

Keywords: glucagon, gut amino acid production, hepatic glucose metabolism, type 1 diabetes
Abstract
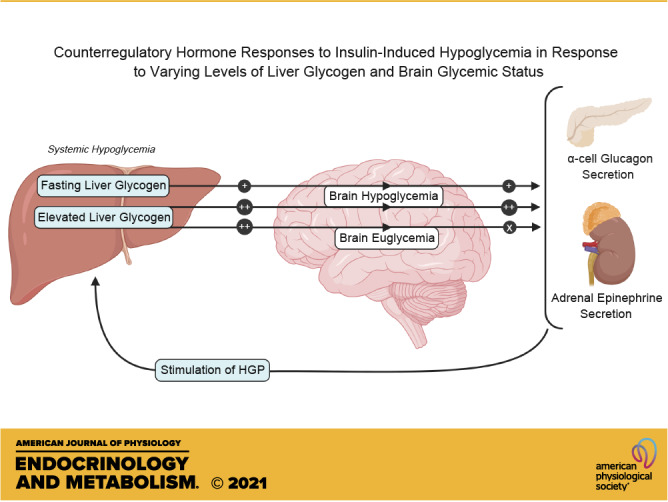
Iatrogenic hypoglycemia is a prominent barrier to achieving optimal glycemic control in patients with diabetes, in part due to dampened counterregulatory hormone responses. It has been demonstrated that elevated liver glycogen content can enhance these hormonal responses through signaling to the brain via afferent nerves, but the role that hypoglycemia in the brain plays in this liver glycogen effect remains unclear. During the first 4 h of each study, the liver glycogen content of dogs was increased by using an intraportal infusion of fructose to stimulate hepatic glucose uptake (HG; n = 13), or glycogen was maintained near fasting levels with a saline infusion (NG; n = 6). After a 2-h control period, during which the fructose/saline infusion was discontinued, insulin was infused intravenously for an additional 2 h to bring about systemic hypoglycemia in all animals, whereas brain euglycemia was maintained in a subset of the HG group by infusing glucose bilaterally into the carotid and vertebral arteries (HG-HeadEu; n = 7). Liver glycogen content was markedly elevated in the two HG groups (43 ± 4, 73 ± 3, and 75 ± 7 mg/g in NG, HG, and HG-HeadEu, respectively). During the hypoglycemic period, arterial plasma glucose levels were indistinguishable between groups (53 ± 2, 52 ± 1, and 51 ± 1 mg/dL, respectively), but jugular vein glucose levels were kept euglycemic (88 ± 5 mg/dL) only in the HG-HeadEu group. Glucagon and epinephrine responses to hypoglycemia were higher in HG compared with NG, whereas despite the increase in liver glycogen, neither increased above basal in HG-HeadEu. These data demonstrate that the enhanced counterregulatory hormone secretion that accompanies increased liver glycogen content requires hypoglycemia in the brain.
NEW & NOTEWORTHY It is well known that iatrogenic hypoglycemia is a barrier to optimal glycemic regulation in patients with diabetes. Our data confirm that increasing liver glycogen content 75% above fasting levels enhances hormonal responses to insulin-induced hypoglycemia and demonstrate that this enhanced hormonal response does not occur in the absence of hypoglycemia in the brain. These data demonstrate that information from the liver regarding glycogen availability is integrated in the brain to optimize the counterregulatory response.
INTRODUCTION
Healthy humans who fast are remarkably capable of maintaining their blood glucose level within a very narrow range of ∼90–100 mg/dL. To achieve this, the liver makes glucose through two different pathways. Approximately one-half of hepatic glucose production (HGP) comes from gluconeogenesis and the other half from the breakdown of glycogen, after which the newly formed glucose is released into the blood at a rate that matches whole body glucose utilization (∼2.2 mg/kg/min; Refs. 1, 2). As a result of this highly efficient process, the supply of glucose to the brain, which accounts for 50–60% of whole body glucose utilization during fasting (3–6), is kept constant and hypoglycemia is extremely rare.
Powerful counterregulatory mechanisms prevent neuroglucopenia in normal people. The first line of defense against a fall in blood glucose is a decrease in endogenous insulin secretion (7), which increases HGP and reduces glucose utilization. Should the fall in blood glucose persist, glucagon is secreted first, followed later by an increase in adrenergic drive (8–13). These well-coordinated counterregulatory responses (CRR) are inversely responsive to the magnitude of the fall in blood glucose (14) and rapidly stimulate HGP by mobilizing liver glycogen stores and stimulating gluconeogenesis, while at the same time inhibiting insulin-induced glucose utilization.
Type 1 diabetes (T1D) is an autoimmune disease that results in insulin deficiency and impaired regulation of blood glucose homeostasis. Treatment of T1D includes the need for self-administration of insulin subcutaneously, which often leads to dose overestimations as a result of misjudging factors such as carbohydrate intake. In that case, insulin levels in the blood cannot be lowered, increasing the risk of iatrogenic hypoglycemia. In addition, the hypoglycemia-induced rise in glucagon secretion is often absent in individuals with T1D and the rise in epinephrine is also often diminished (9, 14–17). This leads to symptomatic hypoglycemia approximately two times per week on average, and severe hypoglycemia (requiring assistance) once a year (18). Such frequent bouts of debilitating hypoglycemia make it understandable why the fear of low blood sugar is among the most prominent barriers to optimal glycemic management in patients with T1D (19, 20).
Given the brain’s dependence on glucose, its exposure to hypoglycemia is known to be an important stimulus of the CRR. In support of this, preserving euglycemia in the brain during whole body insulin-induced hypoglycemia eliminates the glucagon secretory response and severely diminishes epinephrine and cortisol responses when liver glycogen is at overnight fasting levels (21). On the other hand, elegant studies by Donovan and colleagues (22–25) showed that glucose infusion into the portal vein during hypoglycemia can lower the CRR to hypoglycemia via signaling from the portal-mesenteric vein afferents. These data demonstrate that while glucose sensing in the brain is an important component of CRR during hypoglycemia, feedback from the periphery can also be integrated by the brain as the appropriate CRR is orchestrated. In previous work, we showed that a 75% increase in liver glycogen content above morning levels, such as normally occurs over the course of a day (0700–2200 h; Ref. 26), augmented the secretion of glucagon and epinephrine during insulin-induced hypoglycemia (27). Moreover, we showed that under this condition of glycogen repletion, severing the afferent nerves from the liver to the brain eliminated the glycogen-driven increment in the CRR, thereby raising the possibility that this portion of the CRR might be regulated independently of brain hypoglycemia. To this end, the purpose of this study was to determine whether the enhanced signal from the liver to the brain that is induced when liver glycogen levels are high remains capable of regulating the CRR when the brain is kept euglycemic.
DESIGN METHODS
Animals Surgical Procedures
Studies were carried out on healthy, conscious 18-h fasted mongrel dogs of either sex with a mean weight of 21.1 ± 1.7 kg (means ± SD). All animals consumed a diet of meat and chow (34% protein, 14.5% fat, 46% carbohydrate, and 5.5% fiber based on dry weight; ∼1,700 kcal/day). The housing for the animals met American Association for Accreditation of Laboratory Animal Care guidelines, and the study was approved by Vanderbilt’s Institutional Animal Care and Use Committee (Nashville, TN).
Approximately 2 wk before being studied, each dog underwent general anesthesia and a laparotomy to permit placement of catheters for intraportal infusion of hormones and substrates and for blood sampling across the liver (28). At the same time ultrasonic flow probes (Transonic Systems, Ithaca, NY) were placed around the hepatic portal vein and the hepatic artery as described elsewhere (28). One week later, animals from a subset of HG group in which fructose was infused intraportally (the HG-HeadEu group; described later) also had infusion catheters placed bilaterally into both carotid and both vertebral arteries for glucose infusion and a sampling catheter into the jugular vein for periodic sampling as previously described (21).
Two days before each experiment, blood was drawn to measure leukocyte and hematocrit counts for each animal. Animals were only studied if they had a leukocyte count <16,000/mm3, a hematocrit >35%, a good appetite, and normal stools.
On the morning of each experiment, catheters and flow probe leads were exteriorized from subcutaneous pockets under local anesthesia (2% lidocaine, Hospira, Lake Forest, IL). Angiocatheters (Deseret Medical, Becton Dickinson, Sandy, UT) were inserted into a cephalic and a saphenous vein to allow infusions into the peripheral (Pe) vasculature as desired. The animals stood in a Pavlov harness throughout the experiment.
Experimental Design
Each experiment consisted of a 4-h liver glycogen manipulation period (_360 to _120 min; Fig. 1), a 2-h control period (_120 to 0 min), and then a 2-h hypoglycemic period (0 to 120 min). Each experiment was initiated at minute −360 by a leg vein infusion (Pe) of somatostatin (SRIF; 0.8 µg/kg/min; Bachem, Torrance, CA) to disable the endocrine pancreas. Immediately after the SRIF infusion was started, intraportal (Po) infusions of insulin (0.3 mU/kg/min; Eli Lilly & Co., Indianapolis, IN) and then glucagon (0.55 ng/kg/min; Glucagen, Novo Nordisk, Bagsvaerd, Denmark) were started at basal rates. The arterial plasma glucose level was then doubled by a Pe infusion of 50% dextrose as needed. At the same time, either 0.9% saline was infused Po to maintain liver glycogen levels close to a normal, fasting level (NG; n = 6), or fructose was infused Po (1.3 mg/kg/min; HG; n = 13) to markedly stimulate net hepatic glucose uptake (NHGU) and glycogen deposition (27, 29–31), by stimulating the translocation of hepatic glucokinase from the nucleus to the cytosol (where glucose phosphorylation occurs).
Figure 1.

Research design schematic for the metabolic studies. Each animal underwent an 8-h study divided into a 4-h glycogen deposition period, a 2-h control period, and a 2-h hypoglycemic experimental period. During the glycogen deposition period, all animals underwent a pancreatic clamp [using somatostatin (SRIF)], during which hyperglycemia was brought about with a peripheral (Pe) infusion of glucose, and both insulin (INS) and glucagon (GGN) were replaced intraportally (Po) in basal amounts. One group (NG) received Po saline, while HG and HG-HeadEu received a Po infusion of fructose (1.3 mg/kg/min) to increase hepatic glucose uptake and glycogen deposition. This glycogen deposition period was followed by a 2-h control period during which the saline or fructose infusions were discontinued. Then a 2-h hypoglycemic period ensued. During this final period, a Pe infusion of insulin was started and the arterial glucose level was clamped at ∼50 mg/dL in NG and HG. The HG-HeadEu group underwent a similar hypoglycemic challenge, with the exception that glucose was infused into the carotid and vertebral arteries to maintain brain euglycemia.
The 4-h glycogen deposition period was followed by a 2-h control period during which the fructose (or saline) infusion was discontinued, thereby allowing blood fructose concentrations and hepatic glucose metabolism in the HG groups to return to values similar to the NG group. The basal hormone levels were maintained during the control period. At the outset of the final 2-h hypoglycemic experimental period (0–120 min), the SRIF infusion was stopped, thereby restoring normal endogenous glucagon secretion. At the same time, the Po infusions of glucagon and insulin were stopped and a Pe infusion of insulin (5.0 mU/kg/min; 16 times basal) was started to bring about hypoglycemia (∼50 mg/dL) in all animals. Fifty percent dextrose was infused Pe, as required, to clamp the plasma glucose level at ∼50 mg/dL.
To determine the contribution of hypoglycemia in the brain to the increased CRR seen when hepatic glycogen concentrations are elevated, one subgroup of HG animals (HG-HeadEu; n = 7) received a bilateral infusion of 20% dextrose into the carotid and vertebral artery catheters. This made the brain euglycemic during the experimental period, while the rest of the body was hypoglycemic. In the other HG group (HG), the head glucose was allowed to fall. The final sample size for NG, HG, and HG-HeadEu was 6, 6, and 7, respectively. At the conclusion of each study, animals were euthanized with pentobarbital, the abdomen was opened and the positions of the catheter tips were verified. Liver biopsies were freeze clamped, and stored at −80°C for the determination of glycogen content.
Analysis of Samples
Plasma glucose was analyzed using the glucose oxidase method (Analox Instruments; Lunenburg, MA). Insulin and glucagon were measured using commercially available radioimmunoassay kits (Millipore), and cortisol was measured using a radioimmunoassay protocol developed by the Vanderbilt University Medical Center Hormone Assay and Analytical Services core using an in-house antibody (W. Nicholson) and 125I-cortisol (MP Biomedicals). Catecholamines and amino acids (AAs) were measured using high-performance liquid chromatography, and lactate, glycerol, and nonesterified fatty acid (NEFA) concentrations were measured using colorimetric assays. Liver samples were assayed for glycogen using the enzymatic method of Keppler and Decker (32).
Calculations Data Analysis
Hepatic blood flow was measured using ultrasonic flow probes (Transonic Systems; Ithaca, NY). Net hepatic balance of substrates (NHSB) was calculated as NHSB = Loadout – Loadin, where Loadout = (FH × SH) and Loadin = [(FHA × SHA) + (FPV × SPV)], where F and S refer to the blood/plasma flow and substrate concentration, respectively, of the hepatic artery (HA), hepatic portal vein (PV), or hepatic vein (H). Net hepatic gluconeogenesis was calculated as one-half the sum of the net hepatic uptake of gluconeogenic amino acids (serine, threonine, glycine, and alanine), lactate, and glycerol, where one-half the sum is to account for the conversion of these three carbon substrates to six carbon glucose minus 0.2 mg/kg/min to account for the rate of hepatic glucose oxidation. Net hepatic glycogenolysis was calculated as net hepatic glucose balance minus net hepatic gluconeogenesis. Hepatic sinusoidal insulin and glucagon levels were calculated as [(HRMHA × %FHA) + (HRMPV × %FPV)], where HRMHA and HRMPV refer to the hormone concentration in the hepatic artery and hepatic portal vein, respectively, and %FHA and %FPV refer to the percentage of total hepatic blood flow accounted for by the hepatic artery and hepatic portal vein, respectively. Plasma glucose levels were converted to whole blood values for the calculation of NHGB by multiplying by 0.74 (33, 34). The average rate of nonhepatic glucose uptake between two time points (T1 and T2) was calculated as the sum of the average net hepatic glucose balance between T1 and T2 and the average exogenous glucose infusion rate between T1 and T2 minus the change in blood glucose mass from T1 to T2. Prehypoglycemic liver glycogen concentrations were calculated by subtracting net hepatic glycogen breakdown during the 2-h hypoglycemia period from the post-experimental liver glycogen level. While jugular vein glucose levels were measured directly in HG-HeadEu, the levels for NG and HG were calculated to be 83% of arterial values based on the work of Biggers et al. (21).
Statistical Analysis
All data are presented as means ± SE unless stated otherwise, and statistical analyses were performed using SigmaStat (Aspire Software International; Ashburn, VA) software. Data were analyzed using ANOVA with repeated measures, and post hoc comparisons were made using the Student-Newman-Keuls test. Statistical significance was set at P < 0.05.
RESULTS
Glycogen Deposition Period
During the 4-h glycogen deposition period (Fig. 1; min −360 to min −120), arterial plasma glucose levels were two times basal levels and similar in each group, while arterial and hepatic sinusoidal levels of insulin and glucagon were also clamped at basal (Table 1). Net hepatic glucose uptake (NHGU) was low (1.8 ± 0.5 mg/kg/min) in NG because insulin was basal. However, it was fourfold higher in HG and HG-HeadEu as a result of the fructose infusion (Table 1). The resultant liver glycogen concentrations in the two HG groups were ∼70% greater than NG at the start of the hypoglycemic period (P < 0.01 for HG and HG-HeadEu compared with NG; Fig. 2A).
Table 1.
Metabolic parameters at the end of the glycogen manipulation period (i.e., minute −120)
| Group | Arterial Plasma Glucose, mg/dL | Arterial Plasma Insulin, µU/mL | Hepatic Sinusoidal Insulin, µU/mL | Arterial Plasma Glucagon, pg/mL | Hepatic Sinusoidal Glucagon, pg/mL | Total Hepatic Blood Flow, mL/kg/min | Net Hepatic Glucose Uptake, mg/kg/min |
|---|---|---|---|---|---|---|---|
| NG | 210 ± 2 | 7.9 ± 0.4 | 26 ± 2 | 24 ± 3 | 35 ± 3 | 26 ± 2 | 1.8 ± 0.5 |
| HG | 217 ± 3 | 8.1 ± 1.1 | 23 ± 4 | 29 ± 3 | 41 ± 5 | 25 ± 2 | 7.6 ± 0.3* |
| HG-HeadEu | 218 ± 3 | 7.7 ± 0.6 | 21 ± 2 | 23 ± 4 | 36 ± 4 | 26 ± 1 | 8.0 ± 0.7* |
HG, intraportal (Po) infusion of fructose was used to stimulate hepatic glucose uptake; NG, saline was infused Po to maintain liver glycogen levels close to a normal, fasting level; HG-HeadEu, subset of HG group in which brain euglycemia was maintained by infusing glucose bilaterally into the carotid and vertebral arteries. *P < 0.05, compared with NG.
Figure 2.
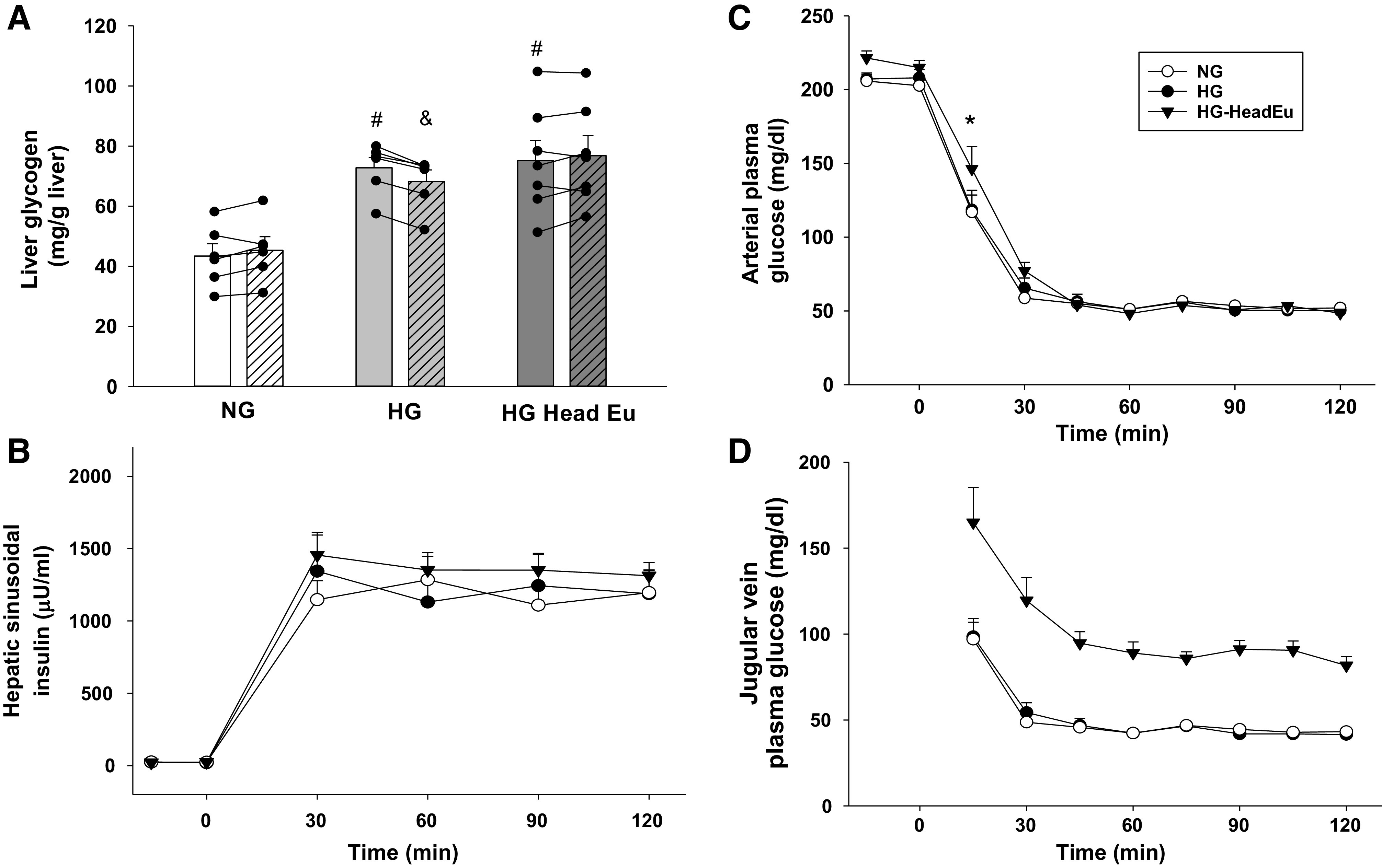
Metabolic control during the hyperinsulinemic/hypoglycemic clamp studies. Prehypoglycemia (open bars) and posthypoglycemia (hatched bars) liver glycogen content (A), hepatic sinusoidal insulin levels (B), arterial plasma glucose levels (C), and jugular vein plasma glucose levels (D) during the experimental period. HG, intraportal (Po) infusion of fructose was used to stimulate hepatic glucose uptake; NG, saline was infused Po to maintain liver glycogen levels close to a normal, fasting level; HG-HeadEu, subset of HG group in which brain euglycemia was maintained by infusing glucose bilaterally into the carotid and vertebral arteries. *P < 0.05, HG-HeadEu compared with NG. #P < 0.05, compared with the same time point in NG. &P < 0.01, compared with the preexperimental value. Data were analyzed using two-way repeated measures ANOVA.
Hypoglycemic Experimental Period
Glucose metabolism.
At the outset of the hypoglycemic experimental period, the somatostatin and glucagon infusions were discontinued and the Pe infusion rate of insulin was increased. As a result, hepatic sinusoidal insulin levels were increased to a similar extent in each group (Fig. 2B), and the plasma glucose level was allowed to fall over a 30-min period to ∼50 mg/dL (Fig. 2C). The slightly higher plasma glucose level at the end of the control period in HG-HeadEu, along with the carotid and vertebral artery glucose infusions, caused their plasma glucose level to be slightly higher at the 15-min time point compared with NG and HG (Fig. 2C; P < 0.05). However, this did not impact the rate of fall of glucose between groups over the first 30 min of the hypoglycemic period (4.8 ± 0.2, 4.8 ± 0.4, and 4.6 ± 0.2 mg/dL/min in NG, HG and HG-HeadEu, respectively; P = NS). Thereafter, the plasma glucose levels became indistinguishable in the three groups. Jugular vein glucose levels were significantly higher in the HG-HeadEu group for the entirety of the hypoglycemic period (Fig. 2D; P< 0.001 for each time point). The jugular vein glucose level in HG-HeadEu averaged 88 ± 5 mg/dL (range at each time point of 82–91 mg/dL).
Increased liver glycogen content resulted in higher glucagon and epinephrine levels in HG compared with NG (Fig. 3, A, B, and D). Despite the same systemic hypoglycemic stimulus, brain euglycemia in HG-HeadEu prevented any increase above basal in the levels of both epinephrine and glucagon during this period, not just the increase caused by elevated liver glycogen content (P > 0.05 at each time point in HG-HeadEu compared with minute 0). Plasma norepinephrine concentrations were also higher in HG compared with NG, especially late in the hypoglycemic period (Fig. 3E). However, as opposed to glucagon and epinephrine, adrenergic drive was only modestly suppressed by euglycemia in the brain (i.e., the response in HG-HeadEu was similar to that of NG; Fig. 3E). The cortisol response to hypoglycemia did not vary based on liver glycogen content but was markedly attenuated by euglycemia in the brain (Fig. 3C).
Figure 3.

Hormonal responses during the hypoglycemic experimental period. Hepatic sinusoidal plasma glucagon (A), arterial glucagon (B), arterial cortisol (C), arterial plasma epinephrine (D), and norepinephrine (norepi; E) levels during the experimental period in NG, HG, and HG-HeadEu groups. HG, intraportal (Po) infusion of fructose was used to stimulate hepatic glucose uptake; NG, saline was infused Po to maintain liver glycogen levels close to a normal, fasting level; HG-HeadEu, subset of HG group in which brain euglycemia was maintained by infusing glucose bilaterally into the carotid and vertebral arteries. *P < 0.05, HG compared with NG. #P < 0.05, HG-HeadEu compared with NG. Data were analyzed using two-way repeated measures ANOVA.
In response to the changing metabolic milieu during the hypoglycemic period, the livers of each group transitioned from taking up glucose at the end of the control period to producing it (Fig. 4A). Net hepatic glucose production was threefold higher in HG compared with NG over the final 90 min of the hypoglycemic period. This increase was accounted for entirely by enhanced glycogen mobilization (Fig. 4B), as net gluconeogenesis increased in both NG and HG but was not different between them (Fig. 4C). As a result of greater glucose output by the liver in HG, the glucose infusion rate required to clamp the glucose at 50 mg/dL was lower compared with NG (Fig. 4D). When liver glycogen content was increased, but brain euglycemia was maintained (HG-HeadEu), lower hormone secretion decreased epinephrine-driven gluconeogenesis (Fig. 4C), but this only had a marginal impact on net hepatic glucose production (Fig. 4A). The lower epinephrine levels in HG-HeadEu also led to a 50% increase in nonhepatic glucose uptake (Table 2), which, when combined with lower net hepatic glucose output, resulted in a greater GIR required to clamp the glucose level (Fig. 4D).
Figure 4.

Metabolic responses during the hypoglycemic experimental period. Net hepatic glucose balance (A), net hepatic glycogenolysis (GLY; B), net hepatic gluconeogenesis (GNG; C), and the peripheral (Pe) glucose infusion rate required to match the glucose levels among the groups (D). HG, intraportal (Po) infusion of fructose was used to stimulate hepatic glucose uptake; NG, saline was infused Po to maintain liver glycogen levels close to a normal, fasting level; HG-HeadEu, subset of HG group in which brain euglycemia was maintained by infusing glucose bilaterally into the carotid and vertebral arteries. *P < 0.05, HG compared with NG. #P < 0.05, HG-HeadEu compared with NG. †P < 0.05, HG-HeadEu compared with NG. ‡P < 0.05, between all 3 groups. Data were analyzed using two-way repeated measures ANOVA.
Table 2.
Total hepatic blood flow, lactate, free fatty acid and glycerol metabolism, and nonhepatic glucose uptake before (−15 to 0 min) and during insulin-induced hypoglycemia (0-120 min)
| Group | Time, min |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control period |
Hypoglycemic experimental period |
|||||||||
| −15 | 0 | 15 | 30 | 45 | 60 | 75 | 90 | 105 | 120 | |
| Total hepatic blood flow, mL/kg/min | ||||||||||
| NG | 27 ± 2 | 27 ± 2 | 32 ± 1 | 32 ± 2 | 38 ± 2 | 44 ± 2 | 45 ± 2 | 46 ± 1 | 47 ± 2 | 44 ± 1 |
| HG | 27 ± 2 | 27 ± 2 | 25 ± 2 | 28 ± 2 | 36 ± 4 | 40 ± 4 | 42 ± 3 | 41 ± 2 | 44 ± 3 | 41 ± 3 |
| HG-HeadEu | 26 ± 1 | 26 ± 1 | 29 ± 2 | 32 ± 2 | 34 ± 1 | 36 ± 3 | 38 ± 3 | 39 ± 3 | 40 ± 4 | 38 ± 3 |
| Arterial blood lactate, µmol/L | ||||||||||
| NG | 681 ± 97 | 723 ± 71 | 843 ± 94 | 979 ± 131 | 1,055 ± 159 | |||||
| HG | 637 ± 79 | 629 ± 118 | 851 ± 81 | 1,113 ± 180 | 1,277 ± 176 | |||||
| HG-HeadEu | 845 ± 57 | 940 ± 99 | 591 ± 36 | 580 ± 62† | 538 ± 80† | |||||
| Net hepatic lactate balance, µmol/kg/min | ||||||||||
| NG | 4.8 ± 1.8 | −0.1 ± 0.6 | −2.5 ± 0.9 | −2.3 ± 1.4 | −2.3 ± 1.3 | |||||
| HG | 6.0 ± 1.1 | 1.5 ± 1.2 | 4.8 ± 2.9* | −2.2 ± 1.6 | −1.7 ± 1.7 | |||||
| HG-HeadEu | 9.0 ± 1.5 | 3.0 ± 1.6 | 2.0 ± 1.4* | 2.8 ± 1.5 | 1.4 ± 1.3 | |||||
| Arterial plasma free fatty acids, µmol/L | ||||||||||
| NG | 417 ± 58 | 144 ± 38 | x405 ± 91 | 521 ± 126 | 404 ± 123 | |||||
| HG | 361 ± 77 | 107 ± 33 | 511 ± 100 | 405 ± 71 | 476 ± 71 | |||||
| HG-HeadEu | 386 ± 58 | 34 ± 15 | 104 ± 53† | 82 ± 24† | 102 ± 42† | |||||
| Net hepatic fee fatty acid balance, µmol/kg/min | ||||||||||
| NG | −1.4 ± 0.2 | −0.1 ± 0.3 | −3.1 ± 1.0 | −2.5 ± 0.9 | −1.5 ± 0.6 | |||||
| HG | −0.7 ± 0.3 | 0.2 ± 0.1 | −1.2 ± 0.4* | −1.0 ± 0.5* | −1.4 ± 0.6 | |||||
| HG-HeadEu | −1.4 ± 0.2 | −0.1 ± 0.3 | −0.7 ± 0.3* | −1.0 ± 0.5 | −0.7 ± 0.7 | |||||
| Arterial blood glycerol, µmol/L | ||||||||||
| NG | 50 ± 5 | 35 ± 6 | 119 ± 12 | 156 ± 19 | 156 ± 26 | |||||
| HG | 53 ± 3 | 45 ± 7 | 156 ± 14 | 192 ± 26 | 189 ± 29 | |||||
| HG-HeadEu | 67 ± 13 | 33 ± 5 | 62 ± 16† | 78 ± 15† | 83 ± 16† | |||||
| Net hepatic glycerol balance, µmol/kg/min | ||||||||||
| NG | −0.8 ± 0.1 | −0.9 ± 0.2 | −3.5 ± 0.4 | −4.6 ± 0.6 | −4.2 ± 1.1 | |||||
| HG | −0.9 ± 0.1 | −0.8 ± 0.2 | −4.1 ± 0.7 | −5.0 ± 0.7 | −4.9 ± 0.9 | |||||
| HG-HeadEu | −0.9 ± 0.2 | −0.5 ± 0.1 | −1.6 ± 0.6† | −2.3 ± 0.5† | −2.3 ± 0.7† | |||||
| Nonhepatic glucose uptake, mg/kg/min | ||||||||||
| NG | 2.5 ± 0.5 | 5.9 ± 0.8 | 7.5 ± 0.9 | 7.3 ± 0.7 | 7.6 ± 0.6 | 7.7 ± 0.6 | 7.2 ± 0.6 | |||
| HG | 2.5 ± 0.9 | 4.7 ± 1.3 | 6.2 ± 1.6 | 7.4 ± 1.2 | 6.4 ± 0.8 | 6.1 ± 1.3 | 5.7 ± 1.1 | |||
| HG-HeadEu | 1.9 ± 0.3 | 8.9 ± 1.0† | 8.8 ± 0.7 | 9.0 ± 0.6 | 9.9 ± 0.7 | 9.5 ± 0.5 | 10.3 ± 0.9† | |||
Positive and negative values for net hepatic substrate balances indicate production and uptake, respectively. HG, intraportal (Po) infusion of fructose was used to stimulate hepatic glucose uptake; NG, saline was infused Po to maintain liver glycogen levels close to a normal, fasting level; HG-HeadEu, subset of HG group in which brain euglycemia was maintained by infusing glucose bilaterally into the carotid and vertebral arteries. *P < 0.05, compared with NG. †P < 0.01, compared with NG and HG.
Hepatic blood flow, and lactate and fat metabolism.
As expected, total hepatic blood flow rose steadily in response to insulin-induced hypoglycemia and was similar in each of the three groups (Table 2). Blood lactate concentrations rose in response to insulin-induced hypoglycemia in NG and HG but fell in HG-HeadEu, due to lower epinephrine concentrations (Table 2). Net hepatic lactate production decreased to a similar extent in all three groups in response to hypoglycemia, with NG and HG transitioning to net lactate uptake, while the HG-HeadEu group continued to release small amounts of lactate throughout the experimental period. After an initial fall due to hyperinsulinemia, plasma free fatty acid and glycerol levels increased in both NG and HG, while each product of lipolysis remained low when euglycemia in the head was maintained. The lower plasma concentrations of free fatty acids and glycerol in HG-HeadEu diminished the uptake of both by the liver, although only the latter was significant (Table 2).
Amino acid metabolism.
In response to the hyperinsulinemic-hypoglycemic challenge, arterial concentrations of the AAs serine, threonine, glycine, and alanine declined similarly in all groups (Table 3). Despite this reduction, their net uptake by the liver increased (Table 3) due to enhanced fractional extraction. As expected, the production of each AA by the gut increased in all three groups during insulin induced hypoglycemia (Table 3). The time effect (where data from all 3 groups were combined) for net gut balance was significant for all four AAs, revealing greater production at min 60, 90, and 120 of hypoglycemia compared with minute 0 (P < 0.05 for each, with the exception of the 90-min data point for threonine, P = 0.10). Net production of serine, glycine, and alanine by the gut was slightly elevated in NG early in the hypoglycemic period (i.e., minute 60), although this difference did not persist throughout the experiment. No other differences in gut AA metabolism were observed between the three groups.
Table 3.
Concentration of amino acids in the blood and their net balance across the liver and gut before (0 min) and during insulin-induced hypoglycemia (0-120 min)
| Group | Time, min |
||||
|---|---|---|---|---|---|
| Control period |
Hypoglycemic experimental period |
||||
| 0 | 30 | 60 | 90 | 120 | |
| Arterial blood serine, µmol/L | |||||
| NG | 89 ± 10 | 76 ± 8 | 84 ± 8 | 73 ± 15 | 58 ± 6 |
| HG | 97 ± 8 | 90 ± 7 | 96 ± 8 | 78 ± 5 | 71 ± 5 |
| HG-HeadEu | 111 ± 9 | 91 ± 8 | 88 ± 6 | 68 ± 10 | 66 ± 5 |
| Net hepatic serine balance, µmol/kg/min | |||||
| NG | −0.5 ± 0.2 | −0.7 ± 0.3 | −1.5 ± 0.4 | −1.3 ± 0.4 | −1.4 ± 0.2 |
| HG | −0.5 ± 0.1 | −0.7 ± 0.1 | −1.3 ± 0.3 | −1.3 ± 0.1 | −1.2 ± 0.2 |
| HG-HeadEu | −0.7 ± 0.1 | −0.9 ± 0.2 | −1.3 ± 0.1 | −1.5 ± 0.1 | −1.5 ± 0.2 |
| Net gut serine balance, µmol/kg/min | |||||
| NG | −0.1 ± 0. 1 | 0.0 ± 0.03 | 1.3 ± 0.2 | 0.5 ± 0.4 | 0. 6 ± 0.1 |
| HG | −0.1 ± 0.1 | 0.0 ± 0.1 | 0.7 ± 0.1 | 0.5 ± 0.2 | 0.7 ± 0.2 |
| HG-HeadEu | −0.3 ± 0.1 | 0.0 ± 0.1 | 0.4 ± 0.2* | 1.4 ± 0.6† | 0.6 ± 0.1 |
| Arterial blood threonine, µmol/L, | |||||
| NG | 123 ± 15 | 100 ± 12 | 105 ± 9 | 84 ± 9 | 70 ± 8 |
| HG | 175 ± 27 | 152 ± 20 | 147 ± 20‡ | 123 ± 16 | 96 ± 15 |
| HG-HeadEu | 159 ± 16 | 118 ± 16 | 94 ± 14 | 98 ± 11 | 77 ± 10 |
| Net hepatic threonine balance, µmol/kg/min | |||||
| NG | 0.0 ± 0.1 | −0.3 ± 0.1 | −0.5 ± 0.2 | −0.5 ± 0.1 | −0.6 ± 0.1 |
| HG | −0.1 ± 0.1 | −0.4 ± 0.1 | −0.2 ± 1.0 | −0.4 ± 0.1 | −0.2 ± 0.3 |
| HG-HeadEu | −0.8 ± 0.6 | −0.5 ± 0.2 | −0.4 ± 0.1 | −0.5 ± 0.1 | −1.0 ± 0.5 |
| Net gut threonine balance, µmol/kg/min | |||||
| NG | −0.3 ± 0.1 | −0.2 ± 0.1 | 0.9 ± 0.2 | 0.4 ± 0.2 | 0.3 ± 0.1 |
| HG | −0.2 ± 0.1 | −0.1 ± 0.1 | 0.1 ± 0.2 | 0.2 ± 0.2 | 1.0 ± 0.6 |
| HG-HeadEu | −0.3 ± 0.1 | −0.2 ± 0.1 | 0.6 ± 0.8 | 0.3 ± 0.3 | 0.7 ± 0.6 |
| Arterial blood glycine, µmol/L | |||||
| NG | 188 ± 23 | 164 ± 19 | 153 ± 15 | 133 ± 16 | 112 ± 10 |
| HG | 214 ± 23 | 191 ± 22 | 179 ± 20 | 144 ± 13 | 126 ± 16 |
| HG-HeadEu | 208 ± 34 | 190 ± 14 | 176 ± 10 | 156 ± 10 | 131 ± 9 |
| Net hepatic glycine balance, µmol/kg/min | |||||
| NG | −0.8 ± 0.1 | −1.3 ± 0.2 | −2.0 ± 0.3 | −2.0 ± 0.3 | −1.7 ± 0.3 |
| HG | −0.8 ± 0.1 | −1.1 ± 0.2 | −1.6 ± 0.3 | −1.7 ± 0.2 | −1.4 ± 0.3 |
| HG-HeadEu | −1.2 ± 0.2 | −1.5 ± 0.2 | −1.7 ± 0.1 | −1.8 ± 0.1 | −2.3 ± 0.5 |
| Net gut glycine balance, µmol/kg/min | |||||
| NG | 0.2 ± 0.1 | 0.3 ± 0.1 | 1.8 ± 0.3 | 0.9 ± 0.4 | 0.8 ± 0.4 |
| HG | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.9 ± 0.2* | 0.7 ± 0.2 | 1.2 ± 0.3 |
| HG-HeadEu | 0.2 ± 0.1 | 0.4 ± 0.2 | 0.6 ± 0.2* | 1.1 ± 0.3 | 1.5 ± 0.6 |
| Arterial blood alanine, µmol/L | |||||
| NG | 477 ± 77‡ | 362 ± 50 | 312 ± 37 | 257 ± 20 | 244 ± 20 |
| HG | 574 ± 51 | 431 ± 32 | 351 ± 28 | 298 ± 16 | 256 ± 11 |
| HG-HeadEu | 691 ± 64 | 395 ± 68 | 336 ± 32 | 268 ± 30 | 207 ± 26 |
| Net hepatic alanine balance, µmol/kg/min | |||||
| NG | −2.8 ± 0.4‡ | −4.0 ± 0.6 | −5.3 ± 0.7 | −5.1 ± 0.5 | −4.3 ± 0.7 |
| HG | −3.0 ± 0.4‡ | −3.4 ± 0.5 | −4.1 ± 0.7 | −4.2 ± 0.3 | −4.2 ± 0.5 |
| HG-HeadEu | −5.4 ± 0.7 | −5.0 ± 0.7 | −4.7 ± 0.3 | −4.9 ± 0.5 | −4.7 ± 0.9 |
| Net gut alanine balance, µmol/kg/min | |||||
| NG | 0.9 ± 0.2 | 1.3 ± 0.3 | 3.9 ± 0.7 | 2.4 ± 0.7 | 1.6 ± 0.8 |
| HG | 1.2 ± 0.5 | 1.1 ± 0.5 | 2.1 ± 0.4* | 1.8 ± 0.5 | 2.5 ± 0.5 |
| HG-HeadEu | 2.4 ± 0.6 | 1.6 ± 0.5 | 1.9 ± 0.8* | 3.3 ± 0.6 | 2.9 ± 0.5 |
Negative values for net hepatic and gut amino acid balances indicate net uptake. HG, intraportal (Po) infusion of fructose was used to stimulate hepatic glucose uptake; NG, saline was infused Po to maintain liver glycogen levels close to a normal, fasting level; HG-HeadEu, subset of HG group in which brain euglycemia was maintained by infusing glucose bilaterally into the carotid and vertebral arteries. *P < 0.05, compared with NG. †P < 0.01, compared with NG and HG. ‡P ≤ 0.05, compared with HG-HeadEu.
DISCUSSION
Fear of iatrogenic hypoglycemia continues to be among the most prominent barriers to effective glycemic control in patients with T1D due to its debilitating effect on key physiological functions, such as cognitive awareness and cardiac rhythm (35). In a previous study (27), we observed that increased liver glycogen content enhanced the CRR to insulin-induced hypoglycemia. Interestingly, when the afferent nerves from the liver to the brain were severed, only the glycogen-driven increment in hormone secretion was lost, not the remainder of the CRR. This raises the possibility that this afferent signal might regulate the CRR independent of those pathways in the brain that regulate the CRR when liver glycogen is at fasting levels. The current results confirm that acutely increasing liver glycogen content augments the glucagon and epinephrine responses to insulin-induced hypoglycemia, thereby resulting in enhanced hepatic glycogen mobilization and a threefold increase in net hepatic glucose output. In contrast, despite an identical systemic hypoglycemic challenge, maintaining euglycemia in the brain when liver glycogen was enhanced completely suppressed the hormonal responses to insulin-induced hypoglycemia. In fact, the glucagon response was absent, while the epinephrine and cortisol levels in the HG-HeadEu group were also markedly diminished. This resulted in hormone levels identical to those reported by Biggers and colleagues (21), who infused insulin into overnight fasted dogs and clamped the head glucose level at 85 mg/dL, similar to our HG-HeadEu group (88 mg/dL). This juxtaposition allows us to conclude that while increased liver glycogen content is capable of augmenting the CRR to hypoglycemia beyond what is seen when liver glycogen is at fasting levels, this signal is not independent and instead is integrated with other hypoglycemia-derived cues by the brain, after which delivery of the optimal CRR occurs. Moreover, in the absence of neuroglucopenia, the brain is fully capable of muting this signal.
As a result of the head glucose infusion, plasma concentrations of glycerol and fatty acids were greatly diminished. Given the importance of adrenergic drive to lipolysis during insulin-induced hypoglycemia (36, 37), the removal of significant metabolic stress in the brain likely contributed to diminished fat mobilization in HG-HeadEu. Importantly, however, lipolysis was driven down even further by the reduction in epinephrine secretion. Fanelli and colleagues (38) demonstrated that inhibiting lipolysis in healthy humans during prolonged insulin-induced hypoglycemia reduced endogenous glucose production and alanine-derived gluconeogenesis despite identical plasma insulin concentrations. This suggests that diminished lipolysis in HG-HeadEu likely contributed to the lower rates of gluconeogenesis seen in this group.
In contrast with their effect on glucose metabolism, neither head glucose infusion nor liver glycogen content impacted amino acid metabolism. Previous elegant studies have shown that over one-half of whole body proteolysis during hypoglycemia is accounted for by the gut, with the AAs being first delivered to the liver via the hepatic portal vein (39). In a follow-up study, the same authors used a head glucose infusion, similar to what we used in the current studies, to test the hypothesis that euglycemia in the brain would diminish leucine mobilization by the gut during insulin-induced hypoglycemia (40). Their use of glucose infusions into the arteries of the head reduced catecholamine and cortisol levels and eliminated the glucagon response to systemic hypoglycemia, resulting in a sixfold lowering of leucine production by the gut and reduced hepatic uptake of the AA. Our results differ somewhat in that the gut production and hepatic uptake of the gluconeogenic AAs serine, threonine, glycine and alanine all increased normally in HG-HeadEu, suggesting that AA mobilization and utilization during hypoglycemia are not mediated by hypoglycemia sensing in the CNS. In this context, important differences between our study design and that of Hourani and colleagues (39) included the duration (3 vs. 2 h in the study of Hourani et al. and current study, respectively) and severity (42 vs. 52 mg/dL in the systemic circulation and 67 vs. 88 mg/dL in the jugular vein, respectively) of the hypoglycemic period, as well as the AAs that were examined. Future studies will be required to reconcile how these differences impact amino acid mobilization during insulin-induced hypoglycemia.
There are numerous ways to manipulate liver glycogen content. In the current studies, we utilized a catalytic intraportal infusion of fructose for a number of reasons. First, and most importantly, fructose infusion resulted in an average (of HG and HG-HeadEu) net hepatic glucose uptake rate of 7.8 mg/kg/min, which is ∼50% higher than what is normally observed during the postprandial state (41). Second, the time required for the enhanced hepatic glucose uptake to subside is minimal (∼1 h), making it an ideal fit for our study design. Third, despite reports to the contrary in rodents (42), we have used this same dose in previous studies and not seen any indication that it impacts lipid metabolism (31) or gluconeogenesis (27). Finally, in support of these points, we have also observed that the use of an intraportal glucagon infusion to lower liver glycogen content reduced the hormonal and hepatic responses to insulin-induced hypoglycemia (27). Collectively, these observations argue against a confounding effect of fructose infusion on whole body and tissue-specific metabolism in our studies.
In the current study, the increase in epinephrine during hypoglycemia was less pronounced in the HG group compared with NG (35% during the final hour), than the doubling we previously reported (27). In that study, insulin was infused intraportally, resulting in much lower arterial insulin levels (∼300 µU/mL) due to insulin extraction by the liver. In the current study, insulin was infused intravenously, resulting in arterial levels of ∼1,300 µU/mL. Diamond and colleagues (43) observed in healthy humans that when the hypoglycemic stimulus is matched, higher insulin levels (429 vs. 44 µU/mL) lowered levels of epinephrine, glucagon and growth hormone. On the other hand, Davis et al. (44–47) observed that higher insulin levels enhance the epinephrine and cortisol responses to hypoglycemia in humans [509 vs. 81 µU/mL (44)] and dogs [660 vs. 76 µU/mL (45)] and that this increment is neurally driven (46, 47). Thus disparate arterial insulin levels may have contributed to the difference in epinephrine responses to increased liver glycogen between the current and former study (27). Consistent with previous studies, there was no difference in cortisol responses to hypoglycemia when liver glycogen levels were elevated. This is a result of different areas of the brain being responsible for the secretion of specific counterregulatory hormones as demonstrated in elegant studies showing that AMPK manipulation in the ventromedial hypothalamus impacts the secretion of glucagon and epinephrine but has no effect on corticosterone (48, 49).
It has been demonstrated that liver glycogen accretion in response to a meal is impaired in patients with T1D (26), and it has also been shown that this population’s ability to mobilize liver glycogen during insulin-induced hypoglycemia is diminished (50). The deficiency in glycogen synthesis most likely arises as a result of subcutaneously delivered insulin, which dilutes the insulin concentration in blood that reaches the liver, thereby inverting the normal 2.5-to-1 Po:Pe distribution of the hormone. At this time, it is possible that the use of drugs such as hepato-selective insulin analogs and glucokinase activators could be used to restore the normal diurnal rhythm of liver glycogen accretion and potentially lower the risk of hypoglycemia in this population. Another consideration is the role that changes in glucose uptake and sensing by the brain play in the CRR to hypoglycemia. For example, patients with T1D that experience recurrent hypoglycemia (RH) have a lower brain glycemic threshold to invoke symptoms and CRR to hypoglycemia (51, 52), which may be a function of enhanced cerebral blood flow and/or brain glucose uptake (53–55). With this in mind, our results suggest that in the presence of RH, elevated liver glycogen concentrations would not be expected to restore the glycemic threshold at which CRR occur, as this is regulated by brain glucose metabolism. On the other hand, in the presence or absence of RH, elevated liver glycogen content could be protective against insulin-induced hypoglycemia by increasing the hormonal responses to a given neuroglucopenic stimulus.
In summary, our data provide further evidence that increased liver glycogen content enhances the hormonal responses to hypoglycemia, which increases hepatic glycogen mobilization and glucose production. Interestingly, this hormone-induced enhancement in substrate flux is ameliorated when euglycemia in the head is maintained. This suggests that during hypoglycemia, the brain gathers input from the liver regarding the status of its glycogen stores, after which it delivers the appropriate CRR in an autonomous fashion. In contrast, AA metabolism by the gut and liver during hypoglycemia were not impacted by liver glycogen content, counterregulatory hormones, or the maintenance of euglycemia in the brain, thereby suggesting that these responses are regulated by the peripheral glycemic level. Future studies will be required to unravel the mechanistic basis for the complex relationship between liver glycogen content, brain glucose metabolism, and the CRR to insulin-induced hypoglycemia, as it could be an accessible avenue to lessen the risk of iatrogenic hypoglycemia in patients with type 1 diabetes.
GRANTS
Funding for these studies was provided by the National Institutes of Diabetes and Digestive and Kidney Diseases Grant DK-106364. All assays were conducted by Vanderbilt University’s Hormone Assay and Analytical Services Core supported by Grants DK-059637 and DK-020593.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.J.W. conceived and designed research; A.M.W., M.S., B.F., D.S.E., and J.J.W. performed experiments; S.O.W., M.S., and J.J.W. analyzed data; S.O.W., A.M.W., Y.D., N.S., D.S.E., and J.J.W. interpreted results of experiments; S.O.W. prepared figures; S.O.W. and J.J.W. drafted manuscript; S.O.W., A.M.W., M.S., B.F., Y.D., N.S., D.S.E., and J.J.W. edited and revised manuscript; S.O.W., A.M.W., M.S., B.F., Y.D., N.S., D.S.E., and J.J.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Alan Cherrington and Phil Williams (Vanderbilt University) for support as these studies were being conducted. J.J.W. is the guarantor of the work. Portions of these data were presented at the 79th ADA Conference in San Francisco, CA in 2019. The graphical abstract was created using biorender.com.
REFERENCES
- 1.Basu R, Chandramouli V, Dicke B, Landau B, Rizza R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes 54: 1942–1948, 2005. doi: 10.2337/diabetes.54.7.1942. [DOI] [PubMed] [Google Scholar]
- 2.Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, Landau BR, Ferrannini E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes 49: 1367–1373, 2000. doi: 10.2337/diabetes.49.8.1367. [DOI] [PubMed] [Google Scholar]
- 3.Boyle PJ, Scott JC, Krentz AJ, Nagy RJ, Comstock E, Hoffman C. Diminished brain glucose metabolism is a significant determinant for falling rates of systemic glucose utilization during sleep in normal humans. J Clin Invest 93: 529–535, 1994. doi: 10.1172/JCI117003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinneen S, Gerich J, Rizza R. Carbohydrate metabolism in non-insulin-dependent diabetes mellitus. N Engl J Med 327: 707–713, 1992. doi: 10.1056/NEJM199209033271007. [DOI] [PubMed] [Google Scholar]
- 5.Reinmuth OM, Scheinberg P, Bourne B. Total cerebral blood flow and metabolism: a new method for the repeated serial measurement of total cerbral blood flow using iodoantipyrine (1131) with a report of determination in normal human beings of blood flow, oxygen consumption, glucose utilization and respiratory quotient of the whole brain. Arch Neurol 12: 49–66, 1965. doi: 10.1001/archneur.1965.00460250053007. [DOI] [Google Scholar]
- 6.Sherwin RS. Role of the liver in glucose homeostasis. Diabetes Care 3: 261–265, 1980. doi: 10.2337/diacare.3.2.261. [DOI] [PubMed] [Google Scholar]
- 7.Boyle PJ, Shah SD, Cryer PE. Insulin, glucagon, and catecholamines in prevention of hypoglycemia during fasting. Am J Physiol Endocrinol Metab 256: E651–E661, 1989. doi: 10.1152/ajpendo.1989.256.5.E651. [DOI] [PubMed] [Google Scholar]
- 8.Bolli GB, Gottesman IS, Cryer PE, Gerich JE. Glucose counterregulation during prolonged hypoglycemia in normal humans. Am J Physiol Endocrino Metab 247: E206–E214, 1984. doi: 10.1152/ajpendo.1984.247.2.E206. [DOI] [PubMed] [Google Scholar]
- 9.Enoksson S, Caprio SK, Rife F, Shulman GI, Tamborlane WV, Sherwin RS. Defective activation of skeletal muscle and adipose tissue lipolysis in type 1 diabetes mellitus during hypoglycemia. J Clin Endocrinol Metab 88: 1503–1511, 2003. doi: 10.1210/jc.2002-021013. [DOI] [PubMed] [Google Scholar]
- 10.Flattem N, Igawa K, Shiota M, Emshwiller MG, Neal DW, Cherrington AD. Alpha- and beta-cell responses to small changes in plasma glucose in the conscious dog. Diabetes 50: 367–375, 2001. doi: 10.2337/diabetes.50.2.367. [DOI] [PubMed] [Google Scholar]
- 11.Frizzell RT, Hendrick GK, Biggers DW, Lacy DB, Donahue DP, Green DR, Carr RK, Williams PE, Stevenson RW, Cherrington AD. Role of gluconeogenesis in sustaining glucose production during hypoglycemia caused by continuous insulin infusion in conscious dogs. Diabetes 37: 749–759, 1988. 10.2337/diabetes.37.6.749, doi: 10.2337/diab.37.6.749. [DOI] [PubMed] [Google Scholar]
- 12.Gerich J, Davis J, Lorenzi M, Rizza R, Bohannon N, Karam J, Lewis S, Kaplan R, Schultz T, Cryer P. Hormonal mechanisms of recovery from insulin-induced hypoglycemia in man. Am J Physiol Endocrinol Metab 236: E380–E385, 1979. doi: 10.1152/ajpendo.1979.236.4.E380. [DOI] [PubMed] [Google Scholar]
- 13.Rivera N, Ramnanan CJ, An Z, Farmer T, Smith M, Farmer B, Irimia JM, Snead W, Lautz M, Roach PJ, Cherrington AD. Insulin-induced hypoglycemia increases hepatic sensitivity to glucagon in dogs. J Clin Invest 120: 4425–4435, 2010. doi: 10.1172/JCI40919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dagogo-Jack SE, Craft S, Cryer PE. Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus. Recent antecedent hypoglycemia reduces autonomic responses to, symptoms of, and defense against subsequent hypoglycemia. J Clin Invest 91: 819–828, 1993. doi: 10.1172/JCI116302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrou Z, Seaquist ER, Robertson RP. Pancreas transplantation in diabetic humans normalizes hepatic glucose production during hypoglycemia. Diabetes 43: 661–666, 1994. doi: 10.2337/diabetes.43.5.661. [DOI] [PubMed] [Google Scholar]
- 16.Diem P, Redmon JB, Abid M, Moran A, Sutherland DE, Halter JB, Robertson RP. Glucagon, catecholamine and pancreatic polypeptide secretion in type I diabetic recipients of pancreas allografts. J Clin Invest 86: 2008–2013, 1990. doi: 10.1172/JCI114936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siafarikas A, Johnston RJ, Bulsara MK, O'Leary P, Jones TW, Davis EA. Early loss of the glucagon response to hypoglycemia in adolescents with type 1 diabetes. Diabetes Care 35: 1757–1762, 2012. doi: 10.2337/dc11-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cryer PE. The barrier of hypoglycemia in diabetes. Diabetes 57: 3169–3176, 2008. doi: 10.2337/db08-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cryer PE. Hypoglycemia: Pathophysiology, Diagnosis, and Treatment. Oxford, UK: Oxford University Press, 1997. [Google Scholar]
- 20.Cryer PE, Fisher JN, Shamoon H. Hypoglycemia. Diabetes Care 17: 734–755, 1994. doi: 10.2337/diacare.17.7.734. [DOI] [PubMed] [Google Scholar]
- 21.Biggers DW, Myers SR, Neal D, Stinson R, Cooper NB, Jaspan JB, Williams PE, Cherrington AD, Frizzell RT. Role of brain in counterregulation of insulin-induced hypoglycemia in dogs. Diabetes 38: 7–16, 1989. doi: 10.2337/diab.38.1.7. [DOI] [PubMed] [Google Scholar]
- 22.Donovan CM, Halter JB, Bergman RN. Importance of hepatic glucoreceptors in sympathoadrenal response to hypoglycemia. Diabetes 40: 155–158, 1991. doi: 10.2337/diab.40.1.155. [DOI] [PubMed] [Google Scholar]
- 23.Donovan CM, Hamilton-Wessler M, Halter JB, Bergman RN. Primacy of liver glucosensors in the sympathetic response to progressive hypoglycemia. Proc Natl Acad Sci U S A 91: 2863–2867, 1994. doi: 10.1073/pnas.91.7.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hevener AL, Bergman RN, Donovan CM. Novel glucosensor for hypoglycemic detection localized to the portal vein. Diabetes 46: 1521–1525, 1997. doi: 10.2337/diabetes.46.9.1521. [DOI] [PubMed] [Google Scholar]
- 25.Saberi M, Bohland M, Donovan CM. The locus for hypoglycemic detection shifts with the rate of fall in glycemia: the role of portal-superior mesenteric vein glucose sensing. Diabetes 57: 1380–1386, 2008. doi: 10.2337/db07-1528. [DOI] [PubMed] [Google Scholar]
- 26.Hwang JH, Perseghin G, Rothman DL, Cline GW, Magnusson I, Petersen KF, Shulman GI. Impaired net hepatic glycogen synthesis in insulin-dependent diabetic subjects during mixed meal ingestion. A 13C nuclear magnetic resonance spectroscopy study. J Clin Invest 95: 783–787, 1995. doi: 10.1172/JCI117727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winnick JJ, Kraft G, Gregory JM, Edgerton DS, Williams P, Hajizadeh IA, Kamal MZ, Smith M, Farmer B, Scott M, Neal D, Donahue EP, Allen E, Cherrington AD. Hepatic glycogen can regulate hypoglycemic counterregulation via a liver-brain axis. J Clin Invest 126: 2236–2248, 2016. doi: 10.1172/JCI79895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers SR, McGuinness OP, Neal DW, Cherrington AD. Intraportal glucose delivery alters the relationship between net hepatic glucose uptake and the insulin concentration. J Clin Invest 87: 930–939, 1991. doi: 10.1172/JCI115100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winnick JJ, An Z, Kraft G, Ramnanan CJ, Irimia JM, Smith M, Lautz M, Roach PJ, Cherrington AD. Liver glycogen loading dampens glycogen synthesis seen in response to either hyperinsulinemia or intraportal glucose infusion. Diabetes 62: 96–101, 2013. doi: 10.2337/db11-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winnick JJ, An Z, Moore MC, Ramnanan CJ, Farmer B, Shiota M, Cherrington AD. A physiological increase in the hepatic glycogen level does not affect the response of net hepatic glucose uptake to insulin. Am J Physiol Endocrinol Metab 297: E358–E366, 2009. doi: 10.1152/ajpendo.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winnick JJ, An Z, Ramnanan CJ, Smith M, Irimia JM, Neal DW, Moore MC, Roach PJ, Cherrington AD. Hepatic glycogen supercompensation activates AMP-activated protein kinase, impairs insulin signaling, and reduces glycogen deposition in the liver. Diabetes 60: 398–407, 2011. doi: 10.2337/db10-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keppler D, Decker K. Glycogen. In: Methods of Enzymatic Analysis, edited by Bergmeyer HU.New York: Verlag Chemie, 1984, p. 11–18.. [Google Scholar]
- 33.Hsieh PS, Moore MC, Neal DW, Venson P, Cherrington AD. Hepatic glucose uptake rapidly decreases after elimination of the portal signal in conscious dogs. Am J Physiol Endocrinol Metab 275: E987–E992, 1998. doi: 10.1152/ajpendo.1998.275.6.E987. [DOI] [PubMed] [Google Scholar]
- 34.Moore MC, Hsieh PS, Flakoll PJ, Neal DW, Cherrington AD. Differential effect of amino acid infusion route on net hepatic glucose uptake in the dog. Am J Physiol Endocrinol Metab 276: E295–E302, 1999. doi: 10.1152/ajpendo.1999.276.2.E295. [DOI] [PubMed] [Google Scholar]
- 35.Martyn-Nemeth P, Schwarz Farabi S, Mihailescu D, Nemeth J, Quinn L. Fear of hypoglycemia in adults with type 1 diabetes: impact of therapeutic advances and strategies for prevention–a review. J Diabetes Complications 30: 167–177, 2016. doi: 10.1016/j.jdiacomp.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Connolly CC, Adkins-Marshall BA, Neal DW, Pugh W, Jaspan JB, Cherrington AD. Relationship between decrements in glucose level and metabolic response to hypoglycemia in absence of counterregulatory hormones in the conscious dog. Diabetes 41: 1308–1319, 1992. doi: 10.2337/diab.41.10.1308. [DOI] [PubMed] [Google Scholar]
- 37.Connolly CC, Ivy RE, Adkins-Marshall BA, Dobbins RL, Neal DW, Williams PE, Cherrington AD. Counterregulation by epinephrine and glucagon during insulin-induced hypoglycemia in the conscious dog. Diabetes Res Clin Pract 31: 45–56, 1996. doi: 10.1016/0168-8227(96)01212-0. [DOI] [PubMed] [Google Scholar]
- 38.Fanelli C, Calderone S, Epifano L, De Vincenzo A, Modarelli F, Pampanelli S, Perriello G, De Feo P, Brunetti P, Gerich JE. and Demonstration of a critical role for free fatty acids in mediating counterregulatory stimulation of gluconeogenesis and suppression of glucose utilization in humans. J Clin Invest 92: 1617–1622, 1993. doi: 10.1172/JCI116746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hourani H, Williams P, Morris JA, May ME, Abumrad NN. Effect of insulin-induced hypoglycemia on protein metabolism in vivo. Am J Physiol Endocrinol Metab 259: E342–E350, 1990. doi: 10.1152/ajpendo.1990.259.3.E342. [DOI] [PubMed] [Google Scholar]
- 40.Hourani H, Lacy B, Eltayeb K, Abumrad NN. The role of the central nervous system in modulating glucose and protein metabolism during insulin-induced hypoglycemia. Brain Res 587: 276–284, 1992. doi: 10.1016/0006-8993(92)91008-3. [DOI] [PubMed] [Google Scholar]
- 41.Moore MC, Cherrington AD, Cline G, Pagliassotti MJ, Jones EM, Neal DW, Badet C, Shulman GI. Sources of carbon for hepatic glycogen synthesis in the conscious dog. J Clin Invest 88: 578–587, 1991. doi: 10.1172/JCI115342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinote A, Faria JA, Roman EA, Solon C, Razolli DS, Ignacio-Souza LM, Sollon CS, Nascimento LF, de Araujo TM, Barbosa AP, Lellis-Santos C, Velloso LA, Bordin S, Anhe GF. Fructose-induced hypothalamic AMPK activation stimulates hepatic PEPCK and gluconeogenesis due to increased corticosterone levels. Endocrinology 153: 3633–3645, 2012. doi: 10.1210/en.2012-1341. [DOI] [PubMed] [Google Scholar]
- 43.Diamond MP, Hallarman L, Starick-Zych K, Jones TW, Connolly-Howard M, Tamborlane WV, Sherwin RS. Suppression of counterregulatory hormone response to hypoglycemia by insulin per se. J Clin Endocrinol Metab 72: 1388–1390, 1991. doi: 10.1210/jcem-72-6-1388. [DOI] [PubMed] [Google Scholar]
- 44.Davis SN, Goldstein RE, Jacobs J, Price L, Wolfe R, Cherrington AD. The effects of differing insulin levels on the hormonal and metabolic response to equivalent hypoglycemia in normal humans. Diabetes 42: 263–272, 1993. doi: 10.2337/diab.42.2.263. [DOI] [PubMed] [Google Scholar]
- 45.Davis SN, Dobbins R, Tarumi C, Colburn C, Neal D, Cherrington AD. Effects of differing insulin levels on response to equivalent hypoglycemia in conscious dogs. Am J Physiol Endocrinol Metab 263: E688–E695, 1992. doi: 10.1152/ajpendo.1992.263.4.E688. [DOI] [PubMed] [Google Scholar]
- 46.Davis SN, Colburn C, Dobbins R, Nadeau S, Neal D, Williams P, Cherrington AD. Evidence that the brain of the conscious dog is insulin sensitive. J Clin Invest 95: 593–602, 1995. doi: 10.1172/JCI117703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis SN, Dunham B, Walmsley K, Shavers C, Neal D, Williams P, Cherrington AD. Brain of the conscious dog is sensitive to physiological changes in circulating insulin. Am J Physiol Endocrinol Metab 272: E567–E575, 1997. doi: 10.1152/ajpendo.1997.272.4.E567. [DOI] [PubMed] [Google Scholar]
- 48.Fan X, Ding Y, Brown S, Zhou L, Shaw M, Vella MC, Cheng H, McNay EC, Sherwin RS, McCrimmon RJ. Hypothalamic AMP-activated protein kinase activation with AICAR amplifies counterregulatory responses to hypoglycemia in a rodent model of type 1 diabetes. Am J Physiol Regul Integr Comp Physiol 296: R1702–R1708, 2009. doi: 10.1152/ajpregu.90600.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCrimmon RJ, Shaw M, Fan X, Cheng H, Ding Y, Vella MC, Zhou L, McNay EC, Sherwin RS. Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes 57: 444–450, 2008. doi: 10.2337/db07-0837. [DOI] [PubMed] [Google Scholar]
- 50.Kishore P, Gabriely I, Cui MH, Di Vito J, Gajavelli S, Hwang JH, Shamoon H. Role of hepatic glycogen breakdown in defective counterregulation of hypoglycemia in intensively treated type 1 diabetes. Diabetes 55: 659–666, 2006. doi: 10.2337/diabetes.55.03.06.db05-0849. [DOI] [PubMed] [Google Scholar]
- 51.Amiel SA, Tamborlane WV, Simonson DC, Sherwin RS. Defective glucose counterregulation after strict glycemic control of insulin-dependent diabetes mellitus. N Engl J Med 316: 1376–1383, 1987. doi: 10.1056/NEJM198705283162205. [DOI] [PubMed] [Google Scholar]
- 52.Cryer PE. Mechanisms of hypoglycemia-associated autonomic failure and its component syndromes in diabetes. Diabetes 54: 3592–3601, 2005. doi: 10.2337/diabetes.54.12.3592. [DOI] [PubMed] [Google Scholar]
- 53.Boyle PJ, Kempers SF, O'Connor AM, Nagy RJ. Brain glucose uptake and unawareness of hypoglycemia in patients with insulin-dependent diabetes mellitus. N Engl J Med 333: 1726–1731, 1995. doi: 10.1056/NEJM199512283332602. [DOI] [PubMed] [Google Scholar]
- 54.Criego AB, Tkac I, Kumar A, Thomas W, Gruetter R, Seaquist ER. Brain glucose concentrations in patients with type 1 diabetes and hypoglycemia unawareness. J Neurosci Res 79: 42–47, 2005. doi: 10.1002/jnr.20296. [DOI] [PubMed] [Google Scholar]
- 55.Wiegers EC, Becker KM, Rooijackers HM, von Samson-Himmelstjerna FC, Tack CJ, Heerschap A, de Galan BE, van der Graaf M. Cerebral blood flow response to hypoglycemia is altered in patients with type 1 diabetes and impaired awareness of hypoglycemia. J Cereb Blood Flow Metab 37: 1994–2001, 2017. doi: 10.1177/0271678X16658914. [DOI] [PMC free article] [PubMed] [Google Scholar]