

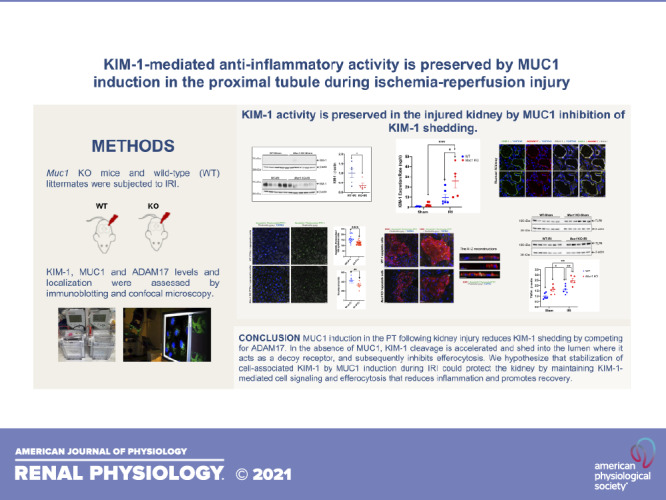
Keywords: acute kidney injury, efferocytosis, ischemia-reperfusion injury, kidney injury molecule-1, mucin 1
Abstract
Cell-associated kidney injury molecule-1 (KIM-1) exerts an anti-inflammatory role following kidney injury by mediating efferocytosis and downregulating the NF-κB pathway. KIM-1 cleavage blunts its anti-inflammatory activities. We reported that mucin 1 (MUC1) is protective in a mouse model of ischemia-reperfusion injury (IRI). As both KIM-1 and MUC1 are induced in the proximal tubule (PT) during IRI and are a disintegrin and metalloprotease 17 (ADAM17) substrates, we tested the hypothesis that MUC1 protects KIM-1 activity. Muc1 knockout (KO) mice and wild-type (WT) littermates were subjected to IRI. KIM-1, MUC1, and ADAM17 levels (and signaling pathways) were assessed by immunoblot analysis. PT localization was assessed by confocal microscopy and an in situ proximity ligation assay. Findings were extended using human kidneys and urine as well as KIM-1-mediated efferocytosis assays in mouse PT cultures. In response to tubular injury in mouse and human kidneys, we observed induction and coexpression of KIM-1 and MUC1 in the PT. Compared with WT mice, Muc1 KO mice had higher urinary KIM-1 and lower kidney KIM-1. KIM-1 was apical in the PT of WT kidneys but predominately with luminal debris in Muc1 KO mice. Efferocytosis was reduced in Muc1 KO PT cultures compared with WT cultures, whereas inflammation was increased in Muc1 KO kidneys compared with WT kidneys. MUC1 was cleaved by ADAM17 in PT cultures and blocked KIM-1 shedding in Madin-Darby canine kidney cells. We conclude that KIM-1-mediated efferocytosis and thus anti-inflammatory activity during IRI is preserved in the injured kidney by MUC1 inhibition of KIM-1 shedding.
NEW & NOTEWORTHY KIM-1 plays a key role in the recovery of the tubule epithelium during renal IRI by mediating efferocytosis and associated signaling that suppresses inflammation. Excessive cleavage of KIM-1 by ADAM17 provides a decoy receptor that aggravates efferocytosis and subsequent signaling. Our data from experiments in mice, patients, and cultured cells show that MUC1 is also induced during IRI and competes with KIM-1 for cleavage by ADAM17. Consequently, MUC1 protects KIM-1 anti-inflammatory activity in the damaged kidney.
INTRODUCTION
Kidney injury molecule-1 (KIM-1) is a type I transmembrane glycoprotein that is rapidly induced at the gene and protein levels after acute kidney injury (AKI) (1, 2). Specifically, KIM-1 is upregulated on the apical surface of surviving proximal tubule (PT) epithelial cells of the kidney following ischemic and toxic AKI (1–3). KIM-1 is also upregulated in chronic kidney disease, renal cell carcinoma, and polycystic kidney disease (4–6). Mechanistically, it has been recently reported that the transcription factor signal transducer and activator of transcription 3 (STAT3) is phosphorylated by checkpoint kinase 1 and/or extracellular signal-regulated kinases 1 and 2 (ERK1/2) following kidney injury, which leads to the upregulation of KIM-1 expression (7, 8). Structurally, KIM-1 contains an extracellular Ig-like domain, a mucin-like domain, one transmembrane domain, and a cytoplasmic domain with a tyrosine kinase phosphorylation motif (9). The ectodomain of human and mouse KIM-1 is cleaved by a disintegrin and metalloproteinase domain (ADAM)17 generating soluble KIM-1 (cleaved form) and thereby constitutively shed into the urine, providing an early and sensitive biomarker for kidney injury (10, 11). There are also reports that cleavage by ADAM10 may contribute to shedding of mouse and human KIM-1 (12).
Cell-associated KIM-1 exhibits an anti-inflammatory function and, thus, plays a protective role after kidney injury by 1) downregulating the proinflammatory NF-κB signaling pathway in a phosphoinositide 3-kinase (PI3K)-dependent manner and 2) acting as a receptor for phosphatidylserine (an “eat me” signal presented by apoptotic cells) (1, 2). The cell-associated form of KIM-1 enables PT epithelial cells to recognize and mediate phagocytosis of apoptotic and necrotic cells (efferocytosis) following kidney injury, which, in turn, prevents inflammation and promotes kidney repair (1, 2). However, accelerated shedding of KIM-1 that is mediated by ADAM17 and regulated by p38 MAPK in a cell culture model competitively inhibits efferocytosis, as the excess soluble KIM-1 acts as a decoy to cell-associated KIM-1 (10, 13).
The involvement of ADAM17 in a variety of processes reflects the wide range of ADAM17 substrates, including the proinflammatory cytokine TNF-α, epidermal growth factor receptor ligand, KIM-1, and mucin 1 (MUC1) (10, 14). Despite the substantial clinical impact of KIM-1 cleavage by ADAM17 during AKI, there are no in vivo data about the regulation of KIM-1 shedding following AKI. It is known that ADAM17 is weakly expressed by the normal kidney, whereas its expression is significantly upregulated in the PT, capillaries, and glomeruli after AKI (15, 16). But, with more than 80 different reported ADAM17 substrates, it is clear that targeting or blocking cleavage of KIM-1 would involve a cell-specific event.
There is emerging evidence that the transmembrane glycoprotein MUC1, expressed on the apical surface of polarized epithelial cells in the kidney, plays novel and important roles under physiological and pathological conditions (17). We have previously reported that MUC1 plays a protective role in a mouse model of ischemia-reperfusion injury (IRI) by stabilizing hypoxia-inducible factor-1α and β-catenin levels and downstream signaling (18, 19). Although MUC1 is normally expressed in the thick ascending limb and distal nephron, we discovered that MUC1 is present at low levels in the PT and significantly induced after ischemic injury (17, 19). There are also reports that the MUC1 ectodomain is shed in response to specific cleavage by ADAM17 but not ADAM10 (14, 20).
As both KIM-1and MUC1 are protective during IRI, although both are induced in the PT, we were intrigued in preliminary studies to find increased KIM-1 during IRI in the urine of Muc1 knockout (KO) mice compared with wild-type (WT) littermates. We initially expected to find increased KIM-1 expression in response to increased injury in the absence of MUC1, but instead, we found reduced KIM-1 levels in the kidney. As activation of upstream pathways regulating KIM-1 expression was no different in Muc1 KO and WT mice, we also assessed p38 MAPK regulation of ADAM17 expression but found no differences. However, when we compared data from mouse kidneys and urine during IRI and cell culture models of KIM-1 and MUC1 shedding, we established that MUC1 stabilizes KIM-1 and its activity in the PT by direct competition as a substrate for cleavage by ADAM17. This study provides new insights into our understanding of the mechanism of KIM-1 shedding in vivo that has a significant clinical impact on kidney recovery following injury. It also provides a novel aspect of MUC1 protection of the kidney during IRI by preserving KIM-1 function.
MATERIALS AND METHODS
Cells and Reagents
Madin-Darby canine kidney (MDCK 2001) cells were obtained from Kai Simons in the European Molecular Biology Laboratory in Heidelberg, Germany. These cells were grown in DMEM-F-12 (D6421) supplemented with 5% FBS (GIBCO/Thermo Fisher Scientific, Grand Island, NY) and maintained at 37°C in 5% CO2. All chemical compounds used in the experiments presented here were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. The sources and other information of primary antibodies are provided in Supplemental Table S1 (all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.14364890). FITC- or rhodamine-labeled antibodies and TO-PRO Cy5 were obtained from Molecular Probes (Thermo Fisher Scientific). Secondary horseradish peroxidase-conjugated anti-rabbit IgG and anti-mouse IgG antibodies were obtained from SeraCare (Milford, MA). Horseradish peroxidase-conjugated anti-β-actin antibodies were obtained from Proteintech (Rosemont, IL).
Preparation of Primary Cultures of PT Cells
Primary cultures of mouse kidney PT cells (PTCs) from Muc1 KO, WT littermates, and transgenic mice expressing human MUC1 were generated as previously described (1, 21, 22). Briefly, the kidney cortex was dissected and minced into pieces (∼1 mm) in prewarmed HBSS (Life Technologies, Grand Island, NY). Tissues were then gently washed to remove debris and digested in HBSS containing 1 mg/mL collagenase type II (Sigma-Aldrich). Tissue dissociation was performed for 60 min in a shaker (200 rpm) at 37°C with intermittent mixing (vortex for 30 s). The collagenase reaction was terminated using 5% BSA, and cell preparations were then washed and passed through a cell strainer (nylon mesh) with 40-µm pores to remove undigested material and glomeruli. Finally, cells were washed twice and resuspended in collagen type I-coated glass coverslips or 12-well plates in a medium composed of equal volumes of DMEM and Ham’s F-12 plus 60 nM sodium selenate, 5 mg/mL transferrin, 2 mM glutamine, 50 nM dexamethasone, 1 nM triiodothyronine, 10 ng/mL epidermal growth factor, 5 mg/mL insulin, 20 mM d-glucose, 2% (vol/vol) FBS, and 20 mM HEPES with antibiotics at pH 7.4 (reagents from Life Technologies and Sigma-Aldrich). Epithelial cells were used in experiments between 6 and 7 days of culture without passaging. All solute concentrations are in wt/vol, unless noted otherwise.
Animals and IRI Protocol
All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh School of Medicine Animal Care and Use Committee. All experiments were conducted using 12- to 16-wk-old male and female C57BL/6J mice. Muc1 global KO mice on a C57BL/6J background were as previously described and have no obvious phenotype under normal physiological conditions other than increased sensitivity to bacterial infections (23, 24). AKI was induced as previously described using the two-kidney hanging-weight model of renal IRI (25). Briefly, each mouse was anesthetized with isoflurane (5% for induction and 1–2% for maintenance) in oxygen and placed in the supine position on a thermostatically controlled heating system connected to a rectal temperature sensor to maintain body temperature at 37°C. Ischemia was induced by passing a 4/0 silk suture between the renal artery and vein of a kidney, and the ends of the suture were passed over a pulley system. Next, the ends were attached to 1-g weights and the weights were allowed to hang, thus compressing the renal artery with the suture. After 20 min of ischemia, the suture was removed, and the procedure was repeated on the opposite kidney followed by reperfusion for 48 h. Sham-operated mice were treated exactly like injured mice except that the weights were not attached to the ends of the suture.
After 48 h of recovery, mean arterial blood pressure and heart rate were measured after insertion of a PE-10 catheter into the carotid artery of an anesthetized mouse and connected to a digital blood pressure analyzer (Micro-Med, Louisville, KY). Renal blood flow was measured by placing a transit-time flow probe (0.5SB with a J-reflector and no handle, custom designed by Transonic Systems, Ithaca, NY) on either the right or the left renal artery of the anesthetized mouse and connected to a model T402-PP Transonic flowmeter.
Glomerular filtration rate (GFR) was estimated as previously described (25). Briefly, a constant rate infusion (10 µL/min) of [14C]inulin (0.0035 µCi/min dissolved in 0.9% saline containing 2.45% albumin) was initiated through the femoral vein catheter. For the collection of urine, a short segment of PE-50 tubing was inserted into silastic tubing, a small hole was made in the rostral end of the bladder using a cautery, and the silastic-covered PE-50 tubing was inserted into the bladder. After completion of surgery, a 1-h rest period was allowed to permit the mouse to stabilize and to let plasma [14C]inulin levels approach steady state. At the end of the 1-h rest period, urine was collected for 1 h, and the mouse was carefully moved to the prone position. [14C]]Inulin was determined in urine and plasma by measuring radioactivity in these samples, and GFR was estimated by calculating the renal clearance of [14C]inulin.
KIM-1 was also measured in urine as a marker for PT injury using a mouse ELISA kit (ab119596, Abcam, Cambridge, MA). As a marker of distal nephron injury, neutrophil gelatinase-associated lipocalin was also measured in urine with a mouse ELISA kit (ab119601, Abcam). At the end of the experiment, a blood sample was collected in heparin from the carotid artery catheter, and serum creatinine was measured at the University of Texas Southwestern Medical Center using the capillary electrophoresis method (26).
Kidney Tissue Preparation and Confocal Immunofluorescence Microscopy
One kidney from each mouse was sliced lengthwise and placed in 4% diethyl pyrocarbonate (DEPC)-treated paraformaldehyde and then methacarn (chloroform/methanol/acetic acid) for histology or snap-frozen in liquid nitrogen and then stored at −80°C for subsequent preparation of homogenates for further analysis (e.g., immunoblot analysis). Subsets of kidneys in 4% paraformaldehyde were sent to the University of Pittsburgh’s pathology laboratory (the P30 O’Brien Core) for preparation of paraffin blocks and for production of kidney slides. Immunofluorescence (IF) labeling was subsequently performed, after removal of paraffin and antigen retrieval, using antibodies against mouse KIM-1 raised in the goat (1:200 dilution), followed by secondary donkey anti-goat antibody coupled to Alexa 488 (1:400 dilution). Tissues were also colabeled with antibody raised in the rabbit against organic anion transporter 1 (OAT1) localized on the PT basolateral surface (1:400 dilution), followed by secondary antibody raised in the donkey coupled to CY3 (1:800 dilution). Mouse kidney tissues were also labeled with monoclonal antibody raised in the Armenian hamster against the MUC1 cytoplasmic tail (CT2; 1:200 dilution), followed by secondary goat anti-Armenian hamster antibody coupled to Alexa 488 (1:400 dilution). Immunolabeled tissues were mounted in VECTASHIELD mounting medium (Vector Laboratories) and imaged in a confocal laser scanning microscope (Leica TCS SP5, model upright DM 6000S, Leica Microsystems, Buffalo Grove, IL) using a ×63 objective with identical laser settings for all samples.
Immunoblot Analysis
These experiments were performed according to previously published methods (19, 27) and adapted as follows. The snap-frozen kidney quarter (∼0.4 mg) was homogenized in ice-cold lysis buffer (all solute concentrations are in wt/vol, unless noted otherwise): 150 mM NaCl, 10 mM Tris (pH 7.4), 1% Triton X-100, 0.5% Igepal, 1 mM EDTA, 1 mM EGTA, and both protease and phosphatase inhibitor cocktails (EMD Millipore, Burlington, MA). Protein samples were then separated by SDS-PAGE on a 4–12% gradient gel (Nu-PAGE, Invitrogen), followed by transfer to nitrocellulose membranes (0.45 µm, Millipore). The blots were then probed with anti-MUC1 CT2 antibody (1:1,000 dilution); antibodies raised in the rabbit against ADAM17 (1:1,000 dilution), phosphorylated (p-)STAT3 (1:1,000 dilution), ERK1/2 (1:5,000 dilution), p-ERK1/2 (1:5,000 dilution), p38 MAPK (1:5,000 dilution), p-p38 MAPK (1:5,000 dilution), NF-κB p65 (1:5,000 dilution), and Toll-like receptor-4 (TLR4; 1:5,000 dilution); antibodies raised in the goat against mouse TIM-1/KIM-1/HAVCR (1:1,000 dilution); mouse antibody against STAT3 (1:1,000 dilution); or anti-β-actin antibody raised in the mouse (1:20,000 dilution) to confirm equal loading of protein. Each probed protein was then visualized and quantified using Bio-Rad Clarity ECL, a Bio-Rad ChemiDoc Imager, and Bio-Rad Quantity One or Image Lab software. Blots of samples from Muc1 KO and WT mouse kidneys were developed side by side for the same time.
Isolation of nuclear and cytoplasmic fractions of soluble proteins from a quarter of a kidney was performed using the NE-PER Nuclear and Cytoplasmic Extraction Kit as per the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). Aliquots of the nuclear and cytoplasmic fractions were subjected to immunoblotting for NF-κB p65 (1:5,000 dilution) and β-actin (cytoplasmic fraction) or histone H3 (nuclear fraction). Blots of samples from Muc1 KO and wild-type mouse kidneys were developed side by side for the same time.
Quantitative Real-Time RT-PCR
Whole kidney mRNA was isolated using TRIzol. One microgram of total RNA was added to a cDNA synthesis kit (Bio-Rad), and cDNA was added to reactions containing SYBR Green master mix and a pairing of the following primers: KIM-1, forward 5′-TTCAGGAAGCTGAGCAAACAT-3′ and reverse 5′-CCCCAACATGTCGTTGTGATT-3′, and 18s, forward 5′-GTAACCCGTTGAACCCCATT-3′ and reverse 5′-CCATCCAATCGGTAGTAGCG-3′. Differences in expression were calculated using the 2−ΔΔCt method (where Ct is cycle threshold).
Human Kidneys with Ischemic Damage
Whole adult human kidneys were obtained from the Center for Organ Recovery and Education (Pittsburgh, PA) through Protocol No. 462 approved by the University of Pittsburgh Committee for Oversight of Research and Clinical Training Involving Decedents. Kidney samples were from donation after cardiac death or brain-dead donors that were not accepted for transplant. Therefore, we anticipated that these kidneys exhibit some degree of ischemic damage, allowing the opportunity to study the expression of KIM-1, the most highly upregulated protein in the PT after kidney injury. Supplemental Table S3 provides the wedge biopsy reports of the samples used in this study. All reports listed some degree of inflammation, tubular injury, or fibrosis (albeit mild) and further supported our anticipation that these kidneys underwent some sort of stress conditions that would induce KIM-1 expression. Kidneys were then processed as previously described (21). For the human kidney, tissues were processed and 5-µm-thick cryosections were prepared as described earlier. IFlabeling was performed using mouse monoclonal antibody B27.29 against human MUC1 tandem repeat (1:1,000 dilution) with goat antibodies against human KIM-1 (1:200 dilution), followed by secondary donkey anti-mouse antibody coupled to CY3 (1:800 dilution) and donkey anti-goat antibody coupled to Alexa 488 (1:400 dilution), respectively. The B27.29 epitope maps to two separate parts of the tandem repeats including the PDTRP peptide and glycosylated Thr* and Ser* (GVT*S*APDTRPAPG), and therefore, affinity can be affected by glycosylation. As human MUC1 has 40–100 tandem repeats, this antibody is much more sensitive for IF staining than the anti-cytoplasmic tail antibody CT2. Immunolabeled tissues were mounted and imaged as described above in Kidney Tissue Preparation and Confocal Immunofluorescence Microscopy.
In Situ Proximity Ligation Assay
Proximity ligation assay (PLA) was performed according to the manufacturer’s instructions (Duolink In Situ Detection Reagent-Red, Sigma-Aldrich). Briefly, kidney tissues were permeabilized with 0.5% Triton X-100 in PBS (pH 7.4) and incubated overnight with mouse monoclonal antibody B27.29 against the MUC1 extracellular domain tandem repeat (1:1,000 dilution) and with rabbit antibodies against ADAM17 (1:100 dilution). Next day, donkey anti-mouse PLAminus and donkey anti-rabbit PLAplus probes were applied, followed by ligation and amplification with Duolink detection reagent Texas red according to the manufacturer’s instructions (Duolink In Situ Detection Reagent-Red, Sigma-Aldrich). Samples were then imaged in a confocal laser scanning microscope (Leica TCS SP5, model upright DM 6000S, Leica Microsystems, Buffalo Grove, IL) using a ×20 objective with identical laser settings for all samples. All PLAs were performed in two independent experiments with a negative control where only ADAM17 antibody was added.
In Vitro Cell Culture Model of KIM-1 Shedding
MDCK cells were transfected with pcDNA 3.1(−) neo mKIM-1 encoding cDNA for mouse KIM-1, and a stable mixed population of MDCK-KIM-1 cells was obtained by growth in G418. MDCK-KIM-1 cells plated in a 12-well dish were transiently transfected the next day with human MUC1 cDNA (pcDNA3.1 MUC1) or empty vector. The next day, cells were incubated for 2 h in serum-free media with either 1.6 µM phorbol-12-myristate-13-acetate in DMSO (PMA; induces ADAM17 metalloprotease-mediated shedding of KIM-1 and MUC1), 10 mM BB-94 in DMSO [Batimastat (Abcam, Cambridge, MA), a broad-spectrum matrix metalloprotease inhibitor], both PMA and BB-94, or vehicle (DMSO). Media were collected on ice and then centrifuged for 5 min at 2,000 g. The supernatant was moved to a clean tube and incubated overnight with 0.5 µg of either goat anti-mouse KIM-1 or mouse anti-MUC1 (B27.29) antibodies and protein G conjugated to Sepharose 4B (Thermo Fisher Scientific). Beads were washed with 0.5 mL of HEPES-buffered saline [10 mM HEPES-HCl (pH 7.4) and 150 mM NaCl] with 1% Triton X-100 and then 0.5 mL of HEPES-buffered saline before being heated in Bio-Rad sample buffer with β-mercaptoethanol. Cells were washed twice with ice-cold Dulbecco’s PBS (Corning Cellgro, Manassas, VA) before extraction with 0.2 mL of detergent solution [100 mM NaCal, 40 mM KCl, 1 mM EDTA, 20 mM HEPES-KCl (pH 7.4), 10% glycerol, 1% Nonidet P-40, and 0.4% deoxycholate] containing protease inhibitors (EMD Millipore, Burlington, MA). Cell extracts (2.5%) or immunoprecipitates of KIM-1 or MUC1 from media were immunoblotted for KIM-1 or MUC1 using SDS-PAGE on a Bio-Rad TGX 4–15% acrylamide gradient gel and transferred to nitrocellulose (0.45 µm, Millipore). Blots were developed with Bio-Rad Clarity Max Western ECL Substrate and imaged with a Bio-Rad Chemidoc and Bio-Rad Image Lab software.
In Vitro Primary Cell Culture Model of MUC1 Shedding
Primary cultures of PTCs from transgenic mice expressing human MUC1 (n = 3) were grown for 1 wk on plastic and then treated with either 3.0 µM PMA or vehicle (DMSO) in serum-free media for 1 h in the presence or absence of protease inhibitors against both ADAM10 and ADAM17 (GW280264, 1.0 µM) or specific for ADAM10 (GI254023, 0.5 µM) (28). Inhibitor concentrations were based on published IC50 values for GW280264 and GI254023 with ADAM17 and ADAM10 (28). Others have reported mouse KIM-1 shedding from human embryonic kidney-293 cells based on inhibition by GI254023 (4 µM) (13), although the IC50 value for ADAM10 is 5 nM (28). Cell extract or media were immunoblotted for MUC1 as described above in Immunoblot Analysis. Transgenic mice expressing human MUC1 were used because no antibodies exist to study mouse extracellular domain shedding. The only available antibody for mouse MUC1 is the Armenian hamster CT2 antibody that recognizes the MUC1 cytoplasmic tail.
Preparation of Apoptotic Cells
Apoptotic thymocytes were prepared as previously described (1, 10, 29). Briefly, primary mouse thymocytes were disrupted into a single-cell suspension by passing through a 40-µm cell strainer. The next day, thymocytes were labeled with carboxyfluorescein succinimidyl ester (CFSE; CellTrace CFSE dye, Invitrogen, Eugene, OR) according to the manufacturer’s instructions and exposed to ultraviolet radiation (254 nm) for 1 min to induce apoptosis before incubation overnight at 37°C in 5% CO2. Using an Annexin V/Dead Cell Apoptosis Kit (Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit with Alexa Fluor 488 annexin V and propidium iodide, Invitrogen), we observed that >90% of exposed cells were deemed to be early apoptotic cells as evidence of annexin V staining (binds to phosphatidylserine: an “eat me” signal), whereas <5% of those were positive for the uptake of propidium iodide.
Phagocytosis Assay
To quantitate phagocytosis of apoptotic cells, we used a previously established method (1, 10, 29). Briefly, primary cultures of mouse kidney PTCs obtained from Muc1 KO and WT littermates were grown on glass coverslips or 12-well plate and then incubated with 2 × 106 CFSE fluorescently labeled (FITC) apoptotic thymocytes for 1 h at 37°C. After being washed vigorously five times with ice-cold PBS (without Mg2+/Ca2+) to remove noningested apoptotic cells, PTC monolayers on glass coverslips were fixed, premetallized, and costained as described above in Kidney Tissue Preparation and Confocal Immunofluorescence Microscopy. To quantify phagocytic uptake of apoptotic cells (efferocytosis), total number of internalized apoptotic cells per total number of epithelial cells was scored by blinded microscopic counting of random fields (5 images/mouse, n = 4 mice). PTC monolayers grown on 12-well plate were solubilized using lysis buffer containing 62.5 mM EDTA (pH 8.0), 50 mM Tris (pH 8.0), 4 mg sodium deoxycholate, and 1% Nonidet P-40 (IGEPAL). Fluorescence of the lysates was then analyzed using a multi-detection plate reader (Promega Glomax Multi Detection System, Madison, WI). PTC monolayers not incubated with thymocytes were considered as a reference control.
Statistical Analyses
All analyses were performed using GraphPad Prism version 9.0.0 (GraphPad Software, San Diego, CA). Comparisons involving only two groups were conducted with an unpaired t test, whereas multiple groups were analyzed using two-way ANOVA with a Tukey post hoc test. P < 0.05 was considered significant and presented with an asterisk(s). All data are reported as means ± SE.
RESULTS
MUC1 Protection of Kidney Function During IRI Is Associated With a Role in the PT
Muc1 KO mice and WT littermates were subject to 20-min ischemia and 48-h recovery using the kidney hanging weight protocol or sham surgery. Although heart rate, mean arterial blood pressure, and renal blood flow were not different in Muc1 KO mice and WT littermates after IRI or sham surgery (see Supplemental Fig. S1, A−C), we did find a significant reduction in GFR in Muc1 KO mice after IRI compared with in Muc1 KO sham mice (Fig. 1A). We also found a significant increase in serum creatinine in Muc1 KO mice after IRI compared with Muc1 KO sham mice or WT mice after IRI or sham surgery (Fig. 1B). Moreover, we found that the PT marker of kidney injury, KIM-1, but not the distal tubule injury marker of kidney injury, neutrophil gelatinase-associated lipocalin, was increased in the urine of Muc1 KO mice after IRI compared with in the urine of Muc1 KO mice sham and WT mice after IRI (Fig. 1, C and D). Taken together, these data indicate that MUC1 protection during IRI is not related to changes in kidney hemodynamics but reflects a role in renal epithelial cells of the PT where MUC1 induction was previously observed (17, 18).
Figure 1.

Mucin 1 (MUC1) protection of kidney function during ischemia-reperfusion injury (IRI) is associated with a role in the proximal tubule. Kidneys of Muc1 knockout (KO) mice (red) and wild-type (WT) littermates (blue) were subjected to 20-min ischemia and 48-h recovery using the kidney hanging weight protocol (IRI) or sham surgery (sham). Before euthanization, mice were assessed for glomerular filtration rate (GFR; A). Blood and urine were collected at euthanization and assayed for serum creatinine (B), urinary kidney injury molecule-1 (KIM-1) excretion rate (C), and urinary neutrophil gelatinase-associated lipocalin (NGAL) excretion rate (D). Values are means ± SE. Significant differences by two-way ANOVA are noted (*P < 0.05; ***P < 0.001). n = 5–7 mice.
Using our established confocal IF microscopy protocols (27, 30), we observed a strong induction of KIM-1 in the PT as confirmed by costaining with OAT1 (PT basolateral marker) in human kidneys with ischemic injury (see Supplemental Fig. S6, top) and in the mouse kidney subjected to 20-min ischemia and 48-h recovery (see Supplemental Fig. S6, bottom). Interestingly, we observed colocalization of KIM-1 and MUC1 in the same PT of WT mouse kidneys subjected to IRI (Fig. 2A). Similar results were observed by costaining sections from human kidneys with ischemic damage (Fig. 2C and Supplemental Fig. S2). Indeed, we observed induction of both KIM-1 and MUC1 in the same PT mainly at the apical membrane with substantial colocalization.
Figure 2.

Kidney injury molecule-1 (KIM-1) and mucin 1 (MUC1) are induced and coexpressed in the proximal tubule in the setting of ischemia. Kidneys of Muc1 knockout (KO) mice and wild-type (WT) littermates were subjected to 20-min ischemia and 48-h recovery [ischemia-reperfusion injury (IRI)] or sham surgery (sham). Kidneys were harvested at euthanization and stained for KIM-1 (green), MUC1 (CT2, red), and nuclei (TO-PRO, blue) (A) or KIM-1 (green), a disintegrin and metalloprotease 17 (ADAM17; red), and nuclei (TO-PRO, blue) (B) as indicated. C: human kidneys with ischemic damage were stained for KIM-1 (green), MUC1 (B27.29, red), and nuclei (TO-PRO, blue) as indicated. MUC1 staining was also observed in tubules not expressing KIM-1, consistent with MUC1 expression in the thick ascending limb, distal convoluted tubule, and collecting duct. Scale bar = 10 µm. n = 3 mice.
Kidney KIM-1 Levels Are Significantly Lower in Muc1 KO Mice Than WT Mice After IRI
The increased urinary KIM-1 observed in the absence of MUC1 during IRI could reflect an increase in KIM-1 expression in the PT in response to more severe injury. Surprisingly, immunoblot analysis of kidney extracts after IRI revealed a significant twofold lower level of KIM-1 in Muc1 KO mice compared with WT mice (Fig. 3A), whereas MUC1 levels increased 40% during IRI (Fig. 3B). Interestingly, we found a significant upregulation of the levels of kidney KIM-1 mRNA in both Muc1 KO and WT littermates following IRI compared with sham surgery, yet we did not observe a significant difference based on genotype (Fig. 3C). We considered several mechanisms for this observation. First, MUC1 could sequester ADAM17 from KIM-1 after injury, but we did not observe a difference in ADAM17 subcellular localization in Muc1 KO mouse kidneys compared with WT littermate kidneys after injury, as we noticed colocalization between KIM-1 and ADAM17 in both WT and Muc1 KO mouse kidneys (Fig. 2B). Second, MUC1 could stimulate the ERK1/2 → STAT3 → KIM-1 pathway reported to increase KIM-1 during IRI, but we found no changes in activation/phosphorylation of ERK1/2 and STAT3 by immunoblot analysis, suggesting posttranslational effects of MUC1 on KIM-1 expression (see Supplemental Fig. S3, A and B). Third, Zhang et al. (11) previously reported that ADAM17-mediated cleavage of KIM-1 is accelerated by p38 MAPK activation, but we found no difference in the levels of active p-p38 MAPK or ADAM17 by immunoblot analysis of kidney extracts from WT or Muc1 KO mice during IRI (see Supplemental Fig. S4, A and B). Our results indicate that the pathway of KIM-1-accelerated shedding was apparently activated in our mouse model of IRI, but this induction of ADAM17 activity was not affected by MUC1 levels.
Figure 3.

Kidney injury molecule-1 (KIM-1) expression is significantly lower in mucin 1 (Muc1) knockout (KO) mice than wild-type (WT) mice after ischemia-reperfusion injury (IRI). Kidneys of Muc1 KO mice (red) and WT littermates (blue) were subjected to 20-min ischemia and 48-h recovery (IRI) or sham surgery (sham). Kidneys were harvested at euthanization, and extracts of homogenates were subjected to immunoblot analysis for KIM-1 and β-actin (A) or MUC1 small subunit (using CT2) and β-actin (B). Levels of KIM-1 or MUC1 were normalized to β-actin, and values for WT-IRI mice were set at 1. Data are presented as means ± SE. Significant differences by Student’s t test are noted (*P < 0.05). n = 5. C: levels of KIM-1 mRNA were assessed by quantitative RT-PCR, and relative levels are presented with WT sham set at 1. Data were analyzed by two-way ANOVA, and significant differences are noted (***P < 0.001; ****P < 0.0001). n = 5–7 mice.
MUC1, ADAM17, and KIM-1 Are in the Same Complex in the Kidney in the Setting of Ischemia
Thathiah et al. (14) previously demonstrated that MUC1 and ADAM17 are in the same protein complex in human uterine epithelial cells. To assess this interaction in the kidney, we used both confocal immunofluorescence microscopy and an in situ PLA (27, 31). First, we observed an induction and immunofluorescence colocalization of both MUC1 and ADAM17 within the same PT of kidneys from WT mice subjected to IRI (Fig. 4A) and human kidneys with ischemic damage (Fig. 4B). Although we could not detect a signal for MUC1 in the glomeruli, we observed a strong staining for ADAM17 in the glomeruli (Fig. 4B, middle), which we used as one of our negative controls for PLA. We also observed a robust PLA signal for MUC1 and ADAM17 in the human kidney with ischemic injury that was completely absent in the negative control lacking the anti-MUC1 antibody and absent in the glomerulus, which lacks MUC1 expression (Fig. 4C). As there is no PLA probe available for the Armenian hamster CT2 antibody that recognizes mouse MUC1, we were not able to assess this interaction in mouse kidneys. Importantly, we also observed colocalization between KIM-1, MUC1, and ADAM17 within the same PT of human kidneys with ischemic damage (Fig. 4D). Taken together, these data further suggest that KIM-1, MUC1, and ADAM17 are in the same protein complex and that ADAM17 likely mediates KIM-1 and MUC1 shedding in the kidney.
Figure 4.

Mucin 1 (MUC1), a disintegrin and metalloprotease 17 (ADAM17), and kidney injury molecule-1 (KIM-1) are in the same complex in the kidney in the setting of ischemia. A: kidneys of wild-type (WT) mice were subjected to 20-min ischemia and 48-h recovery. Kidneys were harvested at euthanization and stained for MUC1 small subunit (CT2, green), ADAM17 (red), and nuclei (TO-PRO, blue) as indicated. B: human kidneys with ischemic damage incubated with no primary antibodies (left) or with antibodies against MUC1 extracellular tandem repeats (B27.29, green), ADAM17 (red), and nuclei (TO-PRO, blue) as indicated. C: in situ proximity ligation assay (PLA) was carried out using proximity probes against MUC1 (mouse B27.29) and ADAM17 (rabbit polyclonal antibody) to visualize the MUC1/ADAM17 complex in human kidney sections. Notice that the PLA signal is absent in the negative control lacking the anti-MUC1 antibody and absent in the glomerulus (GM) that lacks MUC1 expression despite high levels of ADAM17 expression. D: human kidneys with ischemic damage were costained for KIM-1 (green), ADAM17 (red), MUC1 extracellular tandem repeats (B27.29, gray), and nuclei (TO-PRO, blue) as indicated. Scale bar = 10 µm. n = 3 mice. IRI, ischemia-reperfusion injury.
KIM-1 Shedding Is Reduced by MUC1 Expression in a Cell Culture Model
Using primary cultures of PTCs prepared from transgenic mice expressing human MUC1, we did observe ADAM-17-dependent shedding of MUC1 as previously described in human uterine epithelial cells (see Supplemental Fig. S5) (14). To test the hypothesis that MUC1 competes with KIM-1 as a substrate for ADAM17, we used an in vitro model of cultured MDCK cells stably transfected with mouse KIM-1 and transiently transfected with human MUC1 cDNA or empty vector. The following day, cells were incubated in serum-free medium with either 1) PMA (an ADAM17 activator), 2) BB-94 (a broad-spectrum matrix metalloprotease inhibitor), 3) both PMA and BB-94, or 4) vehicle (DMSO). By immunoblot analysis of cell extracts and immunoprecipitates from culture medium, we observed that KIM-1 shedding was significantly enhanced after treatment of the cells with PMA and inhibited after BB-94 treatment (Fig. 5). Levels of KIM-1 shedding (∼1%/h) were similar to a previous study in cultured cells (10). Using two-way ANOVA, we found that KIM-1 shedding was significantly reduced overall by MUC1 expression (P < 0.01; Fig. 5C). We conclude that MUC1 and KIM-1 compete as substrates for ADAM17.
Figure 5.

Kidney injury molecule-1 (KIM-1) shedding is reduced by mucin 1 (MUC1) expression in a cell culture model. Madin–Darby canine kidney (MDCK) cells stably transfected with mouse KIM-1 were transiently transfected with human MUC1 cDNA or empty vector (EV). The next day, cells were incubated for 2 h in serum-free media with either 1) 1.6 mM phorbol-12-myristate-13-acetate (PMA) to induce a disintegrin and metalloprotease 17 (ADAM17) metalloprotease-mediated shedding of KIM-1 and MUC1, 2) 10 mM BB-94 (BB; a broad-spectrum matrix metalloprotease inhibitor), 3) both PMA and BB-94, or 4) vehicle (DMSO). Cell extracts (2.5%; A) or immunoprecipitates (B) of KIM-1 or MUC1 from media were immunoblotted for KIM-1 or MUC1 large subunit (B27.29). C: levels of KIM-1 in the medium are presented as percentages of levels in the same cells transfected with MUC1 (blue) or EV (red). Data were analyzed by two-way ANOVA, and significant differences are noted (*P < 0.05; **P < 0.01). n = 7−12 independent experiments.
Cell Association and Function of KIM-1 Are Reduced After IRI in the Absence of MUC1
Using confocal IF, we observed KIM-1 staining in the WT mouse kidney PT localized primarily to the apical membrane (Fig. 6A), with a low level of KIM-1 staining surrounding cellular debris within the adjacent tubule lumen consistent with efficient efferocytosis (Fig. 6A, inset). In contrast, KIM-1 staining was more diffuse in the Muc1 KO mouse kidney PT following injury (Fig. 6B), and KIM-1 staining was primarily associated with cell debris in the lumen of the PT (Fig. 6B, inset). The data are consistent with enhanced shedding of KIM-1 from the PT after IRI in the absence of MUC1.
Figure 6.

Cell-associated kidney injury molecule-1 (KIM-1) is reduced after ischemia-reperfusion injury (IRI) in the absence of mucin 1 (MUC1). Kidneys of Muc1 knockout (KO) mice and wild-type (WT) littermates were subjected to 20-min ischemia and 48-h recovery (IRI). Kidneys (n = 4 mice) were harvested at euthanization and stained for KIM-1 (green) and nuclei (TO-PRO; blue). Note that KIM-1 in WT kidneys (A) was primarily cell associated or surrounds luminal cell debris (see inset), whereas KIM-1 staining in Muc1 KO kidneys (B) was more diffuse and found primarily with cell debris (see inset).
As KIM-1-mediated efferocytosis is dependent on cell-associated KIM-1, we used previously established assays (1, 2, 29) to measure the uptake of fluorescently labeled (FITC) apoptotic thymocytes by primary cultures of PTCs (Fig. 7). We observed a significant 22% reduction in thymocyte uptake by Muc1 KO PTCs compared with WT PTCs by IF (P < 0.003; Fig. 7, A and B). Using a plate reader, Muc1 KO PTCs growing on plastic exhibited significantly less fluorescence (76%; Fig. 7C) compared with WT PTCs (P < 0.01). Immunoblot analysis of cell extracts revealed lower levels of KIM-1 expression in Muc1 KO PTCs (71%) compared with WT PTCs (Fig. 7, D and E). Thymocytes were also observed in KIM-1-expressing cells but not in KIM-1-negative cells (Fig. 7, F and G). Of note, we observed that 25–30% of PTCs were expressing KIM-1 in the absence of ischemic injury. Taken together, the decreased efferocytosis in Muc1 KO PTCs could be explained, in part, by reduced surface expression of KIM-1 in the absence of MUC1.
Figure 7.

Decreased efferocytosis in primary cultured proximal tubule cells from mucin 1 (Muc1) knockout (KO) compared with wild-type (WT) littermates. Primary cultures of mouse kidney proximal tubule (PT) cells obtained from Muc1 KO (red) and WT littermates (blue) were grown on glass coverslips (A, B, F, and G) or 12-well plates (C−E) and then incubated with carboxyfluorescein succinimidyl ester fluorescently labeled (green) apoptotic thymocytes. A: cocultured PT monolayers with FITC-thymocytes (green) on coverslips were costained for actin (phalloidin; gray) and nuclei (TO-PRO; blue). Scale bar = 10 µm. B: total number of internalized apoptotic cells per total number of epithelial cells was scored by blinded microscopic counting of random fields. C: cocultured PT cell monolayers with FITC-thymocytes on 12-well plates were washed and solubilized, and fluorescence of the lysates was quantitatively assessed by a plate reader. D: PT cell lysates were subjected to immunoblot analysis for kidney injury molecule-1 (KIM-1) and β-actin. Levels of KIM-1 were normalized to β-actin, and values for WT mice were set at 1. E: data from D are presented as means ± SE. Significant differences by Student’s t test are noted (*P < 0.05). n = 4. F: cocultured PT cell monolayers with FITC-thymocytes (green) were costained for KIM-1 (red), phalloidin (gray), and nuclei (TO-PRO; blue). G: confocal reconstruction (X–Z) of KIM-1 and FITC-thymocytes demonstrating KIM-1 localization at the phagocytic cup and surrounding phagocytosed apoptotic cells within PT cells. Scale bar = 10 µm. n = 4 mice. PTC, PT cells.
Cell-associated KIM-1 exhibits an anti-inflammatory function by downregulating TLR4 and its downstream signaling proinflammatory NF-κB pathway following injury (2). By immunoblot analysis of kidney extracts, we observed a significantly higher increase in TLR4 and NF-κB (nuclear and cytoplasmic) during IRI in Muc1 KO mice compared with WT mice (Fig. 8, A−C). Taken together, the data indicate that MUC1 stabilizes cell-associated KIM-1 during IRI, which could protect the kidney by downregulating innate immunity and inflammation (Fig. 9).
Figure 8.

Mucin 1 (MUC1) stabilization of cell-associated kidney injury molecule-1 (KIM-1) correlates with reduced inflammation following ischemia-reperfusion injury (IRI). Kidneys of Muc1 knockout (KO) mice (red) and wild-type (WT) littermates (blue) were subjected to 20-min ischemia and 48-h recovery (IRI) or sham surgery (sham). A: kidneys were harvested at euthanization, and extracts of homogenates were subjected to immunoblot analysis for Toll-like receptor 4 (TLR4) and β-actin. B and C: aliquots of either nuclear extracts (Nuc) or cytoplasmic postnuclear supernatants (Cyto) of mouse kidneys were subjected to immunoblot analysis for NF-κB and histone H3 or β-actin, respectively. TLR4 and NF-κB levels were normalized to β-actin or histone H3 as indicated, and levels for WT-sham were set to 1. Data were analyzed by two-way ANOVA (*P < 0.05; **P < 0.01). n = 5 and 6 mice.
Figure 9.

Kidney injury molecule-1 (KIM-1) activity is preserved in the injured kidney by mucin 1 (MUC1) inhibition of KIM-1 shedding. Left: MUC1 induction in the proximal tubule following kidney injury reduces KIM-1 shedding by competing for a disintegrin and metalloprotease 17 (ADAM17). Right: in the absence of MUC1, KIM-1 cleavage is accelerated and shed into the lumen, where it acts as a decoy receptor, and subsequently inhibits efferocytosis. We hypothesize that stabilization of cell-associated KIM-1 by MUC1 induction during ischemia-reperfusion injury could protect the kidney by maintaining KIM-1-mediated cell signaling and efferocytosis that reduces inflammation and promotes recovery. AKI, acute kidney injury.
DISCUSSION
Using a mouse model of IRI where the renal pedicles were clamped for 19 min, we found that MUC1, predominately found in the thick ascending loop and later segments of the kidney tubule, was actually present and notably induced in the PT over 72-h recovery (17, 18). Kidney damage was worse and recovery was blocked during IRI in Muc1 KO mice compared with congenic sham mice (based on histology and serum creatinine), and we discovered that MUC1 stabilized both hypoxia-inducible factor-1α and β-catenin, their protective signaling pathways, and prevented metabolic stress, including prolonged activation of AMP-activated protein kinase (18, 19). In the present study, we used the hanging weight protocol for renal IRI, which has been shown to be a more clinically realistic model of IRI that provides highly reproducible kidney injury with virtually no tissue trauma or blood vessel congestion by blocking only the renal artery without venous occlusion (32, 33). After 20-min ischemia using this new protocol and 48-h recovery, we found that Muc1 KO mice had more severe kidney injury than littermate WT mice, consistent with our previous work. We also found MUC1 induced in kidney extracts by immunoblot analysis and that MUC1 was induced in all kidney tubules, including the PT, by confocal microscopy. Although we were not surprised to find significantly higher levels of urinary KIM-1 in Muc1 KO mice compared with WT mice due to the more severe kidney injury, we were surprised to find reduced levels of KIM-1 in kidney extracts of Muc1 KO mice despite similar induction of mRNA levels in Muc1 KO and WT kidneys. Our subsequent analysis was, therefore, focused on understanding how the absence of MUC1 could affect KIM-1 expression and consequently its function.
Given that KIM-1 is the most highly upregulated protein in the PT and because of its anti-inflammatory function following kidney injury (1, 2), there has been great effort to identify the upstream regulators of KIM-1. Although the results of two recent studies agree that phosphorylation of STAT3 is key to KIM-1 upregulation, two different kinases were implicated in rat (checkpoint kinase 1) and mouse (ERK1/2) studies (7, 8). However, we found no difference in STAT3 levels or activation between our sham and treated mice and no difference between Muc1 KO and WT mice. Even though we could have missed any acute-phase reactions by focusing on 48-h recovery (7, 8, 34), we also considered whether KIM-1 shedding was affected by the absence of MUC1.
We first asked whether MUC1 could increase KIM-1 shedding by regulating the expression of a relevant protease. ADAM17, also known as TNF-α converting enzyme, is responsible for the ectodomain shedding of many cell surface proteins, including KIM-1 and MUC1, and generation of many diverse active soluble ligands involved in development, regeneration, and inflammation (10, 14, 35, 36). In fact, Tang et al. (37) found increased levels of active p38 MAPK and mature ADAM17 in a liver model of IRI, and Zhang et al. (11) found that KIM-1 shedding was accelerated in a cell culture model by p38 MAPK activation. Although we did find activation of p38 MAPK and increased ADAM17 in our mouse kidney model of IRI compared with sham, we found no differences between Muc1 KO and WT mice.
Alternatively, we asked whether MUC1 could compete with KIM-1 as a substrate for ADAM17. This possibility would be consistent with the increase in urinary levels we observed in the absence of MUC1 during IRI in mice while kidney levels of KIM-1 were reduced. Using IF confocal microscopy of mouse and human kidneys and an in situ PLA in human kidneys, we first established that MUC1, KIM-1, and ADAM17 were colocalized at the apical plasma membrane of PTs. However, after ischemic injury, mouse KIM-1 was primarily associated with cellular debris in the tubule lumen in the absence of MUC1, suggesting that MUC1 blocks shedding of KIM-1. We also found that mouse KIM-1 shedding was stimulated in cultured MDCK cells (canine) by PMA, consistent with induction of ADAM17, and that shedding was consistently reduced by coexpression with MUC1 and/or treatment of MDCK cells with a broad-spectrum metalloprotease inhibitor. Human MUC1 shedding was also reduced in primary cultures of transgenic mouse PTCs by an inhibitor specific for ADAM17 and ADAM10 but not by an inhibitor specific for ADAM10 alone. The consistency of our data implicating MUC1 inhibition of KIM-1 shedding is particularly interesting, as human, murine, and canine ADAM17 have >90% amino acid sequence identity, whereas the transmembrane subunit of human and murine MUC1 exhibits only 65% identity, which could reflect differences between species. Data from the Carson laboratory previously identified a specific site for human ADAM17 cleavage of human MUC1 at the position 58 residues from the transmembrane domain but found no evidence of MUC1 cleavage by ADAM9, ADAM10, ADAM12, or ADAM15 (14, 20). As recent studies have implicated ADAM10 in the cleavage of human and mouse KIM-1 (12, 13), it becomes important to reassess the role of ADAM10 in mouse and human MUC1 shedding in future studies.
More importantly, we observed a difference in KIM-1 functional activity between Muc1 KO mice and WT mice in kidneys during IRI. Cell-associated KIM-1 has an anti-inflammatory role, as it mediates phagocytic clearance of apoptotic and necrotic cells (efferocytosis) following kidney injury, which, in turn, prevents inflammation and promotes kidney repair (1, 29). Furthermore, efferocytosis mediated by cell-associated KIM-1 also triggers KIM-1 signaling and thus downregulation of the proinflammatory NF-κB signaling pathway (2). However, any accelerated shedding of KIM-1 competitively inhibits efferocytosis and its subsequent anti-inflammatory effects, as the excess soluble KIM-1 also acts as a decoy to cell-associated KIM-1 (10). Using confocal microscopy, we observed that KIM-1 was primarily apical (cell associated) in the PT of WT mice where it mediates phagocytosis by interacting directly and partly surrounding the adjacent cellular debris following injury. In contrast, KIM-1 staining is confined mainly to the tubular lumen (cleaved form) together with cell debris even in the surviving PT of Muc1 KO mouse kidneys after injury. Furthermore, using IF staining, we also observed KIM-1 localized at the phagocytic cup and surrounding phagocytosed apoptotic cells within PTCs. More importantly, we noticed a significant difference in KIM-1 functional activity (efferocytosis) between Muc1 KO and WT primary cultured PTCs. Finally, we demonstrated internalization of apoptotic cells within KIM-1-expressing cells but not adjacent KIM-1-negative primary cells.
Our findings are in agreement with a previous report in which authors confirmed the internalization of apoptotic cells by KIM-1-expressing cells (efferocytosis) when they demonstrated a direct contact between cell-associated KIM-1 and apoptotic cells using a confocal microscopy approach (1). Consistent with this distinct cellular distribution of KIM-1 after kidney injury, we found that kidneys of Muc1 KO mice have more than a twofold significant increase in active NF-κB levels (nuclear expression) than WT mice following IRI. Previous work showed a similar result in which greater inflammatory response and kidney dysfunction were reported in mice expressing KIM-1Δmucin (KIM-1 protein missing the mucin domain required for apoptotic cells binding) compared with congenic shams following ischemic AKI (2). Altogether, these findings suggested that stabilizing cell-associated KIM-1 by MUC1 induction during IRI could protect the kidney by maintaining KIM-1-mediated efferocytosis and thus downregulating innate immunity and inflammation (Fig. 9).
Perspectives and Significance
KIM-1 that is rapidly induced in the PT during renal IRI plays a key role in the recovery of the tubule epithelium by serving as a cell surface receptor for clearance of apoptotic cells (efferocytosis) and associated signaling that suppresses inflammation. Although ADAM17 cleavage of KIM-1 yields a urinary biomarker, excessive cleavage provides a decoy receptor that aggravates efferocytosis and subsequent signaling. MUC1 is also induced during IRI and cleaved by ADAM17. Our data from experiments in mice and cultured cells show that MUC1 competes with KIM-1 for cleavage by ADAM17. Consequently, MUC1 protects KIM-1 anti-inflammatory activity in the damaged kidney.
SUPPLEMENTAL DATA
Supplemental Tables S1 and S3 and Supplemental Figs. S1–S6: https://doi.org/10.6084/m9.figshare.14364890.
GRANTS
This work was supported by National Institutes of Health Grants K01DK109038, HL109002, DK091190, HL069846, DK070910, and DK038470; the Kidney Imaging Core of the Pittsburgh Center for Kidney Research (P30DK079307); and Dialysis Clinic Incorporated.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M.A., C.L.K., Z.M., E.K.J., D.R.E., J.A.K., and R.P.H. conceived and designed research; M.M.A., C.L.K., Z.M., E.K.J., S.M.M., D.R.E., and R.P.H. performed experiments; M.M.A., Z.M., E.K.J., S.M.M., J.A.K., and R.P.H. analyzed data; M.M.A., E.K.J., J.A.K., and R.P.H. interpreted results of experiments; M.M.A. and S.M.M. prepared figures; M.M.A. drafted manuscript; M.M.A., C.L.K., Z.M., E.K.J., D.R.E., J.A.K., and R.P.H. edited and revised manuscript; M.M.A., C.L.K., Z.M., E.K.J., D.R.E., J.A.K., and R.P.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Joseph Bonventre (Director, Renal Division, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA) and Dr. Craig Brooks (Department of Cell and Developmental Biology, Vanderbilt School of Medical, Nashville, TN) for providing a cDNA for mouse KIM-1. We thank Dr. Olivera Finn (Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA) for providing kidneys from transgenic MUC1 mice and the Center for Organ Recovery and Education for provision of human material for research (Pittsburgh, PA).
REFERENCES
- 1.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118: 1657–1668, 2008. doi: 10.1172/JCI34487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, Ichimura T, Kuchroo V, Bonventre JV. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest 125: 1620–1636, 2015. doi: 10.1172/JCI75417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ichimura T, Hung CC, Yang SA, Stevens JL, Bonventre JV. Kidney injury molecule-1: a tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Renal Physiol 286: F552–563, 2004. doi: 10.1152/ajprenal.00285.2002. [DOI] [PubMed] [Google Scholar]
- 4.Humphreys BD, Xu F, Sabbisetti V, Grgic I, Movahedi Naini S, Wang N, Chen G, Xiao S, Patel D, Henderson JM, Ichimura T, Mou S, Soeung S, McMahon AP, Kuchroo VK, Bonventre JV. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest 123: 4023–4035, 2013. doi: 10.1172/JCI45361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han WK, Alinani A, Wu CL, Michaelson D, Loda M, McGovern FJ, Thadhani R, Bonventre JV. Human kidney injury molecule-1 is a tissue and urinary tumor marker of renal cell carcinoma. J Am Soc Nephrol 16: 1126–1134, 2005. doi: 10.1681/ASN.2004070530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuehn EW, Park KM, Somlo S, Bonventre JV. Kidney injury molecule-1 expression in murine polycystic kidney disease. Am J Physiol Renal Physiol 283: F1326–F1336, 2002. doi: 10.1152/ajprenal.00166.2002. [DOI] [PubMed] [Google Scholar]
- 7.Collier JB, Schnellmann RG. Extracellular signal-regulated kinase 1/2 regulates mouse kidney injury molecule-1 expression physiologically and following ischemic and septic renal injury. J Pharmacol Exp Ther 363: 419–427, 2017. doi: 10.1124/jpet.117.244152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ajay AK, Kim TM, Ramirez-Gonzalez V, Park PJ, Frank DA, Vaidya VS. A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J Am Soc Nephrol 25: 105–118, 2014. doi: 10.1681/ASN.2013020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han WK, Waikar SS, Johnson A, Betensky RA, Dent CL, Devarajan P, Bonventre JV. Urinary biomarkers in the early diagnosis of acute kidney injury. Kidney Int 73: 863–869, 2008. [Erratum in Kidney Int 76: 348–349, 2009]. doi: 10.1038/sj.ki.5002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandhi R, Yi J, Ha J, Shi H, Ismail O, Nathoo S, Bonventre JV, Zhang X, Gunaratnam L. Accelerated receptor shedding inhibits kidney injury molecule-1 (KIM-1)-mediated efferocytosis. Am J Physiol Renal Physiol 307: F205–F221, 2014. doi: 10.1152/ajprenal.00638.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Humphreys BD, Bonventre JV. Shedding of the urinary biomarker kidney injury molecule-1 (KIM-1) is regulated by MAP kinases and juxtamembrane region. J Am Soc Nephrol 18: 2704–2714, 2007. doi: 10.1681/ASN.2007030325. [DOI] [PubMed] [Google Scholar]
- 12.Schweigert O, Dewitz C, Moller-Hackbarth K, Trad A, Garbers C, Rose-John S, Scheller J. Soluble T cell immunoglobulin and mucin domain (TIM)-1 and -4 generated by a disintegrin and metalloprotease (ADAM)-10 and -17 bind to phosphatidylserine. Biochim Biophys Acta 1843: 275–287, 2014. doi: 10.1016/j.bbamcr.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Sriranganathan S, Tutunea-Fatan E, Abbasi A, Gunaratnam L. Mapping and functional characterization of murine kidney injury molecule-1 proteolytic cleavage site. Mol Cell Biochem 476: 1093–1108, 2021. doi: 10.1007/s11010-020-03975-5. [DOI] [PubMed] [Google Scholar]
- 14.Thathiah A, Blobel CP, Carson DD. Tumor necrosis factor-alpha converting enzyme/ADAM 17 mediates MUC1 shedding. J Biol Chem 278: 3386–3394, 2003. doi: 10.1074/jbc.M208326200. [DOI] [PubMed] [Google Scholar]
- 15.Kefaloyianni E, Muthu ML, Kaeppler J, Sun X, Sabbisetti V, Chalaris A, Rose-John S, Wong E, Sagi I, Waikar SS, Rennke H, Humphreys BD, Bonventre JV, Herrlich A. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 1: e87023, 2016. doi: 10.1172/jci.insight.87023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulder GM, Melenhorst WB, Celie JW, Kloosterhuis NJ, Hillebrands JL, Ploeg RJ, Seelen MA, Visser L, van Dijk MC, van Goor H. ADAM17 up-regulation in renal transplant dysfunction and non-transplant-related renal fibrosis. Nephrol Dial Transplant 27: 2114–2122, 2012. doi: 10.1093/ndt/gfr583. [DOI] [PubMed] [Google Scholar]
- 17.Al-Bataineh MM, Sutton TA, Hughey RP. Novel roles for mucin 1 in the kidney. Curr Opin Nephrol Hypertens 26: 384–391, 2017. doi: 10.1097/MNH.0000000000000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pastor-Soler NM, Sutton TA, Mang HE, Kinlough CL, Gendler SJ, Madsen CS, Bastacky SI, Ho J, Al-Bataineh MM, Hallows KR, Singh S, Monga SP, Kobayashi H, Haase VH, Hughey RP. Muc1 is protective during kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol 308: F1452–F1462, 2015. doi: 10.1152/ajprenal.00066.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Bataineh MM, Kinlough CL, Poland PA, Pastor-Soler NM, Sutton TA, Mang HE, Bastacky SI, Gendler SJ, Madsen CS, Singh S, Monga SP, Hughey RP. Muc1 enhances the β-catenin protective pathway during ischemia-reperfusion injury. Am J Physiol Renal Physiol 310: F569–F579, 2016. doi: 10.1152/ajprenal.00520.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thathiah A, Carson DD. MT1-MMP mediates MUC1 shedding independent of TACE/ADAM17. Biochem J 382: 363–373, 2004. doi: 10.1042/BJ20040513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emlet DR, Pastor-Soler N, Marciszyn A, Wen X, Gomez H, WHt H, Morrisroe S, Volpe JK, Kellum JA. Insulin-like growth factor binding protein 7 and tissue inhibitor of metalloproteinases-2: differential expression and secretion in human kidney tubule cells. Am J Physiol Renal Physiol 312: F284–F296, 2017. doi: 10.1152/ajprenal.00271.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheridan AM, Schwartz JH, Kroshian VM, Tercyak AM, Laraia J, Masino S, Lieberthal W. Renal mouse proximal tubular cells are more susceptible than MDCK cells to chemical anoxia. Am J Physiol Renal Physiol 265: F342–F350, 1993. doi: 10.1152/ajprenal.1993.265.3.F342. [DOI] [PubMed] [Google Scholar]
- 23.Kardon R, Price RE, Julian J, Lagow E, Tseng SC, Gendler SJ, Dd C. Bacterial conjunctivitis in Muc1 null mice. Invest Ophthal Vis Sci 40: 1328–1335, 1999. [PubMed] [Google Scholar]
- 24.DeSouza MM, Surveyor GA, Price RE, Julian J, Kardon R, Zhou X, Gendler S, Hilkens J, Carson DD. MUC1/episialin: a critical barrier in the female reproductive tract. J Reprod Immunol 45: 127–158, 1999. doi: 10.1016/s0165-0378(99)00046-7. [DOI] [PubMed] [Google Scholar]
- 25.Jackson EK, Menshikova EV, Mi Z, Verrier JD, Bansal R, Janesko-Feldman K, Jackson TC, Kochanek PM. Renal 2′,3′-Cyclic nucleotide 3′-phosphodiesterase is an important determinant of AKI severity after ischemia-reperfusion. J Am Soc Nephrol 27: 2069–2081, 2016. doi: 10.1681/ASN.2015040397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zinellu A, Caria MA, Tavera C, Sotgia S, Chessa R, Deiana L, Carru C. Plasma creatinine and creatine quantification by capillary electrophoresis diode array detector. Anal Biochem 342: 186–193, 2005. doi: 10.1016/j.ab.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 27.Al-Bataineh MM, Alzamora R, Ohmi K, Ho PY, Marciszyn AL, Gong F, Li H, Hallows KR, Pastor-Soler NM. Aurora kinase A activates the vacuolar H+-ATPase (V-ATPase) in kidney carcinoma cells. Am J Physiol Renal Physiol 310: F1216–F1228, 2016. doi: 10.1152/ajprenal.00061.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102: 1186–1195, 2003. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 29.Brooks CR, Yeung MY, Brooks YS, Chen H, Ichimura T, Henderson JM, Bonventre JV. KIM-1-/TIM-1-mediated phagocytosis links ATG5-/ULK1-dependent clearance of apoptotic cells to antigen presentation. EMBO J 34: 2441–2464, 2015. doi: 10.15252/embj.201489838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Bataineh MM, Li H, Ohmi K, Gong F, Marciszyn AL, Naveed S, Zhu X, Neumann D, Wu Q, Cheng L, Fenton RA, Pastor-Soler NM, Hallows KR. Activation of the metabolic sensor AMP-activated protein kinase inhibits aquaporin-2 function in kidney principal cells. Am J Physiol Renal Physiol 311: F890–F900, 2016. doi: 10.1152/ajprenal.00308.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45: 227–232, 2008. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Grenz A, Eckle T, Zhang H, Huang DY, Wehrmann M, Kohle C, Unertl K, Osswald H, Eltzschig HK. Use of a hanging-weight system for isolated renal artery occlusion during ischemic preconditioning in mice. Am J Physiol Renal Physiol 292: F475–F485, 2007. doi: 10.1152/ajprenal.00275.2006. [DOI] [PubMed] [Google Scholar]
- 33.Jackson EK, Menshikova EV, Mi Z, Verrier JD, Bansal R, Janesko-Feldman K, Jackson TC, Kochanek PM. Renal 2′,3′-cyclic nucleotide 3′-phosphodiesterase is an important determinant of AKI severity after ischemia-reperfusion. J Am Soc Nephrol 27: 2069–2081, 2016.doi: 10.1681/asn.2015040397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinton LJ, Blahna MT, Jones MR, Allen E, Ferrari JD, Hilliard KL, Zhang X, Sabharwal V, Algul H, Akira S, Schmid RM, Pelton SI, Spira A, Mizgerd JP. Hepatocyte-specific mutation of both NF-κB RelA and STAT3 abrogates the acute phase response in mice. J Clin Invest 122: 1758–1763, 2012. doi: 10.1172/JCI59408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, van Heel DA, Ruschendorf F, Toynbee M, Walne A, O’Toole EA, Martin JE, Lindley K, Vulliamy T, Abrams DJ, MacDonald TT, Harper JI, Kelsell DP. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med 365: 1502–1508, 2011. doi: 10.1056/NEJMoa1100721. [DOI] [PubMed] [Google Scholar]
- 36.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science 282: 1281–1284, 1998. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 37.Tang ZY, Loss G, Carmody I, Cohen AJ. TIMP-3 ameliorates hepatic ischemia/reperfusion injury through inhibition of tumor necrosis factor-alpha-converting enzyme activity in rats. Transplantation 82: 1518–1523, 2006. doi: 10.1097/01.tp.0000243381.41777.c7. [DOI] [PubMed] [Google Scholar]