

Abstract
The success of immunotherapy with immune checkpoint inhibitors (ICIs) in a subset of individuals has been very exciting. However, in many cancers, the responses to current ICIs are modest and seen only in small subsets of patients. Herein, a widely applicable approach that increases the benefit of ICIs is reported. Intratumoral administration of Augmenting Immune Response and Inhibiting Suppressive Environment of tumors – AIRISE-02 nanotherapeutic that co-delivers CpG and STAT3 siRNA results in not only regression of the injected tumor, but also tumors at distant sites in multiple tumor model systems. In particular, three doses of AIRISE-02 in combination with systemic ICIs completely cure both treated and untreated aggressive melanoma tumors in 5 out of 8 mice, while ICIs alone do not cure any mice. Long-term memory immune effect is also reported. AIRISE-02 is effective in breast and colon tumor models, as well. Lastly, AIRISE-02 is well tolerated in mice and non-human primates. This approach combines multiple therapeutic agents into a single nanoconstruct to create whole-body immune responses across multiple cancer types. Being a local therapeutic, AIRISE-02 circumvents regulatory challenges of systemic nanoparticle delivery, facilitating rapid translation to the clinic. AIRISE-02 is under IND-enabling studies, and clinical trials will soon follow.
Keywords: Cancer immunotherapy, melanoma, nanotechnology, intratumoral, translational research
Graphical Abstract
AIRISE-02 is a nano-immunotherapy candidate that co-delivers CpG and STAT3 siRNA to a local tumor, and generates anti-tumor immune response against cancer everywhere in the body. AIRISE-02 inhibits both local and untreated distant tumors in mice. Combination of AIRISE-02 and standard immune checkpoint inhibitors cures 60% of mice with melanoma tumors, while the inhibitors alone cure none.
A new strategy to improve the efficacy and response rate of cancer immunotherapy is reported herein. Cancer immunotherapy has received substantial attention in recent years due to many fast-track FDA approvals supported by strong objective responses in clinical trials. Most notable is a class of immune checkpoint inhibitors (ICIs), such as antibodies against PD-L1/PD-1 and CTLA-4, which have been widely studied and shown promising outcomes in clinics.[1] ICIs release brakes on the patients’ own immune system, increasing responses to tumors and offering immunological memory and long-lasting immunity even after treatment stops. ICIs thus have the potential to provide more durable responses than chemotherapy and targeted therapy. However, ICIs are effective in only a subset of patients even in cancers that respond best to immunotherapy, such as melanoma or non-small cell lung cancer, while having modest to no activity in many other cancer types.[2] Lack of response to ICIs is typically correlated with the absence of pre-existing anti-tumor immunity (e.g., CD8+ anti-tumor T cells)[3] and an immunosuppressive environment of tumors,[4] especially in those pre-treated with chemotherapy or radiation therapy.[5]
In situ tumor vaccination utilizes patients’ own tumors as the depot for tumor antigens to generate antitumor immunity, thus bypassing the need to identify tumor-specific antigens, which vary vastly across tumor types and patients due to cancer heterogeneity. Talimogene laherparepvec (T-VEC) is the first-in-class oncolytic virus to generate an in situ vaccination effect in melanoma.[6] However, T-VEC is not effective in late stages nor in tumors pre-treated with other drugs,[7] because of poor virus uptake and replication in these tumors.[7–8] Viral proteins can also cause ER stress and ROS production,[9] exacerbating the immunosuppressive tumor microenvironment (TME). Immunostimulatory agents (e.g., vaccine adjuvants) have been injected intratumorally to achieve an in situ vaccination effect.[10] Among adjuvants, CpG is a Toll-like receptor 9 (TLR9) agonist that triggers TLR9+ cells (e.g., dendritic cells (DC), macrophages) to mount an innate immune response via the production of Th1 and pro-inflammatory cytokines.[11] Specifically, CpG activates DCs to take up and process antigens to prime T cells.[12] CpG also induces inflammatory monocyte differentiation into anti-tumor M1 macrophages.[13] Furthermore, delivering CpG to cancer cells co-localizes CpG with tumor antigens, allowing effective priming of tumor-specific immune responses. In clinics, intratumorally administered CpG was well tolerated in patients.[14] However, CpG (e.g., CpG 7909, Class B, Promune, Pfizer) has not moved forward[15] due to its poor response in the Phase III clinical trial (e.g., not performing better than chemotherapy).[16] CpG (e.g., SD-101, Class C, Dynavax) was effective in 78% of patients with unresectable melanoma (stage III-IV)[17] naïve to PD-1 inhibitors, but only 12% of non-naïve patients.
Regulating tumor-associated immunosuppressive pathways in the tumor has great potential to amplify in situ vaccination effect. In particular, there is a strong rationale to combine CpG with STAT3 inhibition. STAT3 mediates immunosuppressive functions in multiple tumor-associated immune cells including macrophages, myeloid-derived suppressor cells (MDSCs), and DCs.[18] Depleting STAT3 in DCs improves antigen-presenting activity and enhances adaptive antitumor immune responses.[19] Inhibiting STAT3 can also improve the immunostimulatory effect of CpG in the TME.[20] In addition to immune cells, inhibiting STAT3 in cancer cells enhances the immunogenic cell death and increases many interferon-responsive chemokines that mediate immune cell infiltration.[21] STAT3 is difficult to drug by antibodies or small molecule inhibitors, therefore siRNA or antisense are ideal alternatives for STAT3 inhibition. We hypothesize that co-delivery of CpG and siRNA against STAT3 using our scalable and optimized nanoparticle platform (NP) for oligonucleotide delivery[22] will result in a very effective in situ vaccination effect. Our nanoparticles can protect oligonucleotides (e.g., siRNA and CpG) from enzymatic degradation,[22] retain them in tumors, enhance CpG’s adjuvanticity, and deliver them to both cancer and myeloid cells. The nanoparticle co-delivering CpG and siRNA against STAT3 is termed Augmenting Immune Response and Inhibiting Suppressive Environment of tumors, AIRISE-02. Co-delivery of siRNA and adjuvant by nanoparticle (of any kind), without adding tumor antigens, to trigger in situ tumor vaccination has never been reported before.
Our nanoparticle (NP) platform is based on mesoporous silica nanoparticle (MSNP) coated with bioreducibly crosslinked polyethylenimine (PEI, 13 wt.%) and polyethylene glycol (PEG, 18 wt.%). NP was synthesized and characterized following our standard method.[22] The nanoparticle platform was originally optimized for systemic siRNA delivery.[22] It has also been used effectively for dermal siRNA delivery.[23] Figure 1 shows (A) the MSNP core and (B) surface modification to generate AIRISE-02. CpG and siRNA were loaded on the nanoparticle by electrostatic interaction of PEI polymer layer and the negatively charged phosphodiester backbone of siRNA and phosphorothioate backbone of CpG in a sequence-independent manner. Oligonucleotides are protected under the PEG layer from enzymatic degradation.[22] AIRISE-02 comprises NP that is loaded with 2 wt.% siSTAT3 and 7 wt.% of CpG oligo. Oligonucleotides were loaded in a complete-binding manner in that we detected no unbound oligonucleotide in the supernatant upon centrifuging down AIRISE-02. Further, we have shown that our NP achieves complete binding of siRNA up to 25 wt.%, while maintaining hydrodynamic size of about 100 nm (Table S1). AIRISE-02 has a hydrodynamic size of 106 ± 0.4 nm with a narrow size distribution (PDI of 0.15, Figure 1C). AIRISE-02 is slightly positively charged (zeta potential of 6.5 ± 0.4 mV in 10 mM NaCl). CpG 1826 (a class B CpG for mice)[24] was used throughout mouse studies, while CpG 7909 with proven activity in humans[25] was used in the non-human primate study. The selected siSTAT3 sequence has cross-reactivity in mice, canines, and primates.
Figure 1. MSNP-based nanoconstruct (NP).
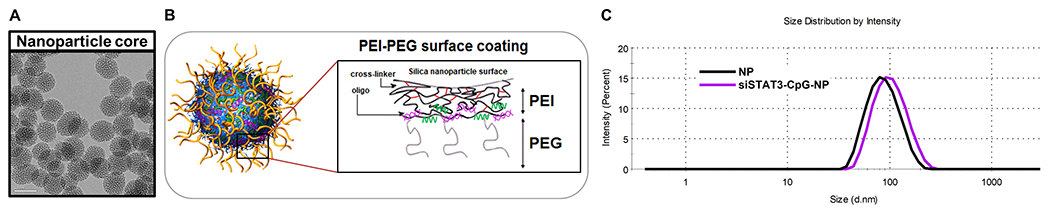
A) TEM image of the 50-nm MSNP core (scale bar = 50 nm). B) Surface coating layer-by-layer with PEI coating and crosslinking, PEG, and oligonucleotides including single-stranded CpG (green) and double-stranded siRNA (purple). C) Hydrodynamic size of NP and AIRISE-02 (siSTAT3-CpG-NP).
We first focus on melanoma, which is the most serious form of skin cancer, accounting for >75% of all skin cancer deaths.[26] It can metastasize to lymph nodes and other organs such as the lung and liver, at which point the disease is life-threatening and rarely curable.[27] The incidence of melanoma has been rising rapidly over the past few decades. It is estimated that there were 1,195,608 Americans living with melanoma in 2016,[28] and there will be 100,350 new cases of melanoma in the US in 2020.[26a] The current first-line immunotherapy for melanoma includes PD-1 antibody (nivolumab or pembrolizumab) alone or in combination with CTLA-4 antibody (ipilimumab). Nevertheless, melanoma remains one of the most aggressive human cancers and only 25% of advanced melanoma patients survive beyond 5 years,[26a] calling for improvements in current therapy.
We found that CpG-loaded nanoparticles (CpG-NP) activated DCs in draining lymph nodes (DLN) significantly better than free CpG in C57BL/6 mice, measured at 24 hours after foot pad injection by flow cytometry analysis for CD11c, MHCII, and CD80 expression of myeloid (CD45+CD3−CD19−) cells (Figure S1). To evaluate the therapeutic effect of CpG-NP in both treated and untreated distant tumors, we exploited a bilateral orthotopic mouse melanoma tumor model (B16F10),[29] as shown in Figure 2A. Eight days after tumor implantation, CpG-NP was injected into the local tumor for a total of three doses, each three days apart, while the distant tumor was left untreated. We found that CpG-NP reduced the size of both treated and untreated tumors and prolonged survival of mice, consistent with the hypothesis that local injection of CpG-NP induces a systemic antitumor immune response (Figure 2B–2D).
Figure 2. Effectiveness of AIRISE-02 (siSTAT3-CpG-NP) in inducing in situ tumor vaccination in mice bearing bilateral syngeneic melanoma tumors.
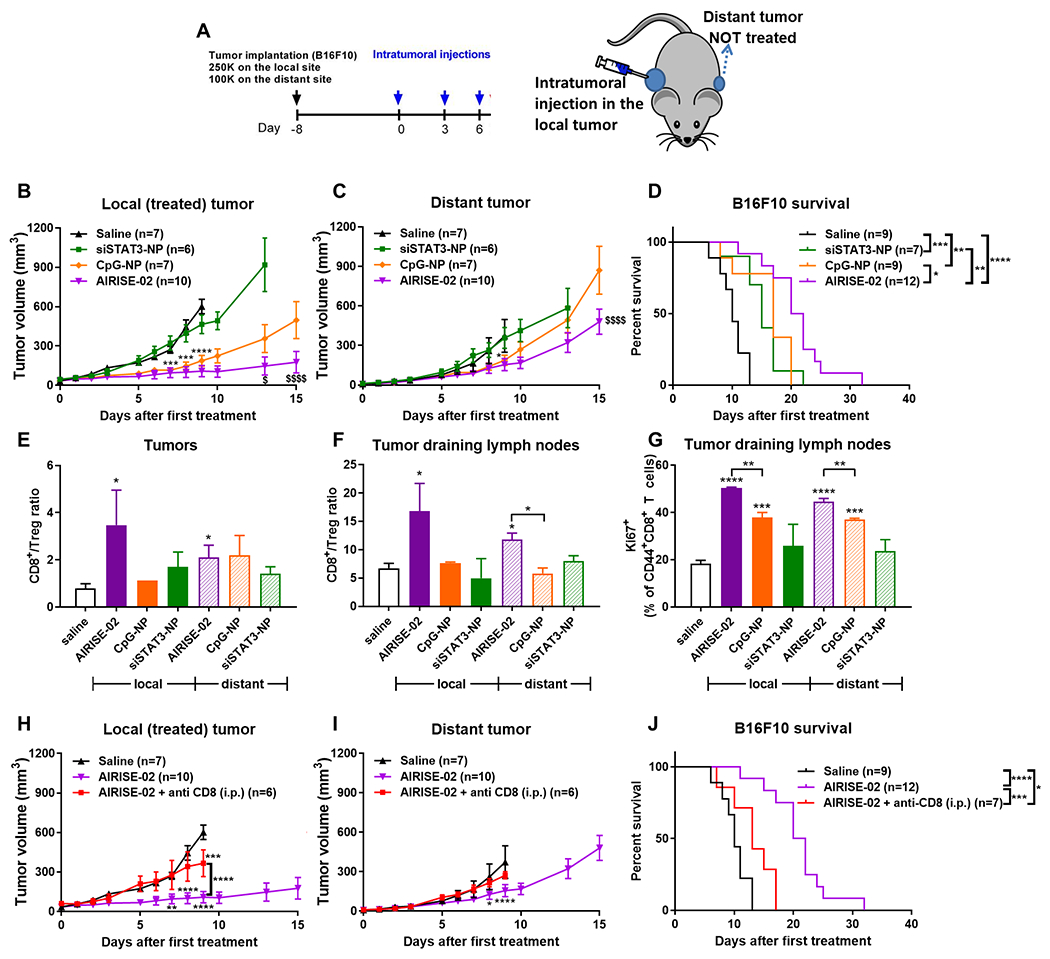
(A) C57BL/6 mice were orthotopically implanted with 250,000 and 100,000 B16F10 melanoma cells on both shoulders to model local (primary) and distant (metastatic) tumors, respectively. Eight days after tumor implantation, treatments were intratumorally injected into the local tumor for a total of three doses over one week, while the distant tumor was left untreated. (B-D) Mice were treated with AIRISE-02 (NP carrying 20 μg CpG and 4 μg siSTAT3 per each injection) and controls. CpG-NP and siSTAT3-NP contain the same amount of CpG and siSTAT3, respectively, as AIRISE-02. Tumor growth curves of B) local treated tumors and C) distant untreated tumors are plotted as mean ± SEM for mice that survived the first 9-15 days (i.e., >75% of all mice per group, each group still has the same average starting tumor size). Spider plots of tumor sizes for all mice without exclusion were reported in Figure S7 in the Supporting Information. D) Survival curve of all mice is reported. E-G) In another set of mice treated in a similar manner as (A), mice (n = 3-4) were sacrificed at day 7. Cells harvested from tumors and associated draining lymph nodes (DLNs) were analyzed to determine the ratio of CD8+ T cells over Treg (CD4+FoxP3+) within the T cell (CD45+CD3+) population of E) local and distant tumors and F) their associated tumor-DLNs, and G) proliferation status (Ki67) of effector T cells (CD44+CD8+) in tumor-DLNs. To confirm immune-mediated therapeutic action, mice were treated with AIRISE-02 (as shown in (A)) with or without CD8 depletion (200 μg anti-CD8 Ab; i.p. twice weekly throughout the entire study), and monitored for the growth of H) local (AIRISE-02-treated) and I) distant (untreated) tumors, along with J) their survival profile. Statistical significance (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001) is between the test group and saline group, unless specified otherwise by brackets. $p<0.05, $$$$p<0.0001 in (B) and (C) denote the statistical significance of AIRISE-02 vs. CpG-NP. Statistical significance of AIRISE-02 versus saline is not shown in (B) and (C), but shown in (H) and (I) for local and distant tumors, respectively. Saline and AIRISE-02 treatments were conducted independently twice (along with other groups in this figure and Figure 3), and the combined data were used for both figures.
In addition to enhancing CpG’s adjuvanticity, our NP can deliver other therapeutic cargos (e.g., siRNA).[30] We assessed STAT3 as a therapeutic target since inhibiting STAT3 can elicit both direct anti-tumor activity and enhance immune activity in the TME.[18] In particular, Figure S2A shows that a single sequence of siSTAT3 delivered with our NP decreased STAT3 levels by >70% in both immune cells (BMDC and J774) and cancer cell lines from various species (mouse B16F10, mouse CT26, mouse MM3MG-HER2Δ16, human HCC1954, canine D-17), indicating that the targeted STAT3 region is well conserved across species. siSTAT3 delivered by a commercial transfection agent (i.e., Dharmafect-1) is also effective at knocking down STAT3 in cancer cells, but less effective in immune cells even with the manufacturer’s recommended formulation (i.e., Dharmafect-4 for J774) (Figure S2B). Importantly, when co-delivering siRNA and CpG, the siRNA-CpG-NP nanoconstructs still elicits effective gene knockdown, while siRNA-CpG-Dharmafect lost most of the knockdown efficacy (Figure S2C). While siRNA-CpG-NP maintains a hydrodynamic size of about 100 nm, the size of oligo-loaded Dharmafect-1 and Dharmafect-4 ranges from 500-900 nm (Figure S2D). When compared with other platforms, our NP required 50 nM siSTAT3 to achieve about 70% STAT3 knock down, much lower than those reported by CpG-conjugated siRNA of 500-1000 nM.[31] In addition, our NP can knock down the target gene more effectively than the siRNA-PEI-polyplex counterpart (75% vs. 0% luciferase knockdown when using equal 30 nM luciferase siRNA, Figure S3), indicating the importance of the silica core. The rigid MSNP core and optimal crosslinked PEI and PEG loading allow high loading of siRNA and CpG without increased size (e.g., hydrodynamic size of 106 nm (Figure 1) vs. 650 nm for siRNA-PEI polyplex (Figure S3) or 500-900 nm for siRNA-Dharmafect (Figure S2D). NP loaded with siRNA and/or CpG also did not decrease viability of immune cells (BMDC and J774, Figure S4).
Figure S5 shows that siRNA-CpG-NP was taken up effectively in a panel of immune cells (J774 and BMDC) and cancer cells (B16F10, CT26, and MM3MG-HER2Δ16) in vitro after 2-h incubation, which is in agreement with knock-down efficacy across multiple immune and cancer cell lines. Also in agreement with the in vitro uptake data, intratumorally injected NP co-delivering siRNA and CpG (siRNA-CpG-NP) was taken up by both cancer and myeloid cells in the TME (Figure S6). This experiment was performed in mice with bilateral tumors as described in Figure 2B, wherein only one tumor was treated. Figure S6 shows that at two hours after intratumoral injection, siRNA-CpG-NPs were taken up by 15% of cells in the treated tumor. Of the cells that took up the siRNA-CpG-NP, 80% were non-hematopoietic cells (CD45−, primarily cancer cells),[32] and 20% were hematopoietic cells (CD45+, primarily immune cells). Out of the immune cells, myeloid cells (CD45+CD3−CD19−) took up the most siRNA-CpG-NP. These include macrophages (F4/80+) and DCs (CD11c+MHCII+). In comparison, after 1-3 hours, CpG-siRNA conjugates were reported to be taken up by 2% of cells in the tumor, which are mainly (>87%) DCs and macrophages (labelled with CD11c and F4/80).[31] This is because the CpG-siRNA conjugate depends on CpG for processing and is primarily taken up and processed by only TLR9+ immune cells (e.g., DCs and macrophages), and not TLR9− cells.[31] Delivery of siSTAT3 and CpG to both cancer and myeloid cells enabled by our NP has the potential to elicit a strong immunotherapeutic effect. We did not detect any NP in the distant untreated tumor (i.e., no cells have fluorescent signal of siRNA-CpG-NP (NP+) above that of tumor cells from saline treated mice, not shown), indicating no significant systemic leakage of the nanoconstruct to the untreated tumor.
As anticipated, using the aforementioned bilateral B16F10 melanoma tumor model, Figure 2 shows that co-delivery of siSTAT3 and CpG on the NP (AIRISE-02; siSTAT3-CpG-NP) intratumorally resulted in a statistically significant reduction in tumor volume of both treated tumors and distant tumors, as well as an improvement in survival compared to either CpG-NP or siSTAT3-NP (Figure 2B–2D). In particular, AIRISE-02 treated tumors were significantly smaller than saline treated tumors starting from day 7 (p<0.0001). The distant tumors were also smaller in the AIRISE-02 treated group starting from day 8 (p<0.05). No statistical comparison with the saline group was performed beyond day 9 when many saline treated mice were sacrificed due to their combined tumor volume exceeding 2000 mm3. CpG-NP exhibited a greater anti-tumor effect than siSTAT3-NP. However, AIRISE-02 was significantly more effective than CpG-NP in inhibiting local tumors (p<0.05 at day 13) and distant tumors (p<0.0001 at day 15).
To determine whether intratumoral therapy altered the immune profile in either the local or distant tumor, we treated tumor-bearing mice with AIRISE-02 in the same manner as Figure 2A. At day 7, tumor and DLNs were collected and subjected to immune profiling with multi-color flow cytometry. Figure 2E–2G show that AIRISE-02 resulted in significantly higher CD8/Treg ratios in both local and distant tumors and DLNs (p<0.05 for AIRISE-02 vs. saline), confirming successful in situ tumor vaccination. CpG-NP or siSTAT3-NP did not significantly increase CD8/Treg in tumors or DLNs at this dose and time point, consistent with its poorer efficacy than AIRISE-02. AIRISE-02 also induced proliferation (assessed by Ki67+) of effector CD8+ T cells in DLNs (associated with both treated and untreated tumors) more than CpG-NP or siSTAT3-NP (Figure 2G).
As discussed earlier, we found no trace of AIRISE-02 in the distant tumors, but the immune profile in these untreated tumors and associated lymph nodes was altered. Thus, the effect of AIRISE-02 in distant tumors is primarily immune mediated rather than directly cytotoxic. To further substantiate the role of adaptive immune response, we performed CD8 depletion in mice using anti-CD8 antibodies and found that the therapeutic response of AIRISE-02 was mostly eliminated (Figure 2H–2J). Specifically, there was no difference in tumor volume of distant tumors in mice treated with AIRISE-02 + anti-CD8 antibodies compared to those of saline treated mice. For local tumors, the difference was not observed until day 9 (p<0.001). AIRISE-02 + anti-CD8 antibodies did not completely eliminate AIRISE-02’s therapeutic action in terms of local tumor volume and survival, suggesting other factors, such as direct cancer cell killing and innate immunity, may be involved. Nevertheless, the therapeutic action of AIRISE-02 is primarily CD8+ T cell-dependent.
Based on these data, the mechanism of how AIRISE-02 manipulates a local tumor into a site that stimulates whole-body antitumor immune response is illustrated in Scheme 1. Upon injection, AIRISE-02 is taken up by both cancer and myeloid cells (e.g., DCs, MDSCs, macrophages) in the tumor. Knocking down STAT3 negates immunosuppressive pathways[18] in both cancer and myeloid cells (Figure S2), but is not toxic to these immune cells (Figure S4). STAT3 also has conserved region such that the same siSTAT3 sequence can knock down STAT3 in murine, canine, and human cells, faciliating direct translation from mice to dogs and humans (Figure S2). Utilizing tumor antigens in situ, intratumoral administration of AIRISE-02 leads to control of both treated and untreated tumors (Figure 2) owing to higher CD8/Treg ratios and enhanced CD8+ T cell proliferation in tumors and tumor draining lymph nodes. Regulatory T cells (Treg) are typically elevated in patients’ tumors and suppress antitumor immune response.[33] Thus, a high CD8/Treg ratio in tumors is desirable and is one indicator of prolonged survival in cancer patients. We also further substantiated that the treatment effect is primarily CD8-dependent.
Scheme 1. In situ tumor vaccination effect with AIRISE-02 (siSTAT3-CpG-NP).
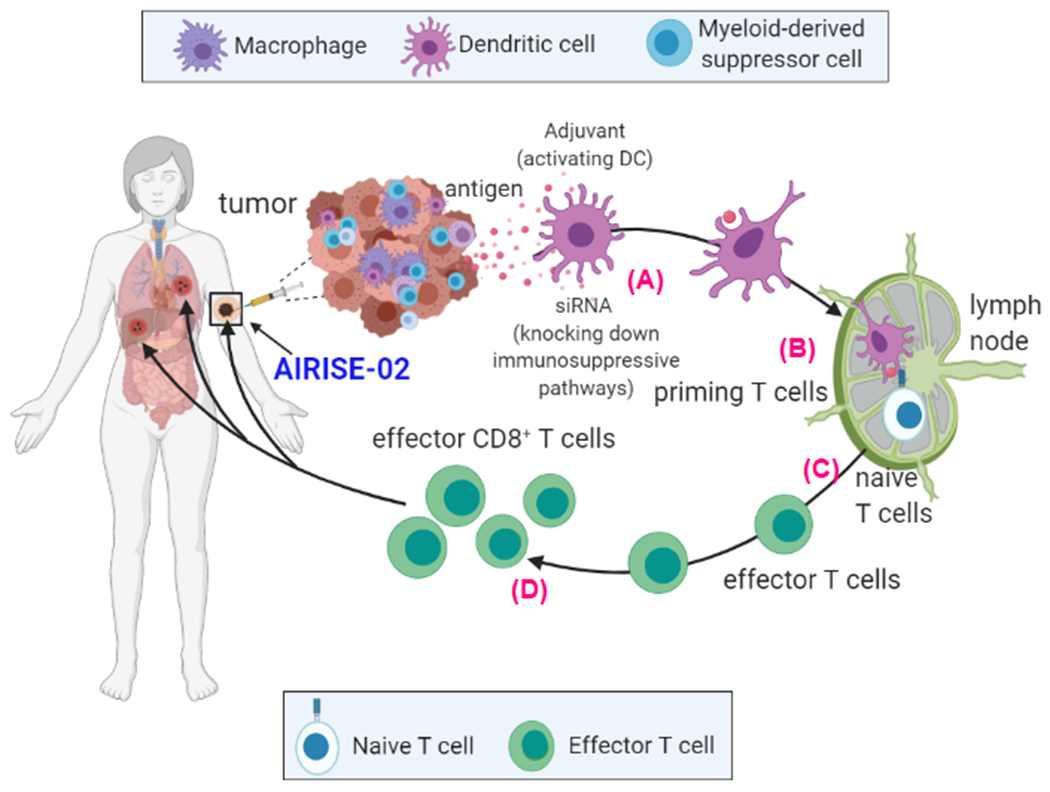
Upon intratumoral injection of AIRISE-02 to the primary tumor, A) CpG activates local antigen-presenting cells (APCs, primarily DCs). siSTAT3 negates immunosuppressive pathways in both cancer and myeloid cells (e.g., DCs, MDSCs, macrophages) in the TME. Tumor antigens are taken up by the AIRISE-02-activated APCs in the TME, which then traffic to tumor-draining lymph nodes. B) APCs then (cross) present these antigens to prime tumor antigen-specific T cells. C) The activated cytotoxic (effector) T cells proliferate and exit the lymph node into systemic circulation. D) They home specifically to tumors sharing the same antigens wherever they are located in the body (e.g., both treated primary tumor and untreated tumors in distant metastatic sites). Death of cancer cells by cytotoxic T cells releases more tumor antigens, amplifying the process of anti-tumor T cell generation in a positive feedback loop. This vaccination induced locally at the tumor site generates systemic antitumor immune response. Figure created with BioRender.com.
Since intratumoral AIRISE-02 boosts systemic CD8+ anti-tumor T cell repertoire, we hypothesized that AIRISE-02 would enhance the efficacy of ICIs. In this tumor model, ICIs against CTLA-4 and PD-1 (given i.p.) markedly decreased the tumor volume of both local and distant tumor sites (Figure 3A–B). When compared with ICIs alone, the addition of AIRISE-02 inhibited the growth of local tumor further (p<0.01), but not significantly in the distant tumor at day 15. However, remarkably, the combination of AIRISE-02 and ICIs appeared to cure 5 out of 8 mice (which remained tumor-free for 17 months), while no mice were cured with AIRISE-02 or ICIs alone (Figure 3C). Curative effect in the B16F10 model is impressive since this model is known to be aggressive, and no curative effect was previously reported with other CpG-based vaccines + ICIs when the tumors had been established. For instance, novel CpG-based nano-vaccines bearing tumor antigens successfully show complete B16F10 tumor rejection in a prophylactic setting,[34] but the vaccines combined with ICIs did not result in long-term cure when treatment started after tumor implantation.[34a]
Figure 3. AIRISE-02 + immune checkpoint inhibitors (ICIs) cured mice bearing B16F10 bilateral tumors.
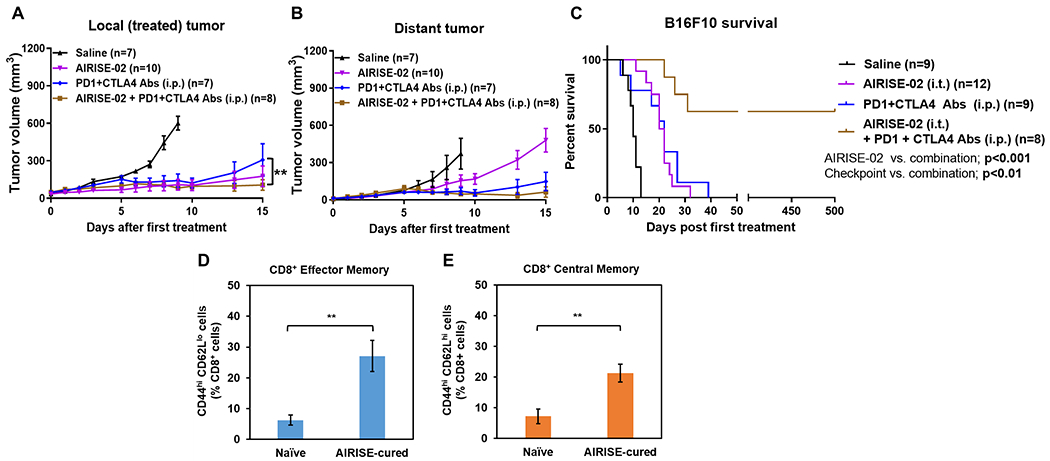
Mice were treated with AIRISE-02 alone (same model and AIRISE-02 treatment as in Figure 2), ICIs alone (200 μg PD-1 Ab + 100 μg CTLA-4 Ab/mouse; i.p. given three times total on the same days as AIRISE-02) or AIRISE-02 + ICIs (given on the same day). A,B) Tumor growth curves of local tumors (A) and distant (untreated with AIRISE-02) tumors (B) are plotted as mean ± SEM for mice that survived the first 9-15 days (i.e., >75% of all mice per group, each group still has the same average starting tumor size). Spider plots of tumor sizes for all mice without exclusion were reported in Figure S7 in the Supporting Information. C) Survival curve of all mice. D,E) At 17 months, blood was collected to investigate the number of circulating effector memory (CD44hi CD62Llo) (D) and central memory (CD44hi CD62Lhi) (E) T cells as percentage of CD8+ T (CD3+CD45+) cells. **p<0.01. Saline and AIRISE-02 treatments were conducted independently twice (along with other groups in Figures 2 and in this figure), and the combined data were used for both figures.
We performed tumor rechallenge by implanting B16F10 cells in the cured mice at 3 months after the last treatment. The implanted B16F10 cells did not develop tumors, suggesting long-lasting anti-tumor immunity (memory T cell effect) in these cured mice. Further, at 17 months, we measured the levels of circulating effector (CD44hiCD62Llo) and central (CD44hiCD62Lhi) memory CD8+ T cells, which differ in proliferative capacity, location, and response to antigens,[35] in blood of the cured mice vs. naïve mice. In agreement with prior studies,[36] we found that the cured mice had both higher effector memory T cells (p<0.01, Figure 3D) and central memory T cells (p<0.01, Figure 3E) compared to naïve mice, supporting the ability of AIRISE-02 + ICIs to induce long-lasting T cell immunity. Elevation of both central and effector memory T cells indicates a robust memory T cell population capable of rapid proliferation, anti-tumor cytokine secretion, and cytolytic activity.[37]
In addition to melanoma, AIRISE-02 was also effective in other tumor models, such as colon cancer (CT26) as shown in Figure 4A–4C. The ICIs (PD-1 and CTLA-4 inhibitors) were highly effective in CT26 tumors; however, tumors continued to grow in 2 out of 6 mice. In contrast, the combination of AIRISE-02 and ICIs induced complete responses in 6 out of 6 mice. Even for the untreated distant tumors, AIRISE-02 + ICIs resulted in smaller tumors than ICIs alone (p<0.05 at day 22). Moreover, AIRISE-02 also improved the effiacy of one ICI (PD-1 antibody) in a HER2+ (MM3MG-HER2Δ16) orthotopic breast tumor model (Figure 4D–4F).[38] In a bilateral MM3MG-HER2Δ16 tumor model, AIRISE-02 + ICI decreased burden of both tumors (e.g., p<0.01 vs. saline for both local and distant tumors on day 20) more effectively than ICI alone. Together, these show the broad efficacy of AIRISE-02 across different tumor types.
Figure 4. AIRISE-02 + ICIs combination treatment is efficacious in syngeneic murine tumor models of colon cancer and breast cancer.
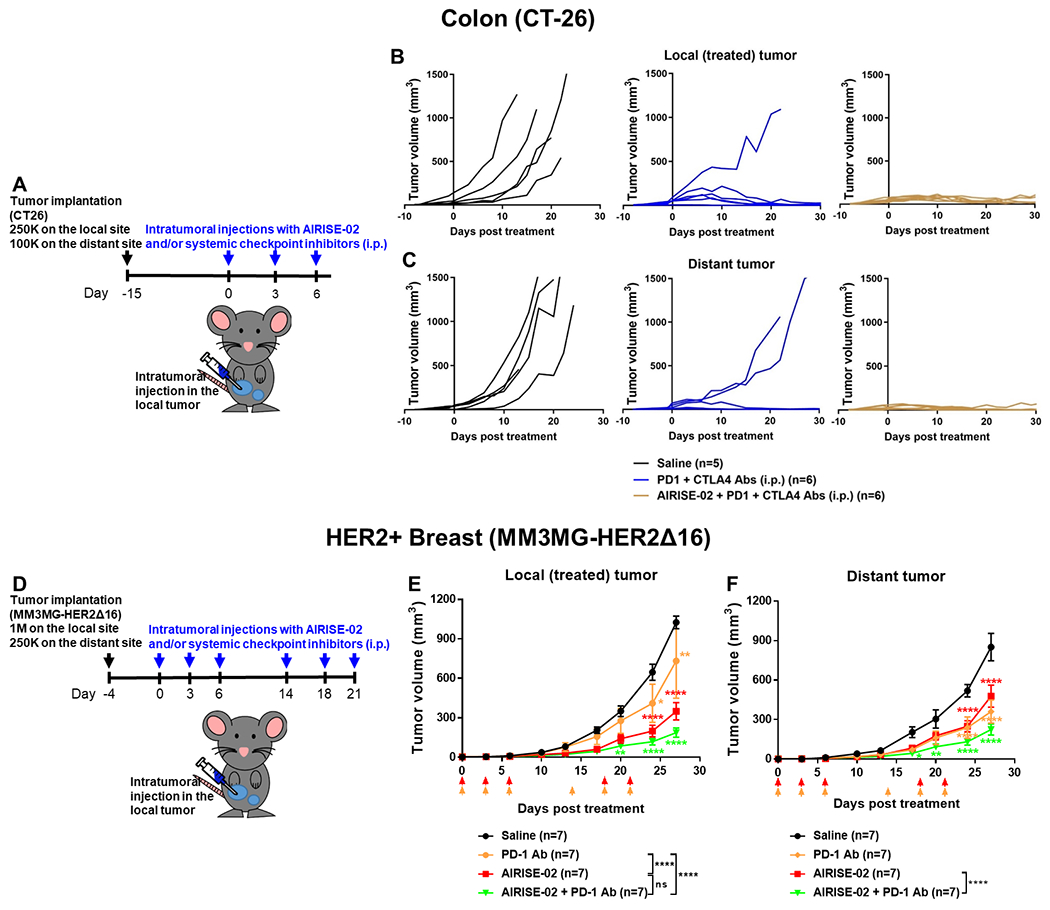
A) Balb/c mice were implanted with 250,000 and 100,000 CT26 cells on bilateral abdomens; 15 days after tumor implantation, mice were treated as described. Mice were treated with ICIs (PD-1 + CTLA-4 antibodies) with or without AIRISE-02. Tumor growth curves of B) local treated tumors and C) distant untreated tumors are plotted as spider plots (each line represents an individual mouse) due to the short survival time of untreated mice and variation in responses. D) Balb/c mice were implanted with 1,000,000 and 250,000 MM3MG-HER2Δ16 cells on bilateral mammary fat pads; 4 days after tumor implantation, mice were treated with ICI (PD-1 antibody) with or without AIRISE-02 as indicated by arrows. Tumor growth curves of E) local treated tumors and F) distant untreated tumors are plotted as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 denote statistical significance between the tested group and saline control at different time points. Statistical significance between each tested group was also evaluated on day 27, and significance levels are shown in figure legends. AIRISE-02 comprises 0.26 mg NP with 7 wt.% CpG and 2 wt.% siRNA per each i.t. injection; dosage of PD-1 Ab is 200 μg/mouse/injection (i.p.); and dosage of CTLA-4 Ab is 100 μg/mouse/injection (i.p.).
In addition to intratumoral (i.t.) route, AIRISE-02 also provided therapeutic effect upon intravenous (i.v., tail-vein) injection (p<0.0001 vs. saline) in the same bilateral B16F10 tumor model (Figure S8). However, intratumorally administered AIRISE-02 yielded superior outcome compared to the i.v. counterpart (p<0.05) in terms of overall survival, even when i.t. dose was three fold lower than the i.v. dose, and only one of the two tumors was i.t. injected. Nevertheless, i.v. AIRISE-02 may be useful for deeper tumors that are not accessible by local injection.
In terms of safety, multiple i.t. doses of AIRISE-02 or AIRISE-02 + PD-1 antibody in mice bearing bilateral MM3MG-HER2Δ16 tumors as described above (Figure 4D) resulted in no observed toxicity (e.g., no body weight change vs. saline, Figure 5A). Biomarkers of mouse serum samples at the study completion also revealed no liver or kidney toxicity (Figure 5A, no statistical significance between the test groups and saline). This was later substantiated by histology grading of key organs in a blinded manner (Mass Histology Service, Inc). Specifically, histopathology performed by a qualified animal histopathologist reported no evidence of systemic adverse effects (e.g., no inflammation, necrosis, thrombosis) in all tissues evaluated (i.e., liver, lung, kidneys, spleen—data not shown). Draize test[39] also revealed no adverse reactions at the injection site of tumor-free mice receiving an efficacious dose of AIRISE-02 (n = 3, time points observed at 0, 24 h, and 72 h) intramuscularly compared to pre-injection (Figure 5B).
Figure 5. AIRISE-02 is safe in mice and monkeys.
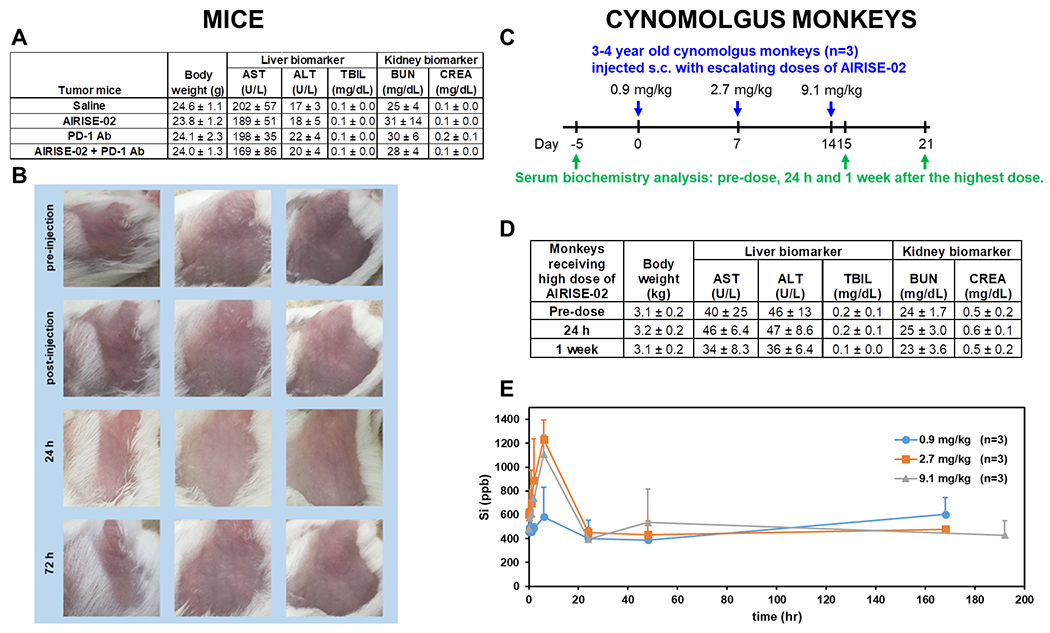
A) Serum biochemistry from mice treated with multiple doses of AIRISE-02 (siSTAT3-CpG-NP: 0.26 mg NP, with 7 wt.% CpG and 2 wt.% siRNA per each injection), PD-1 Ab, AIRISE-02 + PD-1 Ab, or saline at the study endpoint (i.e., mice from efficacy study in Figure 4D). B) 13-week old naïve tumor-free Balb/c mice were injected intramuscularly into the thigh muscle with a dose of AIRISE-02 (same dose as (A)). Injection site was evaluated before and immediately after injection, 24 h, and 72 h after injection for edema and erythema using the standard Draize test criteria. C) Cynomolgus monkeys were injected subcutaneously with 3 escalating doses of AIRISE-02 as indicated by blue arrows. D) Serum biochemistry was evaluated before the treatment, and 24 hours and 1 week after the highest dose as outlined by green arrows in (C). E) Si (silicon) in plasma at different time points after each dose was quantified by inductively coupled plasma mass spectrometry (ICP-MS) and reported as concentration (ppb) in plasma. AST, aspartate aminotransferase; ALT, alanine aminotransferase; TBIL, total bilirubin; BUN, blood urea nitrogen; CREA, creatinine.
Charles River Laboratory (CRL) was contracted to conduct a preliminary safety profile of AIRISE-02 in cynomolgus monkeys. Female monkeys (n=3) were injected subcutaneously with escalating doses of 0.9 (low), 2.7 (mid), and 9.1 mg/kg (high) of AIRISE-02 with a one week washout period between dosing (Figure 5C). The mid dose contains about 0.5 mg CpG per dose, similar to that used in other primate studies[40] and is our anticipated efficacious dose (derived from mouse data). CRL reported that all three doses were safe with no systemic toxicity, and none had reached the maximum tolerated dose. At the mid dose, grade 2-3 edema was reported at 2-3 days post-treatment, but resolved within a week. Some edema (grades 1-3) and erythema (grade 1) were also noted in animals receiving the highest dose but they were not dose limiting according to the CRL toxicologists. Inflammation at the injection site is anticipated due to immunostimulatory effect, and thus should be less concerning with intratumoral treatment than subcutaneous treatment. Figure 5D shows body weight and serum biomarkers from monkeys receiving the highest dose of AIRISE-02, which indicates no systemic toxicity (i.e., no significant difference between pre-treatment and post-treatment). Full datasets from the monkey study including body weight, hematology parameters (e.g., white blood cell count, neutrophils, red blood cell count, platelet count), coagulation parameters (e.g., prothrombin time, activated partial thromboplastin time, fibrinogen), clinical chemistry parameters (e.g., aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, blood urea nitrogen, creatinine, creatine kinase), dermal observation, and cytokine levels (TNF-α, IFN-γ, IL-1b, IL-6, and MCP-1) are provided in the Supplementary Dataset.
We also measured the pharmacokinetic profile of AIRISE-02 in the monkey safety study by monitoring levels of Si (silicon from MSNP) and siSTAT3/CpG in plasma samples collected at various time points after subcutaneous injection in monkeys. Si content was characterized by ICP-MS, and CpG and siSTAT3 by a hybridization-based AEX-HPLC fluorescence method.[41] This method is very sensitive, yielding a lower detection limit of 1.6 nM siSTAT3 in plasma. Across each dose level, we found the peak plasma Si to be 600-1200 ppb, which corresponds to 1-4% of the injected doses at 6 hours post-treatment. Si level returned to baseline at 24 hours (Figure 5E). Even at the highest dose, we did not find detectable levels of siSTAT3 and CpG in the plasma at any time point. All data suggest very low release of AIRISE-02 into the systemic circulation and/or rapid clearance of any excess Si, siSTAT3, and CpG by the body (e.g., via kidneys).
Mesoporous silica nanoparticles are water-soluble at physiological pH (pH 7.4) into benign silicic acid[42] and can be fully excreted from the body within 4 days in urine and feces.[43] In vitro solubility was also reported to agree with the body clearance of mesoporous silica nanoparticles. Likewise, Figure S9 shows that our PEG-PEI coated mesoporous silica nanoparticles (MSNP) are also soluble over time in PBS (pH 7.4); e.g., by >50% in 24 h at 0.2 mg/ml of starting nanoparticle concentration). The solubility is concentration-dependent and has a limit in a closed system (e.g., plateaued at 85% dissolved when starting at 0.2 mg/ml and 25% dissolved when starting at 2.0 mg/ml). In vivo, it is anticipated to be fully dissolved (as the dissolved species is transported from the injection site to circulation) and cleared from the body over time. This is in agreement with our observation in monkeys (Figure 5E) where we found plasma level of Si (silicon) to peak at 6 hours post s.c. injection and level out to baseline plasma Si level. What’s more, silica is first dissolved from inside out because the pore wall is weaker than the outer shell.[44] Accordingly, at 50% solubility, we observe the particle size remains intact initially (see NP sizes in the first 24 hours, Figure S9D). During this time, the siRNA and CpG remain with the particles (since they are bound to the polymers on the surface). The nanoparticle was previously reported to protect 100% of siRNA from enzymatic degradation in serum for at least 24 hours.[22]
Toward clinical applications, AIRISE-02 is under IND enabling studies by contract manufacturing organizations (CMOs), and the stability studies under GMP will be reported in due course. In short, AIRISE-02 is designed as a two-vial formulation; nanoparticles in PBS in one vial and siSTAT3+CpG in PBS in another vial. The NP vial can be terminal-sterilized by e-beam irradiation, and we found that the NP maintains the size and siRNA delivery efficacy after such terminal sterilization (Figures S10A–B). Both vials will be stored at −20 °C, thawed, and mixed together in the clinical pharmacy before injecting to patients. We found in our lab that the nanoparticles are stable at −20 °C for at least 4 months, longest tested to date (Figure S10C–D) and −80 °C for years (not shown) in terms of size and knockdown efficacy. Likewise, siRNA and CpG are known to be stable for years at −20 °C. The stored nanoparticles can bind siRNA and CpG in under a few minutes of simple mixing at room temperature, which achieves complete binding.
In conclusion, nanoparticle-based AIRISE-02 co-delivers CpG and STAT3 siRNA to induce effective whole-body antitumor immune response upon intratumoral treatment, as shown by regression of both treated and untreated tumors, along with curative outcome when combined with ICIs. CpG and STAT3 siRNA have been previously co-delivered as a conjugate in a seminal report by Kortylewski and colleagues.[31] The technology showed promising efficacy in multiple tumor models. However, CpG-siRNA conjugates get processed and elicit gene knockdown only in TLR9+ cells, while AIRISE-02 entered and elicited gene knockdown in both cancer and APCs/myeloid cells regardless of TLR9 status (Figure S2). This is owing to a slight cationic charge and surface roughness of the NP, which promotes cellular uptake[45] and successful gene knockdown in both myeloid and cancer cells independent of TLR9. Nanoparticles also enhance serum stability and tumor retention of oligonucleotides compared to conjugates (whose typical size is less than 10 nm). Furthermore, the siRNA and CpG payloads per uptake event are higher for a nanoparticle than a conjugate. For instance, each of our particles carries 1 x 104 siRNA molecules (calculated from 2.0 wt.% of siRNA (14 kDa) per NP and 8.8 x 1010 NP per mg measured by NanoSight (Malvern, PA)), while a conjugate carries 1-2 siRNAs per conjugate. As a result, knocking down 70% of STAT3 with siRNA delivered on our NP required about 50 nM of siRNA, while the reported conjugates required 500-1000 nM to achieve the knockdown efficacy.[31] In short, higher payload, better tumor retention, better protection against enzymatic degradation, and ability to be taken up by both cancer and APCs lead to high efficacy of AIRISE-02. In particular, while AIRISE-02 required 3 doses at 3 days apart, CpG-siSTAT3 conjugates required 8-10 doses given every other day to achieve similar in vivo efficacy.[31] Nanoparticle technology also provides the opportunity to incorporate multiple siRNAs to simultaneously manipulate TME in subsequent therapeutic pipelines.
AIRISE-02 is a nanoparticle co-delivering an adjuvant and siRNA intratumorally (without adding antigens) to induce antitumor immune response in both locally treated and distant untreated tumors. Co-delivery of tumor antigens and adjuvants (e.g., CpG, R837) with or without siRNA (e.g., siSTAT3, siIL-10) on polymeric nanoparticles has been investigated before for cancer vaccination.[46] AIRISE-02, on the other hand, does not co-deliver antigens, but stimulate recognition of the unique antigens in the treated tumor of each patient (in situ). In situ tumor vaccination approach circumvents the costly and time-consuming process of identifying tumor antigens, generating and incorporating them into a vaccine formulation. In situ tumor vaccination with CpG-loaded peptide-based nanocomplex (6-7 doses) was recently reported to show impressive inhibition of the distal tumor in a bilateral B16F10 tumor mouse model, when compared to naked CpG. The CpG-nanocomplex also improved responsiveness to anti-CTLA-4 antibodies; however, complete regression or cure was not reported.[29c] AIRISE-02 achieved cure in mice when combining with ICIs, suggesting the benefits of siSTAT3 and our nanoparticle delivery.
Not any nanoparticle type will lead to the same efficacy as our NP. Table S2 compares our NP and other nanoparticle benchmarks for potential use in in situ vaccination. siRNA on PEI polyplex and lipid (Dharmafect) carriers have much larger hydrodynamic sizes (500 nm-1 micron) and poorer gene knockdown than that of our NP. The nanoparticle core of AIRISE-02 is mesoporous silica (MSNP), which is inexpensive and biodegradable, and does not trigger oxidative stress[23] that could worsen an immunosuppressive TME. Silicon (Si) is also the third most abundant trace element in the body after iron and zinc.[47] A silica NP with PET tracers was in a clinical trial with favorable safety profile.[48] Layer-by-layer surface modification affords high batch-to-batch reproducibility and the ability to purify and remove unbound reagents after every layer (by centrifuge or tangential flow filtration). Low content and small molecular weight of cationic polymer (PEI) is used with biodegradable cross-linkers, allowing high efficacy without toxicity of high molecular weight PEI.[22] MSNPs are benign and soluble at physiological pH to non-toxic silicic acid for kidney clearance.[42] As a result, AIRISE-02 has been shown to be safe in mice and non-human primates, and is anticipated to be safe in humans.
T-VEC is an FDA approved intralesional therapy for unresectable melanoma that is considered to be a category 1 drug (high-level of evidence).[49] We cannot compare AIRISE-02 and T-VEC in B16F10 tumor model since B16F10 cells lack HSV-1 viral entry receptors, which are required for T-VEC entry and function.[29b] In the CT26 colon model, which has the receptors, 6 out of 10 mice treated with 4 doses of OncoVEXmGM-CSF (mouse version of T-VEC) + anti-CTLA-4 antibodies responded,[50] while 6 out of 6 mice treated with 3 doses of AIRISE-02 + anti-PD-1/CTLA-4 antibodies responded (see Figure 4). Although this is not a side-by-side comparison, the observations show promise for the AIRISE-02 approach. T-VEC relies on virus replication, which can be hindered in late-stage indolent tumors or a highly immunosuppressive TME.[7] On the other hand, nanoparticles do not require virus replication, and siSTAT3 can modulate an immunosuppressive TME. Altogether, data suggest that AIRISE-02 warrants exploration in a clinical setting. In addition, intratumoral injection of AIRISE-02 limits drug exposure to systemic circulation, thus should have lower toxicity concerns and less regulatory (FDA) challenges compared to the systemic counterpart. AIRISE-02 is hence under IND-enabling studies (GMP manufacturing and GLP-compliant toxicology studies), and phase I/IIa clinical trials enrolling patients with melanoma, breast, head and neck cancer as well skin metastatic colon cancer (similar to those of T-VEC)[51] will soon follow. We envision that AIRISE-02 can be applicable to many cutaneous tumors, including breast, melanoma, head and neck cancer, cutaneous lymphoma, along with cancers amendable to local administration, such as ovarian, bladder, cancer with skin (e.g., colorectal, lung, and kidney cancers) and lymph node metastasis (e.g., gastric cancer).
Lastly, siRNA holds great potential since any gene can be modulated precisely and effectively, allowing various immunosuppressive pathways to be tackled. In addition, different specific immune cell populations can also be targeted by conjugating antibodies on our NP as homing moieties.[22] As we understand more about the TME and immune systems, the robust AIRISE nanotechnology could be utilized to rapidly translate new findings into clinical therapeutics.
Experimental Section
Materials:
Cetyltrimethylammonium chloride (CTAC), tri-ethanolamine (TEA), tetraethyl orthosilicate (TEOS), and 2-mercaptoethanol (β-Me) were obtained from Sigma-Aldrich (MO). Branched-PEI (1.8 or 10 kDa) was obtained from Alfa Aesar (MA). Mal-PEG(5 kDa)-NHS was obtained from JenKem Technology USA (TX). PBS pH 7.2, RNase free water, ethanol, hydrochloric acid (HCl), sodium hydroxide (NaOH), dithiobis(succinimidyl propionate) (DSP), DharmaFECT #1, DharmaFECT #4, fetal bovine serum (FBS), RPMI-1640 medium, DMEM medium, IMDM medium, L-glutamine, and penicillin-streptomycin-glutamine were obtained from Thermo Fisher Scientific (MA). ATCC-formulated Eagle’s Minimum Essential Medium, mouse RBC lysis buffer, bovine serum albumin (BSA), FcR (CD16/CD32), HBSS, and Collagenase D were obtained from Fisher Scientific (PA). Anti-mouse PD-1 (clone RMP1-14) and anti-mouse CTLA-4 (clone 9H10) were purchased from BioXcell (NH). GM-CSF and IL-4 were purchased from Peprotech (NJ). Lipopolysaccharides (LPS) was purchased from Invivogen (CA). DNase I was from Roche Diagnostics (Germany). All reagents are of highest purity grade available.
Mouse CpG 1826 and human CpG 7909 were obtained from Invivogen (CA). siRNAs were synthesized by Thermo Fisher Scientific (MA) or Dharmacon (CO). Scrambled siRNA with Alexa Fluor 488 attached to the sense strand were obtained from Qiagen (CA).
The sequences are summarized as follows. Upper case letters denote riboses, and lower case letters denote deoxyriboses.
| siRNA/CpG | Sequence |
|---|---|
| siSTAT3 | Sense: 5’-GGAUCUAGAACAGAAAAUGtt-3’ Antisense: 5’-CAUUUUCUGUUCUAGAUCCtg-3’ |
| siLUC | Sense: 5’-CGGAUUACCAGGGAUUUCAtt-3’ Antisense: 5’-UGAAAUCCCUGGUAAUCCGtt-3’ |
| siSCR | Sense: 5’-UGGUUUACAUGUCGACUAA-3’ Antisense: 5’-UUAGUCGACAUGUAAACCA-3’ |
| CpG 1826* | 5’-tccatgacgttcctgacgtt-3’ |
| CpG 7909 (2006)* | 5’-tcgtcgttttgtcgttttgtcgtt-3’ |
phosphorothioate backbone
Cells (HCC1954, B16F10, J774, CT26, and D-17) were obtained from ATCC, while BMDCs were harvested from Balb/c mice following published protocols.[52] LM2-4luc+H2N was obtained as a gift from Prof. Robert Kerbel (University of Toronto, Canada). MM3MG-HER2Δ16 was obtained as a gift from Prof. Zachary Hartman (Duke University, NC). Cell media recipes are summarized below.
| Cell lines | Cell media recipe |
|---|---|
| HCC1954, CT26 | RPMI-1640 + 10% FBS |
| B16F10, J774 | DMEM + 10% FBS |
| MM3MG-HER2Δ16 | DMEM + 10% FBS + 1X Antibiotic/antimycotic |
| D-17 | EMEM + 10% FBS |
| LM2-4luc+H2N | RPMI-1640 + 5% FBS |
| BMDC | Primary DC media: IMDM + 10% FBS + 3 mM L-glutamine + 100 IU/mL penicillin + 100 mg/mL streptomycin + 50 μM β-Me + 25 ng/mL GM-CSF + 25 ng/mL IL-4 Secondary DC media: primary DC media + 100 ng/mL LPS |
Nanoparticle Synthesis:
Mesoporous Silica Nanoparticle (MSNP) was synthesized and characterized following our published protocol.[22,53] Briefly, CTAC was mixed with TEA in water at 95°C. TEOS was then slowly added to the mixture under vigorous stirring for 1 h. MSNP was then washed and dried overnight. The dried MSNP was resuspended and refluxed in acidic methanol (0.6 M HCl), washed, and dried. To make a functionalized nanoparticle (NP), MSNP (10 mg) was shaken with 2.5 mg branched-PEI in ethanol for 3 h at room temperature, centrifuged, and resuspended in an ethanol solution containing PEI and 0.2 mg DSP as a cross-linker. The mixture was shaken for 40 min, then washed and resuspended in PBS pH 7.2. Mal-PEG(5 kDa)-NHS (10 mg) was conjugated to MSNP-PEI by shaking for 2 h. The MSNP-PEI-PEG was washed with PBS, resuspended, and stored in PBS. The suspension can be stored at −20 °C or −80 °C for long-term use, or lyophilized following our prior report.[53] The optimal NP consists of 68.3 wt.% 50 nm-MSNP, 13.5 wt.% PEI, and 18.2 wt.% PEG by TGA analysis.[22]
siRNA and CpG oligo were added onto the nanoparticles by 10-min mixing in PBS at room temperature, which achieved complete binding. The complete binding was confirmed by no detectable levels of CpG and siRNA in the supernatant after the siRNA-CpG-NPs were pelleted down by centrifugation. The supernatant siRNA and CpG levels were monitored by fluorescent assessment of dye-tagged oligonucleotides along with Nanodrop Spectrophotometry. The oligo bound to the PEI layer was protected under the PEG layer from enzymatic degradation by at least 24 hours in 50% human serum.[22]
Material characterization:
Nanoparticles’ hydrodynamic size and zeta potential (charge) were measured with Zetasizer (ZS-90/Malvern, Westborough, MA). To quantify nanoparticle concentration and polymer loading, 1 mg nanoparticles (MSNP, MSNP-PEI, or MSNP-PEI-PEG) were heated to 950 °C (20 °C/min) with TGA Q50 (TA Instruments, New Castle, DE). Weight/temperature profiles of MSNP, MSNP-PEI, and MSNP-PEI-PEG were compared to determine percent loading of each polymer by weight of silica.
Unbound oligonucleotide in the solution (or the lack thereof) was characterized using gel electrophoresis following our prior report[22] and quantitatively by Nanodrop Spectrophotometry or hybridization-based AEX-HPLC fluorescence.[41]
STAT3 knockdown and viability of cells treated with AIRISE-02:
AIRISE-02 (35 μg/ml; 50 nM siRNA) was evaluated for its STAT3 knockdown efficacy in canine osteosarcoma (D-17), murine melanoma cell (B16F10), murine breast cancer cell (MM3MG-HER2Δ16), murine colorectal cancer cell (CT26), murine bone marrow derived dendritic cells (BMDC), murine macrophage (J774), and human breast cancer cells (HCC1954). STAT3 mRNA knockdown was evaluated at 2 days post-treatment by qRT-PCR, and HPRT was used as a housekeeping gene. RNA was isolated and purified from cell lysate using GeneJet RNA purification kit from Thermo Fisher Scientific (Waltham, MA) following manufacturer’s protocol. RNA concentration was determined using Nanodrop Spectrophotometer. One-Step qRT-PCR was performed on Rotor-Gene Q (Qiagen, Germany), using EXPRESS One-Step Superscript™ qRT-PCR Kit from Invitrogen (CA). Twenty nanograms of RNA was used per reaction. Cycling conditions were 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. TAQMAN gene expression primers: human HPRT mRNA (Hs99999909_m1), human STAT3 mRNA (Hs1051722_s1), mouse HPRT mRNA (Mm00446968_m1), mouse STAT3 mRNA (Mm_01219775_m1), canine HPRT mRNA (Cf02626256_m1), and canine STAT3 mRNA (Cf02666647_m1) were purchased from Thermo Fisher Scientific (MA). Data was analyzed using 2−ΔΔCT method.
The effects were benchmarked with CpG and siSTAT3 delivered by commercial transfection agent Dharmafect (Horizon Discovery, following manufacturer’s protocol). Dharmafect #1 was used for all cancer cells, while Dharmafect #4 was used for immune cells per manufacturer’s recommendation. We found Dharmafect #4 to have slightly better efficacy in J774 (Figure S2B) than Dharmafect #1 (not shown). Cell viability was also evaluated at 2 days post-treatment using CellTiter-Glo® (Promega), following manufacturer’s protocol.
Uptake profile of AIRISE-02 in vitro:
AIRISE-02 was loaded with Alexa488-tagged siRNA to monitor in vitro AIRISE-02 uptake in BMDC, J774, B16F10, CT26, and MM3MG-HER2Δ16 cells. Cells were suspended at 1 million cells/mL and mixed with 100 μg/mL Alexa-488 tagged AIRISE-02. Cells were then placed on a rocker in the cell incubator (37 °C, 5% CO2) for 2 h. Cells were centrifuged at 200 g for 4 minutes at 4°C and washed once with FACS buffer (1% BSA in PBS). Alexa-488 signal on the exterior of cells was quenched in 0.2% Trypan Blue. Cells were analyzed in Guava easyCyte 12 (Burlington, MA).
In vivo mouse study:
All animals were recruited and used under an approved protocol of the Institutional Animal Care and Use Committee (IACUC) of Oregon Health and Science University (Protocol #IS03483). All animal experiments were carried out under the auspices of the OHSU Department of Comparative Medicine.
Flow cytometry and antibodies:
The following fluorescent dye–conjugated antibodies against surface and intracellular antigens were used: CD8 (clone #53-6.7, BD Biosciences, BV650), CD4 (clone #RM4-5, BD Biosciences, BV711), CD44 (clone #IM7, BioLegend, PerCP/Cy5.5), CD3 (clone #17A2, BioLegend, PE or BV650), CD19 (clone #6D5, BioLegend, BV650), CD45 (clone #30-F11, BD Biosciences, APC-Cy7 or FITC or PerCP5.5), CD62L (clone #MEL-14, Invitrogen, APC), MHCII (clone #M5/144.15.2, BioLegend, BV421), CD80 (clone #16-10A1, BioLegend, BV605), CD11b (clone #M1/70, BioLegend, BV711), Ly6C (clone #HK1.4, BioLegend, BV785), CD11c (clone #N418, Invitrogen, PE), F4/80 (clone #BM8, BioLegend, Alexa Fluor 647), FoxP3 (clone # MF-14, BioLegend, Alexa Fluor 647), Ki67 (clone # SolA15, Invitrogen, eFluor 450).
Tumors and/or lymph nodes were harvested and cut into small sections for digestion. Tissues were digested in Digestion Media (1 mg/mL Collagenase D and 0.1 mg/mL DNase I in HBSS) at 37 °C for 30 minutes and mechanically dissociated by passing through 70-μm pore nylon cell strainers. Red blood cells in the sample were lysed by incubating in RBC lysis buffer (Alfa Aesar) at room temperature for 5 minutes. Cells were washed twice with PBS and stained with Live/Dead Fixable Aqua Stain (Thermo Fisher Scientific Cat# L34966) for 15 minutes. Cells were washed twice with FACS buffer (1% BSA in PBS), incubated with FcR blocking solution for 5 minutes, then stained for a select panel of surface-staining antibodies for 10 minutes at room temperature. In some studies, intracellular staining (e.g., for FoxP3 and Ki67) was performed with BD Cytofix/Cytoperm (BD Biosciences), following the manufacturer’s protocol after cell surface staining. Samples were washed twice with FACS buffer and resuspended in FACS buffer for analysis. All data were acquired with a BD LSRFortessa flow cytometer (OHSU’s Flow Cytometry Core), and analyzed using FlowJo Software (TreeStar Inc.). Only live cells (determined by live-dead stain occurring before fixing/permeabilization) were analyzed.
In vivo adjuvanticity of CpG vs CpG-NP:
C57BL/6 mice (n=3/group) were injected via footpad with 4 μg free CpG or 4 μg CpG loaded in the NP (CpG-NP). Twenty-four hours later, local draining lymph nodes (DLN) and non-draining lymph nodes (NDLN) were harvested and analyzed by flow cytometry for CD11c, MHCII, and CD80 expression (of CD45+CD3−CD19−).
B16F10 bilateral orthotopic murine melanoma tumor model:
6 week old female C57BL/6 mice were obtained from Charles River NCI colony (Wilmington, MA). Each mouse was intradermally implanted with B16F10 cells on the left (local, 250,000 cells) and right (distant, 100,000 cells) shoulders. At 8 days post-implantation, the test compound was intratumorally injected to only the left (local) tumor, while the right (distant) tumor was left untreated. Unless otherwise specified, the test compound was given every 3 days for 3 doses. Burden of both local and distant tumors in mice were measured with Vernier Caliper every 1-2 days, and tumor volume was calculated by V = 0.5 x length x width2. Survival was also monitored. Mice were sacrificed when total tumor burden exceeded 2000 mm3. For the study that combined intratumoral treatment with immune checkpoint inhibitors, a cocktail of PD-1 Ab (200 μg/mouse) and CTLA-4 Ab (100 μg/mouse) was given intraperitoneally on the same day (three doses every three days) as intratumoral treatment of our test compounds.
To confirm that the treatment efficacy was immune-mediated, mice treated with AIRISE-02 were injected intraperitoneally with CD8 antibody (200 μg/mouse; clone 2.43, BioXcell), starting one day before the first intratumoral treatment and continuing twice a week throughout the entire study.
CT26 bilateral murine colorectal tumor model:
6 week old female Balb/c mice were obtained from Charles River NCI colony (Wilmington, MA). Each mouse was subcutaneously implanted with CT26 cells on the right (local, 250,000 cells) and left (distant, 100,000 cells) flank. At 15 days post-implantation, AIRISE-02 was intratumorally injected to only the right (local) tumor, while the left (distant) tumor was left untreated. The test compound was given every 3 days for 3 doses. Burden of both local and distant tumors in mice were measured with Vernier Caliper every 1-2 days, and tumor volume was calculated by V = 0.5 x length x width2. Survival was also monitored. Mice were sacrificed when total tumor burden exceeded 2000 mm3. For treatment combined with immune checkpoint inhibitors, a cocktail of PD-1 Ab (200 μg/mouse) and CTLA-4 Ab (100 μg/mouse) was given intraperitoneally on the same day (three doses every three days) as intratumoral treatment of AIRISE-02.
MM3MG-HER2Δ16 bilateral orthotopic murine breast tumor model:
6 week old female Balb/c mice were obtained from Charles River NCI colony (Wilmington, MA). Each mouse was orthotopically implanted with MM3MG-HER2Δ16 cells on the right (local, 1,000,000 cells) and left (distant, 250,000 cells) mammary fat pads. At 4 days post-implantation, test compound was intratumorally injected to only the right (local) tumor, while the left (distant) tumor was left untreated. Test compound was given on days 0, 3, 6, 18, and 21. Burden of both local and distant tumors in mice were measured with Vernier Caliper every 1-2 days, and tumor volume was calculated by V = 0.5 x length x width2. Survival was also monitored. Mice were sacrificed when total tumor burden exceeded 2000 mm3. For treatment combined with immune checkpoint inhibitors, PD-1 Ab (200 μg/mouse) was given intraperitoneally on days 0, 3, 6, 14, 18, and 21.
Uptake profile of AIRISE-02 in various cells in the TME:
AIRISE-02 was loaded with Alexa488-tagged siRNA to monitor AIRISE-02 uptake in different cell populations in tumors at 2 hours post-injection. Tumors were harvested and processed as described in the flow cytometry section. Cells were stained for live/dead, CD45, CD3, CD19, MHC II, CD11c, and F4/80. Only live cells were analyzed. Cells with Alexa-488 signals were identified as having NP (NP+ cells), and were further interrogated for different cell populations. Non-hematopoietic (primarily cancer) cells are identified as CD45−, while hematopoietic (primarily immune) cells are identified as CD45+. Myeloid cells are identified as CD45+CD3−CD19−, which can be further gated to macrophage (F4/80+) and dendritic cells (CD11c+ MHCII+).
Immune cell profiling upon treatment of AIRISE-02:
Tumors and lymph nodes from AIRISE-02-treated and control mice were harvested and processed as described earlier. Cells were stained for CD45, CD3, CD4, CD8, CD44, FoxP3 (intracellular), and Ki67 (intracellular). The population of CD8+ T cells was gated as CD45+CD3+CD8+. The population of Treg was gated as CD45+CD3+CD4+FoxP3+. The activity/status of CD8+ T cells was examined with CD44 and Ki67.
Analyzing memory T cell population in blood:
100-200 ul of blood was collected via tail vein into heparinized tubes from AIRISE-cured (from Figure 3) and naïve control mice. Red blood cells were lysed by incubating whole blood with 2 mL RBC lysis buffer at room temperature for 5 minutes. Lysis reaction was stopped by adding 5 mL PBS and centrifuging cells at 2,000 rpm for 5 minutes. Cells were washed once with PBS, then incubated for 15 minutes with Live/Dead Fixable Aqua Stain (Thermo Fisher Scientific Cat# L34966). Cells were then washed twice with FACS buffer, incubated with FcR blocking solution for 5 minutes, then stained for CD8, CD4, CD44, CD3, CD62L, and CD45. Samples were washed twice with FACS buffer and analyzed using BD LSRFortessa flow cytometer.
In vivo toxicity study
Potential systemic toxicity after repeated administration:
At the study endpoint (Figure 4D), mice treated with multiple doses of AIRISE-02 were euthanized. Organs and serum were collected.
Upon euthanasia, blood was collected by cardiac puncture and left at room temperature to clot for 30 minutes to an hour. The coagulated blood was centrifuged at 1,500 x g for 15 minutes at 4 °C to remove clot. The serum was stored in −20 °C until analysis. Serum clinical chemistry parameters (liver, kidney, and muscle biomarkers) were analyzed with Beckman AU680 instrument, performed by IDEXX BioAnalytics (CA).
Organs were weighed and fixed with at least ten times (w/w) phosphate buffered formalin 10% solution (Fisher Scientific, PA) overnight at 4 °C. The formalin fixed tissue was then embedded in paraffin, and sectioned at Histopathology core (OHSU, OR) for IHC analysis. Histopathology evaluation of H&E stained slides was performed by Mass Histology Service (MA) in a treatment-blinded manner.
AIRISE-02 skin sensitivity test:
To evaluate possible skin sensitivity to AIRISE-02, 13 week-old Balb/c mice were injected intramuscularly into the thigh muscle with 40 μl of AIRISE-02 (0.26 mg NP; efficacious dose). Injection site was evaluated before and immediately after injection, 24 h, and 72 h after injection for edema and erythema using the standard Draize test criteria.[39]
Safety and pharmacokinetic study in cynomolgus monkeys:
Monkey studies were conducted at Charles River Laboratories Ashland (Ashland, OH). Three cynomolgus monkeys (Macaca fascicularis; of Chinese origin) were obtained from the Charles River animal colony. The animals were 3 to 4 years old and weighed between 2.9-3.4 kg before the start of treatment. Animals were returned to the Charles River animal colony after the study. AIRISE-02 was administered in escalating doses into each monkey (n=3) by subcutaneous injection (0.9, 2.7, 9.1 mg/kg AIRISE-02). Injection volume was maintained at 2 mL/kg for each dose. Animals were temporarily restrained and not sedated during the treatment. Each dose was administered in one injection, at a separate injection site in the dorsoscapular region. There was a 1-week washout period between each dose, and a staggered dosing regimen was used in which one animal was dosed and monitored for at least 24 hours before the remaining two animals were dosed.
Clinical observations, body weight, and food consumption were monitored throughout the study. Dermal reactions were evaluated according to the method of Draize,[54] and recorded pre-dose, and 6, 24, 48, and 72 hours post each dose, and also on day 8 for the high dose. Clinical pathology (hematology, coagulation, and clinical chemistry) was evaluated before dosing, 24 hours and one week post each dose. Serum samples for cytokine analysis were taken pre-dose, and 6, 24, and 48 hours after each dose. Cytokine analysis (TNF-α, IFN-γ, IL-1b, IL-6, and MCP-1) was performed by Charles River Mattawan (Mattawan, MI), using multiplex Luminex assay. Plasma samples for pharmacokinetic evaluation were taken pre-dose, 15 min, 1, 2, 6, 24, and 48 hours post each dose, and also day 8 after the last dose. Oligonucleotide concentration in plasma was analyzed by a hybridization-based AEX-HPLC fluorescence method as mentioned previously, and nanoparticle concentration in plasma was analyzed as “Si” by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). ICP-MS analysis was performed by OHSU’s Elemental Analysis Core (Portland, OR).
Stability of nanoparticles:
0.2 or 2.0 mg/ml NP were incubated in PBS (pH 7.4) or 0.2 mM NaOH (pH 11, forced degradation) under shaking condition (150 rpm) at 37°C for a different period of time. At the end of each timepoint, NP concentration (mg/mL) was determined by particle count rates (Zetasizer, ZS-90/Malvern, Westborough, MA). NP hydrodynamic size was also monitored at the same time.
Sterilization and shelf-life of nanoparticles:
NP was terminal sterilized with a single dose of 30 kGy e-beam irradiation by DualBeam X-ray/E-beam processing (Steri-tek, Fresno, CA). Sterilized NP and NP stored under different conditions were evaluated for size (by Zetasizer) and their ability to deliver siRNA effectively by using luciferase siRNA as a model following our previously published protocol.[22]
Statistical analysis:
In vitro experiments were performed in triplicates (experimentally and analytically), and the results are presented as mean ± SD. When in vivo experiments were performed independently twice; data were combined. In vivo data are presented as mean ± SEM. Comparisons of all groups at a single time point were performed after testing for D’Agostino–Pearson omnibus normality tests (GraphPad Prism 6.0). Comparisons of 2 groups were performed either with Student t tests (for normal distribution) or Mann–Whitney test (for nonparametric test, unpaired groups). For comparisons of more than 3 groups, statistical analysis was done with one-way ANOVA with post-hoc Dunnett multiple comparison tests. Two-way repeated-measure ANOVA followed by Sidak’s multiple comparison tests was performed to analyze the treatment effects over time in tumor burden. Survival curves were analyzed using Kaplan–Meier and its multiple comparison tests were analyzed using log-rank test methods with adjusted alpha by Bonferroni correction. GraphPad Prism 6.0 software (GraphPad Software Inc.) was utilized for all statistical analyses. p < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgement
This work was funded by the Wayne D. Kuni & Joan E. Kuni Foundation, NIH/NCI grant# R44CA217534, NIH/NCATS grant# R43TR001906, OHSU Knight Cancer Institute’s Hillcrest Committee Pilot Award, and OHSU Center for Women’s Health Circle of Giving Award. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH and US government. We would like to thank Drs. Michael Templin and Melinda Tyner of Charles River Laboratory and their team for assistance in designing and executing primate studies along with data interpretation. We thank Dr. Zachary Hartman of Duke University for the MM3MG-HER2Δ16 cell line and providing advice on the breast tumor model for immunotherapeutic testing. We appreciate Dr. Jeremy Heidel for his advice on Chemistry, Manufacturing, and Controls (CMC) strategies. We thank Ms. Freda Hu for the assistance with the artwork and compiling primate’s data, and Ms. Alyssa Carlson for assistance with NP count measurement. We appreciate OHSU’s Flow Cytometry Shared Resource, Histopathology Core, and Elemental Analysis Core. Lastly, we would like to thank Dr. Zheng Xia of OHSU’s Department of Molecular Microbiology and Immunology for his independent reviewing of the data in this manuscript as required by OHSU’s conflict of interest guidelines.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Competing Interests
OHSU, J.W.G., and W.Y. have a significant financial interest in PDX Pharmaceuticals, Inc. This potential personal and institutional conflict of interest has been reviewed and managed by OHSU. A patent application has been submitted by OHSU and PDX Pharmaceuticals based on these results with W.Y., A.W.L., W.N., and M.R. as inventors.
Contributor Information
Worapol Ngamcherdtrakul, PDX Pharmaceuticals, Inc., Portland, OR 97239, USA.
Moataz Reda, PDX Pharmaceuticals, Inc., Portland, OR 97239, USA.
Molly A. Nelson, PDX Pharmaceuticals, Inc., Portland, OR 97239, USA
Ruijie Wang, PDX Pharmaceuticals, Inc., Portland, OR 97239, USA.
Husam Y. Zaidan, PDX Pharmaceuticals, Inc., Portland, OR 97239, USA
Daniel S. Bejan, PDX Pharmaceuticals, Inc., Portland, OR 97239, USA
Ngoc Ha Hoang, Department of Biomedical Engineering, Oregon Health and Science University, Portland, OR 97239, USA.
Ryan S. Lane, Department of Cell, Developmental & Cancer Biology, Oregon Health and Science University, Portland, OR 97239, USA
Shiuh-Wen Luoh, VA Portland Health Care System, Portland, OR 97239, USA; Knight Cancer Institute, Oregon Health and Science University, Portland, OR 97239, USA.
Sancy A. Leachman, Department of Dermatology, Oregon Health and Science University, Portland, OR 97239, USA; Knight Cancer Institute, Oregon Health and Science University, Portland, OR 97239, USA
Gordon B. Mills, Knight Cancer Institute, Oregon Health and Science University, Portland, OR 97239, USA
Joe W. Gray, Department of Biomedical Engineering, Oregon Health and Science University, Portland, OR 97239, USA; Knight Cancer Institute, Oregon Health and Science University, Portland, OR 97239, USA
Amanda W. Lund, Department of Cell, Developmental & Cancer Biology, Oregon Health and Science University, Portland, OR 97239, USA
Wassana Yantasee, PDX Pharmaceuticals, Inc., Portland, OR 97239, USA; Department of Biomedical Engineering, Oregon Health and Science University, Portland, OR 97239, USA; Knight Cancer Institute, Oregon Health and Science University, Portland, OR 97239, USA.
References
- [1].(a) Sharon E, Streicher H, Goncalves P, Chen HX, Chinese journal of cancer 2014, 33, 434; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Buchbinder EI, Desai A, American Journal of Clinical Oncology 2016, 39, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].(a) Ribas A, Update on cancer therapeutics 2007, 2, 133; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M, The New England journal of medicine 2012, 366, 2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].(a) Santarpia M, Karachaliou N, Cancer Biology & Medicine 2015, 12, 74; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, Nature 2014, 515, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu D, Jenkins RW, Sullivan RJ, Am J Clin Dermatol 2019, 20, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shevtsov M, Sato H, Multhoff G, Shibata A, Front Oncol 2019, 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rehman H, Silk AW, Kane MP, Kaufman HL, Journal for Immunotherapy of Cancer 2016, 4, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Jr WHM, Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS, Journal of Clinical Oncology 2015, 33, 2780. [DOI] [PubMed] [Google Scholar]
- [8].Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F, Expert Review of Anticancer Therapy 2008, 8, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Inoue H, Tani K, Cell Death and Differentiation 2014, 21, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sheen MR, Fiering S, WIREs Nanomedicine and Nanobiotechnology 2019, 11, e1524. [DOI] [PubMed] [Google Scholar]
- [11].Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM, Expert review of vaccines 2011, 10, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cai Q, Kublo L, Cumberland R, Gooding W, Baar J, Clinical and Translational Science 2009, 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shirota BY, Shirota H, Klinman DM, Journal of Immunology (Baltimore, Md. : 1950) 2012, 188, 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].(a) Hofmann MA, Kors C, Audring H, Walden P, Sterry W, Trefzer U, Journal of immunotherapy (Hagerstown, Md. : 1997) 2008, 31, 520; [DOI] [PubMed] [Google Scholar]; (b) Molenkamp BG, Sluijter BJ, van Leeuwen PA, Santegoets SJ, Meijer S, Wijnands PG, Haanen JB, van den Eertwegh AJ, Scheper RJ, de Gruijl TD, Clinical cancer research : an official journal of the American Association for Cancer Research 2008, 14, 4532; [DOI] [PubMed] [Google Scholar]; (c) Milhem MM, Zarour HM, Gabrail NY, Mauro DJ, Greenberg NM, Slichenmyer WJ, Krieg AM, Journal of Clinical Oncology 2016, 34, TPS9593. [Google Scholar]
- [15].(a) Delannois F, Planty C, Giordano G, Destexhe E, Stanislaus D, Da Silva FT, Stegmann J-U, Thacker K, Reynaud L, Garçon N, Segal L, Reproductive Toxicology 2018, 75, 110; [DOI] [PubMed] [Google Scholar]; (b) Adamus T, Kortylewski M, Contemp Oncol (Pozn) 2018, 22, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].(a) Hirsh V, Paz-Ares L, Boyer M, Rosell R, Middleton G, Eberhardt WE, Szczesna A, Reiterer P, Saleh M, Arrieta O, Bajetta E, Webb RT, Raats J, Benner RJ, Fowst C, Meech SJ, Readett D, Schiller JH, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011, 29, 2667; [DOI] [PubMed] [Google Scholar]; (b) Manegold C, van Zandwijk N, Szczesna A, Zatloukal P, Au JSK, Blasinska-Morawiec M, Serwatowski P, Krzakowski M, Jassem J, Tan EH, Benner RJ, Ingrosso A, Meech SJ, Readett D, Thatcher N, Annals of oncology : official journal of the European Society for Medical Oncology 2012, 23, 72; [DOI] [PubMed] [Google Scholar]; (c) Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, Jassem J, Yoshimura M, Dahabreh J, Nakayama H, Havel L, Kondo H, Mitsudomi T, Zarogoulidis K, Gladkov OA, Udud K, Tada H, Hoffman H, Bugge A, Taylor P, Gonzalez EE, Liao ML, He J, Pujol JL, Louahed J, Debois M, Brichard V, Debruyne C, Therasse P, Altorki N, The Lancet. Oncology 2016, 17, 822; [DOI] [PubMed] [Google Scholar]; (d) Dreno B, Thompson JF, Smithers BM, Santinami M, Jouary T, Gutzmer R, Levchenko E, Rutkowski P, Grob J-J, Korovin S, Drucis K, Grange F, Machet L, Hersey P, Krajsova I, Testori A, Conry R, Guillot B, Kruit WHJ, Demidov L, Thompson JA, Bondarenko I, Jaroszek J, Puig S, Cinat G, Hauschild A, Goeman JJ, van Houwelingen HC, Ulloa-Montoya F, Callegaro A, Dizier B, Spiessens B, Debois M, Brichard VG, Louahed J, Therasse P, Debruyne C, Kirkwood JM, The Lancet Oncology 2018, 19, 916. [DOI] [PubMed] [Google Scholar]
- [17].Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, Barve M, Daniels GA, Wong DJ, Schmidt EV, Candia AF, Coffman RL, Leung ACF, Janssen RS, Cancer Discovery 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].(a) Lee H, Pal SK, Reckamp K, Figlin RA, Yu H, Curr Top Microbiol Immunol 2011, 344, 41; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kortylewski M, Yu H, Current opinion in immunology 2008, 20, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Iwata-Kajihara T, Sumimoto H, Kawamura N, Ueda R, Takahashi T, Mizuguchi H, Miyagishi M, Takeda K, Kawakami Y, J Immunol 2011, 187, 27. [DOI] [PubMed] [Google Scholar]
- [20].Kortylewski M, Kujawski M, Herrmann A, Yang C, Wang L, Liu Y, Salcedo R, Yu H, Cancer research 2009, 69, 2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang H, Yamazaki T, Pietrocola F, Zhou H, Zitvogel L, Ma Y, Kroemer G, Cancer research 2015, 75, 3812. [DOI] [PubMed] [Google Scholar]
- [22].Ngamcherdtrakul W, Morry J, Gu S, Castro DJ, Goodyear SM, Sangvanich T, Reda MM, Lee R, Mihelic SA, Beckman BL, Hu Z, Gray JW, Yantasee W, Advanced Functional Materials 2015, 25, 2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Morry J, Ngamcherdtrakul W, Gu S, Goodyear SM, Castro DJ, Reda MM, Sangvanich T, Yantasee W, Biomaterials 2015, 66, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].(a) Jordan M, Waxman DJ, Cancer letters 2016, 373, 88; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Westwood JA, Potdevin Hunnam TCU, Pegram HJ, Hicks RJ, Darcy PK, Kershaw MH, PLOS ONE 2014, 9, e95847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].(a) Koster BD, van den Hout MFCM, Sluijter BJR, Molenkamp BG, Vuylsteke RJCLM, Baars A, van Leeuwen PAM, Scheper RJ, Petrousjka van den Tol M, van den Eertwegh AJM, de Gruijl TD, Clinical Cancer Research 2017, 23, 5679; [DOI] [PubMed] [Google Scholar]; (b) Millward M, Underhill C, Lobb S, McBurnie J, Meech SJ, Gomez-Navarro J, Marshall MA, Huang B, Mather CB, Br J Cancer 2013, 108, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) American Cancer Society, Cancer Facts & Figures 2020, https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf, accessed: March, 2021;; (b) Gershenwald JE, Guy GP Jr., Journal of the National Cancer Institute 2016, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].(a) PDQ Adult Treatment Editorial Board, Melanoma Treatment (PDQ®)–Health Professional Version, accessed: ; [Google Scholar]; (b) National Library of Medicine, Melanoma - Genetics Home Reference - NIH, accessed: [Google Scholar]
- [28].National Cancer Institute, Cancer Stat Facts: Melanoma of the Skin, accessed: March, 2021
- [29].(a) van Elsas A, Hurwitz AA, Allison JP, J Exp Med 1999, 190, 355; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cooke K, Estrada J, Zhan J, Mitchell P, Bulliard Y, Beltran PJ, presented at AACR 107th Annual Meeting 2016, New Orleans, April2016; [Google Scholar]; (c) Buss CG, Bhatia SN, Proceedings of the National Academy of Sciences 2020, 117, 13428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].(a) Ngamcherdtrakul W, Yantasee W, Translational research : the journal of laboratory and clinical medicine 2019, 214, 105; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ngamcherdtrakul W, Castro DJ, Gu S, Morry J, Reda M, Gray JW, Yantasee W, Cancer treatment reviews 2016, 45, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, Lee H, Scuto A, Liu Y, Yang C, Deng J, Soifer HS, Raubitschek A, Forman S, Rossi JJ, Pardoll DM, Jove R, Yu H, Nature biotechnology 2009, 27, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].(a) Song C, Piva M, Sun L, Hong A, Moriceau G, Kong X, Zhang H, Lomeli S, Qian J, Yu CC, Damoiseaux R, Kelley MC, Dahlman KB, Scumpia PO, Sosman JA, Johnson DB, Ribas A, Hugo W, Lo RS, Cancer Discovery 2017, 7, 1248; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi J, Beaino W, Fecek RJ, Fabian KPL, Laymon CM, Kurland BF, Storkus WJ, Anderson CJ, Journal of Nuclear Medicine 2018, 59, 1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].(a) Antohe M, Nedelcu RI, Nichita L, Popp CG, Cioplea M, Brinzea A, Hodorogea A, Calinescu A, Balaban M, Ion DA, Diaconu C, Bleotu C, Pirici D, Zurac SA, Turcu G, Oncol Lett 2019, 17, 4155; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sideras K, Galjart B, Vasaturo A, Pedroza-Gonzalez A, Biermann K, Mancham S, Nigg AL, Hansen BE, Stoop HA, Zhou G, Verhoef C, Sleijfer S, Sprengers D, Kwekkeboom J, Bruno MJ, Journal of Surgical Oncology 2018, 118, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].(a) Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL, Gao J, Luk BT, Dehaini D, Gao W, Zhang L, Advanced Materials 2017, 29, 1703969; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fan Y, Kuai R, Xu Y, Ochyl LJ, Irvine DJ, Moon JJ, Nano Letters 2017, 17, 7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Roberts AD, Ely KH, Woodland DL, J Exp Med 2005, 202, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].(a) Chauchet X, Hannani D, Djebali S, Laurin D, Polack B, Marvel J, Buffat L, Toussaint B, Le Gouëllec A, Mol Ther Oncolytics 2016, 3, 16033; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Malamas AS, Hammond SA, Schlom J, Hodge JW, Oncotarget 2017, 8, 90825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Martin MD, Badovinac VP, Front Immunol 2018, 9, 2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tsao L-C, Crosby EJ, Trotter TN, Agarwal P, Hwang B-J, Acharya C, Shuptrine CW, Wang T, Wei J, Yang X, Lei G, Liu C-X, Rabiola CA, Chodosh LA, Muller WJ, Lyerly HK, Hartman ZC, JCI Insight 2019, 4, e131882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].DRAIZE JH, WOODARD G, CALVERY HO, Journal of Pharmacology and Experimental Therapeutics 1944, 82, 377. [Google Scholar]
- [40].(a) Klinman DM, Xie H, Little SF, Currie D, Ivins BE, Vaccine 2004, 22, 2881; [DOI] [PubMed] [Google Scholar]; (b) Hartmann G, Weeratna RD, Ballas ZK, Payette P, Blackwell S, Suparto I, Rasmussen WL, Waldschmidt M, Sajuthi D, Purcell RH, Davis HL, Krieg AM, Journal of Immunology 2000, 164, 1617. [DOI] [PubMed] [Google Scholar]
- [41].(a) Tian Q, Rogness J, Meng M, Li Z, Bioanalysis 2017, 9, 861; [DOI] [PubMed] [Google Scholar]; (b) Roehl I, Schuster M, Seiffert S (Axolabs Gmbh), US Patent US 2018/0312909 A1, 2018.
- [42].Tarn D, Ashley CE, Xue M, Carnes EC, Zink JI, Brinker CJ, Accounts of chemical research 2013, 46, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].(a) Lu J, Liong M, Li Z, Zink JI, Tamanoi F, Small 2010, 6, 1794; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bhavsar D, Patel V, Sawant K, Microporous and Mesoporous Materials 2019, 284, 343. [Google Scholar]
- [44].Chen K, Zhang J, Gu H, Journal of Materials Chemistry 2012, 22, 22005. [Google Scholar]
- [45].Nel AE, Mädler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nature Materials 2009, 8, 543. [DOI] [PubMed] [Google Scholar]
- [46].(a) Heo MB, Lim YT, Biomaterials 2014, 35, 590; [DOI] [PubMed] [Google Scholar]; (b) Pradhan P, Qin H, Leleux JA, Gwak D, Sakamaki I, Kwak LW, Roy K, Biomaterials 2014, 35, 5491; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Conniot J, Scomparin A, Peres C, Yeini E, Pozzi S, Matos AI, Kleiner R, Moura LIF, Zupančič E, Viana AS, Doron H, Gois PMP, Erez N, Jung S, Satchi-Fainaro R, Florindo HF, Nature Nanotechnology 2019, 14, 891. [DOI] [PubMed] [Google Scholar]
- [47].Jugdaohsingh R, The journal of nutrition, health & aging 2007, 11, 99. [PMC free article] [PubMed] [Google Scholar]
- [48].Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, Humm J, Gönen M, Kalaigian H, Schöder H, Strauss HW, Larson SM, Wiesner U, Bradbury MS, Science Translational Medicine 2014, 6, 260ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Henderson MA, Melanoma Management 2019, 6, MMT23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Moesta AK, Cooke K, Piasecki J, Mitchell P, Rottman JB, Fitzgerald K, Zhan J, Yang B, Le T, Belmontes B, Ikotun OF, Merriam K, Glaus C, Ganley K, Cordover DH, Boden AM, Ponce R, Beers C, Beltran PJ, Clinical cancer research : an official journal of the American Association for Cancer Research 2017, 23, 6190. [DOI] [PubMed] [Google Scholar]
- [51].Hu JCC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, Harrington KJ, James ND, Love CA, McNeish I, Medley LC, Michael A, Nutting CM, Pandha HS, Shorrock CA, Simpson J, Steiner J, Steven NM, Wright D, Coombes RC, Clinical Cancer Research 2006, 12, 6737. [DOI] [PubMed] [Google Scholar]
- [52].(a) Matheu MP, Sen D, Cahalan MD, Parker I, J Vis Exp 2008, 773; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kimura A, Naka T, Kishimoto T, Proceedings of the National Academy of Sciences 2007, 104, 12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ngamcherdtrakul W, Sangvanich T, Reda M, Gu S, Bejan D, Yantasee W, International journal of nanomedicine 2018, 13, 4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].DRAIZE JH, Dermal Toxicity 1965, 46. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.