

Abstract
DNA methylation plays crucial roles in many biological processes and abnormal DNA methylation patterns are often observed in diseases. Recent studies have shed light on cis-acting DNA elements that regulate locus-specific DNA methylation, which involves transcription factors, histone modification and DNA secondary structures. In addition, several recent studies have surveyed DNA motifs that regulate DNA methylation and suggest potential applications in diagnosis and prognosis. Here, we discuss the current biological foundation for the cis-acting genetic code that regulates DNA methylation. We review the computational models that predict DNA methylation with genetic features and discuss the biological insights revealed from these models. We also provide an in-depth discussion on how to leverage such knowledge in clinical applications, particularly in the context of liquid biopsy for early cancer diagnosis and treatment.
Keywords: DNA methylation, DNA motif, cancer, liquid biopsy, cfDNA
Introduction
DNA methylation in the mammal genomes is the addition of a methyl group to cytosines to form 5-methylcytosine (5mC), primarily at CG and also at CH (CH=CA, CT, CC) sites. DNA methylation in specific loci plays important roles in many biological functions. For example, DNA methylation in promoters represses gene transcription, and DNA methylation in gene bodies is associated with transcription elongation and splicing [1–3],. The synergy between DNA methylation and local histone modification is also locus-specific in development, somatic cell reprogramming and tumorigenesis [4,5]. Understanding how DNA methylation is established, maintained and removed in a particular locus is thus critical.
Locus-specific DNA methylation or demethylation depends on the recruitment of specific enzymes such as TET and DNMTs to the target genomic regions [6–8] (Figure 1A). DNA methylation is catalyzed by DNA methyltransferases (DNMTs) [9]. De novo methylation on both DNA strands involves DNMT3A/3B/3 L. Existing DNA methylation is maintained by a complex of DNMT1 and UHRF1, which recognizes half-methylated DNA strand (hemimethylation) after replication. Removal of the methyl group from cytosines is catalyzed by the ten-eleven-translocation enzymes (TET1/2/3), which can oxidize 5mC to 5-hydroxymethylcytosine (5hmC) and other oxidized cytosines (5-formylcytosine, 5fC, and 5-carboxylcytosine, 5caC), and then demethylate to cytosine through various pathways [10].
Figure 1 .
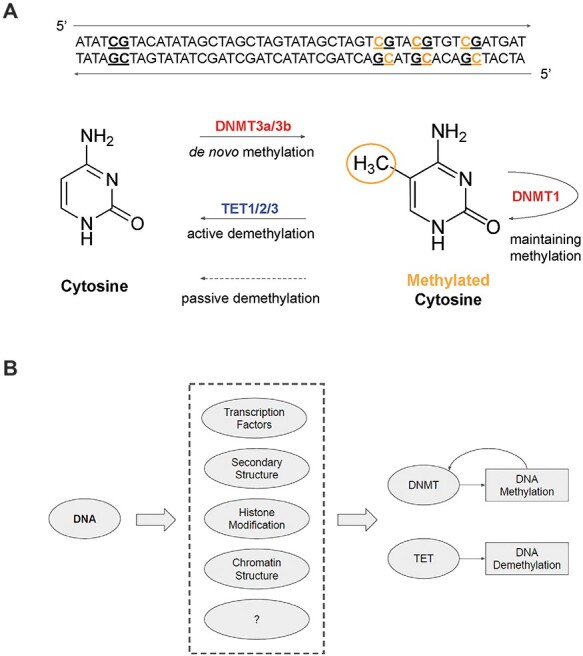
Mechanisms of locus-specific DNA methylation and demethylation.
Emerging evidence has suggested that enzymes like DNMTs and TETs are recruited to specific genomic regions by factors recognizing certain DNA sequences [6,11]. Recently, we have systematically identified 313 DNA motifs that regulate DNA methylation from 34 whole-genome methylomes. We show that these motifs are functional and can be applied to improve cancer prognosis and diagnosis [12]. In this review, we first survey the mechanisms proposed in the literature that orchestrate DNA methylation. We also review machine learning models that derive the genetic features of DNA methylation and discuss the biological insights revealed from these models. Finally, we propose to combine DNA methylation associated motifs and genetic mutations in clinical applications for liquid biopsy and early cancer diagnosis. We show how this approach improves the current paradigm where the discovery of biomarkers is focused on a small number of genes.
The Emerging DNA Features of Locus-specific Methylation
Accumulating evidence has shown that certain DNA sequence patterns are associated with local DNA methylation levels, such as lower GC content, enrichment of short nucleotide combinations (2–6 bp) and longer DNA motifs [13–23]. However, a puzzling observation is that the modifying enzymes including TETs and DNMTs do not have high recognition specificity of DNA motifs [6,11]. While TET1, TET3 and DNMT1 all possess a CXXC domain [24] interacting with DNA sequences, the CXXC domain mainly recognizes unmodified CpG dinucleotide. Recently, Xu et al. [25] identified four groups of DNA sequences bound by CXXC domains, all of which are at the CpG candidates for DNA methylation. Importantly, the DNA preference of the CXXC domains cannot explain how ~80% of the 28 million CpGs in the human genome are methylated (or how ~20% of the CpGs are unmethylated).
Furthermore, numerous reports have confirmed the existence of DNA sequences that dictate where DNA methylation/demethylation occurs. For example, Lienert et al. [26] have identified methylation-determining regions, which mediate de novo methylation and demethylation. Interestingly, these regions contain cis-regulatory motifs that can be recognized by DNA-binding factors (SP1, CTCF, Rfx), and mutating these motifs alters the methylation pattern. Stadler et al. [27] have shown that introducing CTCF motifs is necessary and sufficient to lower methylation of nearby CpGs.
Taken together, these reports suggest the locus-specificity of DNA methylation is encoded in the genomic sequence, recognized, and mediated by locus-specific factors. Here, we review the emerging mechanisms of locus-specific DNA methylation guided by cis-acting DNA sequences, through cross-talks between transcription factors (TFs), DNMTs, TETs, DNA secondary structures and histone modifications (Figure 1B).
TFs recruit TETs for active demethylation
TET1 and TET3 can contain DNA-binding CXXC-zinc finger domain [25]. TETs prefer CpG-rich sequences such as CpG island (CGI) which spans several kilobases [28] and can bind CpG-rich DNA sequences [6] in mammals to maintain stable demethylation [29]. In addition, TET recruitment through locus-specific TF binding has been widely reported. For example, introducing a CTCF binding site at a particular locus leads to TET recruitment and local DNA demethylation [27]. PPARG binds to promoters and recruits TET for demethylation [30]. In a recent study, Suzuki et al. [31] have designed a method to screen for TFs that can facilitate DNA demethylation in a site-directed manner. In particular, they transduced selected TFs in sub-cloned vectors to cells and evaluated the methylation levels using the HumanMethylation450 methylation array near the TF binding sites (estimated by the motif locations) with and without ectopic expression of the TFs. Using this strategy, Suzuki et al. [31] have shown that RUNX1 site-specific binding correlates with demethylation in hematopoietic cells, and they have further confirmed recruitments of critical proteins involved in DNA demethylation, including TET2, TET3, TDG and GADD45, using co-immunoprecipitation. Suzuki et al. [32] further scaled-up this strategy and found that eight (RUNX3, GATA2, CEBPB, MAFB, NR4A2, MYOD1, CEBPA and TBX5) out of 15 (plus NANOG, HNF1A, PAX4, Nkx2–5, SOX2, POU5F1, HNF4A) tested TFs can facilitate demethylation of DNA in a site-directed manner.
TFs block DNMT3s and prevent de novo methylation
Many TFs can maintain low methylation by blocking the access of DNMTs to specific regions. For example, SP1 preferentially binds to CpG-rich promoters, preventing de novo methylation in mice [33,34]. Proteins containing a CXXC domain (CFP1, MLL, KDM2A/2B, IDAX) can bind to unmethylated CpGs to keep the region from being methylated [24,35,36]. Interestingly, DNMT1 has a CXXC domain, which may facilitate its binding to hemimethylated CpGs [37]; TET1 and TET3 also have a CXXC domain, which has been shown to contribute to their locus-specificity [38,39]. However, other studies have shown that the CXXC domain failed to restrain the activity of Dnmt1 on unmethylated CpG sites [40].
TFs recruit DNMTs for de novo methylation
Similarly, many TFs have been reported to facilitate DNA methylation in particular loci. For example, NR6A1 (or GCNF) can silence Oct-3/4 by binding to its promoter and recruit Dnmt3a and Dnmt3b in the mouse, facilitating methylation [41]. Dnmt3a has been reported to interact with Myc and specifically target the promoter of p21Cip1, leading to transcription repression [42]. Dnmt3b is recruited by the TF E2F6 to silence germ-line genes in murine somatic tissues [43].
DNA secondary structure shape DNA methylation
Besides TF-directed locus-specific methylation, DNA secondary structure has also been reported to shape local DNA-methylation. For example, Clark and Smith [44] showed that variable number tandem repeats (VNTR) at a non-B DNA structure contributes to abnormal DNA methylation in human breast cancers. Mao et al. [45] reported G-quadruplex (G4) DNA secondary structures are associated with hypomethylation at the CGI in the human genome. This is because G4 sites are enriched with DNMT1 binding but inhibit DNMT1 enzymatic activity, leading to the inhibition of local CpG methylation. Other studies have shown a certain group of G4 structures play roles in both DNA methylation and histone modification [46]. Meanwhile, G4 secondary structures are characterized by strong telomeric repeats, with cis-acting DNA motifs such as (GGGGCC)(n), TG (4)T(2)G(4) T and GGGCT(4) GGGC [47–49], which are GC-rich motifs that associate recruitment of TETs and hypomethylation [12,13,50]. Taken together, the DNA secondary structure provides another mechanism of how DNA sequence maintains and alters local methylation.
Same factors involved in both methylation and demethylation
Some factors are involved in both site-specific methylation and demethylation. For example, SPI1 can mediate both de novo methylation (by interacting with DNMT3B) and demethylation (by interacting with TET2) in a site-specific manner [51,52]. CTCF is another example that has opposite roles in regulating DNA methylation. CTCF can promote unmethylation through blocking DNMTs. For example, Schoenherr et al. [53] showed that mutating CTCF-binding sites resulted in the recruitment of DNMTs, leading to increased methylation at the imprinting control region of Igf2/H19 locus in mouse. Stadler et al. [27] reported that CTCF binding creates a low methylation region through the presence of TETs. Other studies showed that CTCF facilitates histone modification and open chromatin, although the causality in relation to DNA methylation remains unclear [54–56].
Crosstalk with histone modification
The maintenance of DNA methylation also involves crosstalk with histone modification. For example, studies have established DNA maintenance on Uhrf1, where Dnmt1 and ubiquitination of histone H3 are involved to convert hemimethylated DNA to fully methylated DNA [57]. DNA methylation is also linked to H3K9me3 and H3K27me3, where the H3K9 methyltransferase SETDB1 interacts with DNMT3A and 3B [58,59]. Interestingly, SETDB1 does not bind to DNA but forms a repression complex with TRIM28 and zinc fingers such as ZNF274 to achieve locus-specificity [59,60]. Furthermore, Viré et al. [61] showed that the H3K27 methyltransferase EZH2, a component of the polycomb repressive complex PRC2, can interact with DNMT1, DNMT3A and DNMT3B. A more recent study by Baubec et al. [62] using genome-wide ChIP-seq and methylome measurements confirmed that DNMT3A and DNMT3B are localized to methylated CpG-dense regions in mouse stem cells; notably, they found that the PWWP domain of DNMT3B recognizes the SETD2-mediated H3K36 methylation, leading to DNMT3B preferential binding and methylation of the bodies of actively transcribed genes [62].
DNA methylation can also be co-repressed by TF binding and H3K4 methylation. Cfp1 has been reported to recruit H3K4 methyltransferases to promote H3K4me3, preventing CGI from methylation in mouse embryonic stem cells. However, Cfp1 knockout is insufficient to remove local hypomethylation, suggesting other factors are involved in this process [35,63]. In another study, unmethylated H3K4 tails were shown to interact with the de novo methylation machinery, such as Dnmt3L and Dnmt3a [64]. The association between H3K4 methylation and allele-specific DNA methylation has been shown at imprinted loci as well [65], guided by factors like KDM1B [66].
Cell-type specificity and methylation dynamics
The above-mentioned evidence showed that locus-specific methylation is tied to genetic features. Although the DNA sequences remain unchanged for a given genome, the readout of the motifs is dynamic and dependent upon cellular conditions. For example, the expression of a modifying enzyme (e.g. TETs and DNMTs) or the activity of a DNA-binding regulator and its access to DNA is cell-type/condition-dependent, which leads to the dynamic and cell-type-specific modification of epigenome [67]. Such recognition is similar to the binding of TFs to their motifs: the TF motifs in the promoters and enhancers remain the same, but the transcriptional regulation is tissue-specific and dynamic. However, although we have seen increasing evidence for motif-directed recruitment of effectors that can both promote and inhibit DNA methylation [68,69], further study is required toward a systematic characterization of the relationship between the expression of these effectors and cell-type-specific methylation level of their interacting regions.
The Models: Prediction and Revelation
The molecular mechanisms described above have laid the foundation for many studies that use genetic features to predict local DNA methylation. These studies have shed light on the sequence features of locus-specific methylation and demethylation. Below, we review the development of these studies and discuss the perspectives (Supplementary Table S1).
Earlier methylation studies typically employ enzymatic fractionation assays. For example, McrBC digests methylated sequences while many methylation-sensitive restriction endonucleases remove unmethylated sequences [70]. Due to the limited data coverage and resolution, these studies tend to focus on the methylated CGIs. The CGIs reside in the promoters and their demethylation facilitates the binding of TFs [71]. To distinguish unmethylated CGI (non-CGI) from methylated CGI, a variety of predictive features have been found using machine learning methods. For example, Yamada et al. [19] have determined the methylation status of CGIs (from fully methylated to fully unmethylated) using the HpaII-McrBC PCR method in human peripheral blood leukocytes, and then used Support Vector Machine (SVM) and random forest to identify the enriched nucleotide k-mers. They showed CG, CT and CA are the most predictive dinucleotide features for human CGI states. Similarly, Das et al. [15] have separated methylated and unmethylated CpGIs using methylation-sensitive restriction endonucleases and McrBC in the normal human adult brain, and showed that Alu coverage and certain hexamers are the most predictive (86% accuracy) among ~100 predefined features such as CG content, dinucleotide counts and trinucleotide counts. Performance is further improved when including non-sequence features such as trinucleotide physicochemical properties [16] (i.e. bendability, nucleosome rigidity and nucleosome positioning), histone modification [18,72] and the methylation states of flanking CpGs [22]. Note that while both studies ([72] and [18]) used multiple mammalian tissues and cell lines, the prediction accuracy and selected sequence features generalize well across them, with top predictive features being CpGI properties, DNA sequence composition, DNA structure patterns and histone modification status.
Recent studies take advantage of genome-wide methylation assays, such as 450 K array, RRBS and WGBS. The expanded coverage of methylomes has profoundly changed the locus-specific analysis of DNA methylation in several ways. For example, functional motifs have been found outside of CGIs, extending into non-coding regions [12, 23]. In addition, genomic and epigenomic data from multiple cell lines and tissues have been made available by consortium efforts such as ENCODE [73], ROADMAP [74], TCGA [75] and iHEC [76]. Methylation levels are compared across multiple tissues, cell lines and species to establish variability. For example, Zeng et al. [23] have analyzed 50 RRBS +1 WGBS datasets and established the impact of DNA variants on local methylation. Wang et al. [12] have identified genomic regions and motifs associated with common and variable methylation across 34 WGBS, validated in 32 450 K array data sets. Scala et al. [77] examined variance and aberration of CpGs from various cancer types and blood samples across 450 K data sets in TCGA [75] and the Epic cohort [78], and have identified motifs associated with methylation stability, instability and aberration in cancers. More datasets have also allowed more sophisticated machine learning models, such as neural networks [17, 21, 23], to outperform previously best-performing machine learning models like SVM and random forest [15, 19, 22, 72]. DNA sequence features have shifted from using predefined sequences and short k-mer combinations (usually 2–5 bp) [15, 18, 19, 21, 22, 72] to using longer de novo motifs (>9 bp) [12, 13, 17, 23, 79, 80]. These studies revealed novel perspectives on how certain genetic patterns can play important roles in regulating DNA methylation.
The most fundamental change in methylation motif studies is from making predictions to exploring the functional mechanisms of DNA motifs. A natural first step to illustrate the functions of the found de novo motifs is to match them to known TFs [12, 13, 17, 23, 79, 80]. As a result, while earlier studies have associated hypomethylation with high GC contents [18, 19], recent studies have revealed that the contributing DNA motifs with repeating GC tandems are matched to known TFs associated with TET recruitment, such as CTCF, SP family and WT1 [12, 23]. Furthermore, contrary to the previous belief that methylated regions have aberrant TF binding, some TFs have also been found to preferentially bind to highly methylated regions. For example, Wang et al. [12] have identified 92 motifs associated with high methylation in WGBS with enriched bindings of DNMTs. Xuan-lin et al. [80] cross-referenced ChIP-seq of TFs and WGBS to characterize over 500 known TFBS with cell-type-specific CpG methyl-level in their motifs, and have shown some TFs, such as ZBTB33, have high binding affinity to methylated DNA. Ngo et al. [79] have proposed a high-throughput pipeline that not only revealed de novo methylated motifs but also discovered known TFs like CEBPB, NRF1, CTCF and EGR1 that can bind to highly methylated motif patterns (e.g. [GT]ATT [AG]mCGCAAT for CEBPB) which are sequentially and locationally distinct from their canonical motifs. Whitaker et al. [13] have further provided a computational framework to identify DNA motifs representing cis-acting elements with the site-specific DNA-binding factors that establish and maintain epigenomic modifications, including DNA methylation and six histone modifications, and have shown that motifs like CFP1 are found to prefer the center of DNA methylation valleys, with a specific association to H3K4me3 and H3K27me3 modification. Finally, recent studies have highlighted crosstalk between DNA methylation and histone modification among these motifs, especially between H3K36me3 and methylation motifs, as well as between H3K27ac/H3K4me3 and unmethylation motifs [12, 81]. It is worth noting that many de novo motifs found relevant to DNA methylations do not match any known motif [12] and the mechanisms of these motifs await further investigation.
Along with the mechanistic insights on the shaping of the methylome, recent studies also highlight the functional validation of the identified motifs through DNA variants. For example, Wang et al. [12] have shown motifs with enriched methylation quantitative trait loci (mQTL) and expression quantitative trait loci (eQTL), and somatic mutation on the motifs correlates with altered local CpG methylation. Similarly, Banovich et al. [82] have characterized the mQTL in relation to TF binding and expression, and shown that STAT5 and ZNF274 have positive associations between TF expression and DNA methylation nearby binding sites. Further, Zeng et al. [23] have proposed a deep learning framework, CpGenie, to systematically predict methylation change from sequence variant, given the neighboring methylation and DNA sequences.
Taken together, we have observed explosive growth of computational models that explain DNA methylation based on sequence features, in combination with the traditional usage of physicochemical properties, nearby CpG states, TF occupancies and histone states. The improved model performance and the revealed genetic-epigenetic association have made the clinical application possible.
Clinical Application
DNA methylation is closely linked to development, aging and cancer [83, 84]. A common observation in cancer is that methylation on the promoter of a tumor suppressor gene often results in transcriptional repression and phenotypic alteration [1, 85]. Such DNA methylation patterns can thus be used for diagnosis and prognosis purposes. Notably, the recent development of early cancer diagnosis and treatment guidance has been enabled by liquid biopsy [86, 87], whose successful application depends on differentiating the tumorous circulating tumor DNA (ctDNA) from the ‘normal’ call-free DNA (cfDNA) fragments [86, 87]. However, the major challenge is that ctDNA is a small fraction (0.01%–10%) of the total cfDNA [86–88]. Therefore, to achieve sensitive and selective tumor variant detection, current strategies rely heavily on carefully selecting a collection of features (or biomarkers) combinatorically most predictive of the target phenotypes.
Over the years, the choice of biomarkers has shifted from focusing on genetic mutations on tumor suppressors (such as TP53 [89] and PTEN [90]) to leveraging epigenetics signatures [91]. For example, BRCA1, PTEN, HRK, APC and RASSF1A have been found methylated in cancer, and some related to prognosis and reflect on the efficacy of therapy [92–94]. DNA methylation patterns derived from RRBS have also been used as a predictor for breast cancer dissemination [95]. Other studies have reported success with DNA methylation cfDNA assay outside plasma for specific cancer types, such as urine-based assays for prostate cancer [96, 97] and stool-based assays for colorectal cancers [98]. Guo et al. [99] reported segments of DNA methylation (termed haplotype blocks) from plasma DNA can aid the deconvolution of heterogeneous tissue samples. A more recent study by Grail has successfully mapped and identified tumor origin by cfDNA methylation in 25 human tissues and cells [100]. Notably, Shen et al. [101] have developed an immunoprecipitation-based genome-wide cfDNA methylome screening protocol (cfMeDIP–seq). They showed sensitive tumor detection and classification among several tumor types, using differentially methylated regions and CpGs. Overall, the adoption of cfDNA methylation analysis has greatly improved the diagnosis power in previously low-performing cancer types.
A natural progression is combining both genetic and epigenetic signals to further improve performance and detection limit in early cancer diagnosis and personalized treatment. Indeed, Westesson et al. [102] have recently shown that with combined genomic, methylation and fragmentomic signals in 162 early-stage colorectal cancer patients; they achieved an overall sensitivity of detection at 90.3% (90% Stage I; 88% Stage II; 96% Stage III) and specificity at 96.6%. The rapid adoption of the multi-omics approach evokes an emerging strategy where the knowledge of how cis-acting DNA variants impact disease-associated epigenome leads to improved diagnostics and prognostics. For example, the presence of a single-nucleotide polymorphism (SNP) at the MGMT promoter negates the promoter’s methylation in glioblastoma, correlates with worse temozolomide treatment outcome [103]. An SNP at the CpG site located at the ARPC3 promoter is associated with hypertriglyceridemia in overweight patients [104]. Three CpG-SNP pairs have been reported significant for the prognosis of breast cancer patients [105]. Multiple studies have reported DNA variants are particularly found in the CGI at the promoter of genes related to cancer [106–109]. Zeng et al. [23] have reported a model to accurately quantify how DNA variants can impact local CpG methylation and gene expression. Recently, we have discovered and characterized 313 DNA motifs that regulate DNA methylation and unmethylation and showed that DNA mutation overlapping with these motifs impacts local CpG methylation (Figure 2A). Moreover, we have demonstrated that profiling somatic mutations in cancer patients based on which DNA motifs they overlap, providing a significant performance improvement over using these somatic mutations alone, both for diagnosis and prognosis [12] (Figure 2B). Taken together, these results suggest understanding how non-coding DNA-variants change methylation can improve the re-evaluation of the existing DNA biomarkers and provide new perspectives on biomarker discovery.
Figure 2 .
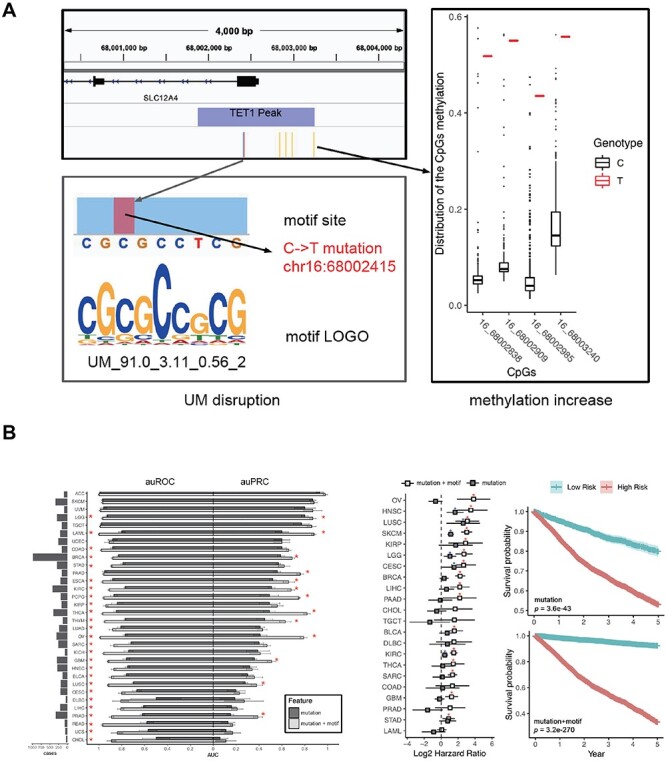
Clinical application. A. An example of altered cis-acting DNA motif changes local methylation level. B. Evidence that combining the prior knowledge of DNA motifs that regulates DNA with somatic mutation significantly improves performance for both cancer diagnosis (left) and prognosis (right). (reprinted from Wang et al. 2019).
Outlook
Current research for liquid biopsy benefits from two contributing factors: the quickly increasing sequencing power [110], and clinical studies linking molecular profile to early pan-cancer diagnosis, as well as treatment outcome of late-stage cancer patients [111–113]. Therefore, while currently available assays have relatively small numbers of features (i.e. 10–100 biomarkers) due to limited variant data [114], future studies can use many more features. Furthermore, the large data set will allow the development of more powerful deep learning models to improve the prediction power. Both trends require a deeper understanding of the interplay between the existing features (i.e. DNA methylation and DNA variant).
Ideally, cancer diagnosis and prognosis could benefit from combining a diverse set of relevant molecular signatures, including DNA variants, methylome, transcriptome, proteome, HLA signature and chromatin structure. However, given the limited resources, the major challenge to distinguish between cancer and normal cfDNA is the limited number of biomarkers, and how to detect them frugally. As a result, we would argue that the most cost-effective strategy is to adopt the prior knowledge of how DNA sequence and methylation interact with each other to further improve accuracy and sensitivity. Recent technological advances have made it possible to simultaneously detect DNA variants and methylation variants on cfDNA [101]. The synergistic interplay between DNA variants and DNA methylation makes using DNA motifs advantageous and versatile in many clinical settings.
In addition to DNA methylation, we have recently discovered DNA motifs that regulate histone modifications [13, 81] and showed that the altered DNA motif leads to abolished histone modification, which is also important in cancer [115]. These cis-acting motifs can be leveraged to reveal information on the state of histones, which is not readily available in cfDNA [87]. Furthermore, DNA patterns are also important in establishing local DNA secondary structures, which have been reported as an epigenetic determinant of cancer genome [116]. Clark et al. [44] have reported a sequence pattern in the DNA secondary structures as a hotspot for DNA methylation in human breast cancer patients.
Taken together, we believe the ever-growing research revealing genetic-epigenetic interplay has opened doors to previously underexplored strategies in biomarker selection and points to new perspectives in characterizing DNA variants in combination with epigenetic signatures.
Key Points
Increasing evidence has shown locus-specific DNA methylation is regulated by cis-acting DNA elements.
Recently, computational models are used to predict genetic features of DNA methylation patterns.
Biological insights have been revealed from these models.
Future application of methylation biomarkers considering liquid biopsy for early cancer diagnosis and treatment are discussed.
Supplementary Material
Acknowledgements
This work was partially supported by The National Institutes of Health (NIH) (R01HG009626) and California Institute for Regenerative Medicine (CIRM) (RB5-07012).
Mengchi Wang is a PhD student at the Bioinformatics and Systems Biology at University of California at San Diego, with current research focus on the genetic basis for DNA methylation.
Vu Ngo is a PhD student at the Bioinformatics and Systems Biology at University of California at San Diego, with current research focus on the genetic basis for histone modification.
Wei Wang is the principle investigator and full professor at the Bioinformatics and Systems Biology, Department of Chemistry and Biochemistry, and Department of Cellular and Molecular Medicine at University of California at San Diego.
Contributor Information
Mengchi Wang, Bioinformatics and Systems Biology at University of California, USA.
Vu Ngo, Bioinformatics and Systems Biology at University of California, USA.
Wei Wang, Bioinformatics and Systems Biology, Department of Chemistry and Biochemistry, and Department of Cellular and Molecular Medicine at University of California, USA.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1.Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev 1991;55:451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ziller MJ, Gu H, Müller F, et al. . Charting a dynamic DNA methylation landscape of the human genome. Nature 2013;500:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maor GL, Yearim A, Ast G. The alternative role of DNA methylation in splicing regulation. Trends Genet 2015;31:274–80. [DOI] [PubMed] [Google Scholar]
- 4.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009;10:295–304. [DOI] [PubMed] [Google Scholar]
- 5.Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta 2014;1839:1362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016;30:733–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem 2013;288:34287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ravichandran M, Jurkowska RZ, Jurkowski TP. Target specificity of mammalian DNA methylation and demethylation machinery. Org Biomol Chem 2018;16:1419–35. [DOI] [PubMed] [Google Scholar]
- 9.Robertson KD. DNA methylation and human disease. Nat Rev Genet 2005;6:597–610. [DOI] [PubMed] [Google Scholar]
- 10.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013;502:472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem 2011;12:206–22. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Zhang K, Ngo V, et al. . Identification of DNA motifs that regulate DNA methylation. Nucleic Acids Res 2019;47:6753–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitaker JW, Chen Z, Wang W. Predicting the human epigenome from DNA motifs. Nat Methods 2015;12:265–727 p following 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu C, Yao S, Li X, et al. . Genome-wide prediction of DNA methylation using DNA composition and sequence complexity in human. Int J Mol Sci 2017;18:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das R, Dimitrova N, Xuan Z, et al. . Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci U S A 2006;103:10713–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng P, Chen W, Lin H. Prediction of CpG island methylation status by integrating DNA physicochemical properties. Genomics 2014;104:229–33. [DOI] [PubMed] [Google Scholar]
- 17.Angermueller C, Lee HJ, Reik W, et al. . DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol 2017;18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards JR, O’Donnell AH, Rollins RA, et al. . Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res 2010;20:972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada Y, Satou K. Prediction of genomic methylation status on CpG islands using DNA sequence features. WSEAS Transactions on Biology and Biomedicine 2008;5:153–62. [Google Scholar]
- 20.Su J, Shao X, Liu H, et al. . Genome-wide dynamic changes of DNA methylation of repetitive elements in human embryonic stem cells and fetal fibroblasts. Genomics 2012;99:10–7. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Liu T, Xu D, et al. . Predicting DNA methylation state of CpG dinucleotide using genome topological features and deep networks. Sci Rep 2016;6:19598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wrzodek C, Büchel F, Hinselmann G, et al. . Linking the epigenome to the genome: correlation of different features to DNA methylation of CpG islands. PLoS One 2012;7:e35327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng H, Gifford DK. Predicting the impact of non-coding variants on DNA methylation. Nucleic Acids Res 2017;45:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long HK, Blackledge NP, Klose RJ. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem Soc Trans 2013;41:727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu C, Liu K, Lei M, et al. . DNA sequence recognition of human CXXC domains and their structural determinants. Structure 2018;26:85, e3–95. [DOI] [PubMed] [Google Scholar]
- 26.Lienert F, Wirbelauer C, Som I, et al. . Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet 2011;43:1091–7. [DOI] [PubMed] [Google Scholar]
- 27.Stadler MB, Murr R, Burger L, et al. . DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011;480:490–5. [DOI] [PubMed] [Google Scholar]
- 28.Elango N, Yi SV. Functional relevance of CpG island length for regulation of gene expression. Genetics 2011;187:1077–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Gu C, Yang L, et al. . The sequence preference of DNA methylation variation in mammalians. PLoS One 2017;12:e0186559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujiki K, Shinoda A, Kano F, et al. . PPARγ-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat Commun 2013;4:2262. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki T, Shimizu Y, Furuhata E, et al. . RUNX1 regulates site specificity of DNA demethylation by recruitment of DNA demethylation machineries in hematopoietic cells. Blood Adv 2017;1:1699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki T, Maeda S, Furuhata E, et al. . A screening system to identify transcription factors that induce binding site-directed DNA demethylation. Epigenetics Chromatin 2017;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brandeis M, Frank D, Keshet I, et al. . Spl elements protect a CpG island from de novo methylation. Nature 1994;371:435–8. [DOI] [PubMed] [Google Scholar]
- 34.Macleod D, Charlton J, Mullins J, et al. . Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev 1994;8:2282–92. [DOI] [PubMed] [Google Scholar]
- 35.Thomson JP, Skene PJ, Selfridge J, et al. . CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010;464:1082–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko M, An J, Bandukwala HS, et al. . Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013;497:122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song J, Rechkoblit O, Bestor TH, et al. . Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011;331:1036–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Zhang X, Clark E, et al. . TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res 2010;20:1390–3. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, Xu C, Kato A, et al. . Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell 2012;151:1200–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frauer C, Rottach A, Meilinger D, et al. . Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One 2011;6:e16627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato N, Kondo M, Arai K-I. The orphan nuclear receptor GCNF recruits DNA methyltransferase for Oct-3/4 silencing. Biochem Biophys Res Commun 2006;344:845–51. [DOI] [PubMed] [Google Scholar]
- 42.Brenner C, Deplus R, Didelot C, et al. . Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J 2005;24:336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velasco G, Hubé F, Rollin J, et al. . Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc Natl Acad Sci U S A 2010;107:9281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark J, Smith SS. Secondary structure at a hot spot for DNA methylation in DNA from human breast cancers. Cancer Genomics Proteomics 2008;5:241–51. [PMC free article] [PubMed] [Google Scholar]
- 45.Mao S-Q, Ghanbarian AT, Spiegel J, et al. . DNA G-quadruplex structures mold the DNA methylome. Nat Struct Mol Biol 2018;25:951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mukherjee AK, Sharma S, Chowdhury S. Non-duplex G-Quadruplex structures emerge as mediators of epigenetic modifications. Trends Genet 2019;35:129–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra SK, Tawani A, Mishra A, et al. . G4IPDB: a database for G-quadruplex structure forming nucleic acid interacting proteins. Sci Rep 2016;6:38144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burge S, Parkinson GN, Hazel P, et al. . Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res 2006;34:5402–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Salvo M, Pinatel E, Talà A, et al. . G4PromFinder: an algorithm for predicting transcription promoters in GC-rich bacterial genomes based on AT-rich elements and G-quadruplex motifs. BMC Bioinformatics 2018;19:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura R, Uno A, Kumagai M, et al. . Hypomethylated domain-enriched DNA motifs prepattern the accessible nucleosome organization in teleosts. Epigenetics Chromatin 2017;10:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki M, Yamada T, Kihara-Negishi F, et al. . Site-specific DNA methylation by a complex of PU. 1 and Dnmt3a/b. Oncogene 2006;25:2477. [DOI] [PubMed] [Google Scholar]
- 52.la RL, Rica L, Rodríguez-Ubreva J, et al. . PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol 2013;14:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schoenherr CJ, Levorse JM, Tilghman SM. CTCF maintains differential methylation at the Igf2/H19 locus. Nat Genet 2003;33:66–9. [DOI] [PubMed] [Google Scholar]
- 54.Rao SSP, Huntley MH, Durand NC, et al. . A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Splinter E, Heath H, Kooren J, et al. . CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 2006;20:2349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weth O, Paprotka C, Günther K, et al. . CTCF induces histone variant incorporation, erases the H3K27me3 histone mark and opens chromatin. Nucleic Acids Res 2014;42:11941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishiyama A, Yamaguchi L, Nakanishi M. Regulation of maintenance DNA methylation via histone ubiquitylation. J Biochem 2016;159:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H, Rauch T, Chen Z-X, et al. . The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem 2006;281:19489–500. [DOI] [PubMed] [Google Scholar]
- 59.Schultz DC. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 2002;16:919–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frietze S, O’Geen H, Blahnik KR, et al. . ZNF274 recruits the histone methyltransferase SETDB1 to the 3′ ends of ZNF genes. PLoS One 2010;5:e15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viré E, Brenner C, Deplus R, et al. . The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006;439:871–4. [DOI] [PubMed] [Google Scholar]
- 62.Baubec T, Colombo DF, Wirbelauer C, et al. . Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015;520:243–7. [DOI] [PubMed] [Google Scholar]
- 63.Clouaire T, Webb S, Skene P, et al. . Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev 2012;26:1714–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010;11:204–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Delaval K, Govin J, Cerqueira F, et al. . Differential histone modifications mark mouse imprinting control regions during spermatogenesis. EMBO J 2007;26:720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ciccone DN, Su H, Hevi S, et al. . KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009;461:415–8. [DOI] [PubMed] [Google Scholar]
- 67.Gu T, Lin X, Cullen SM, et al. . DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol 2018;19:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet 2016;17:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Héberlé É, Bardet AF. Sensitivity of transcription factors to DNA methylation. Essays Biochem 2019;63:727–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rollins RA, Haghighi F, Edwards JR, et al. . Large-scale structure of genomic methylation patterns. Genome Res 2006;16:157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011;25:1010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng H, Wu H, Li J, et al. . CpGIMethPred: computational model for predicting methylation status of CpG islands in human genome. BMC Med Genomics 2013;6(Suppl 1):S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Consortium RE, Kundaje A, Meuleman W, et al. . Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, et al. . The cancer genome atlas pan-cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bujold D, Morais DA de L, Gauthier C, et al. . The international human epigenome Consortium data portal. Cell Syst 2016;3:496–499.e2. [DOI] [PubMed] [Google Scholar]
- 77.Scala G, Federico A, Palumbo D, et al. . DNA sequence context as a marker of CpG methylation instability in normal and cancer tissues. Sci Rep 2020;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Calza S, Specchia C, Frasca G, et al. . EPIC-Italy cohorts and multipurpose national surveys. A comparison of some socio-demographic and life-style characteristics. Tumori 2003;89:615–23. [DOI] [PubMed] [Google Scholar]
- 79.Ngo V, Wang M, Wang W. Finding de novo methylated DNA motifs. Bioinformatics 2019;35:3287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xuan Lin QX, Sian S, An O, et al. . MethMotif: an integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic Acids Res 2019;47:D145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ngo V, Chen Z, Zhang K, et al. . Epigenomic analysis reveals DNA motifs regulating histone modifications in human and mouse. Proc Natl Acad Sci U S A 2019;116:3668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Banovich NE, Lan X, McVicker G, et al. . Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet 2014;10:e1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet 2018;392:777–86. [DOI] [PubMed] [Google Scholar]
- 84.Fardi M, Solali S, Farshdousti Hagh M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis 2018;5:304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics 2009;1:239–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med 2018;379:1754–65. [DOI] [PubMed] [Google Scholar]
- 87.Wan JCM, Massie C, Garcia-Corbacho J, et al. . Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223–38. [DOI] [PubMed] [Google Scholar]
- 88.Hao X, Luo H, Krawczyk M, et al. . DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci U S A 2017;114:7414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ 2019;26:199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene 2008;27:5443–53. [DOI] [PubMed] [Google Scholar]
- 91.Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet 2019;20:109–27. [DOI] [PubMed] [Google Scholar]
- 92.Müller HM, Widschwendter A, Fiegl H, et al. . DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res 2003;63:7641–5. [PubMed] [Google Scholar]
- 93.Fiegl H, Millinger S, Mueller-Holzner E, et al. . Circulating tumor-specific DNA: a marker for monitoring efficacy of adjuvant therapy in cancer patients. Cancer Res 2005;65:1141–5. [DOI] [PubMed] [Google Scholar]
- 94.Fackler MJ, Lopez Bujanda Z, Umbricht C, et al. . Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res 2014;74:2160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Widschwendter M, Evans I, Jones A, et al. . Methylation patterns in serum DNA for early identification of disseminated breast cancer. Genome Med 2017;9:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao F, Olkhov-Mitsel E, Kamdar S, et al. . A urine-based DNA methylation assay, ProCUrE, to identify clinically significant prostate cancer. Clin Epigenetics 2018;10:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brikun I, Nusskern D, Decatus A, et al. . A panel of DNA methylation markers for the detection of prostate cancer from FV and DRE urine DNA. Clin Epigenetics 2018;10:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Han YD, Oh TJ, Chung T-H, et al. . Early detection of colorectal cancer based on presence of methylated syndecan-2 (SDC2) in stool DNA. Clin Epigenetics 2019;11:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guo S, Diep D, Plongthongkum N, et al. . Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet 2017;49:635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moss J, Magenheim J, Neiman D, et al. . Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9:5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shen SY, Singhania R, Fehringer G, et al. . Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563:579–83. [DOI] [PubMed] [Google Scholar]
- 102.Westesson O, Axelrod H, Dean J, et al. . Abstract 2316: integrated genomic and epigenomic cell-free DNA (cfDNA) analysis for the detection of early-stage colorectal cancer. Cancer Res 2020;80:2316–6. [Google Scholar]
- 103.Rapkins RW, Wang F, Nguyen HN, et al. . The MGMT promoter SNP rs16906252 is a risk factor for MGMT methylation in glioblastoma and is predictive of response to temozolomide. Neuro Oncol 2015;17:1589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toro-Martin J, Guenard F, Tchernof A, et al. . A CpG-SNP located within the ARPC3 gene promoter is associated with hypertriglyceridemia in severely obese patients. Ann Nutr Metab 2016;68:203–12. [DOI] [PubMed] [Google Scholar]
- 105.Shilpi A, Bi Y, Jung S, et al. . Identification of genetic and epigenetic variants associated with breast cancer prognosis by integrative bioinformatics analysis. Cancer Inform 2017;16:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fan H, Liu D, Qiu X, et al. . A functional polymorphism in the DNA methyltransferase-3A promoter modifies the susceptibility in gastric cancer but not in esophageal carcinoma. BMC Med 2010;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rakyan VK, Hildmann T, Novik KL, et al. . DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol 2004;2:e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kerkel K, Spadola A, Yuan E, et al. . Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet 2008;40:904–8. [DOI] [PubMed] [Google Scholar]
- 109.Shoemaker R, Deng J, Wang W, et al. . Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res 2010;20:883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Technology MM. Getting Moore from DNA sequencing. Nat Rev Genet 2011;12:586. [DOI] [PubMed] [Google Scholar]
- 111.Sicklick JK, Kato S, Okamura R, et al. . Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med 2019;25:744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Odegaard JI, Vincent JJ, Mortimer S, et al. . Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res 2018;24:3539–49. [DOI] [PubMed] [Google Scholar]
- 113.Fiala C, Diamandis EP. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med 2018;16:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Koch A, Joosten SC, Feng Z, et al. . Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol 2018;15:459–66. [DOI] [PubMed] [Google Scholar]
- 115.Zhang L, Liang Y, Li S, et al. . The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol Cancer 2019;18:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.De S, Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat Struct Mol Biol 2011;18:950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.