

Abstract
We hypothesized that ranolazine-induced adenosine release is responsible for its beneficial effects in ischemic heart disease. Sixteen open-chest anesthetized dogs with noncritical coronary stenosis were studied at rest, during dobutamine stress, and during dobutamine stress with ranolazine. Six additional dogs without stenosis were studied only at rest. Regional myocardial function and perfusion were assessed. Coronary venous blood was drawn. Murine endothelial cells and cardiomyocytes were incubated with ranolazine and adenosine metabolic enzyme inhibitors, and adenosine levels were measured. Cardiomyocytes were also exposed to dobutamine and dobutamine with ranolazine. Modeling was employed to determine whether ranolazine can bind to an enzyme that alters adenosine stores. Ranolazine was associated with increased adenosine levels in the absence (21.7 ± 3.0 vs. 9.4 ± 2.1 ng/mL, P < 0.05) and presence of ischemia (43.1 ± 13.2 vs. 23.4 ± 5.3 ng/mL, P < 0.05). Left ventricular end-systolic wall stress decreased (49.85 ± 4.68 vs. 57.42 ± 3.73 dyn/cm2, P < 0.05) and endocardial-to-epicardial myocardial blood flow ratio tended to normalize (0.89 ± 0.08 vs. 0.76 ± 0.10, P = nonsignificant). Adenosine levels increased in cardiac endothelial cells and cardiomyocytes when incubated with ranolazine that was reversed when cytosolic-5′-nucleotidase (cN-II) was inhibited. Point mutation of cN-II aborted an increase in its specific activity by ranolazine. Similarly, adenosine levels did not increase when cardiomyocytes were incubated with dobutamine. Modeling demonstrated plausible binding of ranolazine to cN-II with a docking energy of −11.7 kcal/mol. We conclude that the anti-adrenergic and cardioprotective effects of ranolazine-induced increase in tissue adenosine levels, likely mediated by increasing cN-II activity, may contribute to its beneficial effects in ischemic heart disease.
NEW & NOTEWORTHY Ranolazine is a drug used for treatment of angina pectoris in patients with ischemic heart disease. We discovered a novel mechanism by which this drug may exhibit its beneficial effects. It increases coronary venous levels of adenosine both at rest and during dobutamine-induced myocardial ischemia. Ranolazine also increases adenosine levels in endothelial cells and cardiomyocytes in vitro, by principally increasing activity of the enzyme cytosolic-5′-nucleotidase. Adenosine has well-known myocardial protective and anti-adrenergic properties that may explain, in part, ranolazine’s beneficial effect in ischemic heart disease.
Keywords: adenosine, coronary microcirculation, myocardial ischemia, ranolazine
INTRODUCTION
Ranolazine, a piperazine derivative, is used for the treatment of chronic angina pectoris (7). The anti-ischemic and anti-anginal effects of this drug have been extensively studied both in laboratory animals (27, 51) and in patients with coronary artery disease (24, 42, 44). Ranolazine, unlike other anti-anginal drugs, is clinically effective at plasma concentrations of 2–6 µM that do not appreciably change the rate-pressure product. Thus it has been proposed that the mechanism of action of ranolazine is independent of its effect on myocardial oxygen consumption (MV̇o2). In these clinical studies, other determinants of MV̇o2, such as cardiac contractility and preload, were not measured.
The current preponderantly held view of the anti-ischemic effect of ranolazine is that it inhibits the late sodium channel, leading to a reduction in the intracellular Na-dependent cellular calcium overload and, consequently, to a decrease in diastolic tension and improved myocardial relaxation (7). However, ranolazine has also been shown to possess α1-, β1-, and β2-adrenergic antagonistic activities (2, 22, 27) that have not been thoroughly studied. Moreover, in our pilot studies, ranolazine was found to increase systemic venous adenosine levels, which had not been previously reported.
Consequently, we hypothesized that ranolazine-induced increase in adenosine myocardial levels may form the basis of its beneficial effects noted in ischemic heart disease. We used an anesthetized open-chest canine model where autonomic regulation is suppressed and anti-adrenergic effects are likely to be unmasked. In addition, murine endothelial cells and cardiomyocytes were incubated with ranolazine in the presence and absence of adenosine metabolic enzyme inhibitors, and adenosine levels were measured in the supernatant. Furthermore, in silico modeling was employed to probe whether ranolazine can putatively increase activity of an enzyme that inhibits adenosine degradation and whether point mutations in the enzyme abort the ability of ranolazine to do so.
METHODS
In Vivo Experiments
Animal preparation.
The study was approved by the Animal Care and Use Committee (IACUC) at Oregon Health & Science University and conformed to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Two groups of adult male dogs (25–35 kg) were studied. Female animals were not used to avoid potential for undetected pregnancy. Group I included 16 animals undergoing the protocol illustrated in Fig. 1, A and B. Group II included six dogs that received ranolazine alone (Fig. 1C). They were intubated and ventilated with room air. Pentobarbital sodium was used to induce (40 mg/kg) and maintain (200 mg/h) anesthesia throughout the experiment. Heart rate, oxygen saturation, end-tidal CO2, and temperature were continuously monitored (Advisor Vital Signs Monitor, Surgivet, Norwell, MA). Catheters (7-Fr) were placed in both the femoral veins for microbubble infusion and administration of fluids and drugs and in the descending aorta to measure pressure.
Fig. 1.

A: stenosis experimental protocol for group I dogs (n) depicting that each animal was studied at baseline and with noncritical stenosis (S), noncritical stenosis with dobutamine stress (S+D), noncritical stenosis with ranolazine (S+R), and noncritical stenosis with dobutamine stress in the presence of ranolazine (S+D+R). B: experimental protocol for obtaining arteriolar blood volume (aBV) during myocardial contrast echocardiography (MCE) in group I dogs at baseline and during various levels of noncritical stenosis without and with ranolazine. C: no stenosis experimental protocol for group II dogs, depicting that each animal was studied before and after ranolazine administration. LAD, left anterior descending; MV̇o2, myocardial oxygen consumption.
A left lateral thoracotomy was performed, and the heart was suspended in a pericardial cradle. A micromanometer-tipped catheter (Millar Instruments, Houston, TX) was inserted into the left ventricular (LV) cavity via the LV apex to measure LV change in pressure over time (dP/dt). The proximal portions of the left anterior descending (LAD) and left circumflex (LCx) coronary arteries were dissected free from surrounding tissue. Time-of-flight ultrasonic flow probes (series SC, Transonics, Ithaca, NY) were placed on both arteries and connected to a digital flow meter (model T206, Transonics) to monitor coronary blood flow (CBF). A 7-Fr catheter was inserted into the left atrium (LA) to measure pressure, and a 20-gauge polyethylene catheter was inserted into the distal LAD to measure pressure as well as collect blood. In group I animals, a custom-designed occluder was placed around the proximal LAD and adjusted to create noncritical stenoses.
In group I dogs, a 22-gauge polyethylene catheter was placed in the great coronary vein adjacent to the LAD, and, in group II dogs, a second catheter was inserted in the LV vein parallel to the LCx. Blood samples were collected from both catheters. Samples for adenosine levels were collected in a tube containing stop solution to prevent adenosine degradation (38).
Hemodynamic measurements and calculations.
Catheters and flow meters were interfaced with a multichannel recorder (PowerLab, AD Instruments, Inc., Colorado Springs, CO). CBF, mean central aortic and LA pressures, and LV dP/dt were acquired digitally and displayed online on a computer system (iMac, Apple, Cupertino, CA). All data were saved and analyzed off-line using LabChart 6.
Cardiac output was calculated from two-dimensional echocardiography-derived stroke volume using the formula [(end-diastolic area − end-systolic area) × 10] × heart rate. Systemic vascular resistance was calculated using the formula: [(mean aortic pressure − right atrial pressure)/cardiac output] × 80.
Assessment of LV function.
Global and regional LV function was measured using the Vivid E9 ultrasound system with a broadband (1.5–4.5 MHz) transducer (GE Medical Systems, Milwaukee, WI). The transducer was affixed to the procedure table. A saline bath acted as an interface between the transducer and the heart. Gel was placed between the bath and the anterior cardiac surface. All images were acquired at the midpapillary muscle short-axis view. LV end-diastolic and end-systolic areas were measured from two-dimensional images. Regional end-systolic wall stress (ESWS) was calculated for the LAD and LCx beds, where curvature represents the inverse of the radius. Therefore, ESWS (dyn/cm) = p/(2t/c), where p = end-systolic pressure, t = wall thickness, and c = curvature (14).
The method for measuring myocardial wall thickening (WT) has been previously described (39). Briefly, the epicardial junction of the RV posterior wall and the LV free wall was identified from which WT measurement was initiated in each frame, allowing for registration of the same endocardial and epicardial points between frames, despite cardiac rotation around its long axis. A dozen targets were then outlined on the endocardium and epicardium in each frame from end-diastole to end-systole, which were then automatically connected using cubic spline interpolation. One-hundred equidistant points were automatically defined on the epicardial outline to which a tangent was drawn and a line perpendicular to the tangent intersected the epicardium and endocardium as the shortest distance between them. The resulting chord length was measured in all frames from end-diastole to end-systole, with the first chord being the reference point. Using overlays derived from myocardial contrast echocardiography (MCE) images, WT was averaged in the central 50% of the LAD and LCx perfusion beds (region not opacified when U.S. contrast was injected into the LAD) and expressed as maximal percent change over the cardiac cycle.
Speckle tracking data were acquired in group I dogs during end expiration (10 s using the respirator) for analysis with a dedicated software package (EchoPAC version BT11; GE Medical Systems). The electrocardiogram was recorded simultaneously. Peak radial strain and strain rate were determined from the short-axis view at the midpapillary muscle level (same level as the LV cavity and ESWS measurements). Frame rates of 70–80·s−1 were used with optimal sector width and depth. The endocardium was manually traced, after which the LV was automatically divided into six equal segments. Optimal tracking of all six segments was ensured. Cardiac systole was measured using pulsed-wave tracings from the LV outflow tract (aortic valve opening to aortic valve closure) (43). Separate investigators made the different echocardiographic measurements in a blinded manner.
Assessment of LV perfusion using myocardial contrast echocardiography.
High mechanical index (MI) ultraharmonic ultrasound (Sonos-5500, Phillips Healthcare, Andover, MA) was used to measure myocardial blood flow (MBF) and arteriolar blood volume (aBV), as previously described (47–49). For MBF measurements, imaging was performed after steady state was achieved during a continuous infusion of diluted Optison (3 mL mixed with 27 mL of normal saline; GE Healthcare, Inc., Princeton, NJ) infused at 90 mL/h. Images were acquired with ultrasound gated to the electrocardiogram at end-systole at pulsing intervals (PI) of 1, 2, 3, 5, 8, 10, and 20 cardiac cycles to allow for progressively greater bubble replenishment of the ultrasound beam elevation (47). At least five images were obtained at baseline (precontrast) and at each PI. Custom-designed software was used for image analysis.
PI versus background-subtracted acoustic density plots were generated from regions of interest (ROI) placed on the LAD and LCx beds (defined on MCE during transient occlusion of each artery). They were fitted to an exponential function, y = A (1 − e−βt), where y = acoustic density at PI time t, A = acoustic density after the ultrasound beam is completely replenished and reflects myocardial blood volume (MBV), and β = rate constant that reflects the mean MBF velocity. The product, A × β, was calculated to derive regional MBF. This noninvasive method of measuring MBF, including its transmural distribution, has been validated against the gold standard techniques of radioactive microspheres (23, 47) and positron emission tomography (45).
For aBV measurements in group I dogs, Optison was administered as a rapid intravenous bolus, followed by a 5 mL saline flush. MI was set at 1.0, image depth at 6 cm to focus on the anterior myocardium, and compression at 51 dB. Gain settings were optimized at the beginning of each study and then held constant throughout. A frame rate of 15 Hz (PI of 67 ms) was used to allow microbubble replenishment of only larger intramyocardial arterioles with high flow velocities. Changes in systolic versus diastolic acoustic intensities from these arterioles reflect changes in aBV in resistance vessels (48, 49). A ROI was placed over the mid-myocardium of the anterior wall on end-systolic and end-diastolic images. The mid-myocardium from all other selected frames was then manually aligned to the same ROI, from which myocardial acoustic densities were automatically derived. The precontrast tissue signal was then subtracted from contrast-enhanced values. The change in aBV was represented as the ratio of background-subtracted systolic-to-diastolic (S/D) acoustic density ratio (48, 49).
Experimental protocol.
Hemodynamics (including CBF), regional WT, LV dimensions, and MBF data were acquired in both group I and II dogs (Fig. 1A). In addition, LV radial strain was acquired in group I dogs, which underwent four stages: noncritical stenosis (S); dobutamine in the presence of noncritical stenosis (S+D), ranolazine in the presence of noncritical stenosis (S+R); and both ranolazine and dobutamine in the presence of noncritical stenosis (S+D+R). After data acquisition during the S+D stage, 30–60 min elapsed to allow for hemodynamic parameters to return to predobutamine state. aBV measurements were also acquired in group I dogs at all stages, except where dobutamine was administered (Fig. 1B). Noncritical stenosis was defined by the absence of reduction in resting CBF in the presence of an aortic-LAD gradient, coupled with an attenuation but not abolishment of the hyperemic response. Ranolazine was administered as a bolus (2 mg/kg) over 30 s, followed by a continuous infusion (120 µg·kg−1·min−1) over 20 min. Data were acquired after steady state was reached. Coronary venous adenosine levels were measured in about one-half the group I and all the group II dogs. In a subset of group I dogs, O2 saturation, lactate level, and coronary venous pH were also measured from the LAD and coronary vein.
In group I dogs, dobutamine was administered as a continuous infusion, 5–20 µg·kg−1·min−1 in the absence of ranolazine and 5–40 µg·kg−1·min−1 in the presence of ranolazine, to achieve a similar heart rate for the two stages. At the conclusion of each study, euthanasia was performed by intravenous bolus administration of potassium chloride (2 mmol/kg) with concurrent continuous intravenous infusion of pentobarbital (200 mg/h). This method conformed to the June 2007 American Veterinary Medical Association Guidelines on Euthanasia and was approved by our IACUC.
In vitro experiments.
Primary murine cardiac endothelial cells were isolated by selection using CD31 and CD102 antibodies. Hearts from 8-wk-old male C57BL6 mice (Charles River Laboratory, Wilmington, MA) were digested in collagenase type 2 (Worthington Biochemical, Lakewood, NJ) and triturated. Cells were pelleted, resuspended in PBS containing 0.1% BSA, and incubated with CD31 antibody-coated (BD PharMingen, San Jose, CA) sheep anti-rat Dynabeads (Invitrogen, Carlsbad, CA) for 40 min at room temperature. The cell-Dynabead suspension was subsequently mounted on a magnetic separator; the CD31 Dynabead-bound cells were plated on collagen type IV-coated (Sigma, St. Louis, MO) flasks in high-glucose Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 20% fetal bovine serum, 50 μg/mL gentamycin, 2 mmol/L glutamine, 100 μg/mL heparin, 100 μg/mL endothelial cell growth supplement, and endothelial mitogen (Biomedical Technologies, Stoughton, MA), whereas nonbound cells were discarded. Once confluent, cells were detached (0.05% trypsin-EDTA, Sigma, St. Louis, MO) and sorted using CD102 antibody-coated (BD PharMingen, San Jose, CA) sheep anti-rat Dynabeads and cultured. Once confluent, cells with bound beads were removed; the remaining cells were plated in 12-well plates and grown to confluence.
Cardiomyocyte isolation and culture.
Ventricular cardiomyocytes from adult mice were isolated and cultured according to adaptation and modification from a previously published protocol (6). Cardiac digestion was achieved by subsequent perfusion with collagenase type II for 25 min. The heart was then submerged in stopping buffer containing 1% bovine serum albumin in calcium-free Krebs-Henseleit perfusion buffer. It was then minced, and cells were gently dispersed to complete cell isolation. Isolated cardiomyocytes were collected by centrifugation, and the fibroblast containing supernatant was discarded. Calcium was reintroduced to a final concentration of 1.2 mM in a three-step process. Finally, cardiomyocytes were plated at a density of 30,000 cells/mL in a laminin-coated 24-well plate with medium 199 and cultured overnight in a CO2 incubator before experiments.
Adenosine metabolism.
Wells were washed three times with PBS, before phenol red-free DMEM containing ranolazine (10 µM) was added, and incubated for 4 h for endothelial cells and 1 h for cardiomyocytes. Following incubation, the culture medium was collected for adenosine analysis and immediately frozen on dry ice.
To assess the mechanism of adenosine production, wells were washed three times with PBS before phenol-red free DMEM containing either 2 mM thymidine 5′-monophosphate (TMP) (Sigma, Burlington, MA), 2.5 µM 3-deazane planocin A (DZNep) (Tocris, Bristol, UK), 10 µM penstostatin (PS) (Tocris, Bristol, UK), 10 µM 4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2,3-d]pyrimidine (ABT 702 dihydrochloride) (Tocris, Bristol, UK), or sodium 2,4-dinitrobenzene sulfonate (DNBS) (Sigma, Burlington, MA). TMP inhibits cytosolic-5′-nucleotidase (cN-II), which decreases adenosine production. DZNep inhibits S-adenosylhomocysteine hydroxylase that plays a role in both adenosine production and degradation. PS inhibits adenosine deaminase, reducing adenosine metabolism to inosine. ABT 702 dihydrochloride inhibits adenosine kinase, which reduces adenosine metabolism to 5′-adenosine monophosphate (5′-AMP) in the intracellular compartment. DNBS inhibits ecto-5′-nucleotidase (CD73), which reduces production of 5′-AMP in the extracellular compartment (Fig. 2). After inhibitor incubation, ranolazine (10 µM) was added, and incubation continued for an additional 4 h. The culture medium was then collected for adenosine analysis and immediately frozen on dry ice. To determine whether dobutamine induced adenosine production in the absence and presence of ranolazine, cardiomyocytes were incubated with dobutamine alone (1.18 μM) (21) for 1 h, as well as dobutamine followed by ranolazine (10 µM) for an additional 1 h. The culture medium was then collected for adenosine analysis and immediately frozen on dry ice.
Fig. 2.

Cytosolic and ectosolic metabolic pathways for adenosine production and consumption and specific enzyme inhibitors. 5′-AMP, 5′-adenosine monophosphate (1).
Adenosine and ranolazine levels.
Coronary venous adenosine and ranolazine levels (group I dogs) and systemic and coronary venous adenosine levels (group II dogs) were measured by liquid chromatography and tandem mass spectroscopy (26, 31, 34). Concentrated standard stocks were diluted to 10 mg/mL, adenosine (Sigma, Burlington, MA) was diluted in 1% formic acid, and 2-chloroadenosine (Sigma, Burlington, MA) was diluted in water. Further dilutions were prepared in mass spectrometry grade water. The standard for canine samples was prepared using naive dog plasma. The standard for culture medium was prepared using high-glucose phenol-red free DMEM. Samples were placed in vials using an injection volume of 10 µL. The lower limit of quantification was 0.125 ng/mL with a precision (relative standard deviation) of <20%. Measurements were performed using a 5500 Q-TRAP hybrid/triple quadrupole linear ion trap mass spectrometer (Applied Biosystems, Framingham, MA) with electrospray ionization in positive mode. Samples were infused individually, and instrument parameters optimized for each reaction.
Ranolazine was quantified against standard curves generated by analysis of blank canine plasma spiked with known amounts of analyte standards. Standard curves were generated from analysis of plasma standards by power fit regression of peak area ratio of analyte to internal standard versus concentration data. The concentration range for ranolazine was 5–2,500 ng/mL.
cN-II ecto-5′-nucleotidase (CD73) activity.
The ability of ranolazine to inhibit cN-II and CD73 hydrolysis of the 5′-phosphate group from its substrate, 5′-AMP, was measured using the Malachite Green Phosphate Detection Kit (R&D Systems, Minneapolis, MN). Recombinant human cN-II, His-tagged (Creative Biomart, Shirley, NY), and CD73 (R&D Systems, Minneapolis, MN), 0.001 µg, were incubated with 35.7 µM 5′-AMP in the presence of 10 µM ranolazine or vehicle for 20 min at 37°C before addition of Malachite green reagents. Microwell plates were read on a Victor3 plate reader (Perkin Elmer, Waltham, MA) at 620 nm. Assays were also repeated using recombinant human cN-II His-tagged, mutant, containing the following mutations: F157A, Y210A, H352A, and I353A (see below).
In Silico Modeling
To understand how ranolazine could interact with cN-II, we performed docking simulations. The cN-II protein structure (PDB ID: 2JC9) (46) was first prepared using Schrodinger suite (Schrödinger, LLC, New York, NY, 2013). The active site-bound sulfate and water molecules were then removed, leaving the Mg2+ ion, and subsequently hydrogen atoms were added. Slight minimization was performed using the optimized potentials for liquid simulations force field to optimize protein structure. Ranolazine structure was sketched and prepared using the LigPrep tool. Initially, the deoxyguanosine monophosphate (dGMP) structure was manually placed into the binding pocket, and the ranolazine structure was aligned to that of dGMP. The resultant ranolazine pose was used to define the cN-II active site. The induced-fit docking (36, 37) protocol was performed in the extended mode, generating more than 80 poses per molecule. The ranolazine molecule and the active site residues were allowed to move to simulate physiologically relevant flexibility. Finally, binding site complimentarity and grid-based ligand docking with energetics (GLIDE) (9, 10, 12) were considered for rank-ordering the predicted binding poses.
To validate the predicted poses, we introduced mutations to disrupt ranolazine binding. The chemical perturbations included F157A/Y210A/H352A/I353A. The quadruple mutations are expected to cause energetic hindrance to the ligand. F157A/Y210A removes the majority of the aromatic interaction required for positioning the methoxybenzene ring. H352A removes hydrogen bond donation by H325 to the amide group, and I353A leads to reduced hydrophobic interaction with the dimethylphenyl group.
Statistical Methods
Comparisons between two stages were performed using a paired Student’s t test. Comparisons between multiple stages were performed using repeated-measures ANOVA. When a difference was found, Bonferroni’s multiple comparison tests were performed for interstage comparisons. A P value of <0.05 (two-sided) was considered statistically significant for all comparisons.
RESULTS
In Vivo Experiments
Group I dogs.
Each dog served as its own control with a baseline stage. None of the animals died before protocol completion. Suboptimal echocardiographic images were excluded from analysis.
hemodynamics and myocardial blood flow.
Table 1 depicts the hemodynamic and MCE-derived MBF data. As expected, heart rate, systolic and mean aortic pressures, and rate-pressure product did not change with placement of a noncritical stenosis. Also as expected, all of these values declined after intravenous administration of ranolazine, indicating its anti-adrenergic effects. The rate-pressure product more than doubled with dobutamine compared with baseline, as did cardiac output, and systemic vascular resistance declined significantly. Combining ranolazine with dobutamine caused a decline in mean and systolic aortic pressures and rate-pressure product compared with dobutamine alone. Heart rate did not change because the dose of dobutamine was increased to oppose the β1 anti-adrenergic effect of ranolazine. Notwithstanding, the rate-pressure product was lower with the addition of ranolazine. The cardiac output and systemic vascular resistance did not change. The mean dobutamine dose in the absence of ranolazine was 13 ± 1 μg·kg−1·min−1 and in the presence of ranolazine was 21 ± 2 μg·kg−1·min−1. The mean coronary venous ranolazine concentration was 6.24 ± 0.91 µM 30 min after intravenous administration.
Table 1.
Hemodynamic and blood flow results in group I dogs
| Parameter | Baseline | Stenosis | Stenosis + Ranolazine | Stenosis + Dobutamine | Stenosis + Dobutamine + Ranolazine |
|---|---|---|---|---|---|
| Heart rate, beats/min | 104 ± 3 (n = 16) |
103 ± 3 (n = 16) |
94 ± 4† (n = 16) |
160 ± 4*†‡ (n = 16) |
155 ± 5*†‡# (n = 16) |
| Mean aortic pressure, mmHg | 105 ± 5 (n = 16) |
102 ± 4 (n = 16) |
84 ± 9*† (n = 16) |
159 ± 9*†‡ (n = 16) |
118 ± 8†‡# (n = 16) |
| Systolic aortic pressure, mmHg | 127 ± 6 (n = 16) |
120 ± 6 (n = 16) |
101 ± 6*† (n = 16) |
190 ± 10*†‡ (n = 16) |
159 ± 9*†‡# (n = 16) |
| RPP, ×1,000 beats·min−1·mmHg−1 | 13.3 ± 0.9 (n = 16) |
12.5 ± 0.8 (n = 16) |
8.6 ± 0.8*† (n = 16) |
30.7 ± 2.1*†‡ (n = 16) |
23.9 ± 2.0*†‡# (n = 16) |
| Cardiac output, L/min | 4.3 ± 0.3 (n = 16) |
4.8 ± 0.2 (n = 16) |
3.5 ± 0.2 (n = 16) |
11.0 ± 0.7*†‡ (n = 16) |
10.2 ± 0.8*†‡ (n = 16) |
| Systemic vascular resistance, dyn·s·cm−5 | 1,870 ± 114 (n = 16) |
1,590 ± 90 (n = 16) |
1,749 ± 170 (n = 16) |
1,103 ± 73*†‡ (n = 16) |
919 ± 90*†‡ (n = 16) |
| LAD pressure, mmHg | 106 ± 5 (n = 16) |
85 ± 3* (n = 16) |
69 ± 4*† (n = 16) |
101 ± 6†‡ (n = 16) |
77 ± 5*# (n = 16) |
| Aortic-LAD pressure gradient, mmHg | 0 ± 0 (n = 16) |
17 ± 3* (n = 16) |
13 ± 1* (n = 16) |
50 ± 6*†‡ (n = 16) |
42 ± 6*†‡ (n = 16) |
| LAD blood flow, mL/min | 25 ± 2 (n = 16) |
21 ± 2 (n = 16) |
21 ± 4 (n = 16) |
47 ± 5*†‡ (n = 16) |
43 ± 5*†‡ (n = 16) |
| LCX blood flow, mL/min | 39 ± 5 (n = 16) |
36 ± 4 (n = 16) |
40 ± 5 (n = 16) |
99 ± 10*†‡ (n = 16) |
93 ± 8*†‡ (n = 16) |
| LAD CVR, mmHg·mL−1·min−1 | 4.1 ± 0.4 (n = 16) |
3.9 ± 0.3 (n = 16) |
3.4 ± 0.3 (n = 16) |
2.3 ± 0.3*†‡ (n = 16) |
2.0 ± 0.2*†‡# (n = 16) |
| LAD transmural MBF, A × β | 49.5 ± 6.8 (n = 14) |
53.9 ± 7.1 (n = 11) |
45.4 ± 7.2 (n = 15) |
60.6 ± 7.1 (n = 14) |
62.0 ± 8.7 (n = 14) |
| LCX transmural MBF, A × β | 46.1 ± 6.3 (n = 12) |
48.5 ± 5.9 (n = 11) |
48.7 ± 5.9 (n = 16) |
118.4 ± 12.9*†‡ (n = 16) |
99.3 ± 14.4*†‡ (n = 14) |
| LAD endocardial-to-epicardial ratio | 1.11 ± 0.05 (n = 14) |
0.85 ± 0.08* (n = 11) |
1.16 ± 0.15 (n = 14) |
0.76 ± 0.10*‡ (n = 16) |
0.89 ± 0.08 (n = 15) |
| LCX endocardial-to-epicardial ratio | 1.15 ± 0.07 (n = 12) |
1.14 ± 0.10 (n = 10) |
1.24 ± 0.12 (n = 14) |
1.04 ± 0.11 (n = 15) |
1.13 ± 0.09 (n = 14) |
Values are means ± SE; n, number of dogs. CVR, coronary vascular resistance (LAD pressure − right trial pressure)/LAD flow; LAD, left anterior descending; LCX, left circumflex; MBF, myocardial blood flow; RPP, rate-pressure product. P < 0.05 vs.
baseline,
stenosis,
stenosis + ranolazine, and
stenosis + dobutamine.
By design, the placement of LAD stenosis resulted in a gradient between the mean aortic and LAD pressures. Since the stenosis was noncritical, LAD flow did not change. By causing a decrease in the mean aortic pressure, ranolazine caused a decrease in the LAD pressure without significantly changing the aortic-LAD pressure gradient. Dobutamine increased the LAD pressure by increasing the mean aortic pressure and resulted in a larger aortic-LAD pressure gradient. Both LAD and LCx CBF did not change with ranolazine, but doubled with dobutamine. Combining dobutamine with ranolazine had the same effect on these parameters as dobutamine alone. Similar results were obtained for transmural MBF.
As expected, the endocardial-to-epicardial MBF ratio in the LAD region showed a mild reversal in the presence of a stenosis, despite no decrease in LAD CBF or transmural MBF. Ranolazine returned this ratio to normal levels. In the presence of dobutamine, the ratio decreased significantly compared with baseline and trended toward improvement when ranolazine was added. The ratio remained unchanged for all stages in the LCx region (Table 1).
global and regional left ventricular function.
Table 2 depicts measurements of global and regional LV function in group I dogs. Neither LV dP/dt nor LA pressure changed after the placement of noncritical stenosis. Ranolazine caused a significant decrease in LV dP/dt without a change in LA pressure because of its anti-adrenergic effects. The mean aortic pressure also declined (Table 1). Dobutamine increased LV dP/dt and LA pressure. Combining dobutamine and ranolazine showed a mild but nonsignificant decline in the two parameters.
Table 2.
Left ventricular function in group I dogs
| Parameter | Baseline | Stenosis | Stenosis + Ranolazine | Stenosis + Dobutamine | Stenosis + Dobutamine + Ranolazine |
|---|---|---|---|---|---|
| Peak LV dP/dt, ×1,000 mmHg/s | 10.5 ± 2.1 (n = 11) |
8.5 ± 1.7 (n = 13) |
6.2 ± 0.9† (n = 14) |
26.1 ± 4.3*†‡ (n = 14) |
21.2 ± 3.7*†‡ (n = 13) |
| Left atrial pressure, mmHg | 15 ± 1 (n = 11) |
16 ± 1 (n = 13) |
15 ± 1 (n = 14) |
18 ± 1‡ (n = 14) |
16 ± 2 (n = 13) |
| ESA, cm2 | 6.4 ± 0.4 (n = 14) |
7.1 ± 0.4 (n = 16) |
8.7 ± 0.5*† (n = 16) |
6.4 ± 0.4‡ (n = 16) |
6.3 ± 0.5‡ (n = 16) |
| EDA, cm2 | 10.6 ± 0.5 (n = 14) |
11.7 ± 0.6 (n = 16) |
12.6 ± 0.6 (n = 16) |
13.2 ± 0.7 (n = 16) |
12.5 ± 0.7 (n = 16) |
| ESWS LAD, dyn/cm2 | 38.46 ± 3.99 (n = 14) |
34.15 ± 2.82 (n = 16) |
28.24 ± 2.86*† (n = 16) |
57.42 ± 3.73*†‡ (n = 16) |
49.85 ± 4.68†‡# (n = 16) |
| ESWS LCX, dyn/cm2 | 27.34 ± 2.35 (n = 14) |
29.52 ± 2.33 (n = 16) |
23.48 ± 1.13†§ (n = 16) |
42.47 ± 3.04*†‡ (n = 16) |
44.63 ± 0.60*†‡ (n = 16) |
| WT LAD, % | 37.6 ± 0.2 (n = 14) |
36.2 ± 0.6§ (n = 16) |
24.8 ± 2.2*†§ (n = 16) |
38.7 ± 2.6‡§ (n = 16) |
35.6 ± 2.7‡§ (n = 16) |
| WT LCX, % | 37.9 ± 0.3 (n = 14) |
38.0 ± 0.3 (n = 16) |
38.0 ± 0.4 (n = 16) |
44.2 ± 0.6*†‡ (n = 16) |
43.4 ± 0.9*†‡ (n = 16) |
| LAD radial strain, % | 21.01 ± 2.66 (n = 13) |
15.39 ± 1.78 (n = 12) |
23.08 ± 3.54 (n = 13) |
24.98 ± 3.68 (n = 13) |
|
| LCX radial strain, % | 21.66 ± 2.43 (n = 13) |
23.53 ± 2.40 (n = 12) |
31.49 ± 4.04† (n = 13) |
31.20 ± 3.10‡ (n = 13) |
Values are means ± SE; n, number of dogs. dP/dt, change in pressure over time; EDA, end-diastolic area; ESA, end-systolic area; ESWS, end-systolic wall stress; LAD, left anterior descending; LCX, left circumflex; LV, left ventricular; WT, wall thickening. P < 0.05 vs.
baseline,
stenosis,
stenosis + ranolazine,
stenosis + dobutamine, and
LCX.
Ranolazine also caused an increase LV end-systolic size, which was associated with a decrease in LAD and LCx wall stress. Dobutamine had the opposite effect, reducing LV end-systolic size and increasing wall stress. When ranolazine was combined with dobutamine, although LV end-systolic size did not change, LAD ESWS decreased consequent to the reduction in aortic pressure (Table 1).
Percent WT was similar between baseline and stenosis stages. Similar to ESWS, radial strain declined in the LAD region with ranolazine. WT and radial strain increased with dobutamine, being lower in the LAD (stenosis supplied) compared with the LCx region. Combining ranolazine with dobutamine had no additional effect. The percent increase in radial strain between rest and stress in the LAD region was greater after administration of ranolazine (2.07 ± 2.74 versus 9.74 ± 4.47%, respectively, P = 0.046).
arterial blood volume and myocardial blood volume.
Tachycardia precluded acquisition of aBV images during the dobutamine stages. The S/D aBV ratio increased after placement of stenosis and increased further after intravenous administration of ranolazine (Table 3). This increase in the S/D ratio implies dilation of intramyocardial arterioles (resistance vessels) because capillary MBV did not increase with either stenosis or ranolazine (not shown).
Table 3.
aBV and MBV in the LAD region in group I dogs
| Parameter | Baseline | Stenosis | Stenosis + Ranolazine | Stenosis + Dobutamine | Stenosis + Dobutamine + Ranolazine |
|---|---|---|---|---|---|
| aBV (S/D) ratio | 0.23 ± 0.02 (n = 10) |
0.30 ± 0.02* (n = 20) |
0.42 ± 0.04*† (n = 17) |
||
| LAD MBV (A) | 73.9 ± 3.6 (n = 14) |
76.4 ± 4.2 (n = 11) |
67.1 ± 5.2 (n = 15) |
69.7 ± 3.9§ (n = 14) |
72.8 ± 4.4 (n = 14) |
| LCX MBV (A) | 68.4 ± 3.8 (n = 12) |
82.2 ± 4.3 (n = 11) |
73.7 ± 5.1 (n = 16) |
74.3 ± 3.3 (n = 16) |
76.0 ± 5.4 (n = 14) |
Values are means ± SE; n, number of dogs. aBV, arteriolar blood volume; LAD, left anterior descending; LCX, left circumflex; MBV, myocardial blood volume; S/D, systolic/diastolic. P < 0.05 vs.
baseline,
stenosis, and
LCX.
cardiac metabolites.
Table 4 depicts cardiac metabolites in a subset of group I dogs. Adenosine level increased significantly when ranolazine was administered in the presence of a stenosis. It was also elevated during dobutamine infusion. Combining ranolazine with dobutamine almost doubled the adenosine level, despite decreasing aortic pressure and the rate-pressure product.
Table 4.
Cardiac metabolites from a subset of group I dogs
| Parameter | Baseline | Stenosis | Stenosis + Ranolazine | Stenosis + Dobutamine | Stenosis + Dobutamine + Ranolazine |
|---|---|---|---|---|---|
| Adenosine level, ng/mL | 9.4 ± 2.1 (n = 9) |
21.7 ± 3.0† (n = 8) |
23.4 ± 5.3† (n = 8) |
43.1 ± 13.2†# (n = 6) |
|
| Coronary venous, pH | 7.39 ± 0.01 (n = 6) |
7.39 ± 0.01 (n = 6) |
7.38 ± 0.02 (n = 6) |
7.31 ± 0.04 (n = 6) |
7.22 ± 0.02*†‡ (n = 6) |
| MV̇o2, mL/min | 17.9 ± 3.4 (n = 5) |
14.4 ± 0.8 (n = 5) |
12.6 ± 2.4 (n = 5) |
44.4 ± 4.7*†‡ (n = 5) |
38.3 ± 3.4*†‡ (n = 5) |
| LAD coronary arterial lactate level, mmol/L | 1.80 ± 0.41 (n = 5) |
1.67 ± 0.26 (n = 6) |
1.75 ± 0.19 (n = 6) |
1.57 ± 0.20 (n = 6) |
1.82 ± 0.27 (n = 6) |
| Great cardiac venous lactate level, mmol/L | 1.68 ± 0.34 (n = 5) |
1.38 ± 0.29 (n = 6) |
1.55 ± 0.22 (n = 6) |
1.93 ± 0.42 (n = 6) |
1.97 ± 0.36 (n = 6) |
| AV lactate difference, mmol/L | 0.12 ± 0.14 (n = 5) |
0.28 ± 0.10 (n = 6) |
0.20 ± 0.12 (n = 6) |
−0.37 ± 0.33 (n = 6) |
−0.15 ± 0.33 (n = 6) |
| LAD bed lactate consumption, mol/min | 3.6 ± 2.4 (n = 5) |
5.4 ± 1.9 (n = 6) |
2.8 ± 1.7 (n = 6) |
22.3 ± 12.1 (n = 6) |
20.7 ± 4.9 (n = 6) |
Values are means ± SE; n, number of dogs. AV, arteriovenous; LAD, left anterior descending; MV̇o2, myocardial oxygen consumption. P < 0.05 vs.
baseline,
stenosis,
stenosis + ranolazine, and
stenosis + dobutamine.
Coronary venous pH did not change during baseline, after placement of stenosis, and when ranolazine or dobutamine were administered individually during stenosis placement. pH decreased compared with the first three stages when dobutamine and ranolazine were both given during stenosis, but was no different from the S+D stage. Similar results were seen for MV̇o2. Although arteriovenous lactate difference decreased when dobutamine and dobutamine plus ranolazine were administered during stenosis, they were not significantly different from other stages. Similarly, whereas LAD bed lactate consumption increased during the last two compared with the first three stages, the increase did not reach statistical significance. Importantly, while there was no difference in any of the metabolites between the last two stages, coronary venous adenosine was nearly twofold higher when ranolazine was added to dobutamine during stenosis compared with dobutamine alone with stenosis.
Group II dogs.
This group was studied to determine whether myocardial adenosine levels increased after ranolazine administration without stenosis. In one dog, we measured adenosine levels from both the inferior vena cava and the coronary veins (great cardiac and LV) 2 h before, as well as 2 and 3 h after, administration of ranolazine. Since coronary venous levels were significantly higher (Fig. 3A) than systemic venous levels, we only measured adenosine levels from the two coronary veins in subsequent dogs and averaged them. Figure 3B shows coronary vein adenosine levels increased 2–3 h after ranolazine administration compared with baseline.
Fig. 3.
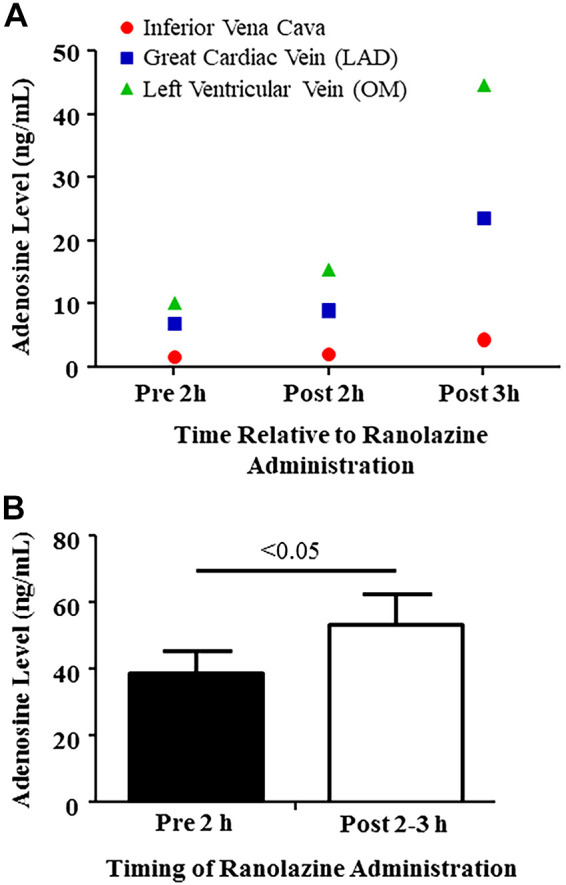
Adenosine levels in group II dogs, 2 h before and 2- and 3-h after ranolazine administration. A: levels from inferior vena cava, great cardiac vein, and left ventricular vein in 1 dog. B: average values from great cardiac and left ventricular veins. Values are means ± SE (Student’s t test). LAD, left anterior descending; OM, obtuse marginal.
Table 5 depicts the hemodynamic, MBF, and regional and global LV function before and after ranolazine administration. Similar to group I dogs, the anti-adrenergic effects of ranolazine were seen in this group as well. Systolic and mean aortic pressures and rate-pressure products declined significantly, whereas cardiac output and systemic vascular resistance did not change. Albeit not significant, there was also a decline in heart rate, LV dP/dt, and ESWS and an increase in LV end-diastolic as well as end-systolic dimensions. Furthermore, MBF tended to increase in both LAD and LCx regions.
Table 5.
Results from group II dogs
| Parameter | Pre-Ranolazine | Post-Ranolazine |
|---|---|---|
| Hemodynamics | ||
| Heart rate, beats/min | 127 ± 12 | 118 ± 12 |
| Systolic blood pressure, mmHg | 123 ± 9 | 101 ± 6* |
| Mean aortic pressure, mmHg | 104 ± 6 | 84 ± 6* |
| RPP, ×1,000 beats·min−1·mmHg−1 | 15.8 ± 1.9 | 12.0 ± 1.6* |
| Cardiac output, L/min | 6.4 ± 0.8 | 5.7 ± 0.6 |
| Systemic vascular resistance, dyn·s·cm−5 | 1,401 ± 313 | 1,256 ± 65 |
| Left atrial pressure, mmHg | 12 ± 2 | 16 ± 1 |
| LAD blood flow, mL/min | 29 ± 3 | 24 ± 3 |
| LCX blood flow, mL/min | 33 ± 2 | 29 ± 4 |
| Myocardial function | ||
| Peak LV dP/dt, ×1,000 mmHg/s | 4.1 ± 2.7 | 3.1 ± 2.0 |
| End-systolic area, cm2 | 6.8 ± 0.6 | 7.3 ± 0.8 |
| End-diastolic area, cm2 | 11.7 ± 0.6 | 12.1 ± 0.7 |
| ESWS LAD, dyn/cm2 | 38.45 ± 4.91 | 25.88 ± 2.66 |
| ESWS LCX, dyn/cm2 | 37.03 ± 4.13 | 30.13 ± 3.04 |
| WT LAD, % | 36.9 ± 0.1 | 37.0 ± 0.2 |
| WT LCX, % | 36.8 ± 0.3 | 37.0 ± 0.3 |
| Myocardial perfusion | ||
| LAD myocardial blood volume (A) | 49.7 ± 5.9 | 54.0 ± 5.3 |
| LAD velocity (β) | 0.54 ± 0.07 | 0.64 ± 0.10 |
| LAD transmural MBF (A × β) | 26.9 ± 5.0 | 35.0 ± 6.9 |
| LAD endocardial-to-epicardial ratio | 0.99 ± 0.16 | 1.10 ± 0.10 |
| LCX myocardial blood volume (A) | 60.5 ± 3.7 | 56.6 ± 5.0 |
| LCX velocity (β) | 0.55 ± 0.05 | 0.83 ± 0.14 |
| LCX transmural MBF (A × β) | 34.5 ± 5.3 | 47.7 ± 8.8 |
| LCX endocardial-to-epicardial ratio | 1.22 ± 0.21 | 1.24 ± 0.17 |
Values are means ± SE; n = 6 dogs. dP/dt, change in pressure over time; ESWS, end systolic wall stress; LAD, left anterior descending; LCX, left circumflex; LV, left ventricular; RPP, rate-pressure product; WT, wall thickening.
P < 0.05 vs. pre-ranolazine.
In Vitro Experiments
Adenosine levels in isolated endothelial cells and cardiomyocytes also increased significantly when exposed to ranolazine (Fig. 4, A and B). However, cardiac myocytes did not exhibit increased adenosine levels when exposed to dobutamine, with or without ranolazine (Fig. 4C). When the cNII inhibitor TMP was added to endothelial cells and cardiomyocytes, adenosine levels did not increase with ranolazine (Fig. 5, A and B). When adenosine deaminase was inhibited with PS, there was an additional rise in adenosine levels that was statistically significant in the cardiomyocytes and showed a similar trend in the endothelial cells (Fig. 6, A and B). Combining DZNep, ABT 702 dihydrochloride, and DNBS with ranolazine had no effect on adenosine levels in either cell type. Since TMP abolished the ability of ranolazine to increase adenosine release from both endothelial cells and cardiomyocytes, we investigated whether ranolazine was able to affect activity of the wild-type and mutant cN-II enzyme. Figure 7A demonstrates that ranolazine increased cN-II activity. Conversely, ranolazine decreased CD73 activity (Fig. 7B). The mutant version of cN-II enzyme aborted the ability of ranolazine to increase its specific activity (Fig. 7C).
Fig. 4.

Adenosine levels increased in the presence of ranolazine (RAN) in isolated murine cardiac endothelial cells (n = 5 dogs; A) and cardiomyocytes (n = 4 dogs; B and C). C: in comparison, adenosine levels did not increase in cardiomyocytes in the presence of dobutamine or in the presence of both dobutamine and ranolazine (n = 7 dogs). Values are means ± SE (Student’s t test, one-way ANOVA, Bonferroni’s multiple-comparison test). DMSO, dimethyl sulfoxide.
Fig. 5.

A: adenosine levels increased when isolated murine cardiac endothelial cells were incubated with ranolazine (RAN) and decreased during incubation with thymidine 5′-monophosphate (TMP), an inhibitor of cytosolic-5′-nucleotidase (n = 6 dogs). B: a similar pattern was observed in murine cardiomyocytes (n = 7 dogs). Values are means ± SE (one-way ANOVA, Bonferroni’s multiple-comparison test).
Fig. 6.

Pentostatin (PS), an inhibitor of adenosine deaminase, augmented ranolazine (RAN)-induced adenosine production in isolated murine cardiac endothelial cells (n = 6 dogs; A) and in isolated murine cardiomyocytes (n = 7 dogs; B). Values are means ± SE (one-way ANOVA, Bonferroni’s multiple-comparison test).
Fig. 7.

Ranolazine (RAN) increased cytosolic 5′-nucleotidase (cN-II) activity (n = 5 dogs; A) and decreased ecto-5′-nucleotidase (CD73) activity (n = 4 dogs; B). C: in comparison, point mutation failed to increase cN-II activity. Values are means ± SE (Student’s t test).
In Silico Modeling
To understand how ranolazine could increase adenosine via cN-II, we performed structure-based molecular docking simulations. The cN-II enzyme acts both as a hydrolase and phosphatase of monophosphate nucleosides, with both activities being dependent on Mg2+. Initially, docking was performed in all potential binding sites (the active and effector sites). However, the predicted binding complementarity and energy associated with each pose suggested that ranolazine binds more tightly at the active site, which is also the binding site for nucleotides (Fig. 8). The docking score for ranolazine against the active site is −11.7 kcal/mol.
Fig. 8.
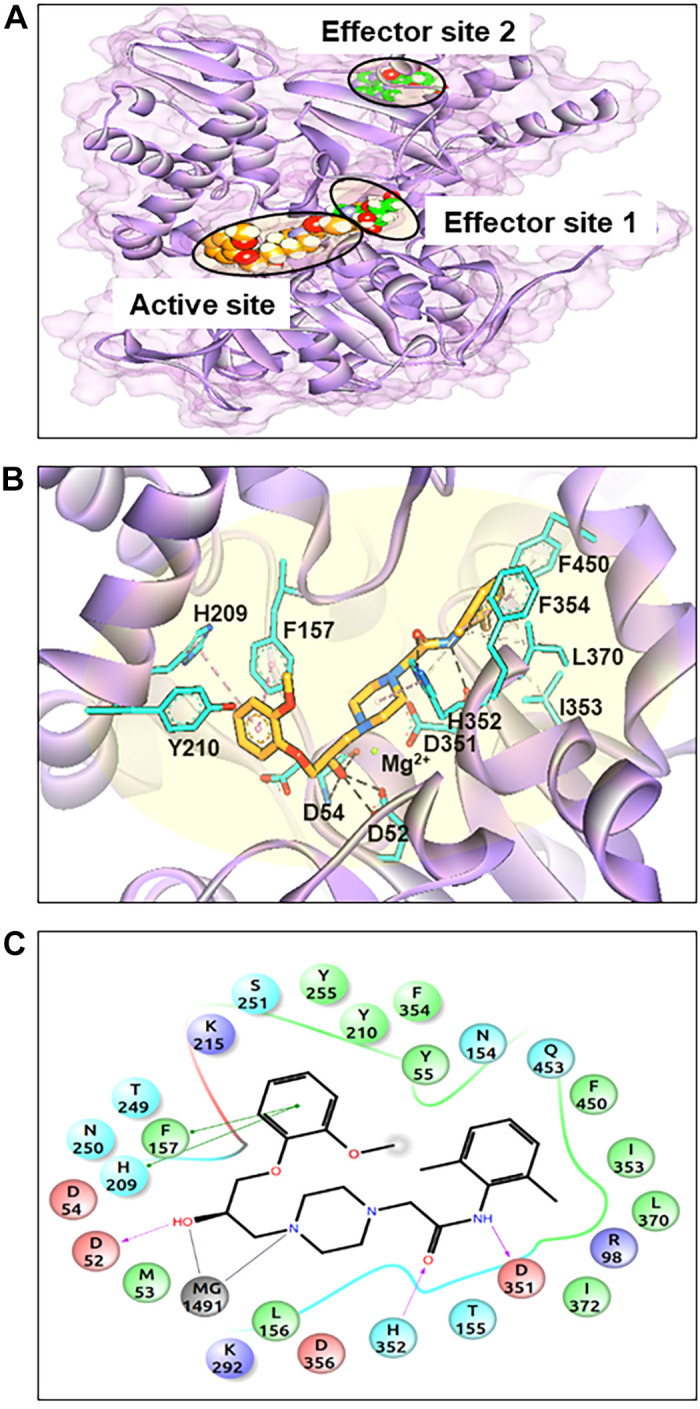
Overall structure and binding pockets of cytosolic 5′-nucleotidase (cN-II). A: location of cN-II binding pockets, including two effector sites fitted with adenosine (green-filled space) and the predicted binding pose of ranolazine (orange-filled space). B: a closer view of ranolazine binding in the active site, with the residues involved in binding shown using a stick model and the Mg2+ ion shown as a green sphere. Hydrogen bonds are depicted as black dashed lines, hydrophobic bonds as gray and pink dashed lines, and metal-ligand coordination as red dashed lines. C: two-dimensional illustration of the predicted binding mode of ranolazine in the substrate binding site of cN-II. Arrows depict predicted ranolazine interactions with cN-II residue.
Analysis of the docking pose indicates that the first part of ranolazine mimics a nucleotide ring, while its hydroxyl group was comparable with the hydroxyl group of furanose. The anisole ring interacts with F517, H209, and Y210. This interaction confirms previous reports (11, 46) demonstrating that the hydroxyl group acts as a hydrogen bond donor to D52 and D54. Furthermore, our model predicts that piperazine nitrogen chelates Mg2+, the amide group establishes hydrogen bonds with H352 and D351, and that dimethylbenzene binds at the hydrophobic subpocket formed by F354, F450, L370, and I353. Accordingly, our structure-based modeling reveals a possible binding site for ranolazine in the cN-II structure.
DISCUSSION
As expected, in the open-chest, anesthetized dogs with coronary stenosis and induced myocardial ischemia, intravenous ranolazine produced profound anti-adrenergic effects. The novel information from our study, however, is that intravenous administration of ranolazine was associated with an increase in coronary venous adenosine levels, even in the absence of myocardial ischemia. Ranolazine seems to interact with intracellular enzyme cN-II and adenosine deaminase and possibly the extracellular enzyme CD73, resulting in elevated adenosine levels in endothelial cells and cardiomyocytes. When ranolazine was administered in the presence of dobutamine-induced myocardial ischemia, adenosine levels increased further and were associated with a decrease in LV ESWS as well as improvement in the endocardial-to-epicardial MBF ratio, despite no change in CBF, transmural MBF, myocardial lactate consumption, MV̇o2, and coronary venous pH. Thus, in addition to its direct anti-adrenergic effects as well as its inhibition of the late sodium channel, resulting in better myocardial relaxation, the anti-adrenergic and cardioprotective effects of ranolazine-induced increase in tissue adenosine levels may contribute to its beneficial effects in ischemic heart disease.
Anti-Adrenergic Effects of Ranolazine
Adenosine has well known anti-adrenergic effects (35). For example, intracoronary administration of prazosin markedly attenuates release of adenosine and severely dampens its hyperemic response through an anti-α1-adrenergic effect (16). This effect may have contributed to the attenuation of hyperemia by prazosin when coadministered with ranolazine, reported previously in a porcine study (27). The effect of prazosin is even more marked during ischemia (16), implying that adrenergic receptor activation is necessary for the vasodilatory effects of adenosine (13). The vasodilatory effect of ranolazine alone in the porcine study (27) could also be explained by the increase in adenosine level that is associated with its administration, as shown in our study. Nonetheless, our results do not refute direct α1-adrenergic receptor inhibition as a possible mechanism of coronary vasodilation by ranolazine, as suggested previously (27).
In the presence of a noncritical coronary stenosis, intravenous administration of ranolazine caused a decrease in aortic pressure without compensatory tachycardia. Rather, the heart rate, peak positive LV dP/dt, and LV wall strain all decreased, while LV size increased. These effects are consistent with the β1-anti-adrenergic effect of ranolazine (22). Previous studies have demonstrated that adenosine antagonizes the stimulatory effects of catecholamines, causing a decrease in myocardial contractility and cyclic AMP (8). This effect, as well as its own effect on the A2a receptor, could explain the reduction in ESWS and improvement in the endocardial-to-epicardial MBF ratio seen with increased adenosine levels.
Extravascular compressive forces cause a change in myocardial impedance that affects small intramyocardial arterioles during systole. This effect is seen predominantly in the endocardium because of the large transmural gradient in systolic compressive forces (17). It is also exaggerated during exercise (simulated by dobutamine administration in our study), especially in the presence of a coronary stenosis (3). Our results indicate that the administration of ranolazine reduces ESWS, resulting in a normalization of the endocardial-to-epicardial MBF ratio during both rest and stress. The increase in the aBV noted in our study suggests that this effect may be mediated by dilation of small intramyocardial arterioles. The beneficial effects of ranolazine in patients with microvascular angina (24, 44) could potentially be explained by this effect. Our laboratory has previously shown that women with syndrome X have abnormalities in their coronary resistance vessels (32).
When ranolazine was administered in the presence of dobutamine, the aortic pressure fell and ESWS in the ischemic region decreased without a change in LV size, despite higher doses of dobutamine used. Radial wall strain in the LAD region was not affected, and peak positive LV dP/dt did not decline significantly. These effects could again be explained by the anti-adrenergic effects of ranolazine (14, 22, 51). The blood levels of adenosine doubled when ranolazine was administered during dobutamine compared with dobutamine alone. Although higher doses of dobutamine were used at this stage, the heart rate was not higher than when dobutamine alone was used. MV̇o2, myocardial lactate consumption, and coronary venous pH were also no different. The requirement for higher doses of dobutamine to reach the same heart rate in the presence compared with the absence of ranolazine is also likely due to the β1-adrenergic antagonistic effects of ranolazine (22).
Potential Role of Adenosine
The high coronary venous level of adenosine (mean of 35–162 mM) produced by ranolazine in our study can by itself have many beneficial effects on the ischemic myocardium through several of its receptors. Adenosine and other A1-receptor agonists have been shown to protect the myocardium when administered before an ischemic event (33, 41). Adenosine also offers cardioprotection through coronary vasodilation mediated through the A2A receptor (5, 28). Although less well-characterized, the A2B receptor has also been identified on coronary endothelium (29) and found to increase both CBF and cardiac contractility (25). The vasodilatory effects of adenosine are seen mainly through its effect on resistance vessels, which exerts its myocardial protective effect by increasing local O2 delivery.
Stimulation of the A2A receptor has been shown to reduce reperfusion injury when administered immediately after the event (15, 19), at which time is has been reported to improve LV systolic function, an effect not seen in the absence of ischemia (30). It has also been implicated in ischemic postconditioning. The A3 receptor has been shown to be cardioprotective, especially when administered before onset of myocardial ischemia (5, 28). Similarly, the A2B receptor can participate in coronary vasodilation when adenosine levels are high. Its stimulation has been reported to be cardioprotective, both before and after the onset of ischemia (40). Finally, there is evidence to suggest that there is interaction between the different adenosine receptors that potentiates cardioprotection (50).
Our in vitro data indicate that the ranolazine augmented adenosine production by increasing cN-II activity in endothelial cells and cardiomyocytes. Point mutation of this enzyme aborted this action of ranolazine. Since the half-life of adenosine is extremely short (26), it is unlikely that all of it was derived from existing stores. Our data suggest that the mechanism of increased adenosine production by ranolazine involves de novo synthesis. Under ischemic conditions, it would be advantageous for myocardial tissue to be equipped with several mechanisms to replete both cytosolic and ectosolic adenosine stores. In the cytosol, we found that ranolazine could potentially bind to the active site of cN-II with docking energy of −11.7 kcal/mol and speculate that this pathway could serve as the primary source of adenosine production by ranolazine. The binding affinity also appears to depend on the availability of magnesium ions at the substrate binding site.
The significance of the apparent inhibition of CD73 by ranolazine is unclear, since adenosine levels in vivo were increased by ranolazine, both at rest and under ischemic conditions. It is possible that, during ischemia, the extracellular concentration of AMP is significantly higher than that of ranolazine, rendering any apparent inhibition by the drug irrelevant due to higher affinity of AMP to CD73 than ranolazine. Extracellular adenosine arises either from active transport of intracellular stores (mainly formed by cN-II) or from breakdown of adenine nucleotides outside the cell initially via CD39, which breaks down ATP to AMP, followed by AMP conversion to adenosine via CD73. In the intracellular space, adenosine can be synthesized de novo during purine biosynthesis or accumulate as a result of ATP breakdown. During episodes of ischemia and increased metabolic demand, when there is a mismatch between ATP synthesis and utilization, some of the ATP breakdown product, AMP, is dephosphorylated to adenosine inside the cell. Adenosine does not freely pass across the cell membrane and requires the use of nucleoside transporters to facilitate the process. In addition to adenosine, ATP itself can be extruded to the outside of the cell, where it undergoes further degradation and conversion to adenosine (via CD39 and CD73) (20). This may explain the very high levels of adenosine when ranolazine was administered during dobutamine-induced ischemia in group I dogs.
The catalytic sites for both CD73 and cN-II are similar, although not identical, because both enzymes use 5′-AMP as a substrate. By mimicking a nucleotide ring, ranolazine can, in theory, fit in both catalytic sites, although we did not investigate potential fit in CD73 using computational modeling. Nonetheless, we speculate that the slight differences in catalytic site structures may account for ranolazine’s stimulatory effect of cN-II, yet inhibitory action on CD73. There is precedence for this concept. For instance, unlike their distant monomeric bacterial 5′-nucleotidases that hydrolyze AMP, ADP, and ATP for purine salvage, the eukaryotic CD73 enzymes are dimeric and specifically hydrolyze extracellular AMP and other nucleotide monophosphates (52). Importantly, ADP and ATP are competitive inhibitors of the eukaryotic e5NT enzymes (18). On the other hand, cN-II is allosterically activated by adenine and guanine nucleotides, with ATP being one of the most potent activators (46).
Arteriolar Blood Volume
MCE utilizes gas-filled microbubbles for assessing myocardial perfusion. After these bubbles are destroyed by ultrasound, they take ~5 s to fill the 5-mm-thick tissue imaged by ultrasound (the beam elevation of ultrasound is ~5 mm thick) (47). Because the blood in myocardial arterioles comprises only a small proportion of MBV, microbubble signals from these vessels are usually negligible when the ultrasound beam is fully replenished. However, if imaging is performed using a short interval between destructive ultrasound pulses, the signals obtained are derived only from vessels that fill in this short period, as neither capillaries nor venules have adequate time to fill. Thus signals representing aBV can be obtained from the myocardium using MCE. Because forward flow in large intramyocardial vessels occurs during diastole, aBV signals on MCE are seen predominantly during diastole (48, 49). We have shown both in an animal model and in humans that the systolic-to-diastolic aBV ratio measured at rest is increased in the presence of stenosis, and that the degree of increase is proportional to coronary stenosis severity (48, 49).
In the present study, as expected, stenosis increased aBV. There was a further increase in aBV when ranolazine was administered. With stenosis placement, the resistance arterioles dilated because of autoregulation, and further dilation of these vessels was caused by ranolazine. This could occur from ranolazine’s direct α1-adrenergic inhibition (2), or by a decrease in intracellular Na-dependent calcium overload of cardiomyocytes, leading to a decrease in myocardial extravascular compressive forces (7). The most likely explanation of the further vasodilation, however, is the additional release of adenosine caused by ranolazine.
Limitations of the Study
Several study limitations need to be addressed. Some are methodological, and some are results based. The first of the methodological considerations was use of intravenous ranolazine rather than the oral drug, which is not the form in which it is administered clinically. Whether similar levels of adenosine will be seen when the oral drug is used is not known. Second, although we used cardiac endothelial cells and cardiomyocytes from a different species in our in vitro studies, we believe our results remain valid because the extensive body of knowledge on adenosine has been generated from various experimental models, including porcine, canine, guinea pig, mouse, and rat. A third limitation is that the cardiomyocytes were not cultured in ischemic conditions, and the endothelial cells and cardiomyocytes were not studied as intact units. Demonstrating increased adenosine availability in an ischemic environment at the cellular level would further strengthen our findings. Fourth, measuring coronary venous adenosine levels does not indicate the source of adenosine (cardiac versus systemic). Thus we measured coronary and systemic venous adenosine levels simultaneously in one group II animal and showed that coronary venous adenosine levels were an order of magnitude higher than systemic venous levels and, therefore, our measurements reflect predominantly myocardial adenosine release. Fifth, we also did not measure cardiac metabolites in all group I dogs. Sixth, ischemia was induced by dobutamine rather than pacing because we wanted to increase the double product, a more likely clinical scenario for angina/ischemia. Finally, we waited 30–60 min between stages to allow hemodynamic variable to return to baseline. Since the half-life of adenosine is very short, it is most unlikely that adenosine levels from one stage affected that of another stage.
Among the results limitations, it could be argued that ranolazine has a direct vasodilatory effect (27), which causes endocardial ischemia in the presence of a stenosis, leading to adenosine release (4). The argument against this possibility is that, associated with the increased level of adenosine, there was a decrease in LV ESWS and normalization of the endocardial-to-epicardial MBF ratio. Thus ischemia is reduced rather than increased with ranolazine. It has been previously reported that the anti-adrenergic effects of ranolazine may be seen in anesthetized animals because of lack of autonomic control, and this effect is not seen in conscious animals (51). Clinical studies seem to support this observation (42). Autonomic control could mask the hypotension and bradycardia in conscious awake animals and humans, but it is unlikely to affect the other anti-adrenergic and direct myocardial effects of adenosine released by the administration of ranolazine, such as cardiac contractility. We noted hypotension in our anesthetized animals with ranolazine, but cardiac output and systemic vascular resistance did not change appreciably. In our in vitro studies, as expected, cardiomyocytes exposed to clinically relevant levels of dobutamine failed to increase adenosine levels. However, we cannot explain why, unlike the in vivo studies, addition of ranolazine to dobutamine did not increase adenosine levels.
Summary
In summary, the anti-adrenergic and cardioprotective effects of ranolazine-induced increase in adenosine levels may contribute to its beneficial effects in ischemic heart disease. Our study is the first to show that ranolazine elevates resting as well as stress-induced myocardial adenosine levels and causes small-vessel vasodilation that can further ameliorate ischemia. Ranolazine could be exerting an anti-ischemic effect through the myocardial protective properties of adenosine involving cN-II and CD73 activity.
GRANTS
This work was supported, in part, by a grant from Gilead Sciences, Foster City, CA, and resources at the Veterans Affairs Portland Health Care System, Portland, OR.
DISCLAIMERS
The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.E.L., C.M.D., K.W., S.N., N.J.A., and S.K. conceived and designed research; D.E.L., C.M.D., K.W., Y.Z., Z.C., M.N., K.L.S., L.L., S.N., and S.K. performed experiments; D.E.L., C.M.D., K.W., Y.Z., Z.C., K.L.S., L.L., S.N., N.J.A., and S.K. analyzed data; D.E.L., C.M.D., K.W., Z.C., S.N., N.J.A., and S.K. interpreted results of experiments; D.E.L., C.M.D., and S.N. prepared figures; D.E.L., C.M.D., K.W., Z.C., S.N., N.J.A., and S.K. drafted manuscript; D.E.L., C.M.D., S.N., N.J.A., and S.K. edited and revised manuscript; D.E.L., C.M.D., S.N., N.J.A., and S.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Dennis Koop and Jenny Luo of the Bioanalytical Shared Resource/Pharmacokinetics Core University Shared Resources for assistance with adenosine measurements. We are most grateful to Dr. Luiz Belardinelli for critical review of the manuscript.
REFERENCES
- 1.Adair TH. Growth regulation of the vascular system: an emerging role for adenosine. Am J Physiol Regul Integr Comp Physiol 289: R283–R296, 2005. doi: 10.1152/ajpregu.00840.2004. [DOI] [PubMed] [Google Scholar]
- 2.Allely MC, Brown CM, Kenny BA, Kilpatrick AT, Martin A, Spedding M. Modulation of alpha 1-adrenoceptors in rat left ventricle by ischaemia and acyl carnitines: protection by ranolazine. J Cardiovasc Pharmacol 21: 869–873, 1993. doi: 10.1097/00005344-199306000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Ball RM, Bache RJ. Distribution of myocardial blood flow in the exercising dog with restricted coronary artery inflow. Circ Res 38: 60–66, 1976. doi: 10.1161/01.RES.38.2.60. [DOI] [PubMed] [Google Scholar]
- 4.Bin JP, Le E, Pelberg RA, Coggins MP, Wei K, Kaul S. Mechanism of inducible regional dysfunction during dipyridamole stress. Circulation 106: 112–117, 2002. doi: 10.1161/01.CIR.0000020223.08390.05. [DOI] [PubMed] [Google Scholar]
- 5.Boucher M, Pesant S, Falcao S, de Montigny C, Schampaert E, Cardinal R, Rousseau G. Post-ischemic cardioprotection by A2A adenosine receptors: dependent of phosphatidylinositol 3-kinase pathway. J Cardiovasc Pharmacol 43: 416–422, 2004. doi: 10.1097/00005344-200403000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Cao Z, Liu L, Van Winkle DM. Met5-enkephalin-induced cardioprotection occurs via transactivation of EGFR and activation of PI3K. Am J Physiol Heart Circ Physiol 288: H1955–H1964, 2005. doi: 10.1152/ajpheart.00256.2004. [DOI] [PubMed] [Google Scholar]
- 7.Chaitman BR. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation 113: 2462–2472, 2006. doi: 10.1161/CIRCULATIONAHA.105.597500. [DOI] [PubMed] [Google Scholar]
- 8.Dobson JG Jr, Schrader J. Role of extracellular and intracellular adenosine in the attenuation of catecholamine evoked responses in guinea pig heart. J Mol Cell Cardiol 16: 813–822, 1984. doi: 10.1016/S0022-2828(84)80005-X. [DOI] [PubMed] [Google Scholar]
- 9.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47: 1739–1749, 2004. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 10.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49: 6177–6196, 2006. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 11.Gallier F, Lallemand P, Meurillon M, Jordheim LP, Dumontet C, Périgaud C, Lionne C, Peyrottes S, Chaloin L. Structural insights into the inhibition of cytosolic 5′-nucleotidase II (cN-II) by ribonucleoside 5′-monophosphate analogues. PLOS Comput Biol 7: e1002295, 2011. doi: 10.1371/journal.pcbi.1002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47: 1750–1759, 2004. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 13.Herrmann SC, Feigl EO. Adrenergic blockade blunts adenosine concentration and coronary vasodilation during hypoxia. Circ Res 70: 1203–1216, 1992. doi: 10.1161/01.RES.70.6.1203. [DOI] [PubMed] [Google Scholar]
- 14.Hood WP Jr, Rackley CE, Rolett EL. Wall stress in the normal and hypertrophied human left ventricle. Am J Cardiol 22: 550–558, 1968. doi: 10.1016/0002-9149(68)90161-6. [DOI] [PubMed] [Google Scholar]
- 15.Kin H, Zatta AJ, Lofye MT, Amerson BS, Halkos ME, Kerendi F, Zhao Z-Q, Guyton RA, Headrick JP, Vinten-Johansen J. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res 67: 124–133, 2005. doi: 10.1016/j.cardiores.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Kitakaze M, Hori M, Tamai J, Iwakura K, Koretsune Y, Kagiya T, Iwai K, Kitabatake A, Inoue M, Kamada T. Alpha 1-adrenoceptor activity regulates release of adenosine from the ischemic myocardium in dogs. Circ Res 60: 631–639, 1987. doi: 10.1161/01.RES.60.5.631. [DOI] [PubMed] [Google Scholar]
- 17.Klocke FJ. Ranolazine and the myocardial demand-supply balance. JACC Cardiovasc Imaging 2: 1310–1312, 2009. doi: 10.1016/j.jcmg.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Knapp K, Zebisch M, Pippel J, El-Tayeb A, Müller CE, Sträter N. Crystal structure of the human ecto-5′-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure 20: 2161–2173, 2012. doi: 10.1016/j.str.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Lasley RD, Jahania MS, Mentzer RM Jr. Beneficial effects of adenosine A(2a) agonist CGS-21680 in infarcted and stunned porcine myocardium. Am J Physiol Heart Circ Physiol 280: H1660–H1666, 2001. doi: 10.1152/ajpheart.2001.280.4.H1660. [DOI] [PubMed] [Google Scholar]
- 20.Layland J, Carrick D, Lee M, Oldroyd K, Berry C. Adenosine: physiology, pharmacology, and clinical applications. JACC Cardiovasc Interv 7: 581–591, 2014. doi: 10.1016/j.jcin.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Leier CV, Unverferth DV, Kates RE. The relationship between plasma dobutamine concentrations and cardiovascular responses in cardiac failure. Am J Med 66: 238–242, 1979. doi: 10.1016/0002-9343(79)90537-0. [DOI] [PubMed] [Google Scholar]
- 22.Létienne R, Vié B, Puech A, Vieu S, Le Grand B, John GW. Evidence that ranolazine behaves as a weak beta1- and beta2-adrenoceptor antagonist in the rat cardiovascular system. Naunyn Schmiedebergs Arch Pharmacol 363: 464–471, 2001. doi: 10.1007/s002100000378. [DOI] [PubMed] [Google Scholar]
- 23.Linka AZ, Sklenar J, Wei K, Jayaweera AR, Skyba DM, Kaul S. Assessment of transmural distribution of myocardial perfusion with contrast echocardiography. Circulation 98: 1912–1920, 1998. doi: 10.1161/01.CIR.98.18.1912. [DOI] [PubMed] [Google Scholar]
- 24.Mehta PK, Goykhman P, Thomson LE, Shufelt C, Wei J, Yang Y, Gill E, Minissian M, Shaw LJ, Slomka PJ, Slivka M, Berman DS, Bairey Merz CN. Ranolazine improves angina in women with evidence of myocardial ischemia but no obstructive coronary artery disease. JACC Cardiovasc Imaging 4: 514–522, 2011. doi: 10.1016/j.jcmg.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrison RR, Talukder MA, Ledent C, Mustafa SJ. Cardiac effects of adenosine in A(2A) receptor knockout hearts: uncovering A(2B) receptors. Am J Physiol Heart Circ Physiol 282: H437–H444, 2002. doi: 10.1152/ajpheart.00723.2001. [DOI] [PubMed] [Google Scholar]
- 26.Möser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol Cell Physiol 256: C799–C806, 1989. doi: 10.1152/ajpcell.1989.256.4.C799. [DOI] [PubMed] [Google Scholar]
- 27.Nieminen T, Tavares CAM, Pegler JR, Belardinelli L, Verrier RL. Ranolazine injection into coronary or femoral arteries exerts marked, transient regional vasodilation without systemic hypotension in an intact porcine model. Circ Cardiovasc Interv 4: 481–487, 2011. doi: 10.1161/CIRCINTERVENTIONS.111.962852. [DOI] [PubMed] [Google Scholar]
- 28.Norton ED, Jackson EK, Turner MB, Virmani R, Forman MB. The effects of intravenous infusions of selective adenosine A1-receptor and A2-receptor agonists on myocardial reperfusion injury. Am Heart J 123: 332–338, 1992. doi: 10.1016/0002-8703(92)90643-A. [DOI] [PubMed] [Google Scholar]
- 29.Olanrewaju HA, Qin W, Feoktistov I, Scemama JL, Mustafa SJ. Adenosine A(2A) and A(2B) receptors in cultured human and porcine coronary artery endothelial cells. Am J Physiol Heart Circ Physiol 279: H650–H656, 2000. doi: 10.1152/ajpheart.2000.279.2.H650. [DOI] [PubMed] [Google Scholar]
- 30.Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res 70: 308–314, 2006. doi: 10.1016/j.cardiores.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 31.Ren J, Mi Z, Jackson EK. Assessment of nerve stimulation-induced release of purines from mouse kidneys by tandem mass spectrometry. J Pharmacol Exp Ther 325: 920–926, 2008. doi: 10.1124/jpet.108.137752. [DOI] [PubMed] [Google Scholar]
- 32.Rinkevich D, Belcik T, Gupta NC, Cannard E, Alkayed NJ, Kaul S. Coronary autoregulation is abnormal in syndrome X: insights using myocardial contrast echocardiography. J Am Soc Echocardiogr 26: 290–296, 2013. doi: 10.1016/j.echo.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Safran N, Shneyvays V, Balas N, Jacobson KA, Nawrath H, Shainberg A. Cardioprotective effects of adenosine A1 and A3 receptor activation during hypoxia in isolated rat cardiac myocytes. Mol Cell Biochem 217: 143–152, 2001. doi: 10.1023/A:1007209321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma K, Singh R, Giri S, Rajagopal S, Mullangi R. Highly sensitive method for the determination of adenosine by LC-MS/MS-ESI: method validation and scope of application to a pharmacokinetic/pharmacodynamic study. Biomed Chromatogr 26: 81–88, 2012. doi: 10.1002/bmc.1629. [DOI] [PubMed] [Google Scholar]
- 35.Shepherd JT, Vanhoutte PM. George E. Brown memorial lecture. Local modulation of adrenergic neurotransmission. Circulation 64: 655–666, 1981. doi: 10.1161/01.CIR.64.4.655. [DOI] [PubMed] [Google Scholar]
- 36.Sherman W, Beard HS, Farid R. Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des 67: 83–84, 2006. doi: 10.1111/j.1747-0285.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 37.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49: 534–553, 2006. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 38.Shryock JC, Boykin MT, Hill JA, Belardinelli L. A new method of sampling blood for measurement of plasma adenosine. Am J Physiol Heart Circ Physiol 258: H1232–H1239, 1990. doi: 10.1152/ajpheart.1990.258.4.H1232. [DOI] [PubMed] [Google Scholar]
- 39.Sklenar J, Jayaweera AR, Kaul S. A computer-aided approach for the quantitation of regional left ventricular function using two-dimensional echocardiography. J Am Soc Echocardiogr 5: 33–40, 1992. doi: 10.1016/S0894-7317(14)80100-4. [DOI] [PubMed] [Google Scholar]
- 40.Solenkova NV, Solodushko V, Cohen MV, Downey JM. Endogenous adenosine protects preconditioned heart during early minutes of reperfusion by activating Akt. Am J Physiol Heart Circ Physiol 290: H441–H449, 2006. doi: 10.1152/ajpheart.00589.2005. [DOI] [PubMed] [Google Scholar]
- 41.Stambaugh K, Jacobson KA, Jiang JL, Liang BT. A novel cardioprotective function of adenosine A1 and A3 receptors during prolonged simulated ischemia. Am J Physiol Heart Circ Physiol 273: H501–H505, 1997. doi: 10.1152/ajpheart.1997.273.1.H501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stone PH, Chaitman BR, Stocke K, Sano J, DeVault A, Koch GG. The anti-ischemic mechanism of action of ranolazine in stable ischemic heart disease. J Am Coll Cardiol 56: 934–942, 2010. doi: 10.1016/j.jacc.2010.04.042. [DOI] [PubMed] [Google Scholar]
- 43.Takemoto Y, Pellikka PA, Wang J, Modesto KM, Cauduro S, Belohlavek M, Seward JB, Thomson HL, Khandheria B, Abraham TP. Analysis of the interaction between segmental relaxation patterns and global diastolic function by strain echocardiography. J Am Soc Echocardiogr 18: 901–906, 2005. doi: 10.1016/j.echo.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 44.Villano A, Di Franco A, Nerla R, Sestito A, Tarzia P, Lamendola P, Di Monaco A, Sarullo FM, Lanza GA, Crea F. Effects of ivabradine and ranolazine in patients with microvascular angina pectoris. Am J Cardiol 112: 8–13, 2013. doi: 10.1016/j.amjcard.2013.02.045. [DOI] [PubMed] [Google Scholar]
- 45.Vogel R, Indermühle A, Reinhardt J, Meier P, Siegrist PT, Namdar M, Kaufmann PA, Seiler C. The quantification of absolute myocardial perfusion in humans by contrast echocardiography: algorithm and validation. J Am Coll Cardiol 45: 754–762, 2005. doi: 10.1016/j.jacc.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 46.Walldén K, Stenmark P, Nyman T, Flodin S, Gräslund S, Loppnau P, Bianchi V, Nordlund P. Crystal structure of human cytosolic 5′-nucleotidase II: insights into allosteric regulation and substrate recognition. J Biol Chem 282: 17828–17836, 2007. doi: 10.1074/jbc.M700917200. [DOI] [PubMed] [Google Scholar]
- 47.Wei K, Jayaweera AR, Firoozan S, Linka A, Skyba DM, Kaul S. Quantification of myocardial blood flow with ultrasound-induced destruction of microbubbles administered as a constant venous infusion. Circulation 97: 473–483, 1998. doi: 10.1161/01.CIR.97.5.473. [DOI] [PubMed] [Google Scholar]
- 48.Wei K, Le E, Bin JP, Coggins M, Goodman NC, Kaul S. Detection of noncritical coronary stenosis at rest without recourse to exercise or pharmacological stress. Circulation 105: 218–223, 2002. doi: 10.1161/hc0202.101986. [DOI] [PubMed] [Google Scholar]
- 49.Wei K, Tong KL, Belcik T, Rafter P, Ragosta M, Wang XQ, Kaul S. Detection of coronary stenoses at rest with myocardial contrast echocardiography. Circulation 112: 1154–1160, 2005. doi: 10.1161/CIRCULATIONAHA.104.513887. [DOI] [PubMed] [Google Scholar]
- 50.Xi J, McIntosh R, Shen X, Lee S, Chanoit G, Criswell H, Zvara DA, Xu Z. Adenosine A2A and A2B receptors work in concert to induce a strong protection against reperfusion injury in rat hearts. J Mol Cell Cardiol 47: 684–690, 2009. doi: 10.1016/j.yjmcc.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao G, Walsh E, Shryock JC, Messina E, Wu Y, Zeng D, Xu X, Ochoa M, Baker SP, Hintze TH, Belardinelli L. Antiadrenergic and hemodynamic effects of ranolazine in conscious dogs. J Cardiovasc Pharmacol 57: 639–647, 2011. doi: 10.1097/FJC.0b013e31821458e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem J 285: 345–365, 1992. doi: 10.1042/bj2850345. [DOI] [PMC free article] [PubMed] [Google Scholar]