

Abstract
The main protease (Mpro) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the cause of coronavirus disease (COVID-19), is an ideal target for pharmaceutical inhibition. Mpro is conserved among coronaviruses and distinct from human proteases. Viral replication depends on cleavage of the viral polyprotein at multiple sites. We present crystal structures of SARS-CoV-2 Mpro bound to two viral substrate peptides. The structures show how Mpro recognizes distinct substrates and how subtle changes in substrate accommodation can drive large changes in catalytic efficiency. One peptide, constituting the junction between viral non-structural proteins 8 and 9 (nsp8/9), has P1′ and P2′ residues that are unique among the SARS-CoV-2 Mpro cleavage sites but conserved among homologous junctions in coronaviruses. Mpro cleaves nsp8/9 inefficiently, and amino acid substitutions at P1′ or P2′ can enhance catalysis. Visualization of Mpro with intact substrates provides new templates for antiviral drug design and suggests that the coronavirus lifecycle selects for finely tuned substrate-dependent catalytic parameters.
Keywords: SARS-CoV-2, Mpro, protease, virology
Graphical Abstract
Developing and stockpiling pan-coronavirus antiviral drugs for pandemic prevention has been a goal since the SARS outbreak of 2003.1, 2 The coronavirus main protease (nsp5 or Mpro) is a conserved drug target and a focus of these efforts. Hundreds of Mpro inhibitors have been reported. Most of these drugs occupy the active site cleft responsible for recognizing the N-terminal fragments of substrate peptides, and many form covalent bonds to the active site cysteine of Mpro (Cys145).3–7 A recent crystal structure of the nsp5/6 acyl-enzyme intermediate provides one template for chemical mimicry of this essential catalytic step.8 We provide evidence that enzyme-substrate contacts on both sides of the Mpro catalytic site affect the rate of formation of the covalent complex, a characteristic that could be exploited by new protease inhibitors.
The nsp8/9 junction is a conserved Mpro substrate (Figure 1A–B). The nearly invariant Asn residues at P1′ and P2′ are unique within a given coronavirus polyprotein; Gly, Ser, or Ala predominate at these positions in the other substrates.9 Cleavage of nsp8/9 is slow but required for replication of the closely-related Murine Hepatitis Virus.10 Indeed, a recently-determined cryo-EM structure shows that the N-terminus of nsp9 contacts nsp12, a core component of the viral RNA polymerase.11 In this context, the nsp8/9 P1′ to P3′ residues contribute to a binding site for a nucleotide that is transferred to the amino terminus of the P1′ residue.12 Therefore, the nsp8/9 junction has evolved to satisfy two evolutionary constraints required for viral replication: it must be cleaved in the Mpro active site, and it must serve as a substrate in a nucleotide monophosphate transfer reaction catalyzed by nsp12. We have used X-ray crystallography to study nsp8/9 and nsp4/5 recognition by Mpro. The structures show unique features of the Mpro nsp8/9 complex and highlight the importance of P1′-P3′ residues in catalysis.
Figure 1.

Viral Mpro substrates. (A) Protein sequence alignment of the 11 SARS-CoV-2 Mpro cleavage sites required for maturation of SARS-CoV2. (B) Protein sequence alignment of nsp8/9 Mpro cleavage sites from representative coronaviruses.
To study Mpro activity, we monitored cleavage of labeled substrate peptides in vitro and derived Michaelis-Menten parameters describing the reactions. Mpro cleavage of nsp4/5 is more efficient than cleavage of nsp8/9 (36-fold difference in kcat/KM; Table 1, Figure S1).13 We sought to understand the influence of the Asn residues at the P1′ and P2′ sites of the nsp8/9 substrate (Table 1). Altering the steric properties by alanine substitution at either position approximately doubled the catalytic efficiency. The P1′ Asn-to-Ala substitution lowered KM and raised kcat, while P2′ substitution only raised kcat. Installation of an isosteric Asp residue at the P1′ position completely abrogated activity, while the analogous Asn-to-Asp substitution at P2′ diminished but did not abrogate activity. We suspect that placing additional negative charge near the active site raises the energetic barrier to attaining the oxyanion transition states.14, 15
Table 1.
Catalytic efficiencies for Mpro substrates and analogs
| Substrate | Sequencea | kcat (S−1) | Km (μM) | kcat/KM (M−1 s−1) | Fold changeb |
|---|---|---|---|---|---|
| nsp4/5 | TSAVLQ/SGFRKM | 0.52 ± 0.07 | 41 ± 9 | 1.3 ± 0.3 × 104 | – |
| nsp8/9 | RVVKLQ/NNELMP | 0.013 ± 0.001 | 36 ± 6 | 3.6 ± 0.7 × 102 | 1.0 |
| nsp8/9 N1′A | RVVKLQ/ANELMP | 0.022 ± 0.001 | 22 ± 3 | 1.0 ± 0.1 × 103 | 2.9 |
| nsp8/9 N2′A | RVVKLQ/NAELMP | 0.034 ± 0.002 | 46 ± 5 | 7.5 ±0.8 × 102 | 2.1 |
| nsp8/9 N1′D | RVVKLQ/DNELMP | – | – | – | – |
| nsp8/9 N2′D | RVVKLQ/NDELMP | 0.0029 ± 0.0001 | 19 ± 1 | 1.6 ± 1.2 × 102 | 0.4 |
Lys-DABCYL and Glu-EDANS and are appended to the N- and C-termini. Residues that differ from the wild-type sequence are bolded.
Fold change = (kcat/KM) nsp8/9 analog /(kcat/KM)nsp8/9
Differences in kcat for the tested substrates dominated the small changes in KM and drove the observed changes in kcat/KM. The P5-P1 residues were constant for nsp8/9 and its derivatives, ruling out acyl-enzyme hydrolysis as the step that determines kcat. Therefore, either formation of the enzyme-substrate complex or conversion to the acyl-enzyme intermediate must limit kcat for the nsp8/9 substrate, and similar KM values imply the latter is true. These data imply kinetic competition among the 11 viral Mpro substrates during virus replication.
To better understand the different cleavage efficiencies, we determined crystal structures of Mpro bound to the nsp4/5 and nsp8/9 substrates (Figure 2). The structures were resolved to 1.84 Å for nsp4/5 and to 1.94 Å for nsp8/9 (Table S1), which enabled detailed interpretation of the atomic contacts between the enzyme and both substrates. The active site Cys145Ala mutation trapped the intact substrates and enabled visualization of the P′ residues (Figure S2). The C145A mutation creates a cavity in the Mpro active site that could influence the position of the scissile bond. At least one previous study has used the Mpro H41A mutation to circumvent this potential problem.10 Nevertheless, the high resolutions of both structures, the unambiguous positions of both scissile bonds, and the apparent specificity of peptide-enzyme contacts permitted detailed analysis of substrate engagement in both cases.
Figure 2.
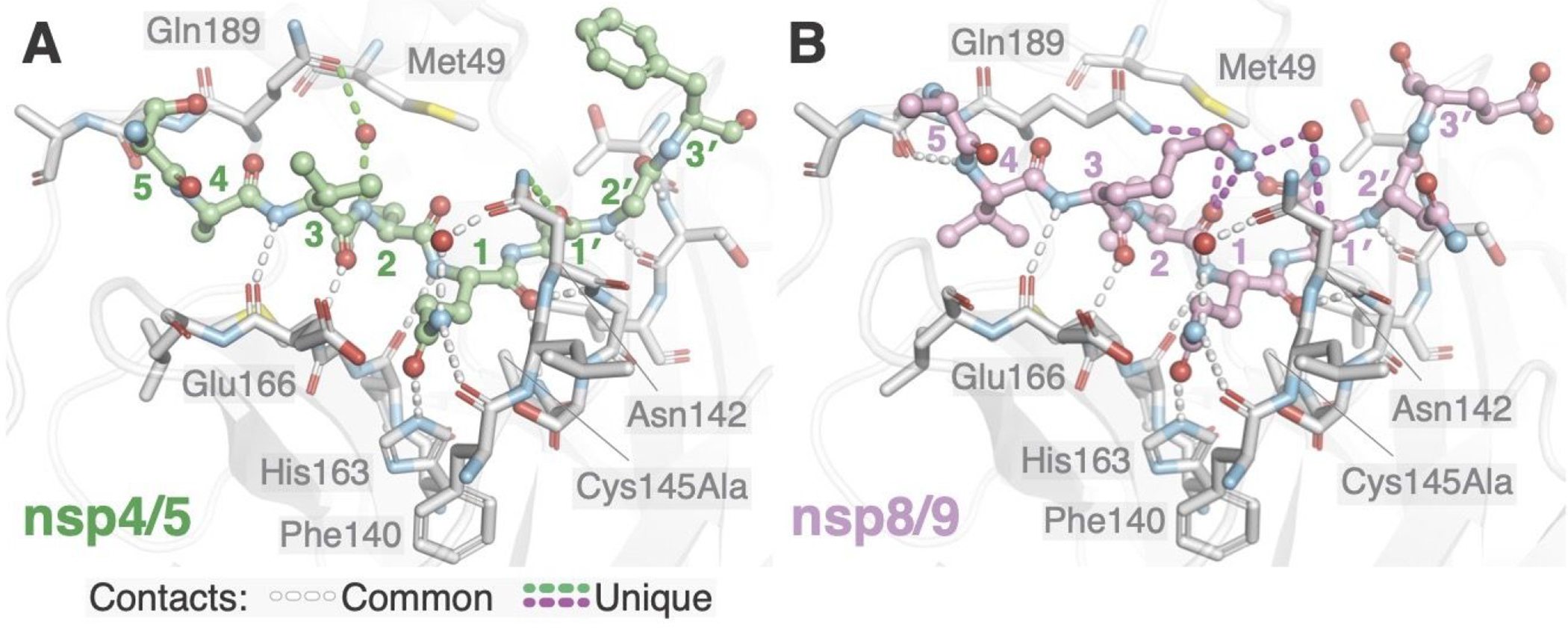
Differential recognition of nsp4/5 and nsp8/9 substrates by Mpro. Identical views of nsp4/5 (A) and nsp8/9 (B) substrates in the MproCys145Ala active site. Substrate peptide P and P′ residues are labeled with colored numbers. Key Mpro residues mentioned in the text are labeled. Conserved hydrogen bonds enabling Mpro recognition of substrate mainchain and P1 Gln side chain atoms are shown as white dashed lines. Hydrogen bonds that differ between the complex with nsp4/5 and that with nsp8/9 are shown in green and magenta, respectively. Mpro Asn142 and Gln189 contact both substrate through bound water molecules, and the resulting networks of hydrogen bonds differ between the two substrates.
11 conserved hydrogen bonds occur between Mpro and each of the substrates (Figure 2, white dashed lines). Eight contacts between the peptide backbones of enzyme and substrate are shared among SARS-CoV nsp4/5, PEDV nsp4/5,16, 17 and the two SARS-CoV-2 peptides reported here. Mpro Gly143 and Ala145 mainchain amides form the oxyanion hole by donating a pair of hydrogen bonds to the scissile P1 carbonyl oxygen, which stabilizes the developing negative charge during covalent catalysis. His163 and the mainchain carbonyl of Phe140 make hydrogen bonds with the invariant sidechain of the P1 Gln, and Asn142 contacts the P1 Gln through a conserved water bridge. Neither SARS-CoV nor PEDV Mpro·nsp4/5 complexes show the hydrogen bonds observed with Asn142 in the SARS-CoV2 substrate complexes.16, 17
Substrate interactions with the Mpro Asn142 and Gln189 sidechains distinguish nsp4/5 and nsp8/9 recognition (Figure 2, green and magenta dashed lines). Mpro Asn142 forms a hydrogen bond with the nsp4/5 P1′ backbone carbonyl oxygen, and Mpro Gln189 forms a water bridge with the nsp4/5 P2 amide nitrogen. In contrast, Mpro Gln189 engages the nsp8/9 P3 and P1′ side chains via an ordered water molecule. In addition to these contacts, the ordered waters found in the nsp8/9-bound structure could donate hydrogen bonds to the P1′ and P2 mainchain carbonyl oxygens. Finally, the nsp8/9 P3 Lys forms a hydrogen bond with the P2 carbonyl.
The peptide recognition described above supports distinct modes of P′ fragment accommodation by Mpro for the nsp4/5 and nsp8/9 substrates. The near-invariant nsp8/9 P1′ and P2′ Asn side chains are bulkier than the P1′ (Ser/Ala) and P2′ (Gly/Ala) side chains of other Mpro substrates, although there is greater tolerance for P2′ diversity.18 The nsp8/9 P1′ Asn projects more deeply into the S1′ subsite than the nsp4/5 P1′ Ser and therefore likely restrains the P′ peptide to a greater degree. Mpro Asn142 and Gly143 coordinate the nsp8/9 P2′ residue through peptide backbone interactions, and similar interactions position the nsp4/5 P2′ Gly. Overall, the bulkier nsp8/9 Asn side chains in the S1′ and S2′ subsites shift nsp8/9 relative to nsp4/5 (Figure 3A–B), and the resulting alignment with the Mpro cysteine nucleophile differs slightly (Figure S3). In addition to this small change in the position of the scissile bond, the nsp8/9 substrate bends away from the enzyme, resulting in a ~1.5 Å displacement of P4 and P4’ Cα positions relative to nsp4/5 (Figure S3) and widening the active site cleft formed between Mpro Met49 and Asn142 (7 Å to 10 Å, Figure 3A–B). These differences provide an explanation for reduced catalytic efficiency for the nsp8/9 substrate. The alanine substitutions discussed above presumably restore catalytic efficiency by enabling nsp8/9 to adopt an overall conformation and position more like nsp4/5.
Figure 3.
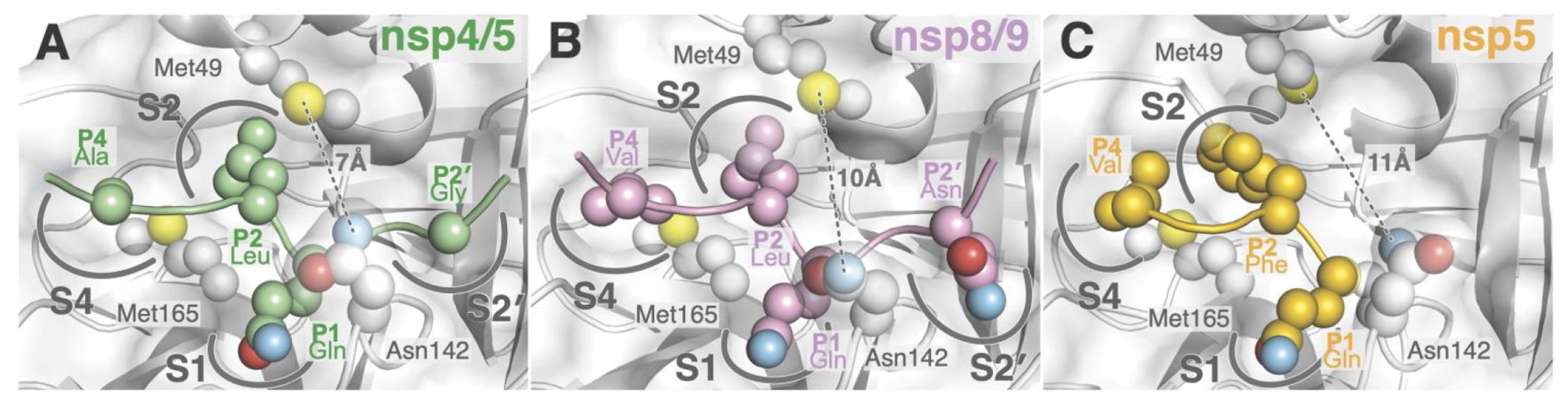
Steric effects that influence substrate recognition and Mpro activity. Spheres show positions of atoms dictating shape complementarity between Mpro subsites and nsp4/5 (A), nsp8/9 (B), and nsp5 (C; acyl-enzyme intermediate, PDB 7KHP). Labels show Mpro subsites and the distance between Mpro Met49 and Asn142 (thioether to amide nitrogen).
Hydrophobic interactions dictate recognition of N-terminal substrate fragments (P residues, excluding the invariant P1 Gln). Mpro Met49 and Met165 define the S2 subsite (Figure 3). The nsp4/5 P4 Ala is smaller than the ns8/9 P3 Val, allowing nsp4/5 to sit more deeply in the S4 subsite (Figure 3A–B). A recent crystal structure shows that the intact nsp5/6 substrate is also shifted relative to nsp4/5 due to a bulky Phe at the P2 position (Figure 3C).8 Indeed, Mpro cleavage is most efficient for peptides bearing P2 Leu and less efficient for those bearing P2 Phe.19 Like nsp8/9, cleavage of SARS nsp5/6 depends more heavily on P′ recognition than does nsp4/5.20, 21
The nsp8/9 P3 Lys might also limit catalysis. Water bridges connect its terminal nitrogen (Nζ) with the nsp8/9 P1′ Asn (mentioned above), and the resulting conformation could slow peptide accommodation to the Mpro active site. Therefore, diverse Mpro-substrate interactions contribute to finely-tuned substrate geometry that results in substrate-specific catalytic efficiency.22
The structures we have determined show how Mpro active site plasticity and substrate evolution can tune catalysis. Slow cleavage of the nsp8/9 junction, which is observed among disparate coronaviruses, might be a selected trait required for coordinated assembly of the RNA replication machinery.9, 13, 23, 24 Distinct kinetic parameters associated with cleavage of the viral substrate could be important for maturation of the viral polyprotein. The need for the nsp8/9 junction to support both Mpro cleavage and nsp12 binding (and subsequent nucleotide monophosphate acceptance) accounts for the near-invariance of the P1-P2′ residues. The sequence is therefore a compromise that satisfies the requirements of two unrelated catalytic mechanisms, and mimicry of the nsp8/9 junction presents a unique opportunity to chemically inhibit both Mpro and the viral polymerase.
The structures also present templates for new protease inhibitor scaffolds. In particular, that the nsp8/9 P3 sidechain can fold back to contact P1′ suggests macrocyclic inhibitors could mimic this interaction. Similar strategies have been pursued for Hepatitis C NS3, HIV-1, and Rhinovirus 3C proteases.25–27 Kinetic analyses of nsp8/9 and its variants suggests that inhibitor P1′ and P2′ site contacts could influence formation of covalent inhibitor-enzyme adducts. While α-ketoamide warheads have been investigated as ligands for Mpro Cys145,3 more comprehensive exploration of this warhead in combination with P′ mimicry could be beneficial.
METHODS
Complete methods are included in the Supporting Information file associated with this Report. A plasmid for recombinant expression of codon-optimized SARS_CoV-2 main protease (Mpro) was a gift from Zhang et al.3 Mpro expressed and purified from E. coli carrying this plasmid was used for peptide cleavage assays. Mpro Cys145Ala was purified from E. coli as a SUMO fusion protein. The N-terminus was generated by Ulp1 cleavage before use in crystallography experiments.
For crystallization, Mpro Cys145Ala was incubated with a 10-fold molar excels of each peptide (nsp4/5 – AVLQSGFRK; nsp8/9 – AVKLQNNEL) before mixing with mother liquor. Crystallization conditions are given in the Supporting Information file. Diffraction data were collected at the Advanced Photon Source on NE-CAT beamline 24-IDC.
Enzyme kinetics were determined using Förster resonance energy transfer (FRET) substrate peptides labeled with N-terminal fluorophore Dabcyl and C-terminal quencher Edans. Increasing concentrations of labeled substrates were incubated with Mpro (0.25 μM for nsp4/5 experiments and 0.4 μM for nsp8/9 experiments), and fluorescence was measured. Absolute product concentrations were determined and used to convert initial velocities to nM/s for triplicate reactions at each substrate concentration. Michaelis-Menten parameters (KM and Kcat) were determined using Prism 6 software.
Supplementary Material
ACKNOWLEDGMENTS
Enzyme assays were performed at the Institute for Chemistry and Chemical Biology (ICCB-Longwood) at Harvard Medical School. We thank Linlin Zhang (Hilgenfeld Laboratory, University of Lubeck) for providing the codon-optimized Mpro expression plasmid. X-ray diffraction data were collected at the Advanced Photon Source (APS) on NE-CAT beamline 24-IDC.
Funding Sources
We acknowledge funding support from the Massachusetts Consortium for Pathogen Readiness (M.N.N.), the Howard Hughes Medical Institute (S.C.H.), and the Nancy Lurie Marks Family Foundation (S.C.H.). S.M.H. is an HHMI Fellow of the Helen Hay Whitney Foundation. NE-CAT is funded by NIH grant P30 GM 124165. APS is operated for the DOE Office of Science by Argonne National Laboratory under contract DE-AC02-06CH11357.
Conflict of Interest
Mark Namchuk is the recipient of funding from AbbVie. The funding did not contribute to this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Supporting Information document contains methods, inhibition experiments, and crystallographic information. Crystallographic data have been deposited at the PDB with associated codes 7MGR (Mpro·nsp8/9) and 7MGS (Mpro·nsp4/5).
REFERENCES
- 1.Anand K; Ziebuhr J; Wadhwani P; Mesters JR; Hilgenfeld R, Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 2.Hilgenfeld R, From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J 2014, 281, 4085–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R, Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lockbaum GJ; Reyes AC; Lee JM; Tilvawala R; Nalivaika EA; Ali A; Kurt Yilmaz N; Thompson PR; Schiffer CA, Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses 2021, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J, Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 2020, 30, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rathnayake AD; Zheng J; Kim Y; Perera KD; Mackin S; Meyerholz DK; Kashipathy MM; Battaile KP; Lovell S; Perlman S; Groutas WC; Chang KO, 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci Transl Med 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai W; Zhang B; Jiang XM; Su H; Li J; Zhao Y; Xie X; Jin Z; Peng J; Liu F; Li C; Li Y; Bai F; Wang H; Cheng X; Cen X; Hu S; Yang X; Wang J; Liu X; Xiao G; Jiang H; Rao Z; Zhang LK; Xu Y; Yang H; Liu H, Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J; Worrall LJ; Vuckovic M; Rosell FI; Gentile F; Ton AT; Caveney NA; Ban F; Cherkasov A; Paetzel M; Strynadka NCJ, Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat Commun 2020, 11, 5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegyi A; Ziebuhr J, Conservation of substrate specificities among coronavirus main proteases. J Gen Virol 2002, 83, 595–599. [DOI] [PubMed] [Google Scholar]
- 10.Deming DJ; Graham RL; Denison MR; Baric RS, Processing of open reading frame 1a replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication. J Virol 2007, 81, 10280–10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan L; Ge J; Zheng L; Zhang Y; Gao Y; Wang T; Huang Y; Yang Y; Gao S; Li M; Liu Z; Wang H; Li Y; Chen Y; Guddat LW; Wang Q; Rao Z; Lou Z, Cryo-EM Structure of an Extended SARS-CoV-2 Replication and Transcription Complex Reveals an Intermediate State in Cap Synthesis. Cell 2021, 184, 184–193 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slanina H; Madhugiri R; Bylapudi G; Schultheiss K; Karl N; Gulyaeva A; Gorbalenya AE; Linne U; Ziebuhr J, Coronavirus replication-transcription complex: Vital and selective NMPylation of a conserved site in nsp9 by the NiRAN-RdRp subunit. Proc Natl Acad Sci U S A 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krichel B; Falke S; Hilgenfeld R; Redecke L; Uetrecht C, Processing of the SARS-CoV pp1a/ab nsp7-10 region. Biochem J 2020, 477, 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertus JD; Kraut J; Alden RA; Birktoft JJ, Subtilisin; a stereochemical mechanism involving transition-state stabilization. Biochemistry 1972, 11, 4293–4303. [DOI] [PubMed] [Google Scholar]
- 15.Simon L; Goodman JM, Enzyme catalysis by hydrogen bonds: the balance between transition state binding and substrate binding in oxyanion holes. J Org Chem 2010, 75, 1831–1840. [DOI] [PubMed] [Google Scholar]
- 16.Xue X; Yu H; Yang H; Xue F; Wu Z; Shen W; Li J; Zhou Z; Ding Y; Zhao Q; Zhang XC; Liao M; Bartlam M; Rao Z, Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol 2008, 82, 2515–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye G; Deng F; Shen Z; Luo R; Zhao L; Xiao S; Fu ZF; Peng G, Structural basis for the dimerization and substrate recognition specificity of porcine epidemic diarrhea virus 3C-like protease. Virology 2016, 494, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koudelka T; Boger J; Henkel A; Schonherr R; Krantz S; Fuchs S; Rodriguez E; Redecke L; Tholey A, N-Terminomics for the Identification of In Vitro Substrates and Cleavage Site Specificity of the SARS-CoV-2 Main Protease. Proteomics 2021, 21, e2000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rut W; Groborz K; Zhang L; Sun X; Zmudzinski M; Pawlik B; Wang X; Jochmans D; Neyts J; Mlynarski W; Hilgenfeld R; Drag M, SARS-CoV-2 M(pro) inhibitors and activity-based probes for patient-sample imaging. Nat Chem Biol 2021, 17, 222–228. [DOI] [PubMed] [Google Scholar]
- 20.Muramatsu T; Takemoto C; Kim YT; Wang H; Nishii W; Terada T; Shirouzu M; Yokoyama S, SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc Natl Acad Sci U S A 2016, 113, 12997–13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue X; Yang H; Shen W; Zhao Q; Li J; Yang K; Chen C; Jin Y; Bartlam M; Rao Z, Production of authentic SARS-CoV M(pro) with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J Mol Biol 2007, 366, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kneller DW; Phillips G; O’Neill HM; Jedrzejczak R; Stols L; Langan P; Joachimiak A; Coates L; Kovalevsky A, Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nat Commun 2020, 11, 3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gildenhuys S, Expanding our understanding of the role polyprotein conformation plays in the coronavirus life cycle. Biochem J 2020, 477, 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snijder EJ; Decroly E; Ziebuhr J, The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv Virus Res 2016, 96, 59–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X; Zhang YK; Liu Y; Ding CZ; Zhou Y; Li Q; Plattner JJ; Baker SJ; Zhang S; Kazmierski WM; Wright LL; Smith GK; Grimes RM; Crosby RM; Creech KL; Carballo LH; Slater MJ; Jarvest RL; Thommes P; Hubbard JA; Convery MA; Nassau PM; McDowell W; Skarzynski TJ; Qian X; Fan D; Liao L; Ni ZJ; Pennicott LE; Zou W; Wright J, Novel macrocyclic HCV NS3 protease inhibitors derived from alpha-amino cyclic boronates. Bioorg Med Chem Lett 2010, 20, 5695–5700. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh AK; Sean Fyvie W; Brindisi M; Steffey M; Agniswamy J; Wang YF; Aoki M; Amano M; Weber IT; Mitsuya H, Design, synthesis, X-ray studies, and biological evaluation of novel macrocyclic HIV-1 protease inhibitors involving the P1′-P2′ ligands. Bioorg Med Chem Lett 2017, 27, 4925–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Namoto K; Sirockin F; Sellner H; Wiesmann C; Villard F; Moreau RJ; Valeur E; Paulding SC; Schleeger S; Schipp K; Loup J; Andrews L; Swale R; Robinson M; Farady CJ, Structure-based design and synthesis of macrocyclic human rhinovirus 3C protease inhibitors. Bioorg Med Chem Lett 2018, 28, 906–909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.