

Abstract
Background
The severe acute respiratory syndrome-2019 has affected more than 190 million people around the world and caused severe crises throughout the globe. Due to rapid mutation in the viral genome, its became important to simultaneously improvise the host immunity while targeting viral proteins to reduce the severity of infection.
Aim
The current computational work focuses on multi-level rigorous screening of 47 medicinal plant-based phytochemicals for discovering effective phytochemical inhibitors against the host and viral targets.
Experimental procedure
A total of 586 phytochemicals were analyzed in detail based on their drug-likeness, pharmacological properties, and structure-based activity against the viral proteins (Spike glycoprotein, Papain-like protease, and Main protease) and host proteins (ACE2, Importin-subunit α-5, and β-1). Phytochemicals showing higher binding affinity with the dual capacity to target both the categories of proteins were further analyzed by profiling of their chemical reactivity using Density-Functional Theory (DFT) based quantum chemical methods. Finally, detailed molecular dynamics simulations were performed to analyze the interactions of the complexes.
Results and conclusion
The results revealed that the selected phytochemicals from Andrographis paniculata, Aconitum heterophyllum, Costus speciosus and Inula racemosa may have the capacity to act with prominent affinity towards the host and viral proteins. Therefore, the combination of active phytochemicals of these plants may prove to be more beneficial and can be used for developing the potential phytotherapeutic intervention.
Keywords: SARS-CoV-2, Host targets, Viral targets, Phytochemicals, Molecular screening
Graphical abstract
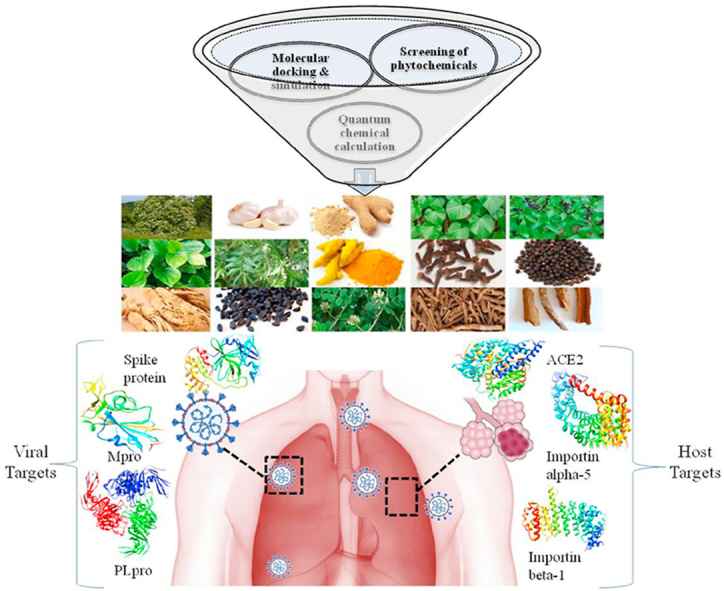
Highlights
-
•
COVID-19 caused severe crisis throughout the globe.
-
•
Current drug discovery efforts are targeting SARS-CoV-2 viral and host proteins using repurposed drugs.
-
•
Screening of 586 phytochemicals from 47 medicinal plants against both the host as well as viral targets.
-
•
Phytochemicals probably acts by inhibiting specific targets, thus help in reducing SARS-CoV-2 infection.
List of abbreviations
- ACE2
Angiotensin-Converting Enzyme 2
- PLpro
Papain-like protease
- Mpro
Main protease
- DFT
Density-Functional Theory
- SARS-CoV-2
Severe Acute Respiratory Syndrome Corona Virus 2
- SARS-CoV
Severe Acute Respiratory Syndrome Corona Virus
- MERS-CoV
Middle-East Respiratory Syndrome Corona virus
- NSPs
Non-Structural Proteins
- PDB
Protein Data Bank
- LGA
Lamarckian Genetic Algorithm
- MD simulations
Molecular dynamics simulations
- RMSD
Root Mean Square Deviation
- RMSF
Root Mean Square Fluctuation
- BBB
Blood-Brain Barrier
1. Introduction
A novel corona virus (SARS-CoV-2) has been associated with infectious respiratory disease, hastily spreading in the human population.1,2 Up to April 2021, this disease has spread in more than 219 countries proving to be a global pandemic.3 Alarmingly, the number of infected patients is rising day by day. At an early stage of infection, a person is associated with a cough, fever, shortness of breath, and headache which further leads to severe pneumonia. The epidemiological studies have revealed that apart from the age factor, an individual's immunity is also severely compromised due to this virus.4 SARS-CoV-2 shares a similar infectious and morphological pattern with Severe Acute Respiratory Syndrome Corona Virus (SARS-CoV) (affected 8098 individuals in 2003) and Middle-East Respiratory Syndrome Corona Virus (MERS-CoV) (affected 2494 individuals in 2012).5,6 The structural analysis of SARS family reveals that SARS-CoV-2 is a single-stranded RNA virus having genes for Spike (S), Envelope (E), Membrane (M), Nucleocapsid (N) and Non-structural proteins (NSPs).7, 8, 9, 10 Once the virus enters the nucleus, replication and packaging of its genetic material begins by using its non-structural proteins. This viral cycle causes respiratory tract infection which eventually leads to respiratory failure, cardiac arrest, and other multi-disorders.
Various therapeutic developments are in progress to discover an effective vaccine and drugs, which can directly target the viral proteins. In this direction, traditional medicines are effective to respond against various viral pathogenesis as well as in the host immunity modulation. Considering the antiviral properties of these medicinal plants, researchers are focusing on various phytochemicals that can act as potent drug components in the treatment of SARS-CoV-2 infection. Currently, more than 100 clinical trials based on medicinal plants are in progress (https://www.clinicaltrials.gov/). A recent work by Maurya et al. revealed a significant binding of Curcumin and Nimbin with Spike protein of SARS-CoV-2 and humane ACE2 receptor.11 The beneficial role of Nictoflorine, Aloenine, and Berberine for the inhibition of protease enzyme was also reported.12 Steroidal phytochemicals of Withania somnifera and triterpenoids of Azadirachta indica can inhibit the SARS-CoV-2 proteins.13,14 These overhead studies are concentrated toward the understanding of the plant-based bioactive compounds to decrease the virulence of SARS-CoV-2. Interestingly, these studies also suggest that medicinal phytochemicals can target both virus-based-proteins and host-based receptors and thus directly impact the viral cycle which is dependent on the host proteins.
Among the viral targets, the most common is the Spike protein which is mainly responsible for the entry of corona virus into the host cell by interacting with the ACE2 receptor.15,16 Spike protein is made up of two subunits (S1 and S2) where a sequence of S1 subunit is the most variable part of the SARS-CoV-2 genome. The emerging variants of concerns such as delta (B1.617.2) and kappa (B1.617.1) are known for their high infectivity rate. It is reported that several double/multiple mutants in the spike protein of SARS-CoV-2 are enhancing the viral host interactions. Sequence analysis defines that variant B1.617.2 and B1.617.1 have two mutations in the Receptor-Binding Domain (RDB) of the spike protein. The key mutation for B1.617.2 (delta variant) is L452R, T478K and L452R, E484Q for B1.617.1 (kappa variants), respectively. These variants are now getting huge consideration because of their antibody neutralizing tendency.17
Inside the host cell, the viral genome shows the expression of PLpro and 3CLpro/Mpro that are responsible for the formation of various non-structural proteins and further utilize the nuclear importins to enter the nucleus of the host cell.15 ACE2, Importin subunit α-5, and Importin subunit β-1 are the few major host proteins that are involved in the viral infection. Drugs like Ivermectin act by targeting the Importin α/β receptor of the host to weaken the viral cycle.18,19 It is also reported that drugs acting as allosteric modulators can decrease the strength of spike and ACE2 complex that will further help in the reduction of viral incubations.20
In this context, identifying medicinal bioactive compounds that can selectively target both virus-based-proteins and host-based-receptors simultaneously and might have the potential to alleviate the effect of SARS-CoV-2 infection.21,22 Therefore, in this investigation, we have selected 586 phytochemicals from 47 medicinal plants which have immune boosting properties and exhibit antiviral activity for different viral infections. The aim of this study was to identify active compounds against the spike glycoprotein, PLpro and Mpro receptors of virus and ACE2, Importin subunit α-5 and Importin subunit β-1 of the host machinery in a cooperative manner.23,24 Different properties of the phytochemicals such as drug likeness, pharmacokinetic parameters are calculated. Their target binding efficiency was also calculated and compared to different drugs reported for the chosen protein targets. Control drugs such as Arbidol (spike), Disulfiram (PLpro), Lopinavir (Mpro) and Hydroxychloroquine (ACE2), Ivermectin (importin-alpha-5 and importin-beta-1) were considered to systematically compare the reactivity of the resulting plant based bioactive compounds.25 Finally, DFT based reactivity properties were calculated and molecular dynamics simulations were also performed to understand the protein-ligand interaction.
2. Material and methods
2.1. Curation of phytochemicals
We have created a library of 586 bioactive phytochemicals having antiviral properties from 47 medicinal plants from the literature (Supplementary Table 1).21,23,24,26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41 The 3D .sdf structures of all the selected phytochemicals were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/).
2.2. Drug-likeness properties and ADME/toxicity prediction
All the selected phytochemicals were screened for their drug-likeness based on Lipinski, Ghose, Veber, Egan & Muegge rules by using SwissADME (http://www.swissadme.ch/). Further, those phytochemicals which fulfilled the criteria of drug-likeness were checked for their Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) properties. The pharmacokinetics and pharmacodynamics properties evaluation were done with the help of admetSAR (http://lmmd.ecust.edu.cn/admetsar1/predict/). After the ADMET analysis, the phytochemicals showing efficient pharmacokinetic parameters were selected for the molecular interaction studies with SARS-COV-2 viral and the host targets.
2.3. Protein and ligand preparation
The key therapeutic targets of virus, as well as the host proteins, were subjected to the screening of phytochemicals. The three-dimensional crystal structure of the selected viral targets for SARS-CoV-2 such as spike glycoprotein (PDB ID: 6LZG), PLpro (PDB ID: 6W9C), Mpro (PDB ID: 6LU7), and the host protein targets that includes ACE2 (PDB ID: 6M0J), Importin subunit α-5 (PDB ID: 2JDQ), and Importin subunit β-1 (PDB ID: 1F59) were chosen from protein data bank (https://www.rcsb.org/) and are presented in Supplementary Fig. 1. Files of the target protein retrieved from the PDB databank were prepared by removing water molecules, heteroatoms, and extra chains. We also included new SARS-CoV-2 variants of ceoncern such as B1.617.1 (Kappa) and B.1.617.2 (Delta) of B.1.617 lineage to address the interaction potential of phytochemicals. These variants of Spike RBD were studied, starting from the thorough literature search of RBD mutations in both the kappa and delta variants and incorporated these respective changes in the Spike RBD sequence (PDB ID:6LZG). These modified sequences are used for modeling of variants 3D protein structures using SWISS-MODEL (https://swissmodel.expasy.org/interactive) along with validation of modeled structure by SAVES v6.0 (https://saves.mbi.ucla.edu/) includes PROCHECK, Verify 3D and ERRAT modules. Top performing phytochemicals from the property calculation were selected to evaluate their interaction with the mentioned protein targets. For this purpose, a systematic comparison was performed with the known drugs namely Arbidol for spike, Disulfiram for PLpro, Lopinavir for Mpro and in case of host protein Hydroxychloroquine for ACE2, Ivermectin for Importin-alpha-5 and Importin-beta-1 acting as a control for the individual targets.19,42, 43, 44, 45, 46, 47, 48, 49, 50 All the chemical structures were obtained from the PubChem database. Further, chemical structures including controls and phytochemicals were converted into .pdb 3D conformation and prepared for molecular docking studies by adding hydrogen atoms through UCSF Chimera.
2.4. Molecular docking studies
The molecular interaction studies were performed by using AutoDock 4.2.51 All the structural proteins targets were set rigid and the ligands were kept flexible. To adopt more than one ligand conformation, 90 × 90 × 90x grid box was prepared around the active site of each target protein. The Lamarckian Genetic Algorithm (LGA) was selected as the search algorithm. All the other parameters and setting of docking were kept as standard. Further, for the generation of grid parameter file and docking parameter file, Cygwin was used. The docked conformations of each ligand were selected on the basis of binding energy and the top-ranked conformations. The docked conformations of complexes were visualized with the help of Discovery Studio Visualizer.52
2.5. Quantum chemical calculations
Phytochemicals in association with their control compounds were optimized to obtain most favorable binding energy by using the Gaussian 16 suite to understand their stability and reactivity nature.53 For the ground state geometry optimization (in gas phase) DFT based Becke's three parameter exchange function (B3) with Lee-Yang-Parr hybrid density functional (LYP) with 6–31G (d, p) basis set were used.54,55 Further Koopman's theorem was applied on optimized geometries for the calculation of highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) and HOMO-LUMO band gap.56,57 Simultaneously, different thermodynamic properties, such as global reactivity parameters (as chemical potential and chemical hardness), heat capacity and entropy were also calculated.58
2.6. Molecular dynamics (MD) simulations
MD simulations of the strongest protein-ligand complexes as compared to the control drug were performed to understand the conformational changes and dynamic properties of the interactions. A total of eight systems were selected and simulated using GROMACS 2015 (Groningen Machine for Chemical Simulations) with GROMOS96 54a7 force field.59 ATB server was used for the generation of ligand parameters associated with the chosen force field. The protein-ligand complexes were solvated in dodecahedron boxes by single point charge (SPC) water molecules and the system was neutralized by adding counter ions.59,60 Further, all the eight systems were subjected to energy minimization by running the steepest descent minimization integrator at 50,000 steps with <10 kJ/mol force convergence. Subsequently, equilibrations of all complexes were performed by using NVT (canonical) and NPT (isothermal-isobaric) ensembles at temperature 300 K. Thereafter, the systems were simulated for the 50 ns and were analyzed by using different parameters such as Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and a number of hydrogen bonds interactions of the protein-ligand complexes.
3. Results and discussion
3.1. Curation, preparation and screening of phytochemicals
Among the evaluated 586 phytochemicals, 161 were found to follow the drug-likeness criteria. These phytochemicals had a molecular weight below five hundred, LogP was less than five, hydrogen bond acceptors count was below ten and hydrogen bond donors counts were under five. Further, these phytochemicals were also screened for the pharmacokinetic properties such as Aqueous Solubility, Blood-Brain Barrier (BBB) penetration, Human Intestinal Absorption (HIA), Cytochrome enzyme inhibitor, Mutagenicity (AMES-Test) and Carcinogenicity. The results represented that, 24 phytochemicals (Table 1) were showed favorable activity in these parameters. The permissible limit criteria of pharmacokinetic parameters for all 24 phytochemicals showed that their solubility was in the range between −6.5 to 0.5, with acceptable BBB limits found to be in between −3.0 to 1.2, and HIA was more than 30% with low Cytochrome enzyme inhibition (Supplementary Table 2).
Table 1.
Structure of phytochemicals following the drug likeness properties obtained after screening of compounds from the 47 medicinal plants.

3.2. Protein modeling and validation
From literature, we found that the Kappa variant mutations (in the residues L452R (452, leucine-to-arginine), E484Q (484, glutamic acid-to-glutamine)) and the Delta variant mutations (at L452R (452, leucine-to-arginine) & T478K (478 is threonine-to-lysine)). These changes are incorporated to the Spike RBD FASTA sequence and used for mutant RBD homology modeling using the SWISS-MODEL. Validation was performed by Ramachandran plot analysis of modeled Kappa variant Spike RBD. PROCHECK shows that 89.3% of residues were in favoured regions, 10.1% residues in additional allowed regions and 0.6% in disallowed regions with 99.44% final protein quality factor by ERRAT plot analysis. For the modeled Delta variant, the Ramachandran plot shows 89.3% of residues in favoured regions, 10.1% residues in additional allowed regions, 0.6% residues were in disallowed regions with a 99.43% quality factor (Supplementary Fig. 7). According to validation analysis both the modeled protein is of good quality and having less error value for further molecular screening studies.
3.3. Molecular docking studies
To find out the most potent phytochemical from amongst the selected 24 phytochemicals, with the highest inhibition capacity, molecular interactions studies were performed. These phytochemicals were further categorized based on their binding energies, hydrogen bonds, hydrophobic interaction and interactive amino acid residues.
The binding energies of all the selected phytochemicals were compared with their chosen control drugs (Supplementary Table 3, Supplementary Figs. 2 and 3). Additionally, the binding energies of all 24 phytochemicals were plotted (ranging between −2.0 kcal/mol to −8.0 kcal/mol) against the host and viral targets (Fig. 1). The plot is depicting that the 14-deoxy-14, 15-didehydroandrographolide shows comparatively high binding energy with all targets except Importin subunits among the chosen chemicals.
Fig. 1.

Spider plot showing relative binding energies of bioactive phytochemicals with the host as well as viral proteins.
Potential inhibitors for the host targets: In the category of host-based targets (ACE2, Importin subunit α-5, and Importin subunit β-1), the binding energies of the screened phytochemicals were compared with those of the respective control drugs as Hydroxychloroquine for ACE2, Ivermectin for Importin subunit α-5 and Importin subunit β-1. All the 24 phytochemicals with ACE2 target had binding energy in the range of −4.42 to −8.46 (kcal/mol) and that for the control compound, it was −5.19 (kcal/mol) (Supplementary Table 4). Hetisinone (CID_101930090) from Aconitum heterophyllum showed the highest binding energy (8.46 (kcal/mol)) among all selected phytochemicals. In case of Importin subunit α-5, and Importin subunit β-1 the binding energies for phytochemicals ranged between −7.22 to −2.0 (kcal/mol) and −7.95 to −4.83 (kcal/mol) for their respective targets (Supplementary Tables 5 and 6). Control compound (Ivermectin) was found to be binding more favorably with the Importin subunit α-5 and Importin subunit β-1 as compared to that of the chosen phytochemical for these targets (Fig. 2).
Fig. 2.

Interaction of the host (a) ACE2 (b) Importin subunit alpha-5 and (c) Importin subunit beta-1 and viral (d) Spike, (d1) Spike-Kappa variant, (d2) Spike-Delta variant, (e) Mpro, and (f) Plpro targets interation with phytochemicals having highest binding energies (kcal/mol).
Potential inhibitors for SARS-CoV-2 (Viral) targets: In the case of viral proteins (spike glycoprotein, PLpro and Mpro) all the selected 24 phytochemicals were also screened to check their interactions by molecular docking. We have also performed the screening of top interacting phytochemicals with the Spike RBD of B1.617.1 (Kappa) and B.1.617.2 (Delta) of B.1.617 lineage. The results showed that the binding energy of the phytochemicals with spike proteins was in the range of −4.65 to −7.76 (kcal/mol) and for the control compound (Arbidol) binding energy with spike protein was −5.9 (kcal/mol) (Supplementary Table 7 & Fig. 2). 14-deoxy-11, 12-didehydroandrographolide (CID_15708351) from Andrographis paniculata was showing maximum binding score −7.76 (kcal/mol), −7.4 with (kcal/mol) and −7.09 (kcal/mol) with Spike protein as well as with both mutants B1.617.1 (Kappa) and B.1.617.2 (Delta), respectively (Supplementary Table 11 & Fig. 3)). For PLpro and Mpro receptors, the binding energies of the phytochemicals ranged between −5.06 to −8.20 (kcal/mol) and −4.17 to −7.40 (kcal/mol) respectively. When compared with their respected control compounds Disulfiram (PLpro), and Lopinavir (Mpro), the obtained energy was found to less favorable as the energies were −3.11 (kcal/mol) and −5.21 (kcal/mol) respectively (Supplementary Tables 8 and 9). The phytochemical Germacranolide (CID_101616641) from Inula racemosa and Costunolide (CID_5281437) from plant Costus speciosus were found to be showed the highest binding score −8.20 and −7.40 kcal/mol for PLpro and Mpro, respectively (Fig. 2 (e, f)).
Fig. 3.

Optimized structure and contour map of phytochemical and control (drug) compounds at minimum energy state.
3.4. Analysis of frontier molecular orbital and reactivity parameters
All the screened and shortlisted phytochemicals were next optimized at their minimum energy state with zero imaginary frequency (Fig. 3) and their molecular properties were computed. All the optimized compounds were found to be stable and non-planar (except disulfiram).
Highest Occupied Molecular Orbital (HOMO), and Lowest Unoccupied Molecular Orbital (LUMO) represent the frontier molecular orbitals (FMOs) of chemicals. The energy of HOMO and LUMO are one of the most significant aspects to describe the chemical/bioactivities of drug components. Higher the value of EHOMO, higher will be its electron donating ability.61 For all selected phytochemicals the energy of HOMO was higher in comparison to that of their respective control compounds. The energy of HOMO for phytochemicals and the control were obtained to lie in between −6.56 eV and −6.02 eV and −5.76 eV to −5.39 respectively. However, the variation in ELUMO reflected the presence of electron giving or extracting groups in the structure (Supplementary Fig. 4).
The chemical reactivity parameters such as chemical hardness (η), and electronic chemical potential (μ), were also calculated for all the phytochemicals and known drug compounds.56 Chemical hardness is a parameter which defines the stability/reactivity of a particular chemicals and lower heat capacity with a positive entropy, is typically associated with hydrophobic interactions and conformational changes of ligand during binding.58 The values of chemical hardness for phytochemicals ranged between 2.43 eV and 3.09 eV and were found to be higher than those of their respective drug compound. The value of the heat capacity (between 64.58 and 92.91) and entropy (127.69–161.08) for phytochemicals was quite low. Therefore, higher the chemical hardness with lower entropy parameter shows the high stability with lower randomness for all phytochemicals than their respective drug compounds62 (Supplementary Table 10 and Fig. 4).
Fig. 4.

Plot (a) Chemical hardness (eV) and (b) Entropy (S) for control compounds (drug) vs phytochemicals with their host as well as viral targets representing that the stability (chemical hardness) of phytochemicals are higher and randomness (Entropy) is lower in comparison to their control compounds.
3.5. Molecular dynamics simulation analysis
The MD simulation helped to understand the stability and conformational variation in the protein-ligand complexes (49). Based upon the molecular docking interaction trends, among the host proteins, ligands interacted most favorably with ACE2 receptor. Thus, in the host category, simulation undertaking Hetisinone phytochemical was performed with ACE2 receptor and compared it with control drug Hydroxychloroquine (HCQ).
The obtained RMSD profile (Fig. 5 (a)) showed that in case of Hetisinone, the stability of the protein backbone was better and remained within the range of 0.2 nm–0.3 nm. However, in the case of HCQ, larger fluctuations were observed around 30 ns, and which continued till 50 ns Thus, the backbone initially showed fluctuation, however, attained equilibrium after 10ns in both the cases. To check the flexibility of interacting residues RMSF was calculated. It was observed that the rate of amino acid fluctuation for Hetisinone was higher in comparison to that of HCQ (Fig. 5(b)). The RMSF value ranged in between 0.2 and 0.8 nm for Hetisinone and for HCQ it ranged between 0.2 and 0.6 nm. The increased value of RMSF shows higher flexibility of the complexes at the time of simulations. Finally, the analysis of intermolecular hydrogen bonds (H-bonds) was performed to check the binding stability of the ligands with their targets (Fig. 5 (c)). Following this, Phytochemical Hetisinone formed 6 H-bonds whereas, HCQ formed 4 H-bonds. Based upon these observations it can be concluded that the hydrogen bond interaction between Hetisinone and ACE2 represented a strong interaction in comparison with control drug molecule. HCQ disrupts the viral-host interaction by binding to an allosteric region of the ACE2 receptor. This allosteric interaction can inhibit the viral invasion inside the human cell. Although Hetisinone exhibited non-specific binding on to the ACE2 receptor, it has the potential to demonstrate better activity specific to SARS-CoV-2.
Fig. 5.

Graphs representing MD simulation for the host ACE2 receptor with phytochemical Hetisinone [Red] and control Hydroxychloroquine [Black] and for viral Spike glycoprotein with 14-deoxy-11, 12-didehydroandrographolide [Red] and control Arbidol [Black] at 50 ns (d) RMSD, (e) RMSF, (f) Hydrogen bond count.
During the SARS-CoV-2 infection, Spike glycoprotein and papain-like proteases (Plpro and Mpro) are the essential proteins for the processing and maturation of viral genome and due to this, these proteins are important therapeutic targets. Phytochemicals like 14-deoxy-11, 12-didehydroandrographolide, Germacranolide and Lopinavir were selected for detailed MD analysis to understand the binding mode with Spike, PLpro, and Mpro.
For Spike protein, as shown in Fig. 5 (d) the RMSD of the backbone revealed that in presence of 14-deoxy-14, 15-didehydroandrographolide fluctuation in protein continued till 35 ns and attained stability, which remained in the range of 0.2 nm–0.3 nm whereas with control Arbidol the RMSD reflected an increment starting from 0.2 nm to 0.45 nm.
The range of fluctuation (RMSF) in Spike protein and control is between 0.3 and 0.8 nm while with phytochemical 14-deoxy-14, 15-didehydroandrographolide it is between 0.3 and 0.6 nm (Fig. 5 (e)). 14-deoxy-11, 12-didehydroandrographolide also formed 3 stable H-bonds with Spike glycoprotein at 50 ns (Fig. 5(f)). It is known that the Arbidol has the capability to target the Spike glycoprotein which may help in blocking mechanism of the trimerization of SARS-CoV-2 spike glycoprotein,63 the observed interaction of 14-deoxy-14, 15-didehydroandrographolide with Spike glycoprotein can possibly lead to inhibition of trimerization of the Spike protein as the binding pattern was found to be similar in both the cases.
Further, for PLpro, RMSD plot of phytochemical Costunolide, was in the range of 0.2 nm–0.4 nm and the value of RMSF was between 0.4 and 0.7 nm (Supplementary Fig. 5 (a, b)). For control molecule RMSD ranges between 0.2 and 0.3 nm and RMSF was in the range of 0.4–0.8 nm. The comparative graph of RMSD for control and phytochemical with PLpro is depicts that the Costunolide is having slightly higher fluctuation with Disulfiram.
The observed range was better than that of the control molecule Disulfiram. Finally, H-bond calculation revealed that at 50 ns, 3 stable H-bonds were found between Plpro and Costunolide whereas no H-bond was observed with Disulfiram (Supplementary Fig. 5 (c)). Since 1951, Disulfiram is used in the treatment of alcohol aversion and is also a competitive inhibitor for SARS-CoV Plpro.64 With another papain-like protease Mpro phytochemical Germacranolide showed the most potential interaction during molecular docking studies in comparison to that of its control drug molecule (Lopinavir).
The comparative molecular dynamics analysis of Germacranolide with Mpro protein revealed fluctuations in RMSD value which lasted till 15 ns and further remained stable until 45 ns On the other hand, Lopinavir showed better stability after crossing 35 ns (Supplementary Fig. 6 (a)). We also found almost a similar RMSF peak 0.2–1 nm in case of both phytochemicals as well as in the control molecule with Mpro (Supplementary Fig. 6 (b)). The H-bond plot showed that Germacranolide created 4 stable H-bonds at 50 ns which were quite higher than that of Lopinavir (Supplementary Fig. 6 (c)). During the SARS-CoV-2 treatment, Lopinavir can inhibit SARS-CoV-2 proteases in combination with other drugs by binding to the active site of Mpro protein.65 Considering the results, it can thus be concluded that Germacranolide can target Mpro a more effective manner based upon its interaction dynamics.
During the SARS-CoV-2 infection, the host protein defense mechanism is activated to suppress the viral invaders and at the same time, SARS-CoV-2 captivates the host cellular mechanism for their replication and protein translation mechanism. This viral attack further leads to tissue damage and hyper inflammation in the host immune system. In this current investigation, we developed a comprehensive approach to reduce COVID-19 infection by targeting conserved viral proteins that are responsible for the entry and replication of SARS-CoV-2 as well as by inhibiting the activity of those host protein which facilitated viral replication inside the body. This strategy advantageous while considering the range of mutation and the effect of SARS-CoV-2 on the host organ system. This study reveals that, for the treatment of COVID-19, combination of active phytochemicals of Andrographis paniculata, Aconitum heterophyllum, Costus speciosus and Inula racemosa may prove to be more beneficial and can be used for developing the potential phytotherapeutic intervention. Multi-scale computational screening of the selected phytochemicals indicate that these compounds can act as cooperative and multi-target allosteric regulators and offer a unique category for drug development against COVID-19 infection. The approach of finding the appropriate phytochemicals targeting virus-host interaction and the host proteins together might be a new regimen for SARS-CoV-2 treatment.
4. Conclusion
SARS-CoV-2 pandemic has become a major challenge to the global health system with an increasing number of infections all over the world. Viral proteins are the key factors of SARS-CoV-2 virulence which allows virus to replicate and affect the host defence mechanisms. So, it is required to decipher the potential therapeutics including active phytochemicals for the treatment of COVID-19 infection. Herein, we screened 586 potential antiviral phytochemicals for the drug-likeness, efficient pharmacokinetics analysis, and excellent binding energy parameters against key viral and the host components. Additionally, our study has also included on evaluating phytochemicals potency against the spike RBD of novel SARS-CoV-2 variants Kappa (B1.617.1) and Delta (B.1.617.2). Overall, our systematic analysis found that 14-deoxy-11,12-didehydroandrographolide, Costunolide, Germacranolide and Hetisinone can act as probable inhibitory phytochemicals that can help in reducing the burden of SARS-CoV-2 infection by acting upon the host system as well as viral targets. This work adds strong support to phytochemical medication as a cooperative multi-target treatment and recommended further in-vitro and in-vivo validation of these chemicals to convert them as clinical drugs for SARS-CoV-2.
Consent for publication
All the authors have read and approved the manuscript in all respects for publication.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are thankful to the Council of Scientific and Industrial Research, New Delhi, India for providing funding under Mission Mode Project HCP-35. The authors also thank CSIR-IITR, Lucknow for providing computational resources. CSIR-IITR manuscript communication number is 3676.
Footnotes
Peer review under responsibility of The Center for Food and Biomolecules, National Taiwan University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jtcme.2021.09.001.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Supplementary data: List of selected phytochemicals, with their PubChem IDs and plant sources along with comprehensive details of computed properties, interactions with viral and the host protein.
References
- 1.Bai Y., Yao L., Wei T., et al. Presumed asymptomatic carrier transmission of COVID-19. Jama. 2020;323(14):1406–1407. doi: 10.1001/jama.2020.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang C., Wang Y., Li X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donnelly C.A., Ghani A.C., Leung G.M., et al. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. The lancet. 2003;361(9371):1761–1766. doi: 10.1016/S0140-6736(03)13410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan R.E., Adab P., Cheng K. British Medical Journal Publishing Group; 2020. Covid-19: Risk Factors for Severe Disease and Death. [DOI] [PubMed] [Google Scholar]
- 5.Drosten C., Günther S., Preiser W., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 6.Zaki A.M., Van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 7.De Haan C.A., Kuo L., Masters P.S., Vennema H., Rottier P.J. Coronavirus particle assembly: primary structure requirements of the membrane protein. J Virol. 1998;72(8):6838–6850. doi: 10.1128/jvi.72.8.6838-6850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woo P.C., Huang Y., Lau S.K., Yuen K.-Y. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2(8):1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu Y., McCauley J. GISAID: global initiative on sharing all influenza data–from vision to reality. Euro Surveill. 2017;22(13):30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaiswal Y.S., Williams L.L. A glimpse of Ayurveda–The forgotten history and principles of Indian traditional medicine. Journal of traditional and complementary medicine. 2017;7(1):50–53. doi: 10.1016/j.jtcme.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maurya V.K., Kumar S., Prasad A.K., Bhatt M.L., Saxena S.K. Structure-based drug designing for potential antiviral activity of selected natural products from Ayurveda against SARS-CoV-2 spike glycoprotein and its cellular receptor. VirusDisease. 2020:1–15. doi: 10.1007/s13337-020-00598-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava A.K., Kumar A., Misra N. 2020. On the Inhibition of COVID-19 Protease by Indian Herbal Plants: An in Silico Investigation. arXiv preprint arXiv:2004.03411. [Google Scholar]
- 13.Parida P.K., Paul D., Chakravorty D. 2020. Nature to Nurture-Identifying Phytochemicals from Indian Medicinal Plants as Prophylactic Medicine by Rational Screening to Be Potent against Multiple Drug Targets of SARS-CoV-2. [Google Scholar]
- 14.Prakrity Singh S.P. Ramakrishnan Parthasarathi Profiling some plant-based immunomodulatory bioactive compounds for COVID-19 prophylaxis and treatment based on Indian traditional medicine. Chem World. 2021 ISBN: 978-1-83916-306-7. [Google Scholar]
- 15.van Kasteren P.B., Bailey-Elkin B.A., James T.W., et al. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc Natl Acad Sci Unit States Am. 2013;110(9):E838–E847. doi: 10.1073/pnas.1218464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79(24):15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kannan, S. R.; Spratt, A. N.; Cohen, A. R., et al, Evolutionary analysis of the delta and delta plus variants of the SARS-CoV-2 viruses. J Autoimmun 2021, 102715. [DOI] [PMC free article] [PubMed]
- 18.Heidary F., Gharebaghi R. Ivermectin: a systematic review from antiviral effects to COVID-19 complementary regimen. The Journal of Antibiotics. 2020:1–10. doi: 10.1038/s41429-020-0336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gil C., Ginex T., Maestro I., et al. COVID-19: drug targets and potential treatments. J Med Chem. 2020;63(21):12359–12386. doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 20.Di Paola L., Giuliani A. 2020. Mapping Active Allosteric Loci SARS-CoV Spike Proteins by Means of Protein Contact Networks. arXiv preprint arXiv:2003.05200. [Google Scholar]
- 21.Deb A., Barua S., Das B. Pharmacological activities of Baheda (Terminalia bellerica): a review. J Pharmacogn Phytochem. 2016;5(1):194. [Google Scholar]
- 22.Verma A.K., Kumar V., Singh S., et al. Repurposing potential of Ayurvedic medicinal plants derived active principles against SARS-CoV-2 associated target proteins revealed by molecular docking, molecular dynamics and MM-PBSA studies. Biomed Pharmacother. 2021;137:111356. doi: 10.1016/j.biopha.2021.111356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma P., Dwivedee B.P., Bisht D., Dash A.K., Kumar D. The chemical constituents and diverse pharmacological importance of Tinospora cordifolia. Heliyon. 2019;5(9) doi: 10.1016/j.heliyon.2019.e02437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao Y. K, Vimalamma G., Rao C.V., Tzeng Y.M. Flavonoids and andrographolides from Andrographis paniculata. Phytochemistry. 2004;65(16):2317–2321. doi: 10.1016/j.phytochem.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Molavi Z., Razi S., Mirmotalebisohi S.A., et al. Identification of FDA approved drugs against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) and 3-chymotrypsin-like protease (3CLpro), drug repurposing approach. Biomed Pharmacother. 2021:111544. doi: 10.1016/j.biopha.2021.111544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makihara H., Koike Y., Ohta M., et al. Gallic acid, the active ingredient of Terminalia bellirica, enhances adipocyte differentiation and adiponectin secretion. Biol Pharm Bull. 2016;39(7):1137–1143. doi: 10.1248/bpb.b16-00064. [DOI] [PubMed] [Google Scholar]
- 27.Kumar V., Van Staden J. A review of Swertia chirayita (Gentianaceae) as a traditional medicinal plant. Front Pharmacol. 2016;6:308. doi: 10.3389/fphar.2015.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel J.J., Acharya S.R., Acharya N.S. Clerodendrum serratum (L.) Moon.–A review on traditional uses, phytochemistry and pharmacological activities. J Ethnopharmacol. 2014;154(2):268–285. doi: 10.1016/j.jep.2014.03.071. [DOI] [PubMed] [Google Scholar]
- 29.Ju Y., Xiao B. Chemical constituents of Cyperus rotundus L. and their inhibitory effects on uterine fibroids. Afr Health Sci. 2016;16(4):1000–1006. doi: 10.4314/ahs.v16i4.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonwa M.M., König W.A. Chemical study of the essential oil of Cyperus rotundus. Phytochemistry. 2001;58(5):799–810. doi: 10.1016/s0031-9422(01)00301-6. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed K., Shaheen G., Asif H. Zingiber officinale Roscoe (pharmacological activity) J Med Plants Res. 2011;5(3):344–348. [Google Scholar]
- 32.Bhowmik D., Kumar K.S., Yadav A., Srivastava S., Paswan S., Dutta A.S. Recent trends in Indian traditional herbs Syzygium aromaticum and its health benefits. J Pharmacogn Phytochem. 2012;1(1):13–22. [Google Scholar]
- 33.Reddy B.S., Rao N.R., Vijeepallam K., Pandy V. Phytochemical, pharmacological and biological profiles of Tragia species (family: euphorbiaceae) Afr J Tradit, Complementary Altern Med. 2017;14(3):105–112. doi: 10.21010/ajtcam.v14i3.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sethiya N.K., Ahmed N.M., Shekh R.M., Kumar V., Singh P.K., Kumar V. Ethnomedicinal, phytochemical and pharmacological updates on Hygrophila auriculata (Schum.) Hiene: an overview. Journal of integrative medicine. 2018;16(5):299–311. doi: 10.1016/j.joim.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Afshari A.R., Sadeghnia H.R., Mollazadeh H. A review on potential mechanisms of Terminalia chebula in Alzheimer's disease. Advances in Pharmacological Sciences. 2016;2016 doi: 10.1155/2016/8964849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.H El-Far A., A Badria F., M Shaheen H. Possible anticancer mechanisms of some Costus speciosus active ingredients concerning drug discovery. Curr Drug Discov Technol. 2016;13(3):123–143. doi: 10.2174/1570163813666160802154403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chao W.-W., Lin B.-F. Isolation and identification of bioactive compounds in Andrographis paniculata (Chuanxinlian) Chin Med. 2010;5(1):17. doi: 10.1186/1749-8546-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bagalkotkar G., Sagineedu S., Saad M., Stanslas J. Phytochemicals from Phyllanthus niruri Linn. and their pharmacological properties: a review. J Pharm Pharmacol. 2006;58(12):1559–1570. doi: 10.1211/jpp.58.12.0001. [DOI] [PubMed] [Google Scholar]
- 39.Joshi D.R., Shrestha A.C., Adhikari N. A review on diversified use of the king of spices: piper nigrum (Black Pepper) Int J Pharmaceut Sci Res. 2018;9(10):4089–4101. [Google Scholar]
- 40.Fiore C., Eisenhut M., Krausse R., et al. Antiviral effects of Glycyrrhiza species. Phytother Res: An International Journal Devoted to Pharmacological and Toxicological Evaluation of Natural Product Derivatives. 2008;22(2):141–148. doi: 10.1002/ptr.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohanraj K., Karthikeyan B.S., Vivek-Ananth R., et al. IMPPAT: a curated database of I ndian M edicinal P lants, P hytochemistry A nd T herapeutics. Sci Rep. 2018;8(1):1–17. doi: 10.1038/s41598-018-22631-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Z., Lu Z., Xu T., et al. Arbidol monotherapy is superior to lopinavir/ritonavir in treating COVID-19. J Infect. 2020;81(1):e21–e23. doi: 10.1016/j.jinf.2020.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin M.-H., Moses D.C., Hsieh C.-H., et al. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antivir Res. 2018;150:155–163. doi: 10.1016/j.antiviral.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou D., Dai S.-M., Tong Q. COVID-19: a recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J Antimicrob Chemother. 2020;75(7):1667–1670. doi: 10.1093/jac/dkaa114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hashem A.M., Alghamdi B.S., Algaissi A.A., et al. Therapeutic use of chloroquine and hydroxychloroquine in COVID-19 and other viral infections: a narrative review. Trav Med Infect Dis. 2020:101735. doi: 10.1016/j.tmaid.2020.101735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagstaff K.M., Sivakumaran H., Heaton S.M., Harrich D., Jans D.A. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem J. 2012;443(3):851–856. doi: 10.1042/BJ20120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caly L., Wagstaff K.M., Jans D.A. Nuclear trafficking of proteins from RNA viruses: potential target for antivirals? Antivir Res. 2012;95(3):202–206. doi: 10.1016/j.antiviral.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 48.Caly L., Druce J.D., Catton M.G., Jans D.A., Wagstaff K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir Res. 2020:104787. doi: 10.1016/j.antiviral.2020.104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang S.N., Atkinson S.C., Wang C., et al. The broad spectrum antiviral ivermectin targets the host nuclear transport importin α/β1 heterodimer. Antivir Res. 2020:104760. doi: 10.1016/j.antiviral.2020.104760. [DOI] [PubMed] [Google Scholar]
- 50.Lim J., Jeon S., Shin H.-Y., et al. Case of the index patient who caused tertiary transmission of COVID-19 infection in Korea: the application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by quantitative RT-PCR. J Kor Med Sci. 2020;35(6) doi: 10.3346/jkms.2020.35.e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morris G.M., Huey R., Lindstrom W., et al. AutoDock 4 and AutoDockTools 4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Biovia D.S. 2016. Discovery Studio Modeling Environment, Release 2017, San Diego: DassaultSystèmes, 2016. Available from. [Google Scholar]
- 53.M. Frisch, et al. Gaussian 16, Gaussian, Inc., Wallingford, CT: (2016).
- 54.Kohn W., Sham L.J. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140(4A):A1133. [Google Scholar]
- 55.Zhao Y., Truhlar D.G. Density functionals with broad applicability in chemistry. Acc Chem Res. 2008;41(2):157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 56.Parr R.G., Yang W. Density functional approach to the frontier-electron theory of chemical reactivity. J Am Chem Soc. 1984;106(14):4049–4050. [Google Scholar]
- 57.Bharadwaj S., Dubey A., Yadava U., Mishra S.K., Kang S.G., Dwivedi V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Briefings Bioinf. 2021;22(2):1361–1377. doi: 10.1093/bib/bbaa382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reed J.L. Electronegativity: chemical hardness I. J Phys Chem. 1997;101(40):7396–7400. [Google Scholar]
- 59.Berendsen H.J., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1-3):43–56. [Google Scholar]
- 60.Van Aalten D., Amadei A., Linssen A., Eijsink V., Vriend G., Berendsen H. The essential dynamics of thermolysin: confirmation of the hinge-bending motion and comparison of simulations in vacuum and water. Proteins: Structure, Function, and Bioinformatics. 1995;22(1):45–54. doi: 10.1002/prot.340220107. [DOI] [PubMed] [Google Scholar]
- 61.Mabkhot Y.N., Aldawsari F.D., Al-Showiman S.S., et al. Novel enaminone derived from thieno [2, 3-b] thiene: synthesis, x-ray crystal structure, HOMO, LUMO, NBO analyses and biological activity. Chem Cent J. 2015;9(1):24. doi: 10.1186/s13065-015-0100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garbett N.C., Chaires J.B. Thermodynamic studies for drug design and screening. Expet Opin Drug Discov. 2012;7(4):299–314. doi: 10.1517/17460441.2012.666235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vankadari N. Arbidol: a potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int J Antimicrob Agents. 2020;56(2):105998. doi: 10.1016/j.ijantimicag.2020.105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma C., Hu Y., Townsend J.A., et al. Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are non-specific promiscuous SARS-CoV-2 main protease inhibitors. bioRxiv. 2020;3(6):1265–1277. doi: 10.1021/acsptsci.0c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumar Y., Singh H., Patel C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J Infect Public Health. 2020;13(9):1210–1223. doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.