support the assumption that, i.e., HRT following surgery cannot adversely affect the non-progression and total survival of EOC cases. A retrospective study that included 77 cases supported the assumption that HRT following surgery failed to not significantly influence non-progression and total survival in cases suffering from EOC. Likewise, various HRTs (an estrogen-tibolone integration, tibolone independently, or estrogen independently) failed to notably affect the prognosis of patients with EOC (275). This finding was validated in another meta-analysis that covered 419 EOC survivors using HRT and 1,029 non-users (276). In a recent meta-analysis that included 350 cases suffering from EOC, the authors concluded that HRT may slightly improve the total survival of cases who had undergone surgical treatment, and there may be a minimal or no effect of HRT use on non-progression survival (24).
8. Conclusions and future perspectives
In summary, OC is a malignancy depending on hormones in which steroid hormones and the relevant receptor critically affect its advancement. Although hormone therapy is effective in cases with advanced or recurrent OC, and has a low profile of toxicity, studies evaluating the therapeutic value of hormone therapy in OC have not been conclusive due to small sample sizes, different pathological types of OC, different hormone receptor-expressing states within OC cells, and the lack of molecular markers. In addition, the lack of corresponding NCT numbers for a number of clinical trials also does not facilitate the readers' understanding of the trials and may affect the authenticity of the trial results. Therefore, multicenter, prospective, randomized trial studies are required to confirm its efficacy. In addition, further studies are warranted to determine whether the combination of hormones and chemotherapeutic drugs can improve the effectiveness of chemotherapy, and whether this can be used as therapy for OC, as well as to identify the molecular markers of hormone therapy. In cases suffering from ER-positive breast carcinoma, hormone therapy has been used as part of systemic cancer treatment, and in endometrial cancer, hormone therapy functions as a second-line treatment option in terms of cancer metastasis. The ovary as an endocrine organ and the application of hormone therapy in OC patients, is worthy of further study. In future clinical treatment, hormone receptor expression should be routinely tested, and the expression should be standardized and classified into treatment considerations. In addition, different treatment options should be adopted according to the various pathological types of OC.
Acknowledgements
Not applicable.
Funding Statement
The present study was supported by the National Major Scientific and Technological Special Project for the ‘Significant New Drugs Development’ (grant no. 2018ZX09201018-013) and the National Natural Science Foundation of China (grant no. 81821002).
Funding
The present study was supported by the National Major Scientific and Technological Special Project for the ‘Significant New Drugs Development’ (grant no. 2018ZX09201018-013) and the National Natural Science Foundation of China (grant no. 81821002).
Availability of data and materials
Not applicable.
Authors' contributions
HL and YL wrote the initial manuscript and confirm the authenticity of all the raw data. YW created the figures and contributed to writing the material and providing new ideas. XZ and XQ revised the manuscript and approved the final version. All authors (HL, YL, YW, XZ, XQ) read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: A review. Cancer Biol Med. 2017;14:9–32. doi: 10.20892/j.issn.2095-3941.2016.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen VW, Ruiz B, Killeen JL, Coté TR, Wu XC, Correa CN. Pathology and classification of ovarian tumors. Cancer. 2003;97(Suppl 10):S2631–S2642. doi: 10.1002/cncr.11345. [DOI] [PubMed] [Google Scholar]
- 5.Prat J. New insights into ovarian cancer pathology. Ann Oncol. 2012;23(Suppl 10):x111–x117. doi: 10.1093/annonc/mds300. [DOI] [PubMed] [Google Scholar]
- 6.Peres LC, Cushing-Haugen KL, Anglesio M, Wicklund K, Bentley R, Berchuck A, Kelemen LE, Nazeran TM, Gilks CB, Harris HR, et al. Histotype classification of ovarian carcinoma: A comparison of approaches. Gynecol Oncol. 2018;151:53–60. doi: 10.1016/j.ygyno.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duska LR, Kohn EC. The new classifications of ovarian, fallopian tube, and primary peritoneal cancer and their clinical implications. Ann Oncol. 2017;28(Suppl 8):viii8–viii12. doi: 10.1093/annonc/mdx445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meinhold-Heerlein I, Fotopoulou C, Harter P, Kurzeder C, Mustea A, Wimberger P, Hauptmann S, Sehouli J. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch Gynecol Obstet. 2016;293:695–700. doi: 10.1007/s00404-016-4073-2. [DOI] [PubMed] [Google Scholar]
- 9.Bowtell DD, Böhm S, Ahmed AA, Aspuria PJ, Bast RC, Jr, Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668–679. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 11.Hunter SM, Anglesio MS, Ryland GL, Sharma R, Chiew YE, Rowley SM, Doyle MA, Li J, Gilks CB, Moss P, et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget. 2015;6:37663–37677. doi: 10.18632/oncotarget.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilbur MA, Shih IM, Segars JH, Fader AN. Cancer implications for patients with endometriosis. Semin Reprod Med. 2017;35:110–116. doi: 10.1055/s-0036-1597120. [DOI] [PubMed] [Google Scholar]
- 13.Munksgaard PS, Blaakaer J. The association between endometriosis and gynecological cancers and breast cancer: A review of epidemiological data. Gynecol Oncol. 2011;123:157–163. doi: 10.1016/j.ygyno.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 14.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayhan A, Mao TL, Seckin T, Wu CH, Guan B, Ogawa H, Futagami M, Mizukami H, Yokoyama Y, Kurman RJ, Shih IeM. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int J Gynecol Cancer. 2012;22:1310–1315. doi: 10.1097/IGC.0b013e31826b5dcc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollis RL, Gourley C. Genetic and molecular changes in ovarian cancer. Cancer Biol Med. 2016;13:236–247. doi: 10.20892/j.issn.2095-3941.2016.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yarmolinsky J, Relton CL, Lophatananon A, Muir K, Menon U, Gentry-Maharaj A, Walther A, Zheng J, Fasching P, Zheng W, et al. Appraising the role of previously reported risk factors in epithelial ovarian cancer risk: A Mendelian randomization analysis. PLoS Med. 2019;16:e1002893. doi: 10.1371/journal.pmed.1002893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Vecchia C. Ovarian cancer: Epidemiology and risk factors. Eur J Cancer Prev. 2017;26:55–62. doi: 10.1097/CEJ.0000000000000217. [DOI] [PubMed] [Google Scholar]
- 19.Liao Y, Tu C, Song X, Cai L. Case report: Analysis of BRCA1 and BRCA2 gene mutations in a hereditary ovarian cancer family. J Assist Reprod Genet. 2020;37:1489–1495. doi: 10.1007/s10815-020-01783-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daly MB, Pal T, Berry MP, Buys SS, Dickson P, Domchek SM, Elkhanany A, Friedman S, Goggins M, Hutton ML, et al. Genetic/Familial high-risk assessment: Breast, ovarian, and pancreatic, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2021;19:77–102. doi: 10.6004/jnccn.2021.0001. [DOI] [PubMed] [Google Scholar]
- 21.Onwude J. Hormone therapy and ovarian cancer. Lancet. 2015;386:1037–1038. doi: 10.1016/S0140-6736(15)00137-3. [DOI] [PubMed] [Google Scholar]
- 22.Mørch LS, Løkkegaard E, Andreasen AH, Krüger-Kjaer S, Lidegaard O. Hormone therapy and ovarian cancer. JAMA. 2009;302:298–305. doi: 10.1001/jama.2009.1052. [DOI] [PubMed] [Google Scholar]
- 23.Colombo N, Sessa C, du Bois A, Ledermann J, McCluggage WG, McNeish I, Morice P, Pignata S, Ray-Coquard I, Vergote I, et al. ESMO-ESGO consensus conference recommendations on ovarian cancer: Pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease†. Ann Oncol. 2019;30:672–705. doi: 10.1093/annonc/mdz062. [DOI] [PubMed] [Google Scholar]
- 24.Saeaib N, Peeyananjarassri K, Liabsuetrakul T, Buhachat R, Myriokefalitaki E. Hormone replacement therapy after surgery for epithelial ovarian cancer. Cochrane Database Syst Rev. 2020;1:CD012559. doi: 10.1002/14651858.CD012559.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sieh W, Köbel M, Longacre TA, Bowtell DD, deFazio A, Goodman MT, Høgdall E, Deen S, Wentzensen N, Moysich KB, et al. Hormone-receptor expression and ovarian cancer survival: An ovarian tumor tissue analysis consortium study. Lancet Oncol. 2013;14:853–862. doi: 10.1016/S1470-2045(13)70253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paleari L, DeCensi A. Endocrine therapy in ovarian cancer: Where do we stand? Curr Opin Obstet Gynecol. 2018;30:17–22. doi: 10.1097/GCO.0000000000000423. [DOI] [PubMed] [Google Scholar]
- 27.Cramer DW, Welch WR. Determinants of ovarian cancer risk. II. Inferences regarding pathogenesis. J Natl Cancer Inst. 1983;71:717–721. [PubMed] [Google Scholar]
- 28.Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Weiderpass E, Persson IR. Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J Natl Cancer Inst. 2002;94:497–504. doi: 10.1093/jnci/94.7.497. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez C, Patel AV, Calle EE, Jacob EJ, Thun MJ. Estrogen replacement therapy and ovarian cancer mortality in a large prospective study of US women. JAMA. 2001;285:1460–1465. doi: 10.1001/jama.285.11.1460. [DOI] [PubMed] [Google Scholar]
- 30.Risch HA. Hormonal etiology of epithelial ovarian cancer, with a hypothesis concerning the role of androgens and progesterone. J Natl Cancer Inst. 1998;90:1774–1786. doi: 10.1093/jnci/90.23.1774. [DOI] [PubMed] [Google Scholar]
- 31.Konishi I, Kuroda H, Mandai M. Review: Gonadotropins and development of ovarian cancer. Oncology. 1999;57(Suppl 2):S45–S48. doi: 10.1159/000055274. [DOI] [PubMed] [Google Scholar]
- 32.Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C. Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol. 1996;88:554–559. doi: 10.1016/0029-7844(96)00226-8. [DOI] [PubMed] [Google Scholar]
- 33.Goulis DG, Mantzoros CS. Reproductive endocrinology: Novel insights into pathophysiology and clinical management. Metabolism. 2018;86:1–2. doi: 10.1016/j.metabol.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 34.Cardenas C, Alvero AB, Yun BS, Mor G. Redefining the origin and evolution of ovarian cancer: A hormonal connection. Endocr Relat Cancer. 2016;23:R411–R422. doi: 10.1530/ERC-16-0209. [DOI] [PubMed] [Google Scholar]
- 35.Trabert B, Coburn SB, Falk RT, Manson JE, Brinton LA, Gass ML, Kuller LH, Rohan TE, Pfeiffer RM, Qi L, et al. Circulating estrogens and postmenopausal ovarian and endometrial cancer risk among current hormone users in the Women's Health Initiative Observational Study. Cancer Causes Control. 2019;30:1201–1211. doi: 10.1007/s10552-019-01233-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webb PM, Jordan SJ. Epidemiology of epithelial ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 2017;41:3–14. doi: 10.1016/j.bpobgyn.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Risch HA, Marrett LD, Jain M, Howe GR. Differences in risk factors for epithelial ovarian cancer by histologic type. Results of a case-control study. Am J Epidemiol. 1996;144:363–372. doi: 10.1093/oxfordjournals.aje.a008937. [DOI] [PubMed] [Google Scholar]
- 38.Hankinson SE, Colditz GA, Hunter DJ, Willett WC, Stampfer MJ, Rosner B, Hennekens CH, Speizer FE. A prospective study of reproductive factors and risk of epithelial ovarian cancer. Cancer. 1995;76:284–290. doi: 10.1002/1097-0142(19950715)76:2<284::AID-CNCR2820760219>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 39.Moorman PG, Havrilesky LJ, Gierisch JM, Coeytaux RR, Lowery WJ, Peragallo Urrutia R, Dinan M, McBroom AJ, Hasselblad V, Sanders GD, Myers ER. Oral contraceptives and risk of ovarian cancer and breast cancer among high-risk women: A systematic review and meta-analysis. J Clin Oncol. 2013;31:4188–4198. doi: 10.1200/JCO.2013.48.9021. [DOI] [PubMed] [Google Scholar]
- 40.Rzepka-Górska I, Chudecka-Głaz A, Kosmowska B. FSH and LH serum/tumor fluid ratios and malignant tumors of the ovary. Endocr Relat Cancer. 2004;11:315–321. doi: 10.1677/erc.0.0110315. [DOI] [PubMed] [Google Scholar]
- 41.Halperin R, Pansky M, Vaknin Z, Zehavi S, Bukovsky I, Schneider D, Sackler Faculty of Medicine, Tel Aviv University, Israel Luteinizing hormone in peritoneal and ovarian cyst fluids: A predictor of ovarian carcinoma. Eur J Obstet Gynecol Reprod Biol. 2003;110:207–210. doi: 10.1016/S0301-2115(03)00122-2. [DOI] [PubMed] [Google Scholar]
- 42.Chen FC, Oskay-Ozcelik G, Bühling KJ, Köpstein U, Mentze M, Lichtenegger W, Sehouli J. Prognostic value of serum and ascites levels of estradiol, FSH, LH and prolactin in ovarian cancer. Anticancer Res. 2009;29:1575–1578. [PubMed] [Google Scholar]
- 43.Chudecka-Głaz A, Rzepka-Górska I. Concentrations of follicle stimulating hormone are increased in ovarian tumor fluid: Implications for the management of ovarian cancer. Eur J Gynaecol Oncol. 2008;29:37–42. [PubMed] [Google Scholar]
- 44.Fathalla MF. Incessant ovulation-a factor in ovarian neoplasia? Lancet. 1971;2:163. doi: 10.1016/S0140-6736(71)92335-X. [DOI] [PubMed] [Google Scholar]
- 45.Fleming JS, Beaugié CR, Haviv I, Chenevix-Trench G, Tan OL. Incessant ovulation, inflammation and epithelial ovarian carcinogenesis: Revisiting old hypotheses. Mol Cell Endocrinol. 2006;247:4–21. doi: 10.1016/j.mce.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 46.Fathalla MF. Incessant ovulation and ovarian cancer-a hypothesis re-visited. Facts Views Vis Obgyn. 2013;5:292–297. [PMC free article] [PubMed] [Google Scholar]
- 47.Peres LC, Moorman PG, Alberg AJ, Bandera EV, Barnholtz-Sloan J, Bondy M, Cote ML, Funkhouser E, Peters ES, Schwartz AG, et al. Lifetime number of ovulatory cycles and epithelial ovarian cancer risk in African American women. Cancer Causes Control. 2017;28:405–414. doi: 10.1007/s10552-017-0853-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang HP, Murphy KR, Pfeiffer RM, George N, Garcia-Closas M, Lissowska J, Brinton LA, Wentzensen N. Lifetime number of ovulatory cycles and risks of ovarian and endometrial cancer among postmenopausal women. Am J Epidemiol. 2016;183:800–814. doi: 10.1093/aje/kwv308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mizushima T, Miyamoto H. The role of androgen receptor signaling in ovarian cancer. Cells. 2019;8:176. doi: 10.3390/cells8020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gibson DA, Simitsidellis I, Collins F, Saunders PT. Evidence of androgen action in endometrial and ovarian cancers. Endocr Relat Cancer. 2014;21:T203–T218. doi: 10.1530/ERC-13-0551. [DOI] [PubMed] [Google Scholar]
- 51.Kollara A, Shathasivam P, Park S, Ringuette MJ, Brown TJ. Increased androgen receptor levels and signaling in ovarian cancer cells by VEPH1 associated with suppression of SMAD3 and AKT activation. J Steroid Biochem Mol Biol. 2020;196:105498. doi: 10.1016/j.jsbmb.2019.105498. [DOI] [PubMed] [Google Scholar]
- 52.Huang SL, Chang TC, Chao CCK, Sun NK. Role of the TLR4-androgen receptor axis and genistein in taxol-resistant ovarian cancer cells. Biochem Pharmacol. 2020;177:113965. doi: 10.1016/j.bcp.2020.113965. [DOI] [PubMed] [Google Scholar]
- 53.Feng D, Zhao T, Yan K, Liang H, Liang J, Zhou Y, Zhao W, Ling B. Gonadotropins promote human ovarian cancer cell migration and invasion via a cyclooxygenase 2-dependent pathway. Oncol Rep. 2017;38:1091–1098. doi: 10.3892/or.2017.5784. [DOI] [PubMed] [Google Scholar]
- 54.Parrott JA, Doraiswamy V, Kim G, Mosher R, Skinner MK. Expression and actions of both the follicle stimulating hormone receptor and the luteinizing hormone receptor in normal ovarian surface epithelium and ovarian cancer. Mol Cell Endocrinol. 2001;172:213–222. doi: 10.1016/S0303-7207(00)00340-3. [DOI] [PubMed] [Google Scholar]
- 55.Zheng W, Magid MS, Kramer EE, Chen YT. Follicle-stimulating hormone receptor is expressed in human ovarian surface epithelium and fallopian tube. Am J Pathol. 1996;148:47–53. [PMC free article] [PubMed] [Google Scholar]
- 56.Chambers GM, Sullivan EA, Ishihara O, Chapman MG, Adamson GD. The economic impact of assisted reproductive technology: A review of selected developed countries. Fertil Steril. 2009;91:2281–2294. doi: 10.1016/j.fertnstert.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 57.Pawlikowski Łódź M. Expression of follicle stimulating hormone receptors in intra-tumoral vasculature and in tumoral cells-the involvement in tumour progression and the perspectives of application in cancer diagnosis and therapy. Endokrynol Pol. 2018;69:192–198. doi: 10.5603/EP.2018.0022. [DOI] [PubMed] [Google Scholar]
- 58.Robin B, Planeix F, Sastre-Garau X, Pichon C, Olesen TK, Gogusev J, Ghinea N. Follicle-stimulating hormone receptor expression in endometriotic lesions and the associated vasculature: An immunohistochemical study. Reprod Sci. 2016;23:885–891. doi: 10.1177/1933719115623647. [DOI] [PubMed] [Google Scholar]
- 59.Lenhard M, Lennerová T, Ditsch N, Kahlert S, Friese K, Mayr D, Jeschke U. Opposed roles of follicle-stimulating hormone and luteinizing hormone receptors in ovarian cancer survival. Histopathology. 2011;58:990–994. doi: 10.1111/j.1365-2559.2011.03848.x. [DOI] [PubMed] [Google Scholar]
- 60.Choi JH, Choi KC, Auersperg N, Leung PC. Overexpression of follicle-stimulating hormone receptor activates oncogenic pathways in preneoplastic ovarian surface epithelial cells. J Clin Endocrinol Metab. 2004;89:5508–5516. doi: 10.1210/jc.2004-0044. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Z, Jia L, Feng Y, Zheng W. Overexpression of follicle-stimulating hormone receptor facilitates the development of ovarian epithelial cancer. Cancer Lett. 2009;278:56–64. doi: 10.1016/j.canlet.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 62.Deuster E, Mayr D, Hester A, Kolben T, Zeder-Göß C, Burges A, Mahner S, Jeschke U, Trillsch F, Czogalla B. Correlation of the aryl hydrocarbon receptor with FSHR in ovarian cancer patients. Int J Mol Sci. 2019;20:2862. doi: 10.3390/ijms20122862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheung J, Lokman NA, Abraham RD, Macpherson AM, Lee E, Grutzner F, Ghinea N, Oehler MK, Ricciardelli C. Reduced gonadotrophin receptor expression is associated with a more aggressive ovarian cancer phenotype. Int J Mol Sci. 2020;22:71. doi: 10.3390/ijms22010071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lau MT, Wong AS, Leung PC. Gonadotropins induce tumor cell migration and invasion by increasing cyclooxygenases expression and prostaglandin E(2) production in human ovarian cancer cells. Endocrinology. 2010;151:2985–2993. doi: 10.1210/en.2009-1318. [DOI] [PubMed] [Google Scholar]
- 65.Song K, Dai L, Long X, Wang W, Di W. Follicle-stimulating hormone promotes the proliferation of epithelial ovarian cancer cells by activating sphingosine kinase. Sci Rep. 2020;10:13834. doi: 10.1038/s41598-020-70896-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Warrenfeltz SW, Lott SA, Palmer TM, Gray JC, Puett D. Luteinizing hormone-induced up-regulation of ErbB-2 is insufficient stimulant of growth and invasion in ovarian cancer cells. Mol Cancer Res. 2008;6:1775–1785. doi: 10.1158/1541-7786.MCR-08-0214. [DOI] [PubMed] [Google Scholar]
- 67.Liao H, Zhou Q, Gu Y, Duan T, Feng Y. Luteinizing hormone facilitates angiogenesis in ovarian epithelial tumor cells and metformin inhibits the effect through the mTOR signaling pathway. Oncol Rep. 2012;27:1873–1878. doi: 10.3892/or.2012.1745. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Z, Liao H, Chen X, Zheng Y, Liu Y, Tao X, Gu C, Dong L, Duan T, Yang Y, et al. Luteinizing hormone upregulates survivin and inhibits apoptosis in ovarian epithelial tumors. Eur J Obstet Gynecol Reprod Biol. 2011;155:69–74. doi: 10.1016/j.ejogrb.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 69.Sakai T, Shiraishi A, Kawada T, Matsubara S, Aoyama M, Satake H. Invertebrate Gonadotropin-releasing hormone-related peptides and their receptors: An update. Front Endocrinol (Lausanne) 2017;8:217. doi: 10.3389/fendo.2017.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jankowska AG, Andrusiewicz M, Fischer N, Warchol PJ. Expression of hCG and GnRHs and their receptors in endometrial carcinoma and hyperplasia. Int J Gynecol Cancer. 2010;20:92–101. doi: 10.1111/IGC.0b013e3181bbe933. [DOI] [PubMed] [Google Scholar]
- 71.Gründker C, Schlotawa L, Viereck V, Eicke N, Horst A, Kairies B, Emons G. Antiproliferative effects of the GnRH antagonist cetrorelix and of GnRH-II on human endometrial and ovarian cancer cells are not mediated through the GnRH type I receptor. Eur J Endocrinol. 2004;151:141–149. doi: 10.1530/eje.0.1510141. [DOI] [PubMed] [Google Scholar]
- 72.Kang SK, Tai CJ, Cheng KW, Leung PC. Gonadotropin- releasing hormone activates mitogen-activated protein kinase in human ovarian and placental cells. Mol Cell Endocrinol. 2000;170:143–151. doi: 10.1016/S0303-7207(00)00320-8. [DOI] [PubMed] [Google Scholar]
- 73.Gründker C, Günthert AR, Westphalen S, Emons G. Biology of the gonadotropin-releasing hormone system in gynecological cancers. Eur J Endocrinol. 2002;146:1–14. doi: 10.1530/eje.0.1460001. [DOI] [PubMed] [Google Scholar]
- 74.Wilkinson SJ, Kucukmetin A, Cross P, Darby S, Gnanapragasam VJ, Calvert AH, Robson CN, Edmondson RJ. Expression of gonadotrophin releasing hormone receptor I is a favorable prognostic factor in epithelial ovarian cancer. Hum Pathol. 2008;39:1197–1204. doi: 10.1016/j.humpath.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 75.Peng C, Fan NC, Ligier M, Väänänen J, Leung PC. Expression and regulation of gonadotropin-releasing hormone (GnRH) and GnRH receptor messenger ribonucleic acids in human granulosa-luteal cells. Endocrinology. 1994;135:1740–1746. doi: 10.1210/endo.135.5.7956897. [DOI] [PubMed] [Google Scholar]
- 76.Choi KC, Auersperg N, Leung PC. Expression and antiproliferative effect of a second form of gonadotropin-releasing hormone in normal and neoplastic ovarian surface epithelial cells. J Clin Endocrinol Metab. 2001;86:5075–5078. doi: 10.1210/jcem.86.10.8100. [DOI] [PubMed] [Google Scholar]
- 77.Choi JH, Choi KC, Auersperg N, Leung PC. Differential regulation of two forms of gonadotropin-releasing hormone messenger ribonucleic acid by gonadotropins in human immortalized ovarian surface epithelium and ovarian cancer cells. Endocr Relat Cancer. 2006;13:641–651. doi: 10.1677/erc.1.01057. [DOI] [PubMed] [Google Scholar]
- 78.Kim KY, Choi KC, Auersperg N, Leung PC. Mechanism of gonadotropin-releasing hormone (GnRH)-I and -II-induced cell growth inhibition in ovarian cancer cells: Role of the GnRH-I receptor and protein kinase C pathway. Endocr Relat Cancer. 2006;13:211–220. doi: 10.1677/erc.1.01033. [DOI] [PubMed] [Google Scholar]
- 79.Padula AM. GnRH analogues-agonists and antagonists. Anim Reprod Sci. 2005;88:115–126. doi: 10.1016/j.anireprosci.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 80.Limonta P, Marelli MM, Moretti R, Marzagalli M, Fontana F, Maggi R. GnRH in the human female reproductive axis. Vitam Horm. 2018;107:27–66. doi: 10.1016/bs.vh.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 81.Newton CL, Riekert C, Millar RP. Gonadotropin-releasing hormone analog therapeutics. Minerva Ginecol. 2018;70:497–515. doi: 10.23736/S0026-4784.18.04316-2. [DOI] [PubMed] [Google Scholar]
- 82.Newton CL, Anderson RC, Millar RP. Therapeutic neuroendocrine agonist and antagonist analogs of hypothalamic neuropeptides as modulators of the hypothalamic-pituitary- gonadal axis. Endocr Dev. 2016;30:106–129. doi: 10.1159/000439337. [DOI] [PubMed] [Google Scholar]
- 83.Wuntakal R, Seshadri S, Montes A, Lane G. Luteinising hormone releasing hormone (LHRH) agonists for the treatment of relapsed epithelial ovarian cancer. Cochrane Database Syst Rev. 2016;2016:CD011322. doi: 10.1002/14651858.CD011322.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Limonta P, Montagnani Marelli M, Mai S, Motta M, Martini L, Moretti RM. GnRH receptors in cancer: From cell biology to novel targeted therapeutic strategies. Endocr Rev. 2012;33:784–811. doi: 10.1210/er.2012-1014. [DOI] [PubMed] [Google Scholar]
- 85.Gründker C, Emons G. The role of gonadotropin-releasing hormone in cancer cell proliferation and metastasis. Front Endocrinol (Lausanne) 2017;8:187. doi: 10.3389/fendo.2017.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Y, Lu ZY, Yan XH, Wang JJ, Feng WW, Ding JX, Hua KQ. Study on the effects of gonadotropin-releasing hormone analogues in the inhibition of ovarian cancer transplanted tumors and in the protection of ovarian function after chemotherapy on nude mice. Zhonghua Fu Chan Ke Za Zhi. 2011;46:892–897. (In Chinese) [PubMed] [Google Scholar]
- 87.Völker P, Gründker C, Schmidt O, Schulz KD, Emons G. Expression of receptors for luteinizing hormone-releasing hormone in human ovarian and endometrial cancers: Frequency, autoregulation, and correlation with direct antiproliferative activity of luteinizing hormone-releasing hormone analogues. Am J Obstet Gynecol. 2002;186:171–179. doi: 10.1067/mob.2002.119633. [DOI] [PubMed] [Google Scholar]
- 88.Gründker C, Völker P, Emons G. Antiproliferative signaling of luteinizing hormone-releasing hormone in human endometrial and ovarian cancer cells through G protein alpha(I)-mediated activation of phosphotyrosine phosphatase. Endocrinology. 2001;142:2369–2380. doi: 10.1210/endo.142.6.8190. [DOI] [PubMed] [Google Scholar]
- 89.Gründker C, Völker P, Schulz KD, Emons G. Luteinizing hormone-releasing hormone agonist triptorelin and antagonist cetrorelix inhibit EGF-induced c-fos expression in human gynecological cancers. Gynecol Oncol. 2000;78:194–202. doi: 10.1006/gyno.2000.5863. [DOI] [PubMed] [Google Scholar]
- 90.Engel JB, Hahne JC, Häusler SF, Meyer S, Segerer SE, Diessner J, Dietl J, Honig A. Peptidomimetic GnRH antagonist AEZS-115 inhibits the growth of ovarian and endometrial cancer cells. Anticancer Res. 2012;32:2063–2068. [PubMed] [Google Scholar]
- 91.Günthert AR, Gründker C, Hollmann K, Emons G. Luteinizing hormone-releasing hormone induces JunD-DNA binding and extends cell cycle in human ovarian cancer cells. Biochem Biophys Res Commun. 2002;294:11–15. doi: 10.1016/S0006-291X(02)00427-8. [DOI] [PubMed] [Google Scholar]
- 92.Imai A, Takagi A, Horibe S, Takagi H, Tamaya T. Fas and Fas ligand system may mediate antiproliferative activity of gonadotropin-releasing hormone receptor in endometrial cancer cells. Int J Oncol. 1998;13:97–100. doi: 10.3892/ijo.13.1.97. [DOI] [PubMed] [Google Scholar]
- 93.Imai A, Takagi A, Horibe S, Takagi H, Tamaya T. Evidence for tight coupling of gonadotropin-releasing hormone receptor to stimulated Fas ligand expression in reproductive tract tumors: Possible mechanism for hormonal control of apoptotic cell death. J Clin Endocrinol Metab. 1998;83:427–431. doi: 10.1210/jc.83.2.427. [DOI] [PubMed] [Google Scholar]
- 94.Meyer C, Sims AH, Morgan K, Harrison B, Muir M, Bai J, Faratian D, Millar RP, Langdon SP. Transcript and protein profiling identifies signaling, growth arrest, apoptosis, and NF-κB survival signatures following GNRH receptor activation. Endocr Relat Cancer. 2013;20:123–136. doi: 10.1530/ERC-12-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mo Y, Peng P, Zhou R, He Z, Huang L, Yang D. Regulation of gonadotropin-releasing hormone (GnRH) receptor-I expression in the pituitary and ovary by a GnRH agonist and antagonist. Reprod Sci. 2010;17:68–77. doi: 10.1177/1933719109348026. [DOI] [PubMed] [Google Scholar]
- 96.Gründker C, Schulz K, Günthert AR, Emons G. Luteinizing hormone-releasing hormone induces nuclear factor kappaB-activation and inhibits apoptosis in ovarian cancer cells. J Clin Endocrinol Metab. 2000;85:3815–3820. doi: 10.1210/jcem.85.10.6859. [DOI] [PubMed] [Google Scholar]
- 97.Gründker C, Günthert AR, Millar RP, Emons G. Expression of gonadotropin-releasing hormone II (GnRH-II) receptor in human endometrial and ovarian cancer cells and effects of GnRH-II on tumor cell proliferation. J Clin Endocrinol Metab. 2002;87:1427–1430. doi: 10.1210/jcem.87.3.8437. [DOI] [PubMed] [Google Scholar]
- 98.Fister S, Günthert AR, Aicher B, Paulini KW, Emons G, Gründker C. GnRH-II antagonists induce apoptosis in human endometrial, ovarian, and breast cancer cells via activation of stress-induced MAPKs p38 and JNK and proapoptotic protein Bax. Cancer Res. 2009;69:6473–6481. doi: 10.1158/0008-5472.CAN-08-4657. [DOI] [PubMed] [Google Scholar]
- 99.Morgan K, Conklin D, Pawson AJ, Sellar R, Ott TR, Millar RP. A transcriptionally active human type II gonadotropin-releasing hormone receptor gene homolog overlaps two genes in the antisense orientation on chromosome 1q.12. Endocrinology. 2003;144:423–436. doi: 10.1210/en.2002-220622. [DOI] [PubMed] [Google Scholar]
- 100.Neill JD, Musgrove LC, Duck LW. Newly recognized GnRH receptors: Function and relative role. Trends Endocrinol Metab. 2004;15:383–392. doi: 10.1016/j.tem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 101.Gründker C, Huschmand Nia A, Emons G. Gonadotropin- releasing hormone receptor-targeted gene therapy of gynecologic cancers. Mol Cancer Ther. 2005;4:225–231. [PubMed] [Google Scholar]
- 102.Parmar H, Phillips RH, Rustin G, Lightman SL, Schally AV. Therapy of advanced ovarian cancer with D-Trp-6-LH-RH (decapeptyl) microcapsules. Biomed Pharmacother. 1988;42:531–538. [PubMed] [Google Scholar]
- 103.Jäger W, Wildt L, Lang N. Some observations on the effect of a GnRH analog in ovarian cancer. Eur J Obstet Gynecol Reprod Biol. 1989;32:137–148. doi: 10.1016/0028-2243(89)90195-0. [DOI] [PubMed] [Google Scholar]
- 104.Carnino F, Iskra L, Fuda G, Foglia G, Odicino F, Bruzzone M, Chiara S, Gadducci A, Ragni N. The treatment of progressive ovarian carcinoma with D-Trp-LHRH (Decapeptyl). Gruppo Oncologico Nord ovest (GONO) Eur J Cancer. 1994;30A:1903–1904. doi: 10.1016/0959-8049(94)00272-7. [DOI] [PubMed] [Google Scholar]
- 105.Ron IG, Wigler N, Merimsky O, Inbar MJ, Chaitchik S. A phase II trial of D-Trp-6-LHRH (decapeptyl) in pretreated patients with advanced epithelial ovarian cancer. Cancer Invest. 1995;13:272–275. doi: 10.3109/07357909509094461. [DOI] [PubMed] [Google Scholar]
- 106.Duffaud F, van der Burg ME, Namer M, Vergote I, Willemse PH, ten Bokkel Huinink W, Guastalla JP, Nooij, Kerbrat P, Piccart M, et al. D-TRP-6-LHRH (Triptorelin) is not effective in ovarian carcinoma: An EORTC gynaecological cancer Co-operative group study. Anticancer Drugs. 2001;12:159–162. doi: 10.1097/00001813-200102000-00010. [DOI] [PubMed] [Google Scholar]
- 107.Emons G, Ortmann O, Teichert HM, Fassl H, Löhrs U, Kullander S, Kauppila A, Ayalon D, Schally A, Oberheuser F. Luteinizing hormone-releasing hormone agonist triptorelin in combination with cytotoxic chemotherapy in patients with advanced ovarian carcinoma. A prospective double blind randomized trial. Decapeptyl Ovarian Cancer Study Group. Cancer. 1996;78:1452–1460. doi: 10.1002/(SICI)1097-0142(19961001)78:7<1452::AID-CNCR12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 108.Jager W, Sauerbrei W, Beck E, Maassen V, Stumpfe M, Meier W, Kuhn W, Janicke F. A randomized comparison of triptorelin and tamoxifen as treatment of progressive ovarian cancer. Anticancer Res. 1995;15:2639–2642. [PubMed] [Google Scholar]
- 109.Sevelda P, Vavra N, Fitz R, Barrada M, Salzer H, Baur M, Dittrich C. Goserelin a GnRH-analogue as third-line therapy of refractory epithelial ovarian cancer. Int J Gynecol Cancer. 1992;2:160–162. doi: 10.1046/j.1525-1438.1992.02030160.x. [DOI] [PubMed] [Google Scholar]
- 110.Lind MJ, Cantwell BM, Millward MJ, Robinson A, Proctor M, Simmons D, Carmichael J, Harris AL. A phase II trial of goserelin (Zoladex) in relapsed epithelial ovarian cancer. Br J Cancer. 1992;65:621–623. doi: 10.1038/bjc.1992.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hasan J, Ton N, Mullamitha S, Clamp A, McNeilly A, Marshall E, Jayson GC. Phase II trial of tamoxifen and goserelin in recurrent epithelial ovarian cancer. Br J Cancer. 2005;93:647–651. doi: 10.1038/sj.bjc.6602752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marinaccio M, D'Addario V, Serratì A, Pinto V, Cagnazzo G. Leuprolide acetate as a salvage-therapy in relapsed epithelial ovarian cancer. Eur J Gynaecol Oncol. 1996;17:286–288. [PubMed] [Google Scholar]
- 113.Paskeviciute L, Roed H, Engelholm S. No rules without exception: Long-term complete remission observed in a study using a LH-RH agonist in platinum-refractory ovarian cancer. Gynecol Oncol. 2002;86:297–301. doi: 10.1006/gyno.2002.6778. [DOI] [PubMed] [Google Scholar]
- 114.du BA, Meier W, Lück HJ, Emon G, Moebus V, Schroeder W, Costa S, Bauknecht T, Olbricht S, Jackisch C, et al. Chemotherapy versus hormonal treatment in platinum- and Paclitaxel-refractory ovarian cancer: A randomised trial of the german arbeitsgemeinschaft gynaekologische onkologie (AGO) study group ovarian cancer. Ann Oncol. 2002;13:251–257. doi: 10.1093/annonc/mdf038. [DOI] [PubMed] [Google Scholar]
- 115.Balbi G, Piano LD, Cardone A, Cirelli G. Second-line therapy of advanced ovarian cancer with GnRH analogs. Int J Gynecol Cancer. 2004;14:799–803. doi: 10.1111/j.1048-891X.2004.014511.x. [DOI] [PubMed] [Google Scholar]
- 116.Kavanagh JJ, Roberts W, Townsend P, Hewitt S. Leuprolide acetate in the treatment of refractory or persistent epithelial ovarian cancer. J Clin Oncol. 1989;7:115–118. doi: 10.1200/JCO.1989.7.1.115. [DOI] [PubMed] [Google Scholar]
- 117.Bruckner HW, Motwani BT. Treatment of advanced refractory ovarian carcinoma with a gonadotropin-releasing hormone analogue. Am J Obstet Gynecol. 1989;161:1216–1218. doi: 10.1016/0002-9378(89)90669-8. [DOI] [PubMed] [Google Scholar]
- 118.Miller DS, Brady MF, Barrett RJ. A phase II trial of leuprolide acetate in patients with advanced epithelial ovarian carcinoma. A Gynecologic Oncology Group study. Am J Clin Oncol. 1992;15:125–128. doi: 10.1097/00000421-199204000-00006. [DOI] [PubMed] [Google Scholar]
- 119.Verschraegen CF, Westphalen S, Hu W, Loyer E, Kudelka A, Völker P, Kavanagh J, Steger M, Schulz KD, Emons G. Phase II study of cetrorelix, a luteinizing hormone-releasing hormone antagonist in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2003;90:552–559. doi: 10.1016/S0090-8258(03)00408-6. [DOI] [PubMed] [Google Scholar]
- 120.Parmar H, Rustin G, Lightman SL, Phillips RH, Hanham IW, Schally AV. Response to D-Trp-6-luteinising hormone releasing hormone (Decapeptyl) microcapsules in advanced ovarian cancer. Br Med J (Clin Res Ed) 1988;296:1229. doi: 10.1136/bmj.296.6631.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Emons G, Gorchev G, Sehouli J, Wimberger P, Stähle A, Hanker L, Hilpert F, Sindermann H, Gründker C, Harter P. Efficacy and safety of AEZS-108 (INN: Zoptarelin doxorubicin acetate) an LHRH agonist linked to doxorubicin in women with platinum refractory or resistant ovarian cancer expressing LHRH receptors: A multicenter phase II trial of the ago-study group (AGO GYN 5) Gynecol Oncol. 2014;133:427–432. doi: 10.1016/j.ygyno.2014.03.576. [DOI] [PubMed] [Google Scholar]
- 122.Medl M, Peters-Engel C, Fuchs G, Leodolter S. Triptorelin (D-Trp-6-LHRH) in combination with carboplatin-containing polychemotherapy for advanced ovarian cancer: A pilot study. Anticancer Res. 1993;13:2373–2376. [PubMed] [Google Scholar]
- 123.Falkson CI, Falkson HC, Falkson G. Cisplatin versus cisplatin plus D-Trp-6-LHRH in the treatment of ovarian cancer: A pilot trial to investigate the effect of the addition of a GnRH analogue to cisplatin. Oncology. 1996;53:313–317. doi: 10.1159/000227579. [DOI] [PubMed] [Google Scholar]
- 124.Rzepka-Górska I, Chudecka-Glaz A, Kosmider M, Malecha J. GnRH analogues as an adjuvant therapy for ovarian cancer patients. Int J Gynaecol Obstet. 2003;81:199–205. doi: 10.1016/S0020-7292(03)00029-8. [DOI] [PubMed] [Google Scholar]
- 125.Mizushima T, Tirador KA, Miyamoto H. Androgen receptor activation: A prospective therapeutic target for bladder cancer? Expert Opin Ther Targets. 2017;21:249–257. doi: 10.1080/14728222.2017.1280468. [DOI] [PubMed] [Google Scholar]
- 126.Fujii S, Kagechika H. Androgen receptor modulators: A review of recent patents and reports (2012–2018) Expert Opin Ther Pat. 2019;29:439–453. doi: 10.1080/13543776.2019.1618831. [DOI] [PubMed] [Google Scholar]
- 127.Gucalp A, Traina TA. The androgen receptor: Is it a promising target? Ann Surg Oncol. 2017;24:2876–2880. doi: 10.1245/s10434-017-5961-9. [DOI] [PubMed] [Google Scholar]
- 128.Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem. 2002;277:38087–38094. doi: 10.1074/jbc.M203313200. [DOI] [PubMed] [Google Scholar]
- 129.Pagliarulo V. Androgen deprivation therapy for prostate cancer. Adv Exp Med Biol. 2018;1096:1–30. doi: 10.1007/978-3-319-99286-0_1. [DOI] [PubMed] [Google Scholar]
- 130.Rahim B, O'Regan R. AR Signaling in breast cancer. Cancers (Basel) 2017;9:21. doi: 10.3390/cancers9030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Antonarakis ES. AR Signaling in human malignancies: Prostate cancer and beyond. Cancers (Basel) 2018;10:22. doi: 10.3390/cancers10010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hamilton TC, Davies P, Griffiths K. Androgen and oestrogen binding in cytosols of human ovarian tumours. J Endocrinol. 1981;90:421–431. doi: 10.1677/joe.0.0900421. [DOI] [PubMed] [Google Scholar]
- 133.Kühnel R, de Graaff J, Rao BR, Stolk JG. Androgen receptor predominance in human ovarian carcinoma. J Steroid Biochem. 1987;26:393–397. doi: 10.1016/0022-4731(87)90106-3. [DOI] [PubMed] [Google Scholar]
- 134.Chadha S, Rao BR, Slotman BJ, van Vroonhoven CC, van der Kwast TH. An immunohistochemical evaluation of androgen and progesterone receptors in ovarian tumors. Hum Pathol. 1993;24:90–95. doi: 10.1016/0046-8177(93)90067-Q. [DOI] [PubMed] [Google Scholar]
- 135.Sheach LA, Adeney EM, Kucukmetin A, Wilkinson SJ, Fisher AD, Elattar A, Robson CN, Edmondson RJ. Androgen-related expression of G-proteins in ovarian cancer. Br J Cancer. 2009;101:498–503. doi: 10.1038/sj.bjc.6605153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cardillo MR, Petrangeli E, Aliotta N, Salvatori L, Ravenna L, Chang C, Castagna G. Androgen receptors in ovarian tumors: Correlation with oestrogen and progesterone receptors in an immunohistochemical and semiquantitative image analysis study. J Exp Clin Cancer Res. 1998;17:231–237. [PubMed] [Google Scholar]
- 137.Elattar A, Warburton KG, Mukhopadhyay A, Freer RM, Shaheen F, Cross P, Plummer ER, Robson CN, Edmondson RJ. Androgen receptor expression is a biological marker for androgen sensitivity in high grade serous epithelial ovarian cancer. Gynecol Oncol. 2012;124:142–147. doi: 10.1016/j.ygyno.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 138.de Toledo MC, Sarian LO, Sallum LF, Andrade LL, Vassallo J, de Paiva Silva GR, Pinto GA, Soares FA, Fonseca CD, Derchain SF. Analysis of the contribution of immunologically-detectable HER2, steroid receptors and of the ‘triple-negative’ tumor status to disease-free and overall survival of women with epithelial ovarian cancer. Acta Histochem. 2014;116:440–447. doi: 10.1016/j.acthis.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 139.Jönsson JM, Arildsen NS, Malander S, Måsbäck A, Hartman L, Nilbert M, Hedenfalk I. Sex Steroid hormone receptor expression affects ovarian cancer survival. Transl Oncol. 2015;8:424–433. doi: 10.1016/j.tranon.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Butler MS, Ricciardelli C, Tilley WD, Hickey TE. Androgen receptor protein levels are significantly reduced in serous ovarian carcinomas compared with benign or borderline disease but are not altered by cancer stage or metastatic progression. Horm Cancer. 2013;4:154–164. doi: 10.1007/s12672-013-0135-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Edmondson RJ, Monaghan JM, Davies BR. The human ovarian surface epithelium is an androgen responsive tissue. Br J Cancer. 2002;86:879–885. doi: 10.1038/sj.bjc.6600154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Syed V, Ulinski G, Mok SC, Yiu GK, Ho SM. Expression of gonadotropin receptor and growth responses to key reproductive hormones in normal and malignant human ovarian surface epithelial cells. Cancer Res. 2001;61:6768–6776. [PubMed] [Google Scholar]
- 143.Nourbakhsh M, Golestani A, Zahrai M, Modarressi MH, Malekpour Z, Karami-Tehrani F. Androgens stimulate telomerase expression, activity and phosphorylation in ovarian adenocarcinoma cells. Mol Cell Endocrinol. 2010;330:10–16. doi: 10.1016/j.mce.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 144.Ligr M, Patwa RR, Daniels G, Pan L, Wu X, Li Y, Tian L, Wang Z, Xu R, Wu J, et al. Expression and function of androgen receptor coactivator p44/Mep50/WDR77 in ovarian cancer. PLoS One. 2011;6:e26250. doi: 10.1371/journal.pone.0026250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Silva EG, Tornos C, Fritsche HA, Jr, el-Naggar A, Gray K, Ordonez NG, Luna M, Gershenson D. The induction of benign epithelial neoplasms of the ovaries of guinea pigs by testosterone stimulation: A potential animal model. Mod Pathol. 1997;10:879–883. [PubMed] [Google Scholar]
- 146.Sawada M, Terada N, Wada A, Mori Y, Yamasaki M, Saga T, Endo K. Estrogen- and androgen-responsive growth of human ovarian adenocarcinoma heterotransplanted into nude mice. Int J Cancer. 1990;45:359–363. doi: 10.1002/ijc.2910450225. [DOI] [PubMed] [Google Scholar]
- 147.Park BY, Grisham RN, den Hollander B, Thapi D, Berman T, de Stanchina E, Zhou Q, Iyer G, Aghajanian C, Spriggs DR. Tumor Inhibition by Enzalutamide in a xenograft model of ovarian cancer. Cancer Invest. 2016;34:517–520. doi: 10.1080/07357907.2016.1242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Proverbs-Singh T, Feldman JL, Morris MJ, Autio KA, Traina TA. Targeting the androgen receptor in prostate and breast cancer: Several new agents in development. Endocr Relat Cancer. 2015;22:R87–R106. doi: 10.1530/ERC-14-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sun NK, Huang SL, Chang PY, Lu HP, Chao CC. Transcriptomic profiling of taxol-resistant ovarian cancer cells identifies FKBP5 and the androgen receptor as critical markers of chemotherapeutic response. Oncotarget. 2014;5:11939–11956. doi: 10.18632/oncotarget.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sun NK, Huang SL, Lu HP, Chang TC, Chao CC. Integrative transcriptomics-based identification of cryptic drivers of taxol-resistance genes in ovarian carcinoma cells: Analysis of the androgen receptor. Oncotarget. 2015;6:27065–27082. doi: 10.18632/oncotarget.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tumolo S, Rao BR, van der Burg ME, Guastalla JP, Renard J, Vermorken JB. Phase II trial of flutamide in advanced ovarian cancer: An EORTC Gynaecological Cancer Cooperative Group study. Eur J Cancer. 1994;30A:911–914. doi: 10.1016/0959-8049(94)90112-0. [DOI] [PubMed] [Google Scholar]
- 152.Vassilomanolakis M, Koumakis G, Barbounis V, Hajichristou H, Tsousis S, Efremidis A. A phase II study of flutamide in ovarian cancer. Oncology. 1997;54:199–202. doi: 10.1159/000227688. [DOI] [PubMed] [Google Scholar]
- 153.Gruessner C, Gruessner A, Glaser K, AbuShahin N, Zhou Y, Laughren C, Wright H, Pinkerton S, Yi X, Stoffer J, et al. Flutamide and biomarkers in women at high risk for ovarian cancer: Preclinical and clinical evidence. Cancer Prev Res (Phila) 2014;7:896–905. doi: 10.1158/1940-6207.CAPR-13-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Levine D, Park K, Juretzka M, Esch J, Hensley M, Aghajanian C, Lewin S, Konner J, Derosa F, Spriggs D, et al. A phase II evaluation of goserelin and bicalutamide in patients with ovarian cancer in second or higher complete clinical disease remission. Cancer. 2007;110:2448–2456. doi: 10.1002/cncr.23072. [DOI] [PubMed] [Google Scholar]
- 155.Grisham RN, Giri DD, Iasonos A, Zhou Q, Girshman J, McGrath SP, O'Cearbhaill RE, Sabbatini P, Tew WP, Michael Hyman D, et al. A phase II trial of enzalutamide in patients with androgen receptor positive (AR+) ovarian, primary peritoneal or fallopian tube cancer and one, two, or three prior therapies. J Clin Oncol. 2017;35(Suppl 15):TPS5610. doi: 10.1200/JCO.2017.35.15_suppl.TPS5610. [DOI] [Google Scholar]
- 156.Miyamoto H, Messing EM, Chang C. Androgen deprivation therapy for prostate cancer: Current status and future prospects. Prostate. 2004;61:332–353. doi: 10.1002/pros.20115. [DOI] [PubMed] [Google Scholar]
- 157.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135–170. doi: 10.1016/bs.apcsb.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Björnström L, Sjöberg M. Estrogen receptor-dependent activation of AP-1 via non-genomic signalling. Nucl Recept. 2004;2:3. doi: 10.1186/1478-1336-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kumar A, Foster TC. G Protein-Coupled estrogen receptor: Rapid effects on hippocampal-dependent spatial memory and synaptic plasticity. Front Endocrinol (Lausanne) 2020;11:385. doi: 10.3389/fendo.2020.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.O'Donnell AJ, Macleod KG, Burns DJ, Smyth JF, Langdon SP. Estrogen receptor-alpha mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr Relat Cancer. 2005;12:851–866. doi: 10.1677/erc.1.01039. [DOI] [PubMed] [Google Scholar]
- 162.Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, Fontaine C, Gourdy P, Chambon P, Katzenellenbogen B, Katzenellenbogen J. Membrane and nuclear estrogen receptor alpha actions: From tissue specificity to medical implications. Physiol Rev. 2017;97:1045–1087. doi: 10.1152/physrev.00024.2016. [DOI] [PubMed] [Google Scholar]
- 163.Ghasemi A, Saeidi J, Mohtashami M, Hashemy SI. Estrogen-independent role of ERα in ovarian cancer progression induced by leptin/Ob-Rb axis. Mol Cell Biochem. 2019;458:207–217. doi: 10.1007/s11010-019-03544-5. [DOI] [PubMed] [Google Scholar]
- 164.Bogush TA, Basharina AA, Bogush EA, Ryabinina OM, Tjulandina AS, Tjulandin SA. Estrogen receptors alpha and beta in ovarian cancer: Expression level and prognosis. Dokl Biochem Biophys. 2018;482:249–251. doi: 10.1134/S1607672918050058. [DOI] [PubMed] [Google Scholar]
- 165.Andersen CL, Sikora MJ, Boisen MM, Ma T, Christie A, Tseng G, Park Y, Luthra S, Chandran U, Haluska P, et al. Active estrogen receptor-alpha signaling in ovarian cancer models and clinical specimens. Clin Cancer Res. 2017;23:3802–3812. doi: 10.1158/1078-0432.CCR-16-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chan KK, Wei N, Liu SS, Xiao-Yun L, Cheung AN, Ngan HY. Estrogen receptor subtypes in ovarian cancer: A clinical correlation. Obstet Gynecol. 2008;111:144–151. doi: 10.1097/01.AOG.0000296715.07705.e9. [DOI] [PubMed] [Google Scholar]
- 167.Bossard C, Busson M, Vindrieux D, Gaudin F, Machelon V, Brigitte M, Jacquard C, Pillon A, Balaguer P, Balabanian K, Lazennec G. Potential role of estrogen receptor beta as a tumor suppressor of epithelial ovarian cancer. PLoS One. 2012;7:e44787. doi: 10.1371/journal.pone.0044787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rutherford T, Brown WD, Sapi E, Aschkenazi S, Muñoz A, Mor G. Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet Gynecol. 2000;96:417–421. doi: 10.1016/S0029-7844(00)00917-0. [DOI] [PubMed] [Google Scholar]
- 169.Hoffmann M, Gogola J, Ptak A. Apelin abrogates the stimulatory effects of 17β-estradiol and insulin-like growth factor-1 on proliferation of epithelial and granulosa ovarian cancer cell lines via crosstalk between APLNR and ERα/IGF1R. Mol Biol Rep. 2019;46:6325–6338. doi: 10.1007/s11033-019-05073-2. [DOI] [PubMed] [Google Scholar]
- 170.Liu H, Yan Y, Wen H, Jiang X, Cao X, Zhang G, Liu G. A novel estrogen receptor GPER mediates proliferation induced by 17β-estradiol and selective GPER agonist G-1 in estrogen receptor α (ERα)-negative ovarian cancer cells. Cell Biol Int. 2014;38:631–638. doi: 10.1002/cbin.10243. [DOI] [PubMed] [Google Scholar]
- 171.Chan KK, Leung TH, Chan DW, Wei N, Lau GT, Liu SS, Siu MK, Ngan HY. Targeting estrogen receptor subtypes (ERalpha and ERbeta) with selective ER modulators in ovarian cancer. J Endocrinol. 2014;221:325–336. doi: 10.1530/JOE-13-0500. [DOI] [PubMed] [Google Scholar]
- 172.Park SH, Cheung LW, Wong AS, Leung PC. Estrogen regulates Snail and Slug in the down-regulation of E-cadherin and induces metastatic potential of ovarian cancer cells through estrogen receptor alpha. Mol Endocrinol. 2008;22:2085–2098. doi: 10.1210/me.2007-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Moll F, Katsaros D, Lazennec G, Hellio N, Roger P, Giacalone PL, Chalbos D, Maudelonde T, Rochefort H, Pujol P. Estrogen induction and overexpression of fibulin-1C mRNA in ovarian cancer cells. Oncogene. 2002;21:1097–1107. doi: 10.1038/sj.onc.1205171. [DOI] [PubMed] [Google Scholar]
- 174.Ciucci A, Zannoni GF, Travaglia D, Scambia G, Gallo D. Mitochondrial estrogen receptor β2 drives antiapoptotic pathways in advanced serous ovarian cancer. Hum Pathol. 2015;46:1138–1146. doi: 10.1016/j.humpath.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 175.Kyriakidis I, Papaioannidou P. Estrogen receptor beta and ovarian cancer: A key to pathogenesis and response to therapy. Arch Gynecol Obstet. 2016;293:1161–1168. doi: 10.1007/s00404-016-4027-8. [DOI] [PubMed] [Google Scholar]
- 176.Lazennec G. Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett. 2006;231:151–157. doi: 10.1016/j.canlet.2005.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Treeck O, Pfeiler G, Mitter D, Lattrich C, Piendl G, Ortmann O. Estrogen receptor {beta}1 exerts antitumoral effects on SK-OV-3 ovarian cancer cells. J Endocrinol. 2007;193:421–433. doi: 10.1677/JOE-07-0087. [DOI] [PubMed] [Google Scholar]
- 178.Liu J, Viswanadhapalli S, Garcia L, Zhou M, Nair BC, Kost E, Rao Tekmal R, Li R, Rao MK, Curiel T, et al. Therapeutic utility of natural estrogen receptor beta agonists on ovarian cancer. Oncotarget. 2017;8:50002–50014. doi: 10.18632/oncotarget.18442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chan KKL, Siu MKY, Jiang YX, Wang JJ, Wang Y, Leung THY, Liu SS, Cheung ANY, Ngan HYS. Differential expression of estrogen receptor subtypes and variants in ovarian cancer: Effects on cell invasion, proliferation and prognosis. BMC Cancer. 2017;17:606. doi: 10.1186/s12885-017-3601-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chan KKL, Siu MKY, Jiang YX, Wang JJ, Leung THY, Ngan HYS. Estrogen receptor modulators genistein, daidzein and ERB-041 inhibit cell migration, invasion, proliferation and sphere formation via modulation of FAK and PI3K/AKT signaling in ovarian cancer. Cancer Cell Int. 2018;18:65. doi: 10.1186/s12935-018-0559-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fekete T, Rásó E, Pete I, Tegze B, Liko I, Munkácsy G, Sipos N, Rigó J, Jr, Györffy B. Meta-analysis of gene expression profiles associated with histological classification and survival in 829 ovarian cancer samples. Int J Cancer. 2012;131:95–105. doi: 10.1002/ijc.26364. [DOI] [PubMed] [Google Scholar]
- 182.Schüler-Toprak S, Weber F, Skrzypczak M, Ortmann O, Treeck O. Estrogen receptor β is associated with expression of cancer associated genes and survival in ovarian cancer. BMC Cancer. 2018;18:981. doi: 10.1186/s12885-018-4898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fujimoto J, Hirose R, Sakaguchi H, Tamaya T. Clinical significance of expression of estrogen receptor alpha and beta mRNAs in ovarian cancers. Oncology. 2000;58:334–341. doi: 10.1159/000012121. [DOI] [PubMed] [Google Scholar]
- 184.van Kruchten M, van der Marel P, de Munck L, Hollema H, Arts H, Timmer-Bosscha H, de Vries E, Hospers G, Reyners A. Hormone receptors as a marker of poor survival in epithelial ovarian cancer. Gynecol Oncol. 2015;138:634–639. doi: 10.1016/j.ygyno.2015.06.032. [DOI] [PubMed] [Google Scholar]
- 185.van der Vange N, Greggi S, Burger CW, Kenemans P, Vermorken JB. Experience with hormonal therapy in advanced epithelial ovarian cancer. Acta Oncol. 1995;34:813–820. doi: 10.3109/02841869509127191. [DOI] [PubMed] [Google Scholar]
- 186.Rolski J, Pawlicki M. Evaluation of efficacy and toxicity of tamoxifen in patients with advanced chemotherapy resistant ovarian cancer. Ginekol Pol. 1998;69:586–589. (In Polish) [PubMed] [Google Scholar]
- 187.Tropé C, Marth C, Kaern J. Tamoxifen in the treatment of recurrent ovarian carcinoma. Eur J Cancer. 2000;36(Suppl 4):S59–S61. doi: 10.1016/S0959-8049(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 188.Van Der Velden J, Gitsch G, Wain GV, Friedlander ML, Hacker NF. Tamoxifen in patients with advanced epithelial ovarian cancer. Int J Gynecol Cancer. 1995;5:301–305. doi: 10.1046/j.1525-1438.1995.05040301.x. [DOI] [PubMed] [Google Scholar]
- 189.Ahlgren JD, Ellison NM, Gottlieb RJ, Laluna F, Lokich JJ, Sinclair PR, Ueno W, Wampler GL, Yeung KY, Alt D, et al. Hormonal palliation of chemoresistant ovarian cancer: Three consecutive phase II trials of the Mid-atlantic oncology program. J Clin Oncol. 1993;11:1957–1968. doi: 10.1200/JCO.1993.11.10.1957. [DOI] [PubMed] [Google Scholar]
- 190.Jakobsen A, Bertelsen K, Sell A. Cyclic hormonal treatment in ovarian cancer. A phase-II trial. Eur J Cancer Clin Oncol. 1987;23:915–916. doi: 10.1016/0277-5379(87)90335-X. [DOI] [PubMed] [Google Scholar]
- 191.Pagel J RC, Thorpe S. Treatment of advanced ovarian carcinoma with tamoxifen: A phase II trial. Proc 2nd Eur Conf Clin Oncol. 1983 May 29; (Epub ahead of print) [Google Scholar]
- 192.Osborne RJ, Malik ST, Slevin ML, Harvey VJ, Spona J, Salzer H, Williams CJ. Tamoxifen in refractory ovarian cancer: The use of a loading dose schedule. Br J Cancer. 1988;57:115–116. doi: 10.1038/bjc.1988.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Slevin ML, Harvey VJ, Osborne RJ, Shepherd JH, Williams CJ, Mead GM. A phase II study of tamoxifen in ovarian cancer. Eur J Cancer Clin Oncol. 1986;22:309–312. doi: 10.1016/0277-5379(86)90396-2. [DOI] [PubMed] [Google Scholar]
- 194.Shirey DR, Kavanagh JJ, Jr, Gershenson DM, Freedman RS, Copeland LJ, Jones LA. Tamoxifen therapy of epithelial ovarian cancer. Obstet Gynecol. 1985;66:575–578. [PubMed] [Google Scholar]
- 195.Hatch KD, Beecham JB, Blessing JA, Creasman WT. Responsiveness of patients with advanced ovarian carcinoma to tamoxifen. A Gynecologic Oncology Group study of second-line therapy in 105 patients. Cancer. 1991;68:269–271. doi: 10.1002/1097-0142(19910715)68:2<269::AID-CNCR2820680209>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 196.Weiner SA, Alberts DS, Surwit EA, Davis J, Grosso D. Tamoxifen therapy in recurrent epithelial ovarian carcinoma. Gynecol Oncol. 1987;27:208–213. doi: 10.1016/0090-8258(87)90294-0. [DOI] [PubMed] [Google Scholar]
- 197.Schwartz PE, Keating G, MacLusky N, Naftolin F, Eisenfeld A. Tamoxifen therapy for advanced ovarian cancer. Obstet Gynecol. 1982;59:583–588. [PubMed] [Google Scholar]
- 198.George A, McLachlan J, Tunariu N, Della Pepa C, Migali C, Gore M, Kaye S, Banerjee S. The role of hormonal therapy in patients with relapsed high-grade ovarian carcinoma: A retrospective series of tamoxifen and letrozole. BMC Cancer. 2017;17:456. doi: 10.1186/s12885-017-3440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hurteau JA, Brady MF, Darcy KM, McGuire WP, Edmonds P, Pearl ML, Ivanov I, Tewari KS, Mannel RS, Zanotti K, Benbrook DM. Randomized phase III trial of tamoxifen versus thalidomide in women with biochemical-recurrent-only epithelial ovarian, fallopian tube or primary peritoneal carcinoma after a complete response to first-line platinum/taxane chemotherapy with an evaluation of serum vascular endothelial growth factor (VEGF): A Gynecologic Oncology Group Study. Gynecol Oncol. 2010;119:444–450. doi: 10.1016/j.ygyno.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 200.Schwartz PE, Chambers JT, Kohorn EI, Chambers SK, Weitzman H, Voynick IM, MacLusky N, Naftolin F. Tamoxifen in combination with cytotoxic chemotherapy in advanced epithelial ovarian cancer. A prospective randomized trial. Cancer. 1989;63:1074–1078. doi: 10.1002/1097-0142(19890315)63:6<1074::AID-CNCR2820630606>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 201.Panici PB, Greggi S, Amoroso M, Scambia G, Battaglia FA, Gebbia V, Salerno G, Paratore MP, Mancuso S. A combination of platinum and tamoxifen in advanced ovarian cancer failing platinum-based chemotherapy: Results of a Phase II study. Int J Gynecol Cancer. 2001;11:438–444. doi: 10.1046/j.1525-1438.2001.01059.x. [DOI] [PubMed] [Google Scholar]
- 202.Markman M, Webster K, Zanotti K, Peterson G, Kulp B, Belinson J. Phase 2 trial of carboplatin plus tamoxifen in platinum-resistant ovarian cancer and primary carcinoma of the peritoneum. Gynecol Oncol. 2004;94:404–408. doi: 10.1016/j.ygyno.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 203.Wagner U, du Bois A, Pfisterer J, Huober J, Loibl S, Lück HJ, Sehouli J, Gropp M, Stähle A, Schmalfeldt B, et al. Gefitinib in combination with tamoxifen in patients with ovarian cancer refractory or resistant to platinum-taxane based therapy-a phase II trial of the AGO Ovarian Cancer Study Group (AGO-OVAR 2.6) Gynecol Oncol. 2007;105:132–137. doi: 10.1016/j.ygyno.2006.10.053. [DOI] [PubMed] [Google Scholar]
- 204.Bowman A, Gabra H, Langdon SP, Lessells A, Stewart M, Young A, Smyth JF. CA125 response is associated with estrogen receptor expression in a phase II trial of letrozole in ovarian cancer: Identification of an endocrine-sensitive subgroup. Clin Cancer Res. 2002;8:2233–2239. [PubMed] [Google Scholar]
- 205.Colon-Otero G, Weroha SJ, Foster NR, Haluska P, Hou X, Wahner-Hendrickson AE, Jatoi A, Block MS, Dinh TA, Robertson MW, Copland JA. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol Oncol. 2017;146:64–68. doi: 10.1016/j.ygyno.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 206.del Carmen MG, Fuller AF, Matulonis U, Horick NK, Goodman A, Duska LR, Penson R, Campos S, Roche M, Seiden MV. Phase II trial of anastrozole in women with asymptomatic müllerian cancer. Gynecol Oncol. 2003;91:596–602. doi: 10.1016/j.ygyno.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 207.Kavanagh JJ, Hu W, Fu S, Deavers M, Moore C, Coleman RL, Levenback CF, Shen D, Zheng HG, YF Y, et al. Anti-tumor activity of letrozole in patients with recurrent advanced low malignant potential or low-grade serous ovarian tumors. J Clin Oncol. 2007;25(Suppl 18):S5582. doi: 10.1200/jco.2007.25.18_suppl.5582. [DOI] [Google Scholar]
- 208.Krasner CN, Debernardo RL, Findley M, Penson R, Matulonis U, Atkinson T, Roche M, Seiden MV. Phase II trial of anastrazole in combination with gefitinib in women with asymptomatic mullerian cancer. J Clin Oncol. 2005;23(Suppl 16):S5063. doi: 10.1200/jco.2005.23.16_suppl.5063. [DOI] [Google Scholar]
- 209.Papadimitriou CA, Markaki S, Siapkaras J, Vlachos G, Efstathiou E, Grimani I, Hamilos G, Zorzou M, Dimopoulos MA. Hormonal therapy with letrozole for relapsed epithelial ovarian cancer. Long-term results of a phase II study. Oncology. 2004;66:112–117. doi: 10.1159/000077436. [DOI] [PubMed] [Google Scholar]
- 210.Ramirez PT, Schmeler KM, Milam MR, Slomovitz BM, Smith JA, Kavanagh JJ, Deavers M, Levenback C, Coleman RL, Gershenson DM. Efficacy of letrozole in the treatment of recurrent platinum- and taxane-resistant high-grade cancer of the ovary or peritoneum. Gynecol Oncol. 2008;110:56–59. doi: 10.1016/j.ygyno.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 211.Smyth JF, Gourley C, Walker G, MacKean MJ, Stevenson A, Williams AR, Nafussi AA, Rye T, Rye R, Stewart M, et al. Antiestrogen therapy is active in selected ovarian cancer cases: The use of letrozole in estrogen receptor-positive patients. Clin Cancer Res. 2007;13:3617–3622. doi: 10.1158/1078-0432.CCR-06-2878. [DOI] [PubMed] [Google Scholar]
- 212.Tchekmedyian NS, Liem AK, Quan ET, Burtzo DM, Ucar K. Aromatase inhibitor therapy for estrogen receptor positive ovarian cancer. J Clin Oncol. 2006;24(Suppl 18):S15038. doi: 10.1200/jco.2006.24.18_suppl.15038. [DOI] [Google Scholar]
- 213.Verma S, Alhayki M, Le T, Baines K, Rambout L, Hopkins L, Fung Kee Fung M. Phase II study of exemestane (E) in refractory ovarian cancer (ROC) J Clin Oncol. 2006;24(Suppl 18):S5026. doi: 10.1200/jco.2006.24.18_suppl.5026. [DOI] [Google Scholar]
- 214.Williams C, Simera I, Bryant A. Tamoxifen for relapse of ovarian cancer. Cochrane Database Syst Rev. 2010;2010:CD001034. doi: 10.1002/14651858.CD001034.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Perez-Gracia JL, Carrasco EM. Tamoxifen therapy for ovarian cancer in the adjuvant and advanced settings: Systematic review of the literature and implications for future research. Gynecol Oncol. 2002;84:201–209. doi: 10.1006/gyno.2001.6489. [DOI] [PubMed] [Google Scholar]
- 216.Paleari L, Gandini S, Provinciali N, Puntoni M, Colombo N, DeCensi A. Clinical benefit and risk of death with endocrine therapy in ovarian cancer: A comprehensive review and meta-analysis. Gynecol Oncol. 2017;146:504–513. doi: 10.1016/j.ygyno.2017.06.036. [DOI] [PubMed] [Google Scholar]
- 217.Kok PS, Beale P, O'Connell RL, Grant P, Bonaventura T, Scurry J, Antill Y, Goh J, Sjoquist K, DeFazio A, et al. PARAGON (ANZGOG-0903): A phase 2 study of anastrozole in asymptomatic patients with estrogen and progesterone receptor-positive recurrent ovarian cancer and CA125 progression. J Gynecol Oncol. 2019;30:e86. doi: 10.3802/jgo.2019.30.e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Stanley B, Hollis RL, Nunes H, Towler JD, Yan X, Rye T, Dawson C, Mackean MJ, Nussey F, Churchman M, et al. Endocrine treatment of high grade serous ovarian carcinoma; quantification of efficacy and identification of response predictors. Gynecol Oncol. 2019;152:278–285. doi: 10.1016/j.ygyno.2018.11.030. [DOI] [PubMed] [Google Scholar]
- 219.Gershenson DM, Bodurka DC, Coleman RL, Lu KH, Malpica A, Sun CC. Hormonal maintenance therapy for women with low-grade serous cancer of the ovary or peritoneum. J Clin Oncol. 2017;35:1103–1111. doi: 10.1200/JCO.2016.71.0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fader AN, Bergstrom J, Jernigan A, Tanner EJ, III, Roche KL, Stone RL, Levinson KL, Ricci S, Wethingon S, Wang TL, et al. Primary cytoreductive surgery and adjuvant hormonal monotherapy in women with advanced low-grade serous ovarian carcinoma: Reducing overtreatment without compromising survival? Gynecol Oncol. 2017;147:85–91. doi: 10.1016/j.ygyno.2017.07.127. [DOI] [PubMed] [Google Scholar]
- 221.Slomovitz B, Gourley C, Carey MS, Malpica A, Shih IM, Huntsman D, Fader AN, Grisham RN, Schlumbrecht M, Sun CC, et al. Low-grade serous ovarian cancer: State of the science. Gynecol Oncol. 2020;156:715–725. doi: 10.1016/j.ygyno.2019.12.033. [DOI] [PubMed] [Google Scholar]
- 222.Yang A, Curtin J, Muggia F. Ovarian adult-type granulosa cell tumor: Focusing on endocrine-based therapies. Int J Endocrine Oncol. 2018:5. doi: 10.2217/ije-2017-0021. [Google Scholar]
- 223.van Meurs HS, van Lonkhuijzen LR, Limpens J, van der Velden J, Buist MR. Hormone therapy in ovarian granulosa cell tumors: A systematic review. Gynecol Oncol. 2014;134:196–205. doi: 10.1016/j.ygyno.2014.03.573. [DOI] [PubMed] [Google Scholar]
- 224.Graham JD, Clarke CL. Physiological action of progesterone in target tissues. Endocr Rev. 1997;18:502–519. doi: 10.1210/er.18.4.502. [DOI] [PubMed] [Google Scholar]
- 225.Diep CH, Daniel AR, Mauro LJ, Knutson TP, Lange CA. Progesterone action in breast, uterine, and ovarian cancers. J Mol Endocrinol. 2015;54:R31–R53. doi: 10.1530/JME-14-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Mesiano S, Wang Y, Norwitz ER. Progesterone receptors in the human pregnancy uterus: Do they hold the key to birth timing? Reprod Sci. 2011;18:6–19. doi: 10.1177/1933719110382922. [DOI] [PubMed] [Google Scholar]
- 227.Conneely OM, Mulac-Jericevic B, Lydon JP. Progesterone- dependent regulation of female reproductive activity by two distinct progesterone receptor isoforms. Steroids. 2003;68:771–778. doi: 10.1016/S0039-128X(03)00126-0. [DOI] [PubMed] [Google Scholar]
- 228.Diep CH, Charles NJ, Gilks CB, Kalloger SE, Argenta PA, Lange CA. Progesterone receptors induce FOXO1-dependent senescence in ovarian cancer cells. Cell Cycle. 2013;12:1433–1449. doi: 10.4161/cc.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Akahira J, Inoue T, Suzuki T, Ito K, Konno R, Sato S, Moriya T, Okamura K, Yajima A, Sasano H. Progesterone receptor isoforms A and B in human epithelial ovarian carcinoma: Immunohistochemical and RT-PCR studies. Br J Cancer. 2000;83:1488–1494. doi: 10.1054/bjoc.2000.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Akahira J, Suzuki T, Ito K, Kaneko C, Darnel AD, Moriya T, Okamura K, Yaegashi N, Sasano H. Differential expression of progesterone receptor isoforms A and B in the normal ovary, and in benign, borderline, and malignant ovarian tumors. Jpn J Cancer Res. 2002;93:807–815. doi: 10.1111/j.1349-7006.2002.tb01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Lenhard M, Tereza L, Heublein S, Ditsch N, Himsl I, Mayr D, Friese K, Jeschke U. Steroid hormone receptor expression in ovarian cancer: Progesterone receptor B as prognostic marker for patient survival. BMC Cancer. 2012;12:553. doi: 10.1186/1471-2407-12-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Havrilesky LJ, Moorman PG, Lowery WJ, Gierisch JM, Coeytaux RR, Urrutia RP, Dinan M, McBroom AJ, Hasselblad V, Sanders GD, Myers ER. Oral contraceptive pills as primary prevention for ovarian cancer: A systematic review and meta-analysis. Obstet Gynecol. 2013;122:139–147. doi: 10.1097/AOG.0b013e318291c235. [DOI] [PubMed] [Google Scholar]
- 233.Troisi R, Bjørge T, Gissler M, Grotmol T, Kitahara CM, Myrtveit Saether SM, Sköld C, Sørensen HT, Trabert B, Glimelius I. The role of pregnancy, perinatal factors and hormones in maternal cancer risk: A review of the evidence. J Intern Med. 2018;283:430–445. doi: 10.1111/joim.12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Han KH, Kim MK, Kim HS, Chung HH, Song YS. Protective effect of progesterone during pregnancy against ovarian cancer. J Cancer Prev. 2013;18:113–122. doi: 10.15430/JCP.2013.18.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Eisenhofer G, Peitzsch M, Kaden D, Langton K, Pamporaki C, Masjkur J, Tsatsaronis G, Mangelis A, Williams TA, Reincke M, et al. Reference intervals for plasma concentrations of adrenal steroids measured by LC-MS/MS: Impact of gender, age, oral contraceptives, body mass index and blood pressure status. Clin Chim Acta. 2017;470:115–124. doi: 10.1016/j.cca.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ji J, Försti A, Sundquist J, Hemminki K. Risks of breast, endometrial, and ovarian cancers after twin births. Endocr Relat Cancer. 2007;14:703–711. doi: 10.1677/ERC-07-0088. [DOI] [PubMed] [Google Scholar]
- 237.Peluso JJ. Progesterone signaling mediated through progesterone receptor membrane component-1 in ovarian cells with special emphasis on ovarian cancer. Steroids. 2011;76:903–909. doi: 10.1016/j.steroids.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wu NY, Huang HS, Chao TH, Chou HM, Fang C, Qin CZ, Lin CY, Chu TY, Zhou HH. Progesterone prevents high-grade serous ovarian cancer by inducing necroptosis of p53-defective fallopian tube epithelial cells. Cell Rep. 2017;18:2557–2565. doi: 10.1016/j.celrep.2017.02.049. [DOI] [PubMed] [Google Scholar]
- 239.Pelegrina LT, de Los Ángeles Sanhueza M, Ramona Cáceres AR, Cuello-Carrión D, Rodriguez CE, Laconi MR. Effect of progesterone and first evidence about allopregnanolone action on the progression of epithelial human ovarian cancer cell lines. J Steroid Biochem Mol Biol. 2020;196:105492. doi: 10.1016/j.jsbmb.2019.105492. [DOI] [PubMed] [Google Scholar]
- 240.McGlorthan L, Paucarmayta A, Casablanca Y, Maxwell GL, Syed V. Progesterone induces apoptosis by activation of caspase-8 and calcitriol via activation of caspase-9 pathways in ovarian and endometrial cancer cells in vitro. Apoptosis. 2021;26:184–194. doi: 10.1007/s10495-021-01657-1. [DOI] [PubMed] [Google Scholar]
- 241.Xie YL, Yang YJ, Tang C, Sheng HJ, Jiang Y, Han K, Ding LJ. Estrogen combined with progesterone decreases cell proliferation and inhibits the expression of Bcl-2 via microRNA let-7a and miR-34b in ovarian cancer cells. Clin Transl Oncol. 2014;16:898–905. doi: 10.1007/s12094-014-1166-x. [DOI] [PubMed] [Google Scholar]
- 242.Lee JY, Shin JY, Kim HS, Heo JI, Kho YJ, Kang HJ, Park SH, Lee JY. Effect of combined treatment with progesterone and tamoxifen on the growth and apoptosis of human ovarian cancer cells. Oncol Rep. 2012;27:87–93. doi: 10.3892/or.2011.1460. [DOI] [PubMed] [Google Scholar]
- 243.Shin YY, Kang EJ, Jeong JS, Kim MJ, Jung EM, Jeung EB, An BS. Pregnenolone as a potential candidate for hormone therapy for female reproductive disorders targeting ERβ. Mol Reprod Dev. 2019;86:109–117. doi: 10.1002/mrd.23086. [DOI] [PubMed] [Google Scholar]
- 244.Pedernera E, Gómora MJ, Morales-Vásquez F, Pérez-Montiel D, Mendez C. Progesterone reduces cell survival in primary cultures of endometrioid ovarian cancer. J Ovarian Res. 2019;12:15. doi: 10.1186/s13048-019-0486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Peluso JJ, Liu X, Saunders MM, Claffey KP, Phoenix K. Regulation of ovarian cancer cell viability and sensitivity to cisplatin by progesterone receptor membrane component-1. J Clin Endocrinol Metab. 2008;93:1592–1599. doi: 10.1210/jc.2007-2771. [DOI] [PubMed] [Google Scholar]
- 246.Gamarra-Luques CD, Goyeneche AA, Hapon MB, Telleria CM. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer. 2012;12:200. doi: 10.1186/1471-2407-12-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gamarra-Luques CD, Hapon MB, Goyeneche AA, Telleria CM. Resistance to cisplatin and paclitaxel does not affect the sensitivity of human ovarian cancer cells to antiprogestin-induced cytotoxicity. J Ovarian Res. 2014;7:45. doi: 10.1186/1757-2215-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Telleria CM. Repopulation of ovarian cancer cells after chemotherapy. Cancer Growth Metastasis. 2013;6:15–21. doi: 10.4137/CGM.S11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Mangioni C, Franceschi S, Vecchia CL, D'Incalci M. High-dose medroxyprogesterone acetate (MPA) in advanced epithelial ovarian cancer resistant to first- or second-line chemotherapy. Gynecol Oncol. 1981;12:314–318. doi: 10.1016/0090-8258(81)90131-1. [DOI] [PubMed] [Google Scholar]
- 250.Slayton RE, Pagano M, Creech RH. Progestin therapy for advanced ovarian cancer: A phase II Eastern Cooperative Oncology Group trial. Cancer Treat Rep. 1981;65:895–896. [PubMed] [Google Scholar]
- 251.Aabo K, Pedersen AG, Haid I, Dombernowsky P. High-dose medroxyprogesterone acetate (MPA) in advanced chemotherapy-resistant ovarian carcinoma: A phase II study. Cancer Treat Rep. 1982;66:407–408. [PubMed] [Google Scholar]
- 252.Tropé C, Johnsson JE, Sigurdsson K, Simonsen E. High-dose medroxyprogesterone acetate for the treatment of advanced ovarian carcinoma. Cancer Treat Rep. 1982;66:1441–1443. [PubMed] [Google Scholar]
- 253.Hamerlynck JV, Maskens AP, Mangioni C, van der Burg ME, Wils JA, Vermorken JB, Rotmensz N. Phase II trial of medroxyprogesterone acetate in advanced ovarian cancer: An EORTC Gynecological Cancer Cooperative Group Study. Gynecol Oncol. 1985;22:313–316. doi: 10.1016/0090-8258(85)90045-9. [DOI] [PubMed] [Google Scholar]
- 254.Malfetano J, Beecham JB, Bundy BN, Hatch KD. A phase II trial of medroxyprogesterone acetate in epithelial ovarian cancers. A Gynecologic Oncology Group study. Am J Clin Oncol. 1993;16:149–151. doi: 10.1097/00000421-199304000-00014. [DOI] [PubMed] [Google Scholar]
- 255.Geisler HE. The use of high-dose megestrol acetate in the treatment of ovarian adenocarcinoma. Semin Oncol. 1985;12(Suppl 1):S20–S22. [PubMed] [Google Scholar]
- 256.Sikic BI, Scudder SA, Ballon SC, Soriero OM, Christman JE, Suey L, Ehsan MN, Brandt AE, Evans TL. High-dose megestrol acetate therapy of ovarian carcinoma: A phase II study by the Northern California Oncology Group. Semin Oncol. 1986;13(Suppl 4):S26–S32. [PubMed] [Google Scholar]
- 257.Belinson JL, McClure M, Badger G. Randomized trial of megestrol acetate vs. megestrol acetate/tamoxifen for the management of progressive or recurrent epithelial ovarian carcinoma. Gynecol Oncol. 1987;28:151–155. doi: 10.1016/0090-8258(87)90208-3. [DOI] [PubMed] [Google Scholar]
- 258.Veenhof CH, van der Burg ME, Nooy M, Aalders JG, Pecorelli S, Oliveira CF, Rotmensz N, Vermorken JB. Phase II study of high-dose megestrol acetate in patients with advanced ovarian carcinoma. Eur J Cancer. 1994;30A:697–698. doi: 10.1016/0959-8049(94)90548-7. [DOI] [PubMed] [Google Scholar]
- 259.Wiernik PH, Greenwald ES, Ball H, Young JA, Vogl S. High-dose megestrol acetate in the treatment of patients with ovarian cancer who have undergone previous treatment: Eastern Cooperative Oncology Group Study PD884. Am J Clin Oncol. 1998;21:565–567. doi: 10.1097/00000421-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 260.Wilailak S, Linasmita V, Srisupundit S. Phase II study of high-dose megestrol acetate in platinum-refractory epithelial ovarian cancer. Anticancer Drugs. 2001;12:719–724. doi: 10.1097/00001813-200110000-00002. [DOI] [PubMed] [Google Scholar]
- 261.Rocereto TF, Saul HM, Aikins JA, Jr, Paulson J. Phase II study of mifepristone (RU486) in refractory ovarian cancer. Gynecol Oncol. 2000;77:429–432. doi: 10.1006/gyno.2000.5789. [DOI] [PubMed] [Google Scholar]
- 262.Rocereto TF, Brady WE, Shahin MS, Hoffman JS, Small L, Rotmensch J, Mannel RS. A phase II evaluation of mifepristone in the treatment of recurrent or persistent epithelial ovarian, fallopian or primary peritoneal cancer: A gynecologic oncology group study. Gynecol Oncol. 2010;116:332–334. doi: 10.1016/j.ygyno.2009.10.071. [DOI] [PubMed] [Google Scholar]
- 263.Niwa K, Onogi K, Wu Y, Mori H, Harrigan RC, Tamaya T. Clinical implication of medroxyprogesterone acetate against advanced ovarian carcinoma: A pilot study. Eur J Gynaecol Oncol. 2008;29:252–255. [PubMed] [Google Scholar]
- 264.Zheng H, Kavanagh JJ, Hu W, Liao Q, Fu S. Hormonal therapy in ovarian cancer. Int J Gynecol Cancer. 2007;17:325–338. doi: 10.1111/j.1525-1438.2006.00749.x. [DOI] [PubMed] [Google Scholar]
- 265.Laurelli G, Di Vagno G, Scaffa C, Losito S, Del Giudice M, Greggi S. Conservative treatment of early endometrial cancer: Preliminary results of a pilot study. Gynecol Oncol. 2011;120:43–46. doi: 10.1016/j.ygyno.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 266.Bonaventura A, O'Connell RL, Mapagu C, Beale PJ, McNally OM, Mileshkin LR, Grant PT, Hadley AM, Goh JCH, Sjoquist KM, et al. Paragon (ANZGOG-0903): Phase 2 study of anastrozole in women with estrogen or progesterone receptor-positive platinum-resistant or -refractory recurrent ovarian cancer. Int J Gynecol Cancer. 2017;27:900–906. doi: 10.1097/IGC.0000000000000978. [DOI] [PubMed] [Google Scholar]
- 267.Freedman RS, Saul PB, Edwards CL, Jolles CJ, Gershenson DM, Jones LA, Atkinson EN, Dana WJ. Ethinyl estradiol and medroxyprogesterone acetate in patients with epithelial ovarian carcinoma: A phase II study. Cancer Treat Rep. 1986;70:369–373. [PubMed] [Google Scholar]
- 268.Greiser CM, Greiser EM, Dören M. Menopausal hormone therapy and risk of ovarian cancer: Systematic review and meta-analysis. Hum Reprod Update. 2007;13:453–463. doi: 10.1093/humupd/dmm012. [DOI] [PubMed] [Google Scholar]
- 269.Beral V, Bull D, Green J, Reeves G. Ovarian cancer and hormone replacement therapy in the Million Women Study. Lancet. 2007;369:1703–1710. doi: 10.1016/S0140-6736(07)60534-0. [DOI] [PubMed] [Google Scholar]
- 270.Koskela-Niska V, Lyytinen H, Riska A, Pukkala E, Ylikorkala O. Ovarian cancer risk in postmenopausal women using estradiol-progestin therapy-a nationwide study. Climacteric. 2013;16:48–53. doi: 10.3109/13697137.2012.663818. [DOI] [PubMed] [Google Scholar]
- 271.Trabert B, Wentzensen N, Yang HP, Sherman ME, Hollenbeck A, Danforth KN, Park Y, Brinton LA. Ovarian cancer and menopausal hormone therapy in the NIH-AARP diet and health study. Br J Cancer. 2012;107:1181–1187. doi: 10.1038/bjc.2012.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Morch LS, Lokkegaard E, Andreasen AH, Kjaer SK, Lidegaard O. Hormone therapy and different ovarian cancers: A national cohort study. Am J Epidemiol. 2012;175:1234–1242. doi: 10.1093/aje/kwr446. [DOI] [PubMed] [Google Scholar]
- 273.Liu Y, Ma L, Yang X, Bie J, Li D, Sun C, Zhang J, Meng Y, Lin J. Menopausal hormone replacement therapy and the risk of ovarian cancer: A meta-analysis. Front Endocrinol (Lausanne) 2019;10:801. doi: 10.3389/fendo.2019.00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Beral V, Gaitskell K, Hermon C, Moser K, Reeves G, Peto R. Menopausal hormone use and ovarian cancer risk: Individual participant meta-analysis of 52 epidemiological studies. Lancet. 2015;385:1835–1842. doi: 10.1016/S0140-6736(14)61687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wen Y, Huang H, Huang H, Wu M, Shen K, Pan L. The safety of postoperative hormone replacement therapy in epithelial ovarian cancer patients in China. Climacteric. 2013;16:673–681. doi: 10.3109/13697137.2013.806472. [DOI] [PubMed] [Google Scholar]
- 276.Li D, Ding CY, Qiu LH. Postoperative hormone replacement therapy for epithelial ovarian cancer patients: A systematic review and meta-analysis. Gynecol Oncol. 2015;139:355–362. doi: 10.1016/j.ygyno.2015.07.109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.