

Keywords: cancer metabolism, glycolysis, lipid metabolism, metabolic reprogramming, mTOR
Abstract
Cells metabolize nutrients for biosynthetic and bioenergetic needs to fuel growth and proliferation. The uptake of nutrients from the environment and their intracellular metabolism is a highly controlled process that involves cross talk between growth signaling and metabolic pathways. Despite constant fluctuations in nutrient availability and environmental signals, normal cells restore metabolic homeostasis to maintain cellular functions and prevent disease. A central signaling molecule that integrates growth with metabolism is the mechanistic target of rapamycin (mTOR). mTOR is a protein kinase that responds to levels of nutrients and growth signals. mTOR forms two protein complexes, mTORC1, which is sensitive to rapamycin, and mTORC2, which is not directly inhibited by this drug. Rapamycin has facilitated the discovery of the various functions of mTORC1 in metabolism. Genetic models that disrupt either mTORC1 or mTORC2 have expanded our knowledge of their cellular, tissue, as well as systemic functions in metabolism. Nevertheless, our knowledge of the regulation and functions of mTORC2, particularly in metabolism, has lagged behind. Since mTOR is an important target for cancer, aging, and other metabolism-related pathologies, understanding the distinct and overlapping regulation and functions of the two mTOR complexes is vital for the development of more effective therapeutic strategies. This review discusses the key discoveries and recent findings on the regulation and metabolic functions of the mTOR complexes. We highlight findings from cancer models but also discuss other examples of the mTOR-mediated metabolic reprogramming occurring in stem and immune cells, type 2 diabetes/obesity, neurodegenerative disorders, and aging.
CLINICAL HIGHLIGHTS
The mechanistic target of rapamycin (mTOR) controls cellular metabolism by integrating signals from nutrients, growth factors, and other environmental signals.
mTOR is part of two protein complexes, mTORC1 and mTORC2, that are evolutionarily conserved from yeast to human. mTORC1 is the direct target of the clinically important drug rapamycin, which allosterically inhibits mTORC1, whereas it only indirectly inhibits mTORC2. Rapamycin and its drug analogs are currently in use or are undergoing clinical trials to treat a variety of diseases including cancer, neurodegenerative disorders, immune-related diseases, and metabolic disorders. It is also promising as an antiaging compound and could improve health span in humans and animals.
Understanding mTOR-mediated metabolic reprogramming of target cells (e.g., tumors) and other cells of the microenvironment (e.g., immune cells) will facilitate the development of more effective and specific therapeutic strategies.
Optimal activity of mTORC1 and mTORC2 is necessary to maintain metabolic homeostasis, prevent disease, and prolong health span.
1. INTRODUCTION
In the early part of the twentieth century, the metabolic pathways that organisms utilize to synthesize and break down nutrients were mapped, ushering in exploration of the molecular underpinnings of metabolic diseases including diabetes and cancer. It was also around this period that the hormone insulin was discovered, another breakthrough that saved the lives of people suffering from diabetes. Despite the knowledge that insulin controls glucose metabolism, the molecular mechanisms underlying the control of metabolism by hormones were obscure then. Furthermore, how nutrients are sensed and how cells respond to fluctuating nutrient levels remained poorly understood. In the early 1990s, the identification of mTOR (mechanistic target of rapamycin) and the elucidation of phosphatidylinositol 3-kinase (PI3K)/Akt signaling, which was found to mediate insulin signals, set the stage for delineating the molecular control of metabolism by signaling molecules (1, 2). By the beginning of the twenty-first century, it was apparent that mTOR integrates nutrient and growth signals to control metabolism.
Although the basic architecture of metabolic pathways is largely conserved from prokaryotes to eukaryotes, eukaryotes have devised more elaborate ways to control metabolism. The evolution of these control mechanisms allowed eukaryotes, including the unicellular yeast, to respond dynamically not just to the presence but more importantly to the absence of nutrients and control growth. Cells have adapted mechanisms to maintain quiescence to conserve energy while maintaining the capacity to undergo proliferation when conditions become more favorable (3). In addition to acquiring nutrients from the environment, proliferating cells are also capable of mobilizing intracellular nutrient sources by reprogramming metabolism toward recycling or catabolic processes. In multicellular organisms, there is an additional need to coordinate growth of different tissues in response to nutrient availability. This is fulfilled by signals from hormones and growth factors. mTOR has emerged as a key signaling protein that responds not just to nutrients or metabolites but also to other growth signals, thereby orchestrating the control of cellular metabolic processes.
mTOR has conserved structure and functions from yeast to human. It is an atypical protein kinase that shares homology with lipid kinases but phosphorylates proteins. Notably, the activation of mTOR and subsequent phosphorylation of its substrates largely occur in membrane compartments as discussed below. mTOR is allosterically inhibited by a complex formed by the natural compound rapamycin and the prolyl isomerase FKBP12 (4–8). Using rapamycin to inhibit mTOR activity, early genetic studies revealed that yeast TOR promotes protein synthesis under favorable nutrient conditions (9). Rapamycin arrests yeast and mammalian cells in G1 phase of the cell cycle, eliciting a phenotype characteristic of starved cells (4, 9–11). Moreover, rapamycin induces a starvation response termed autophagy, which promotes the degradation and recycling of cellular components (12, 13). In mammals, rapamycin or amino acid starvation diminishes phosphorylation and activity of the translation regulator p70 S6K (14–17). In addition, another translation regulator, 4E-BP1, has shown some sensitivity to rapamycin, further supporting mTOR’s role in translation (18–20). Together, these early findings in yeast and in Drosophila provide substantial evidence that TOR participates not only in nutrient sensing but also in conducting the symphony of metabolic activities to control protein synthesis and cell growth (9, 11, 21, 22).
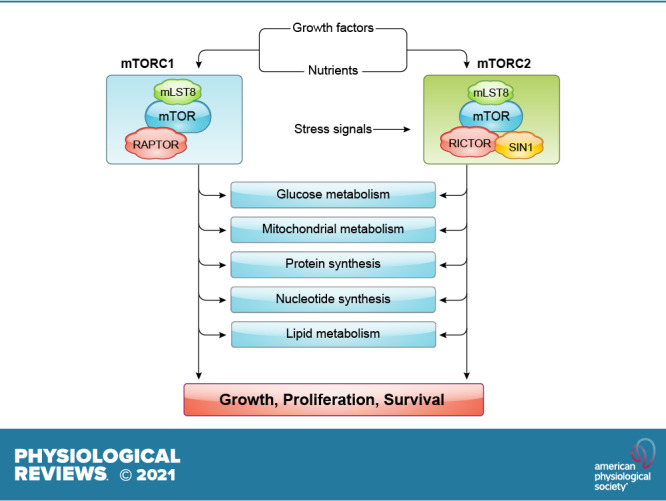
Previous studies have shown that mTOR plays a role in trafficking and regulation of expression of nutrient transporters. In addition, studies showing that the mTOR pathway is affected by ATP levels further strengthened the notion that mTOR is a component of the nutrient signaling cascade (23–26). Investigations in different model organisms have revealed that TOR/mTOR controls cell growth or increase in cell mass that is distinct from the control of cell proliferation or division (27). Further inquiries using rapamycin and whole genome screening revealed that TOR proteins mediate the expression of genes involved in nutrient metabolism (11, 21). Findings in yeast that TOR has both rapamycin-sensitive and -insensitive function suggested that TOR forms distinct protein complexes (28, 29). Biochemical studies led to the purification and identification of two distinct mTOR protein complexes in both yeast and mammals (30–37). These distinct mTOR complexes were the rapamycin-sensitive mTORC1 and the rapamycin-insensitive mTORC2. Raptor and mLST8 associate with mTOR to comprise the rapamycin-sensitive mTORC1, and rictor (mAVO3), SIN1, and mLST8 bind mTOR to form the rapamycin-insensitive mTORC2. Interestingly, SIN1 also interacts with Ras/MAPK signaling molecules. Because of alternative splicing, SIN1 has four different isoforms that may form distinct mTOR complexes (38, 39). The functions of these isoforms remain poorly understood. In addition to conserved partners, mTORCs associate with other distinct, less well-conserved proteins that regulate its activity and function (40).
Since rapamycin allosterically inhibits mTORC1 but not mTORC2, most of the studies on mTOR pertain to mTORC1. As discussed below, there is vast information on how mTORC1 is regulated and its role in metabolism. In contrast, deciphering the biology behind mTORC2 has been more challenging because of a lack of specific pharmacological inhibitors and the seemingly more complex regulation of mTORC2. Nevertheless, as we summarize below, we are gaining more insight into the distinct and overlapping functions of mTORC1 and mTORC2 in metabolism with genetic models. Most of our discussion of the regulation and functions of the mTOR complexes in metabolism has been extracted from studies using cancer models. In addition, we include discussion of stem and immune cells, metabolic disorders such as type 2 diabetes (T2D) and obesity, neurodegenerative disorders, and aging, since they also serve as relevant paradigms to understand the role of mTOR in metabolic reprogramming under normal and pathological conditions.
2. mTORC1 ACTIVATION
As a central regulator of growth and metabolism, mTOR responds to the presence of nutrients. In particular, mTORC1 is positively regulated by amino acids; conversely, when amino acids are low or become limiting, mTORC1 activity declines. mTORC1 activation is also influenced by the levels of other nutrients such as glucose and lipids, but the mechanisms for such regulation, whether it occurs directly or indirectly via other signaling molecules, remains to be elucidated. In multicellular organisms, in addition to nutrients, other extracellular inputs—e.g., growth factors and hormones—control mTORC1 activity, highlighting the crucial role of mTORC1 in coordinating the cell growth, tissue growth, and thus organismal growth of higher organisms.
The activation of mTORC1 occurs in membrane compartments. Several amino acid transporters that are present in the plasma membrane have been linked to mTORC1 activation, including the transporters for glutamine (SLC1A5/ASCT2) and leucine (SLC7A5/LAT1, which imports Leu in exchange for Gln efflux by SLC3A2/CD98/4F2hc) (41–44). Oftentimes, overexpression of these transporters is associated with malignancies. Although mTORC1 may be activated by nutrients in distinct membrane compartments, the most well-characterized mode of mTORC1 activation is its recruitment to the surface of the lysosomes, where it is then activated in a manner involving an army of regulatory molecules. The lysosomes serve as depots for intracellular nutrients owing to their function in degrading and recycling cellular macromolecules. Earlier studies have elucidated how mTORC1 is activated via Ras-related GTP binding proteins (Rags) (45–47). RagA/B is bound to GTP, whereas RagC/D is GDP bound under amino acid sufficiency. The Rag heterodimers (composed of Rag A or B with Rag C or D) interact with raptor and facilitate translocation of mTORC1 to the lysosomal surface. A pentameric complex named Ragulator, consisting of Lamtor1–5 (which are also known as p18, p14, MP1, C7orf59, and HBXIP, respectively) facilitates localization of the Rag GTPases to the lysosomal surface. Ragulator also serves as a guanine nucleotide exchange factor (GEF) for RagA/B (48). The NH2-terminal region of Lamtor1 is myristylated and palmitoylated and anchors Ragulator to the lysosomal surface (49). The signaling adaptor p62 also associates with mTOR and raptor in an amino acid-dependent manner (50). It is required for the interaction of mTOR with Rag GTPases and for localization of mTORC1 to the lysosome. The counteracting GTPase-activating protein (GAP) is a multiprotein complex termed GATOR1, which consists of DEP domain containing 5 (DEPDC5), nitrogen permease regulator 2-like protein (NPRL2), and NPRL3 (51). GATOR1 is engaged to the lysosomes via another large protein complex, KICSTOR, comprised of KPTN1, ITGF2, C12orf66, and SZT2 (52, 53). Another protein complex, GATOR2, consisting of Mios, WD repeat-containing protein 24 (WDR24), WDR59, SEH1-like nucleoporin, and SEC13, indirectly activates mTORC1 by binding to and blocking GATOR1 activity. GPR137B, a lysosomal G protein-coupled receptor (GPCR)-like protein, also regulates the GTP loading state of RagA and localization of Rag and mTORC1 to lysosomes (54). Notably, even in the absence of amino acids, elevated GPR137B expression levels can increase mTORC1 activation. RagC/D is also regulated by a GAP, consisting of Folliculin (FLCN) and its interacting protein folliculin interacting proteins 1 and 2 (FNIP1 and FNIP2). RagC also undergoes phosphorylation at three conserved sites that are growth factor responsive (55). Their phosphorylation is required for growth factor- and amino acid-induced activation of mTORC1 and suppresses starvation-induced autophagy. At least one of these sites, Ser21, is via mTORC1.
Distinct amino acids induce mTORC1 activation through different mechanisms that involve Rag-dependent mechanisms. Recent findings also highlight Rag-independent mechanisms of mTORC1 activation by amino acids. The amino acids leucine and arginine bind to sestrin2 and CASTOR1, respectively, which mediate activation of mTORC1 (56). In the presence of leucine, the inhibition of GATOR1 by GATOR2 is relieved by the sestrin2/leucine interaction, ultimately leading to the activation of RagA/B and mTORC1. Independently of sestrin2, leucine can signal through its catabolite acetyl-coenzyme A (acetyl-CoA) and activate mTORC1 via EP-300-mediated acetylation of raptor at K1097 (57). So far, it appears that this mode of mTORC1 activation is cell type specific. Arginine binding to CASTOR1 also derepresses the inhibition of GATOR2 by CASTOR1, thereby activating mTORC1. While CASTOR1 serves as an Arg sensor, SLC38A9, an Arg-gated amino acid transporter, acts as another Arg sensor that regulates mTORC1 activity. SLC38A9 changes the RagA/RagB nucleotide state by stimulating GDP release from RagA when activated by arginine and thus serves as another mechanism for controlling mTORC1 activation (58–61). Methionine is sensed as S-adenosylmethionine (SAM) via SAMTOR. The increased association of SAMTOR with SAM impedes the SAMTOR-GATOR1 binding, thus enhancing mTORC1 activation (62, 63). Glutamine is utilized as a nitrogen source and alternative carbon source for nutrient/metabolite biosynthesis. It is also used to transport essential amino acids such as leucine. Hence, maintaining sufficient glutamine levels is likely crucial to sustaining mTORC1 activity, and its fluctuations modulate mTORC1. Glutamine uptake via SLC1A5 (ASCT2), a glutamine transporter, and its efflux, which is coupled to leucine import via the CD98 (a complex of SLC7A5/SLC3A2), regulate mTORC1 activity (44). Metabolism of glutamine during glutaminolysis that increases α-ketoglutarate (αKG) production also increases mTORC1 activity. Glutaminolysis is linked to inhibition of autophagy and is also essential for RagB GTP loading (62). Glutamine depletion that can occur as an off-target effect of asparaginase treatment inhibits mTORC1 activity (64). Its depletion likely impacts various glutamine-requiring biosynthetic pathways and import of leucine, thus downregulating mTORC1. Although these findings suggest that glutamine indirectly activates mTORC1, it may also have a more direct role in its activation, but the mechanisms remain to be investigated. So far, it has been shown that glutamine and asparagine promote mTORC1 translocation to the lysosomal surface and its activation via the ADP ribosylation factor (Arf-1) GTPase, independent of Rag (65–67). Hence, different key amino acids or their metabolites modulate mTORC1 through regulatory molecules that impact mTORC1 activation and/or translocation to the lysosomes.
The activation of mTORC1 by amino acids is potentiated by signals from growth factors. Two major growth signaling pathways, phosphatidylinositol 3-kinase (PI3K)-Akt and Ras-ERK, regulate mTORC1 activity through functional inhibition of the tumor suppressor tuberous sclerosis complex (TSC1, TSC2, and TBC1D7) via several phosphorylation events triggered by growth factor-dependent kinases (68–70). TSC2 functions as a GAP that inhibits Rheb GTPase (71–73). The presence of growth stimuli causes inactivation of the TSC complex, allowing Rheb to remain in its GTP-bound state and activate mTORC1. Rheb binds to and activates mTORC1 through realignment of residues in the mTOR active site to enhance catalytic activity (73). Hence, cells that have loss-of-function or inactivating mutations in TSC1 or TSC2 tumor suppressor genes are marked by increased mTORC1 activity. Multiple signals converge on TSC1/TSC2 to modulate mTORC1. The binding of growth factors such as insulin to receptor tyrosine kinases triggers PI3K activation. Insulin/PI3K signals are coupled to the recruitment of Akt to the plasma membrane, where Akt is then phosphorylated within its activation loop at Thr308 by phosphoinositide-dependent kinase 1 (PDK1). Akt-mediated activation of mTORC1 occurs through its multisite phosphorylation of TSC2 at Ser939, Ser981, and Thr1462, which leads to the functional inhibition of the TSC complex and subsequently increased mTORC1 activation (70, 74, 75). This phosphorylation event in response to insulin/PI3K signaling excludes TSC from the lysosomes, thus relieving the negative regulation of mTORC1 (76, 77). Notably, Akt is only partially activated by PDK1 at its Thr308 phosphorylation site, but its activation is sufficient for its TSC2 phosphorylation activity. Akt requires a second phosphorylation event at Ser473 by mTORC2 for optimal activation. When fully activated, Akt phosphorylates a more diverse set of substrates (proteins containing an RXXS/T motif). (33). Hence, mTORC2 can promote mTORC1 signaling via Akt-mediated phosphorylation of TSC2 and other substrates involved in cell growth and metabolism, thus potentiating mTOR signaling (70, 74, 75). Additionally, mTORC2 enhances mTORC1 activation by repression of the class II PI3KC2-β. PI3KC2-β negatively regulates mTORC1 activity at the lysosomes by promoting synthesis of phosphatidylinositol 3,4-bisphosphate during starvation. In the presence of growth factors, mTORC2 phosphorylates PKN2 at Thr958, which in turn phosphorylates PI3KC2-β (78). PKN2-mediated PI3KC2-β phosphorylation promotes binding of the latter to 14-3-3, thus preventing PI3KC2-β function and consequently relieving suppression of mTORC1 activity. Insulin/PI3K signals also activate mTORC1 through other AGC kinases. In response to translation inhibitors, another AGC kinase, PKC-δ, becomes activated and phosphorylates TSC2 at Ser932/939, leading to upregulation of mTORC1 activity (79). PKG also phosphorylates TSC2 at Ser1364/1365 to modulate mTORC1 activity for cardiac protection against pressure overload (80).
Under control of the Ras-ERK pathway, mTORC1 is also regulated through inhibitory phosphorylation of TSC2 on specific sites. Growth factor binding, such as epidermal growth factor, activates the Ras GTPases and triggers a cascade of events leading to activation of mitogen-activated protein kinases (MAPKs) such as ERK. ERK1/2 phosphorylates TSC2 Ser540 and Ser664 (81). ERK also indirectly activates mTORC1 via regulation of another AGC kinase, p90 ribosomal S6 kinase (RSK), which phosphorylates TSC2 at Ser1798 to promote mTORC1 signaling to S6K1 (82).
mTORC1 activation is not limited to the lysosomal surface but can also occur on the Golgi. Amino acids at the surface of the Golgi activate mTORC1 via the small GTPase Rab1A, which promotes the interaction of mTORC1 with Rheb at the Golgi apparatus, thereby activating mTORC1 (83). In this compartment, the amino acid transporter PAT4 (SLC36A4) interacts with mTORC1 and Rab1A (41). Future studies should investigate whether amino acids or other metabolites in different cellular compartments could activate mTORC1 and evaluate the mechanisms by which mTORC1 becomes activated (84).
To ensure that mTORC1 activity is dampened during unfavorable growth conditions, such as during nutrient limitation or the presence of other environmental stress, mTORC1 is negatively modulated by different mechanisms. As discussed above, the TSC1/TSC2 complex serves to negatively modulate mTORC1 during reduced growth factor/PI3K signals. Additionally, the proline-rich Akt substrate of 40 kDa (PRAS40) binds to raptor (85, 86). In the presence of growth factors, PRAS40 is phosphorylated by Akt and mTOR and dissociates from mTORC1, thus relieving mTORC1 inhibition. Another protein, DEPTOR, interacts with mTORC1 (as well as mTORC2) and binds to the FAT domain of mTOR, inhibiting both mTORC1 and mTORC2 (87). It negatively regulates mTORC1 but indirectly controls mTORC2 activation via feedback modulation of PI3K signals (88). Under energy-depleted conditions, AMPK modulates mTORC1 via phosphorylation of raptor and indirectly via phosphorylation of TSC2 (69, 89).
In response to stress conditions including hypoxia, iron depletion, DNA damage, and amino acid starvation, mTORC1 is inhibited through TSC1/TSC2 complex and REDD1 (Regulated in Development and DNA damage response 1). REDD1 inhibits mTORC1 by promoting dephosphorylation of Akt via PP2A and sequesters 14-3-3 away from TSC2 (90, 91). Recently, mTORC1 has been shown to be inhibited by GPCR signaling (92). This inhibition occurs via Gαs proteins, which increase adenosine 3′,5′-cyclic monophosphate (cAMP), leading to the activation of PKA. PKA phosphorylates Ser791 of raptor, consequently diminishing mTORC1 activity; however, the mechanism of this inhibition of mTORC1 remains unclear. It is noteworthy that the PKA-mediated phosphorylation at Ser791 also promotes mTORC1 activation in adipocytes in response to β-adrenergic stimulation (93). As discussed further below, raptor is phosphorylated at multiple sites to modulate mTORC1 activity either positively or negatively.
While we have expanded our understanding of how mTORC1 is activated by nutrients, particularly amino acids, at the surface of the lysosomes, there are emerging studies demonstrating that mTORC1 can respond to other nutrients and metabolites such as purines (94) and phosphatidic acid (95). In addition to nutrients, extracellular signals such as growth factors, hormones, and cytokines affect the activation of this complex. The cellular compartments and the mechanisms involved in mTORC1 activation by different nutrients and other growth signals await further investigation.
3. mTORC2 ACTIVATION
The signals activating mTORC2 and the mechanisms involved in its activation are relatively less understood compared with mTORC1. Nutrients and growth factors also affect mTORC2 activation, but the context of how they modulate mTORC2 appears to be more complicated than mTORC1.
In mammals, increased growth factor/PI3K signaling augments mTORC2 activity (FIGURE 1B). The product of PI3K activation, phosphatidylinositol 3,4,5-trisphosphate (PIP3), tethers signaling molecules to the membrane compartment through binding to the pleckstrin homology (PH) domain in target proteins. The Akt PH domain facilitates membrane recruitment in response to PI3K signaling. Akt is phosphorylated at Ser473 of the hydrophobic motif (HM) by mTORC2 upon PI3K activation, and this phosphorylation is often used as a hallmark of mTORC2 activation in experimental studies. PIP3 also binds SIN1 via its PH domain (96). This binding releases the mTOR kinase domain from SIN1-PH inhibition and allows for mTORC2 activation (97). As mTORC2 is activated by PIP3 at the membrane, it is likely that mTORC2 is modulated in distinct membrane compartments. Studies have shown that endosomal pools of mTORC2 appear to require PI3K signaling (98) and that insulin/PI3K increases mTORC2 activation and association with ribosomes (99). Interestingly, pools of mTORC2 have delayed reactivation upon serum restimulation due to perinuclear positioning of lysosomes, further suggesting that compartmental localization is critical in modulating its activity in response to growth factor signaling (100).
FIGURE 1.
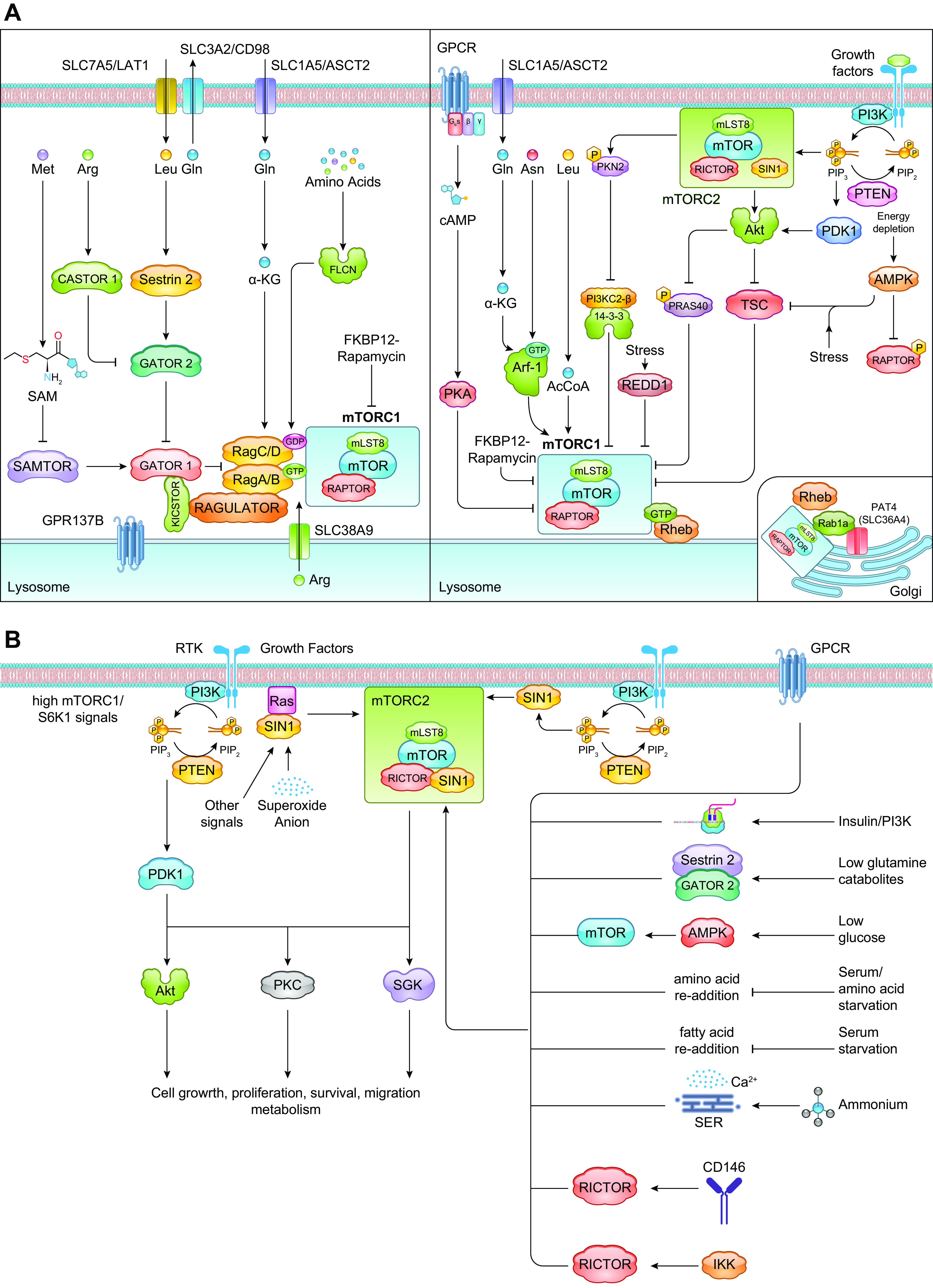
mTORC1 and mTORC2 signaling pathways. A: the activation of mTORC1 occurs via distinct mechanisms that are either Rag dependent or -independent. When nutrients are present, mTORC1 is activated via Rag heterodimers (left). Ragulator serves as a GEF for RagA/B and facilitates localization of the Rag GTPases to the lysosomal surface. GATOR1 is the counteracting GAP and is engaged to the lysosomes via another large protein complex, KICSTOR. GATOR2 indirectly activates mTORC1 by binding to and blocking GATOR1 activity. By binding to regulatory proteins that modulate Rag activation, different amino acids promote mTORC1 activation. mTORC1 is also regulated by Rag-independent mechanisms (right). Glutamine and asparagine promote mTORC1 activation on the lysosomal surface via Arf-1 GTPase. mTORC1 activation is also subject to negative regulation to modulate its activity depending on levels of growth signals. TSC and PRAS40 are modulated by growth factor and energy signals. REDD1 negatively modulates mTORC1 in response to stress conditions. Signals from GPCRs also negatively regulate mTORC1 via PKA. mTORC1 is also modulated by amino acids at the surface of the Golgi via Rab1a (inset). B: mTORC2 is activated by growth factors and fluctuations in nutrient/metabolite levels. In response to growth factors, the increased PI3K signaling enhances mTORC2 activation, leading to phosphorylation of AGC kinase family members such as Akt, PKC, and SGK. mTORC2 allosterically activates these protein kinases, which have numerous cellular functions. mTORC2 activation is also enhanced by other signals including from the GPCR, translating ribosomes, nutrient fluctuations (including withdrawal or readdition), and association of mTORC2 components with other signaling molecules such as Ras, CD146, or IKK. See glossary for abbreviations.
mTORC2 is also responsive to intracellular nutrient or metabolite conditions. mTORC2 activation is enhanced by withdrawal of amino acids (e.g., glutamine) and by glucose starvation in the presence of serum (101). This activation of mTORC2 during glutamine or glucose starvation is essential to maintain flux through a glucose- and glutamine-requiring biosynthetic pathway, the hexosamine biosynthesis pathway. During glucose depletion, AMPK directly activates mTORC2 through phosphorylation of mTOR and possibly rictor in the presence of minimal PI3K signals (102). The mechanisms of mTORC2 activation during glutamine withdrawal remain to be elucidated; however, sestrin2 may have a role in this process. The expression of sestrin2 is upregulated in lung cancer cells during glutamine deprivation, and it associates with mTORC2 (103). The sestrin2-mTORC2 association is accompanied by a reduction in mTORC1 activity, suggesting that mTORC2 activation may be linked to repression of mTORC1 via sestrin2 during glutamine starvation as a prosurvival mechanism. Sestrin2 upregulates the catalytic activity of mTORC2, leading to Akt Ser473 phosphorylation. Sestrin2 promotes Akt phosphorylation via Sestrin/GATOR2 and GATOR2/mTORC2 interaction (104). Thus, mTORC2 activation during glutamine starvation could be linked to repression of mTORC1 via sestrin2. Sestrin3 also associates with rictor and enhances mTORC2 activation in response to insulin and nutrient stimulation (105). The addition of amino acids (a mix of essential and nonessential amino acids) to cells that were starved of serum and all amino acids has also been shown to increase Akt Thr308/Ser473 phosphorylation, suggesting that mTORC2 is also activated by increased levels of amino acids (83). In fission yeast, glucose activates TORC2 via the Rab family GTPase Ryh1 (106). Supplementation of serum-starved cells with fatty acids, specifically unsaturated oleic acid, also increases mTORC2 (as well as mTORC1) activation as indicated by Akt Ser473 phosphorylation (95). mTORC2 activity is also enhanced by ammonium and release of calcium from the ER (107). Thus, mTORC2 activation responds to intracellular fluctuations of nutrient and metabolite levels that can occur during stimulation with growth signals as well as nutrient deprivation or other stress conditions.
mTORC2 activation may also occur independent of PI3K signals. mTORC2 phosphorylates the Akt turn motif (TM) site (Thr450) and PKC TM and HM sites in a constitutive manner and occurs independently of PI3K (108, 109). Phosphorylation of these sites occurs during translation (110). mTORC2 associates with translating ribosomes, and its activity is enhanced under these conditions. Currently, it is not known whether mTORC2-mediated cotranslational phosphorylation can occur in response to levels of nutrients or metabolites. However, Akt Thr450 phosphorylation has been shown to be sensitive to glucose deprivation and acute ATP depletion, thus providing some insights into mTORC2 activity under these conditions (111). mTORC2 is also activated independently of PI3K by association of rictor with CD146 through the juxtamembrane KKGK motif of this cell adhesion molecule [also known as melanoma cell adhesion molecule (MCAM)] (112). This association protects rictor from ubiquitin-mediated degradation and sustains mTORC2 activation.
Increased mTORC2 activation also occurs via association of its components with other signaling molecules. There is accumulating evidence that there is an interaction between SIN1 and Ras and that the interaction of mTORC2 with other Ras-related proteins including Rac1, Rho, Rap1, Rit, and Ryh1 regulates mTORC2 activity (106, 113–119). Superoxide anions enhance mTORC2 activation via association of SIN1 with Ras at the membrane (116). Disruption of this association prevents mTORC2 activation. Other signals that promote mTORC2 activation by the Ras family proteins remains to be investigated. Rictor also physically interacts with the inhibitor of nuclear factor-κB kinase (IKK) (120). Pharmacological depletion or inhibition of IKK decreases phosphorylation of the mTORC2 targets Akt and PKC-α (120, 121). Precisely how the Ras family proteins and IKK signaling modulate mTORC2 activity warrants further investigation.
Feedback signals from mTORC1 also modulate mTORC2 activation. Increased mTORC1 signals elevate S6K1 activity, which promotes phosphorylation of negative regulatory sites of the insulin receptor substrate-1 (IRS-1). Phosphorylation of IRS-1 at these sites dampens downstream signals, including mTORC2 (122). Inhibiting mTORC1 with rapamycin increases PI3K signaling via mitigating S6K1 activity toward IRS-1. mTORC1 also phosphorylates growth factor receptor-bound protein 10 (Grb10), which also negatively feeds back to insulin signaling (123). Inhibiting mTOR by rapamycin also increases the expression of growth factor receptors such as IGF1R, thus further enhancing PI3K/mTORC2 signaling (124). Elevated mTORC2 signaling feeds back to dampen insulin signals via mTORC2-mediated regulation of IRS-1 turnover, thereby subsequently downregulating mTORC2 (125).
β- and α-adrenergic signaling through GPCR modulates mTORC2 (126). β-Adrenergic stimulation of mTORC2 signaling in brown adipocytes in vivo is necessary for cold-induced stimulation of glucose uptake and metabolism to maintain temperature homeostasis (127). In skeletal muscle, β-adrenergic signaling via mTORC2 and mTORC1 promotes protein synthesis and glucose uptake (128). α-Adrenergic signaling to mTORC2 also promotes glucose uptake in cardiomyocytes (129). Adrenergic signaling to mTORC2 is mediated via cAMP and Epac1 (127, 130). Whereas these findings support an anabolic function for the mTOR complexes in response to adrenergic signaling, there are conflicting findings as to how catabolic signals (such as during lipolysis) triggered by stimulation of adrenergic receptors affect both mTORC1 and mTORC2 (93, 131). In particular, mTORC2 seems to be modulated positively by both anabolic and catabolic signals. Although it is strongly activated by anabolic insulin/PI3K signals, the finding that AMPK positively modulates mTORC2 during glucose starvation supports the notion that it is activated by catabolic signals (102, 132). It is likely that cells maintain a basal level of mTORC2 activation and that mTORC2 activity is augmented in response to nutrient/metabolite fluctuations. Modulating mTORC2 activation levels could allow cells to restore mTORC1 activation during nutrient-limiting conditions and reestablish metabolic homeostasis during stress conditions. Future studies should address how mTORC2 could be distinctly modulated by anabolic versus catabolic signals.
4. STRUCTURE
Recent structural and computational studies have provided additional insight into mTOR complexes and their mode of activation. Cocrystal structures of truncated mTOR and mLST8 reveal a highly recessed active site (133). The FKBP12/rapamycin binding (FRB) domain, which is adjacent to the kinase domain of mTOR, serves as a gatekeeper, and the rapamycin-binding site interacts with substrates and allows access to the active site. Binding of FKBP12-rapamycin to the FRB blocks substrate recruitment. By combining cryogenic electron microscopy (cryo-EM) with crystallographic studies, the conserved amino-terminal domain of raptor was shown to be juxtaposed to the kinase active site, consistent with its role in substrate recognition and presentation (134). The raptor α-solenoid detects RagA nucleotide state, whereas the raptor “claw” detects that of RagC (135). As discussed above, Rag/Raptor binding is critical for mTORC1 localization to the lysosome and signaling. With cryo-EM, Rheb was demonstrated to bind mTOR distally from the kinase active site, and this binding allows the inhibitory FAT domain to disengage from the N-lobe to adopt the active conformational state (73). This process is likely mimicked by cancer-associated mutations (73). In contrast to Rheb-mediated mTORC1 activation, PRAS40 inhibits mTORC1 activity by binding substrate recognition sites, the TOR signaling (TOS) motif docking site and the rapamycin-binding site at the entrance of the catalytic cleft, as well as another site on mLST8 (73). Structural analysis of the GATOR-Rag GTPase complex reveals that this complex forms at least two binding modes (136). There is an inhibitory mode between GATOR1 and Rag GTPases that may act to prevent the hyperactivation of GATOR1. The inhibitory interaction is characterized by a strong binding affinity between the Rag GTPases and DEPDC5 and low GAP activity. The second mode includes weaker interactions between the NPRL2-NPRL3 heterodimer and RagA that promote GAP activity.
By computational analysis using structural databases, rictor was identified to contain HEAT and WD40 domains, which could serve as a common interacting motif with raptor for binding to mTOR. Rictor may also contain a PH domain that supports membrane localization. There is also a region that is homologous to 50S protein L17 and 39S protein L17, which can serve as a ribosome binding domain (137). The structure of mammalian mTORC2 by cryo-EM parallels that of the yeast TORC2 (138–140). mTOR forms a dimer, and Rictor and mSIN1 hinder the FKBP12-rapamycin binding site. Rictor forms helical repeat clusters and contacts mTOR at multiple sites. In a recent study, a comparatively higher-resolution cryo-EM study of mTORC2 revealed that rictor is composed of three interacting stacks of α-helical repeats (referred to as the ARM domain), the HEAT-like domain (HD), and the COOH-terminal domain (CD) (141). The CD blocks the binding region of FKBP12-rapamycin in mTORC1, and thus explains the rapamycin insensitivity of mTORC2. The binding of rictor and raptor to mTOR is also mutually exclusive. SIN1 binds to the mTOR/rictor complex via its region spanning amino acids 100–240. This site is located close to the FRB domain and catalytic site of mTOR and serves to recruit mTORC2 substrates (138, 142). SIN1 harbors Ras-binding and PH domains and could thus engage mTORC2 targets to the membrane compartment (97, 143). Cryo-EM structures of mTORC2 reveal that SIN1 barricades the mTOR active site. SIN1 connects rictor with mLST8, suggesting a role for stabilizing mTORC2 (141). SIN1 also positions its substrate-recruiting CRIM domain via mLST8. There are five SIN1 isoforms, and many of the mTORC2-related functions have been ascribed to the longer isoforms (SIN1.1/SIN1.2 or SIN1α/SIN1β) (30, 33). A short isoform, SIN1γ, does not seem to mediate mTORC2 activity, although it associates with this complex (39). Distinct functions for these isoforms have yet to be investigated.
Structural analysis also reveals that the assembly of mTORCs is mediated by the chaperonin CCT complex (144). By cryo-EM, CCT was demonstrated to mediate the folding of the β-propellers of mLST8 and raptor. mLST8 binds CCT within the folding chamber between the two CCT rings. It is not known whether CCT could also mediate folding of rictor and/or SIN1, but it is interesting to note that CCTβ is deregulated in mTORC2-disrupted cells (145), suggesting possible reciprocal regulation of CCT and mTORC2. mTOR folding and mTORC1 dimer assembly is linked to the Hsp90-TTT (Tel2-Tti1-Tti2)-RUVBL cochaperone complex (146, 147). This chaperone complex, which is sensitive to the energy state, is also required for mTORC1-Rag1 interaction and localization to the lysosomes. A more recent cryo-EM analysis revealed two ligand binding sites in mTORC2. The I site in mTOR, present in both mTOR complexes, binds inositol hexakisphosphate and could function in mTOR folding or assembly (141). There is an A site in rictor that binds ATP and may function to link mTORC2-specific partner protein interactions to cellular concentrations of nucleotide triphosphate. Future studies should address precisely how the cellular metabolic state affects these chaperone complexes and thus influences mTORC assembly and signaling.
5. REGULATION OF COMPLEX COMPONENTS
As discussed above, the mTOR complexes interact with a large network of regulators that can induce conformational changes or permit recruitment and transport to distinct cellular compartments for activation of the complex. In this section, we review how each of the main complex components undergoes posttranslational modifications and how their expression levels can be regulated in response to various stimuli or cellular conditions. mTOR is phosphorylated at Ser2448 in response to growth factors. This phosphorylation is rapamycin sensitive and attenuated during amino acid starvation, consistent with a role for the mTORC1 target, S6K1, in phosphorylating this site (148–150). mTOR is also autophosphorylated at Ser2481 in a growth signal-dependent manner (151). Phosphorylation at this site occurs in both mTORC1 and mTORC2, although it is not detected in mTORC2-disrupted cells (110, 152, 153). How phosphorylation at this site affects mTOR activity toward specific substrates in vivo remains poorly understood. mTOR phosphorylation also occurs at Ser2159 and Thr2164, and this phosphorylation promotes mTORC1 kinase activity by modulating the mTOR-raptor interaction (154). In response to insulin, mTOR is phosphorylated at Ser1261, which enhances its catalytic activity (155). This phosphorylation is required for Rheb-driven mTOR Ser2481 autophosphorylation, supporting the idea that Ser1261 phosphorylation is implicated in regulation of mTORC1 catalytic activity. With the exception of Ser 2446 that is phosphorylated during nutrient-deprived conditions (156), most of the identified phosphorylation sites on mTOR positively regulate its activity.
In addition to phosphorylation, mTOR undergoes ubiquitination at K777/K782/K784 (157). The TNF receptor-associated factor 6 (TRAF6), which interacts with p62, is required for K63 ubiquitination of mTOR. Ubiquitination at these sites promotes mTORC1 activation by amino acids (157). mTOR activity is also regulated by malonylation. mTOR is malonylated at K1218 when fatty acid synthase (FASN) is inhibited (158). Blockade of FASN elevates malonyl-CoA levels and increases mTOR malonylation, consequently decreasing mTORC1 kinase activity. The malonylation of mTOR has been observed in FASN knockdown in endothelial cells, leading to decreased protein synthesis and impaired angiogenesis. Hence, malonylation of mTOR at K1218 negatively regulates its anabolic function. mTOR signaling can also be modulated by control of its protein levels. mTOR is ubiquitinated by the tumor suppressor FBXW7, which binds to mTOR at the consensus CDC phosphodegron (CPD) sequence, located at the HEAT domain of mTOR (159). This ubiquitination promotes mTOR degradation. Tumor cell lines with FBXW7 deletions or mutations are particularly sensitive to rapamycin treatment because of increased mTOR protein levels. mTOR expression is also regulated at the level of mRNA. In injured axons, mTOR mRNA is transported into axons via the cell size-regulating RNA-binding protein nucleolin, thus enhancing mRNA translation locally (160). Deletion of the 3′-UTR of mTOR that is involved in axonal localization decreases mTOR translation and impairs proprioceptive neuronal survival after nerve injury. Together, these findings reveal that mTOR protein expression and/or activity is regulated via phosphorylation, malonylation, and ubiquitination of specific amino acid residues and that localized expression is regulated at the level of mRNA.
Raptor contains multiple phosphorylation sites that can either positively or negatively regulate mTORC1 activity in response to various stimuli. In response to mitogen stimulation, RSK mediates raptor phosphorylation at Ser719/721/722 (161, 162). Phosphorylation at these sites enhances mTORC1 activation. Two clusters of phosphorylation sites also occur in raptor (Ser696/Thr706 and Ser855/Ser859/Ser863/Ser877). Ser696/Thr706 are phosphorylated by the cyclin-dependent kinase cdc2/CDK1 during mitosis (163). Among these sites, Ser863 is responsive to insulin and other growth signals that activate mTORC1. mTORC1 phosphorylation is also likely mediated by mTORC1 itself (164) and is required for subsequent phosphorylation at Ser855/Ser859 (162). Although phosphorylation of Ser863 by the stress-regulated kinases JNK and p38 appears to be linked to increased mTORC1 activation (165, 166), phosphorylation of this site by the Nemo-like kinase (NLK) in response to osmotic and oxidative stress signals suppresses mTORC1 by inhibiting its lysosomal localization (167). In addition to Ser863, Ser696 and Ser706 are also phosphorylated in a JNK-dependent manner during osmotic stress. Whether these stress signals promote or inhibit mTORC1 activation via raptor phosphorylation remains to be further investigated. There are several reports on the negative regulation of mTORC1 via raptor phosphorylation during metabolic stress. Raptor is phosphorylated by AMPK at Ser722/792 during energetic stress. This phosphorylation inhibits mTORC1 function and mediates the metabolic checkpoint function of AMPK during conditions of low glucose or after treatment with mitochondrial and glycolytic inhibitors or AMP mimetics (89). Raptor is also phosphorylated at Ser606 by the LATS1 and LATS2 kinases, which are the main modulators of the Hippo pathway (168). Phosphorylation of raptor by LATS inactivates mTORC1 by preventing the interaction of Raptor with Rheb. Knockin mutant mice expressing the phosphomimetic Raptor-Ser606Asp have smaller liver and heart, highlighting the role of the mTORC1 and Hippo in growth regulation. Raptor is phosphorylated by the TANK-binding kinase (TBK1) at Ser877 (169). This phosphorylation corresponds to decreased mTORC1 activity. TBK1 increases phosphorylation at this site in response to pathogen-associated molecular patterns (PAMPS) such as lipopolysaccharides (LPS). Taken together, these studies support that raptor phosphosite regulation is associated with how mTORC1 integrates signals from stress, nutrient, and growth factor signals; therefore, the amplitude and duration of raptor phosphorylation at one or more of these phosphosites can determine mTORC1 response.
In addition to phosphorylation, raptor also becomes acetylated. Raptor is acetylated at K1097 via the acetyltransferase EP300 to enhance mTORC1 activity (57). This occurs during leucine catabolism, which generates acetyl-CoA, the metabolite used for acetylation reactions. Raptor is also modulated at the levels of mRNA and protein expression. Some solid tumor types are marked by upregulated raptor expression. Elevated expression levels of endogenous raptor mRNA and protein in colorectal cancer tumors are found to upregulate the ribosome assembly factor URB1 (170). In renal cancer cells, elevated raptor expression at the mRNA and protein levels contributes to PI3K/mTOR inhibitor resistance (171). The mechanisms involved in increased raptor expression and treatment resistance remain to be investigated. Insights from such studies could also reveal predictive markers of therapeutic response to mTORC1 inhibitors in different cancers.
mTORC2 activity is regulated by posttranslational modifications of its components rictor and SIN1, through activating or inactivating phosphorylation and acetylation at distinct sites. Approximately 37 phosphorylation sites have been identified by tandem mass spectrometry (MS/MS) analysis or predicted via motif analysis for rictor (172, 173). These phosphorylation sites cluster around the COOH-terminal region, which is conserved only in vertebrates. In response to growth factors, Thr1135 is phosphorylated on rictor by S6K (172). A phosphodeficient mutant at this site increases Akt phosphorylation but has no effect on SGK and PKC-α or mTORC2 kinase activity (172–174). Increased Thr1135 phosphorylation promotes binding of rictor to 14-3-3 proteins. Rictor also undergoes phosphorylation at Thr1695 in a GSK3-dependent manner. This phosphorylation site is in a putative CDC4 phosphodegron motif. Mutation of this site prevents association of rictor with FBXW7, which mediates ubiquitination and proteasomal degradation of rictor (175). GSK3β also phosphorylates rictor at Ser1235 in response to osmotic stress and ER stress. Phosphorylation at this site blocks the binding of Akt to mTORC2 and curtails mTORC2 signaling (176). Hence, rictor phosphorylation at distinct sites could either increase or decrease mTORC2 activity. Characterization of the remaining putative phosphosites awaits further investigation.
Rictor is acetylated at multiple sites (177). A deletion construct that abolishes the Lys residues K1116, K1119, and K1125 decreases mTORC2 activity (178). p300 mediates acetylation of rictor and increases mTORC2 activity toward Akt. Thyroid hormone treatment of HepG2 cells promotes rictor deacetylation, which occurs in a Sirt1-dependent manner (179). The deacetylation of rictor inhibits mTORC2 while promoting hepatic FoxO1-target gene expression. Rictor acetylation is also mediated by GCN5L1, a regulator of protein acetylation and mitochondrial energy metabolism (180). In GCN5L1-depleted cardiac cells, restoring rictor acetylation mitigates the generation of mitochondrial reactive oxygen species (ROS) and increases cell survival in response to hypoxia and reoxygenation. Hence, these findings so far reveal that rictor acetylation positively impacts while its deacetylation negatively modulates mTORC2 activation.
Rictor gene amplification is found in several cancer types, and its high expression correlates with lower overall survival (181). Increased rictor mRNA and protein expression has been found in a number of cancers and could drive tumor progression and therapeutic resistance (182–186). Translational control of rictor expression has been demonstrated in several studies, and these findings may provide insights into the oncogenic effects of rictor overamplification. The 3′-UTR of rictor is relatively long and contains several cis-regulatory elements that are implicated in mRNA turnover and translational control. Four consensus binding sites for HuR, which is linked to mRNA stability and translation, were found in the 3′-UTR of rictor mRNA. The heat shock transcription factor 1 (HSF) becomes activated by mTORC2 signaling, which induces HuR activity, resulting in a feedforward loop regulation of mTORC2 signaling and growth in glioblastoma (187). Several micro-RNAs (miRs) have been implicated in the regulation of rictor expression. miR-497 inhibits carcinogenesis of hepatocellular carcinoma by targeting rictor and the Akt pathway (188). miR-218 also directly targets rictor to inhibit its expression and prostate cancer angiogenesis via a mechanism involving VEGFA (189). miR-153 also targets rictor to inhibit glioma cells (190). miR-196b, whose expression is increased during loss of DNA methyltransferase 3B (DNMT3B), targets rictor and decreases its expression, thus preventing melanoma growth (191). miR-142-3p inhibits rictor expression in nasal NK cell lymphoma (192). miR-188 targets rictor to modulate the age-related switch between osteogenesis and adipogenesis of bone marrow mesenchymal stem cells (193). Together, these findings reveal how rictor gene expression is extensively regulated and likely acts as a major mechanism to modulate mTORC2. Targeting rictor expression levels could thus serve as an effective therapeutic strategy to specifically block mTORC2 functions.
SIN1 modifications affect mTORC2 activity. SIN1 is phosphorylated at Thr86 and Thr398, leading to SIN1 dissociation from the mTORC2 complex (194). A patient-derived SIN1-R81T mutation that prevents Thr86 phosphorylation promotes mTORC2 hyperactivation (194). Phosphorylation of this site is mainly mediated by Akt (195). Whether this phosphorylation has a positive or negative effect on mTORC2 activation is controversial (195). In response to angiotensin II stimulation of conventional PKC, SIN1 is also phosphorylated at Ser128, Ser315, and Ser356 (196). Although phosphorylation at these sites was not required for the mTORC2-mediated phosphorylation of SGK1 at Ser422 or Akt Ser473, phosphorylation at Ser128 may potentiate SIN1 phosphorylation at Thr86 and thereby enhance mTORC2 activity. The 5′-UTR of SIN1 is modulated by programmed cell death 4 protein (Pdcd4) by suppression of eIF4A (197). In colon cancer cells, loss of Pdcd4 increases SIN1 protein but not mRNA levels, indicating the regulation of translation of SIN1 by Pdcd4.
Although mLST8 is a shared subunit of both mTORC1 and mTORC2, studies point to its specific role in mTORC2 assembly and function. mLST8 loss blocks mTORC2 assembly without affecting mTORC1 (198). mLST8 also strongly interacts with SIN1 (140). Among the mTOR complex components, mLST8 is more highly ubiquitinated. Within the seventh of the WD40 repeats (WD7) of mLST8, K305 and K313 undergo K63-linked ubiquitination. mLST8 undergoes K63-linked polyubiquitination via the TRAF2 E3 ubiquitin ligase, disrupting its interaction with SIN1 (199). OTUD7B deubiquitinase removes the polyubiquitin chains from mLST8, thus enhancing interaction with SIN1 and increasing mTORC2 assembly and signaling.
Collectively, the mTORCs are modulated at several levels including their gene and protein expression, complex assembly, and modification of complex components. Future investigations will unravel how nutrient/growth conditions impact their regulation and how we can specifically target their activity by exploiting particular regulatory mechanisms that may have tissue-specificity, oncogenic, or metabolic vulnerability.
6. SUBSTRATES
As a protein kinase, mTOR phosphorylates its targets at Ser or Thr residues. mTORC2 has also been reported to have tyrosine kinase activity (200). Phosphoproteomic studies uncovered numerous putative mTOR targets (123, 201, 202). Several of these targets play roles in signaling, metabolic pathways, and protein synthesis. Among the most well-characterized substrates of mTOR, as part of either mTORC1 or mTORC2, are the members of the AGC [protein kinase A (PKA), PKG, PKC] kinase family that also includes Akt (PKB) and S6K (203). mTOR phosphorylates the AGC kinases at the conserved HM, which allosterically activates the AGC kinase. mTORC1 phosphorylates S6K1 at the HM site, Thr389. Another conserved motif in AGC kinases is the turn motif (TM), and several sites in the TM of S6K1 are rapamycin sensitive. The well-characterized function of mTOR in regulating the translation regulators S6K1 and 4E-BP1 is mediated by mTORC1. Raptor serves to present substrates to mTOR via their TOS motifs. The TOS motif is a 5-amino acid peptide that was identified in rapamycin-sensitive targets of mTOR (FDL/IDL in S6K1; FEMDI in 4EBP1) (204). The NH2-terminal domain of raptor interacts with the central region of 4E-BP1 (205). Raptor also interacts with the NH2-terminal region of HIF1α, which harbors the TOS motif (206). During insulin stimulation, mTOR directly phosphorylates PRAS40 on Ser183, Ser212, and Ser221. This phosphorylation promotes activation of mTORC1 by derepression of PRAS40 (207, 208). mTORC1 also phosphorylates AMPK at α1Ser347/α2Ser345, which is linked to reduced phosphorylation of Thr172 at the activation loop (209). This phosphorylation downregulates AMPK signaling. mTORC1 negatively regulates autophagy by phosphorylation of Atg13, ULK1 (Ser758), and DAP1 (Ser3/Ser51) (210, 211).
mTORC2 phosphorylates several AGC kinases including Akt, PKC, and SGK1 at their HM and TM sites. Whereas the phosphorylation of the Akt HM site is induced by PI3K signals, the phosphorylation of the Akt TM site, as well as the HM/TM sites of PKC and SGK, is not. SIN1 interacts with mTOR substrates such as SGK1 to allow its phosphorylation and activation of epithelial sodium channel (212). mTORC2 phosphorylates Akt and PKC at the TM that is critical for folding and stabilization of the kinase domain (108–110). mTORC2 is involved in the phosphorylation of the TM of the PKC superfamily member PKN, but whether this is via direct or indirect phosphorylation remains unclear (213). mTORC2 also phosphorylates the ubiquitin ligase subunit Fbw8 at Ser86 to allow Fbw8-mediated degradation of IRS-1 (125). mTORC2 could phosphorylate its substrates under different stimulatory conditions via subcellular compartmentalization (98, 196). In addition, mTORC2 modulates another AGC kinase member, RSK, although this regulation does not appear to require mTOR kinase activity but could instead serve as a scaffold to allow RSK phosphorylation by ERK at the membrane (145). The fact that most of the identified mTORC2 substrates or effectors such as RSK localize to the membrane supports the notion that mTORC2 modulates its targets in this compartment.
Despite the numerous functions of mTORC1 and mTORC2 in metabolism, our knowledge of their direct substrates remains limited. We further discuss some of the metabolic substrates of mTOR below.
7. METABOLIC REPROGRAMMING
Studies in cancer cells have provided important insights into how metabolic processes are reprogrammed during cell proliferation. Oncogenic mutations that drive unrestrained proliferation subsequently rewire metabolism in order to sustain the production of macromolecules and energy that fuel growth and division. By rewiring signaling circuits, cancer cells are able to route metabolites generated from nutrient catabolism toward macromolecular synthesis. In addition, the increased uptake of glucose enhances flux through glycolysis, and the high glycolytic flux not only produces abundant ATP but also generates many intermediates utilized for biosynthetic pathways (214). This enhanced glycolysis in the presence of oxygen is called aerobic glycolysis or the Warburg effect (215). Although Warburg thought that this aberrant behavior of cancer cells was due to defective mitochondria, more recent studies have provided insights on the complex metabolic processes adapted by cancer cells to sustain their growth and proliferation. Depending on the genetic mutations, tissue type, or the tissue microenvironment, cancer cells upregulate the acquisition of other nutrients in addition to glucose. If a particular nutrient is limiting, cancer cells can generate such nutrient de novo by increasing flux through the corresponding biosynthetic pathways. Cells also have mechanisms to recycle redundant macromolecules to favor synthesis of essential ones. Hence, genetic mutations that allow cells to upregulate de novo biosynthesis and/or catabolize macromolecules are common in cancers. Normal cells also reprogram their metabolism depending on nutrient availability, environmental signals, and intrinsic mechanisms. For normal cells, such metabolic rewiring is tightly regulated. The mTOR signaling pathway plays a central role in orchestrating metabolic reprogramming. Since the discovery of mTOR as a regulator of protein synthesis in response to nutrients, numerous studies through the years have unraveled that mTOR controls many aspects of anabolic and catabolic metabolism. Below, we review how mTORC1 and mTORC2 are involved in different biosynthetic pathways and metabolic processes. We then discuss how such functions of the mTORCs control metabolic reprogramming in healthy states, using stem cells and immune cells as paradigms. We also provide an overview of how deregulation of mTOR metabolic functions plays a role in aging and pathological conditions such as cancer, type 2 diabetes/obesity, and neurodegenerative disorders.
8. GLUCOSE METABOLISM
Glucose metabolism generates energy in the form of ATP. It also produces metabolic intermediates that are utilized for macromolecule synthesis. The uptake and metabolism of glucose are controlled by growth factors such as insulin, which regulates PI3K and mTOR signaling (216). Normal differentiated cells primarily metabolize glucose to pyruvate while generating ATP and reducing the cofactor nicotinamide adenine dinucleotide (NAD+) to NADH (FIGURE 2A). This is followed by complete oxidation of pyruvate to CO2 through the tricarboxylic acid (TCA) cycle by oxidative phosphorylation/respiration (OXPHOS) in the mitochondria under normoxic conditions. However, in oxygen-depleted conditions, pyruvate is metabolized to lactate in the cytosol by lactate dehydrogenase (LDH) (215). In tumors and other proliferating cells, there is an enhanced rate of glucose uptake and glycolytic activity resulting in an increased production of pyruvate at a higher rate than can be metabolized by the mitochondria. Despite oxygen availability, the excess pyruvate is metabolized predominantly to lactate in the cytosol. This switch to aerobic glycolysis, termed the Warburg effect, is a hallmark of cancer cells (217) and occurs in other highly proliferating cells, including stem cells and activated immune cells. Compared with OXPHOS, glycolysis generates less ATP; however, this switch supports ATP production at a higher rate and generates readily available intermediates that serve as carbon sources for biosynthetic pathways branching from the glycolytic pathway, such as de novo generation of nucleotides, lipids, and amino acids. Enhanced growth factor-PI3K/Akt/mTOR pathway signaling in highly proliferating cells drives the aerobic glycolytic switch and reprograms glucose metabolism for increased cell growth and survival. Along the glycolytic pathway, mTORC1 and mTORC2 directly and indirectly control the expression of glucose transporters, glycolytic enzymes, and transcription factors that induce gene expression of various glycolytic effectors (215).
FIGURE 2.
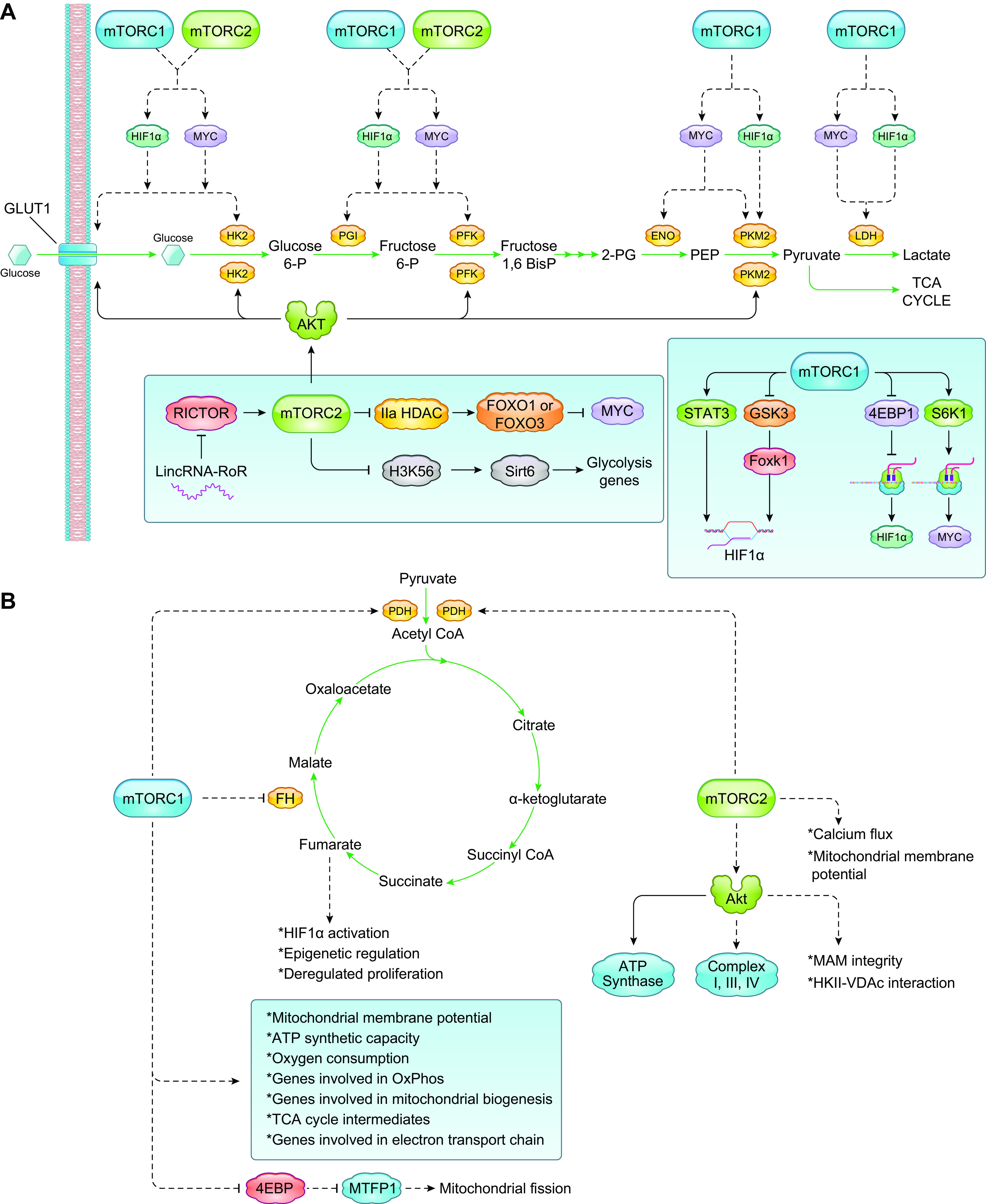
Control of glycolysis and mitochondrial metabolism by mTORC1 and mTORC2. A: in highly proliferating cells there is a switch to aerobic glycolysis, termed the Warburg effect. The enhanced rate of glucose uptake and glycolytic activity results in an increased production of pyruvate, which is preferentially metabolized to lactate in the cytosol by LDH. mTORC1 and mTORC2 directly and indirectly reprogram glucose metabolism for increased cell growth and survival by control of the expression of transcription factors (HIF1α, Myc) that induce gene expression of glycolytic effectors and glucose transporters. mTORC2 also controls membrane trafficking of glucose transporters (GLUT1), expression and/or activity of glycolytic enzymes. B: mTORC1 controls several aspects of mitochondrial metabolism including oxygen consumption, membrane potential, and mitochondrial biogenesis. The role of mTORC2 in oxidative and mitochondrial metabolism is less understood, although its substrate Akt has been shown to modulate different components and processes involved in mitochondrial metabolism. See glossary for abbreviations.
8.1. mTORC1 Regulation of Glycolysis
mTORC1 promotes glycolysis through induction and regulation of two critical transcription factors, HIF1α and Myc. mTORC1 increases the transcription and translation of HIF1α. The heterodimeric HIF1 complex is composed of an HIF1α subunit (gene expression of HIF1α is normally elevated under hypoxic conditions) and a constitutively expressed HIF1β nuclear subunit (218). In hypoxic conditions, HIF1α protein levels are stabilized and accumulate, thus enhancing transcriptional induction of genes that encode glycolytic enzymes and effectors, consequently upregulating glycolysis (219). HIF1α stability and transcriptional activity can be augmented because of oncogenic mutations that promote upregulation of the PI3K/Akt/mTOR signaling pathway. Inappropriate activation of HIF1α under normoxia causes a metabolic shift toward aerobic glycolysis (220). Inhibition of mTOR by rapamycin decreases HIF1α stabilization and transcriptional activity under hypoxic conditions and blocks the growth factor- and mitogen-induced HIF1α expression (221, 222). Activation of mTORC1 drives HIF1α transcription. There are several mechanisms by which mTORC1 regulates HIF1α mRNA transcription. mTORC1 indirectly enhances the transcription of HIF1α mRNA via phosphorylation of its downstream target, STAT3 (223). In insulin-stimulated HEK293 cells, STAT3 is directly phosphorylated by mTORC1 on Ser727 during hypoxia, promoting HIF1α mRNA transcription. mTORC1 also regulates HIF1α mRNA transcription by modulating Foxk1, which is a transcriptional regulator of HIF1α. By directly suppressing the activity of the Foxk1 kinase, GSK3, mTORC1 prevents the phosphorylation and nuclear exclusion of Foxk1 (224).
Activation of mTORC1 also increases HIF1α mRNA translation. In TSC2−/− cells, which exhibit constitutively active mTORC1 signaling, there is increased translation of HIF1α mRNA (225), whereas treatment with rapamycin decreases HIF1α mRNA levels (226, 227). mTORC1 promotes HIF1α synthesis through enhanced translation via the regulation of its two substrates, 4E-BP1 and S6K1, for cap-dependent translation initiation of HIF1 mRNA (223, 228). Knockdown of S6K1 in PTEN-deficient cells decreases HIF1α expression and glycolysis (229).
mTORC1 also promotes glycolytic gene expression via the transcriptional regulator Myc (230, 231). Myc is deregulated in >50% of human cancers (232), and its amplification is often linked to increased aerobic glycolysis and glutaminolysis (233). mTORC1 activation of S6K and 4EBP1 is required for Myc-driven tumorigenesis (234). Several rapamycin-sensitive genes are regulated by Myc (228). mTORC1 modulates Myc expression. By regulation of eIF4B through S6K1 and 4E-BP1, translational repression by the structured 5′-UTR of Myc mRNA is relieved, thus enhancing Myc translation (231, 235).
By modulating HIF and Myc, mTORC1 regulates the expression of genes whose products are involved in different aspects of glycolysis including glucose transport and metabolism. Transcriptional profiling of rapamycin-treated lymphocytes revealed altered glycolytic gene expression in these cells (21, 236). Results of gene expression arrays from TSC2−/− mouse embryonic fibroblasts also confirmed the involvement of mTORC1 in inducing a HIF1α-dependent transcriptional program to promote glycolysis (228).
mTOR controls glucose uptake in glycolysis through regulation of gene expression and membrane trafficking of glucose transporters. mTOR signaling is frequently activated in several types of malignant tumors that are marked by an increased expression of glucose transporter-1 (GLUT1), such as pancreatic, hepatocellular carcinoma, breast, lung, and colorectal cancer cells (237). In macrophages and T cells, the overexpression of GLUT1 also promotes glycolysis and a proinflammatory phenotype (238, 239). The increase in GLUT1 expression via mTORC1 occurs through HIF1α and Myc (240–242). Interestingly, hyperactivation of mTORC1 such as in liver-specific Tsc1 mutant mice was accompanied by a reduction of glucose uptake (243). The mTORC1/S6K1-mediated negative feedback loop that downregulates the PI3K/Akt pathway, which plays a role in glucose transport, could account for this (122).
mTORC1 also modulates the metabolic enzymes that control key steps in glycolysis. Hexokinase 2 (HK2), which phosphorylates glucose in the first step of glycolysis, is often upregulated in cancer cells to enhance glycolysis (244, 245). Increased mTORC1 signaling enhances HK2 expression via HIF1α and Myc transcription factor activity. Increased mTORC1 signaling due to TSC2 loss also specifically upregulates the phosphofructokinase-2/fructose-2,6-bisphosphatase B3 isotype (PFKFB3) in AML (246). Increased PFKFB3 levels are dependent on HIF1α and are important for AML survival. mTORC1 also regulates the mRNA and protein expression of pyruvate kinase M2 (PKM2), which is exclusively expressed in proliferating and tumor cells (247). PKM2 is an alternatively spliced product of PKM pre-mRNA, and the expression of this isoform is increased during the switch to aerobic glycolysis. Pyruvate kinase catalyzes the formation of pyruvate in the final step of glycolysis. The expression of this rate-limiting glycolytic enzyme is regulated transcriptionally by mTORC1 via HIF1α and Myc (248). However, PKM2 is also a transcription coactivator that increases HIF1α target gene binding. In this way, PMK2 amplifies mTORC1 signaling in cancer cells through interaction with HIF1 transcription complex to coregulate HIF1α transcriptional activity and its own expression (249). PKM2 sustains glycolysis in cancer cells and promotes cell proliferation by driving glycolysis (248). Myc upregulates PKM2 expression to sustain glycolytic flux even under normoxia in an mTORC1-dependent manner through the alternative splicing repressors heterogeneous nuclear ribonucleoproteins (hnRNPs) (248). In turn, PKM2 activates mTORC1 through phosphorylation of the mTORC1 inhibitor proline-rich Akt substrate 1 (PRAS40) (250). Phosphorylated PRAS40 dissociates from raptor, resulting in activation of mTORC1. mTORC1, via HIF1α and Myc, also regulates the gene that encodes lactate dehydrogenase (LDH), a tetrameric enzyme composed of a combination of the subunits LDHA and LDHB. LDH converts pyruvate to lactate. In glycolytic cancer cells producing high amounts of pyruvate, increased LDH allows cancer cells to sustain glycolysis (251). mTORC1 inhibition by rapamycin downregulates LDHA gene expression and other metabolic effectors in prostate cancer cell lines (252). LDHB gene expression is upregulated in an mTOR-dependent manner in murine embryonic fibroblasts (MEFs) that have deficiency in TSC1, TSC2, or PTEN and with activated Akt. Enhanced LDHB levels are critical for hyperactive mTOR-mediated tumorigenesis (253).
Disruption of mTORC1 in other nontumorigenic tissues further highlights its role in glucose metabolism (see also sects. 18 and 20). In raptor-deficient muscle, glucose metabolism is downregulated. These mice are lean and resistant to insulin and high-fat diet and have increased energy expenditure (254). When TSC1 is abrogated, leading to increased mTORC1 activity, the mice are also lean and glucose intolerant and develop insulin resistance, similar to the raptor knockout. Hence, optimal mTORC1 activation in the muscle is crucial for whole body metabolism. Conditional disruption of raptor in developing B cells blocks development at the pre-B stage and is accompanied by decreased glycolysis and oxidative phosphorylation (255). In macrophages, deficiency in raptor or pharmacological inhibition of mTORC1 suppresses HK1 protein expression and glycolysis and thus prevents activation of an immune response (256).
Animal studies that knock out the genes involved in negative regulation of mTORC1 further uncover how enhanced mTORC1 activation reprograms metabolism. Knockout of the GATOR1 component NPRL2 in skeletal muscle increases pyruvate metabolism to lactate while minimizing its entry into the TCA cycle (257). A compensatory rise in anaplerotic reactions occurs while levels of amino acids such as aspartate and glutamine decrease, likely because of their utilization for anaplerosis. In the absence of TSC2, which leads to mTORC1 hyperactivation, glycogen synthesis also increases (258). Abnormal glycogen storage is caused by both mTOR-dependent and -independent mechanisms. The underlying mechanism involves impaired autophagic degradation of glycogen due to deregulation of proteins involved in the autophagy-lysosome pathway. This defect can be rescued by mTORC1 or Akt inhibitors. Loss of the TSC complex also enhances gluconeogenesis. The sustained mTORC1 activation prevents CpG methylation and silencing of the delta-like homolog 1 (Dlk1)-deiodinase iodothyronine type III (Dio3) cluster, thus enhancing the miRNA transcription, and leads to increased blood glucose levels (259). Overall, these findings provide insights into how mTORC1 activation levels are tightly regulated to maintain glucose homeostasis. Several diseases characterized by hyperactivation of mTORC1, such as during loss of TSC function, could benefit from treatment with rapamycin or its analogs and possibly restore normal glucose metabolism.
8.2. mTORC2 Regulation of Glycolysis
mTORC2 controls glycolysis via Akt-dependent and -independent mechanisms. Akt has long been studied as a central regulator of glucose metabolism. It modulates glycolytic enzymes such as hexokinase (260) and PFK2 (261) as well as the glucose transporter GLUT1 (262, 263). Akt activation is sufficient to increase the rate of glucose metabolism (264) and the induction of glycolytic genes via HIF1α (265). Furthermore, downregulation of Akt and mTOR expression that is linked to decreased HIF1α during fasting in colorectal cancer (CRC) cells also inhibited aerobic glycolysis and proliferation in CRC (266). Interestingly, this blockade of Akt/mTOR signaling was due to upregulation of the cholesterogenic enzyme Fdft1 (farnesyl-diphosphate farnesyltransferase 1) that is increased during fasting, suggesting that Fdft1 negatively regulates Akt/mTOR. In glioblastoma, wherein Akt activity is elevated, the increase in aerobic glycolysis is linked to decreased expression of the large intergenic noncoding RNA-RoR (LincRNA-RoR). Increased expression of LincRNA-RoR decreases rictor expression, mTORC2 activity, and expression of glycolytic effectors (267). Together, these findings support that mTORC2 and Akt expression/activity upregulate aerobic glycolysis in cancer.
mTORC2 also controls glycolytic metabolism via Akt-independent mechanisms. In glioblastoma mTORC2 enhances aerobic glycolysis through regulation of c-Myc levels. mTORC2 promotes phosphorylation of class IIa HDACs such as HDAC4 and HDAC5/7, thereby inactivating them, which then upregulates acetylation of FoxO1 and FoxO3, resulting in derepression of c-Myc (268). mTORC2 also increases acetylation levels of the histone H3K56 in glioma, which influence glycolytic gene expression due to enhanced recruitment of Sirt6 in the promoter of these genes (269).
Several studies in other noncancer models have also demonstrated the role of mTORC2 in glycolysis (see also sects. 18 and 20). In developing double-negative (CD4−CD8−) thymocytes, SIN1/mTORC2 promotes the expression of PKM2 via an Akt-dependent nuclear translocation of peroxisome proliferator-activated receptor (PPAR)γ, a transcriptional activator of PKM2 (270). SIN1 deficiency in these developing thymocytes decreased proliferation and glycolysis due to downregulation of a number of genes involved in glycolysis and oxidative metabolism. SIN1 also controls the expression and stability of c-Myc protein and maintains the activity of mTORC1 through the Akt-dependent inactivation of GSK3 and TSC1/2, respectively. SIN1 coordinates the activation of mTORC2 and mTORC1 to control B cell growth and metabolism (271). Investigations utilizing liver-specific rictor-knockout mice unraveled the function of mTORC2 in systemic glucose metabolism. In the liver of these mice, glycolysis was impaired, glucokinase activity reduced, and Akt phosphorylation abrogated. However, the glucose flux in these mice was rescued by the expression of a constitutively active Akt or glucokinase (272). These findings provide strong evidence that mTORC2 regulates glycolysis in the liver via Akt. In the muscle, rictor/mTORC2 regulates glucose uptake during exercise in the mice (273). Expression of the mTORC2 effectors PKC-α and NDRG1 is also increased with exercise. mTORC2/rictor also mediates the α1A-adrenoceptor (AR) stimulation of glucose uptake in cardiomyocytes (129). mTORC2 mediates aerobic glycolysis induced by WNT3A/LRP5 signals during osteoblast differentiation (274). In this scenario, mTORC2 activation occurs via RAC1 and results in upregulation of key glycolytic enzymes. Additionally, in the brain, when rictor is specifically deleted in proopiomelanocortin (POMC)-expressing neurons, obesity, hyperphagia, fasting hyperglycemia, and profound glucose intolerance occur (275). Together these findings support that mTORC2 is involved in the central regulation of energy and glucose metabolism.
9. TCA CYCLE/MITOCHONDRIAL METABOLISM
Pyruvate enters the mitochondria, where it is metabolized to generate maximal amounts of ATP via the tricarboxylic acid (TCA) cycle, The TCA cycle also generates intermediates that are utilized as biosynthetic precursors in highly proliferating cells (215). Early studies using rapamycin linked mTORC1 to mitochondrial metabolism. Rapamycin lowers mitochondrial membrane potential, ATP synthetic capacity, and oxygen consumption and alters the mitochondrial phosphoproteome (276) (FIGURE 2B). Raptor deficiency reduces the rate of oxygen consumption, impairs oxidative capacity, diminishes expression of genes involved in oxidative phosphorylation and mitochondrial biogenesis, and reduces amounts of TCA cycle intermediates (277–279). mTORC1 inhibition via raptor silencing downregulates the expression of genes encoding electron transport chain (ETC) proteins, demonstrating that reduced mTORC1 activity can impair mitochondrial respiration (280). Several studies have investigated mTORC1-mediated transcriptional control of mitochondrial oxidative function through interactions with key regulators. In skeletal muscle tissue and cells, rapamycin treatment reduces the expression of mitochondrial transcriptional regulators such as PGC-1α (281). mTOR and raptor also interact with the mitochondrial gene transcriptional regulator yin yang 1 (YY1). Raptor and mTOR are required for YY1 coactivation by PGC-1α and therefore control mitochondrial gene expression. Further studies have shown that mTOR can directly control mitochondrial function (282). A significant fraction of cellular mTOR localizes at the outer mitochondrial membrane (283), where it associates with the apoptotic protein Bcl-xL and substrate transport protein VDAC1 that are both present in this compartment (282, 284). mTOR phosphorylates Ser62 of Bcl-xL in vitro. Phosphorylation at this site regulates Bcl-xL activity. It is not known whether VDAC is also phosphorylated by mTOR or needed for its involvement in substrate transport. Although mTOR-mediated phosphorylation of VDAC has not been demonstrated, inhibition of VDAC2 in Jurkat cells generated changes to the metabolic profile that were reminiscent of rapamycin treatment, including increased production of lactate, glycerol, and upstream glycolytic intermediates (282). Phenylalanine and tyrosine levels are enhanced in response to mTOR inhibition. These amino acids can produce fumarate, an important TCA cycle intermediate, after the catabolism of their carbon skeletons. These findings suggest that mTORC1 inhibition could limit mitochondrial substrate availability, which can in turn promote diversion from mitochondrial respiration to aerobic glycolysis and result in the accumulation of glycolytic intermediates. mTORC1-mediated accumulation of the TCA cycle oncometabolite fumarate is directly associated with tumorigenesis in renal cell carcinoma (285). In kidney-specific TSC1-null renal cell carcinoma mouse models, upregulated mTORC1 activity caused an mTOR-dependent downregulation of fumarate hydratase. Increased levels of fumarate promote oncogenesis via activation of HIF1α, epigenetic and posttranslational modifications. How mTOR regulates fumarate hydratase remains to be investigated. The activity of mTOR can also be modulated by a TCA cycle oncometabolite, 2-hydroxyglutarate (2-HG). This metabolite is generated from α-KG due to a gain-of-function mutation in the isocitrate dehydrogenase (IDH1/2) that occurs in tumors such as gliomas. The activation of mTOR by 2-HG occurs via a decrease in DEPTOR protein stability as a consequence of the inactivation of KDM4A, an α-KG-dependent enzyme of the Jumonji family of lysine demethylases that associates with DEPTOR (286). Hence, mTOR also responds to levels of TCA cycle metabolites and could serve as a modulator of mitochondrial metabolism.
mTORC1 also controls genes involved in mitochondrial function and dynamics at the level of translation. mTORC1, but not mTORC2, induces the expression of nucleus-encoded mitochondrial proteins (278). mTORC1 performs this function via inhibition of 4E-BP1. mTORC1 also regulates mitochondrial dynamics, the balance between fission and fusion, via the mitochondrial fission process protein 1 (MTFP1). The translation of MTFP1 is negatively modulated by 4E-BP1, whereas the other mTORC1 target, S6K1, does not seem to be involved in mediating mitochondrial respiration or glucose flux to pyruvate and lactate (278). Furthermore, S6K1 knockdown did not result in any significant decreases of mitochondrial gene expression (281). Therefore, 4E-BP plays a more predominant role in modulating the translation of genes involved in mitochondrial metabolism.
The role of mTORC2 in oxidative and mitochondrial metabolism is poorly understood. Findings from studies using mTOR catalytic inhibitors suggest that mTORC2 is involved in mitochondrial metabolism, although part of the inhibitor’s effects may be due to mTORC1 blockade. For example, mRNAs involved in mitochondrial transport and function were among a set of genes that could be translationally suppressed by an mTOR catalytic inhibitor but not rapamycin, suggesting an mTORC2-dependent role in translation (287, 288). Incubation of pancreatic cancer cells with an mTOR catalytic inhibitor decreases glycolytic and TCA cycle metabolites, including NAD+, flavin adenine dinucleotide (FAD), and ATP (289). PI3K/mTOR inhibition decreases oxygen consumption rate via increased phosphorylation of pyruvate dehydrogenase complex subunit E1a, thus diminishing mitochondrial respiration (290). In PTEN-deficient MEFs, there is increased activity of the respiratory complexes I, III, and IV that are sensitive to PI3K or Akt inhibition. Interestingly, protein expression of the respiratory complexes is dependent on 4E-BP1. Active Akt phosphorylates mitochondrion-resident proteins including the ATP synthase subunit, resulting in enhanced complex activity (291). Transformed cells that are mTORC2 addicted are also highly dependent on mitochondrial functions (292). However, knockdown of rictor stimulates mitochondrial respiration, suggesting that mTORC2 could have a negative regulatory role (278). Since depletion of rictor also decreases α-KG levels (278), the role of mTORC2 in TCA anaplerosis would need to be further examined. Compartmental localization may influence the role of mTORC2 in mitochondrial metabolism. In response to growth factor stimulation, mTORC2 localizes to the mitochondria-associated endoplasmic reticulum (ER) membrane (MAM), where it functions in controlling MAM integrity, calcium flux, and mitochondrial membrane potential. (293) These mTORC2 functions are mediated by Akt, which regulates the expression of hexokinase II and other proteins involved in MAM integrity. However, Akt also has related functions at the outer mitochondrial membrane, where it regulates the hexokinase-VDAC interaction (260). Akt could facilitate enhanced hexokinase activity and interaction with VDAC at the outer mitochondrial membrane, thus promoting coupling of glucose metabolism to oxidative phosphorylation. Although mTORC2 localization at MAM is associated with ribosomes, suggesting it is active in this compartment (99, 293), there is insufficient understanding of Akt-independent functions of mTORC2 in this compartment.
Overall, although we have sufficient evidence supporting the role of mTORC1 in the TCA cycle and mitochondrial metabolism, more studies are needed to elucidate how mTORC2 is involved in these metabolic processes.
10. GLUTAMINE METABOLISM
Glutamine is the most abundant nonessential amino acid in the plasma and contributes its carbon and nitrogen for energy production and biosynthetic reactions (294) (FIGURE 3). It is important for amino acid biosynthesis (295). It also donates its nitrogen for the de novo synthesis of purines and pyrimidines. Glutamine is used for the production of hexosamines. Importantly, glutamine serves to replenish TCA intermediates via glutaminolysis, which generates α-ketoglutarate (α-KG). Incorporation of α-KG into the TCA is the major anaplerotic step in proliferating cells and subsequently generates oxaloacetate, which reacts with acetyl-CoA to produce citrate. Glutaminolysis consists of two steps: first, the conversion of glutamine to glutamate, a reaction catalyzed by glutaminase (GLS), and second, the conversion of glutamate to α-KG, which is catalyzed by glutamate dehydrogenase (GDH). Because of the requirement for increased carbon and nitrogen sources by cancer cells, glutamine uptake and metabolism are often upregulated by oncogenic signals (296, 297). As with glucose metabolism, mTOR signaling impinges on several aspects of glutamine metabolism. Here, we discuss the role of mTOR in glutaminolysis as it relates to TCA anaplerosis.
FIGURE 3.
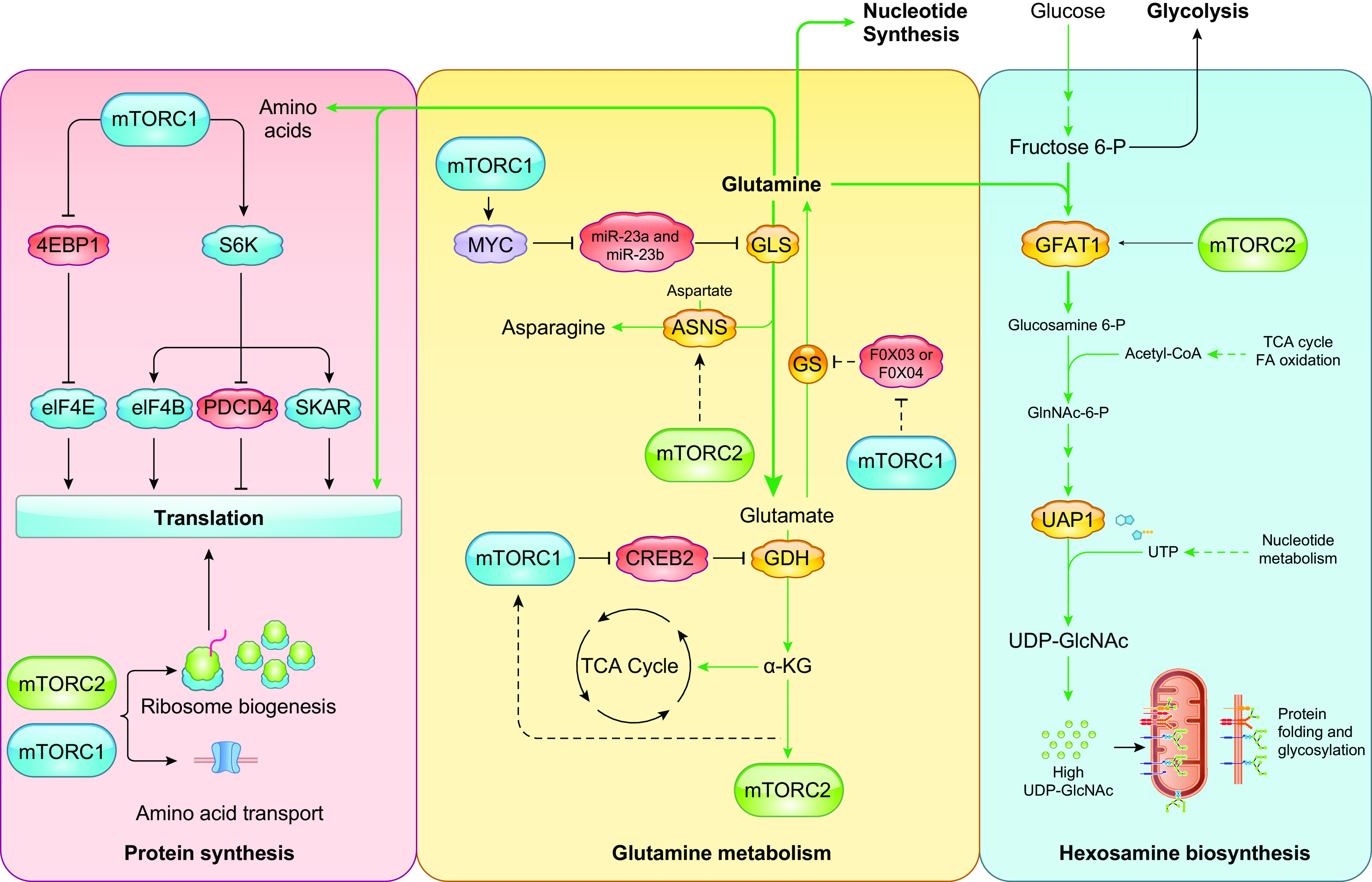
mTORC1 and mTORC2 control metabolic processes important for protein synthesis and folding. mTORC1 controls protein synthesis via phosphorylation of the translation regulators S6K and 4EBP1. Both mTORC1 and mTORC2 are also involved in ribosome biogenesis and transport of amino acids that are required for translation. The amino acid glutamine is a major alternative carbon source and nitrogen source for a number of metabolic reactions including TCA cycle anaplerosis, nucleotide synthesis, and hexosamine biosynthesis. Multiple aspects of glutamine metabolism are controlled by both mTORC1 and mTORC2. mTORC2 has been shown to modulate hexosamine biosynthesis via GFAT1. See glossary for abbreviations.
mTORC1 stimulates glutamine metabolism by regulating transcription factors such as Myc that are involved in expression of glutaminolysis-related genes. Transcription and translation of glutaminase 2, along with other enzymes in the glutamine catabolic pathway, are dependent on Myc in activated T cells (298). There is a regulatory loop wherein mTORC1 enhances glutaminolysis via Myc while glutaminolysis further activates mTORC1 (62, 231, 235). Another way in which mTORC1 promotes glutaminolysis is through regulation of the transcription factor CREB2 (cAMP-responsive element binding 2). mTORC1 promotes the proteasome-mediated degradation of CREB2. Because CREB2 contains a recognition motif in the promoter region of SIRT4, mTORC1-mediated degradation of CREB2 also represses SIRT4 transcription (299). SIRT4, which is localized in the mitochondria, is a member of the sirtuin family of NAD-dependent deacetylases (300). SIRT4 also has NAD-dependent ADP-ribosyltransferase activity and negatively regulates GDH by ADP-ribosylation. By inhibiting GDH, SIRT4 prevents anaplerotic conversion of glutamine to α-KG, thus sparing glutamine in the mitochondria (301). These findings indicate that mTORC1 promotes glutamine metabolism via negative regulation of CREB2, which ultimately leads to activation of GDH.
mTORC2 is also emerging as a crucial player in glutamine metabolism, possibly via its role as a regulator of Myc expression (268). Knockdown of rictor decreases levels of α-KG that are likely derived from glutaminolysis, suggesting that mTORC2 may regulate this process (278). Although earlier studies suggested that the mTORC2 target Akt may not play a role in glutamine consumption or metabolism (62, 297), more recent reports indicate a link between increased mTORC2-mediated Akt phosphorylation and glutamine metabolism. First, mTORC2 is sensitive to intracellular levels of glutamine and/or its metabolites (101). Withdrawal of glutamine increases mTORC2 activation, as indicated by Akt phosphorylation. Second, in lung cancer cells, glutamine depletion decreases mTORC1 activity while increasing mTORC2 activity and sestrin2 stability (103). The inhibition of both mTORC1 and mTORC2 upregulates glutaminolysis, suggesting more complex feedback mechanisms involved, likely to ensure TCA anaplerosis when mTOR signals decrease. Combination treatment using the glutaminase inhibitor CB-839 and mTOR ATP-competitive inhibitors can thus be exploited for more effective therapy in cancers that may be addicted to glutamine metabolism (302, 303).
11. PROTEIN SYNTHESIS AND AMINO ACID METABOLISM
mTOR controls multiple aspects of protein synthesis (FIGURE 3). Much of what we know about this mTOR function relates to mTORC1. In the presence of sufficient nutrients, mTORC1 is active to conduct protein synthesis. mTORC1 phosphorylates two key effectors of translation, S6K1 and 4E-BP1, as discussed above. S6K1 phosphorylates several targets that participate in protein synthesis, such as eIF4B, PDCD4, eIF3, and SKAR. S6K1 directly phosphorylates eIF4B, leading to eIF4B interaction with eIF4G and the mRNA helicase eIF4A. Additionally, S6K1 indirectly controls eIF4B by phosphorylating PDCD4, engendering its degradation and promotion of eIF4A and eIF4G interaction (304). S6K1 phosphorylates eIF3 to enhance PABP-interacting protein (Paip1)-eIF3 association during nutrient replete conditions, thus promoting cap-dependent translation (305). S6K1 also enhances translation efficiency of spliced mRNAs through its phosphorylation of SKAR, a component of exon-junction complexes. Several other S6K1 phosphorylation targets that are involved in metabolism are discussed further below. mTORC1 also regulates translation through its phosphorylation of 4E-BP1. mTORC1 phosphorylates 4E-BP1 at multiple sites, causing the dissociation of 4E-BP1 from eIF4E, which permits eIF4F complex assembly and promotes 5′ cap-dependent mRNA translation. Hence, mTORC1 controls multiple aspects of translation via its direct regulation of S6K1 and 4E-BP1. Whether it can modulate other translation regulators more directly or via other protein kinases requires further investigation. The role of mTORC2 in controlling translational regulators also remains to be explored.
mTOR also controls protein synthesis via its role in promoting the biogenesis of ribosomes, which are the engines of protein synthesis. mTOR positively regulates several processes involved in ribosome biogenesis, including rRNA transcription, synthesis of ribosomal proteins and components of ribosome assembly, and the assembly of preribosomal complexes (306–308). mTORC1, via S6K1, phosphorylates UBF, a nucleolar transcription factor that promotes production of 47S rRNA via RNA polymerase I-mediated transcription (309). mTORC1 also phosphorylates the transcription factor TIF-IA to promote RNA polymerase I transcription (310) and MAF, a repressor of RNA polymerase III, to promote RNA Pol III-dependent transcription of 5S rDNA (311). The synthesis of ribosomal proteins is also modulated by mTOR signaling via phosphorylation of La-related proteins (LARPs) to promote translation of 5′ TOP mRNAs that include genes encoding ribosomal proteins. mTORC1 phosphorylates LARP1, whereas Akt phosphorylates LARP6 (312, 313). Rictor is recruited to the nucleolar compartment during epithelial-to-mesenchymal transition (EMT)-associated ribosome biogenesis (314). Further research is needed to understand the specific role of rictor and mTORC2 in this compartment. Studies have demonstrated that active mTORC2 associates with ribosomes and that it phosphorylates nascent peptides such as Akt and PKC (99, 110). Tumors are characterized by high rates of ribosome biogenesis, as they require higher rates of protein synthesis to support growth and proliferation. Inhibition of RNA polymerase I, either alone or in combination with mTOR inhibitors, is under preclinical and clinical investigation as a potential anticancer strategy (315, 316).
Protein synthesis relies on sufficient amounts of building blocks such as amino acids. Transport of essential and nonessential amino acids is tightly regulated, particularly in highly proliferating cells. mTOR regulates the amounts of amino acid transporters. In genomic studies, rapamycin treatment was associated with a reduction in neutral amino acid transporters (21). Silencing of either raptor or rictor diminishes, while their combined knockdown completely blocks, system A (alanine preferring) and L (leucine preferring) transport activity (317). mTORC1 may control trafficking of these transporters via inhibition of the ubiquitin ligase Nedd4-2, which promotes ubiquitination and decreased expression of these transporters (318). Inhibition of mTOR with Torin1 also downregulates transcription of genes encoding amino acid transporters and other enzymes involved in amino acid metabolism (319). The transcription factor ATF4, which is induced during the integrated stress response to enhance expression of genes that allow cells to withstand amino acid limitation, also mediates this function of mTOR. mTORC2 is also directly involved in amino acid transport via phosphorylation of Ser26 of the cystine-glutamate antiporter xCT, which restricts the efflux of glutamate and influx of cystine (320). Together, these findings support that both mTORC1 and mTORC2 modulate protein synthesis via control of amino acid transporter levels and/or activity.
Amino acids also serve as important metabolic precursors. For humans, essential amino acids must be acquired from the diet, whereas nonessential amino acids can be generated intracellularly. Amino acid-depleting compounds such as asparaginase are used as anticancer drugs because of their ability to block cell proliferation. Asparaginase depletes the serum of asparagine by catalyzing the hydrolysis of this nonessential amino acid into aspartic acid and ammonia. Malignant cells that have diminished asparagine synthase activity (asparagine auxotrophy) are highly sensitive to asparaginase, which further impairs protein synthesis and prevents cell growth. mTOR regulates the expression of asparagine synthetase (ASNS) (21, 282, 321). In non-small cell lung cancer expressing oncogenic KRAS, increased ASNS expression occurs via Akt- and Nrf2-mediated induction of ATF4. The inhibition of Akt together with depletion of extracellular asparagine prevents tumor growth (322). Rapamycin increases gene expression of argininosuccinate synthetase-1 (ASS1), the rate-limiting enzyme for arginine biosynthesis (21). In arginine-auxotrophic tumors, such as melanoma and hepatocellular carcinoma (323), resistance to therapy using arginine deiminase, which degrades extracellular arginine, develops because of elevation of ASS. These resistant tumors become particularly sensitive to PI3K/Akt inhibitors (324), thus implicating a role for mTORC2 in arginine biosynthesis. De novo serine-glycine biosynthesis is also amplified by mTORC1 via ATF4 (325). This occurs during myofibroblast differentiation, wherein there is increased collagen biosynthesis. TGFβ1, in cooperation with mTORC1, upregulates ATF4, which in turn promotes the expression of genes involved in serine-glycine biosynthesis. Finally, the mTORCs may be indirectly involved in amino acid biosynthesis through their role in other metabolic pathways. Many of the intermediates from glycolysis, the pentose phosphate pathway (PPP), and the TCA cycle are metabolized to produce nonessential amino acids in human cells. Future investigations should reveal how the mTORCs coordinate flux through these pathways with the generation of amino acids to maintain metabolic homeostasis.
Amino acids are also catabolized to generate metabolites that feed into biosynthetic pathways. Although how levels of amino acids such as branched-chain amino acids (leucine, isoleucine, valine) are sensed by mTOR, it is well characterized, little is known on how mTOR controls catabolism of these amino acids (326). There is some evidence that inhibition of mTOR by rapamycin promotes amino acid catabolism. In a mouse model of Leigh syndrome, characterized by mitochondrial dysfunction, rapamycin mitigates the buildup of glycolytic intermediates by shifting metabolism toward amino acid catabolism (327). Such treatment enhances survival and prevents disease progression, but the precise mechanisms of rescue by mTOR inhibition remain to be further investigated.
12. PENTOSE PHOSPHATE PATHWAY AND NUCLEOTIDE SYNTHESIS
The pentose phosphate pathway (PPP) produces ribose-5-phosphate, the backbone for the synthesis of nucleic acids. It also generates reducing equivalents in the form of NADPH (FIGURE 4). About 5–30% of glucose fluxes through the PPP. The rate-limiting reaction is catalyzed by glucose-6-phosphate dehydrogenase (G6PD) (328). The PPP bifurcates into an oxidative and a nonoxidative branch, which are irreversible and reversible, respectively. Glucose-6-phosphate (G6P) is oxidized by G6PD to produce NADPH in the oxidative arm. In contrast, a series of reversible reactions convert glycolytic intermediates into ribose-5-phosphate in the nonoxidative branch. Both pathways subsequently produce phosphoribosyl pyrophosphate (PRPP), the precursor for nucleotide synthesis. Because of the increased need for nucleotides, rapidly dividing cells upregulate PPP activity. Concomitant generation of NADPH by the PPP ensures supply of this reducing agent for several synthetic steps of fatty acid, cholesterol, and steroid hormone generation, along with detoxification reactions. mTORC1 promotes flux through the oxidative branch via regulation of the transcription of genes encoding enzymes that drive the PPP (228, 329). mTORC1 also enhances translation of the mRNA encoding ribose-5-phosphate isomerase A (RPIA), which isomerizes ribulose-5-phosphate to ribose-5-phosphate (330). The mTORC1-mediated increase of RPIA occurs in cells deficient of the tumor suppressor p16, a cell cycle inhibitor, thus providing an example of how tumorigenesis is dependent on upregulation of the PPP via mTORC1.
FIGURE 4.
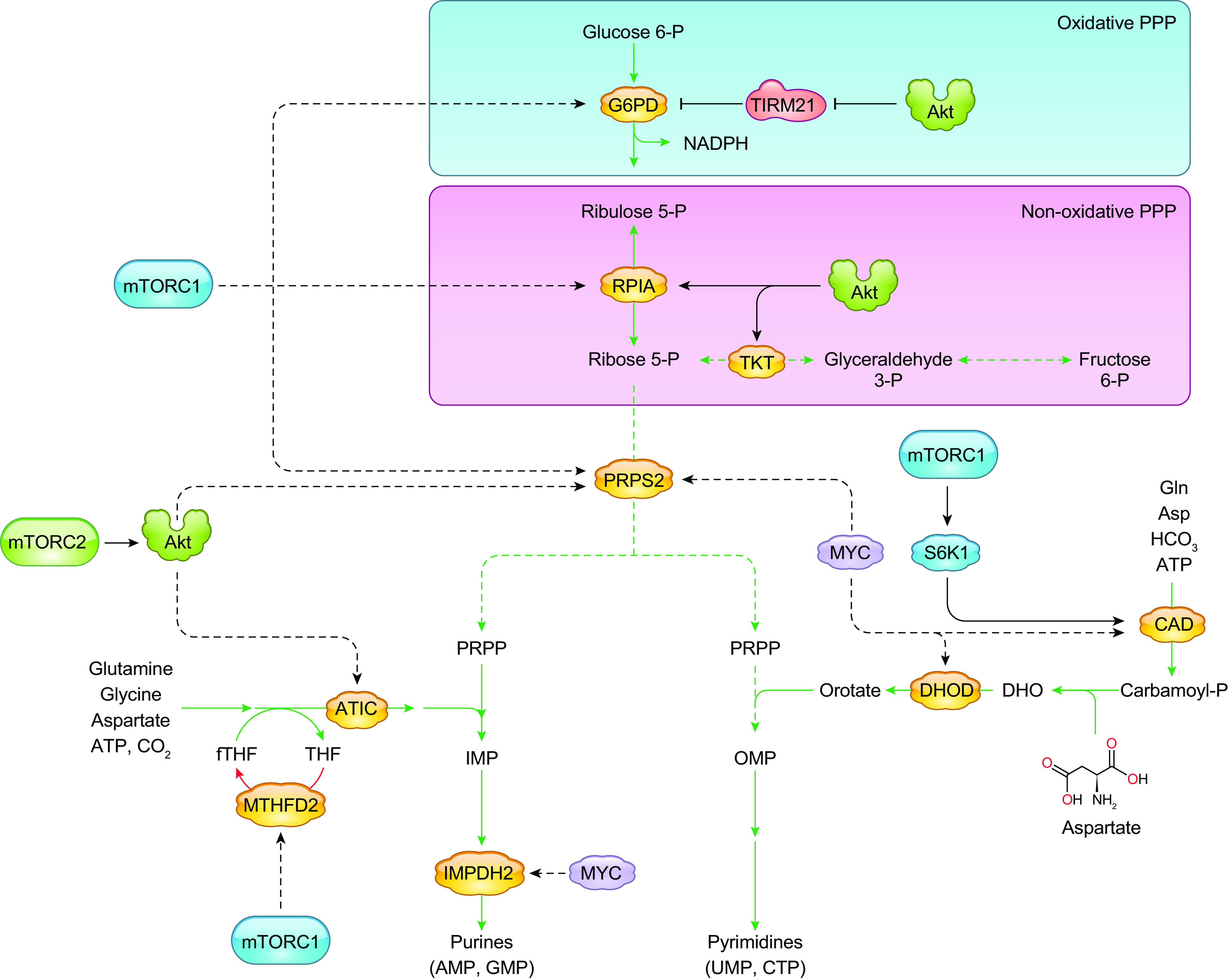
The pentose phosphate pathway (PPP) and nucleotide metabolism are modulated by mTORC1 and mTORC2. The PPP generates the reducing equivalent, NADPH, and ribose-5-phosphate, which serves as a backbone for the synthesis of purines and pyrimidines. mTORC1 and the mTORC2 substrate Akt have been linked to the regulation of the key enzymes involved in the PPP. mTORC1 also controls the key enzyme, CAD, involved in pyrimidine synthesis. Both mTORC1 and mTORC2 have been linked to the regulation of purine synthesis. See glossary for abbreviations.
Proliferating cells have increased demand for nucleotides, which are needed for DNA and RNA synthesis and for ribosome biogenesis. Cellular nucleotide pools are replenished via the salvage of nucleotides from degradation intermediates and through de novo synthesis, which assembles complex nucleotides from basic molecules. mTORC1 regulates the de novo synthesis of pyrimidines via activation of CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydro-orotase) (202, 331). CAD catalyzes the initial steps of pyrimidine synthesis by utilizing glutamine, bicarbonate, and aspartic acid to generate pyrimidine rings. It interacts with mLST8, and its activity decreases during amino acid or serum depletion (332). The mTORC1 target S6K1 directly phosphorylates CAD on Ser1859 and enhances its oligomerization (202). mTORC1 and S6K promote the de novo pyrimidine synthesis in response to growth factor or amino acid stimulation, although they are not required for de novo synthesis per se (331). Inhibition of mTOR in tumors with upregulated CAD activity/pyrimidine synthesis due to deficiency of ASS1, a urea cycle enzyme that converts nitrogen from ammonia and aspartate to urea, decreases proliferation of tumors (333). These studies underscore the role of mTOR in CAD regulation and how inhibiting its activity could be effective for tumors with ASS1 deficiency.
mTORC1 enhances metabolic flux through de novo purine synthesis via control of transcription of enzymes involved in this process, including mRNA and protein expression of methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), which is mediated by ATF4 (334). mTORC1 also senses purine nucleotide levels in a manner similar to its sensing of amino acids (94). mTORC1 senses adenylate via the TSC and Rheb, but independently of AMPK. Interestingly, mTORC1 is not acutely sensitive to guanylate levels, but prolonged depletion leads to decreased Rheb levels.
mTORC2 is also linked to the regulation of the PPP and nucleotide metabolism through the PI3K/Akt signaling axis, in part through the mTORC2 substrate Akt. In an insulin-driven model of hepatocellular carcinoma, Akt promotes upregulation of the PPP through several mechanisms, including via increase of G6PD and RPIA expression and activity, as well as through driving glycolysis (335). The activation of PI3K/Akt also stabilizes the rate-limiting enzyme of the PPP, G6PD, via inhibition of the E3 ligase TIRM21 (336). mTORC2, via Akt, regulates phosphorylation of transketolase (TKT), a key enzyme of the nonoxidative arm of the PPP (337). This phosphorylation of TKT increases purine synthesis. Additionally, the PI3K/Akt signaling axis modulates the nonoxidative PPP. It regulates PRPP synthesis and also modulates the activity of aminoimidazole-carboxamide ribonucleotide transformylase IMP cyclohydrolase (ATIC), thus ensuring regulation of both the early and later steps of this pathway (338). Findings in yeast further support a role for mTORC2 in the PPP, since the proteins involved in this pathway physically associate with TORC2 (339). Furthermore, metabolic intermediates such as 6-phospho-D-gluconate (6PG) and ribose-5-phosphate are diminished upon yeast TOR2 inhibition. Although mTORC2 has a clear role in controlling the PPP and nucleotide metabolism, future investigations are needed to determine whether mTORC2 has functions independently of Akt.
13. HEXOSAMINE BIOSYNTHESIS
Proteins and lipids undergo glycosylation, a modification that results in the production of proteoglycans, glycoproteins, and glycolipids. Glycosylation modulates diverse cellular processes by regulation of trafficking, stability, structure, and activity of proteins and lipids (340). The hexosamine biosynthesis pathway (HBP) provides the main metabolite for glycosylation, uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) (FIGURE 3). The first reaction is catalyzed by GFAT1, which transfers the amino group from glutamine to the glycolytic metabolite fructose-6-phosphate. Subsequent steps along the HBP ultimately produce UDP-GlcNAc. Although only 2–3% of fructose-6-phosphate enters the HBP under physiological conditions, this pathway serves as a central hub to integrate metabolites from major biosynthetic pathways. In proteins, glycosylation occurs co- or posttranslationally, involving the addition of oligosaccharides in O (linked to Ser/Thr in polypeptides) or N (linked to Asn with the motif NXS/T) linkages to many cytosolic, secretory, and membrane proteins. As many cell surface proteins, including growth factor receptors and transporters, are dependent on glycosylation for proper expression, the HBP plays a crucial role in coupling metabolic conditions with cell signaling. Chronic activation of the HBP is linked to enhanced growth and mTOR signaling (341). Deprivation of glucose, which results in decreased fructose-6-phosphate, downregulates the HBP and eventually decreases expression of growth factor receptors. Exogenous metabolites that enter the HBP such as glucosamine and N-acetylglucosamine during glucose starvation rescue receptor expression and mTOR signaling (342, 343). Several cell signaling proteins also undergo O-GlcNAcylation, a modification that is catalyzed by the O-GlcNAc transferase (OGT). Expression of OGT and total O-GlcNAcylation are regulated by mTOR, Akt, and c-MYC (344–346). mTOR, as part of mTORC2, controls mRNA and protein expression as well as phosphorylation of the rate-limiting enzyme of the HBP, glutamine:fructose-6-phosphate amidotransferase 1 (GFAT1), and thereby controls flux through the HBP in response to nutrient levels (101, 347). As cells are deprived of nutrients, such as glucose or glutamine, mTORC2 activation increases. mTORC2 then promotes the expression of GFAT1 in order to maintain flux through the HBP. When glucose is limiting, the transcription factor spliced Xbp1 (Xbp1s) promotes transcription of GFAT1 to enhance its expression, in an mTORC2-dependent manner (101). Under glutamine deprivation, mTORC2 maintains the phosphorylation of GFAT1 at Ser243 to increase its stability and sustain production of UDP-GlcNAc despite nutrient starvation (347). The modulation of the HBP by mTORC2 is linked to TCA cycle anaplerosis, since conditions that trigger enhanced anaplerosis increase mTORC2 activation while maintaining or enhancing GFAT1 expression. By controlling the HBP, with the main metabolite, UDP-GlcNAc, being crucial for protein glycosylation, mTORC2 may couple protein synthesis and folding. The role of mTORC1 in regulating the HBP deserves to be examined.
14. LIPID METABOLISM
Proliferating cells require an ample supply of lipids for biosynthesis of membranes and signaling molecules. Cell membrane lipids, composed of phospholipids, sterols, sphingolipids, and lysophospholipids, are derived in part from acetyl-CoA (FIGURE 5). Acetyl-CoA is generated from citrate or acetate by ATP citrate lyase (ACL) or acyl-CoA synthetase short-chain (ACSS) family members, respectively. When citrate is abundant, it is exported from the mitochondria to the cytosol, where it is processed by ACL to generate cytosolic acetyl-CoA, which serves as a precursor for endogenous synthesis of acyl groups and sterols. ACL mRNA and protein expression are elevated in a number of cancers, underscoring the greater demand for lipids during increased proliferation (348–350). Fatty acid synthase (FASN) then catalyzes synthesis of fatty acids (FA) using acetyl-CoA carboxylase (ACC). Palmitate is generated and is used to produce a number of products including longer FA, unsaturated FA via stearoyl-CoA desaturase 1 (SCD1), and phospholipids. Cholesterol biosynthesis requires hydroxymethylglutaryl-CoA synthase (HMGCS), followed by a series of reactions catalyzed by a series of enzymes that are often overexpressed in cancer (351, 352). A major transcriptional regulator of lipid metabolism-related genes is the sterol regulatory element binding protein (SREBP) family of transcription factors. During lipid depletion, SREBP is processed and translocates to the nucleus to induce transcription of target genes.
FIGURE 5.
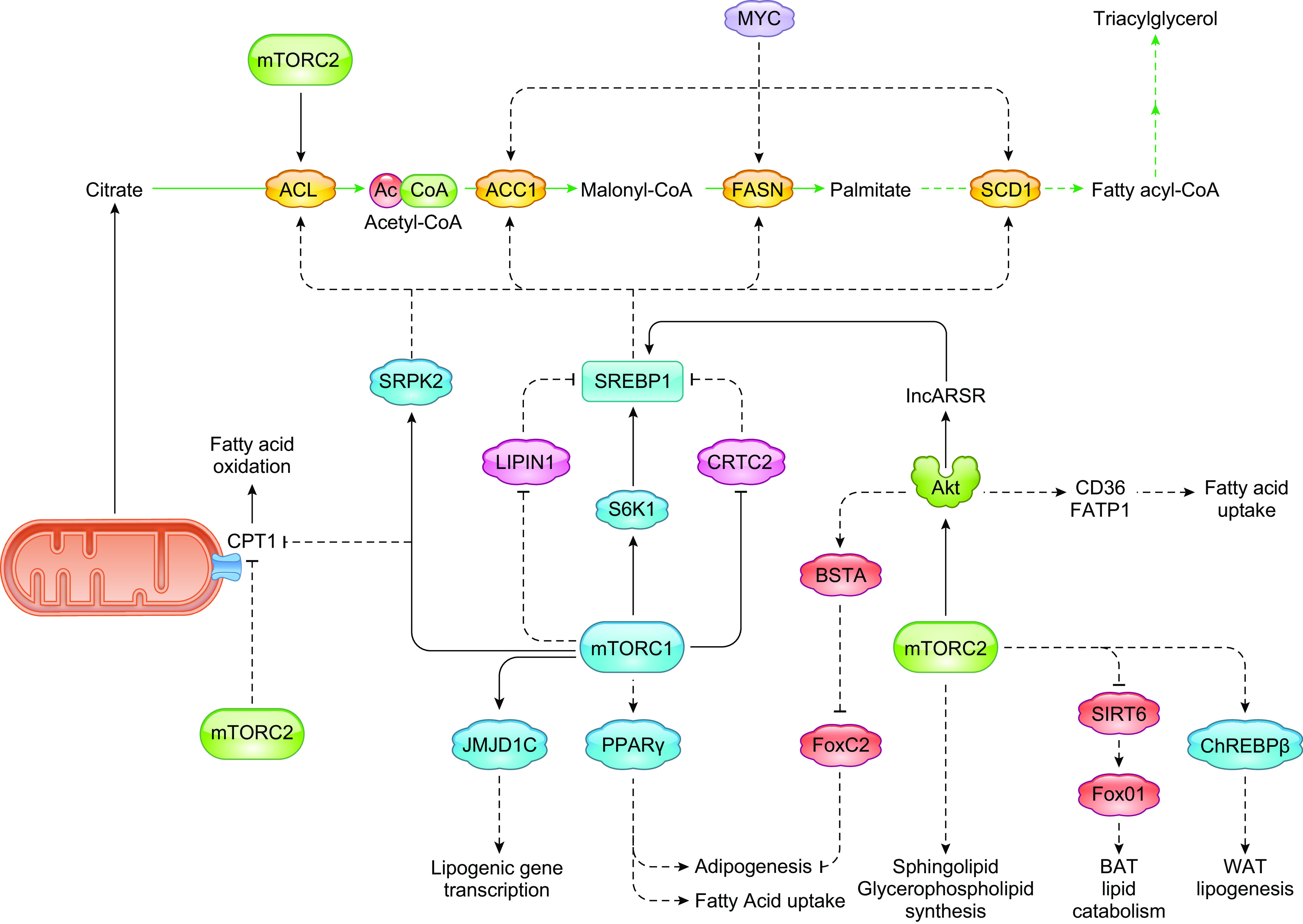
mTORC1 and mTORC2 control lipid metabolism. In proliferating cells, the production of fatty acids and cholesterol is enhanced for the biosynthesis of signaling molecules and membrane lipids. The function of mTORC1 in lipid metabolism is mediated via the transcription factor SREBP1, which is a master regulator of lipid metabolism-related genes. mTORC1 also modulates other proteins that contribute to lipid biosynthesis such as the histone demethylase JMJD1C and SRPK2, which regulates lipogenic mRNAs. mTORC1 also controls adipogenesis and fatty acid uptake via PPARγ. mTORC2 promotes de novo fatty acid and lipid synthesis (sphingolipid and glycerophospholipids) via transcriptional regulation independent of Akt. mTORC2 is also involved in adipogenesis via negative regulation of FoxC2. It promotes white adipose tissue lipogenesis via the transcription factor ChREBPβ and prevents brown adipose tissue lipid catabolism via repression of Fox01. See glossary for abbreviations.
mTORC1 activation is linked to increased lipogenesis, whereas its inhibition promotes lipolysis and fatty acid oxidation (FAO). Studies using cells with elevated mTOR signals support the role of mTORC1 in lipogenesis. In cells expressing active Akt, the induction of lipid synthesis is dependent on mTORC1 (353). TSC2-deficient fibroblasts with hyperactive mTORC1 signals display rapamycin-sensitive increase in de novo lipid biosynthesis (228). Likewise, expression of constitutively active Rheb in adipocytes increases de novo lipogenesis while suppressing lipolysis (354). Disruption of mTORC1 via ablation of raptor in the liver strongly supports the requirement for mTORC1 in lipogenesis (355). Several studies demonstrate that the function of mTORC1 in lipid metabolism is mediated by SREBP. The expression of lipogenic enzymes such as ACC, FASN, and SCD1 is controlled by mTORC1 via the transcription factor SREBP (21, 356, 357). In TSC-deficient cells, rapamycin-sensitive genes are enriched with DNA binding elements that recognize SREBP (228). Three isoforms of SREBP in mammalian cells, SREBP-1a, SREBP-1c, and SREBP-2, regulate distinct, yet overlapping, lipogenic transcriptional programs. In hepatocytes, the insulin-mediated increase in SREBP-1c mRNA is mTORC1 dependent but S6K independent (355, 358, 359). In raptor-deficient T cells, protein expression of both SREBP-1 and SREBP-2 is diminished (279). Thus, mTORC1 controls lipogenesis by regulation of SREBP at the level of transcription and protein expression. In the endoplasmic reticulum, SREBPs are synthesized as inactive precursors, followed by processing in the Golgi and translocation to the nucleus, where they induce transcription of SRE-containing target genes. The processing step is sensitive to sterol levels and modulated by mTORC1. Increased processing of SREBP-1 occurs in TSC-deficient cells, and the mTORC1 target S6K1 regulates SREBP processing (228, 359). S6K1 phosphorylates SREBP-1c at Ser418, Ser419, and Ser422 in an insulin-induced manner and prevents its proteasomal degradation (360). mTOR also modulates the processing of SREBP-1 via phosphorylation of the CREB-regulated transcription coactivator 2 (CRTC2). During feeding, mTOR phosphorylates CRTC2 at Ser136 to attenuate its suppression of COPII-dependent SREBP-1 processing (361). This phosphorylation is rapamycin sensitive, suggesting it is mediated by mTORC1. The stability of the processed active form has also been linked to Akt regulation (353). SREBPs are required to meet the increased lipid requirement of proliferating cells. For example, activated CD8+ T cells require SREBP during clonal expansion of effector cells (362). Another way in which mTORC1 regulates SREBP is through modulation of lipin1 nuclear localization. Lipin1 is a phosphatidic acid phosphatase that represses SREBP activity (363). mTORC1-mediated lipin phosphorylation excludes it from the nucleus, thus allowing SREBP activity.
mTORC1 modulates epigenetic and posttranscriptional regulators of lipid metabolism. In response to feeding/insulin, mTORC1 phosphorylates the liver histone demethylase JMJD1C at Thr505, leading to its demethylation of H3K9me2 (364). Demethylation of H3K9me2 promotes chromatin accessibility and induction of lipogenic genes. Phosphorylation of JMJD1c enhances interaction with USF and recruitment to lipogenic gene promoters. mTORC1 also promotes the splicing of lipogenic genes by S6K1-mediated phosphorylation of SRPK2 (serine/arginine-rich protein kinase 2) (365). Inhibition of SRPK2 phosphorylation leads to intron retention and destabilization of lipogenic mRNAs.
Consistent with the role of mTORC1 in cellular lipid metabolism, it also controls adipocyte differentiation. The upregulation of the mTORC1 pathway, such as by inactivation of TSC2, and the increased phosphorylation of 4E-BP1 and S6K can in part mediate mTORC1 adipogenic activity. mTORC1 also regulates the activity of peroxisome proliferator-activated receptor γ (PPARγ), a key regulator of adipogenesis (366). mTORC1 also increases the activity of lipin 1, which promotes triglyceride synthesis and enhances the PPARγ adipogenic activity (367). In other cell types such as T cells, mTORC1 promotes the induction of FA uptake via PPARγ during activation of CD4+ T cells (368).
Whereas mTORC1 positively regulates lipogenesis, it negatively regulates lipolysis (354). In adipose tissue, mTORC1 inhibits lipolysis by repression of adipose triglyceride lipase (ATGL), which catalyzes the initial reaction in triglyceride hydrolysis. mTORC1 diminishes ATGL expression via promoting the expression of Egr1, a transcriptional repressor of ATGL (369). mTORC1 inhibition increases fatty acid oxidation (FAO) and activity of carnitine palmitoyltransferase I (CPT1), a critical regulatory enzyme of the FAO pathway (370). In Myc-hypomorphic mice that have reduced mTORC1 signaling, there is increased FAO and upregulation of CPT1A (371). Hence, mTORC1 coordinately regulates lipid anabolism and catabolism.
The role of mTORC2 in lipid metabolism has been unveiled in studies using mTOR catalytic inhibitors and in knockout/knockdown models. In breast cancer cells with increased PI3K activity, pharmacological blockade of mTORC1 and mTORC2 reduces lipogenesis due to inhibition of glucose-to-lipid conversion via mTORC2. This is also accompanied by increased lipid catabolism and levels of CPT1A (372). Increased lipid catabolism during mTORC1/mTORC2 inhibition is dependent on autophagy and phospholipase A2 (PLA2), which allow generation of free FA (373). In several cancer cells, silencing of rictor or SIN1 reduces the mature forms of SREBP-1c, leading to diminished expression of the target genes ACC and FASN. mTOR kinase inhibitors or rictor knockdown in these cells induces GSK3/FBXW7-dependent degradation of SREBP-1c, suggesting that mTORC2 is involved in modulating SREBP-1c stability (374). Mice with liver-specific rictor knockout have a marked decrease in lipogenesis resulting from loss of SREBP-1c activity (272, 375). These mice retain functional mTORC1 activity, which suggests that there is an mTORC2-specific role for lipid synthesis. The expression of constitutively active Akt2 in hepatocytes with rictor knockout is able to restore de novo lipogenesis and SREBP-1c activity (272). Akt signaling controls the expression of SREBP-1c via the long non-coding RNA lncARSR in hepatic lipogenesis (376). Akt also phosphorylates ACL (377). Under conditions where Akt signaling is downregulated, mTORC1 activation is not sufficient to stimulate lipogenesis (329, 355). Although this evidence further supports that Akt is required for lipogenesis, a separate study demonstrated that the absence of mTORC2 in the liver prevents Akt-mediated lipogenesis. Akt activation is unable to promote hepatic lipogenesis in mice with liver-specific rictor knockout (375). Therefore, these findings underscore the role of mTORC2 in lipogenesis. They also suggest that it has additional role(s) independent of Akt. Knockdown of rictor in bovine mammary epithelial cells decreases expression of the transcription factor PPARγ and several lipogenic genes. The intracellular levels of triacylglycerol and FA and accumulation of lipids in the culture medium are also reduced (378). In the double-knockout mouse model of Tsc1 and PTEN in the liver, the mice display hepatosteatosis that progress to hepatocellular carcinoma (379). This progression is largely due to mTORC2 promotion of de novo FA and lipid synthesis. Specifically, mTORC2 increases transcription of genes involved in the synthesis of sphingolipid and glycerophospholipids. Enhanced levels of SREBP-1c and its target genes also occur. The pharmacological or genetic disruption of mTORC2 reduces de novo FA synthesis. mTORC1 may also contribute to lipid synthesis in this mouse model via upregulation of pyrimidine biosynthesis, whose products UDP and CDP are required for lipid synthesis. mTORC2, via Akt, also promotes the uptake of FA via promoting cell surface translocation of transporters such as CD36 and FATP1 (380). Thus, mTORC2, like mTORC1, controls several aspects of lipogenesis.
Adipocyte-specific mTORC2 activity has been shown to promote adipogenesis, de novo lipogenesis, and control insulin-stimulated glucose uptake. The role of mTORC2 in adipogenesis is partly attributed to its regulation of Akt. mTORC2 phosphorylates the BSD domain-containing protein BSTA and induces its association with Akt for BSTA-Akt complex formation. This interaction enhances mTORC2-mediated phosphorylation and activation of Akt, leading to suppression of expression of FoxC2, a transcription factor that negatively regulates white adipocyte differentiation (381). Conditional deletion of rictor in adipose tissue leads to defective white adipose tissue (WAT) de novo lipogenesis and insulin resistance (382). Rictor promotes the expression of the lipogenic transcription factor ChREBPβ partly by promoting glucose uptake. mTORC2 is also essential for the development of brown adipose tissue (BAT). Loss of rictor in Myf5 precursors prevents BAT growth and reprograms their metabolism to a more oxidative and less lipogenic state (383). The absence of mTORC2 prevents obesity and metabolic disease in these mice. Conditional deletion of rictor in brown adipocytes inhibits lipid synthesis while promoting lipid catabolism and thermogenesis. The function of rictor in preventing lipid catabolism in brown adipocytes is independent of Akt but involves suppression of FoxO1 deacetylation via SIRT6, which then represses UCP1 mRNA and protein expression (384). Taken together, these findings support the role of mTORC2 in promoting lipid synthesis while preventing lipid catabolism.
How mTOR controls lipid metabolism by sensing lipid levels is still poorly understood. Phosphatidic acid (PA), the product of the phospholipase D-catalyzed conversion of phosphatidylcholine to choline and PA, binds to the FRB domain of mTOR (385, 386). PA binds to both mTORC1 and mTORC2 to modulate their stability and activity (387). Exogenously added fatty acids stimulate mTOR via de novo PA synthesis (95). Even in the absence of amino acids, PA mediates the translocation of mTORC1 to the lysosome (388). Hence, levels of PA may positively modulate mTOR localization to the lysosome and activity. As mentioned in sect. 5, mTOR also undergoes malonylation as malonyl-CoA levels rise. This modification negatively regulates mTOR activity (158). Decreased levels of acetyl-CoA due to deficiency in acyl-CoA oxidase 1 (Acox1), the enzyme that catalyzes the first step in peroxisomal beta oxidation, diminish raptor acetylation, thus lowering mTORC1 activity (389). These findings suggest that acetyl-CoA levels may also be sensed by mTORC1 components to regulate lipid metabolism. Identification of other modifications that occur on mTOR and its partners in response to lipids and lipid metabolites should shed light on how the mTORCs are modulated by this nutrient.
15. ONE-CARBON AND METHIONINE METABOLISM
One-carbon units (also called methyl groups) are used for the synthesis of cellular components such as nucleotides and to support the maintenance of intracellular pools of metabolites including ATP, NADPH, and SAM (S-adenosylmethionine). A carbon unit from serine or glycine is transferred to tetrahydrofolate (THF) to form 5,10-methylene-THF. mTORC1 has been linked to one-carbon metabolism. In oncogenic mouse models with KRAS activation and loss of LKB1, isolated pancreatic epithelial cells have increased mTOR signaling that promotes enhanced serine/one-carbon metabolism (390). During M-CSF-induced myelopoiesis, the absence of raptor decreases levels of metabolites involved in this metabolic pathway (391). In raptor−/− cells, the accumulation of serine is consistent with a possible defect in its catabolism in the mitochondria, a process that depends on one-carbon metabolism. Serine and one-carbon pathways are also upregulated via mTORC1-ATF4 during deficiency of PKC-λ/ι in neuroendocrine prostate cancer (392). PKC-λ/ι, acting as tumor suppressor, represses mTORC1 activity by phosphorylating LAMTOR2, a component of the Ragulator complex that controls mTORC1 localization to the lysosomes. THF is also utilized by methionine synthase to convert homocysteine to methionine. Methionine can be converted to SAM by methionine adenosyltransferase (MAT). SAM is utilized for methylation reactions by methyltransferases. The SAM sensor SAMTOR binds GATOR1, the GAP for RagA/B (393). Methionine starvation decreases SAM levels and promotes the association of SAMTOR with GATOR1, leading to inhibition of mTORC1 activity. In turn, mTORC1 regulates methionine metabolism. In a brain-specific TSC2-knockout mouse model, the resulting constitutive mTORC1 activation leads to a marked reduction in the levels of methionine, SAM, and other metabolites involved in transmethylation. This is likely due to the increased demand for methionine (394). The transport of folate is also dependent on both mTORC1 and mTORC2 (395). The expression of the folate transporter Folate Receptor-a (FR-a) and Reduced Folate Carrier (RFC) are decreased in cultured human trophoblasts when either rictor or raptor expression is silenced.
16. NAD METABOLISM
The levels of NAD+ are tightly regulated by de novo or salvage pathways. NAD+ is used as a cofactor in a number of metabolic reactions including glycolysis, TCA cycle, and redox reactions. It also serves as a substrate for several enzymes such as sirtuins. NAD+ is used to generate NADP+, which is further reduced to NADPH, an important cofactor for reductive metabolism. Conversion of NAD+ to NADP+ occurs via a reaction catalyzed by NADK (nicotinamide adenine dinucleotide kinase). A decline in NAD+ levels is associated with aging. Nicotinamide phosphoribosyl transferase (NAMPT) is the rate-limiting enzyme of the salvage pathway for NAD generation. NAMPT utilizes nicotinamide (NAM) to generate nicotinamide mononucleotide (NMN), one of the precursors of NAD synthesis. In the de novo pathway, tryptophan undergoes conversion via a series of steps that generate nicotinic acid mononucleotide and subsequently NAD+. Inhibition of NAMPT with pharmacological inhibitors blocks mTOR signaling in pancreatic ductal adenocarcinoma and hepatocarcinoma cells (396, 397). Blocking of NAMPT in multiple myeloma also inhibits mTORC1 signaling and promotes autophagy (398). NAMPT inhibition leads to decreased translation via 4EBP1 and S6K1 signaling (397, 399). Inhibition of NAMPT by KPT-9274, a dual inhibitor of NAMPT and PAK4, downregulates the mTORC2 pathway in triple-negative breast cancer (TNBC) (400). Dual inhibition of PAK4 and NAMPT in combination with mTOR inhibitors also inhibits the growth of pancreatic neuroendocrine tumors (401). These tumors have increased expression of PAK4 and NAMPT, and their inhibition decreases the NAD pool and ATP levels. NAMPT is epigenetically regulated by the long noncoding antisense transcript of NAMPT (NAMPT-AS), thus enhancing NAMPT levels in TNBC. The NAMPT-AS/NAMPT promotes the tumor progression of TNBC via mTOR (402). Blocking NAD+ decline by inhibition of the NADase CD38 also attenuates mTORC1/S6K signaling (403). Together, these findings reveal how mTOR signaling is modulated by NAD levels. How the mTORCs could in turn modulate NAD metabolism via regulation of the enzymes involved in this metabolic pathway needs further investigation. As of yet, the mTORC2 target Akt has been demonstrated to phosphorylate the NADK at three phosphosites, Ser44, Ser46, and Ser48, within an amino-terminal domain, stimulating NADK activity by derepression of the autoinhibitory domain (404).
17. STEM CELLS
The balance between glycolysis and mitochondrial OXPHOS is essential for stem cell function, such that metabolic perturbations affect the capacity for self-renewal and differentiation (FIGURE 6A). High glycolytic flux is needed to retain the potential for unlimited proliferation and to maintain pluripotency. Like the Warburg effect in cancer cells, pluripotent stem cells (PSCs) rely on glycolysis for ATP despite the presence of oxygen. However, as stem cells mature and differentiate, they reprogram their metabolism to shift toward OXPHOS (405–410). A switch from OXPHOS to glycolysis reprograms somatic cells to induced pluripotent stem cells (iPSCs) (406, 411). Inhibition of mTOR activity with rapamycin has been shown to promote somatic cell reprogramming to iPSCs, potentially due to induction of autophagy and clearance of damaged mitochondria (mitophagy) (412–414). Optimal levels of mTOR activation are critical, since hyperactivation of mTORC1 suppresses iPSC reprogramming. During the early phases of reprogramming, mTOR activity is likely decreased because of repression of its expression by SOX2, one of the reprogramming factors (415). Several factors enhance somatic cell reprogramming to iPSCs. These factors support the glycolytic phenotype, including hypoxic conditions, production of high levels of lactate, and expression of genes encoding glycolytic enzymes (416). HIF1α improves iPSC reprogramming efficiency by increasing glycolysis and lactate production through activation of target genes encoding glycolytic enzymes, such as PDK and PKM2 (417). Reprogramming efficiency is also enhanced by increased Akt activity, which suggests a role for mTORC2 (418). Indeed, rictor is involved in differentiation of cardiomyocytes from mouse embryonic stem cells (419). Hence, the proper activation of both mTORC1 and mTORC2 is required to maintain the glycolytic metabolism of PSC and that attenuation of mTORC1 activity promotes the metabolic shift toward OXPHOS during differentiation. How the fine-tuned mTORC activity affects different metabolic processes during reprogramming of stem cells awaits further investigation.
FIGURE 6.
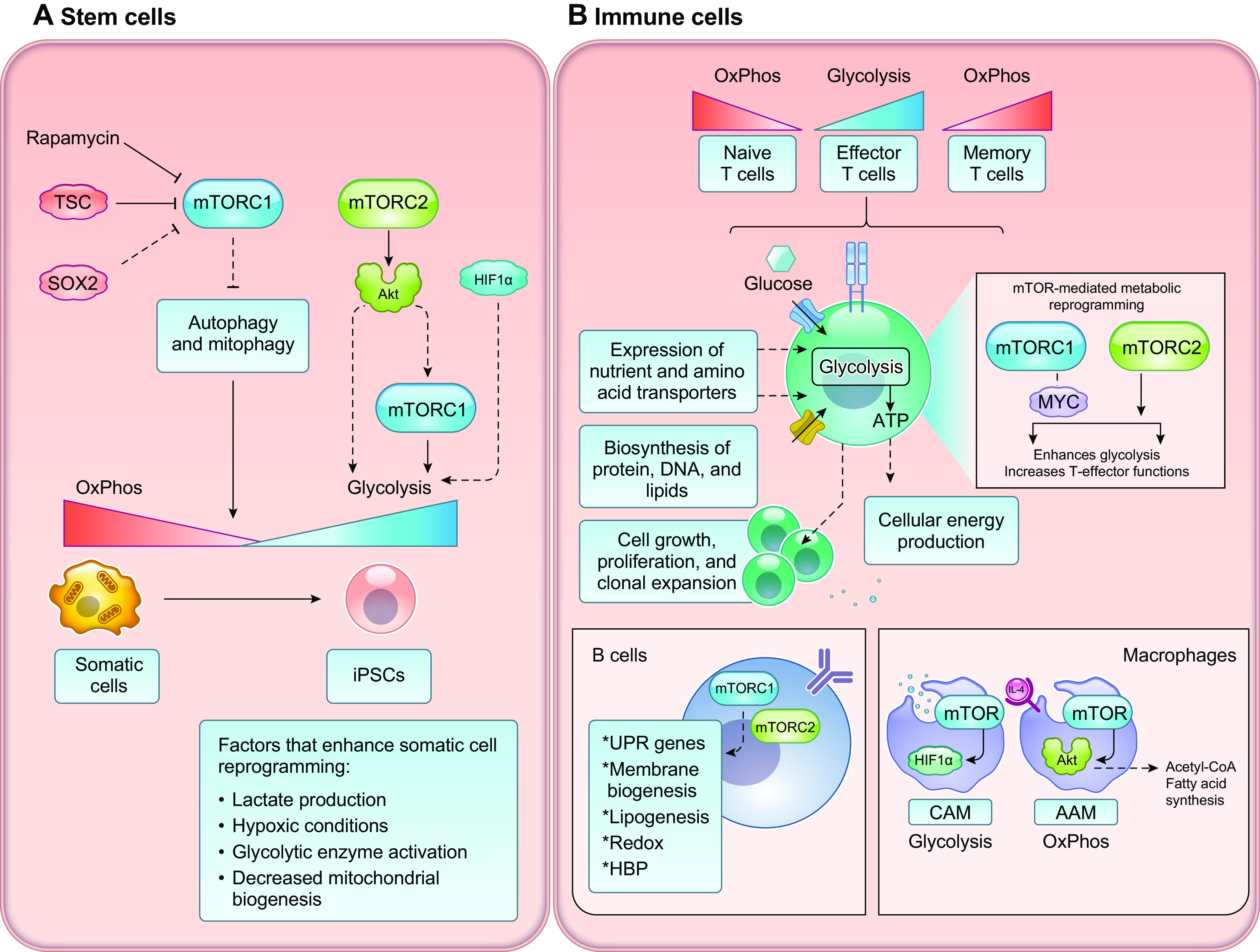
mTOR mediates metabolic reprogramming of stem and immune cells. A: somatic cells switch from OxPhos to glycolysis during reprogramming to become iPSCs, enhanced by factors associated with the glycolytic phenotype (e.g., HIF1α, hypoxic conditions, lactate production, glycolytic enzyme expression). Reprogramming efficiency is also enhanced by increased Akt activity, which suggests a role for mTORC2. Hyperactivation of mTORC1 such as during TSC deficiency suppresses iPSC reprogramming. The proper activation of mTORC1 and mTORC2 is required to maintain the glycolytic metabolism of PSCs. B: upon antigen encounter, naive T cells switch from OxPhos to a glycolytic metabolism. This allows for the synthesis of cytokines and expansion of effector cells to promote pathogen elimination. Both mTOR complexes are required for this metabolic reprogramming. After proliferation of effector cells and elimination of pathogens, T memory cells persist and revert to a naivelike OxPhos metabolism, where mTOR activity is reduced. B cell activation is characterized by synthesis of immunoglobulins and the generation of antibody-secreting plasma cells. To facilitate antibody production and secretion, B cells increase glucose uptake but glucose catabolism is diverted to the PPP to generate sufficient NADPH to maintain redox homeostasis. Activated B cells also enhance de novo lipogenesis and hexosamine biosynthesis. Both mTORC1 and mTORC2 promote B cell proliferation and antibody responses. In macrophages, blocking mTOR activity or glycolysis diminishes inflammatory cytokine production and bacterial elimination. In response to microbial stimuli (e.g., β-glucan), CAMs increase aerobic glycolysis via mTORC1 modulating HIF1α. In response to IL-4, Akt and mTORC1 are upregulated, leading to increased glucose metabolism and lipid synthesis in AAMs. See glossary for abbreviations.
18. IMMUNE CELLS
Adaptive immune cells, including T and B lymphocytes and innate immune cells such as macrophages, reprogram their metabolism upon encountering pathogens to launch a vigorous immune response (FIGURE 6B). This metabolic reprogramming provides ATP and generates biosynthetic precursors to meet the demands for growth, proliferation, clonal expansion, and functional activities of effector populations (420, 421). The maintenance of metabolic homeostasis is critical for preventing autoimmunity and malignancy. Immune cells are dependent on nutrient uptake from the environment. The inflammatory state of the surrounding microenvironment influences their metabolic and immune response. T cells, which are central regulators of an immune response, develop in the thymus. Their development in this compartment from double-negative (CD4−CD8−) to naive single-positive (CD4+ or CD8+) cells expressing antigen-specific T-cell receptor (TCR) is characterized by metabolic reprogramming and involves both mTOR complexes (422, 423). When these naive T cells encounter pathogens in the periphery, they mount a robust immune response that is characterized by a shift from oxidative metabolism to glycolytic metabolism. mTOR activity has been shown to regulate T cell differentiation and function through control of metabolic programming (424–427). Naive T cells have low rates of nutrient uptake and protein synthesis, and they are more metabolically quiescent than immune-activated T cells (279). mTOR is integral for the metabolic reprogramming of T cells to increase membrane expression of nutrient transporters, which augment uptake of nutrients that are utilized for energy production and macromolecule synthesis (428, 429). mTORC1-dependent metabolic reprogramming that drives the exit of T cells from quiescence is partly mediated by Myc. mTORC1 inhibition diminishes TCR-induced Myc expression, which is accompanied by reduction in glycolytic activity (298). In raptor-deficient CD4+ T cells, there is attenuated protein expression of Myc, accompanied by attenuated mRNA expression of glycolytic enzymes (279). These cells also display defective de novo lipid synthesis and fail to induce genes involved in lipogenic pathways upon TCR stimulation (279). Whereas deletion of mTORC1 inhibits glycolysis and reduces effector functions, deletion of mTORC2 enhances glycolysis and increases T effector functions (430). After proliferation of effector cells and elimination of pathogens, T memory cells revert to a naivelike oxidative phosphorylation metabolism. mTOR activity is reduced in memory T cells (431, 432). T regulatory cells (Treg) that have immunosuppressive roles have distinct metabolism as effector cells. Treg proliferation and upregulation of mediators important for suppressive function rely on mTORC1-mediated cholesterol and lipid metabolism, in particular the mevalonate pathway (427).
B cell activation increases glucose uptake via PI3K/Akt signaling, but glucose catabolism is shunted from glycolysis to the PPP to generate sufficient NADPH necessary to maintain redox homeostasis (433–435). The robust activation of B cells is characterized by the synthesis of immunoglobulins (Igs) and the generation of antibody-secreting plasma cells. Both mTORC1 and mTORC2 promote B cell proliferation and antibody responses (271, 436). mTORC1 is involved in early transcription of genes mediating the unfolded protein response (UPR) in activated B cells, before antibody secretion (437). The increased antibody trafficking through the endomembrane system requires enhanced membrane biogenesis. Hence, B cells enhance de novo lipogenesis and become more oxidative (438, 439). As a result, they have an increased need to mitigate oxidative stress via upregulation of the redox system and increased expression of cytosolic and mitochondrial chaperones and other proteins involved in maintaining proteostasis. There is also increased glucose flux through the hexosamine pathway to promote antibody glycosylation and processing (440).
Classically activated macrophages (CAMs), which release inflammatory cytokines and antimicrobial peptides, reprogram metabolism during infection (441). CAMs increase aerobic glycolysis, wherein mTORC1 modulates HIF1α in response to stimuli such as the microbial ligand β-glucan (442). Blocking mTOR activity or glycolysis diminishes inflammatory cytokine production and bacterial elimination. Another population of macrophages called alternatively activated macrophages (AAMs), which enhance tissue repair and other activities during infection in response to IL-4, rely on increased oxidative metabolism. IL-4 upregulates Akt and mTORC1, leading to increased glucose metabolism. Increased OxPhos enhances the production of acetyl-CoA, which is utilized for fatty acid synthesis. IL-4, via Akt/mTORC1, controls the activity of ACL (ACLY), which catalyzes the generation of acetyl-CoA from citrate (443).
19. CANCER
Cancer cells are dependent on abundant supply of macromolecules and energy in order to provide the building blocks and fuel rapid proliferation. A hallmark of cancer is aberrant metabolism (444). Enhanced aerobic glycolysis, or the Warburg effect, allows cancer cells to not only generate ATP but also maintain reduction-oxidation (redox) balance and produce metabolites necessary for macromolecule synthesis. Despite the common need to acquire more nutrients and reprogram metabolism, cancer cells display heterogeneous metabolic vulnerabilities, influenced by specific oncogenic mutations, tissue type, the tissue microenvironment, and other genetic, epigenetic, and environmental factors. Specific metabolic vulnerabilities could thus be exploited for more effective cancer therapy. Tumor metabolic reprogramming is often accompanied by the upregulation of mTOR signaling. mTOR promotes increased nutrient acquisition to generate metabolites needed to support growth and proliferation. Mutations or alterations in the effectors of the PI3K/mTOR signaling are common in malignancies. Several mTOR inhibitors, including rapalogs, mTOR kinase inhibitors, and dual PI3K/mTOR inhibitors, are undergoing preclinical and clinical investigation, and FDA-approved agents are being used in the clinic for a number of diverse solid tumors and hematological malignancies (445, 446). Results from clinical trials that investigated the safety and efficacy of rapalogs reveal how tumors that may have particular metabolic signatures due to mTORC1 activation, such as mutations in TSC, are more sensitive to rapalog monotherapy. The identification of predictive biomarkers may allow for a personalized treatment approach based on tumor stratification and may improve response outcomes for mTOR inhibitor treatments. In addition to genomic and proteomic changes, metabolic alterations may be effective in predicting responses to mTOR and metabolic inhibitors. Identification of mutations in mTOR and its effectors could also inform on sensitivity to mTOR inhibitors. Hyperactivating mTOR mutations have been identified in human cancers (447–452) and may serve as biomarkers to identify tumor types that predict sensitivity to mTOR inhibitors, as well as confer resistance to mTOR inhibition. It is important to understand nutrient requirements, metabolic vulnerabilities, and symbiotic signaling networks among primary tumor cells and neighboring cells, including immune and stromal cells, in the tumor microenvironment. This information will shed light on improving targeted and immunotherapeutic strategies as well as dietary manipulations that can further boost efficacy of such therapies in combination.
20. TYPE 2 DIABETES AND OBESITY
mTOR signaling coordinates systemic energy status. It modulates nutrient and insulin signaling to maintain metabolic homeostasis. Deregulation of mTOR signaling leads to aberrant glucose and lipid metabolism and development of insulin resistance and thus occurs in metabolic disorders including diabetes and obesity. Chronic hyperactivation of mTORC1 signaling occurs during overnutrition, type 2 diabetes (T2D), and obesity (453, 454). The excess in nutrients that elevates mTORC1 signaling could potentially promote oxidative stress, ER stress, and inflammation, thus impairing tissue homeostasis. For example, pancreatic beta cells, which release insulin, become dysfunctional over time in diabetes because of chronic exposure to nutrient overload (455). Insulin-responsive tissues also develop insulin resistance, blunting PI3K/mTORC2 signaling. Furthermore, systemic downregulation of mTORC1 signaling, such as by deletion of S6K1 in mice, provides protection from diet-induced obesity and insulin resistance (454). Generating optimal mTORC1 and mTORC2 signaling is therefore critical to maintaining energy/metabolic homeostasis. At the cellular level, there is cross talk between increased mTORC1 signaling and the insulin/PI3K signaling that results in downregulation of the latter. Disruption of the insulin/mTORC2/Akt pathway, which could be a consequence of hyperactivation of mTORC1/S6K signaling in insulin-sensitive tissues, may contribute to peripheral insulin resistance. The insulin receptor substrate-1 (IRS-1) undergoes serine phosphorylation by S6K1 and mTORC1, leading to IRS-1 downregulation (456, 457). It is also phosphorylated by GRB10 via mTORC1, leading to disruption of IR/IRS-1 (123, 201). Increased mTORC2 activity also promotes insulin resistance due to mTORC2-mediated regulation of the ubiquitin ligase substrate-targeting subunit Fbw8, which stimulates IRS-1 degradation (125).
The role of mTOR complexes in metabolic disorders is underscored by their functions in key metabolic tissues including the liver, adipose tissue, and skeletal muscle as discussed above and highlighted in several recent review articles (458–460). Hepatic insulin resistance is a major contributor to impaired glucose homeostasis and leads to defective hepatic glucose output observed in T2D. Increased glucose production is consistently observed in T2D and can be attributed to increased gluconeogenesis. In the liver, Raptor and Rictor-depleted mice have impaired lipogenesis, gluconeogenesis, and glycolysis (272, 375). Expression of constitutively active Akt2 in hepatocytes with Rictor knockout restores glucose flux and lipogenesis with glucokinase and SREBP-1c activity (272). Chronic rapamycin treatment, which inhibits not just mTORC1 but also mTORC2 activity, contributes to insulin resistance due to inability to activate fatty acid β-oxidation and ketogenesis, leading to an imbalance in lipid metabolism (461, 462). Prolonged rapamycin treatment also leads to decreased β-cell viability and decreased insulin secretion, likely via the inhibition of mTORC2 (463). Although these studies caution against the use of mTOR inhibitors for T2D treatment, some studies reveal that, depending on the mouse model and duration of treatment, rapamycin may have beneficial effects on insulin sensitivity and glucose metabolism. In mice fed a high-fat diet, weekly treatments with lower-dose rapamycin inhibit mTORC1, protect against insulin resistance, and maintain mTORC2 activity (464). Low-dose rapamycin treatment also did not inhibit mTORC2 in a murine model of diet-induced obesity subjected to high-fat, high-sucrose diet for 20 wk. Mice demonstrated reduced plasma glucose and improved insulin sensitivity, with reductions in triglycerides, weight gain, and adiposity (465). In humans, the rapalog everolimus induced hyperglycemia occurring within 3–8 wk after treatment by decreasing insulin production (466). Nevertheless, there was no effect on insulin sensitivity in the liver, skeletal muscle, or adipose tissue. mTORC1 activity is also potently suppressed by metformin, an AMPK-activating agent and a first-line treatment for T2D that suppresses hepatic glucose production and improves systemic glycemic control (467). mTORC2 signaling is enhanced by metformin in the liver in vivo and in primary hepatocytes by AMPK-mediated phosphorylation of mTOR and Rictor, which is sufficient to increase mTORC2 catalytic activity toward Akt and downstream signaling (102).
mTOR signaling also regulates the growth and proliferation of pancreatic β-cells and their ability to secrete insulin, which affects glucose homeostasis (272, 468). The progressive loss of functional β-cell mass in T2D leads to β-cell failure, insulin insufficiency, and failure to meet metabolic demands. Pancreatic islets isolated from human patients with T2D are marked by increased mTORC1 activity and decreased mTORC2 activity (469). Genetic or chemical inhibition of mTORC1-S6K1 signaling restores insulin secretion as well as mTORC2-Akt signaling, suggesting that elevated mTORC1 impairs β cell function, likely due to a negative feedback loop from mTORC1-S6K1 to IRS-1/2-PI3K and mTORC2 (469). Mice with β cell-specific deletion of Rictor provide further insight into the role of mTORC2 in the regulation of insulin secretion, β-cell adaptation, and glucose homeostasis (470, 471). Rictor knockout in pancreatic β-cells of mice fed a high-fat diet showed glucose intolerance and impaired glucose-stimulated insulin secretion compared with control mice on the same diet (471). In another study of Rictor-null mice, mild hyperglycemia and glucose intolerance occurred because of reduction in β-cell mass, β-cell proliferation, pancreatic insulin content, and glucose-stimulated insulin secretion (470). Diabetic β-cell-specific Raptor-deficient mice have β-cell dedifferentiation that corresponds with downregulation of key transcriptional factors and gene transcripts critical for β-cell function, response to glucose, insulin secretion, glucose metabolism, and protein secretory pathway (472). In a human study, there was no difference in mTORC1 activity in human pancreatic islets isolated from patients with T2D and nondiabetic subjects; however, mTORC1 activity was higher from those with impaired fasting glucose compared with nondiabetic subjects (473). Collectively, these studies support the notion that proper activation of mTORC1 and mTORC2 is critical for pancreatic β-cell function.
In addition, mTOR coordinates regulation of peripheral insulin target tissues and the central nervous system (CNS), specifically the hypothalamus, which senses and integrates signals from fluctuating hormonal and nutrient levels and thus controls food intake, glucose, and lipid homeostasis. The activation of mTORC1/S6K is important for leptin- and insulin-mediated anorexigenic effects, leading to reduction in food intake and increase in energy expenditure (474, 475). Leptin and reactive oxygen species (ROS) require mTORC1 activity in the proopiomelanocortin (POMC) neurons to decrease food intake (476). In contrast, diminishing S6K1 activity increases food intake and body weight (474). Although these findings support that increased mTORC1 activity in the hypothalamus correlates with decreased food intake, and may have benefit for improved glucose homeostasis and insulin sensitivity, some studies indicate that there is a contrary relationship between hypothalamic mTORC1 activity and metabolic response. Increased hypothalamic S6K1 activity impairs suppression of hepatic glucose production and induces systemic insulin resistance (477, 478). Overexpression of DEPTOR, the mTORC1 (and mTORC2) negative regulator, specifically in the mediobasal hypothalamus prevents high-fat diet-induced obesity and improves glucose metabolism (479). When DEPTOR is overexpressed specifically in POMC neurons, it does not affect energy balance regulation but mildly elevates fasting blood glucose and insulin resistance (480). Genetic deletion of S6K1 in POMC neurons also has no effect on food intake or body weight but induces insulin resistance (481). Hence, the neuronal compartment and temporal regulation of mTORC1 in the hypothalamus and CNS in general warrants further investigation.
The role of mTORC2 in energy metabolism in the CNS is also emerging. Neuron-specific disruption of rictor leads to a profound increase in fat composition and adiposity and impairs glucose tolerance and behavioral leptin resistance (275). When rictor was deleted specifically in the POMC neurons, the mice demonstrated obesity and hyperphagia, fasting hyperglycemia, and dramatic glucose intolerance, whereas AgRP-specific deletion did not affect energy balance but only caused mild glucose intolerance. The role of rictor in the regulation of food intake is likely via Akt/FoxO1, since FoxO1 enhances the orexigenic neuropeptide AGRP via GPR17 and decreases the anorexigenic neuropeptide POMC via carboxypeptidase E (482, 483). These studies unravel a role for mTORC2 in hypothalamic regulation of energy balance. Future studies should reveal how the mTORC1/mTORC2 signaling-mediated regulation of food intake is wired to modulation of peripheral energy homeostasis and how we can manipulate these signals to treat type 2 diabetes and obesity.
21. NEURODEGENERATIVE DISORDERS
Defects in mTOR signaling underlie the pathogenesis of several neurodegenerative disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) (484, 485) (FIGURE 7). Hyperactivation of the mTOR pathway is found in AD patients, and its inhibition has beneficial effects in several mouse models of AD (486, 487). Treatment with rapamycin or genetic disruption of mTOR signaling reduces amyloid-β (Aβ) deposition, pathogenic tau phosphorylation, and neurofibrillary tangles (NFTs) in these mouse models, and the mice show attenuation of neuronal loss and improved cognitive function (488). These effects could be partly due to promotion of autophagy, a process that is normally downregulated via mTORC1 and becomes dysfunctional in AD (489). AD is also associated with metabolic abnormalities. Defects in branched-chain amino acid (BCAA) catabolism leading to increased BCAA levels enhance tau phosphorylation in an mTOR-dependent manner in AD (490). Abnormalities in insulin signaling and glucose metabolism, increased oxidative stress, and mitochondrial dysfunction are also linked to AD (491, 492). In line with such defects, mTORC2 is also implicated in AD. In mice lacking PTEN in the brain, deletion of rictor, but not raptor, prolongs life span, prevents seizures, and rescues AD-like behaviors and long-term memory, as well as normalizes metabolic changes in the brain (493). However, in a neuronal culture model of AD overexpression of rictor fully reverses the effects of Aβ on insulin resistance and toxicity (494). As dual PI3K/mTOR inhibitors are now being investigated as possible AD therapies (495), the above findings suggest that the efficacy of such treatments would rely on achieving optimal or finely tuned activity of mTORC1 and mTORC2 signaling.
FIGURE 7.
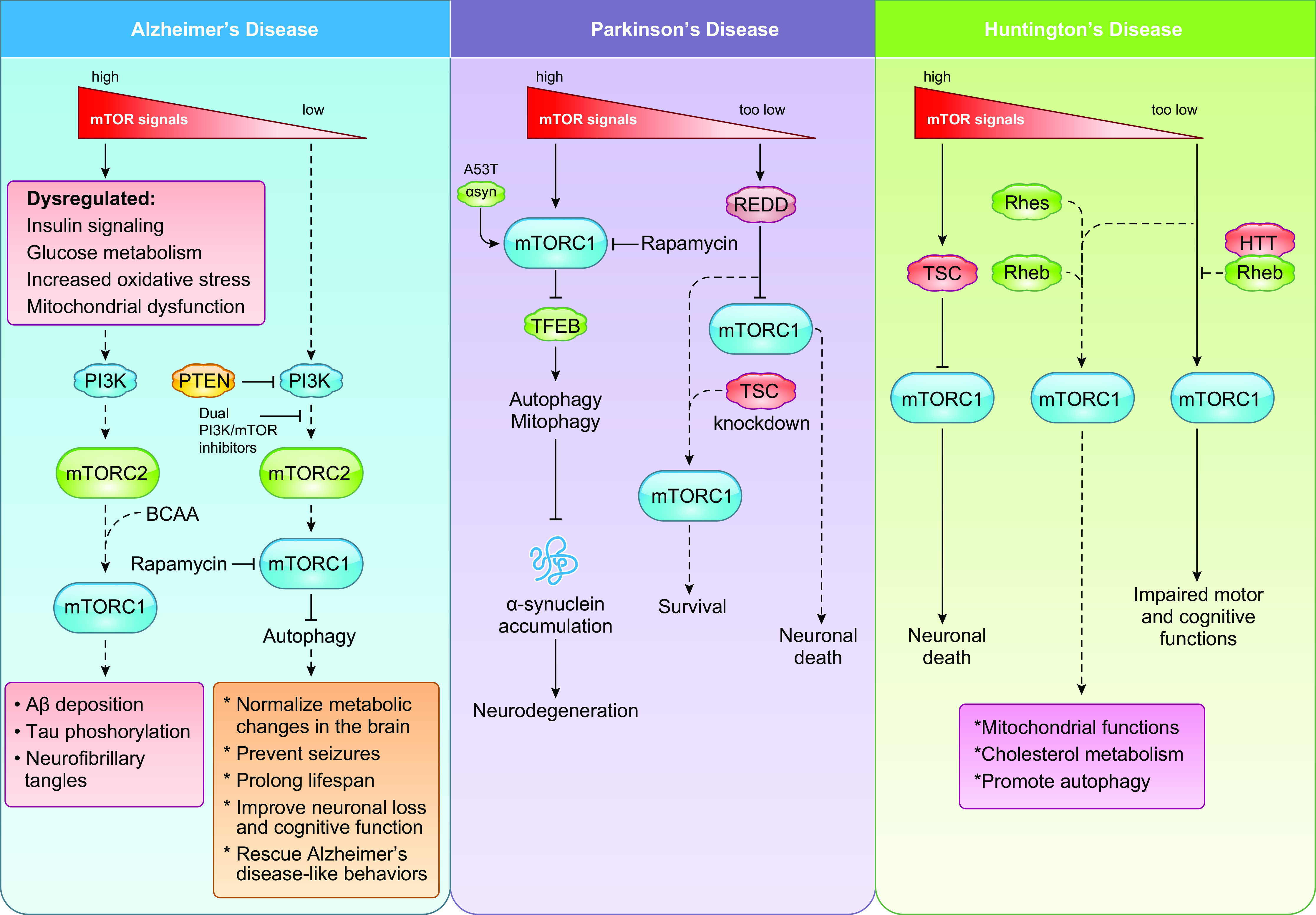
Defects in mTOR signaling underlie the pathogenesis of several neurodegenerative disorders. In Alzheimer’s disease (AD), inhibition of hyperactive mTOR signaling reduces Aβ deposition, pathogenic tau phosphorylation, and neurofibrillary tangles, leading to improvements in neuronal loss and cognitive function potentially due to promoting autophagy. Defects in branched-chain amino acid (BCAA) catabolism leading to increased BCAA levels enhance tau phosphorylation in an mTOR-dependent manner in AD. Deletion of brain-specific mTORC2, but not mTORC1, prolonged life span, prevented seizures, rescued AD-like behaviors and long-term memory, and normalized metabolic changes in the brain. In Parkinson’s disease (PD), insulin/mTOR signaling pathway is deregulated. Accumulation of α-synuclein is accompanied by increased mTOR activity and autophagy dysfunction. Rapamycin and depletion of mTOR mitigates the increased mTOR activity caused by elevated α-synuclein and induces autophagy and clearance of A53T α-synuclein. In models of PD with low mTORC1 activity due to elevated REDD1 levels, depletion of TSC2 enhances mTORC1 activity and is neuroprotective. In Huntington’s disease, enhancing mTORC1 activity by increasing expression of Rheb or Rhes mitigates mitochondrial dysfunction and abnormal cholesterol metabolism and increases autophagy. See glossary for abbreviations.
The insulin/mTOR signaling pathway is also deregulated in PD, a neurodegenerative disorder characterized by mitochondrial dysfunction and accumulation of α-synuclein-containing inclusions termed Lewy bodies. Rapamycin prevents mitophagy defects in PD models harboring a mutation in PARKIN, an E3 ubiquitin ligase involved in promoting mitophagy (496). This effect of rapamycin requires the presence of TFEB, which regulates genes involved in autophagy. Accumulation of α-synuclein is accompanied by increased mTOR activity and autophagy dysfunction. Rapamycin mitigates increased mTOR activity caused by elevated α-synuclein (497). A53T α-synuclein is a common mutation in PD, which upregulates mTOR/S6K1 signaling and impairs autophagy, thus further contributing to the aggregation of toxic A53T α-synuclein. Depletion of mTOR results in the induction of autophagy, leading to clearance of A53T α-synuclein (498). However, enhancing mTOR activity may also have beneficial effects in PD. The negative regulator of mTORC1 activity REDD1 is upregulated in dopaminergic neurons in PD patients and in cellular models of PD. When REDD1 levels are elevated, reconstituting mTOR activity by depletion of TSC2 has neuroprotective effects (499). Hence, maintaining optimal levels of mTOR activation is likely crucial to prevention of PD.
The role of mTOR activity in the pathogenesis of HD is also not simple. Conditions that enhance mTORC1 activity, such as deletion of TSC1, accelerate death in an HD mouse model (500). However, in murine models of HD, expression of a constitutively active Rheb, an mTORC1 regulator, mitigates mitochondrial dysfunction and abnormal cholesterol metabolism and increases autophagy in HD (501). Huntingtin (Htt), which forms an expanded polyglutamine (poly-Q) tract resulting in impaired motor and cognitive functions in HD, interacts with Rheb in response to amino acids. Exogenous expression of another Rheb-related protein, Rhes, which activates mTORC1, also rescues motor deficits and enhances mTORC1 signals in HD mice that have low endogenous Rhes levels (501). In a cell culture model of HD, phosphorylation of ATG14 by ULK1, which promotes autophagy and is defective in HD, is inhibited by Torin1 (mTOR inhibitor) or by amino acid starvation, underscoring mTOR-dependent control of autophagy (502). Thus, like PD, future studies should unravel how optimal activation of mTORC1 and mTORC2 could be achieved for the treatment of HD. Although the roles of several metabolic processes in the pathogenesis of neurodegenerative disorders have been recognized (503), understanding how mTOR complexes mediate the control of such processes will be important to more effectively manipulate mTOR signaling as a therapeutic strategy for these diseases.
22. AGING
Aging is characterized by metabolic alterations that accumulate over time, leading to reduced biological fitness. The central role of mTOR in metabolism makes mTOR an ideal target for antiaging therapeutics. mTOR is part of the growth factor signaling pathway insulin/insulin-like growth factor signaling (IIS) that controls glucose metabolism in multicellular organisms. Reduced signaling through this pathway brought about by downregulation of mTOR by rapamycin, caloric restriction, or genetic mutations of components of this pathway is linked to longevity (FIGURE 8). Dampening mTOR activity with rapamycin extends life span or improves health span in several organisms including humans (461, 504–507). Furthermore, mice with deficient S6K1 have increased longevity (508). The increased longevity seen in Myc-hypomorphic mice is also characterized by decreased mTORC1 activity due to impaired glutamine transport (371). There is substantial evidence supporting that downregulating mTOR could counteract aging hallmarks including deregulated nutrient sensing, mitochondrial dysfunction, loss of proteostasis, cellular senescence, and stem cell exhaustion (509). For example, rapamycin treatment reverses age-dependent cardiac proteome remodeling, including effects on decreased abundance of proteins involved in mitochondrial function, while increasing abundance of proteins involved in glycolysis and oxidative stress response (510). mTOR is an important regulator of oxidative stress by promoting mitochondrial biogenesis and oxidative metabolism. mTORC1 inhibition reduces mitochondrial content and prevents senescence phenotype in the aging mouse liver (511). Attenuating mTORC1 also promotes autophagy, which is involved in promoting proteostasis and elimination of damaged proteins and organelles. In muscle stem cells, age-associated deficits in autophagy lead to the accumulation of intracellular components and damaged organelles such as mitochondria, thereby promoting ROS generation that drives cellular transition from quiescence into senescence in aged satellite cells. Targeting mTOR activity with rapamycin restores autophagy in geriatric satellite cells, which allows for their proliferative expansion while preventing senescence entry, thus enabling new skeletal muscle fiber formation (512). Attenuation of mTORC1 activity is also beneficial to prevent immunosenescence, the decline in immune function during aging. In mouse models, rapamycin restores self-renewal and hematopoiesis of hematopoietic stem cells (HSCs) and increases immune activity against both viral and bacterial pathogens (513, 514). In elderly humans, treatment with rapalogs enhances the response to the influenza vaccine (515, 516). Collectively, mTORC1 inhibition in various tissues could slow aging by inhibiting the senescence phenotype, stimulating autophagy, stimulating clearance of old and dysfunctional mitochondria, preserving the stem cell pool, and maintaining self-renewal.
FIGURE 8.
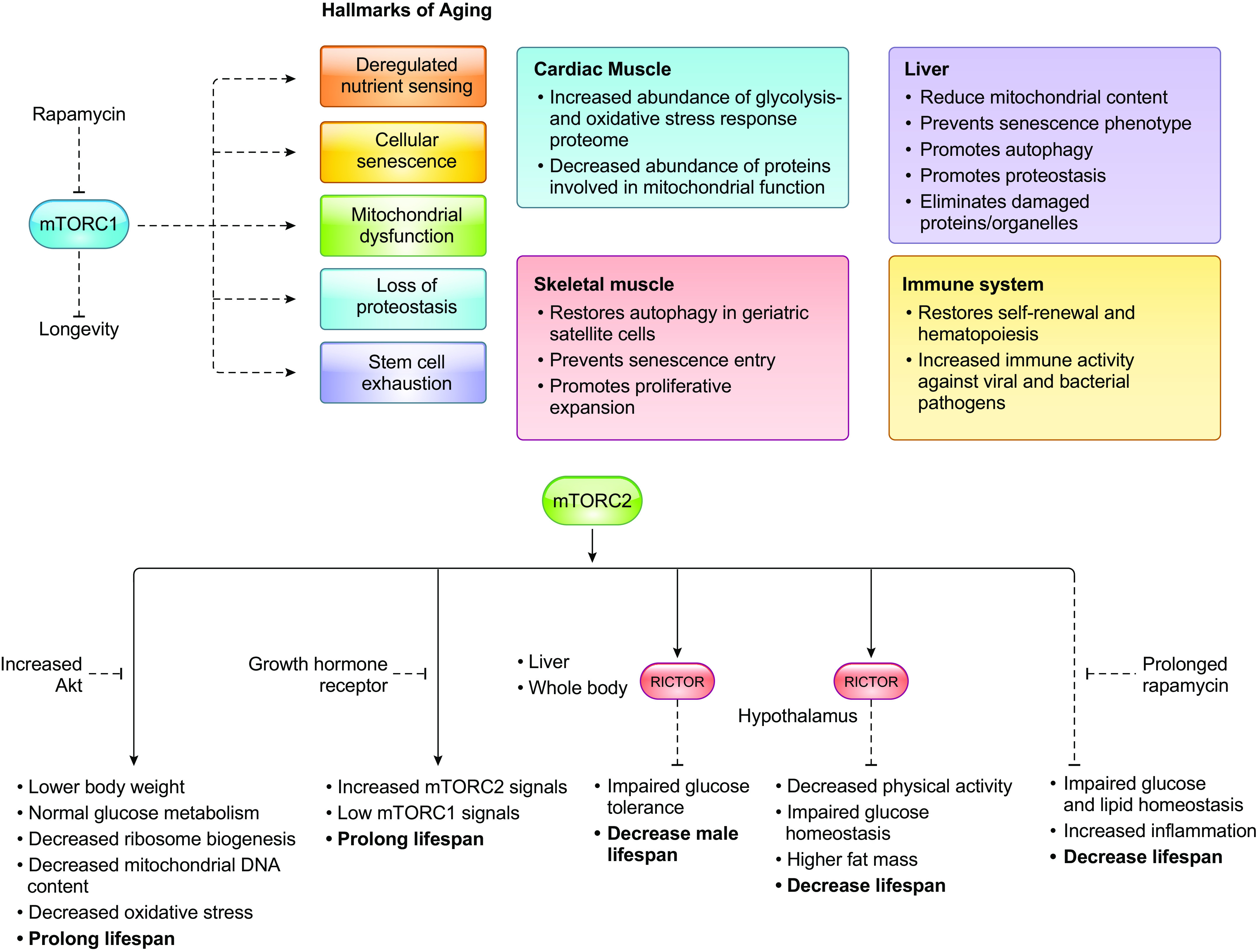
Modulating mTORC1 and mTORC2 signaling is important to prolong health span. A: downregulating mTORC1 in specific tissues and in the organism could counteract aging hallmarks such as deregulated nutrient sensing, mitochondrial dysfunction, loss of proteostasis, cellular senescence, and stem cell exhaustion. B: mTORC2 may have tissue-specific functions that could impact longevity. Decreased Akt signaling and growth hormone receptor prolong life span, while deficiency of rictor in the liver, hypothalamus, and the whole body or inhibition of mTORC2 via prolonged rapamycin treatment decreases life span. See glossary for abbreviations.
mTORC2 is also part of the IIS pathway. Haploinsufficiency of Akt1, an mTORC2 substrate and mediator of IIS signals, extends life span in mice (517). Decreased Akt is accompanied by lower body weight but normal glucose metabolism, decreased ribosome biogenesis, decreased mitochondrial DNA content, and oxidative stress. However, it remains unclear whether inhibition of mTORC2 itself would have beneficial effects on longevity. Inhibition of mTORC2 signaling by depletion of rictor either specifically in the liver or in the whole body decreases male but not female life span, suggesting that mTORC2 activity has a sex-specific positive effect on longevity (518). In agreement with a positive role for mTORC2 in longevity, deletion of rictor in the hypothalamus also reduces life span (519). The loss of rictor decreases physical activity level and confers susceptibility to obesity. These mice also have impaired glucose homeostasis and have higher fat mass. In mice with deficiency of growth hormone receptor, the prolonged life span correlates with increased mTORC2 signaling and low mTORC1 signaling. Furthermore, prolonged treatment with rapamycin, which leads to decreased mTORC2 signaling, impairs glucose and lipid homeostasis, increases inflammation, and decreases life span (520). Since mTORC2 mediates insulin signals, it is possible that the function of mTOR in longevity is due to its positive role in promoting insulin sensitivity and glucose homeostasis. Interestingly, mice with rictor deficiency in adipose tissue that were exposed to a calorically restricted diet remained long-lived despite insulin resistance (521). Although these findings reveal that caloric restriction-induced insulin sensitivity may not be required for longevity, mTORC2 may have tissue-specific functions that could impact longevity.
23. CONCLUSIONS AND FUTURE PERSPECTIVES
mTOR complexes control metabolic pathways at different levels, from transcription and translation to posttranslational mechanisms. mTOR controls its metabolic targets either directly via phosphorylation or via its well-characterized AGC kinase substrates. Most of the mTORC metabolic targets that have been characterized and described here appear to be indirectly regulated by mTOR. Although some transcriptional targets involved in metabolism have been identified as downstream mTORC1/2 effectors, it remains unclear how mTORCs regulate transcription in a more direct manner. In some cases, the function of mTORCs is mediated by canonical substrates such as Akt and S6K1. The mTOR complexes may also spatially regulate metabolic targets. Localization of mTORCs and association with specific regulators and metabolic enzymes in cellular compartments may serve to not only modulate mTORC activity but also recruit other signaling and metabolic effectors and thus trigger a cascade of events that ultimately induce gene expression of metabolic effectors. Since the discovery of mTOR complex components, we have now gained a better understanding of how mTOR activity is controlled. Amino acid modifications, including phosphorylation, ubiquitination, acetylation, and malonylation of complex components alter mTOR activity. Nutrient availability and metabolic state of cells impact the availability of the metabolites that are utilized for such modifications. Thus, mTOR controls metabolic processes but at the same time responds to the products of metabolism. Nutrients that mTOR responds to come from the extracellular environment and from intracellular sources, such as autophagy-mediated recycling of nutrients and catabolism of macronutrients. The mechanisms involved in sensing various amino acids and other nutrients/metabolites are the subject of intense investigation in mTORC1 signaling. We are only beginning to understand how mTORC2 senses nutrient levels in addition to its regulation by growth factors. mTORC1 has now been linked to the regulation of numerous anabolic processes, including protein, nucleotide, lipid, and carbohydrate metabolism. mTORC2 has overlapping as well as distinct functions in the control of these metabolic processes. Whether there are specific stimulatory conditions or tissue specificity that determine mTORC1- or mTORC2-controlled metabolic reprogramming warrants further investigation. It is notable that there is overwhelming evidence that mTORC1 is activated by anabolic signals. In contrast, mTORC2 activation is responsive to both anabolic and catabolic signals. mTORC2 is robustly activated by a surge in growth signals, such as addition of growth factors, as well as by limiting amounts of nutrients. The downstream targets of mTORC2 such as Akt promote cell proliferation in the presence of adequate nutrients. They also enhance cell survival in response to various stress conditions including nutrient limitation. Hence, mTORC2 likely functions to restore metabolic homeostasis in response to nutrient fluctuations. The mTORC2 component SIN1 links mTORC2 signaling to the stress-regulated Ras/MAPK pathway (115, 116). Some studies have also uncovered association of mTOR with other proteins, independently of mTORC1 or mTORC2 (522, 523). Future studies should further define the cross talk between mTOR and other signaling pathways to reprogram metabolism.
We are also beginning to understand how genetic mutations can trigger metabolic reprogramming and the role that mTOR plays in these metabolic processes. The microenvironment, which is composed of different cell types, also influences nutrient availability and thus cellular metabolism. Future studies should further delineate how mTORCs reprogram metabolic and biosynthetic pathways under specific genetic mutations and microenvironmental pressures. mTOR inhibitors such as rapamycin analogs and catalytic inhibitors that suppress both mTOR complexes are now being used in the clinic or are undergoing clinical trials to treat a variety of conditions. Rapamycin is used to prevent organ transplant rejection. It is also used to treat a rare lung disease, lymphangioleiomyomatosis (LAM). Rapalogs have been approved for the treatment of specific types of cancer, such as advanced renal cell carcinoma, subependymal giant cell astrocytoma, renal angiomyolipoma, and pancreatic neuroendocrine tumors. Tumors with specific upregulation of mTORC1 due to genetic mutations (e.g., TSC1/2) seem to be particularly sensitive to rapalogs. Rapamycin is also promising for the treatment of neurodegenerative diseases and neurological disorders such as tuberous sclerosis complex, epilepsy, autism, traumatic brain injury, and stroke (524). An exciting application of rapamycin is for improving health span and prevention of age-related pathologies. Patients with autoimmune disorders could also benefit from mTOR inhibitors given the important role of mTOR in immune cell metabolism. Rapalogs and ATP-competitive inhibitors targeting both mTOR complexes and those that inhibit both PI3K and mTOR (dual PI3K/mTOR inhibitors) are undergoing clinical trials to treat cancer. Early results have been disappointing because of toxicity and undesirable side effects. Rapalogs and catalytic inhibitors are also being used in combination with other chemotherapy and metabolic inhibitors to attain better efficacy but less toxicity. A better understanding of mTOR-mediated metabolic reprogramming in the tumor microenvironment that consists of the target cells (e.g., tumors) and their neighboring cells (e.g., immune cells) will facilitate design of more effective therapeutic strategies. Finally, as metabolism is intimately linked to nutrition, therapeutic strategies to target mTOR signaling may also be improved when combined with diet manipulation (525, 526).
GLOSSARY
- α-KG
α-Ketoglutarate
- 2-HG
2-Hydroxyglutarate
- 3-PG
3-Phosphoglycerate
- 4EBP1
4E-binding protein 1
- 6PG
6-Phospho-D-gluconate
- AAM
Alternatively activated macrophage
- Aβ
Amyloid-β
- ACC
Acetyl-CoA carboxylase
- Ac-CoA
Acetyl-coenzyme A
- ACL
ATP citrate lyase
- ACSS
Acyl-CoA synthetase short chain
- AD
Alzheimer’s disease
- AGC kinases
Protein kinase A (PKA), PKG, PKC
- AML
Acute myeloid leukemia
- AMP
Adenosine monophosphate
- AMPK
AMP-activated protein kinase
- AR
α1A-adrenoceptor
- Arf1
ADP-ribosylation factor 1
- ASNS
Asparagine synthetase
- ASS1
Argininosuccinate synthetase-1
- ATF4
Activating Transcription Factor 4
- ATG13/ATG14
Autophagy related
- ATIC
Aminoimidazole-carboxamide ribonucleotide transformylase IMP cyclohydrolase
- BAT
Brown adipose tissue
- BCAA
Branched-chain amino acids (isoleucine, leucine, and valine)
- Bcl-xL
B-cell lymphoma-extra large
- CAD
Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydro-orotase
- CAM
Classically activated macrophage
- cAMP
Adenosine 3′5′-cyclic monophosphate
- CCT
Chaperonin containing tailless complex polypeptide 1
- CPT1
Carnitine palmitoyltransferase I
- CR
Caloric restriction
- CREB2
cAMP-responsive element binding 2
- CRTC2
CREB regulated transcription coactivator 2
- Cryo-EM
Cryogenic electron microscopy
- CTP
Cytidine triphosphate
- DAP1
Death associated protein 1
- DEPDC5
DEP domain containing 5
- DEPTOR
DEP domain-containing mTOR-interacting protein
- Dlk1-Dio3
Delta-like homolog 1 deiodinase iodothyronine type III cluster
- DNMT3B
DNA methyltransferase 3B
- EMT
Epithelial-to-mesenchymal transition
- ENO
Enolase
- EP300
E1A-associated protein p300
- ER
Endoplasmic reticulum
- ERK
Extracellular signal-regulated kinase
- ETC
Electron transport chain
- FA
Fatty acids
- FAD
Flavin adenine dinucleotide
- FAO
Fatty acid oxidation
- FASN
Fatty acid synthase
- FKBP12
12-kDa FK506-binding protein
- FLCN
Folliculin
- FNIP1 and FNIP2
Folliculin interacting protein 1 and 2
- FOXK1
Forkhead box K1
- FOXO
Forkhead box O
- FR-α
Folate receptor-alpha
- FRB domain
FKBP12/rapamycin binding domain
- G6P
Glucose-6-phosphate
- G6PD
Glucose-6-phosphate dehydrogenase
- GA 3-P
Glyceraldehyde 3-phosphate
- GAP
GTPase-activating protein
- GDH
Glutamate dehydrogenase
- GDP
Guanosine diphosphate
- GEF
Guanine nucleotide exchange factor
- GFAT1
Glutamine:fructose-6-phosphate amidotransferase 1
- GlcNAc
N-acetylglucosamine
- GLS
Glutaminase
- GLUT1
Glucose transporter 1
- GMP
Guanosine monophosphate
- GPCR
G protein-coupled receptor
- Grb10
Growth factor receptor-bound protein 10
- GSK3β
Glycogen synthase kinase 3beta
- GTP
Guanosine-5′-triphosphate
- HBP
Hexosamine biosynthesis pathway
- HD
Huntington’s disease
- HDAC
Histone deacetylase
- HIF
Hypoxia-inducible factor
- HIF1α
Hypoxia-inducible factor 1 alpha
- HK2
Hexokinase 2
- HM
Hydrophobic motif
- HMGCS
Hydroxymethylglutaryl-CoA synthase
- hnRNPs
Heterogeneous nuclear ribonucleoprotein
- HSC
Hematopoietic stem cell
- HSF
Heat-shock transcription factor 1
- HTT
Huntingtin
- HuR
Human antigen R
- IDH1/2
Isocitrate dehydrogenase
- IGF1R
Insulin-like growth factor 1 receptor
- IIS
Insulin/insulin-like growth factor signaling
- IKK
Inhibitor of nuclear factor-κB kinase
- IMP
Inosine monophosphate
- IMPDH2
Inosine-5′-monophosphate dehydrogenase 2
- iPSC
Induced pluripotent stem cell
- IRS-1
Insulin receptor substrate-1
- LAM
Lymphangioleiomyomatosis
- LAMTOR2
Late Endosomal/Lysosomal Adaptor, MAPK and mTOR Activator 2
- LATS1/LATS2
Large tumor suppressor 1,2
- LDH
Lactate dehydrogenase
- LincRNA-RoR
Large intergenic noncoding RNA-RoR
- lncARSR
lncRNA Regulator of Akt Signaling Associated with HCC and RCC
- MAM
Mitochondria-associated ER membrane
- MAPK
Mitogen-activated protein kinase
- MAT
Methionine adenosyltransferase
- MCAM
Melanoma cell adhesion molecule
- MEF
Murine embryonic fibroblast
- miR
MicroRNA
- MTFP1
Mitochondrial fission process protein 1
- MTHFD2
Methylenetetrahydrofolate dehydrogenase 2
- mTOR
Mechanistic target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- NAD
Nicotinamide adenine dinucleotide
- NADK
Nicotinamide adenine dinucleotide kinase
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NAM
Nicotinamide
- NAMPT
Nicotinamide phosphoribosyltransferase
- NAMPT-AS
Long noncoding antisense transcript of NAMPT
- NFT
Neurofibrillary tangle
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NK cells
Natural killer cells
- NLK
Nemo-like kinase
- NMN
Nicotinamide mononucleotide
- NPRL2
Nitrogen permease regulator 2-like protein
- OGT
O-GlcNAc transferase
- OXPHOS
Oxidative phosphorylation
- PAMPS
Pathogen-associated molecular patterns
- PAT4/SLC36A4
SLC36/proton dependent amino acid transporter 4
- PD
Parkinson’s disease
- Pdcd4
Programmed cell death 4 protein
- PDK
Pyruvate dehydrogenase kinase
- PDK1
Phosphoinositide-dependent kinase 1
- PDK3
Pyruvate dehydrogenase kinase 3
- PEP
Phosphoenolpyruvate
- PFK
Phosphofructokinase-2
- PFKFB3
Phosphofructokinase-2/fructose-2,6-bisphosphatase B3 isotype
- PGI
Phosphoglucose isomerase
- PH
Pleckstrin homology domain
- PI3K
Phosphatidylinositol 3-kinase
- PIP3
Phosphatidylinositol 3,4,5-trisphosphate
- PKC
Protein kinase C
- PKM2
Pyruvate kinase isoform M2
- PKN2
Protein kinase N2
- PLA2
Phospholipase A2
- Poly-Q
Polyglutamine
- POMC
Proopiomelanocortin
- PP2A
Protein phosphatase 2A
- PPARγ
Peroxisome proliferator-activated receptor γ
- PPP
Pentose phosphate pathway
- PRAS40
Proline-rich Akt substrate of 40 kDa
- PRPP
Phosphoribosyl pyrophosphate
- PRPS2
Phosphoribosyl pyrophosphate synthetase 2
- PTEN
Phosphatase and tensin homolog
- Rag
Ras-related GTP binding protein
- RAPTOR
Regulatory-associated protein of mTOR
- REDD1
Regulated in development and DNA damage response 1
- RFC
Reduced folate carrier
- Rheb
Ras homolog enriched in brain
- ROS
Reactive oxygen species
- RPIA
Ribose-5-phosphate isomerase A
- RSK
p90 Ribosomal S6 kinase
- RTK
Receptor tyrosine kinase
- S6K
Ribosomal protein S6 kinase
- SAM
S-adenosylmethionine
- SAMTOR
SAM sensor upstream of mTORC1
- SCD1
Stearoyl-CoA desaturase 1
- SIN1
Stress-activated protein kinase interacting protein 1
- SIRT1
Sirtuin 1
- SOX2
SRY-box transcription factor 2
- SREBP
Sterol regulatory element binding protein
- SRPK2
Serine/arginine-rich protein kinase 2
- TBK1
TANK-binding kinase
- TCA
Tricarboxylic acid
- TCR
T-cell receptor
- TGFβ1
Transforming growth factor beta 1
- THF
Tetrahydrofolate
- TKT
Transketolase
- TM
Turn motif
- TNBC
Triple-negative breast cancer
- TOS
TOR signaling motif
- TRAF6
TNF receptor associated factor 6
- Treg
T regulatory cell
- TSC
Tuberous sclerosis complex
- UDP-GlcNAc
Uridine-5-diphosphate-N-acetylglucosamine
- ULK1
unc-51 Like Autophagy Activating Kinase 1
- UMP
Uridine monophosphate
- UPR
Unfolded protein response
- URB1
URB1 Ribosome Biogenesis Homolog
- UTR
Untranslated region
- VEGFA
Vascular endothelial growth factor A
- VDAC2
Voltage Dependent Anion-Selective Channel 2
- WAT
White adipose tissue
- WD40
Trp Asp repeats
- WDR24
WD repeat-containing protein 24
- YY1
yin yang 1
GRANTS
The authors gratefully acknowledge funding from the NJ Commission on Cancer Research (DFHS18CRF008) and NIH (GM-079176, CA-154674, GM-137493).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.S. and E.J. conceived and designed research; A.S., E.K., and E.J. prepared figures; A.S., E.K., and E.J. drafted manuscript; A.S., E.K., and E.J. edited and revised manuscript; E.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Bonnie Firestein and the members of the Jacinto lab for helpful comments and discussions. We apologize to our colleagues in the field for not being able to cite important contributions due to space limitations. Figures were created using BioRender.com.
REFERENCES
- 1.Lorberg A, Hall MN. TOR: the first 10 years. Curr Top Microbiol Immunol 279: 1–18, 2004. doi: 10.1007/978-3-642-18930-2_1. [DOI] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol 13: 195–203, 2012. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 3.Valcourt JR, Lemons JM, Haley EM, Kojima M, Demuren OO, Coller HA. Staying alive: metabolic adaptations to quiescence. Cell Cycle 11: 1680–1696, 2012. doi: 10.4161/cc.19879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369: 756–758, 1994. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 5.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253: 905–909, 1991. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 6.Helliwell SB, Wagner P, Kunz J, Deuter-Reinhard M, Henriquez R, Hall MN. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol Biol Cell 5: 105–118, 1994. doi: 10.1091/mbc.5.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 73: 585–596, 1993. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- 8.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin- dependent fashion and is homologous to yeast TORs. Cell 78: 35–43, 1994. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 9.Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell 7: 25–42, 1996. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 13: 3271–3279, 1999. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardwick JS, Kuruvilla FG, Tong JK, Shamji AF, Schreiber SL. Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc Natl Acad Sci USA 96: 14866–14870, 1999. doi: 10.1073/pnas.96.26.14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 270: 2320–2326, 1995. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 13.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273: 3963–3966, 1998. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 14.Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69: 1227–1236, 1992. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- 15.Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 273: 14484–14494, 1998. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- 16.Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 358: 70–73, 1992. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- 17.Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 257: 973–977, 1992. doi: 10.1126/science.1380182. [DOI] [PubMed] [Google Scholar]
- 18.Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap- dependent initiation of translation. EMBO J 15: 658–664, 1996. doi: 10.1002/j.1460-2075.1996.tb00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci USA 105: 17414–17419, 2008. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graves LM, Bornfeldt KE, Argast GM, Krebs EG, Kong X, Lin TA, Lawrence JC Jr.. cAMP- and rapamycin-sensitive regulation of the association of eukaryotic initiation factor 4E and the translational regulator PHAS-I in aortic smooth muscle cells. Proc Natl Acad Sci USA 92: 7222–7226, 1995. doi: 10.1073/pnas.92.16.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng T, Golub TR, Sabatini DM. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol 22: 5575–5584, 2002. doi: 10.1128/mcb.22.15.5575-5584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 14: 2712–2724, 2000. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient- regulated transcription factors. Nature 402: 689–692, 1999. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- 24.Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science 294: 1102–1105, 2001. doi: 10.1126/science.1063518. [DOI] [PubMed] [Google Scholar]
- 25.Edinger AL, Linardic CM, Chiang GG, Thompson CB, Abraham RT. Differential effects of rapamycin on mammalian target of rapamycin signaling functions in mammalian cells. Cancer Res 63: 8451–8460, 2003. [PubMed] [Google Scholar]
- 26.Schmidt A, Beck T, Koller A, Kunz J, Hall MN. The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J 17: 6924–6931, 1998. doi: 10.1093/emboj/17.23.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell 103: 253–262, 2000. doi: 10.1016/S0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt A, Kunz J, Hall MN. TOR2 is required for organization of the actin cytoskeleton in yeast. Proc Natl Acad Sci USA 93: 13780–13785, 1996. doi: 10.1073/pnas.93.24.13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng XF, Florentino D, Chen J, Crabtree GR, Schreiber SL. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell 82: 121–130, 1995. doi: 10.1016/0092-8674(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 30.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol 16: 1865–1870, 2006. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177–189, 2002. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 32.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6: 1122–1128, 2004. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 33.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127: 125–137, 2006. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 34.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163–175, 2002. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 35.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10: 457–468, 2002. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 36.Sarbassov D, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14: 1296–1302, 2004. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 37.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev 20: 2820–2832, 2006. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schroder W, Cloonan N, Bushell G, Sculley T. Alternative polyadenylation and splicing of mRNAs transcribed from the human Sin1 gene. Gene 339: 17–23, 2004. doi: 10.1016/j.gene.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Yuan Y, Pan B, Sun H, Chen G, Su B, Huang Y. Characterization of Sin1 isoforms reveals an mTOR-dependent and independent function of Sin1gamma. PLoS One 10: e0135017, 2015. doi: 10.1371/journal.pone.0135017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu CC, Chou P, Jacinto E. The target of rapamycin: structure and functions. In: Protein Kinases, edited by Da Silva Xavier G. London: IntechOpen, 2012, p. 1–40. [Google Scholar]
- 41.Fan SJ, Snell C, Turley H, Li JL, McCormick R, Perera SM, Heublein S, Kazi S, Azad A, Wilson C, Harris AL, Goberdhan DC. PAT4 levels control amino-acid sensitivity of rapamycin-resistant mTORC1 from the Golgi and affect clinical outcome in colorectal cancer. Oncogene 35: 3004–3015, 2016. doi: 10.1038/onc.2015.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol 15: 254–266, 2005. doi: 10.1016/j.semcancer.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Kim J, Guan KL. Amino acid signaling in TOR activation. Annu Rev Biochem 80: 1001–1032, 2011. doi: 10.1146/annurev-biochem-062209-094414. [DOI] [PubMed] [Google Scholar]
- 44.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534, 2009. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10: 935–945, 2008. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501, 2008. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290–303, 2010. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150: 1196–1208, 2012. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nada S, Hondo A, Kasai A, Koike M, Saito K, Uchiyama Y, Okada M. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J 28: 477–489, 2009. doi: 10.1038/emboj.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44: 134–146, 2011. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340: 1100–1106, 2013. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peng M, Yin N, Li MO. SZT2 dictates GATOR control of mTORC1 signalling. Nature 543: 433–437, 2017. doi: 10.1038/nature21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, Abu-Remaileh M, Frankel WN, Sabatini DM. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543: 438–442, 2017. doi: 10.1038/nature21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gan L, Seki A, Shen K, Iyer H, Han K, Hayer A, Wollman R, Ge X, Lin JR, Dey G, Talbot WS, Meyer T. The lysosomal GPCR-like protein GPR137B regulates Rag and mTORC1 localization and activity. Nat Cell Biol 21: 614–626, 2019. doi: 10.1038/s41556-019-0321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang G, Humphrey SJ, Murashige DS, Francis D, Wang QP, Cooke KC, Neely GG, James DE. RagC phosphorylation autoregulates mTOR complex 1. EMBO J 38: e99548, 2019. doi: 10.15252/embj.201899548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolfson RL, Sabatini DM. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab 26: 301–309, 2017. doi: 10.1016/j.cmet.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Son SM, Park SJ, Lee H, Siddiqi F, Lee JE, Menzies FM, Rubinsztein DC. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab 29: 192–201.e7, 2019. doi: 10.1016/j.cmet.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jung J, Genau HM, Behrends C. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol 35: 2479–2494, 2015. doi: 10.1128/MCB.00125-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, Scorzoni S, Filipek PA, Huber KV, Bigenzahn JW, Heinz LX, Kraft C, Bennett KL, Indiveri C, Huber LA, Superti-Furga G. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519: 477–481, 2015. doi: 10.1038/nature14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, Wang T, Bar-Peled L, Zoncu R, Straub C, Kim C, Park J, Sabatini BL, Sabatini DM. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347: 188–194, 2015. doi: 10.1126/science.1257132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, Sabatini DM. mTORC1 Activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171: 642–654.e12, 2017. doi: 10.1016/j.cell.2017.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 47: 349–358, 2012. doi: 10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 63.Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol 21: 63–71, 2019. doi: 10.1038/s41556-018-0205-1. [DOI] [PubMed] [Google Scholar]
- 64.Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, Schmitt A, Poulain L, Green AS, Uzunov M, Kosmider O, Radford-Weiss I, Moura IC, Auberger P, Ifrah N, Bardet V, Chapuis N, Lacombe C, Mayeux P, Tamburini J, Bouscary D. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 122: 3521–3532, 2013. doi: 10.1182/blood-2013-03-493163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347: 194–198, 2015. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meng D, Yang Q, Wang H, Melick CH, Navlani R, Frank AR, Jewell JL. Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J Biol Chem 295: 2890–2899, 2020. doi: 10.1074/jbc.AC119.011578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stracka D, Jozefczuk S, Rudroff F, Sauer U, Hall MN. Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J Biol Chem 289: 25010–25020, 2014. doi: 10.1074/jbc.M114.574335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, Finan PM, Kwiatkowski DJ, Murphy LO, Manning BD. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 47: 535–546, 2012. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590, 2003. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 70.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151–162, 2002. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 71.Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11: 1457–1466, 2003. doi: 10.1016/S1097-2765(03)00220-X. [DOI] [PubMed] [Google Scholar]
- 72.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13: 1259–1268, 2003. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 73.Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, Pavletich NP. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552: 368–373, 2017. doi: 10.1038/nature25023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657, 2002. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 75.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol 4: 658–665, 2002. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 76.Demetriades C, Doumpas N, Teleman AA. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156: 786–799, 2014. doi: 10.1016/j.cell.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156: 771–785, 2014. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wallroth A, Koch PA, Marat AL, Krause E, Haucke V. Protein kinase N controls a lysosomal lipid switch to facilitate nutrient signalling via mTORC1. Nat Cell Biol 21: 1093–1101, 2019. doi: 10.1038/s41556-019-0377-3. [DOI] [PubMed] [Google Scholar]
- 79.Zhan J, Chitta RK, Harwood FC, Grosveld GC. Phosphorylation of TSC2 by PKC-delta reveals a novel signaling pathway that couples protein synthesis to mTORC1 activity. Mol Cell Biochem 456: 123–134, 2019. doi: 10.1007/s11010-019-03498-8. [DOI] [PubMed] [Google Scholar]
- 80.Ranek MJ, Kokkonen-Simon KM, Chen A, Dunkerly-Eyring BL, Vera MP, Oeing CU, Patel CH, Nakamura T, Zhu G, Bedja D, Sasaki M, Holewinski RJ, Van Eyk JE, Powell JD, Lee DI, Kass DA. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 566: 264–269, 2019. doi: 10.1038/s41586-019-0895-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121: 179–193, 2005. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 82.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA 101: 13489–13494, 2004. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tato I, Bartrons R, Ventura F, Rosa JL. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem 286: 6128–6142, 2011. doi: 10.1074/jbc.M110.166991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goberdhan DC, Wilson C, Harris AL. Amino acid sensing by mTORC1: intracellular transporters mark the spot. Cell Metab 23: 580–589, 2016. doi: 10.1016/j.cmet.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem 282: 24514–24524, 2007. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- 86.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9: 316–323, 2007. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 87.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137: 873–886, 2009. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Caron A, Briscoe DM, Richard D, Laplante M. DEPTOR at the nexus of cancer, metabolism, and immunity. Physiol Rev 98: 1765–1803, 2018. doi: 10.1152/physrev.00064.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–226, 2008. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dennis MD, Coleman CS, Berg A, Jefferson LS, Kimball SR. REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci Signal 7: ra68, 2014. doi: 10.1126/scisignal.2005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22: 239–251, 2008. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jewell JL, Fu V, Hong AW, Yu FX, Meng D, Melick CH, Wang H, Lam WM, Yuan HX, Taylor SS, Guan KL. GPCR signaling inhibits mTORC1 via PKA phosphorylation of Raptor. eLife 8: e43038, 2019. doi: 10.7554/eLife.43038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu D, Bordicchia M, Zhang C, Fang H, Wei W, Li J, Guilherme A, Guntur K, Czech M, Collins S. Activation of mTORC1 is essential for beta-adrenergic stimulation of adipose browning. J Clin Invest 126: 1704–1716, 2016. doi: 10.1172/JCI83532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, Ben-Sahra I, Manning BD. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep 21: 1331–1346, 2017. doi: 10.1016/j.celrep.2017.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Menon D, Salloum D, Bernfeld E, Gorodetsky E, Akselrod A, Frias MA, Sudderth J, Chen PH, DeBerardinis R, Foster DA. Lipid sensing by mTOR complexes via de novo synthesis of phosphatidic acid. J Biol Chem 292: 6303–6311, 2017. doi: 10.1074/jbc.M116.772988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gan X, Wang J, Su B, Wu D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 286: 10998–11002, 2011. doi: 10.1074/jbc.M110.195016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, Wang B, Blenis J, Cantley LC, Toker A, Su B, Wei W. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov 5: 1194–1209, 2015. doi: 10.1158/2159-8290.CD-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ebner M, Sinkovics B, Szczygieł M, Ribeiro DW, Yudushkin I. Localization of mTORC2 activity inside cells. J Cell Biol 216: 343–353, 2017. doi: 10.1083/jcb.201610060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell 144: 757–768, 2011. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 100.Jia R, Bonifacino JS. Lysosome positioning influences mTORC2 and AKT signaling. Mol Cell 75: 26–38.e3, 2019. doi: 10.1016/j.molcel.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moloughney JG, Kim PK, Vega-Cotto NM, Wu CC, Zhang S, Adlam M, Lynch T, Chou PC, Rabinowitz JD, Werlen G, Jacinto E. mTORC2 responds to glutamine catabolite levels to modulate the hexosamine biosynthesis enzyme GFAT1. Mol Cell 63: 811–826, 2016. doi: 10.1016/j.molcel.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kazyken D, Magnuson B, Bodur C, Acosta-Jaquez HA, Zhang D, Tong X, Barnes TM, Steinl GK, Patterson NE, Altheim CH, Sharma N, Inoki K, Cartee GD, Bridges D, Yin L, Riddle SM, Fingar DC. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci Signal 12: eaav3249, 2019. doi: 10.1126/scisignal.aav3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Byun JK, Choi YK, Kim JH, Jeong JY, Jeon HJ, Kim MK, Hwang I, Lee SY, Lee YM, Lee IK, Park KG. A positive feedback loop between Sestrin2 and mTORC2 is required for the survival of glutamine-depleted lung cancer cells. Cell Rep. 20: 586–599, 2017. doi: 10.1016/j.celrep.2017.06.066. [DOI] [PubMed] [Google Scholar]
- 104.Kowalsky AH, Namkoong S, Mettetal E, Park HW, Kazyken D, Fingar DC, Lee JH. The GATOR2-mTORC2 axis mediates Sestrin2-induced AKT Ser/Thr kinase activation. J Biol Chem 295: 1769–1780, 2020. doi: 10.1074/jbc.RA119.010857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tao R, Xiong X, Liangpunsakul S, Dong XC. Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes 64: 1211–1223, 2015. doi: 10.2337/db14-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hatano T, Morigasaki S, Tatebe H, Ikeda K, Shiozaki K. Fission yeast Ryh1 GTPase activates TOR Complex 2 in response to glucose. Cell Cycle 14: 848–856, 2015. doi: 10.1080/15384101.2014.1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Merhi A, Delrée P, Marini AM. The metabolic waste ammonium regulates mTORC2 and mTORC1 signaling. Sci Rep 7: 44602, 2017. doi: 10.1038/srep44602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J 27: 1932–1943, 2008. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 27: 1919–1931, 2008. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, Roux PP, Su B, Jacinto E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J 29: 3939–3951, 2010. doi: 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen CH, Kiyan V, Zhylkibayev AA, Kazyken D, Bulgakova O, Page KE, Bersimbaev RI, Spooner E, Sarbassov DD. Autoregulation of the mechanistic target of rapamycin (mTOR) complex 2 integrity is controlled by an ATP-dependent mechanism. J Biol Chem 288: 27019–27030, 2013. doi: 10.1074/jbc.M113.498055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu W, Hua H, Chiu YH, Li G, Zhi H, Yu Z, Ren F, Luo Y, Cui W. CD146 regulates growth factor-induced mTORC2 activity independent of the PI3K and mTORC1 pathways. Cell Rep 29: 1311–1322.e5, 2019. doi: 10.1016/j.celrep.2019.09.047. [DOI] [PubMed] [Google Scholar]
- 113.Cai W, Andres DA. mTORC2 is required for rit-mediated oxidative stress resistance. PLoS One 9: e115602, 2014. doi: 10.1371/journal.pone.0115602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Khanna A, Lotfi P, Chavan AJ, Montaño NM, Bolourani P, Weeks G, Shen Z, Briggs SP, Pots H, Van Haastert PJ, Kortholt A, Charest PG. The small GTPases Ras and Rap1 bind to and control TORC2 activity. Sci Rep 6: 25823, 2016. doi: 10.1038/srep25823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG, Khavari PA. The functional proximal proteome of oncogenic ras includes mTORC2. Mol Cell 73: 830–844.e12, 2019. doi: 10.1016/j.molcel.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lone MU, Miyan J, Asif M, Malik SA, Dubey P, Singh V, Singh K, Mitra K, Pandey D, Haq W, Amita H, Singh PK, Kiess W, Kaessner F, Garten A, Bhadauria S. Direct physical interaction of active Ras with mSIN1 regulates mTORC2 signaling. BMC Cancer 19: 1236, 2019. doi: 10.1186/s12885-019-6422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell 42: 50–61, 2011. doi: 10.1016/j.molcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Senoo H, Kamimura Y, Kimura R, Nakajima A, Sawai S, Sesaki H, Iijima M. Phosphorylated Rho-GDP directly activates mTORC2 kinase towards AKT through dimerization with Ras-GTP to regulate cell migration. Nat Cell Biol 21: 867–878, 2019. doi: 10.1038/s41556-019-0348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yao CA, Ortiz-Vega S, Sun YY, Chien CT, Chuang JH, Lin Y. Association of mSin1 with mTORC2 Ras and Akt reveals a crucial domain on mSin1 involved in Akt phosphorylation. Oncotarget 8: 63392–63404, 2017. doi: 10.18632/oncotarget.18818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu Y, Lai E, Liu J, Lin J, Yang C, Jia C, Li Y, Bai X, Li M. IKK interacts with rictor and regulates mTORC2. Cell Signal 25: 2239–2245, 2013. doi: 10.1016/j.cellsig.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 121.Dan HC, Antonia RJ, Baldwin AS. PI3K/Akt promotes feedforward mTORC2 activation through IKKalpha. Oncotarget 7: 21064–21075, 2016. doi: 10.18632/oncotarget.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci 30: 35–42, 2005. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 123.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villén J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332: 1322–1326, 2011. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66: 1500–1508, 2006. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim SJ, DeStefano MA, Oh WJ, Wu CC, Vega-Cotto NM, Finlan M, Liu D, Su B, Jacinto E. mTOR complex 2 regulates proper turnover of insulin receptor substrate-1 via the ubiquitin ligase subunit Fbw8. Mol Cell 48: 875–887, 2012. doi: 10.1016/j.molcel.2012.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chia LY, Evans BA, Mukaida S, Bengtsson T, Hutchinson DS, Sato M. Adrenoceptor regulation of the mechanistic target of rapamycin in muscle and adipose tissue. Br J Pharmacol 176: 2433–2448, 2019. doi: 10.1111/bph.14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Albert V, Svensson K, Shimobayashi M, Colombi M, Muñoz S, Jimenez V, Handschin C, Bosch F, Hall MN. mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol Med 8: 232–246, 2016. doi: 10.15252/emmm.201505610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sato M, Dehvari N, Oberg AI, Dallner OS, Sandstrom AL, Olsen JM, Csikasz RI, Summers RJ, Hutchinson DS, Bengtsson T. Improving type 2 diabetes through a distinct adrenergic signaling pathway involving mTORC2 that mediates glucose uptake in skeletal muscle. Diabetes 63: 4115–4129, 2014. doi: 10.2337/db13-1860. [DOI] [PubMed] [Google Scholar]
- 129.Sato M, Evans BA, Sandström AL, Chia LY, Mukaida S, Thai BS, Nguyen A, Lim L, Tan CY, Baltos JA, White PJ, May LT, Hutchinson DS, Summers RJ, Bengtsson T. alpha1A-adrenoceptors activate mTOR signalling and glucose uptake in cardiomyocytes. Biochem Pharmacol 148: 27–40, 2018. [P MC]doi: 10.1016/j.bcp.2017.11.016. [DOI] [PubMed] [Google Scholar]
- 130.Misra UK, Pizzo SV. Upregulation of mTORC2 activation by the selective agonist of EPAC, 8-CPT-2Me-cAMP, in prostate cancer cells: assembly of a multiprotein signaling complex. J Cell Biochem 113: 1488–1500, 2012. doi: 10.1002/jcb.24018. [DOI] [PubMed] [Google Scholar]
- 131.Mullins GR, Wang L, Raje V, Sherwood SG, Grande RC, Boroda S, Eaton JM, Blancquaert S, Roger PP, Leitinger N, Harris TE. Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proc Natl Acad Sci USA 111: 17450–17455, 2014. doi: 10.1073/pnas.1410530111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jacinto E. Amplifying mTORC2 signals through AMPK during energetic stress. Sci Signal 12: eaax5855, 2019. doi: 10.1126/scisignal.aax5855. [DOI] [PubMed] [Google Scholar]
- 133.Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature 497: 217–223, 2013. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Aylett CH, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, Maier T. Architecture of human mTOR complex 1. Science 351: 48–52, 2016. doi: 10.1126/science.aaa3870. [DOI] [PubMed] [Google Scholar]
- 135.Rogala KB, Gu X, Kedir JF, Abu-Remaileh M, Bianchi LF, Bottino AM, Dueholm R, Niehaus A, Overwijn D, Fils AP, Zhou SX, Leary D, Laqtom NN, Brignole EJ, Sabatini DM. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 366: 468–475, 2019. doi: 10.1126/science.aay0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shen K, Huang RK, Brignole EJ, Condon KJ, Valenstein ML, Chantranupong L, Bomaliyamu A, Choe A, Hong C, Yu Z, Sabatini DM. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 556: 64–69, 2018. doi: 10.1038/nature26158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhou P, Zhang N, Nussinov R, Ma B. Defining the domain arrangement of the mammalian target of rapamycin complex component Rictor protein. J Comput Biol 22: 876–886, 2015. doi: 10.1089/cmb.2015.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen X, Liu M, Tian Y, Li J, Qi Y, Zhao D, Wu Z, Huang M, Wong CCL, Wang HW, Wang J, Yang H, Xu Y. Cryo-EM structure of human mTOR complex 2. Cell Res 28: 518–528, 2018. doi: 10.1038/s41422-018-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Karuppasamy M, Kusmider B, Oliveira TM, Gaubitz C, Prouteau M, Loewith R, Schaffitzel C. Cryo-EM structure of Saccharomyces cerevisiae target of rapamycin complex 2. Nat Commun 8: 1729, 2017. doi: 10.1038/s41467-017-01862-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stuttfeld E, Aylett CH, Imseng S, Boehringer D, Scaiola A, Sauer E, Hall MN, Maier T, Ban N. Architecture of the human mTORC2 core complex. eLife 7: e33101, 2018. doi: 10.7554/eLife.33101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Scaiola A, Mangia F, Imseng S, Boehringer D, Berneiser K, Shimobayashi M, Stuttfeld E, Hall MN, Ban N, Maier T. The 3.2-Å resolution structure of human mTORC2. Sci Adv 6: eabc1251, 2020. doi: 10.1126/sciadv.abc1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cameron AJ, Linch MD, Saurin AT, Escribano C, Parker PJ. mTORC2 targets AGC kinases through Sin1-dependent recruitment. Biochem J 439: 287–297, 2011. doi: 10.1042/BJ20110678. [DOI] [PubMed] [Google Scholar]
- 143.Schroder WA, Buck M, Cloonan N, Hancock JF, Suhrbier A, Sculley T, Bushell G. Human Sin1 contains Ras-binding and pleckstrin homology domains and suppresses Ras signalling. Cell Signal 19: 1279–1289, 2007. doi: 10.1016/j.cellsig.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 144.Cuéllar J, Ludlam WG, Tensmeyer NC, Aoba T, Dhavale M, Santiago C, Bueno-Carrasco MT, Mann MJ, Plimpton RL, Makaju A, Franklin S, Willardson BM, Valpuesta JM. Structural and functional analysis of the role of the chaperonin CCT in mTOR complex assembly. Nat Commun 10: 2865, 2019. doi: 10.1038/s41467-019-10781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chou PC, Rajput S, Zhao X, Patel C, Albaciete D, Oh WJ, Daguplo HQ, Patel N, Su B, Werlen G, Jacinto E. mTORC2 is involved in the induction of RSK phosphorylation by serum or nutrient starvation. Cells 9: 1567, 2020. doi: 10.3390/cells9071567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim SG, Hoffman GR, Poulogiannis G, Buel GR, Jang YJ, Lee KW, Kim BY, Erikson RL, Cantley LC, Choo AY, Blenis J. Metabolic stress controls mTORC1 lysosomal localization and dimerization by regulating the TTT-RUVBL1/2 complex. Mol Cell 49: 172–185, 2013. doi: 10.1016/j.molcel.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Takai H, Xie Y, de Lange T, Pavletich NP. Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev 24: 2019–2030, 2010. doi: 10.1101/gad.1956410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem 280: 26089–26093, 2005. doi: 10.1074/jbc.M504045200. [DOI] [PubMed] [Google Scholar]
- 149.Navé BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 344: 427–431, 1999. doi: 10.1042/bj3440427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reynolds TH, Bodine SC, Lawrence JC Jr.. Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem 277: 17657–17662, 2002. doi: 10.1074/jbc.M201142200. [DOI] [PubMed] [Google Scholar]
- 151.Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem 275: 7416–7423, 2000. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- 152.Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res 69: 1821–1827, 2009. doi: 10.1158/0008-5472.CAN-08-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem 285: 7866–7879, 2010. doi: 10.1074/jbc.M109.096222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ekim B, Magnuson B, Acosta-Jaquez HA, Keller JA, Feener EP, Fingar DC. mTOR kinase domain phosphorylation promotes mTORC1 signaling, cell growth, and cell cycle progression. Mol Cell Biol 31: 2787–2801, 2011. doi: 10.1128/MCB.05437-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Acosta-Jaquez HA, Keller JA, Foster KG, Ekim B, Soliman GA, Feener EP, Ballif BA, Fingar DC. Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol Cell Biol 29: 4308–4324, 2009. doi: 10.1128/MCB.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cheng SW, Fryer LG, Carling D, Shepherd PR. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem 279: 15719–15722, 2004. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- 157.Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 51: 283–296, 2013. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KCS, Dubois C, Cantelmo AR, Chen R, Loroch S, Timmerman E, Caixeta V, Bloch K, Conradi LC, Treps L, Staes A, Gevaert K, Tee A, Dewerchin M, Semenkovich CF, Impens F, Schilling B, Verdin E, Swinnen JV, Meier JL, Kulkarni RA, Sickmann A, Ghesquière B, Schoonjans L, Li X, Mazzone M, Carmeliet P. Impairment of angiogenesis by fatty acid synthase inhibition involves mTOR malonylation. Cell Metab 28: 866–880.e15, 2018. doi: 10.1016/j.cmet.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321: 1499–1502, 2008. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Terenzio M, Koley S, Samra N, Rishal I, Zhao Q, Sahoo PK, Urisman A, Marvaldi L, Oses-Prieto JA, Forester C, Gomes C, Kalinski AL, Di Pizio A, Doron-Mandel E, Perry RB, Koppel I, Twiss JL, Burlingame AL, Fainzilber M. Locally translated mTOR controls axonal local translation in nerve injury. Science 359: 1416–1421, 2018. doi: 10.1126/science.aan1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Carrière A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, Roux PP. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol 18: 1269–1277, 2008. doi: 10.1016/j.cub.2008.07.078. [DOI] [PubMed] [Google Scholar]
- 162.Foster KG, Acosta-Jaquez HA, Romeo Y, Ekim B, Soliman GA, Carriere A, Roux PP, Ballif BA, Fingar DC. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J Biol Chem 285: 80–94, 2010. doi: 10.1074/jbc.M109.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gwinn DM, Asara JM, Shaw RJ. Raptor is phosphorylated by cdc2 during mitosis. PLoS One 5: e9197, 2010. doi: 10.1371/journal.pone.0009197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang L, Lawrence JC Jr, Sturgill TW, Harris TE. Mammalian target of rapamycin complex 1 (mTORC1) activity is associated with phosphorylation of raptor by mTOR. J Biol Chem 284: 14693–14697, 2009. doi: 10.1074/jbc.C109.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kwak D, Choi S, Jeong H, Jang JH, Lee Y, Jeon H, Lee MN, Noh J, Cho K, Yoo JS, Hwang D, Suh PG, Ryu SH. Osmotic stress regulates mammalian target of rapamycin (mTOR) complex 1 via c-Jun N-terminal kinase (JNK)-mediated Raptor protein phosphorylation. J Biol Chem 287: 18398–18407, 2012. doi: 10.1074/jbc.M111.326538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wu XN, Wang XK, Wu SQ, Lu J, Zheng M, Wang YH, Zhou H, Zhang H, Han J. Phosphorylation of Raptor by p38beta participates in arsenite-induced mammalian target of rapamycin complex 1 (mTORC1) activation. J Biol Chem 286: 31501–31511, 2011. doi: 10.1074/jbc.M111.233122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yuan HX, Wang Z, Yu FX, Li F, Russell RC, Jewell JL, Guan KL. NLK phosphorylates Raptor to mediate stress-induced mTORC1 inhibition. Genes Dev 29: 2362–2376, 2015. doi: 10.1101/gad.265116.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gan W, Dai X, Dai X, Xie J, Yin S, Zhu J, Wang C, Liu Y, Guo J, Wang M, Liu J, Hu J, Quinton RJ, Ganem NJ, Liu P, Asara JM, Pandolfi PP, Yang Y, He Z, Gao G, Wei W. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat Cell Biol 22: 246–256, 2020. doi: 10.1038/s41556-020-0463-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Antonia RJ, Castillo J, Herring LE, Serafin DS, Liu P, Graves LM, Baldwin AS, Hagan RS. TBK1 limits mTORC1 by promoting phosphorylation of Raptor Ser877. Sci Rep 9: 13470, 2019. doi: 10.1038/s41598-019-49707-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang T, Zhang WS, Wang ZX, Wu ZW, Du BB, Li LY, Chen YF, Yang XF, Hao XY, Guo TK. RAPTOR promotes colorectal cancer proliferation by inducing mTORC1 and upregulating ribosome assembly factor URB1. Cancer Med 9: 1529–1543, 2020. doi: 10.1002/cam4.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Earwaker P, Anderson C, Willenbrock F, Harris AL, Protheroe AS, Macaulay VM. RAPTOR up-regulation contributes to resistance of renal cancer cells to PI3K-mTOR inhibition. PLoS One 13: e0191890, 2018. doi: 10.1371/journal.pone.0191890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol 29: 5657–5670, 2009. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol 30: 908–921, 2010. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Treins C, Warne PH, Magnuson MA, Pende M, Downward J. Rictor is a novel target of p70 S6 kinase-1. Oncogene 29: 1003–1016, 2010. doi: 10.1038/onc.2009.401. [DOI] [PubMed] [Google Scholar]
- 175.Koo J, Wu X, Mao Z, Khuri FR, Sun SY. Rictor undergoes glycogen synthase kinase 3 (GSK3)-dependent, FBXW7-mediated ubiquitination and proteasomal degradation. J Biol Chem 290: 14120–14129, 2015. doi: 10.1074/jbc.M114.633057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, Wu J, Lin HK, Sarbassov dos D. ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of Rictor. Sci Signal 4: ra10, 2011. doi: 10.1126/scisignal.2001731. [DOI] [PubMed] [Google Scholar]
- 177.Glidden EJ, Gray LG, Vemuru S, Li D, Harris TE, Mayo MW. Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J Biol Chem 287: 581–588, 2012. doi: 10.1074/jbc.M111.304337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Masui K, Tanaka K, Ikegami S, Villa GR, Yang H, Yong WH, Cloughesy TF, Yamagata K, Arai N, Cavenee WK, Mischel PS. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc Natl Acad Sci USA 112: 9406–9411, 2015. doi: 10.1073/pnas.1511759112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Singh BK, Sinha RA, Zhou J, Tripathi M, Ohba K, Wang ME, Astapova I, Ghosh S, Hollenberg AN, Gauthier K, Yen PM. Hepatic FOXO1 target genes are co-regulated by thyroid hormone via RICTOR Protein deacetylation and MTORC2-AKT protein inhibition. J Biol Chem 291: 198–214, 2016. doi: 10.1074/jbc.M115.668673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Manning JR, Thapa D, Zhang M, Stoner MW, Traba J, Corey C, Shiva S, Sack MN, Scott I. Loss of GCN5L1 in cardiac cells disrupts glucose metabolism and promotes cell death via reduced Akt/mTORC2 signaling. Biochem J 476: 1713–1724, 2019. doi: 10.1042/BCJ20190302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhao D, Jiang M, Zhang X, Hou H. The role of RICTOR amplification in targeted therapy and drug resistance. Mol Med 26: 20, 2020. doi: 10.1186/s10020-020-0146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cheng H, Zou Y, Ross JS, Wang K, Liu X, Halmos B, Ali SM, Liu H, Verma A, Montagna C, Chachoua A, Goel S, Schwartz EL, Zhu C, Shan J, Yu Y, Gritsman K, Yelensky R, Lipson D, Otto G, Hawryluk M, Stephens PJ, Miller VA, Piperdi B, Perez-Soler R. RICTOR amplification defines a novel subset of patients with lung cancer who may benefit from treatment with mTORC1/2 inhibitors. Cancer Discov 5: 1262–1270, 2015. doi: 10.1158/2159-8290.CD-14-0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jiang WJ, Feng RX, Liu JT, Fan LL, Wang H, Sun GP. RICTOR expression in esophageal squamous cell carcinoma and its clinical significance. Med Oncol 34: 32, 2017. doi: 10.1007/s12032-017-0894-5. [DOI] [PubMed] [Google Scholar]
- 184.Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res 67: 11712–11720, 2007. doi: 10.1158/0008-5472.CAN-07-2223. [DOI] [PubMed] [Google Scholar]
- 185.Morrison Joly M, Hicks DJ, Jones B, Sanchez V, Estrada MV, Young C, Williams M, Rexer BN, Sarbassov dos D, Muller WJ, Brantley-Sieders D, Cook RS. Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Res 76: 4752–4764, 2016. doi: 10.1158/0008-5472.CAN-15-3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang L, Qi J, Yu J, Chen H, Zou Z, Lin X, Guo L. Overexpression of Rictor protein in colorectal cancer is correlated with tumor progression and prognosis. Oncol Lett 14: 6198–6202, 2017. doi: 10.3892/ol.2017.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Holmes B, Benavides-Serrato A, Freeman RS, Landon KA, Bashir T, Nishimura RN, Gera J. mTORC2/AKT/HSF1/HuR constitute a feed-forward loop regulating Rictor expression and tumor growth in glioblastoma. Oncogene 37: 732–743, 2018. doi: 10.1038/onc.2017.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhang M, Wu J, Zhang R, Yang J, Zhang Q, Liu B. miR-497 inhibits the carcinogenesis of hepatocellular carcinoma by targeting the Rictor/Akt signal pathway. Int J Clin Exp Pathol 12: 1992–2000, 2019. [PMC free article] [PubMed] [Google Scholar]
- 189.Guan B, Wu K, Zeng J, Xu S, Mu L, Gao Y, Wang K, Ma Z, Tian J, Shi Q, Guo P, Wang X, He D, Du Y. Tumor-suppressive microRNA-218 inhibits tumor angiogenesis via targeting the mTOR component RICTOR in prostate cancer. Oncotarget 8: 8162–8172, 2017. doi: 10.18632/oncotarget.14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cui Y, Zhao J, Yi L, Jiang Y. microRNA-153 targets mTORC2 component Rictor to inhibit glioma cells. PLoS One 11: e0156915, 2016. doi: 10.1371/journal.pone.0156915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Micevic G, Muthusamy V, Damsky W, Theodosakis N, Liu X, Meeth K, Wingrove E, Santhanakrishnan M, Bosenberg M. DNMT3b modulates melanoma growth by controlling levels of mTORC2 component RICTOR. Cell Rep 14: 2180–2192, 2016. doi: 10.1016/j.celrep.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chen HH, Huang WT, Yang LW, Lin CW. The PTEN-AKT-mTOR/RICTOR pathway in nasal natural killer cell lymphoma is activated by miR-494-3p via PTEN but inhibited by miR-142-3p via RICTOR. Am J Pathol 185: 1487–1499, 2015. doi: 10.1016/j.ajpath.2015.01.025. [DOI] [PubMed] [Google Scholar]
- 193.Li CJ, Cheng P, Liang MK, Chen YS, Lu Q, Wang JY, Xia ZY, Zhou HD, Cao X, Xie H, Liao EY, Luo XH. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J Clin Invest 125: 1509–1522, 2015. doi: 10.1172/JCI77716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, Liu D, Wan L, Zhai B, Yu Y, Yuan M, Kim BM, Shaik S, Menon S, Gygi SP, Lee TH, Asara JM, Manning BD, Blenis J, Su B, Wei W. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol 15: 1340–1350, 2013. doi: 10.1038/ncb2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yang G, Murashige DS, Humphrey SJ, James DE. A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep 12: 937–943, 2015. doi: 10.1016/j.celrep.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 196.Gleason CE, Oses-Prieto JA, Li KH, Saha B, Situ G, Burlingame AL, Pearce D. Phosphorylation at distinct subcellular locations underlies specificity in mTORC2-mediated activation of SGK1 and Akt. J Cell Sci 132: jcs224931, 2019. doi: 10.1242/jcs.224931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang Q, Zhu J, Wang YW, Dai Y, Wang YL, Wang C, Liu J, Baker A, Colburn NH, Yang HS. Tumor suppressor Pdcd4 attenuates Sin1 translation to inhibit invasion in colon carcinoma. Oncogene 36: 6225–6234, 2017. doi: 10.1038/onc.2017.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hwang Y, Kim LC, Song W, Edwards DN, Cook RS, Chen J. Disruption of the scaffolding function of mLST8 selectively inhibits mTORC2 assembly and function and suppresses mTORC2-dependent tumor growth in vivo. Cancer Res 79: 3178–3184, 2019. doi: 10.1158/0008-5472.CAN-18-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang B, Jie Z, Joo D, Ordureau A, Liu P, Gan W, Guo J, Zhang J, North BJ, Dai X, Cheng X, Bian X, Zhang L, Harper JW, Sun SC, Wei W. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 545: 365–369, 2017. doi: 10.1038/nature22344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yin Y, Hua H, Li M, Liu S, Kong Q, Shao T, Wang J, Luo Y, Wang Q, Luo T, Jiang Y. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res 26: 46–65, 2016. doi: 10.1038/cr.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332: 1317–1322, 2011. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339: 1320–1323, 2013. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- 203.Jacinto E, Lorberg A. TOR regulation of AGC kinases in yeast and mammals. Biochem J 410: 19–37, 2008. doi: 10.1042/BJ20071518. [DOI] [PubMed] [Google Scholar]
- 204.Schalm SS, Blenis J. Identification of a conserved motif required for mTOR signaling. Curr Biol 12: 632–639, 2002. doi: 10.1016/s0960-9822(02)00762-5. [DOI] [PubMed] [Google Scholar]
- 205.Coffman K, Yang B, Lu J, Tetlow AL, Pelliccio E, Lu S, Guo DC, Tang C, Dong MQ, Tamanoi F. Characterization of the Raptor/4E-BP1 interaction by chemical cross-linking coupled with mass spectrometry analysis. J Biol Chem 289: 4723–4734, 2014. doi: 10.1074/jbc.M113.482067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem 282: 20534–20543, 2007. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 207.Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, Kikkawa U, Yonezawa K. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem 282: 20329–20339, 2007. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wang L, Harris TE, Roth RA, Lawrence JC Jr.. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem 282: 20036–20044, 2007. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 209.Ling NX, Kaczmarek A, Hoque A, Davie E, Ngoei KR, Morrison KR, Smiles WJ, Forte GM, Wang T, Lie S, Dite TA, Langendorf CG, Scott JW, Oakhill JS, Petersen J. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat Metab 2: 41–49, 2020. doi: 10.1038/s42255-019-0157-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141, 2011. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Koren I, Reem E, Kimchi A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr Biol 20: 1093–1098, 2010. doi: 10.1016/j.cub.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 212.Lu M, Wang J, Ives HE, Pearce D. mSIN1 protein mediates SGK1 protein interaction with mTORC2 protein complex and is required for selective activation of the epithelial sodium channel. J Biol Chem 286: 30647–30654, 2011. doi: 10.1074/jbc.M111.257592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yang CS, Melhuish TA, Spencer A, Ni L, Hao Y, Jividen K, Harris TE, Snow C, Frierson HF Jr, Wotton D, Paschal BM. The protein kinase C super-family member PKN is regulated by mTOR and influences differentiation during prostate cancer progression. Prostate 77: 1452–1467, 2017. doi: 10.1002/pros.23400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20, 2008. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 215.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033, 2009. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hopkins BD, Goncalves MD, Cantley LC. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol 16: 276–283, 2020. doi: 10.1038/s41574-020-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41: 211–218, 2016. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 123: 3664–3671, 2013. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 23: 9361–9374, 2003. doi: 10.1128/mcb.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 277: 23111–23115, 2002. doi: 10.1074/jbc.M202487200. [DOI] [PubMed] [Google Scholar]
- 221.Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 22: 7004–7014, 2002. doi: 10.1128/mcb.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60: 1541–1545, 2000. [PubMed] [Google Scholar]
- 223.Dodd KM, Yang J, Shen MH, Sampson JR, Tee AR. mTORC1 drives HIF-1alpha and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 34: 2239–2250, 2015. doi: 10.1038/onc.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.He L, Gomes AP, Wang X, Yoon SO, Lee G, Nagiec MJ, Cho S, Chavez A, Islam T, Yu Y, Asara JM, Kim BY, Blenis J. mTORC1 promotes metabolic reprogramming by the suppression of GSK3-dependent Foxk1 phosphorylation. Mol Cell 70: 949–960.e4, 2018. doi: 10.1016/j.molcel.2018.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG Jr.. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4: 147–158, 2003. doi: 10.1016/S1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 226.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 21: 3995–4004, 2001. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med 12: 122–127, 2006. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 228.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39: 171–183, 2010. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tandon P, Gallo CA, Khatri S, Barger JF, Yepiskoposyan H, Plas DR. Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc Natl Acad Sci USA 108: 2361–2365, 2011. doi: 10.1073/pnas.1013629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12: 108–113, 2007. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene 17: 769–780, 1998. doi: 10.1038/sj.onc.1201990. [DOI] [PubMed] [Google Scholar]
- 232.Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, Tamachi A, Tu WB, Penn LZ. MYC deregulation in primary human cancers. Genes (Basel) 8: 151, 2017. doi: 10.3390/genes8060151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Goetzman ES, Prochownik EV. The role for Myc in coordinating glycolysis, oxidative phosphorylation, glutaminolysis, and fatty acid metabolism in normal and neoplastic tissues. Front Endocrinol (Lausanne) 9: 129, 2018. doi: 10.3389/fendo.2018.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci USA 110: 11988–11993, 2013. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, Fendt SM, Roberts TM, Blenis J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol 24: 2274–2280, 2014. doi: 10.1016/j.cub.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Grolleau A, Bowman J, Pradet-Balade B, Puravs E, Hanash S, Garcia-Sanz JA, Beretta L. Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J Biol Chem 277: 22175–22184, 2002. doi: 10.1074/jbc.M202014200. [DOI] [PubMed] [Google Scholar]
- 237.Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, et al. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell 31: 820–832.e3, 2017. doi: 10.1016/j.ccell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem 289: 7884–7896, 2014. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol 180: 4476–4486, 2008. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, Inoki K, Guan KL, Brosius FC 3rd.. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol 295: C836–C843, 2008. doi: 10.1152/ajpcell.00554.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208: 1367–1376, 2011. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, Kater AP, Weinberg JB, Rathmell JC. Suppression of Glut1 and glucose metabolism by decreased Akt/mTORC1 signaling drives t cell impairment in B cell leukemia. J Immunol 197: 2532–2540, 2016. doi: 10.4049/jimmunol.1502464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jiang X, Kenerson H, Aicher L, Miyaoka R, Eary J, Bissler J, Yeung RS. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am J Pathol 172: 1748–1756, 2008. doi: 10.2353/ajpath.2008.070958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, Koochekpour S, Saleem M, Huang H, Lu J, Deng Y. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep 8: 1461–1474, 2014. doi: 10.1016/j.celrep.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C, Guha A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med 208: 313–326, 2011. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Feng Y, Wu L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem Biophys Res Commun 483: 897–903, 2017. doi: 10.1016/j.bbrc.2017.01.031. [DOI] [PubMed] [Google Scholar]
- 247.Wang C, Jiang J, Ji J, Cai Q, Chen X, Yu Y, Zhu Z, Zhang J. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the malignant development of gastric cancer. Sci Rep 7: 2886, 2017. doi: 10.1038/s41598-017-03031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang MR, Lu Y, You H, Kwiatkowski D, Zhang H. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA 108: 4129–4134, 2011. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145: 732–744, 2011. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.He CL, Bian YY, Xue Y, Liu ZX, Zhou KQ, Yao CF, Lin Y, Zou HF, Luo FX, Qu YY, Zhao JY, Ye ML, Zhao SM, Xu W. Pyruvate kinase M2 activates mTORC1 by phosphorylating AKT1S1. Sci Rep 6: 21524, 2016. doi: 10.1038/srep21524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tran Q, Lee H, Park J, Kim SH, Park J. Targeting cancer metabolism—revisiting the Warburg effects. Toxicol Res 32: 177–193, 2016. doi: 10.5487/TR.2016.32.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.van der Poel HG, Hanrahan C, Zhong H, Simons JW. Rapamycin induces Smad activity in prostate cancer cell lines. Urol Res 30: 380–386, 2003. doi: 10.1007/s00240-002-0282-1. [DOI] [PubMed] [Google Scholar]
- 253.Zha X, Wang F, Wang Y, He S, Jing Y, Wu X, Zhang H. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res 71: 13–18, 2011. doi: 10.1158/0008-5472.CAN-10-1668. [DOI] [PubMed] [Google Scholar]
- 254.Guridi M, Kupr B, Romanino K, Lin S, Falcetta D, Tintignac L, Rüegg MA. Alterations to mTORC1 signaling in the skeletal muscle differentially affect whole-body metabolism. Skelet Muscle 6: 13, 2016. doi: 10.1186/s13395-016-0084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Iwata TN, Ramírez JA, Tsang M, Park H, Margineantu DH, Hockenbery DM, Iritani BM. Conditional disruption of Raptor reveals an essential role for mTORC1 in B cell development, survival, and metabolism. J Immunol 197: 2250–2260, 2016. doi: 10.4049/jimmunol.1600492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K, Choi AM. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep 12: 102–115, 2015. doi: 10.1016/j.celrep.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 257.Dutchak PA, Estill-Terpack SJ, Plec AA, Zhao X, Yang C, Chen J, Ko B, Deberardinis RJ, Yu Y, Tu BP. Loss of a negative regulator of mTORC1 induces aerobic glycolysis and altered fiber composition in skeletal muscle. Cell Rep 23: 1907–1914, 2018. doi: 10.1016/j.celrep.2018.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Pal R, Xiong Y, Sardiello M. Abnormal glycogen storage in tuberous sclerosis complex caused by impairment of mTORC1-dependent and -independent signaling pathways. Proc Natl Acad Sci USA 116: 2977–2986, 2019. doi: 10.1073/pnas.1812943116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Liko D, Rzepiela A, Vukojevic V, Zavolan M, Hall MN. Loss of TSC complex enhances gluconeogenesis via upregulation of Dlk1-Dio3 locus miRNAs. Proc Natl Acad Sci USA 117: 1524–1532, 2020. doi: 10.1073/pnas.1918931117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 15: 1406–1418, 2001. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem 272: 17269–17275, 1997. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- 262.Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem 274: 20281–20286, 1999. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- 263.Robey RB, Hay N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol 19: 25–31, 2009. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: 3892–3899, 2004. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 265.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med 10: 594–601, 2004. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 266.Weng ML, Chen WK, Chen XY, Lu H, Sun ZR, Yu Q, Sun PF, Xu YJ, Zhu MM, Jiang N, Zhang J, Zhang JP, Song YL, Ma D, Zhang XP, Miao CH. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1alpha pathway suppression. Nat Commun 11: 1869, 2020. doi: 10.1038/s41467-020-15795-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Li Y, He ZC, Liu Q, Zhou K, Shi Y, Yao XH, Zhang X, Kung HF, Ping YF, Bian XW. Large intergenic non-coding RNA-RoR inhibits aerobic glycolysis of glioblastoma cells via Akt pathway. J Cancer 9: 880–889, 2018. doi: 10.7150/jca.20869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, Yong WH, Cloughesy TF, Mellinghoff IK, Cavenee WK, Shaw RJ, Mischel PS. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab 18: 726–739, 2013. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Vadla R, Haldar D. Mammalian target of rapamycin complex 2 (mTORC2) controls glycolytic gene expression by regulating Histone H3 Lysine 56 acetylation. Cell Cycle 17: 110–123, 2018. doi: 10.1080/15384101.2017.1404207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ouyang X, Han Y, Qu G, Li M, Wu N, Liu H, Arojo O, Sun H, Liu X, Liu D, Chen L, Zou Q, Su B. Metabolic regulation of T cell development by Sin1-mTORC2 is mediated by pyruvate kinase M2. J Mol Cell Biol 11: 93–106, 2019. doi: 10.1093/jmcb/mjy065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Li M, Lazorchak AS, Ouyang X, Zhang H, Liu H, Arojo OA, Yan L, Jin J, Han Y, Qu G, Fu Y, Xu X, Liu X, Zhang W, Yang Z, Ruan C, Wang Q, Liu D, Huang C, Lu L, Jiang S, Li F, Su B. Sin1/mTORC2 regulate B cell growth and metabolism by activating mTORC1 and Myc. Cell Mol Immunol 16: 757–769, 2019. doi: 10.1038/s41423-018-0185-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Rüegg MA, Hall MN. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15: 725–738, 2012. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 273.Kleinert M, Parker BL, Fritzen AM, Knudsen JR, Jensen TE, Kjøbsted R, Sylow L, Ruegg M, James DE, Richter EA. Mammalian target of rapamycin complex 2 regulates muscle glucose uptake during exercise in mice. J Physiol 595: 4845–4855, 2017. doi: 10.1113/JP274203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab 17: 745–755, 2013. doi: 10.1016/j.cmet.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Kocalis HE, Hagan SL, George L, Turney MK, Siuta MA, Laryea GN, Morris LC, Muglia LJ, Printz RL, Stanwood GD, Niswender KD. Rictor/mTORC2 facilitates central regulation of energy and glucose homeostasis. Mol Metab 3: 394–407, 2014. doi: 10.1016/j.molmet.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Schieke SM, Phillips D, McCoy JP Jr, Aponte AM, Shen RF, Balaban RS, Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem 281: 27643–27652, 2006. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 277.Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Rüegg MA. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 8: 411–424, 2008. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 278.Morita M, Gravel SP, Chénard V, Sikström K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St-Pierre J, Topisirovic I, Sonenberg N. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 18: 698–711, 2013. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 279.Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, Guertin DA, Lamb RF, Chi H. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 39: 1043–1056, 2013. doi: 10.1016/j.immuni.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Rosario FJ, Gupta MB, Myatt L, Powell TL, Glenn JP, Cox L, Jansson T. Mechanistic target of rapamycin complex 1 promotes the expression of genes encoding electron transport chain proteins and stimulates oxidative phosphorylation in primary human trophoblast cells by regulating mitochondrial biogenesis. Sci Rep 9: 246, 2019. doi: 10.1038/s41598-018-36265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450: 736–740, 2007. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 282.Ramanathan A, Schreiber SL. Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci USA 106: 22229–22232, 2009. doi: 10.1073/pnas.0912074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci USA 99: 4319–4324, 2002. doi: 10.1073/pnas.261702698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Arif T, Paul A, Krelin Y, Shteinfer-Kuzmine A, Shoshan-Barmatz V. Mitochondrial VDAC1 silencing leads to metabolic rewiring and the reprogramming of tumour cells into advanced differentiated states. Cancers (Basel) 10: 499, 2018. doi: 10.3390/cancers10120499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Drusian L, Nigro EA, Mannella V, Pagliarini R, Pema M, Costa ASH, Benigni F, Larcher A, Chiaravalli M, Gaude E, Montorsi F, Capitanio U, Musco G, Frezza C, Boletta A. mTORC1 upregulation leads to accumulation of the oncometabolite fumarate in a mouse model of renal cell carcinoma. Cell Rep 24: 1093–1104.e6, 2018. doi: 10.1016/j.celrep.2018.06.106. [DOI] [PubMed] [Google Scholar]
- 286.Carbonneau M, M Gagné L, Lalonde ME, Germain MA, Motorina A, Guiot MC, Secco B, Vincent EE, Tumber A, Hulea L, Bergeman J, Oppermann U, Jones RG, Laplante M, Topisirovic I, Petrecca K, Huot MÉ, Mallette FA. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat Commun 7: 12700, 2016. doi: 10.1038/ncomms12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gandin V, Masvidal L, Hulea L, Gravel SP, Cargnello M, McLaughlan S, Cai Y, Balanathan P, Morita M, Rajakumar A, Furic L, Pollak M, Porco JA Jr, St-Pierre J, Pelletier J, Larsson O, Topisirovic I. nanoCAGE reveals 5' UTR features that define specific modes of translation of functionally related MTOR-sensitive mRNAs. Genome Res 26: 636–648, 2016. doi: 10.1101/gr.197566.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Larsson O, Morita M, Topisirovic I, Alain T, Blouin MJ, Pollak M, Sonenberg N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc Natl Acad Sci USA 109: 8977–8982, 2012. doi: 10.1073/pnas.1201689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Soliman GA, Steenson SM, Etekpo AH. Effects of metformin and a mammalian target of rapamycin (mTOR) ATP-competitive inhibitor on targeted metabolomics in pancreatic cancer cell line. Metabolomics (Los Angel). 6: 183, 2016. doi: 10.4172/2153-0769.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Cerniglia GJ, Dey S, Gallagher-Colombo SM, Daurio NA, Tuttle S, Busch TM, Lin A, Sun R, Esipova TV, Vinogradov SA, Denko N, Koumenis C, Maity A. The PI3K/Akt pathway regulates oxygen metabolism via pyruvate dehydrogenase (PDH)-E1alpha phosphorylation. Mol Cancer Ther 14: 1928–1938, 2015. doi: 10.1158/1535-7163.MCT-14-0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Li C, Li Y, He L, Agarwal AR, Zeng N, Cadenas E, Stiles BL. PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free Radic Biol Med 60: 29–40, 2013. doi: 10.1016/j.freeradbiomed.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Colombi M, Molle KD, Benjamin D, Rattenbacher-Kiser K, Schaefer C, Betz C, Thiemeyer A, Regenass U, Hall MN, Moroni C. Genome-wide shRNA screen reveals increased mitochondrial dependence upon mTORC2 addiction. Oncogene 30: 1551–1565, 2011. doi: 10.1038/onc.2010.539. [DOI] [PubMed] [Google Scholar]
- 293.Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 110: 12526–12534, 2013. doi: 10.1073/pnas.1302455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol 23: 362–369, 2012. doi: 10.1016/j.semcdb.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 295.Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR, Vander Heiden MG. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell 36: 540–549, 2016. doi: 10.1016/j.devcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765, 2009. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105: 18782–18787, 2008. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35: 871–882, 2011. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, Stephanopoulos G, Yu J, Blenis J. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153: 840–854, 2013. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126: 941–954, 2006. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 301.Shaw E, Talwadekar M, Rashida Z, Mohan N, Acharya A, Khatri S, Laxman S, Kolthur-Seetharam U. Anabolic SIRT4 exerts retrograde control over TORC1 signaling by glutamine sparing in the mitochondria. Mol Cell Biol 40: e00212, 2020. doi: 10.1128/MCB.00212-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Guo L, Zhou B, Liu Z, Xu Y, Lu H, Xia M, Guo E, Shan W, Chen G, Wang C. Blockage of glutaminolysis enhances the sensitivity of ovarian cancer cells to PI3K/mTOR inhibition involvement of STAT3 signaling. Tumour Biol 37: 11007–11015, 2016. doi: 10.1007/s13277-016-4984-3. [DOI] [PubMed] [Google Scholar]
- 303.Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, Graeber TG, Parlati F, Demo S, Li R, Walser TC, Gricowski M, Shuman R, Ibarra J, Fridman D, Phelps ME, Badran K, St John M, Bernthal NM, Federman N, Yanagawa J, Dubinett SM, Sadeghi S, Christofk HR, Shackelford DB. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 33: 905–921.e5, 2018. doi: 10.1016/j.ccell.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Dennis MD, Jefferson LS, Kimball SR. Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4 proteins in the regulation of protein synthesis. J Biol Chem 287: 42890–42899, 2012. doi: 10.1074/jbc.M112.404822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Martineau Y, Wang X, Alain T, Petroulakis E, Shahbazian D, Fabre B, Bousquet-Dubouch MP, Monsarrat B, Pyronnet S, Sonenberg N. Control of Paip1-eukaryotic translation initiation factor 3 interaction by amino acids through S6 kinase. Mol Cell Biol 34: 1046–1053, 2014. doi: 10.1128/MCB.01079-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Abetov DA, Kiyan VS, Zhylkibayev AA, Sarbassova DA, Alybayev SD, Spooner E, Song MS, Bersimbaev RI, Sarbassov DD. Formation of mammalian pre-ribosomes proceeds from intermediate to composed state during ribosome maturation. J Biol Chem 294: 10746–10757, 2019. doi: 10.1074/jbc.AC119.008378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Gentilella A, Kozma SC, Thomas G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim Biophys Acta 1849: 812–820, 2015. doi: 10.1016/j.bbagrm.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.He J, Yang Y, Zhang J, Chen J, Wei X, He J, Luo L. Ribosome biogenesis protein Urb1 acts downstream of mTOR complex 1 to modulate digestive organ development in zebrafish. J Genet Genomics 44: 567–576, 2017. doi: 10.1016/j.jgg.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 309.Hannan KM, Brandenburger Y, Jenkins A, Sharkey K, Cavanaugh A, Rothblum L, Moss T, Poortinga G, McArthur GA, Pearson RB, Hannan RD. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol 23: 8862–8877, 2003. doi: 10.1128/MCB.23.23.8862-8877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Mayer C, Zhao J, Yuan X, Grummt I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev 18: 423–434, 2004. doi: 10.1101/gad.285504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, Follettie M, Yu K. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J Biol Chem 285: 15380–15392, 2010. doi: 10.1074/jbc.M109.071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Jia JJ, Lahr RM, Solgaard MT, Moraes BJ, Pointet R, Yang AD, Celucci G, Graber TE, Hoang HD, Niklaus MR, Pena IA, Hollensen AK, Smith EM, Chaker-Margot M, Anton L, Dajadian C, Livingstone M, Hearnden J, Wang XD, Yu Y, Maier T, Damgaard CK, Berman AJ, Alain T, Fonseca BD. mTORC1 promotes TOP mRNA translation through site-specific phosphorylation of LARP1. Nucleic Acids Res 49: 3461–3489, 2021. doi: 10.1093/nar/gkaa1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Zhang Y, Stefanovic B. Akt mediated phosphorylation of LARP6; critical step in biosynthesis of type I collagen. Sci Rep 6: 22597, 2016. doi: 10.1038/srep22597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Prakash V, Carson BB, Feenstra JM, Dass RA, Sekyrova P, Hoshino A, Petersen J, Guo Y, Parks MM, Kurylo CM, Batchelder JE, Haller K, Hashimoto A, Rundqivst H, Condeelis JS, Allis CD, Drygin D, Nieto MA, Andäng M, Percipalle P, Bergh J, Adameyko I, Farrants AÖ, Hartman J, Lyden D, Pietras K, Blanchard SC, Vincent CT. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat Commun 10: 2110, 2019. doi: 10.1038/s41467-019-10100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, Huser N, Proffitt C, Bliesath J, Haddach M, Schwaebe MK, Ryckman DM, Rice WG, Schmitt C, Lowe SW, Johnstone RW, Pearson RB, McArthur GA, Hannan RD. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 22: 51–65, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Devlin JR, Hannan KM, Hein N, Cullinane C, Kusnadi E, Ng PY, George AJ, Shortt J, Bywater MJ, Poortinga G, Sanij E, Kang J, Drygin D, O’Brien S, Johnstone RW, McArthur GA, Hannan RD, Pearson RB. Combination therapy targeting ribosome biogenesis and mRNA translation synergistically extends survival in MYC-driven lymphoma. Cancer Discov 6: 59–70, 2016. doi: 10.1158/2159-8290.CD-14-0673. [DOI] [PubMed] [Google Scholar]
- 317.Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol 591: 609–625, 2013. doi: 10.1113/jphysiol.2012.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Rosario FJ, Dimasuay KG, Kanai Y, Powell TL, Jansson T. Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4-2. Clin Sci (Lond) 130: 499–512, 2016. doi: 10.1042/CS20150554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Park Y, Reyna-Neyra A, Philippe L, Thoreen CC. mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep 19: 1083–1090, 2017. doi: 10.1016/j.celrep.2017.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, Ikegami S, Masui K, Gini B, Yang H, Gahman TC, Shiau AK, Cloughesy TF, Christofk HR, Zhou H, Guan KL, Mischel PS. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell 67: 128–138.e7, 2017. doi: 10.1016/j.molcel.2017.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Toda K, Kawada K, Iwamoto M, Inamoto S, Sasazuki T, Shirasawa S, Hasegawa S, Sakai Y. Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia 18: 654–665, 2016. doi: 10.1016/j.neo.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, Sweet-Cordero EA. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell 33: 91–107.e6, 2018. doi: 10.1016/j.ccell.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Kuo MT, Savaraj N, Feun LG. Targeted cellular metabolism for cancer chemotherapy with recombinant arginine-degrading enzymes. Oncotarget 1: 246–251, 2010. doi: 10.18632/oncotarget.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Long Y, Tsai WB, Wangpaichitr M, Tsukamoto T, Savaraj N, Feun LG, Kuo MT. Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Mol Cancer Ther 12: 2581–2590, 2013. doi: 10.1158/1535-7163.MCT-13-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Selvarajah B, Azuelos I, Platé M, Guillotin D, Forty EJ, Contento G, Woodcock HV, Redding M, Taylor A, Brunori G, Durrenberger PF, Ronzoni R, Blanchard AD, Mercer PF, Anastasiou D, Chambers RC. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-beta1-induced collagen biosynthesis. Sci Signal 12: eaav3048, 2019. doi: 10.1126/scisignal.aav3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Sivanand S, Vander Heiden MG. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell 37: 147–156, 2020. doi: 10.1016/j.ccell.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, Kaeberlein M. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342: 1524–1528, 2013. doi: 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 64: 362–369, 2012. doi: 10.1002/iub.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, Manning BD. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 14: 21–32, 2011. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Buj R, Chen CW, Dahl ES, Leon KE, Kuskovsky R, Maglakelidze N, Navaratnarajah M, Zhang G, Doan MT, Jiang H, Zaleski M, Kutzler L, Lacko H, Lu Y, Mills GB, Gowda R, Robertson GP, Warrick JI, Herlyn M, Imamura Y, Kimball SR, DeGraff DJ, Snyder NW, Aird KM. Suppression of p16 induces mTORC1-mediated nucleotide metabolic reprogramming. Cell Rep 28: 1971–1980.e8, 2019. doi: 10.1016/j.celrep.2019.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339: 1323–1328, 2013. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Nakashima A, Kawanishi I, Eguchi S, Yu EH, Eguchi S, Oshiro N, Yoshino K, Kikkawa U, Yonezawa K. Association of CAD, a multifunctional protein involved in pyrimidine synthesis, with mLST8, a component of the mTOR complexes. J Biomed Sci 20: 24, 2013. doi: 10.1186/1423-0127-20-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D, Korman S, Itzkovitz S, Dimmock D, Ulitsky I, Nagamani SC, Ruppin E, Erez A. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527: 379–383, 2015. doi: 10.1038/nature15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Ben-Sahra I, Hoxhaj G, Ricoult SJ, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351: 728–733, 2016. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Evert M, Calvisi DF, Evert K, De Murtas V, Gasparetti G, Mattu S, Destefanis G, Ladu S, Zimmermann A, Delogu S, Thiel S, Thiele A, Ribback S, Dombrowski F. V-AKT murine thymoma viral oncogene homolog/mammalian target of rapamycin activation induces a module of metabolic changes contributing to growth in insulin-induced hepatocarcinogenesis. Hepatology 55: 1473–1484, 2012. doi: 10.1002/hep.25600. [DOI] [PubMed] [Google Scholar]
- 336.Cheng J, Huang Y, Zhang X, Yu Y, Wu S, Jiao J, Tran L, Zhang W, Liu R, Zhang L, Wang M, Wang M, Yan W, Wu Y, Chi F, Jiang P, Zhang X, Wu H. TRIM21 and PHLDA3 negatively regulate the crosstalk between the PI3K/AKT pathway and PPP metabolism. Nat Commun 11: 1880, 2020. doi: 10.1038/s41467-020-15819-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Saha A, Connelly S, Jiang J, Zhuang S, Amador DT, Phan T, Pilz RB, Boss GR. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol Cell 55: 264–276, 2014. doi: 10.1016/j.molcel.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Wang W, Fridman A, Blackledge W, Connelly S, Wilson IA, Pilz RB, Boss GR. The phosphatidylinositol 3-kinase/akt cassette regulates purine nucleotide synthesis. J Biol Chem 284: 3521–3528, 2009. doi: 10.1074/jbc.M806707200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Kliegman JI, Fiedler D, Ryan CJ, Xu YF, Su XY, Thomas D, Caccese MC, Cheng A, Shales M, Rabinowitz JD, Krogan NJ, Shokat KM. Chemical genetics of rapamycin-insensitive TORC2 in S. cerevisiae. Cell Rep 5: 1725–1736, 2013. doi: 10.1016/j.celrep.2013.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Dennis JW, Nabi IR, Demetriou M. Metabolism, cell surface organization, and disease. Cell 139: 1229–1241, 2009. doi: 10.1016/j.cell.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Tran DH, May HI, Li Q, Luo X, Huang J, Zhang G, Niewold E, Wang X, Gillette TG, Deng Y, Wang ZV. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat Commun 11: 1771, 2020. doi: 10.1038/s41467-020-15640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Jones DR, Keune WJ, Anderson KE, Stephens LR, Hawkins PT, Divecha N. The hexosamine biosynthesis pathway and O-GlcNAcylation maintain insulin-stimulated PI3K-PKB phosphorylation and tumour cell growth after short-term glucose deprivation. FEBS J 281: 3591–3608, 2014. doi: 10.1111/febs.12879. [DOI] [PubMed] [Google Scholar]
- 343.Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 24: 2784–2799, 2010. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Park S, Pak J, Jang I, Cho JW. Inhibition of mTOR affects protein stability of OGT. Biochem Biophys Res Commun 453: 208–212, 2014. doi: 10.1016/j.bbrc.2014.05.047. [DOI] [PubMed] [Google Scholar]
- 345.Sodi VL, Khaku S, Krutilina R, Schwab LP, Vocadlo DJ, Seagroves TN, Reginato MJ. mTOR/MYC axis regulates O-GlcNAc transferase expression and O-GlcNAcylation in breast cancer. Mol Cancer Res 13: 923–933, 2015. doi: 10.1158/1541-7786.MCR-14-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Very N, Steenackers A, Dubuquoy C, Vermuse J, Dubuquoy L, Lefebvre T, El Yazidi-Belkoura I. Cross regulation between mTOR signaling and O-GlcNAcylation. J Bioenerg Biomembr 50: 213–222, 2018. doi: 10.1007/s10863-018-9747-y. [DOI] [PubMed] [Google Scholar]
- 347.Moloughney JG, Vega-Cotto NM, Liu S, Patel C, Kim PK, Wu CC, Albaciete D, Magaway C, Chang A, Rajput S, Su X, Werlen G, Jacinto E. mTORC2 modulates the amplitude and duration of GFAT1 Ser-243 phosphorylation to maintain flux through the hexosamine pathway during starvation. J Biol Chem 293: 16464–16478, 2018. doi: 10.1074/jbc.RA118.003991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Beckner ME, Fellows-Mayle W, Zhang Z, Agostino NR, Kant JA, Day BW, Pollack IF. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int J Cancer 126: 2282–2295, 2010. doi: 10.1002/ijc.24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res 68: 8547–8554, 2008. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- 350.Wang Y, Wang Y, Shen L, Pang Y, Qiao Z, Liu P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol Rep 27: 1156–1162, 2012. doi: 10.3892/or.2012.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr.. Cellular fatty acid metabolism and cancer. Cell Metab 18: 153–161, 2013. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7: 763–777, 2007. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 353.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8: 224–236, 2008. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Chakrabarti P, English T, Shi J, Smas CM, Kandror KV. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes 59: 775–781, 2010. doi: 10.2337/db09-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR, Kaestner KH, Sabatini DM, Birnbaum MJ. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab 14: 516–527, 2011. doi: 10.1016/j.cmet.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Brown NF, Stefanovic-Racic M, Sipula IJ, Perdomo G. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism 56: 1500–1507, 2007. doi: 10.1016/j.metabol.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 357.Mauvoisin D, Rocque G, Arfa O, Radenne A, Boissier P, Mounier C. Role of the PI3-kinase/mTor pathway in the regulation of the stearoyl CoA desaturase (SCD1) gene expression by insulin in liver. J Cell Commun Signal 1: 113–125, 2007. doi: 10.1007/s12079-007-0011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA 107: 3441–3446, 2010. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, Goldstein JL, Brown MS. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci USA 109: 16184–16189, 2012. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Dong Q, Majumdar G, O’Meally RN, Cole RN, Elam MB, Raghow R. Insulin-induced de novo lipid synthesis occurs mainly via mTOR-dependent regulation of proteostasis of SREBP-1c. Mol Cell Biochem 463: 13–31, 2020. doi: 10.1007/s11010-019-03625-5. [DOI] [PubMed] [Google Scholar]
- 361.Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, Deng H, Wang Y. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 524: 243–246, 2015. doi: 10.1038/nature14557. [DOI] [PubMed] [Google Scholar]
- 362.Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, Marbois BN, Komisopoulou E, Wilson EB, Osborne TF, Graeber TG, Reue K, Brooks DG, Bensinger SJ. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 14: 489–499, 2013. doi: 10.1038/ni.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146: 408–420, 2011. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Viscarra JA, Wang Y, Nguyen HP, Choi YG, Sul HS. Histone demethylase JMJD1C is phosphorylated by mTOR to activate de novo lipogenesis. Nat Commun 11: 796, 2020. doi: 10.1038/s41467-020-14617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, Yu Y, Liu X, He L, Cavaliere PM, Chavez A, Zhang E, Isik M, Couvillon A, Dephoure NE, Blackwell TK, Yu JJ, Rabinowitz JD, Cantley LC, Blenis J. Post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell 171: 1545–1558.e18, 2017. doi: 10.1016/j.cell.2017.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Zhang HH, Huang J, Düvel K, Boback B, Wu S, Squillace RM, Wu CL, Manning BD. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE 4: e6189, 2009. doi: 10.1371/journal.pone.0006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Huffman TA, Mothe-Satney I, Lawrence JC Jr.. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc Natl Acad Sci USA 99: 1047–1052, 2002. doi: 10.1073/pnas.022634399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, Yokote K, Nakayama T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun 7: 13683, 2016. doi: 10.1038/ncomms13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Singh M, Shin YK, Yang X, Zehr B, Chakrabarti P, Kandror KV. 4E-BPs control fat storage by regulating the expression of Egr1 and ATGL. J Biol Chem 290: 17331–17338, 2015. doi: 10.1074/jbc.M114.631895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Sipula IJ, Brown NF, Perdomo G. Rapamycin-mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism 55: 1637–1644, 2006. doi: 10.1016/j.metabol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 371.Zhao X, Petrashen AP, Sanders JA, Peterson AL, Sedivy JM. SLC1A5 glutamine transporter is a target of MYC and mediates reduced mTORC1 signaling and increased fatty acid oxidation in long-lived Myc hypomorphic mice. Aging Cell 18: e12947, 2019. doi: 10.1111/acel.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Qian J, Chen Y, Meng T, Ma L, Meng L, Wang X, Yu T, Zask A, Shen J, Yu K. Molecular regulation of apoptotic machinery and lipid metabolism by mTORC1/mTORC2 dual inhibitors in preclinical models of HER2+/PIK3CAmut breast cancer. Oncotarget 7: 67071–67086, 2016. doi: 10.18632/oncotarget.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Lue HW, Podolak J, Kolahi K, Cheng L, Rao S, Garg D, Xue CH, Rantala JK, Tyner JW, Thornburg KL, Martinez-Acevedo A, Liu JJ, Amling CL, Truillet C, Louie SM, Anderson KE, Evans MJ, O’Donnell VB, Nomura DK, Drake JM, Ritz A, Thomas GV. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev 31: 2067–2084, 2017. doi: 10.1101/gad.305292.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Li S, Oh YT, Yue P, Khuri FR, Sun SY. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene 35: 642–650, 2016. doi: 10.1038/onc.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Yuan M, Pino E, Wu L, Kacergis M, Soukas AA. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem 287: 29579–29588, 2012. doi: 10.1074/jbc.M112.386854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Zhang M, Chi X, Qu N, Wang C. Long noncoding RNA lncARSR promotes hepatic lipogenesis via Akt/SREBP-1c pathway and contributes to the pathogenesis of nonalcoholic steatohepatitis. Biochem Biophys Res Commun 499: 66–70, 2018. doi: 10.1016/j.bbrc.2018.03.127. [DOI] [PubMed] [Google Scholar]
- 377.Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem 277: 33895–33900, 2002. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- 378.Guo Z, Zhao K, Feng X, Yan D, Yao R, Chen Y, Bao L, Wang Z. mTORC2 regulates lipogenic gene expression through PPARgamma to control lipid synthesis in bovine mammary epithelial cells. BioMed Res Int 2019: 5196028, 2019. doi: 10.1155/2019/5196028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, Jenoe P, Heim MH, Riezman I, Riezman H, Hall MN. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 32: 807–823.e12, 2017. doi: 10.1016/j.ccell.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 380.Khalifeh-Soltani A, McKleroy W, Sakuma S, Cheung YY, Tharp K, Qiu Y, Turner SM, Chawla A, Stahl A, Atabai K. Mfge8 promotes obesity by mediating the uptake of dietary fats and serum fatty acids. Nat Med 20: 175–183, 2014. doi: 10.1038/nm.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Yao Y, Suraokar M, Darnay BG, Hollier BG, Shaiken TE, Asano T, Chen CH, Chang BH, Lu Y, Mills GB, Sarbassov D, Mani SA, Abbruzzese JL, Reddy SA. BSTA promotes mTORC2-mediated phosphorylation of Akt1 to suppress expression of FoxC2 and stimulate adipocyte differentiation. Sci Signal 6: ra2, 2013. doi: 10.1126/scisignal.2003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Tang Y, Wallace M, Sanchez-Gurmaches J, Hsiao WY, Li H, Lee PL, Vernia S, Metallo CM, Guertin DA. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat Commun 7: 11365, 2016. doi: 10.1038/ncomms11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Hung CM, Calejman CM, Sanchez-Gurmaches J, Li H, Clish CB, Hettmer S, Wagers AJ, Guertin DA. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep 8: 256–271, 2014. doi: 10.1016/j.celrep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Jung SM, Hung CM, Hildebrand SR, Sanchez-Gurmaches J, Martinez-Pastor B, Gengatharan JM, Wallace M, Mukhopadhyay D, Martinez Calejman C, Luciano AK, Hsiao WY, Tang Y, Li H, Daniels DL, Mostoslavsky R, Metallo CM, Guertin DA. Non-canonical mTORC2 signaling regulates brown adipocyte lipid catabolism through SIRT6-FoxO1. Mol Cell 75: 807–822 e8, 2019. doi: 10.1016/j.molcel.2019.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294: 1942–1945, 2001. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 386.Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, Carr MD. Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene 27: 585–595, 2008. doi: 10.1038/sj.onc.1210693. [DOI] [PubMed] [Google Scholar]
- 387.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol 29: 1411–1420, 2009. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Frias MA, Mukhopadhyay S, Lehman E, Walasek A, Utter M, Menon D, Foster DA. Phosphatidic acid drives mTORC1 lysosomal translocation in the absence of amino acids. J Biol Chem 295: 263–274, 2020. doi: 10.1074/jbc.RA119.010892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.He A, Chen X, Tan M, Chen Y, Lu D, Zhang X, Dean JM, Razani B, Lodhi IJ. Acetyl-CoA derived from hepatic peroxisomal beta-oxidation inhibits autophagy and promotes steatosis via mTORC1 activation. Mol Cell 79: 30–42.e4, 2020. doi: 10.1016/j.molcel.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, Liesa M, Hammerman PS, Wong KK, Hayes DN, Shirihai OS, Dyson NJ, Haas W, Meissner A, Bardeesy N. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539: 390–395, 2016. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Karmaus PW, Herrada AA, Guy C, Neale G, Dhungana Y, Long L, Vogel P, Avila J, Clish CB, Chi H. Critical roles of mTORC1 signaling and metabolic reprogramming for M-CSF-mediated myelopoiesis. J Exp Med 214: 2629–2647, 2017. doi: 10.1084/jem.20161855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, Bhangoo MS, Thorson PK, Richards A, Rooslid T, Garcia-Olmo DC, Nam-Cha SY, Salinas-Sanchez AS, Eng K, Beltran H, Scott DA, Metallo CM, Moscat J, Diaz-Meco MT. Increased serine and one-carbon pathway metabolism by PKClambda/iota deficiency promotes neuroendocrine prostate cancer. Cancer Cell 35: 385–400.e9, 2019. doi: 10.1016/j.ccell.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, Scaria SM, Harper JW, Gygi SP, Sabatini DM. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358: 813–818, 2017. doi: 10.1126/science.aao3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.McKenna J 3rd, Kapfhamer D, Kinchen JM, Wasek B, Dunworth M, Murray-Stewart T, Bottiglieri T, Casero RA Jr, Gambello MJ. Metabolomic studies identify changes in transmethylation and polyamine metabolism in a brain-specific mouse model of tuberous sclerosis complex. Hum Mol Genet 27: 2113–2124, 2018. doi: 10.1093/hmg/ddy118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Rosario FJ, Powell TL, Jansson T. Mechanistic target of rapamycin (mTOR) regulates trophoblast folate uptake by modulating the cell surface expression of FR-alpha and the RFC. Sci Rep 6: 31705, 2016. doi: 10.1038/srep31705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Espindola-Netto JM, Chini CC, Tarragó M, Wang E, Dutta S, Pal K, Mukhopadhyay D, Sola-Penna M, Chini EN. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget 8: 85054–85067, 2017. doi: 10.18632/oncotarget.18841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Schuster S, Penke M, Gorski T, Gebhardt R, Weiss TS, Kiess W, Garten A. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem Biophys Res Commun 458: 334–340, 2015. doi: 10.1016/j.bbrc.2015.01.111. [DOI] [PubMed] [Google Scholar]
- 398.Cea M, Cagnetta A, Fulciniti M, Tai YT, Hideshima T, Chauhan D, Roccaro A, Sacco A, Calimeri T, Cottini F, Jakubikova J, Kong SY, Patrone F, Nencioni A, Gobbi M, Richardson P, Munshi N, Anderson KC. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 120: 3519–3529, 2012. doi: 10.1182/blood-2012-03-416776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Zucal C, D'Agostino VG, Casini A, Mantelli B, Thongon N, Soncini D, Caffa I, Cea M, Ballestrero A, Quattrone A, Indraccolo S, Nencioni A, Provenzani A. EIF2A-dependent translational arrest protects leukemia cells from the energetic stress induced by NAMPT inhibition. BMC Cancer 15: 855, 2015. doi: 10.1186/s12885-015-1845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Cordover E, Wei J, Patel C, Shan NL, Gionco J, Sargsyan D, Wu R, Cai L, Kong AN, Jacinto E, Minden A. KPT-9274, an inhibitor of PAK4 and NAMPT, leads to downregulation of mTORC2 in triple negative breast cancer cells. Chem Res Toxicol 33: 482–491, 2020. doi: 10.1021/acs.chemrestox.9b00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Mpilla G, Aboukameel A, Muqbil I, Kim S, Beydoun R, Philip PA, Mohammad RM, Kamgar M, Shidham V, Senapedis W, Baloglu E, Li J, Dyson G, Xue Y, El-Rayes B, Azmi AS. PAK4-NAMPT dual inhibition as a novel strategy for therapy resistant pancreatic neuroendocrine tumors. Cancers (Basel) 11: 1902, 2019. doi: 10.3390/cancers11121902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Zhang H, Zhang N, Liu Y, Su P, Liang Y, Li Y, Wang X, Chen T, Song X, Sang Y, Duan Y, Zhang J, Wang L, Chen B, Zhao W, Guo H, Liu Z, Hu G, Yang Q. Epigenetic regulation of NAMPT by NAMPT-AS drives metastatic progression in triple-negative breast cancer. Cancer Res 79: 3347–3359, 2019. doi: 10.1158/0008-5472.CAN-18-3418. [DOI] [PubMed] [Google Scholar]
- 403.Tarragó MG, Chini CC, Kanamori KS, Warner GM, Caride A, de Oliveira GC, Rud M, Samani A, Hein KZ, Huang R, Jurk D, Cho DS, Boslett JJ, Miller JD, Zweier JL, Passos JF, Doles JD, Becherer DJ, Chini EN. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab 27: 1081–1095.e10, 2018. doi: 10.1016/j.cmet.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Hoxhaj G, Ben-Sahra I, Lockwood SE, Timson RC, Byles V, Henning GT, Gao P, Selfors LM, Asara JM, Manning BD. Direct stimulation of NADP+ synthesis through Akt-mediated phosphorylation of NAD kinase. Science 363: 1088–1092, 2019. doi: 10.1126/science.aau3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Agostini M, Romeo F, Inoue S, Niklison-Chirou MV, Elia AJ, Dinsdale D, Morone N, Knight RA, Mak TW, Melino G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ 23: 1502–1514, 2016. doi: 10.1038/cdd.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14: 264–271, 2011. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Kim H, Jang H, Kim TW, Kang BH, Lee SE, Jeon YK, Chung DH, Choi J, Shin J, Cho EJ, Youn HD. Core pluripotency factors directly regulate metabolism in embryonic stem cell to maintain pluripotency. Stem Cells 33: 2699–2711, 2015. doi: 10.1002/stem.2073. [DOI] [PubMed] [Google Scholar]
- 408.Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, Gil J, Beach D. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal 9: 293–299, 2007. doi: 10.1089/ars.2006.1467. [DOI] [PubMed] [Google Scholar]
- 409.Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, Ramalho-Santos J, Van Houten B, Schatten G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 6: e20914, 2011. doi: 10.1371/journal.pone.0020914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, Ma L, Hamm M, Gage FH, Hunter T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 5: e13374, 2016. doi: 10.7554/eLife.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Zhang C, Skamagki M, Liu Z, Ananthanarayanan A, Zhao R, Li H, Kim K. Biological significance of the suppression of oxidative phosphorylation in induced pluripotent stem cells. Cell Rep 21: 2058–2065, 2017. doi: 10.1016/j.celrep.2017.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Chen T, Shen L, Yu J, Wan H, Guo A, Chen J, Long Y, Zhao J, Pei G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 10: 908–911, 2011. doi: 10.1111/j.1474-9726.2011.00722.x. [DOI] [PubMed] [Google Scholar]
- 413.Nishimura K, Fukuda A, Hisatake K. Mechanisms of the metabolic shift during somatic cell reprogramming. Int J Mol Sci 20: 2254, 2019. doi: 10.3390/ijms20092254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Wu Y, Li Y, Zhang H, Huang Y, Zhao P, Tang Y, Qiu X, Ying Y, Li W, Ni S, Zhang M, Liu L, Xu Y, Zhuang Q, Luo Z, Benda C, Song H, Liu B, Lai L, Liu X, Tse HF, Bao X, Chan WY, Esteban MA, Qin B, Pei D. Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat Cell Biol 17: 715–725, 2015. doi: 10.1038/ncb3172. [DOI] [PubMed] [Google Scholar]
- 415.Wang S, Xia P, Ye B, Huang G, Liu J, Fan Z. Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 13: 617–625, 2013. doi: 10.1016/j.stem.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 416.Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, Ruohola-Baker H. HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 31: 2103–2116, 2012. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, Bukowiecki R, Blümlein K, Wanker EE, Ralser M, Cramer T, Adjaye J. HIF1alpha modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells 32: 364–376, 2014. doi: 10.1002/stem.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Yu Y, Liang D, Tian Q, Chen X, Jiang B, Chou BK, Hu P, Cheng L, Gao P, Li J, Wang G. Stimulation of somatic cell reprogramming by ERas-Akt-FoxO1 signaling axis. Stem Cells 32: 349–363, 2014. doi: 10.1002/stem.1447. [DOI] [PubMed] [Google Scholar]
- 419.Zheng B, Wang J, Tang L, Tan C, Zhao Z, Xiao Y, Ge R, Zhu D. Involvement of Rictor/mTORC2 in cardiomyocyte differentiation of mouse embryonic stem cells in vitro. Int J Biol Sci 13: 110–121, 2017. doi: 10.7150/ijbs.16312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev 249: 27–42, 2012. doi: 10.1111/j.1600-065X.2012.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol 13: 907–915, 2012. doi: 10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- 422.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12: 325–338, 2012. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Jacinto E, Werlen G. mTOR: the mTOR complexes in T cell development and immunity. In: Encyclopedia of Inflammatory Diseases, edited by Parnham M. Basel: Birkhauser, 2015. [Google Scholar]
- 424.Chapman NM, Chi H. mTOR links environmental signals to T cell fate decisions. Front Immunol 5: 686, 2014. doi: 10.3389/fimmu.2014.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12: 295–303, 2011. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev 249: 43–58, 2012. doi: 10.1111/j.1600-065X.2012.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature 499: 485–490, 2013. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, Yong C, Surh N, Marie JC, Huehn J, Zimmermann V, Kinet S, Dardalhon V, Taylor N. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 8: ra97, 2015. doi: 10.1126/scisignal.aab2610. [DOI] [PubMed] [Google Scholar]
- 429.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14: 500–508, 2013. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, Delgoffe GM, Powell JD. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J Clin Invest 125: 2090–2108, 2015. doi: 10.1172/JCI77746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature 460: 108–112, 2009. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Salmond RJ. mTOR regulation of glycolytic metabolism in T cells. Front Cell Dev Biol 6: 122, 2018. doi: 10.3389/fcell.2018.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, Chiles TC. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 107: 4458–4465, 2006. doi: 10.1182/blood-2005-12-4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Woodland RT, Fox CJ, Schmidt MR, Hammerman PS, Opferman JT, Korsmeyer SJ, Hilbert DM, Thompson CB. Multiple signaling pathways promote B lymphocyte stimulator dependent B-cell growth and survival. Blood 111: 750–760, 2008. doi: 10.1182/blood-2007-03-077222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Xiao G, Chan LN, Klemm L, Braas D, Chen Z, Geng H, Zhang QC, Aghajanirefah A, Cosgun KN, Sadras T, Lee J, Mirzapoiazova T, Salgia R, Ernst T, Hochhaus A, Jumaa H, Jiang X, Weinstock DM, Graeber TG, Müschen M. B-cell-specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell 173: 470–484.e18, 2018. doi: 10.1016/j.cell.2018.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Chiu H, Jackson LV, Oh KI, Mai A, Ronai ZA, Ruggero D, Fruman DA. The mTORC1/4E-BP/eIF4E axis promotes antibody class switching in B lymphocytes. J Immunol 202: 579–590, 2019. doi: 10.4049/jimmunol.1800602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Gaudette BT, Jones DD, Bortnick A, Argon Y, Allman D. mTORC1 coordinates an immediate unfolded protein response-related transcriptome in activated B cells preceding antibody secretion. Nat Commun 11: 723, 2020. doi: 10.1038/s41467-019-14032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Bertolotti M, Yim SH, Garcia-Manteiga JM, Masciarelli S, Kim YJ, Kang MH, Iuchi Y, Fujii J, Vené R, Rubartelli A, Rhee SG, Sitia R. B- to plasma-cell terminal differentiation entails oxidative stress and profound reshaping of the antioxidant responses. Antioxid Redox Signal 13: 1133–1144, 2010. doi: 10.1089/ars.2009.3079. [DOI] [PubMed] [Google Scholar]
- 439.Dufort FJ, Gumina MR, Ta NL, Tao Y, Heyse SA, Scott DA, Richardson AD, Seyfried TN, Chiles TC. Glucose-dependent de novo lipogenesis in B lymphocytes: a requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. J Biol Chem 289: 7011–7024, 2014. doi: 10.1074/jbc.M114.551051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, McCommis KS, Fahrmann J, Pizzato HA, Nunley RM, Lee J, Wolfgang MJ, Patti GJ, Finck BN, Pearce EL, Bhattacharya D. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity 45: 60–73, 2016. doi: 10.1016/j.immuni.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Jung J, Zeng H, Horng T. Metabolism as a guiding force for immunity. Nat Cell Biol 21: 85–93, 2019. doi: 10.1038/s41556-018-0217-x. [DOI] [PubMed] [Google Scholar]
- 442.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, Manjeri GR, Li Y, Ifrim DC, Arts RJ, van der Veer BM, van der Meer BM, Deen PM, Logie C, O’Neill LA, Willems P, van de Veerdonk FL, van der Meer JW, Ng A, Joosten LA, Wijmenga C, Stunnenberg HG, Xavier RJ, Netea MG. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345: 1250684, 2014. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, Espinosa EC, Lamming D, Manning BD, Zhang Y, Blair IA, Horng T. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 5: e11612, 2016. doi: 10.7554/eLife.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 445.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 10: 868–880, 2011. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 446.Magaway C, Kim E, Jacinto E. Targeting mTOR and metabolism in cancer: lessons and innovations. Cells 8: 1584, 2019. doi: 10.3390/cells8121584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 4: 554–563, 2014. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Kong Y, Si L, Li Y, Wu X, Xu X, Dai J, Tang H, Ma M, Chi Z, Sheng X, Cui C, Guo J. Analysis of mTOR gene aberrations in melanoma patients and evaluation of their sensitivity to PI3K-AKT-mTOR pathway inhibitors. Clin Cancer Res 22: 1018–1027, 2016. doi: 10.1158/1078-0432.CCR-15-1110. [DOI] [PubMed] [Google Scholar]
- 449.Murugan AK, Liu R, Xing M. Identification and characterization of two novel oncogenic mTOR mutations. Oncogene 38: 5211–5226, 2019. doi: 10.1038/s41388-019-0787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 29: 2746–2752, 2010. doi: 10.1038/onc.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Wagle N, Grabiner BC, Van Allen EM, Hodis E, Jacobus S, Supko JG, Stewart M, Choueiri TK, Gandhi L, Cleary JM, Elfiky AA, Taplin ME, Stack EC, Signoretti S, Loda M, Shapiro GI, Sabatini DM, Lander ES, Gabriel SB, Kantoff PW, Garraway LA, Rosenberg JE. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov 4: 546–553, 2014. doi: 10.1158/2159-8290.CD-13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Yamaguchi H, Kawazu M, Yasuda T, Soda M, Ueno T, Kojima S, Yashiro M, Yoshino I, Ishikawa Y, Sai E, Mano H. Transforming somatic mutations of mammalian target of rapamycin kinase in human cancer. Cancer Sci 106: 1687–1692, 2015. doi: 10.1111/cas.12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146: 1473–1481, 2005. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- 454.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431: 200–205, 2004. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 455.Ardestani A, Lupse B, Kido Y, Leibowitz G, Maedler K. mTORC1 signaling: a double-edged sword in diabetic beta cells. Cell Metab 27: 314–331, 2018. doi: 10.1016/j.cmet.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 456.Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol 26: 63–76, 2006. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Yoneyama Y, Inamitsu T, Chida K, Iemura SI, Natsume T, Maeda T, Hakuno F, Takahashi SI. Serine phosphorylation by mTORC1 promotes IRS-1 degradation through SCFbeta-TRCP E3 ubiquitin ligase. iScience 5: 1–18, 2018. doi: 10.1016/j.isci.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol 33: 55–66, 2015. doi: 10.1016/j.ceb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 459.Chakrabarti P, Kandror KV. The role of mTOR in lipid homeostasis and diabetes progression. Curr Opin Endocrinol Diabetes Obes 22: 340–346, 2015. doi: 10.1097/MED.0000000000000187. [DOI] [PubMed] [Google Scholar]
- 460.Lee PL, Jung SM, Guertin DA. The complex roles of mechanistic target of rapamycin in adipocytes and beyond. Trends Endocrinol Metab 28: 319–339, 2017. doi: 10.1016/j.tem.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335: 1638–1643, 2012. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Yu Z, Wang R, Fok WC, Coles A, Salmon AB, Pérez VI. Rapamycin and dietary restriction induce metabolically distinctive changes in mouse liver. J Gerontol A Biol Sci Med Sci 70: 410–420, 2015. doi: 10.1093/gerona/glu053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Barlow AD, Xie J, Moore CE, Campbell SC, Shaw JA, Nicholson ML, Herbert TP. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2). Diabetologia 55: 1355–1365, 2012. doi: 10.1007/s00125-012-2475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Makki K, Taront S, Molendi-Coste O, Bouchaert E, Neve B, Eury E, Lobbens S, Labalette M, Duez H, Staels B, Dombrowicz D, Froguel P, Wolowczuk I. Beneficial metabolic effects of rapamycin are associated with enhanced regulatory cells in diet-induced obese mice. PLoS One 9: e92684, 2014. doi: 10.1371/journal.pone.0092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.den Hartigh LJ, Goodspeed L, Wang SA, Kenerson HL, Omer M, O'Brien KD, Ladiges W, Yeung R, Subramanian S. Chronic oral rapamycin decreases adiposity, hepatic triglycerides and insulin resistance in male mice fed a diet high in sucrose and saturated fat. Exp Physiol 103: 1469–1480, 2018. doi: 10.1113/EP087207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Tanimura J, Nakagawa H, Tanaka T, Kikuchi A, Osada S, Tanaka Y, Tokuyama K, Takamura T. The clinical course and potential underlying mechanisms of everolimus-induced hyperglycemia. Endocr J 66: 615–620, 2019. doi: 10.1507/endocrj.EJ18-0542. [DOI] [PubMed] [Google Scholar]
- 467.Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, Manning BD. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab 25: 463–471, 2017. doi: 10.1016/j.cmet.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, Mirmira RG, Sonenberg N, Liu M, Rüegg MA, Hall MN, Bernal-Mizrachi E. Loss of mTORC1 signalling impairs beta-cell homeostasis and insulin processing. Nat Commun 8: 16014, 2017. doi: 10.1038/ncomms16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Yuan T, Rafizadeh S, Gorrepati KD, Lupse B, Oberholzer J, Maedler K, Ardestani A. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia 60: 668–678, 2017. doi: 10.1007/s00125-016-4188-9. [DOI] [PubMed] [Google Scholar]
- 470.Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes 60: 827–837, 2011. doi: 10.2337/db10-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Xie Y, Cui C, Nie A, Wang Y, Ni Q, Liu Y, Yin Q, Zhang H, Li Y, Wang Q, Gu Y, Ning G. The mTORC2/PKC pathway sustains compensatory insulin secretion of pancreatic beta cells in response to metabolic stress. Biochim Biophys Acta Gen Subj 1861: 2039–2047, 2017. doi: 10.1016/j.bbagen.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 472.Yin Q, Ni Q, Wang Y, Zhang H, Li W, Nie A, Wang S, Gu Y, Wang Q, Ning G. Raptor determines beta-cell identity and plasticity independent of hyperglycemia in mice. Nat Commun 11: 2538, 2020. doi: 10.1038/s41467-020-15935-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Ni Q, Song J, Wang Y, Sun J, Xie J, Zhang J, Ning G, Wang W, Wang Q. Proper mTORC1 activity is required for glucose sensing and early adaptation in human pancreatic beta cell. J Clin Endocrinol Metab 106: e562–e572, 2021. doi: 10.1210/clinem/dgaa786. [DOI] [PubMed] [Google Scholar]
- 474.Blouet C, Ono H, Schwartz GJ. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab 8: 459–467, 2008. doi: 10.1016/j.cmet.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science 312: 927–930, 2006. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 476.Haissaguerre M, Ferrière A, Simon V, Saucisse N, Dupuy N, André C, Clark S, Guzman-Quevedo O, Tabarin A, Cota D. mTORC1-dependent increase in oxidative metabolism in POMC neurons regulates food intake and action of leptin. Mol Metab 12: 98–106, 2018. doi: 10.1016/j.molmet.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM, Schwartz GJ, Rossetti L. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest 118: 2959–2968, 2008. doi: 10.1172/JCI34277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Tavares MR, Lemes SF, de Fante T, Saenz de Miera C, Pavan IC, Bezerra RM, Prada PO, Torsoni MA, Elias CF, Simabuco FM. Modulation of hypothalamic S6K1 and S6K2 alters feeding behavior and systemic glucose metabolism. J Endocrinol 244: 71–82, 2020. doi: 10.1530/JOE-19-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Caron A, Labbé SM, Lanfray D, Blanchard PG, Villot R, Roy C, Sabatini DM, Richard D, Laplante M. Mediobasal hypothalamic overexpression of DEPTOR protects against high-fat diet-induced obesity. Mol Metab 5: 102–112, 2016. doi: 10.1016/j.molmet.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Caron A, Labbé SM, Mouchiroud M, Huard R, Lanfray D, Richard D, Laplante M. DEPTOR in POMC neurons affects liver metabolism but is dispensable for the regulation of energy balance. Am J Physiol Regul Integr Comp Physiol 310: R1322–R1331, 2016. doi: 10.1152/ajpregu.00549.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Smith MA, Katsouri L, Irvine EE, Hankir MK, Pedroni SM, Voshol PJ, Gordon MW, Choudhury AI, Woods A, Vidal-Puig A, Carling D, Withers DJ. Ribosomal S6K1 in POMC and AgRP neurons regulates glucose homeostasis but not feeding behavior in mice. Cell Rep 11: 335–343, 2015. doi: 10.1016/j.celrep.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, Kim AJ, Cawley NX, Paik JH, Loh YP, DePinho RA, Wardlaw SL, Accili D. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med 15: 1195–1201, 2009. doi: 10.1038/nm.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Ren H, Orozco IJ, Su Y, Suyama S, Gutiérrez-Juárez R, Horvath TL, Wardlaw SL, Plum L, Arancio O, Accili D. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 149: 1314–1326, 2012. doi: 10.1016/j.cell.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.de Mello NP, Orellana AM, Mazucanti CH, de Morais Lima G, Scavone C, Kawamoto EM. Insulin and autophagy in neurodegeneration. Front Neurosci 13: 491, 2019. doi: 10.3389/fnins.2019.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Shafei MA, Harris M, Conway ME. Divergent metabolic regulation of autophagy and mTORC1—early events in Alzheimer’s disease? Front Aging Neurosci 9: 173, 2017. doi: 10.3389/fnagi.2017.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Kaeberlein M, Galvan V. Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci Transl Med 11: eaar4289, 2019. doi: 10.1126/scitranslmed.aar4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Sun YX, Ji X, Mao X, Xie L, Jia J, Galvan V, Greenberg DA, Jin K. Differential activation of mTOR complex 1 signaling in human brain with mild to severe Alzheimer’s disease. J Alzheimers Dis 38: 437–444, 2014. doi: 10.3233/JAD-131124. [DOI] [PubMed] [Google Scholar]
- 488.Caccamo A, Belfiore R, Oddo S. Genetically reducing mTOR signaling rescues central insulin dysregulation in a mouse model of Alzheimer’s disease. Neurobiol Aging 68: 1, 2018. doi: 10.1016/j.neurobiolaging.2018.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 489.Uddin MS, Mamun AA, Labu ZK, Hidalgo-Lanussa O, Barreto GE, Ashraf GM. Autophagic dysfunction in Alzheimer’s disease: cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J Cell Physiol 234: 8094–8112, 2019. doi: 10.1002/jcp.27588. [DOI] [PubMed] [Google Scholar]
- 490.Li H, Ye D, Xie W, Hua F, Yang Y, Wu J, Gu A, Ren Y, Mao K. Defect of branched-chain amino acid metabolism promotes the development of Alzheimer’s disease by targeting the mTOR signaling. Biosci Rep 38: BSR20180127, 2018. doi: 10.1042/BSR20180127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Neth BJ, Craft S. Insulin resistance and Alzheimer’s disease: bioenergetic linkages. Front Aging Neurosci 9: 345, 2017. doi: 10.3389/fnagi.2017.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Sun Y, Ma C, Sun H, Wang H, Peng W, Zhou Z, Wang H, Pi C, Shi Y, He X. Metabolism: a novel shared link between diabetes mellitus and Alzheimer’s disease. J Diabetes Res 2020: 4981814, 2020. doi: 10.1155/2020/4981814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Chen CJ, Sgritta M, Mays J, Zhou H, Lucero R, Park J, Wang IC, Park JH, Kaipparettu BA, Stoica L, Jafar-Nejad P, Rigo F, Chin J, Noebels JL, Costa-Mattioli M. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat Med 25: 1684–1690, 2019. doi: 10.1038/s41591-019-0608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Lee HK, Kwon B, Lemere CA, de la Monte S, Itamura K, Ha AY, Querfurth HW. mTORC2 (Rictor) in Alzheimer’s disease and reversal of amyloid-beta expression-induced insulin resistance and toxicity in rat primary cortical neurons. J Alzheimers Dis 56: 1015–1036, 2017. doi: 10.3233/JAD-161029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Bellozi PM, Gomes GF, de Oliveira LR, Olmo IG, Vieira EL, Ribeiro FM, Fiebich BL, de Oliveira AC. NVP-BEZ235 (dactolisib) has protective effects in a transgenic mouse model of Alzheimer’s disease. Front Pharmacol 10: 1345, 2019. doi: 10.3389/fphar.2019.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA, Lithgow GJ, Andersen JK. Mitochondrial quality control via the PGC1alpha-TFEB signaling pathway is compromised by Parkin Q311X mutation but independently restored by rapamycin. J Neurosci 35: 12833–12844, 2015. doi: 10.1523/JNEUROSCI.0109-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Gao S, Duan C, Gao G, Wang X, Yang H. Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int J Biochem Cell Biol 64: 25–33, 2015. doi: 10.1016/j.biocel.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 498.Jiang TF, Zhang YJ, Zhou HY, Wang HM, Tian LP, Liu J, Ding JQ, Chen SD. Curcumin ameliorates the neurodegenerative pathology in A53T alpha-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J Neuroimmune Pharmacol 8: 356–369, 2013. doi: 10.1007/s11481-012-9431-7. [DOI] [PubMed] [Google Scholar]
- 499.Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci 26: 9996–10005, 2006. doi: 10.1523/JNEUROSCI.3292-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Pryor WM, Biagioli M, Shahani N, Swarnkar S, Huang WC, Page DT, MacDonald ME, Subramaniam S. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci Signal 7: ra103, 2014. doi: 10.1126/scisignal.2005633. [DOI] [PubMed] [Google Scholar]
- 501.Lee JH, Tecedor L, Chen YH, Monteys AM, Sowada MJ, Thompson LM, Davidson BL. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 85: 303–315, 2015. doi: 10.1016/j.neuron.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Wold MS, Lim J, Lachance V, Deng Z, Yue Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol Neurodegener 11: 76, 2016. doi: 10.1186/s13024-016-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Sasaki Y. Metabolic aspects of neuronal degeneration: from a NAD+ point of view. Neurosci Res 139: 9–20, 2019. doi: 10.1016/j.neures.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Bitto A, Ito TK, Pineda VV, LeTexier NJ, Huang HZ, Sutlief E, Tung H, Vizzini N, Chen B, Smith K, Meza D, Yajima M, Beyer RP, Kerr KF, Davis DJ, Gillespie CH, Snyder JM, Treuting PM, Kaeberlein M. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. eLife 5: e16351, 2016. doi: 10.7554/eLife.16351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395, 2009. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Leontieva OV, Paszkiewicz GM, Blagosklonny MV. Weekly administration of rapamycin improves survival and biomarkers in obese male mice on high-fat diet. Aging Cell 13: 616–622, 2014. doi: 10.1111/acel.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Swindell WR. Meta-analysis of 29 experiments evaluating the effects of rapamycin on life span in the laboratory mouse. J Gerontol A Biol Sci Med Sci 72: 1024–1032, 2017. doi: 10.1093/gerona/glw153. [DOI] [PubMed] [Google Scholar]
- 508.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326: 140–144, 2009. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 153: 1194–1217, 2013. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu H, Djukovic D, Raftery D, Beyer RP, MacCoss MJ, Rabinovitch PS. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13: 529–539, 2014. doi: 10.1111/acel.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly-Y M, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, von Zglinicki T, Korolchuk VI, Passos JF. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35: 724–742, 2016. doi: 10.15252/embj.201592862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Muñoz-Cánoves P. Autophagy maintains stemness by preventing senescence. Nature 529: 37–42, 2016. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 513.Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2: ra75, 2009. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Hurez V, Dao V, Liu A, Pandeswara S, Gelfond J, Sun L, Bergman M, Orihuela CJ, Galvan V, Padrón Á, Drerup J, Liu Y, Hasty P, Sharp ZD, Curiel TJ. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 14: 945–956, 2015. doi: 10.1111/acel.12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB. mTOR inhibition improves immune function in the elderly. Sci Transl Med 6: 268ra179, 2014. doi: 10.1126/scitranslmed.3009892. [DOI] [PubMed] [Google Scholar]
- 516.Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, Klickstein LB. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med 10: eaaq1564, 2018. doi: 10.1126/scitranslmed.aaq1564. [DOI] [PubMed] [Google Scholar]
- 517.Nojima A, Yamashita M, Yoshida Y, Shimizu I, Ichimiya H, Kamimura N, Kobayashi Y, Ohta S, Ishii N, Minamino T. Haploinsufficiency of akt1 prolongs the lifespan of mice. PLoS One 8: e69178, 2013. doi: 10.1371/journal.pone.0069178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Lamming DW, Mihaylova MM, Katajisto P, Baar EL, Yilmaz OH, Hutchins A, Gultekin Y, Gaither R, Sabatini DM. Depletion of Rictor, an essential protein component of mTORC2, decreases male lifespan. Aging Cell 13: 911–917, 2014. doi: 10.1111/acel.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Chellappa K, Brinkman JA, Mukherjee S, Morrison M, Alotaibi MI, Carbajal KA, Alhadeff AL, Perron IJ, Yao R, Purdy CS, DeFelice DM, Wakai MH, Tomasiewicz J, Lin A, Meyer E, Peng Y, Arriola Apelo SI, Puglielli L, Betley JN, Paschos GK, Baur JA, Lamming DW. Hypothalamic mTORC2 is essential for metabolic health and longevity. Aging Cell 18: e13014, 2019. doi: 10.1111/acel.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Fang Y, Hill CM, Darcy J, Reyes-Ordoñez A, Arauz E, McFadden S, Zhang C, Osland J, Gao J, Zhang T, Frank SJ, Javors MA, Yuan R, Kopchick JJ, Sun LY, Chen J, Bartke A. Effects of rapamycin on growth hormone receptor knockout mice. Proc Natl Acad Sci USA 115: E1495–E1503, 2018. doi: 10.1073/pnas.1717065115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Yu D, Tomasiewicz JL, Yang SE, Miller BR, Wakai MH, Sherman DS, Cummings NE, Baar EL, Brinkman JA, Syed FA, Lamming DW. Calorie-restriction-induced insulin sensitivity is mediated by adipose mTORC2 and not required for lifespan extension. Cell Rep 29: 236–248.e3, 2019. doi: 10.1016/j.celrep.2019.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Nguyen JT, Ray C, Fox AL, Mendonça DB, Kim JK, Krebsbach PH. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci Adv 4: eaao5838, 2018. doi: 10.1126/sciadv.aao5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Smithson LJ, Gutmann DH. Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev 30: 1383–1388, 2016. doi: 10.1101/gad.279661.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol 12: 379–392, 2016. doi: 10.1038/nrneurol.2016.81. [DOI] [PubMed] [Google Scholar]
- 525.Bose S, Allen AE, Locasale JW. The molecular link from diet to cancer cell metabolism. Mol Cell 78: 1034–1044, 2020. doi: 10.1016/j.molcel.2020.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, Bareja R, Ma Y, Harris EM, Sboner A, Beltran H, Rubin MA, Mukherjee S, Cantley LC. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560: 499–503, 2018. doi: 10.1038/s41586-018-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]