

Keywords: intercalated cells, large-conductance potassium channel, potassium secretion, WNK1 kinase
Abstract
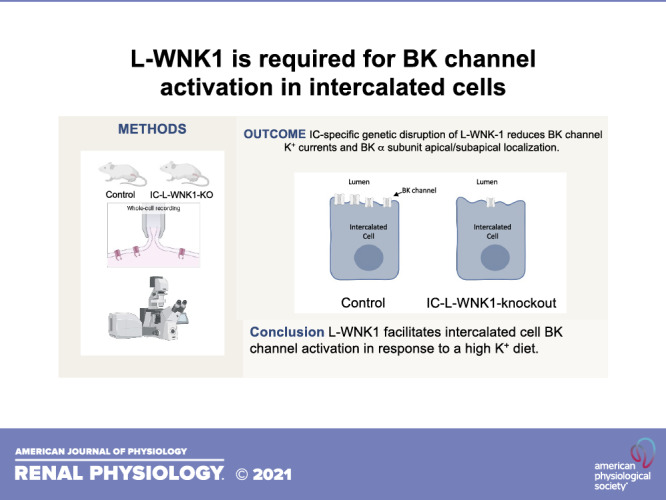
Large-conductance K+ (BK) channels expressed in intercalated cells (ICs) in the aldosterone-sensitive distal nephron (ASDN) mediate flow-induced K+ secretion. In the ASDN of mice and rabbits, IC BK channel expression and activity increase with a high-K+ diet. In cell culture, the long isoform of with-no-lysine kinase 1 (L-WNK1) increases BK channel expression and activity. Apical L-WNK1 expression is selectively enhanced in ICs in the ASDN of rabbits on a high-K+ diet, suggesting that L-WNK1 contributes to BK channel regulation by dietary K+. We examined the role of IC L-WNK1 expression in enhancing BK channel activity in response to a high-K+ diet. Mice with IC-selective deletion of L-WNK1 (IC-L-WNK1-KO) and littermate control mice were placed on a high-K+ (5% K+, as KCl) diet for 10 or more days. IC-L-WNK1-KO mice exhibited reduced IC apical + subapical α-subunit expression and BK channel-dependent whole cell currents compared with controls. Six-hour urinary K+ excretion in response a saline load was similar in IC-L-WNK1-KO mice and controls. The observations that IC-L-WNK1-KO mice on a high-K+ diet have higher blood K+ concentration and reduced IC BK channel activity are consistent with impaired urinary K+ secretion, demonstrating that IC L-WNK1 has a role in the renal adaptation to a high-K+ diet.
NEW & NOTEWORTHY When mice are placed on a high-K+ diet, genetic disruption of the long form of with no lysine kinase 1 (L-WNK1) in intercalated cells reduced relative apical + subapical localization of the large-conductance K+ channel, blunted large-conductance K+ channel currents in intercalated cells, and increased blood K+ concentration. These data confirm an in vivo role of L-WNK1 in intercalated cells in adaptation to a high-K+ diet.
INTRODUCTION
A moderate daily consumption of K+ in humans (∼100 mmol) exceeds the total extracellular K+ content of an adult (∼70 mmol) (1). Life-threatening hyperkalemia would certainly occur if not for the initial rapid intracellular buffering of excess K+ that enters the extracellular space and the eventual urinary excretion of K+. In the context of a high-K+ diet, the aldosterone-sensitive distal nephron (ASDN) is a key site of secretion of K+ into the tubular lumen, leading to net urinary K+ excretion. High flow rates of tubular fluid further enhance K+ secretion in the ASDN, increasing urinary K+ excretion (2–7).
An iberiotoxin-sensitive, large-conductance, Ca2+-, and/or stretch-activated K+ channel, referred to as BK or KCa1.1, has a critical role in flow-induced K+ secretion (FIKS) in the ASDN (6–10). A high-K+ environment (in salamanders) or high-K+ diet (in rabbits and mice) stimulated apical expression of this channel and BK channel-mediated K+ secretion in the collecting duct (CD) (6, 7, 9–12). Global knockout (KO) of the mouse BK α- or β1-subunit provided support for the role of BK channels in renal K+ secretion (13, 14).
Epithelial Na+ channel (ENaC)-dependent Na+ absorption in the ASDN has an important role in K+ secretion and FIKS (15, 16), although ENaC-independent K+ secretion has also been observed (17). BK channels are expressed in both principal cells (PCs) and intercalated cells (ICs) (6, 9, 12, 18, 19). The question of whether BK channels in PCs, ICs, or both cell types are responsible for FIKS has been addressed by several groups. There is a growing body of evidence supporting a key role of IC BK channels in FIKS. BK α-subunit expression in PCs is concentrated within the cells’ single apical cilium (12). Chemical deciliation of PCs did not impair FIKS in isolated, perfused cortical CDs (CCDs), suggesting that PCs do not have a central role in FIKS. Transition of rabbits to a high-K+ diet increases BK α-subunit expression primarily in ICs, rather than PCs (12). In mice, IC-selective KO of the BK α-subunit abolished FIKS in isolated, perfused CCDs from these animals and was associated with an increased blood K+ concentration, at least in male mice on a high-K+ diet (9).
Mechanisms by which increases in dietary K+ enhance IC BK α-subunit expression and BK channel activity remain unclear. Previous studies have suggested that members of the with-no-lysine (WNK) kinase family are intermediaries in this process. WNK4 inhibited protein and functional expression of the BK α-subunit in human embryonic kidney-293 cells (20–22). In contrast, both the long form of WNK1 (L-WNK1) and a truncated, kidney-specific isoform of WNK1 (KS-WNK1) augmented expression of the BK α-subunit in human embryonic kidney-293 cells (23, 24). Furthermore, rabbits fed a high-K+ diet exhibited enhanced apical expression of L-WNK1 selectively in ICs (24). As L-WNK1 inhibits renal outer medullary K+ channels (ROMK) (25, 26), a key channel mediating K+ secretion in PCs, an IC-selective increase in the expression of L-WNK1 has been proposed to have an important role in the renal response to a high-K+ diet (24). These observations led us to examine the role of L-WNK1 in ICs in the renal adaptive response to a high-K+ diet. We generated and characterized a mouse model with IC-specific disruption of L-WNK1 (IC-L-WNK1-KO). We found that in mice on a high-K+ diet, genetic disruption of L-WNK-1 in ICs reduced relative apical + subapical localization of the BK α-subunit, blunted charybdotoxin (ChTX)-sensitive K+ currents in ICs, and increased blood K+ concentration. These data confirm an in vivo role for IC L-WNK-1 in adaptation to a high-K+ diet.
METHODS
Animals
All animal breeding, housing, and protocols were approved by the Institutional Animal Care and Use Committees of the University of Pittsburgh, Icahn School of Medicine at Mount Sinai, and New York Medical College. Mouse facilities were accredited by the American Association for the Accreditation of Laboratory Animal Care. Animals were euthanized in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Generation of IC-Specific L-WNK1-KO Mice
Mice with loxP sites flanking exon 2 of WNK1 (L-WNK1fl/fl) in a C57Bl/6 background (Fig. 1) (28) and mice expressing Cre recombinase under control of the promoter for Atp6v1b1, encoding the vacuolar ATPase B1 subunit (B1:Cre mice, C57Bl/6 background) (29) have been previously characterized. Deletion of exon 2 leaves KS-WNK1 intact while ablating L-WNK1 expression in cells expressing Cre recombinase.
Figure 1.

Generation of knockout mice with intercalated cell-selective deletion of the long form of with no lysine kinase 1 (L-WNK1) (IC-L-WNK1-KO mice). A: primers used for genotyping and confirmation of L-WNK1 exon 2 excision (not to scale). Sequences are as previously reported (27). B: primers cF2 and cR1 amplify the wild-type (WT; nonfloxed) WNK1 locus into a ∼350-bp PCR product. cF2 and cR2 generate a 180-bp product from the floxed allele. C: the cF1/cR1 primer pair amplified the excised allele in the kidney but not in the heart or tail. PCR was optimized for amplification of short sequences; thus, no larger band was seen (e.g., from nonrecombinant tissues). PCR, polymerase chain reaction.
To generate IC-L-WNK1-KO mice, male homozygous floxed L-WNK1 mice were bred with female B1:Cre-expressing mice. Genotyping was performed before weaning by PCR amplification, using the primer scheme shown in Fig. 1 [primer details in Xie et al. (28)]. Female offspring hemizygous for B1:Cre and heterozygous for floxed L-WNK1 were bred with male homozygous floxed L-WNK1 mice to obtain mice homozygous for floxed L-WNK1 and hemizygous for B1:Cre (IC-L-WNK1-KO). Sex-matched littermates that were homozygous for floxed L-WNK1, but without Cre, were used as controls, and mice of both sexes were used for all experiments. After weaning, IC-L-WNK1-KO mice failed to thrive on solid food but achieved normal weights on a water-softened diet (see results). The “standard” diet consisted of water-softened chow (Prolab 5P76 Isopro RMH 3000, 0.94% K+ supplemented with ∼15% DietGel from ClearH2O). The gel “high-K+” diet (KCl) was prepared using 3 g of agar dissolved in 250 mL of boiling H2O. The solution was cooled, 125 g of high-K+ diet powder (5.2% K+ D.09075, Envigo) was incorporated, and the slurry was aliquoted into dishes to cool and harden before being placed in cages.
Quantitative RT-PCR
Kidney quarters, including the cortex and medulla, from animals on a high-K+ diet were flash frozen in liquid nitrogen at the time of euthanization and stored at −80°C until use. After being thawed on ice, samples were homogenized in Invitrogen TRIzol Reagent. Complementary DNA (cDNA) was generated using a Bio-Rad iScript cDNA synthesis kit. Quantitative PCR was performed using SYBR Green Supermix (Bio-RAD) on an Applied Biosystems 7900HT Fast Real-Time PCR System (Life Technologies). L-WNK1, KS-WNK1, and WNK4 cDNA primers were used as previously described (30). Other primers included the following: GAPDH, forward 5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse 5′-TGTAGACCATGTAGTTGAGGTCA-3′; 18 s, forward 5′-TTGATTAAGTCCCTGCCCTTT-3′ and reverse 5′-CAAGTTCGACCGTCTTCTCAG-3′; and β-actin, forward 5′-CAGCTGAGAGGGAAATCGTG-3′ and reverse 5′- CGTTGCCAATAGTGATGACC-3′.
Blood and Urine Metabolite Measurements
In mice that received a standard diet, whole blood was collected from a cardiac ventricle while the animal was anesthetized with 1−3% isoflurane. Electrolytes were measured using an i-STAT handheld analyzer (Abbot Point-of-Care). In mice that received a high-K+ diet for at least 10 days, urinary K+ excretion and urinary Na+ excretion were determined by placing animals in a metabolic cage for ∼1 h. Mice were encouraged to void and then given 10% (vol/wt) sterile 0.9% saline via intraperitoneal injection. Mice were replaced in the metabolic cage, and urine was collected over 6 h. Urine Na+ and K+ concentrations were measured via flame photometry (IL 943, Instrumentation Laboratory, Bedford, MA). Mice continued to receive a high-K+ diet. The next day, animals were euthanized, blood was collected, and electrolytes were measured as aforementioned. Plasma aldosterone levels were measured using an aldosterone enzyme-linked immunosorbent assay (ELISA) kit (Enzo Life Sciences, Farmingdale, NY), which we have previously confirmed to yield expected results with changes in dietary Na+ (31).
Immunofluorescence Microscopy
Mouse kidneys were prepared for immunostaining by sectioning and placement in 4% paraformaldehyde in phosphate-buffered saline (PBS) overnight at 4°C and then transferred to 30% sucrose in PBS with 0.02% azide.
Mice hemizygous for B1:Cre were crossed with Rosa26tdTomato Cre reporter mice to confirm cell-specific Cre expression (9). To examine tdTomato staining (to confirm cell-specific Cre expression) and to quantify WNK1 staining intensity in IC-L-WNK1-KO and control kidneys, 5 µm cryosections were prepared as previously described (32). Immunofluorescence labeling was performed with goat anti-aquaporin-2 (Aqp2) antibody (0.2 μg/mL, sc-9882, Santa Cruz Biotechnology) and a rabbit antibody raised against amino acids 2,031−2,117 of the human L-WNK1 COOH-terminus (3 µg/mL, HPA059157, Atlas) (33), followed by secondary donkey anti-rabbit Ig antibody coupled to Cy3 (0.5 µg/mL, Jackson ImmunoResearch Laboratories, West Grove, PA). TO-PRO-3 was obtained from Molecular Probes (Thermo Fisher Scientific). Immunolabeled tissues were mounted with Vectashield mounting medium (H-1200, Vector Labs) and imaged with a confocal laser scanning microscope (Leica TCS SP5, Model upright DM 6000S, Leica Microsystems, Buffalo Grove, IL) using a ×40 objective (WNK1-stained IC-L-WNK1-KO and control kidneys) or a ×63 objective (Rosa26tdTomato kidneys) with identical laser settings for control and IC-L-WNK1-KO samples. The Atp6v1b1 promoter selectively drives expression of Cre in Aqp2-negative tubular cells of Rosa26tdTomato kidneys (Fig. 2). Occasional staining in Aqp2-positive tubular cells was seen, as previously reported (29).
Figure 2.
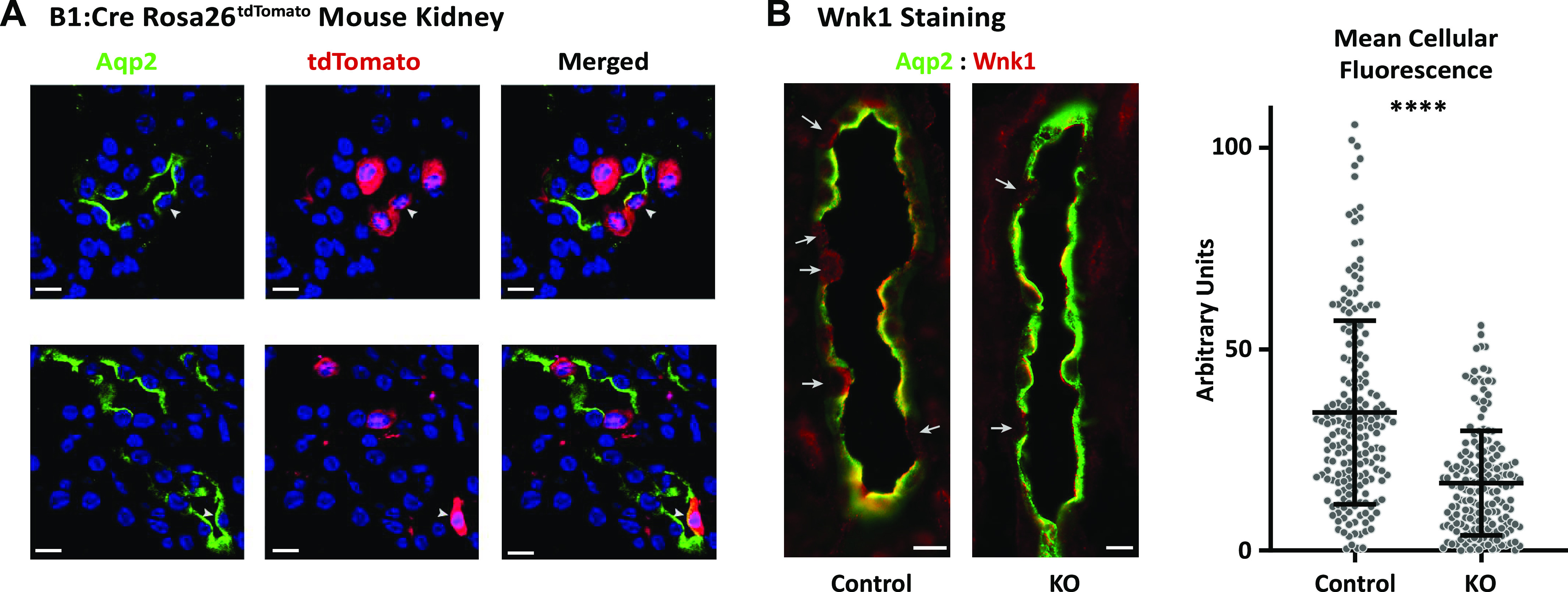
The vacuolar ATPase B1 subunit promoter drives Cre expression and reduces long form of with no lysine kinase 1 (L-WNK1) expression predominantly in aquaporin-2 (Aqp2)-negative cells. A: Cre recombinase is preferentially expressed in intercalated cells (ICs) in B1:Cre mice. Kidney sections are shown from two B1:Cre, Rosa26tdTomato reporter mice, which express tdTomato primarily in ICs expressing the V-ATPase B1 subunit. Green indicates Aqp2 staining. tdTomato is shown in red. Nuclei are highlighted with TO-PRO-3 staining. Most Aqp2-negative cells expressed tdTomato. Occasional Aqp2-expressing cells also expressed tdTomato (arrowheads). Images are representative of two mice. B: WNK1 expression was reduced in Aqp2-negative cells. Quantitative immunofluorescence was performed in kidney sections from knockout (KO) mice with IC-selective deletion of the L-WNK1 (IC-L-WNK1 KO) and control mice maintained on a high-K+ diet for 10 days using an antibody directed against the COOH-terminus of WNK1, which recognizes both L-WNK1 and a truncated, kidney-specific isoform of WNK1 (KS-WNK1) (5). Immunodetectable WNK1 expression was reduced in Aqp2-negative cells in collecting ducts from IC-L-WNK1 KO mice compared with controls. In many cells, the signal was not absent, possibly reflecting expression of KS-WNK1 in ICs. Quantitative comparison of the mean cellular fluorescence intensity in Aqp2-negative cells from IC-L-WNK1 KO mice (n = 196 cells, N = 5 mice) and controls (n = 183 cells, N = 4 mice) revealed that IC-specific KO mice exhibited a significant reduction in WNK1 signal (****P < 0.0001 by an unpaired t test). Scale bars = 14 µm.
Quantification of the WNK1 signal in CD cells was performed using the Fiji processing package for ImageJ. Regions of interest (ROIs) were drawn around AQP2-negative cells. The WNK1 channel was isolated, the background immunofluorescence signal was subtracted, and ROIs were overlayed on the WNK1 channel. Mean fluorescence intensities within ROIs were measured. Control mice consisted of one L-WNK1fl/fl littermate and three C57Bl/6 mice; no significant differences in the WNK1 immunofluorescence signal in K+ loaded L-WNK1fl/fl controls and C57Bl/6 controls were noted. Sections were analyzed from a total of four control mice and five IC-L-WNK1-KO mice.
To examine BK α-subunit localization within the cell, 20 µm sections were cut and pretreated with 10 mM sodium citrate buffer (10 mM, 90°C, 30 min) for antigen retrieval, rinsed with 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4 (pH 7.4) (PBS), permeabilized with 0.3% Triton X-100 in PBS (10 min), and blocked with a solution containing 1% BSA, 10% normal donkey serum, and 0.1% Triton X-100 in PBS (room temperature for 1 h). Sections were then incubated sequentially with chicken IgG anti-BK α-subunit antibody directed against a COOH-terminal epitope (6) (3.5 μg/mL) overnight at 4°C and A488 donkey IgG anti-chicken IgY (3 μg/mL, no. 703-545-155, Jackson ImmunoResearch) for 1 h at room temperature.
To identify PCs and ICs, slices were stained with rat anti-Aqp2 antibody (2.5 µg/mL, no. 20102rs, BiCell Scientific) followed by CF633 donkey IgG anti-rat IgG secondary antibody (10 µg/mL, SAB4600133, Sigma-Aldrich), rabbit anti-V-ATPase B1 subunit antibody (2.0 μg/mL, sc-20943, Santa Cruz Biotechnology) followed by A546 donkey IgG anti-rabbit IgG (4 µg/mL, A10040, Thermo Fisher), or rabbit anti-pendrin antibody (2.5 μg/mL, no. 2051, BiCell Scientific) followed by A546 donkey IgG anti-rabbit IgG (4 µg/mL, A10040, Thermo Fisher Scientific). Immunolabeled sections were mounted with Vectashield mounting medium (H-1200, Vector Labs). Digital images (pinhole: 90.0 μM, step size: 1.0 μM) were collected using a ×63 oil immersion plan-Apochromat objective (numerical aperture: 1.4). Fluorescence intensity within apical + subapical and cytoplasmic ROIs was measured using Leica Application Suite software (LAS X v. 3.7.1.21655, Leica Microsystems) in Aqp2-expressing cells (PCs), cells with apical V-ATPase B1 subunit expression (type A ICs), and cells with apical pendrin expression (type B ICs). Data are reported as the apical + subapical/cytoplasmic intensity quotient for 40−65 individual cells from each of four control mice and five IC-WNK1-KO mice.
Patch-Clamp Electrophysiology
BK channel activity was assessed via perforated whole cell recording of ICs in microdissected, split-open CCDs from IC-L-WNK1-KO and littermate control mice on a high-K+ diet for 10 days, as previously described (9). The bath solution contained (in mM) 130 K+ gluconate, 10 KCl, 0.5 MgCl2, 1.5 CaCl2, and 10 HEPES (pH 7.4), whereas the pipette solution contained (in mM) 130 K+ gluconate, 10 KCl, 1 MgCl2, 8.72 CaCl2, 10 EGTA (1 μM free Ca2+), and 5 HEPES (pH 7.4). After a high-resistance seal (>2 GΩ) had been formed, membrane capacitance was monitored until the whole cell patch configuration was formed. IC membrane capacitance ranged between 12.5 and 13.5 pF; currents were normalized to a membrane capacitance of 13 pF per cell. Cells were clamped at +60 mV and outward BK currents were identified by adding 100 nM ChTx to the bath solution. Currents, recorded using an Axon 200 A patch-clamp amplifier, were low-pass filtered at 1 KHz and digitized by an Axon interface (Digidata 1440 A, Molecular Devices) (21, 34). Data were analyzed using pCLAMP Software System 9 (Molecular Devices).
Data Analysis
Data were analyzed in Graphpad Prism (v. 8.0.2). Removal of outliers was performed using the ROUT method with a coefficient of 1%. The reported errors and error bars shown all represent SDs. Experimental and control data sets were analyzed via a two-tailed Student’s t test. A P value of <0.05 was considered statistically significant. In Figs. 2 and 3, statistical analyses were based on the number of cells analyzed.
Figure 3.
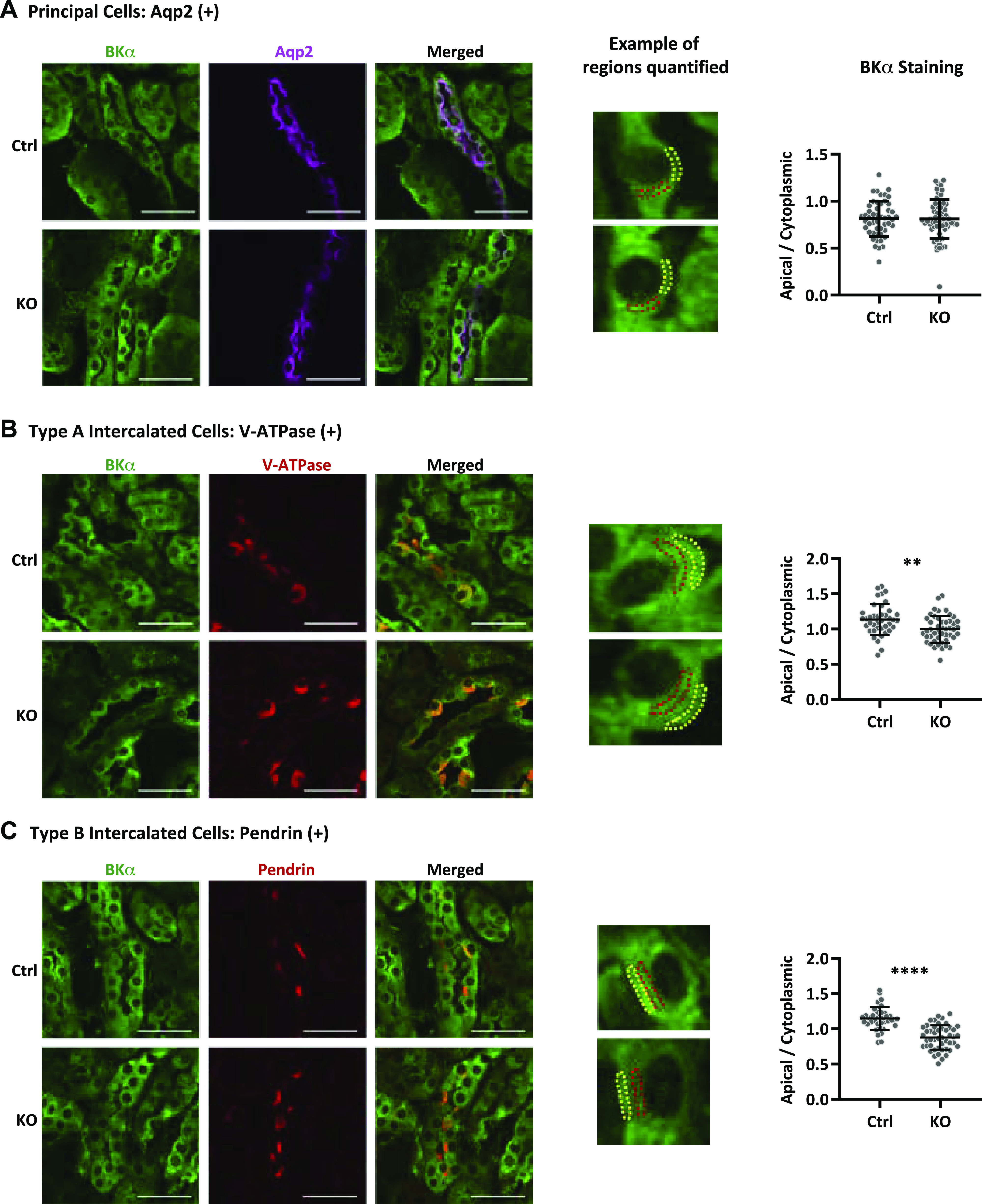
Apical + subapical expression of the large-conductance (BK) K+ channel α-subunit is reduced in intercalated cells (ICs) from knockout (KO) mice with IC-selective deletion of the long form of with no lysine kinase 1 (IC-L-WNK1-KO mice). A: aquaporin-2 (Aqp2)-positive cells (magenta) exhibited similar ratios of apical + subapical to cytoplasmic BK α-subunit staining (green) in control (Ctrl; 0.814 ± 0.188, n = 50 cells) and IC-L-WNK1-KO kidneys (0.810 ± 0.209, n = 65 cells). B: type A ICs, identified by apical V-ATPase staining (red), exhibited reduced BK α-subunit apical + subapical/cytoplasmic staining in IC-L-WNK1-KO mice (0.997 ± 0.191, n = 47 cells) compared with Ctrl mice (1.136 ± 0.219, n = 40 cells, **P < 0.01). C: type B ICs, identified by apical pendrin staining (red), also exhibited reduced BK α-subunit apical + subapicacytoplasmic staining in IC-L-WNK1-KO kidneys (0.878 ± 0.173, n = 47 cells) compared with Ctrl kidneys (1.150 ± 0.160, n = 38 cells, ****P < 0.0001). Representative apical regions are shown in yellow; cytoplasmic regions are shown in red. Scale bars = 40 µm. Sections were analyzed from a total of four Ctrl mice and five IC-L-WNK1-KO mice. Data were analyzed using a Student’s unpaired t test.
RESULTS
IC-L-WNK1-KO Mice
To clearly define the role of L-WNK1 in enhancing IC-dependent BK expression and facilitating the adaptation to a high-K+ diet, we generated mice in which L-WNK1 was selectively deleted from ICs (IC-L-WNK1-KO mice) and littermate controls. Genotyping was performed as previously described (28), and recombination between L-WNK1 loxP sites flanking exon 2 was confirmed by PCR (Fig. 1). A recombinant band was observed in the kidney but not in the tail or heart of IC-L-WNK1-KO mice. IC-L-WNK1-KO mice exhibited no gross morphological abnormalities but did experience feeding difficulty. When given water-softened chow (as described in methods), they developed normally and achieved adult weights similar to control mice (Table 1).
Table 1.
Standard K+ diet
| L-WNK1fl/fl Control | IC-L-WNK1-KO | P Value | |
|---|---|---|---|
| Blood | |||
| K+, mmol/L | 4.3 ± 0.6 (9) | 4.4 ± 0.7 (23) | NS |
| Na+, mmol/L | 146 ± 1.3 (9) | 147 ± 1.2 (23) | NS |
| Cl−, mmol/L | 114 ± 3 (9) | 114 ± 2 (23) | NS |
| Total CO2, mmol/L | 21.8 ± 2.0 (9) | 22.5 ± 2.5 (23) | NS |
| Ionized Ca2+, mmol/L | 1.3 ± 0.1 (9) | 1.3 ± 0.1 (23) | NS |
| Urea nitrogen, mg/dL | 19 ± 4 (9) | 21 ± 3 (23) | NS |
| Hemoglobin, mg/dL | 13 ± 0.6 (9) | 13 ± 0.5 (23) | NS |
| Plasma | |||
| Aldosterone, pg/mL | 442 ± 77 (7) | 598 ± 294 (22) | NS |
| Mouse weights | |||
| Female mice, g | 20.8 ± 1.0 (4) | 22.0 ± 3.4 (14) | NS |
| Male mice, g | 25.9 ± 1.0 (5) | 24.2 ± 1.6 (8) | NS |
Numbers in parentheses are numbers of mice. Measurements were obtained in knockout mice with intercalated cell-selective deletion of the long form of with no lysine kinase 1 (IC-L-WNK1-KO mice) and littermate control mice upon euthanization at 10–14 wk of age, as described in methods. Differences were assessed using a two-tailed Student’s t test. NS, not significant.
Expression of Cre recombinase in Aqp2-negative tubular epithelial cells was confirmed by crossing B1:Cre mice with tdTomato-expressing Cre reporter mice (35). Tubules from these mice demonstrated robust expression of tdTomato in Aqp2-negative cells within Aqp2-expressing tubules (Fig. 2A). A minor fraction of Aqp2-expressing cells also expressed tdTomato, consistent with previous work suggesting that B1 promoter-driven Cre expression was occasionally seen in PCs (29).
Quantitative RT-PCR was performed on whole kidneys from IC-L-WNK1-KO and control mice on a high-K+ diet. L-WNK1 message, normalized to GAPDH, was similar in IC-L-WNK1-KO and control animals (0.03 ± 0.01, N = 11, vs. 0.02 ± 0.01, N = 11, P = not significant; not shown). Findings were similar when L-WNK1 transcript levels were normalized to the ribosomal 18 s subunit or to mRNA encoding β-actin (not shown). The inability to detect reduced L-WNK1 expression in kidneys from IC-L-WNK1-KO mice is not surprising, given that ICs represent a small minority of L-WNK1-expressing kidney cells (36, 37).
To evaluate differences in WNK1 protein expression, we performed quantitative immunofluorescence experiments in kidney sections from IC-L-WNK1-KO and control mice maintained on a high-K+ diet for 10 days. Available antibodies directed specifically against L-WNK1 are nonspecific in mouse kidney tissue. Therefore, we used a previously validated antibody directed against the COOH-terminus of WNK1, recognizing L-WNK1 and KS-WNK1 (33). Immunodetectable total WNK1 expression was reduced in Aqp2-negative cells in CDs from IC-L-WNK1-KO mice compared with controls. However, in many cells the signal was not completely absent, possibly reflecting expression of KS-WNK1 in ICs (Fig. 2B, two left images). Quantitative comparison of the mean cellular fluorescence intensity in Aqp2-negative cells in connecting tubules/CDs from IC-L-WNK1-KO mice (n = 196 cells, N = 5 mice) and control mice (n = 183 cells, N = 4 mice) revealed that IC-L-WNK1-KO mice exhibited a significant reduction in WNK1 signal (P < 0.0001 by an unpaired t test; Fig. 2B, right).
Because immunofluorescence data could not distinguish L-WNK1 from KS-WNK1 expression, we performed quantitative RT-PCR to determine whether deletion of L-WNK1 altered KS-WNK1 expression. KS-WNK1 is expressed in a relatively small subset of kidney cells (36, 37); therefore, changes in KS-WNK1 transcription should be reflected in changes in whole kidney KS-WNK1 message. Using previously validated KS-WNK1 primers (30), we found that transcript levels for KS-WNK1/GAPDH were similar in control and IC-L-WNK1-KO animals (0.21 ± 0.1, N = 11, vs. 0.20 ± 0.1, N = 11, P = not significant; not shown). Results were similar when KS-WNK1 was normalized against β-actin or the 18 s ribosomal subunit. Thus, we were unable to find any evidence that KS-WNK1 expression changed in IC-L-WNK1-KO mice.
On a standard (0.94% K+) diet, blood electrolyte levels, including K+ and total CO2, were similar in control and IC-L-WNK1-KO mice (Table 1). To evaluate whether mice were compensating for impaired K+ excretion by increasing aldosterone levels, we measured plasma aldosterone. IC-L-WNK1-KO mice exhibited a higher point estimate for plasma aldosterone concentration, but the increase was not statistically significant (Table 1).
IC BK Channel Apical + Subapical Localization and Activity Are Reduced in IC-L-WNK1-KO Mice
We asked whether apical + subapical localization of the BK α-subunit was altered in IC-L-WNK1-KO mice on a high-K+ diet. As expected, in PCs, the BK α-subunit apical+ subapical/cytoplasmic staining ratio did not differ in kidneys from control versus IC-L-WNK1-KO mice (0.814 ± 0.188, n = 50, vs. 0.810 ± 0.209, n = 65, P = not significant; Fig. 3A). In contrast, the BK α-subunit apical + subapical/cytoplasmic staining ratio was significantly reduced in type A ICs from IC-L-WNK1-KO mice (0.997 ± 0.191, n = 47) compared with controls (1.136 ± 0.219, n = 40, P < 0.01; Fig. 3B). The BK α-subunit apical + subapical/cytoplasmic staining quotient was also reduced in IC-L-WNK1-KO type B ICs (0.878 ± 0.173, n = 47) compared with controls (1.150 ± 0.160, n = 38, P < 0.0001; Fig. 3C). These data confirm that L-WNK1 influences BK channel expression.
We next evaluated whether the reduction in WNK1 expression and BK α-subunit apical + subapical localization was associated with reduced BK channel activity in IC-L-WNK1-KO mice. Perforated whole cell patch-clamp recordings of K+ currents were performed on ICs clamped at +60 mV in isolated tubules from IC-L-WNK1-KO and littermate control mice on a high-K+ diet for 10 or more days. Currents were normalized to a membrane capacitance of 13 pF per cell. ChTx-sensitive currents, reflecting BK channel-mediated currents (9), were significantly reduced in ICs from IC-L-WNK1-KO mice (452 ± 73 pA, n = 8) compared with littermate controls (691 ± 91 pA, n = 8, P < 0.001 by a two-tailed Student’s t test; Fig. 4).
Figure 4.

Charybdotoxin-sensitive currents (indicative of large-conductance K+ channel activity) were reduced in intercalated cells (ICs) from knockout (KO) mice with IC-selective deletion of the long form of with-no-lysine kinase 1 (IC-L-WNK1-KO mice) on a high-K+ diet. A: perforated whole cell current tracings were collected from ICs in microdissected tubules from IC-L-WNK1-KO or littermate control (Ctrl) mice on a high-K+ diet. B: means ± SD are shown (n = 7). ****P < 0.0001, IC-L-WNK1-KO vs. littermate Ctrl mice by an unpaired Student’s t test.
Previous reports have shown that, in cell culture, another WNK family member, WNK4, decreases cell surface localization and activity of the BK channel (20–22). To evaluate whether WNK4 expression is altered in kidneys from IC-L-WNK1-KO mice, potentially contributing to the decrease in BK channel activity observed, we assessed relative WNK4 transcript levels by quantitative RT-PCR. WNK4/GAPDH transcript levels were similar in IC-L-WNK1-KO and control animals (0.05 ± 0.03, N = 11, vs. 0.05 ± 0.03, N = 11, P = not significant; not shown). The results were similar when WNK4 message was normalized to either the 18 s ribosomal subunit or β-actin.
IC-L-WNK1-KO Mice Exhibited an Elevated Blood K+ Concentration on a High-K+ Diet
In rabbits, increased dietary K+ enhances apical membrane expression of the BK channel (6). Given the reduced cell surface expression and activity of the BK channel in IC-L-WNK1-KO mice, we examined whether these mice exhibited higher blood K+ levels on a high-K+ diet. Mice at the age of 10−13 wk were given a high-K+ (5.2% K+) diet for at least 10 days. Blood K+ was significantly higher in IC-L-WNK1-KO mice (5.4 ± 1.0 mM, n = 26) compared with littermate controls (4.8 ± 0.6 mM, n = 19, P < 0.05; Table 2). Similar levels of blood Na+, Cl−, total CO2, Ca2+, urea nitrogen, and hemoglobin were observed in IC-L-WNK1-KO mice and littermate controls on a high-K+ diet. Aldosterone levels were also not significantly different on a high-K+ diet (Table 2). When analyzed by sex, significant differences in blood metabolites were not observed (not shown).
Table 2.
High-K+ diet
| L-WNK1fl/fl Control | IC-L-WNK1-KO | P Value | |
|---|---|---|---|
| Blood | |||
| K+, mmol/L | 4.8 ± 0.6 (19) | 5.4 ± 1.0 (26) | 0.035 |
| Na+, mmol/L | 146 ± 2 (19) | 146 ± 2 (26) | NS |
| Cl−, mmol/L | 115 ± 4 (19) | 116 ± 4 (26) | NS |
| Total CO2, mmol/L | 21.2 ± 2.1 (19) | 20.3 ± 2.6 (25) | NS |
| Ionized Ca2+, mmol/L | 1.26 ± 0.12 (19) | 1.26 ± 0.11 (26) | NS |
| Urea nitrogen, mg/dL | 20 ± 4 (19) | 22 ± 4 (26) | NS |
| Hemoglobin, mg/dL | 12.7 ± 1.0 (19) | 12.4 ± 1.1 (26) | NS |
| Plasma | |||
| Aldosterone, pg/mL | 1969 ± 2165 (16) | 1779 ± 1978 (16) | NS |
| 6-h Urine | |||
| Total volume, mL | 1.68 ± 0.58 (15) | 1.83 ± 0.55 (19) | NS |
| Urinary Na+ excretion, µmol/h | 29.6 ± 8.2 (15) | 32.2 ± 9.7 (19) | NS |
| Urinary K+ excretion, µmol/h | 26.5 ± 11.6 (15) | 28.1 ± 9.2 (19) | NS |
Numbers in parentheses are numbers of mice. Blood and urine measurements were obtained in knockout mice with intercalated cell-selective deletion of the long form of with no lysine kinase 1 (IC-L-WNK1-KO mice) and littermate control mice after 10 days on a high-K+ diet, as described in methods. Differences were assessed using a two-tailed Student’s t test. NS, not significant.
Blood total CO2 levels were not significantly different between control and IC-L-WNK1-KO mice on either the standard or high-K+ diet. However, in post hoc analysis comparing data from animals on the standard versus high-K+ diet, IC-L-WNK1-KO mice exhibited significantly lower total CO2 on the high-K+ diet (20.3 ± 2.6, N = 25) than on the standard K+ diet (22.5 ± 2.5, N = 23, P < 0.01 via a Student’s t test). In contrast, control mice exhibited similar blood total CO2 on a high-K+ diet (21.2 ± 2.1 meq/L, N = 19) versus the standard K+ diet (21.8 ± 2.0 meq/L, N = 9, P = 0.47 via a Student’s t test). These data suggest that IC-L-WNK1-KO mice may be predisposed to metabolic acidosis.
Urinary Na+ and K+ Excretion
As BK channels in ICs mediate FIKS, we examined whether high K+-fed IC-L-WNK1-KO mice exhibited diminished urinary K+ excretion in the context of an intraperitoneally administered bolus of normal saline (10% of body weight) compared with littermate controls. Urine was collected over a 6-h period, and urinary K+ excretion and urinary Na+ excretion were determined. IC-L-WNK1-KO and littermate control mice exhibited similar urinary K+ excretion (controls: 26.5 ± 11.6 µmol/h, n = 15, and IC-L-WNK1-KO: 28.1 ± 9.2 µmol/h, n = 19, P = not significant) and similar urinary Na+ excretion (controls: 29.6 ± 8.2 µmol/h, n = 15, and IC-L-WNK1-KO: 32.2 ± 9.7 µmol/h, n = 19, P = not significant; Table 2).
DISCUSSION
The WNK family of protein kinases has a key role in coordinating the regulated absorption of Na+ and secretion of K+ in the ASDN (38, 39). In the distal convoluted tubule, both L-WNK1 and WNK4 activate a signaling cascade involving STE20-related proline-alanine-rich kinase (SPAK) and oxidative stress responsive kinase-1 (OSR1) to eventually activate the thiazide-sensitive NaCl cotransporter (NCC), enhancing Na+ and Cl− absorption (38, 39). In more distal aspects of the nephron, WNK4 has been shown to inhibit ENaC (40–42), whereas KS-WNK1 activates ENaC (43, 44). Interestingly, both WNK4 and L-WNK1 activate serum/glucocorticoid-regulated kinase 1 (SGK1) (45), a well-characterized activator of ENaC (46). Furthermore, SPAK has been shown to activate ENaC (47). Most of this work has been performed in heterologous expression systems. Studies of mouse models where specific WNKs have been genetically deleted in PCs are needed to clarify how specific WNKs modulate ENaC in vivo.
WNKs have differing roles in regulating K+ secretory channels. ROMK is inhibited by L-WNK1 and WNK4 (25, 26, 41, 48–50). In heterologous expression systems, WNK4 inhibits BK channels (20–22). In contrast, both L-WNK1 and KS-WNK1 increase surface and functional expression of BK channels, and L-WNK1 kinase activity is not required for BK channel activation (23, 24). Previous work has suggested that L-WNK1 activates BK channels by inhibiting ERK1/2-dependent channel degradation (23). Thus, it appears that WNKs are intimately associated with K+ handling, but with systemic effects that are not easily predictable.
The role of IC L-WNK1 in contributing to K+ secretion through enhancement of BK channel activity appears at odds with a role for L-WNK1 in reducing ASDN K+ secretion by stimulating Na+ reabsorption in the distal convoluted tubule (DCT), as enhanced DCT Na+ reabsorption could reduce Na+ delivery to the collecting tubule/collecting duct for ENaC-dependent Na+ absorption, which facilitates K+ excretion (50). However, increased NCC activity in specific mouse models does not cause hyperkalemia, complicating our understanding of how L-WNK1 activity in the DCT influences tubular K+ handling (61). Regardless, it is not unreasonable to expect that effects of L-WNK1 activity in ICs could differ from its effects in other tubular segments.
BK channels in the ASDN are responsible for FIKS (6–10). Although BK channels are expressed in both PCs and ICs, our recent work suggests that BK channels in ICs mediate FIKS (9). Our current work suggests that L-WNK1 has a role in activating BK channels in ICs in mice fed a high-KCl diet. Specifically, deletion of L-WNK1 in ICs shifted BK channel α-subunit localization from the apical region toward the cytoplasmic region, suggesting reduced cell surface expression. Consistent with this interpretation, in mice fed a high-K+ diet, whole IC BK currents, as measured by the ChTx-sensitive component of K+ current, were significantly reduced. Moreover, blood K+ concentration was higher in IC-L-WNK1-KO mice compared with controls. We did not observe a sex difference regarding blood K+ concentration, as we observed with IC BK α-subunit KO mice, where male mice selectively exhibited increased blood K+ concentrations on a high-K+ diet (9). Interestingly, the magnitude of kaliuresis that we observed in response to volume expansion was similar in IC-L-WNK1-KO and control mice, suggesting compensatory mechanisms in IC-L-WNK1-KO mice were present to maintain K+ homeostasis. These mechanisms remain to be determined.
We have previously found that L-WNK1 expression is selectively enhanced in ICs in response to a high-K+ diet (24). How increased L-WNK1 promotes BK channel activity in ICs remains unclear. A number of cellular factors could influence L-WNK1 expression and activity in response to an increase in dietary K+. For example, L-WNK1 variants express PY motifs that are binding sites for the ubiquitin ligase neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2), targeting L-WNK1 for ubiquitination and degradation (30). Aldosterone-dependent activation of SGK1 in ICs could prevent Nedd4-2-dependent L-WNK1 degradation. WNKs are inhibited by increases in intracellular Cl− concentration (51, 52), although it is not known whether a high-K+ diet is associated with a change in intracellular Cl− concentration in ICs that could affect L-WNK1 activity. It is interesting that aldosterone binding and subsequent signaling in ICs are dependent on dephosphorylation of Ser483 on the mineralocorticoid receptor, and hyperkalemia enhances Ser483 phosphorylation via unc-51-like kinase 1 (53, 54). Previous work suggests that WNK1 reduces expression of unc-51-like kinase 1 (55), raising the possibility of a positive feedback loop. IC mineralocorticoid receptor KO mice exhibit hyperkalemia (56), proposed to be due to reduced ENaC activity, in turn reducing the driving force for K+ secretion. It could also reflect dampened BK channel-mediated K+ excretion, as administration of the mineralocorticoid receptor antagonist spironolactone to rats reduced BK α-subunit abundance (57).
IC apical BK channel expression and currents were moderately reduced in IC-L-WNK1-KO mice compared with the virtual absence of BK channel-mediated currents in ICs from mice with a genetic deletion of BK α-subunits in ICs (9). These observations are consistent with studies that have identified cellular mechanisms and pathways, in addition to L-WNK1, by which a high-K+ diet activates BK channel expression and activity. For example, a high-K+ diet stimulates the expression of cytochrome P-450 2C23 and increases 11,12-epoxyeicosatrienoic acid concentrations in the CCD, which activate BK channels and FIKS (58). A high-K+ diet increases expression and activity of heme oxygenase-1, and carbon monoxide generated by heme oxygenase-1 activates BK channels in the CCD by nitric oxide synthase- and protein kinase G-independent mechanisms (59).
The importance of L-WNK1 expression in ICs suggests that L-WNK1 could also influence acid-base handling. For example, a high-KCl diet represents a systemic acid load (27). Acidosis represents a stimulus for increased activity of H+-K+-ATPase in type A ICs (60). However, increased H+-K+-ATPase activity would exacerbate hyperkalemia if not for an apical K+ channel to allow K+ recycling. BK channel activity may contribute to K+ efflux, indirectly facilitating H+-K+-ATPase activity. Consistent with this model, we observed that IC-L-WNK1-KO mice on a high-K+ diet have reduced blood total CO2 compared with IC-L-WNK1-KO mice on a standard diet. Control mice exhibited no difference in total CO2 on the two diets. Further experiments will be required to confirm this observation and to determine whether it occurs solely as a consequence of reduced BK channel-dependent K+ recycling or because L-WNK1 participates in other aspects of IC physiology and acid-base transport.
In summary, our results demonstrate that L-WNK1 has a role in BK channel activation in ICs in response to a high-K+ diet. Furthermore, the increased blood K+ concentration in IC-L-WNK1-KO mice suggests that this signaling pathway has an important role in the adaptation to a high-K+ diet.
GRANTS
This work was supported by grants from the National Institutes of Health, including DK038470 (to T.R.K. and L.M.S.), DK110332 (to E.C.R.), DK109038 (to M.A.B.), DK054983 (to W.H.W.), DK111542 (to C.L.H.), DK098145 and DK119252 (to A.R.S.), and P30DK079307 (to T.R.K., A.R.S., and L.M.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.C.R., T.R.K., and L.M.S. conceived and designed research; E.C.R., R.C.-G, M.A.-B., A.L.M., L.J.N., J.C., A.W., S.G., T.R.L., D.F., and P.W. performed experiments; E.C.R., R.C.-G., M.A.-B., A.L.M., W.H.W., A.R.S., T.R.K., and L.M.S. analyzed data; E.C.R., R.C-G, M.A.-B., W.H.W., A.R.S., T.R.K., and L.M.S. interpreted results of experiments; E.C.R., R.C.-G., M.A.-B., and WH.W. prepared figures; E.C.R., R.C.-G., T.R.K., and L.M.S. drafted manuscript; E.C.R., R.C.-G, M.A.-B., A.L.M., WH.W., A.R.S., T.R.K., and L.M.S. edited and revised manuscript; E.C.R., R.C.-G, M.A-B., A.L.M., L.J.N., J.C., A.W., S.G., T.R.L., D.F., P.W., W.H.W., C.-LH., A.R.S., T.R.K., and L.M.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Donald Kohan and Dr. Raoul Nelson at the University of Utah for B1:Cre mice.
REFERENCES
- 1.Giebisch G. Renal potassium transport: mechanisms and regulation. Am J Physiol Renal Physiol 274: F817–F833, 1998. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- 2.Engbretson BG, Stoner LC. Flow-dependent potassium secretion by rabbit cortical collecting tubule in vitro. Am J Physiol Renal Physiol 253: F896–F903, 1987. doi: 10.1152/ajprenal.1987.253.5.F896. [DOI] [PubMed] [Google Scholar]
- 3.Khuri RN, Strieder WN, Giebisch G. Effects of flow rate and potassium intake on distal tubular potassium transfer. Am J Physiol 228: 1249–1261, 1975. doi: 10.1152/ajplegacy.1975.228.4.1249. [DOI] [PubMed] [Google Scholar]
- 4.Liu W, Schreck C, Coleman RA, Wade JB, Hernandez Y, Zavilowitz B, Warth R, Kleyman TR, Satlin LM. Role of NKCC in BK channel-mediated net K+ secretion in the CCD. Am J Physiol Renal Physiol 301: F1088–F1097, 2011. doi: 10.1152/ajprenal.00347.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malnic G, Berliner RW, Giebisch G. Flow dependence of K+ secretion in cortical distal tubules of the rat. Am J Physiol Renal Physiol 256: F932–F941, 1989. doi: 10.1152/ajprenal.1989.256.5.F932. [DOI] [PubMed] [Google Scholar]
- 6.Najjar F, Zhou H, Morimoto T, Bruns JB, Li HS, Liu W, Kleyman TR, Satlin LM. Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am J Physiol Renal Physiol 289: F922–F932, 2005. doi: 10.1152/ajprenal.00057.2005. [DOI] [PubMed] [Google Scholar]
- 7.Woda CB, Bragin A, Kleyman TR, Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol 280: F786–F793, 2001. doi: 10.1152/ajprenal.2001.280.5.F786. [DOI] [PubMed] [Google Scholar]
- 8.Carrisoza-Gaytan R, Carattino MD, Kleyman TR, Satlin LM. An unexpected journey: conceptual evolution of mechanoregulated potassium transport in the distal nephron. Am J Physiol Cell Physiol 310: C243–C259, 2016. doi: 10.1152/ajpcell.00328.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrisoza-Gaytan R, Ray EC, Flores D, Marciszyn AL, Wu P, Liu L, Subramanya AR, Wang W, Sheng S, Nkashama LJ, Chen J, Jackson EK, Mutchler SM, Heja S, Kohan DE, Satlin LM, Kleyman TR. Intercalated cell BKalpha subunit is required for flow-induced K+ secretion. JCI Insight 5: e130553, 2020. doi: 10.1172/jci.insight.130553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoner LC, Viggiano SC. Environmental KCl causes an upregulation of apical membrane maxi K and ENaC channels in everted Ambystoma collecting tubule. J Membr Biol 162: 107–116, 1998. doi: 10.1007/s002329900348. [DOI] [PubMed] [Google Scholar]
- 11.Bailey MA, Cantone A, Yan Q, MacGregor GG, Leng Q, Amorim JB, Wang T, Hebert SC, Giebisch G, Malnic G. Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of type II Bartter's syndrome and in adaptation to a high-K diet. Kidney Int 70: 51–59, 2006. doi: 10.1038/sj.ki.5000388. [DOI] [PubMed] [Google Scholar]
- 12.Carrisoza-Gaytan R, Wang L, Schreck C, Kleyman TR, Wang WH, Satlin LM. The mechanosensitive BKα/β1 channel localizes to cilia of principal cells in rabbit cortical collecting duct (CCD). Am J Physiol Renal Physiol 312: F143–F156, 2017. doi: 10.1152/ajprenal.00256.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pluznick JL, Wei P, Carmines PK, Sansom SC. Renal fluid and electrolyte handling in BKCa-β1−/− mice. Am J Physiol Renal Physiol 284: F1274–F1279, 2003. doi: 10.1152/ajprenal.00010.2003. [DOI] [PubMed] [Google Scholar]
- 14.Rieg T, Vallon V, Sausbier M, Sausbier U, Kaissling B, Ruth P, Osswald H. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int 72: 566–573, 2007. doi: 10.1038/sj.ki.5002369. [DOI] [PubMed] [Google Scholar]
- 15.Liu W, Morimoto T, Woda C, Kleyman TR, Satlin LM. Ca2+ dependence of flow-stimulated K secretion in the mammalian cortical collecting duct. Am J Physiol Renal Physiol 293: F227–F235, 2007. doi: 10.1152/ajprenal.00057.2007. [DOI] [PubMed] [Google Scholar]
- 16.O'Neil RG, Boulpaep EL. Effect of amiloride on the apical cell membrane cation channels of a sodium-absorbing, potassium-secreting renal epithelium. J Membr Biol 50: 365–387, 1979. doi: 10.1007/BF01868898. [DOI] [PubMed] [Google Scholar]
- 17.Frindt G, Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol 297: F389–F396, 2009. doi: 10.1152/ajprenal.90528.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frindt G, Palmer LG. Apical potassium channels in the rat connecting tubule. Am J Physiol Renal Physiol 287: F1030–F1037, 2004. doi: 10.1152/ajprenal.00169.2004. [DOI] [PubMed] [Google Scholar]
- 19.Palmer LG, Frindt G. High-conductance K channels in intercalated cells of the rat distal nephron. Am J Physiol Renal Physiol 292: F966–F973, 2007. doi: 10.1152/ajprenal.00191.2006. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Subramanya AR, Satlin LM, Pastor-Soler NM, Carattino MD, Kleyman TR. Regulation of large-conductance Ca2+-activated K+ channels by WNK4 kinase. Am J Physiol Cell Physiol 305: C846–C853, 2013. doi: 10.1152/ajpcell.00133.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue P, Zhang C, Lin D-H, Sun P, Wang W-H. WNK4 inhibits Ca2+-activated big-conductance potassium channels (BK) via mitogen-activated protein kinase-dependent pathway. Biochim Biophys Acta 1833: 2101–2110, 2013. doi: 10.1016/j.bbamcr.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhuang J, Zhang X, Wang D, Li J, Zhou B, Shi Z, Gu D, Denson DD, Eaton DC, Cai H. WNK4 kinase inhibits maxi K channel activity by a kinase-dependent mechanism. Am J Physiol Renal Physiol 301: F410–F419, 2011. doi: 10.1152/ajprenal.00518.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Song X, Shi Y, Shi Z, Niu W, Feng X, Gu D, Bao HF, Ma HP, Eaton DC, Zhuang J, Cai H. WNK1 activates large-conductance Ca2+-activated K+ channels through modulation of ERK1/2 signaling. J Am Soc Nephrol 26: 844–854, 2015. doi: 10.1681/ASN.2014020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webb TN, Carrisoza-Gaytan R, Montalbetti N, Rued A, Roy A, Socovich AM, Subramanya AR, Satlin LM, Kleyman TR, Carattino MD. Cell-specific regulation of L-WNK1 by dietary K. Am J Physiol Renal Physiol 310: F15–F26, 2016. doi: 10.1152/ajprenal.00226.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang L, Garuti R, Kim BY, Wade JB, Welling PA. The ARH adaptor protein regulates endocytosis of the ROMK potassium secretory channel in mouse kidney. J Clin Invest 119: 3278–3289, 2009. doi: 10.1172/JCI37950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazrak A, Liu Z, Huang CL. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci USA 103: 1615–1620, 2006. doi: 10.1073/pnas.0510609103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyd-Shiwarski CR, Weaver CJ, Beacham RT, Shiwarski DJ, Connolly KA, Nkashama LJ, Mutchler SM, Griffiths SE, Knoell SA, Sebastiani RS, Ray EC, Marciszyn AL, Subramanya AR. Effects of extreme potassium stress on blood pressure and renal tubular sodium transport. Am J Physiol Renal Physiol 318: F1341–F1356, 2020. doi: 10.1152/ajprenal.00527.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie J, Wu T, Xu K, Huang IK, Cleaver O, Huang CL. Endothelial-specific expression of WNK1 kinase is essential for angiogenesis and heart development in mice. Am J Pathol 175: 1315–1327, 2009. doi: 10.2353/ajpath.2009.090094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller RL, Lucero OM, Riemondy KA, Baumgartner BK, Brown D, Breton S, Nelson RD. The V-ATPase B1-subunit promoter drives expression of Cre recombinase in intercalated cells of the kidney. Kidney Int 75: 435–439, 2009. doi: 10.1038/ki.2008.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy A, Al-Qusairi L, Donnelly BF, Ronzaud C, Marciszyn AL, Gong F, Chang YP, Butterworth MB, Pastor-Soler NM, Hallows KR, Staub O, Subramanya AR. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest 125: 3433–3448, 2015. doi: 10.1172/JCI75245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan XP, Wu P, Zhang DD, Gao ZX, Xiao Y, Ray EC, Wang WH, Lin DH. Deletion of Kir5.1 abolishes the effect of high-Na+ intake on Kir4.1 and Na+-Cl− cotransporter. Am J Physiol Renal Physiol 320: F1045–F1058, 2021. doi: 10.1152/ajprenal.00004.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouley R, Pastor-Soler N, Cohen O, McLaughlin M, Breton S, Brown D. Stimulation of AQP2 membrane insertion in renal epithelial cells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra). Am J Physiol Renal Physiol 288: F1103–F1112, 2005. doi: 10.1152/ajprenal.00337.2004. [DOI] [PubMed] [Google Scholar]
- 33.Boyd-Shiwarski CR, Shiwarski DJ, Roy A, Namboodiri HN, Nkashama LJ, Xie J, McClain KL, Marciszyn A, Kleyman TR, Tan RJ, Stolz DB, Puthenveedu MA, Huang CL, Subramanya AR. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol Biol Cell 29: 499–509, 2018. doi: 10.1091/mbc.E17-08-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu W, Wei Y, Sun P, Wang WH, Kleyman TR, Satlin LM. Mechanoregulation of BK channel activity in the mammalian cortical collecting duct: role of protein kinases A and C. Am J Physiol Renal Physiol 297: F904–F915, 2009. doi: 10.1152/ajprenal.90685.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng HA. Robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140, 2010. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delaloy C, Lu J, Houot A-M, Disse-Nicodeme S, Gasc J-M, Corvol P, Jeunemaitre X. Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform. Mol Cell Biol 23: 9208–9221, 2003. doi: 10.1128/MCB.23.24.9208-9221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Reilly M, Marshall E, Speirs HJ, Brown RW. WNK1, a gene within a novel blood pressure control pathway, tissue-specifically generates radically different isoforms with and without a kinase domain. J Am Soc Nephrol 14: 2447–2456, 2003. doi: 10.1097/01.asn.0000089830.97681.3b. [DOI] [PubMed] [Google Scholar]
- 38.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 39.Hoorn EJ, Gritter M, Cuevas CA, Fenton RA. Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol Rev 100: 321–356, 2020. doi: 10.1152/physrev.00044.2018. [DOI] [PubMed] [Google Scholar]
- 40.Ring AM, Cheng SX, Leng Q, Kahle KT, Rinehart J, Lalioti MD, Volkman HM, Wilson FH, Hebert SC, Lifton RP. WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc Natl Acad Sci USA 104: 4020–4024, 2007. doi: 10.1073/pnas.0611727104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci USA 104: 4025–4029, 2007. doi: 10.1073/pnas.0611728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu L, Cai H, Yue Q, Alli AA, Wang D, Al-Khalili O, Bao HF, Eaton DC. WNK4 inhibition of ENaC is independent of Nedd4-2-mediated ENaC ubiquitination. Am J Physiol Renal Physiol 305: F31–F41, 2013. doi: 10.1152/ajprenal.00652.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hadchouel J, Soukaseum C, Busst C, Zhou XO, Baudrie V, Zurrer T, Cambillau M, Elghozi JL, Lifton RP, Loffing J, Jeunemaitre X. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci USA 107: 18109–18114, 2010. doi: 10.1073/pnas.1006128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naray-Fejes-Toth A, Snyder PM, Fejes-Tóth G. The kidney-specific WNK1 isoform is induced by aldosterone and stimulates epithelial sodium channel-mediated Na+ transport. Proc Natl Acad Sci USA 101: 17434–17439, 2004. doi: 10.1073/pnas.0408146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heise CJ, Xu BE, Deaton SL, Cha SK, Cheng CJ, Earnest S, Sengupta S, Juang YC, Stippec S, Xu Y, Zhao Y, Huang CL, Cobb MH. Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J Biol Chem 285: 25161–25167, 2010. doi: 10.1074/jbc.M110.103432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhalla V, Soundararajan R, Pao AC, Li H, Pearce D. Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol 291: F714–F721, 2006. doi: 10.1152/ajprenal.00061.2006. [DOI] [PubMed] [Google Scholar]
- 47.Ahmed M, Salker MS, Elvira B, Umbach AT, Fakhri H, Saeed AM, Shumilina E, Hosseinzadeh Z, Lang F. SPAK sensitive regulation of the epithelial Na channel ENaC. Kidney Blood Press Res 40: 335–343, 2015. doi: 10.1159/000368509. [DOI] [PubMed] [Google Scholar]
- 48.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O'Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet 35: 372–376, 2003. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 49.Lin DH, Yue P, Rinehart J, Sun P, Wang Z, Lifton R, Wang WH. Protein phosphatase 1 modulates the inhibitory effect of with-no-lysine kinase 4 on ROMK channels. Am J Physiol Renal Physiol 303: F110–F119, 2012. doi: 10.1152/ajprenal.00676.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, Doucet A, Jeunemaitre X, Hadchouel J. WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci USA 110: 14366–14371, 2013. doi: 10.1073/pnas.1304230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen JC, Lo YF, Lin YW, Lin SH, Huang CL, Cheng CJ. WNK4 kinase is a physiological intracellular chloride sensor. Proc Natl Acad Sci USA 116: 4502–4507, 2019. doi: 10.1073/pnas.1817220116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shibata S, Ishizawa K, Wang Q, Xu N, Fujita T, Uchida S, Lifton RP. ULK1 phosphorylates and regulates mineralocorticoid receptor. Cell Rep 24: 569–576, 2018. doi: 10.1016/j.celrep.2018.06.072. [DOI] [PubMed] [Google Scholar]
- 54.Shibata S, Rinehart J, Zhang J, Moeckel G, Castaneda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, Lifton RP. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013. doi: 10.1016/j.cmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gallolu Kankanamalage S, Lee AY, Wichaidit C, Lorente-Rodriguez A, Shah AM, Stippec S, Whitehurst AW, Cobb MH. Multistep regulation of autophagy by WNK1. Proc Natl Acad Sci USA 113: 14342–14347, 2016. doi: 10.1073/pnas.1617649113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pham TD, Verlander JW, Wang Y, Romero CA, Yue Q, Chen C, Thumova M, Eaton DC, Lazo-Fernandez Y, Wall SM. Aldosterone regulates pendrin and epithelial sodium channel activity through intercalated cell mineralocorticoid receptor-dependent and -independent mechanisms over a wide range in serum potassium. J Am Soc Nephrol 31: 483–499, 2020. doi: 10.1681/ASN.2019050551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wen D, Cornelius RJ, Yuan Y, Sansom SC. Regulation of BK-α expression in the distal nephron by aldosterone and urine pH. Am J Physiol Renal Physiol 305: F463–F476, 2013. doi: 10.1152/ajprenal.00171.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun P, Liu W, Lin DH, Yue P, Kemp R, Satlin LM, Wang WH. Epoxyeicosatrienoic acid activates BK channels in the cortical collecting duct. J Am Soc Nephrol 20: 513–523, 2009. doi: 10.1681/ASN.2008040427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, Yue P, Lin DH, Wang WH. Carbon monoxide stimulates Ca2+-dependent big-conductance K channels in the cortical collecting duct. Am J Physiol Renal Physiol 304: F543–F552, 2013. doi: 10.1152/ajprenal.00530.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol 298: F12–F21, 2010. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCormick JA, Nelson JH, Yang C-L, Curry JN, Ellison DH. Overexpression of the sodium chloride cotransporter is not sufficient to cause familial hyperkalemic hypertension. Hypertension 58: 888–894, 2011. doi: 10.1161/HYPERTENSIONAHA.110.167809. [DOI] [PMC free article] [PubMed] [Google Scholar]