

Abstract
Cell-penetrating peptides (CPPs) are useful tools for the delivery of a wide variety of cargo into cells. Our lab has developed two classes of CPPs based on β-hairpin sequences, one that folds at the surface of cell membranes and the other that is intrinsically disordered. Although these peptides can effectively deliver different types of cargo, their use in protein delivery has been hindered due to the presence of non-natural D-proline within the central turn region of both sequences, which prohibits functionalizing proteins with the CPPs via standard expression protocols. In this work, we describe new CPPs that replace the non-natural turn region with natural turn motifs amenable to protein expression. We first investigate how these changes within the turn affect various CPP-related properties in the absence of protein cargo, and then generate protein fusions for intracellular delivery.
Keywords: cell-penetrating peptides, protein delivery, β-hairpin
1 |. INTRODUCTION
Cell-penetrating peptides (CPPs) are promising compounds that can deliver membrane-impermeable therapeutics into cells for improved efficacy.[1–6] The mechanism in which CPPs enter cells can be affected by both the peptide sequence and the type of attached cargo, with common modes of transport involving direct translocation through the outer membrane or various forms of endocytosis.[4,7–9] Although they have different intended functions, lytic anticancer and antimicrobial peptides often share many features with CPPs, such as amphiphilicity, high positive charge content, and the ability to adopt secondary structure when engaging with negatively charged surfaces.[10–16] For example, our lab previously developed the anticancer peptide SVS1, which acts through a lytic mechanism dependent on the peptide’s ability to bind to negatively charged components of cell surfaces and fold into β-hairpin-rich structures that disrupt the membrane.[17–19] Later studies demonstrated that when administered below its cytotoxic concentration, SVS1 could act as a CPP and effectively deliver small molecule cargo,[20] gold nanocolloids,[21] and quantum dots[22] into cells. Furthermore, when SVS1 was conjugated to the chemotherapeutic Paclitaxel, it provided improved in vivo antitumor activity over treatment with free drug.[20] The cell entry mechanism for SVS1 was similar to many common CPPs, involving a mixture of endocytosis and physical translocation. Continued efforts led to the development of a new class of intrinsically disordered CPPs.[23] The optimized peptide CLIP6 had several improved features over SVS1, such as reduced cytotoxicity and a mechanism for small molecule delivery based solely on direct translocation across the cell membrane. The key component of CLIP6 that lead to these improvements was the replacement of a valine residue with a negatively charged glutamate, which disrupted an otherwise perfect hydrophobic face within an amphiphilic β-hairpin sequence and prevented the peptide from folding at the cell membrane.[24] While this glutamate substitution afforded several benefits, the introduction of a negatively charged residue reduced CLIP6’s overall cell uptake, indicating that further improvement within this class of CPP would be desirable. In addition to their intriguing uptake properties, hairpin-based CPPs often have improved stability toward proteolysis over linear sequences,[20,23,25] which would be advantageous for in vivo delivery applications.
In this report, we use our family of CPPs to bring proteins into cells. Many, if not all protein therapeutics on the market today interact with extracellular targets. Although these types of therapies have had tremendous success, the ability to target intracellular interactions efficiently would be a transformative accomplishment and we wondered whether SVS1 or CLIP6 could be useful starting points in designing CPPs capable of delivering proteins. For practical purposes, we sought to develop a system where the CPP would be fused to a protein of interest via standard protein expression rather than using chemical,[26] enzymatic,[27,28] or NCL-based conjugation methods,[29,30] which all add additional synthetic steps that require optimization. However, both SVS1 and CLIP6 contain a non-natural D-residue within their central β-turn regions that prohibit standard expression. Therefore, we designed analogs of these CPPs that replaced the original turn residues with naturally occurring turn motifs (Figure 1).[31] We first synthesized the peptides by solid phase synthesis to evaluate how the turn residues affect CPP-related properties in the absence of protein cargo. Gratifyingly, these expression-capable SVS1 and CLIP6 analogs (termed XS and XC, respectively) retained CPP activity and demonstrated a diverse range of cytotoxicity and uptake behavior dependent on their capabilities to fold into β-hairpin conformations. We then fused the CPPs with EGFP via bacterial expression and demonstrated that intracellular protein delivery was possible.
FIGURE 1.
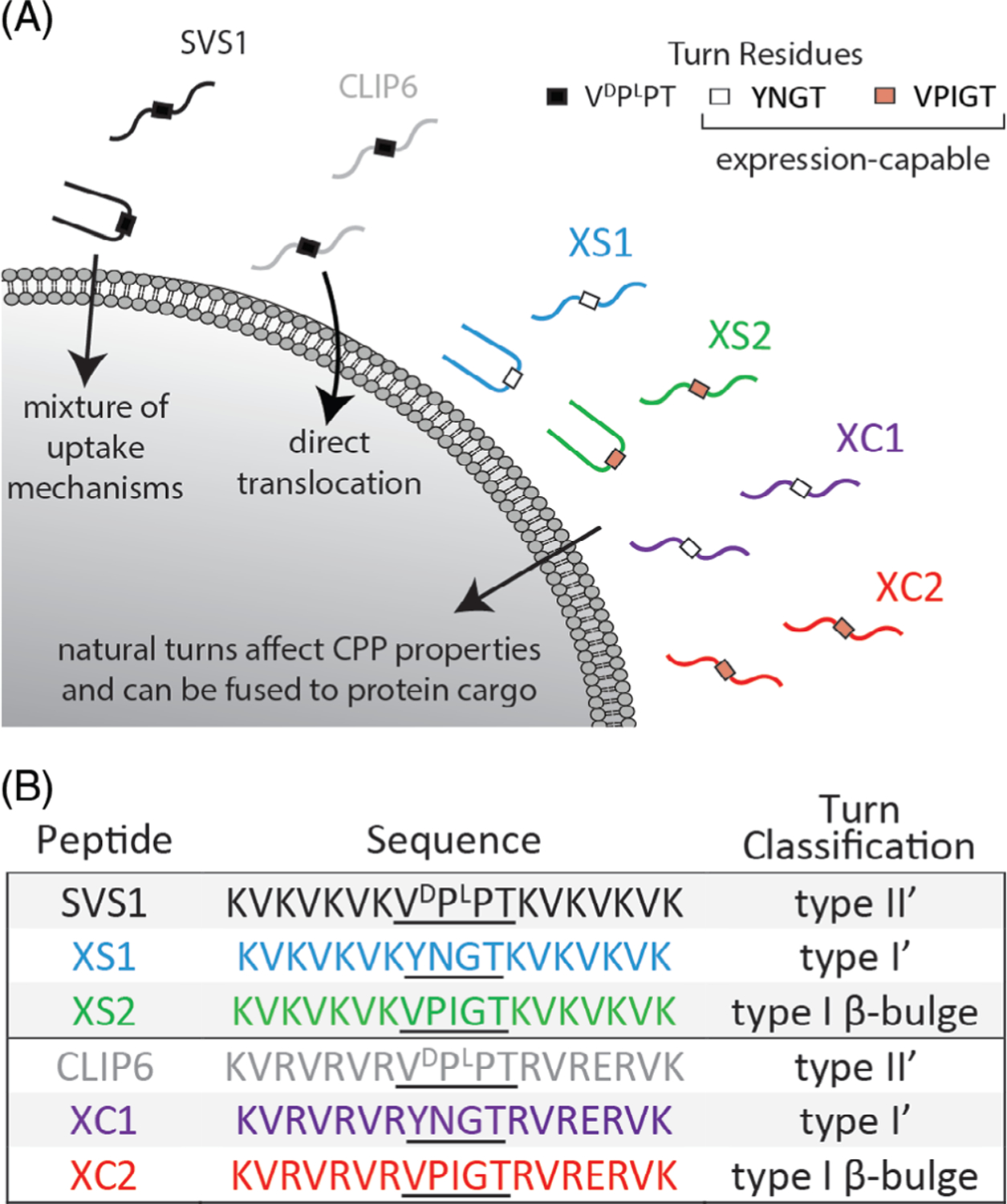
Expression-capable analogs of SVS1 (XS) and CLIP6 (XC). A, SVS1 folds at cell surfaces and enters through a mixture of uptake mechanisms, but can be lytic. CLIP6 is intrinsically disordered, is less toxic, and delivers small molecules through direct translocation. Analogs of SVS1 and CLIP6, utilizing natural turn residues, have altered CPP properties and can be fused to protein cargo through expression. B, Sequences for SVS1, CLIP6, and natural turn containing analogs
2 |. MATERIALS AND METHODS
2.1 |. Materials
All chemicals, reagents, buffers, and media were obtained from commercial vendors. POPC and POPS lipids were purchased from Avanti Polar Lipids. Gateway BP/LR cloning kits and DH5α subcloning efficiency cells were purchased from Invitrogen. Rosetta 2 (DE3) pLysS was purchased from EMD Millipore. DNA primers were purchased from Integrated DNA Technologies. PCR kits and minipreps were purchased from Qiagen. The initial EGFP-containing plasmid and the empty pDonr253, pDest527, and pDest566 vectors were obtained from the Protein Expression Laboratory of the Frederick National Laboratory for Cancer Research.
2.2 |. Peptide synthesis and purification
All peptides were synthesized on an ABI 433a automated peptide synthesizer using rink amide resin at a 0.25 mmol scale. Fmoc deprotection was performed using 19% piperidine/1% DBU in NMP for 5 minutes cycles and was monitored to completion through conductivity measurements after each cycle. Fmoc amino acids (5 eq) were dissolved in NMP, activated by HCTU (4.5 eq) in DMF and DIEA (10 eq), and coupled to the resin for 30 minutes with constant vortexing. At the end of the synthesis, aliquots of resin were removed (~0.025 mmol) and reacted with NHS-fluorescein (2 eq) in DMF on a manual wrist-action shaker overnight. After washing and drying the peptides on resin, they were cleaved from resin for 3 hours (95% TFA, 2.5% TIPS, and 2.5% H2O), after which the resin was filtered off, and the collected filtrate was concentrated. Following ether precipitation, crude solids were dried under vacuum. All peptides were purified by prep-HPLC. Crude peptides were dissolved in Std A (0.1% TFA in Milli-Q water; 4 mg/mL for unlabeled peptides, 2 mg/mL for fluorescently-labeled peptides) and 5 mL per run was injected into a Waters 600 system, equipped with a Waters 2489 UV Detector and a Vydac 218TP column (250 × 22 mm, 10 μm, heated to 40 ○C). A gradient of 1% Std B (80% MeCN, 0.1% TFA, and 19.9% Milli-Q water) per min was used. The UV trace at 220 nm was monitored and fractions containing the major peptide peak were combined and lyophilized. Purity was assessed by LCMS (Shimadzu LCMS 2020) and analytical HPLC (Agilent 1200 series, Vydac 218TP column, 250 × 4.6 mm, 5 μm, heated to 40 ○C) using gradients of 1% Std B per min.
2.3 |. Plasmid generation for EGFP and CPP-fused analogs
Primers encoding N-terminal cleavage domain (either FXa or TEV protease sites to remove His tag) and C-terminal CPP domains underwent overlap-extension PCR using the Qiagen Fast Cycling PCR kit with an EGFP-containing plasmid as the template. PCR products were purified using the QIAquick PCR purification kit, then subjected to Gateway BP cloning using the pDonr253 donor vector, transformed into DH5α sub-cloning efficiency cells, and plated on LB agar plates containing 50 μg/mL spectinomycin. The plates were grown overnight at 37 ○C. Single colonies were picked and grown overnight in 10 mL LB media with 50 μg/mL spectinomycin at 37 ○C, 250 RPM. Minipreps were performed to isolate amplified entry vectors. Once the sequences were validated (using Macrogen standard sequencing), the vectors were subjected to Gateway LR cloning using the pDest527 (N-terminal His tag) or pDest566 (N-terminal His-MBP tag) vector, transformed into DH5α subcloning efficiency cells, and plated on LB agar plates containing 100 μg/mL ampicillin. The plates were grown overnight at 37 ○C. Single colonies were picked and grown overnight in 10 mL LB media with 100 μg/mL ampicillin at 37 ○C, 250 RPM. Minipreps were performed to isolate amplified expression vectors and the sequences were validated prior to expression. When not in use, vectors were stored at −20 ○C.
2.4 |. Protein expression, purification, and His-tag removal
Expression vectors were transformed into Rosetta 2 (DE3) pLysS competent cells and plated on LB agar containing 100 μg/mL carbenicillin and 34 μg/mL chloramphenicol. The plates were grown overnight at 37 ○C. Single colonies were picked and grown overnight in 20 mL LB media, containing 100 μg/mL carbenicillin and 34 μg/mL chloramphenicol, at 37 ○C, 250 RPM. 5 mL of overnight culture was used to seed 500 mL of LB (100 μg/mL carbenicillin and 34 μg/mL chloramphenicol), and the culture was shaken at 37 ○C, 250 RPM. OD600 was monitored until reaching a value of 0.6–0.8, usually around 2–2.5 hours. At this point, the incubator temperature was reduced to 17 ○C and the flasks were cooled for 15 minutes. To induce expression, IPTG was added to a final concentration of 0.5 mM and the flasks were shaken at 17 ○C, 250 RPM for 20 hours. Each flask was split into 2 × 250 mL centrifuge bottles and spun down for 20 minutes at 7000 g. The supernatant was removed, and the pellets were stored at −80 ○C until purification. Pilot expressions had first been conducted to optimize conditions on a small scale at varied temperature/time (37/17 ○C for 6/20 hours respectively) and IPTG concentration (1/0.5/0.25 mM), resulting in the conditions used above.
Pellets (from 500 mL culture) were resuspended in 20 mL lysis buffer (50 mM sodium phosphate pH 8.0, 100 mM NaCl, 10 mM imidazole, 1x Bug Buster, and 1 EDTA-free protease inhibitor tablet) and subjected to sonication (amplitude setting of 1, 1 second on, 1 second off, 1.5 minutes total on). The lysate was then centrifuged at 4 ○C, 7000 g for 20 minutes and the supernatant was syringe filtered (0.45 μm). Using a GE P-960 sample pump, the lysate was injected into a GE ATKA FPLC system equipped with a 5 mL Talon resin column. Purification was carried out using a step gradient of 0/20/60/100% B (Buffer A: 50 mM sodium phosphate pH 8.0, 100 mM NaCl, 10 mM imidazole; Buffer B: 50 mM sodium phosphate pH 8.0, 100 mM NaCl, 100 mM imidazole). Fractions were monitored for absorbance at 280/488 nm (EGFP), typically eluting at 60 and/or 100% B. Purity was confirmed by SDS PAGE gel, with the pure fractions combined and subjected to several rounds of buffer exchanges into 50 mM sodium phosphate pH 8.0, 100 mM NaCl using Amicon Ultra-15 centrifugal filters (MWCO 10 kDa) to achieve sub-micromolar imidazole concentrations. The concentrated protein was diluted to 1.5 mL, and the concentration was determined by UV (EGFP: ε = 56 000 M−1 cm−1 at 488 nm). Typical yields for His-FXa-EGFP were over 50 mg per L culture, whereas the His-TEV-EGFP-XC1/2 analogs had yields up to 10 mg per L culture. The His-TEV-EGFP-XS1/2 sequences had pIs (~8) close to the pH needed for optimal purification, which made them difficult to solubilize after cell lysis. Additionally, the XS1 analogs did not produce fluorescent pellets. Therefore, His-MBP-TEV-EGFP-XS1/2 constructs were made to improve solubility and activity. His-MBP-TEV-EGFP-XS2 led to yields of ~10 mg per L culture. Unfortunately, the XS1 analog still did not produce a fluorescent pellet indicating that folded and functional protein was not produced.
For removal of the N-terminal His-6 tag encoded in the pDest527 expression vector, FXa or TEV proteases were employed. For the generation of free EGFP, FXa cleavage was used. A typical experiment would use 5 mg (determined by UV) of His-FXa-EGFP in 20 mL of cleavage buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 2 mM CaCl2) and 100 μg of FXa (protein:enzyme weight ratio of ~50:1). The reaction was monitored by SDS PAGE gel and was typically completed within 7 hours. The reaction solution was added to prewashed p-aminobenzamidine functionalized resin to bind FXa, allowed to rock at RT for 30 minutes, and then the resin was filtered using an empty polypropylene column. EGFP was exchanged in to Milli-Q water using Amicon Ultra-15 filters (MWCO 10 kDa) and typically achieved near quantitative yields (~4.5 mg from 5 mg NH6 fusion). The CPP domains were susceptible to nonspecific cleavage by FXa, and a TEV cleavage site was installed to circumvent this. The protocol for cleavage by Halo-TEV protease (Promega) followed that of the manufacturer. A typical experiment would use 3 mg (determined by UV) of NH6-TEV-ID#-EGFP/mRuby3 in 3 mL buffer (50 mM phosphate, pH 8.0, 100 mM NaCl), 1 mM DTT, and 10 μL HaloTEV protease. The reactions were monitored by SDS PAGE gel and were typically completed under 7 hours. The completed reaction solution was added to prewashed HaloLink resin for HaloTEV removal. After slowly rocking at RT for 30 minutes, the resin was filtered using an empty polypropylene column. The EGFP-CPPs were exchanged into buffer (50 mM phosphate, pH 8.0, 100 mM NaCl) using Amicon Ultra-15 filters to reduce DTT concentration to sub-micromolar levels and to remove the low molecular weight cleavage byproduct. Yields of around 80% were typically achieved (~2.0 mg). After the cleavage of His-MBP-TEV-EGFP-XS2, the solution was also passed through 0.5 mL Talon resin to remove the cleaved His-MBP domain, as this byproduct is too large for removal by dialysis. Additional buffer exchanges were necessary to remove excess imidazole from EGFP-XS2.
2.5 |. Circular dichroism (CD) spectroscopy to assess peptide folding
CD spectra were collected on an Aviv Model 420 Spectrometer, using 50 μM peptide in either buffer alone (50 mM BTP, 150 mM KF, pH 7.4), buffer containing POPC liposomes, or a 1:1 ratio of POPC: POPS liposomes (lipid:peptide ratio of 50:1). Liposomes were prepared and extruded to ~100 nm as previously described.[17] The sample, in a 1 mm pathlength cuvette, was placed into the instrument’s pre-heated sample holder (37 ○C) and equilibrated for 5 minutes. Wavelength scans were then carried out at 37 ○C between 260 and 200 nm at 1 nm intervals, with an averaging time of 3 seconds at each wavelength. A background spectrum of blank buffer was subtracted from the sample spectra and the mean residue ellipticity [θ] was calculated from the equation [θ]= (θobs/10lc)/r, where θobs is the measured ellipticity in millidegrees, l is the length of the cell in centimeters (0.1 cm), c is the molar concentration (0.00005 M), and r is the number of peptide bonds (18).
2.6 |. Cell viability experiments in the presence of CPPs
A549 cells were grown in T75 flasks using complete RPMI media supplemented with 10% FBS, and incubated at 37 ○C with 5% CO2 and humidity control. Prior to experiments, cells were trypsinized with 3 mL 0.25% trypsin-EDTA solution for 5 minutes, diluted with media, pelleted by centrifugation, and reconstituted in media. The viable cells were counted using trypan blue staining and the cells were diluted to 5 × 104 cells/mL. 100 μL of diluted cells was pipetted into wells of a clear 96 well plate (5000 cells per well), then grown overnight (~16 hours). Serial dilutions of each peptide were made in serum-free RPMI. The overnight culture media was removed from the cells and replaced with the peptide-containing media and incubated for 24 hours. Cells were also incubated in media without peptide and media with 20% DMSO, as negative and positive controls, respectively. After a day, the media was removed and the cells were washed with 100 μL media. After removal of the wash, 100 μL media was added to cells, plus 10 μL of MTT solution (5 mg/mL in Milli-Q water). The samples were incubated for 3 hours, after which the media was removed and 150 μL of spectroscopic grade DMSO was added. The plates were incubated at 37 ○C with light rocking for 15 minutes, then read at 540 nm using a UV plate reader (Biotek). The absorbance of the positive controls was subtracted from each sample as a blank, and percent viability was calculated using the equation: (Abstreated/ Absuntreated) × 100. IC50 values were computed using Graphpad Prism 8 software and represented the average of three independent experiments, each in triplicate (n = 9).
2.7 |. Cellular internalization of fluorescently-labeled peptides and EGFP-CPPs
For flow cytometry, A549 cells were diluted to 5 × 105 cells/mL and 0.5 mL was added to clear 24 well plates (2.5 × 105 cells per well) and incubated overnight (~16 hours) at 37 ○C with 5% CO2. Stocks of fluorescently-labeled peptide or protein were made in buffer and the concentrations were determined by UV absorbance (fluorescein: ε494 = 70 000 M−1; EGFP: ε488 = 56 000 M−1) and diluted to 5 μM in serum-free media. After removal of the overnight culture media, 0.5 mL peptide/protein solution was added to the wells and incubated for 1 hour. The cells were then washed with media and incubated with 150 μL of 0.25% trypsin-EDTA solution for 15 minutes. The cells were diluted with 0.5 mL cold PBS, transferred to 12 × 75 mm polystyrene tubes, placed on ice, and then analyzed by flow cytometry using a BD FACSCalibur instrument. To assess the role of energy dependent uptake mechanisms and clathrin-mediated endocytosis, experiments were also conducted under ATP depletion and hyperosmolar sucrose conditions, respectively. Prior to peptide/protein incubation, cells were pretreated with either with ATP depletion (glucose-free/serum-free RPMI, 10 mM sodium azide, and 50 mM 2-deoxy-D-glucose, 30 minutes) or hyperosmolar sucrose (serum-free RPMI, 0.45 M sucrose, 1 hour), followed by peptide/protein incubation in respective media. Mean fluorescence statistics for each peptide/protein were computed using Graphpad Prism 8 and significance values were determined by applying two-way ANOVA analysis.
For live cell imaging experiments, A549 cells were to diluted to 5 × 104 cells/mL in media and 2 mL was added to circular glass slides (1 × 105 cells per slide). The cells were incubated overnight at 37 ○C with 5% CO2. Stocks of EGFP and EGFP-XS2 were made, with concentrations determined by UV absorbance (ε488 = 56 000 M−1). Both proteins were diluted to 20 μM in serum-free media. After removal of the overnight culture media, 2 mL of the protein solutions were added to the slides and incubated for 5 hours. After 4.5 hours of incubation, 2 μL of 1 mg/mL Hoescht 33 342 dye was added to the slides, the solution was gently aspirated, and the samples were incubated for an additional 30 minutes. The cells were then washed with media and imaged using a Zeiss LSM710 Confocal Microscope with an environmental chamber to maintain 37 ○C and 5% CO2. Microscopy images were processed using ImageJ.
3 |. RESULTS AND DISCUSSION
3.1 |. Altering the turn residues of SVS1 and CLIP6 affect folding, cytotoxicity, and cell uptake mechanisms
SVS1 is a designed sequence with a strong propensity to fold into a β-hairpin conformation when bound to negatively charged surfaces, such as negatively charged membranes or the glycocalyx.[17,19] It contains two perfectly amphiphilic β-strands connected by a four-residue sequence –VDPPT– (Figure 1B), which is known to avidly adopt a type II׳ reverse turn and drive hairpin folding when an appropriate surface is engaged. Previous crystallographic studies using short model peptides have shown that this turn’s preferred dihedral angles result in the formation of a very regular β-hairpin.[32] CLIP6 also contains the central –VDPPT– sequence, but the introduction of a glutamate residue on the hydrophobic face prohibits its ability to adopt a hairpin conformation.[23] For XS1 and XC1, the non-natural turn was replaced by the four-residue sequence –YNGT–,[33] which is known to form a type I0 reverse turn that is somewhat less effective at nucleating hairpin structure than D-proline-containing turns.[34] XS2 and XC2 contain a five-residue sequence known to form a type I β-bulge reverse turn in proteins (Figure 1B), but this turn is less entropically favored in the context of short peptides when compared to the four-residue turns.[35] After synthesizing the peptides by solid phase synthesis (Figure S1), we first performed CD experiments to assess how the different turn motifs affect the secondary structure of the new CPPs. Studies were performed in buffer as well as in the presence of model liposomes to assess how membrane surface charge might influence folding. As we have seen previously with SVS1 and CLIP6,[17,23] all sequences adopt unfolded conformations in physiological buffer and in the presence of neutral POPC liposomes (Figure S2). However, when incubated with liposomes containing the negatively charged lipid POPS, SVS1 showed a spectrum consistent with a β-sheet-rich structure (Figure 2A). Many cancer cells have increased negative charge at their outer surface compared to noncancerous cells and this charge-induced folding behavior could impart selectivity over which cells the CPPs prefer to engage.[17] Both XS1 and XS2 adopt β-sheet structure, but afford slightly different spectra compared to SVS1. These differences are most likely due to both the differential contribution of the specific reverse turn structures to the far UV-CD and the ability of each turn to promote the folding of a canonical β-hairpin. For example, compared to SVS1, XS1 exhibits a spectrum that contains a degree of random coil contribution, which is even more prominent for XS2 (compare [θ] at 200 nm for each). As will be shown below, the different turn types not only influence hairpin structure but also function. With respect to CLIP6, XC1, and XC2, these peptides remain unfolded in the presence of negatively charged liposomes. Again, each of these sequences contains a glutamate on what would be an otherwise perfect hydrophobic face of a hairpin conformation if the peptides were to fold. The inclusion of this charged residue represents a significant energetic barrier to folding even under solution conditions that normally promote the formation of structure. Thus, the natural turn motifs did not alter the conformational bias of the glutamate-containing peptides and remain unfolded.
FIGURE 2.
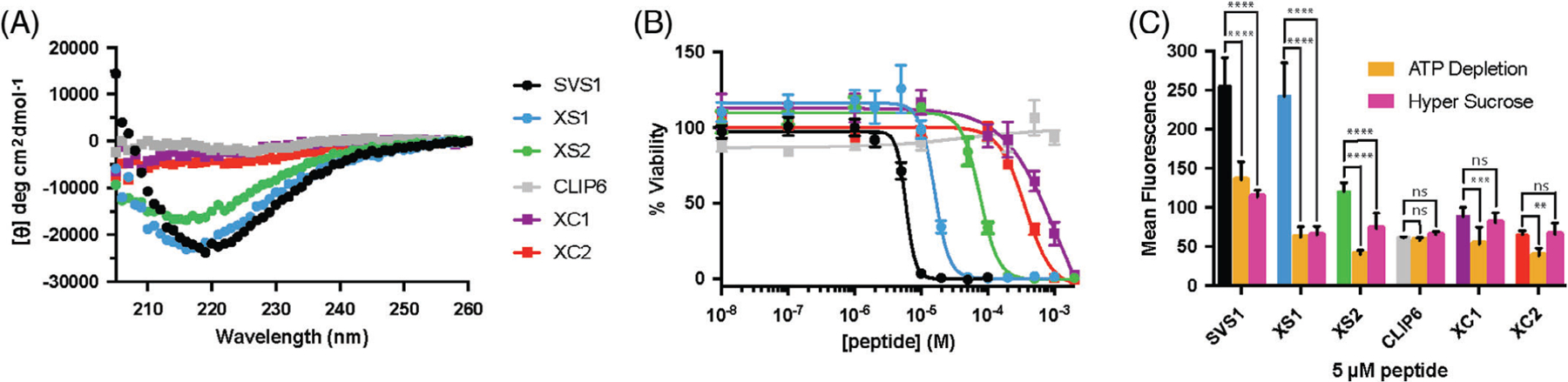
Turn residues affect folding and activity. A, CD spectra in the presence of negatively charged POPC/POPS liposomes. B, Cytotoxicity toward A549 cells. C, Uptake of fluorescein-tagged peptides in A549, with and without the presence of cell-uptake inhibitors. ****P < .0001; ***P < .001; **P < .01; ns = not significant. For interpeptide P values, see Table S1
Next, we investigated how the different turns affected the bioactivity of the peptides toward human lung adenocarcinoma (A549) cells. Most membrane-active peptides have the capacity to function as both cell penetrating and lytic agents, with peptide concentration dictating which function is enacted.[10] Cationic amphiphilic peptides can penetrate cells at low concentrations, but lyse membranes at higher concentrations. We first studied the lytic potential of the peptides in viability experiments, which demonstrated that cytotoxicity was closely linked to peptide conformation (Figure 2B). SVS1, which has the highest propensity to fold, was the most toxic (IC50 = 5 μM). XS1 and XS2 showed stepwise decreases in cytotoxicity (17 and 80 μM, respectively), in-line with their decreased propensities to adopt structure. Interestingly, although XC1 and XC2 were less toxic than SVS1 and its derivatives as expected due to their inability to fold (675 and 349 μM, respectively), they were still capable of killing cells at higher concentrations. This is in contrast to CLIP6, which was essentially nontoxic at the concentrations studied (>1 mM). This is intriguing because on one hand, the –VDPPT– turn motif was responsible for the increased toxicity of well-folded SVS1 compared to its lesser folded natural turn analogs, but that same turn sequence when incorporated into intrinsically disordered CLIP6 endeared nontoxic behavior relative to XC1 and XC2. Thus, it seems as if the glutamate-containing peptides, although all unfolded, still have the capacity to kill cells in a manner dependent on the residues within the turn region of the peptide. It may be that the ensemble of disordered states defining unfolded CLIP6 may be quite different than those of XC1 and XC2, perhaps due to the conformational bias imposed by the D-proline/L-proline motif, though this is only speculative at this point.
We next studied the cell penetrating properties of the peptides at concentrations below their lytic IC50 values. Flow cytometry experiments were conducted with fluorescently-labeled peptides to assess cellular uptake (Figure 2C, Table S1). In general, the SVS1 analogs had greater overall uptake than the CLIP6 analogs. This demonstrates that a well-folded conformation, which is more likely to engage with cell surfaces, also leads to more efficient CPPs. Within the same family of peptides, the –YNGT– turns (XS1/XC1) had greater uptake than the – VPIGT– turns (XS2/XC2), indicating that the local conformation imparted by the YNGT motif was more favorable for cell entry. Additionally, the different turns resulted in changes to the mechanisms of cell uptake, which were probed using ATP depletion[36] and hyperosmolar sucrose[37] conditions. ATP depletion shuts down energy-dependent processes involved in uptake, such as all forms of endocytosis and some forms of ion channels or transporters, whereas hyperosmolar sucrose conditions shut down clathrin-mediated endocytosis. XS1 had similar overall uptake as SVS1 in the absence of inhibitors, but showed an even greater reliance on clathrin-mediated uptake. XS2, which had lower uptake than both SVS1 and XS1, still had a noticeable portion of its uptake requiring energy-dependent processes, but had a lesser contribution from clathrin-mediated endocytosis. For the intrinsically disordered sequences, both XC1 and XC2 had modest increases in overall uptake compared to CLIP6, but required some energy-dependent process for cell entry that was not clathrin-mediated. Again, this signifies something unique about turn residues of CLIP6. In addition to being less toxic than its natural turn-containing counterparts, CLIP6 has energy-independent cell entry for small molecule delivery that is indicative of direct translocation across the cell membrane. This further supports the notion that the turn of CLIP6 is imparting some local conformational bias that is affecting activity. Nonetheless, the overall general trend appears to be that peptides capable of folding at the cell surface can better enter cells, but this same folding potential also increases their lytic behavior. Less-folded peptides are less toxic, but as CPPs they enter cells less efficiently and are more able to evade clathrin-mediated endocytosis, at least with respect to the delivery of small molecule cargo (such as fluorescein).
3.2 |. EGFP-CPP fusions can be generated and can enter cells
After evaluating the CPP properties of the natural turn-containing peptides in the absence of protein cargo, we then directed our efforts toward intracellular protein delivery using EGFP derivatives that contain these new sequences fused to their C-terminus (Figures 3A and S3). Fluorescent proteins are useful models for intracellular delivery because their uptake efficiency and localization can be easily monitored by flow cytometry and confocal microscopy.[26,38–40] The initial unmodified EGFP vector we obtained contained a noncleavable His-tag at the protein’s N-terminus (EGFP within the pDest527 vector). Since poly-His has been shown to have mild CPP behavior,[41] His-tag removal would allow for a more accurate assessment of our CPPs for mediating protein uptake. Therefore, in addition to the C-terminal CPP domains, we also incorporated an N-terminal protease cleavage site for His-tag removal. Pilot expressions were conducted to find optimized conditions, with reduced temperature (17 ○C) and IPTG concentrations (0.5 mM) typically leading to the greatest yields of fluorescently active protein. The expression of His-EGFP-XC analogs led to purified yields of up to 10 mg per L culture. The EGFP-XS analogs required further optimization because the initial vector used for expression (pDest527) resulted in His-tagged constructs with pIs close to the optimal pH (~8) needed for immobilized metal ion affinity chromatography (IMAC). This prohibitively reduced the EGFP-XS solubility after bacterial cell lysis (Figure S4A). To circumvent this issue, we then cloned the XS sequences into a His-MBP-containing vector (pDest566) for improved solubility (Figures S3 and S4B), which afforded His-MBP-EGFP-XS2 in yields of ~10 mg per L culture. However, the EGFP-XS1 constructs were unable to produce acceptable yields of soluble, fluorescently active material. We hypothesize that this is due to XS1’s increased folding capability and subsequent lytic behavior when compared to the other CPPs containing natural turns. Since fusions containing lytic peptide domains can hinder bacterial cell growth and overall protein production, these troublesome sequences often require post-expression conjugation for attaching to proteins of interest.[29,42,43] Additionally, EGFP folding and maturation is a relatively slow process.[44,45] It is possible that after protein expression, sequences with strong β-sheet propensities like XS1 could interpolate into the fluorescent protein domain and disrupt formation of the proper β-barrel structure. This would not only inhibit chromophore maturation and fluorescence, but could also lead to aggregation induced insolubility. Although we could not generate EGFP-XS1, milligram quantities of protein were readily obtained for the other EGFP-CPP conjugates and all underwent efficient His-tag removal (Figure 3B).
FIGURE 3.
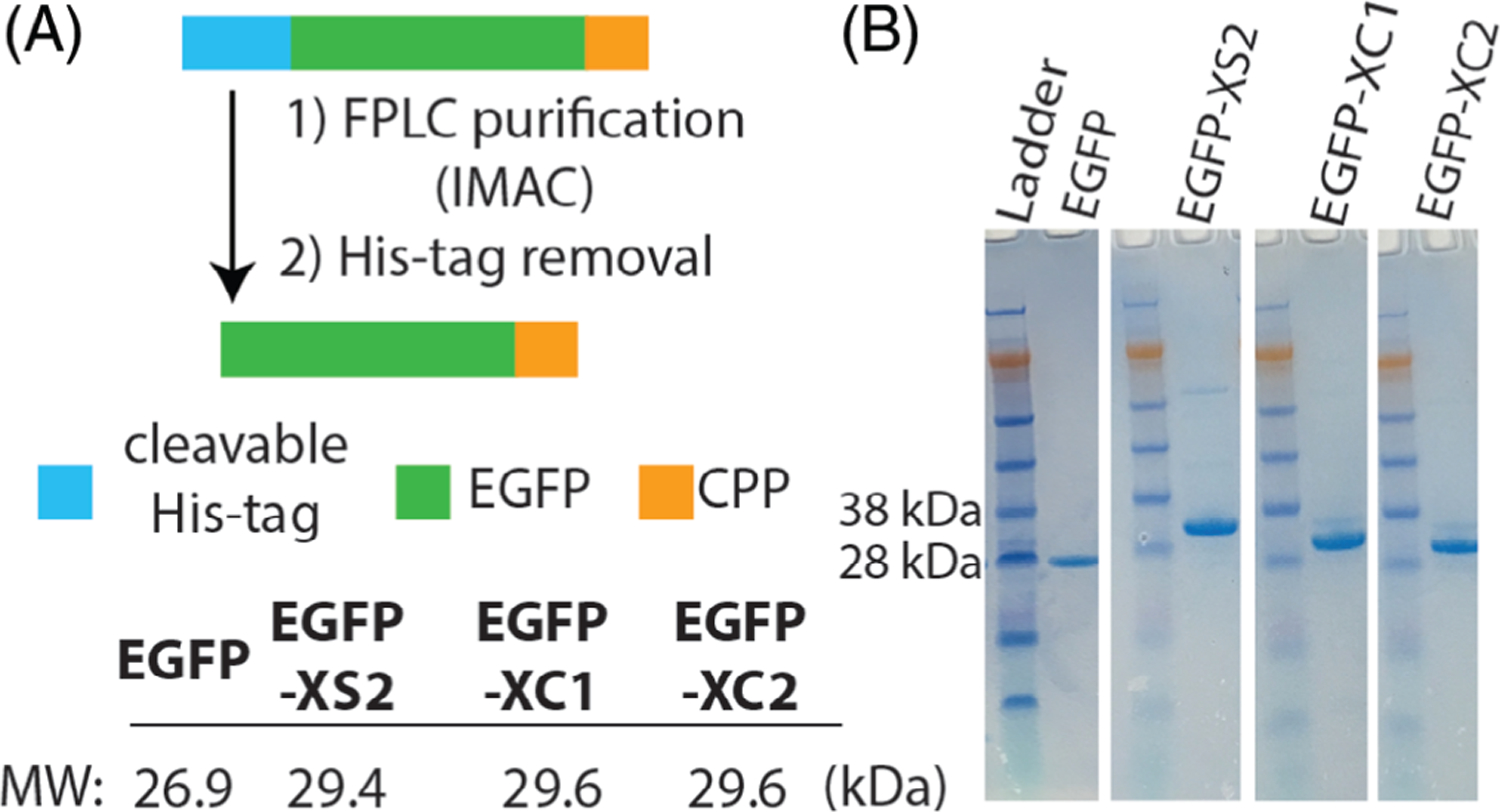
Generation of EGFP-CPP analogs. A, Proteins are expressed from E. coli with an N-terminal His-tag, that is cleaved following immobilized metal ion affinity chromatography (IMAC). B, SDS-PAGE gels of purified proteins after His-tag removal
We then conducted flow cytometry to quantify CPP-mediated protein delivery into A549 cells. XS2 was significantly better at delivering EGFP than either of the two XC analogs (Figure 4A, Table S2). This suggests that within our family of β-hairpin-inspired CPPs, sequences that adopt folded conformations and that more readily engage the cell surface are most suited for macromolecular delivery.[21,22] All of the EGFP-CPP conjugates were more dependent on clathrin-mediated endocytosis than the analogous peptides tagged with a small molecule fluorescent dye (Figure 2C), which highlights the major hurdle for therapeutic translation of many CPPs with respect to macromolecular delivery. Intracellular protein delivery was studied further for EGFP-XS2 using confocal microscopy (Figure 4B). Substantial amounts of protein were localized within punctate vesicles, indicative of endocytic uptake, with weakly diffuse fluorescence demonstrating that some protein reached the cytoplasm. In contrast, EGFP void of the CPP fusion was not taken up by the cells, indicating that the CPP was crucial for delivery. These results show that even though endosomal entrapment is a concern, intracellular protein delivery can be accomplished with our β-hairpin-inspired CPPs and lays the foundation for future designs that could improve these up efforts.
FIGURE 4.
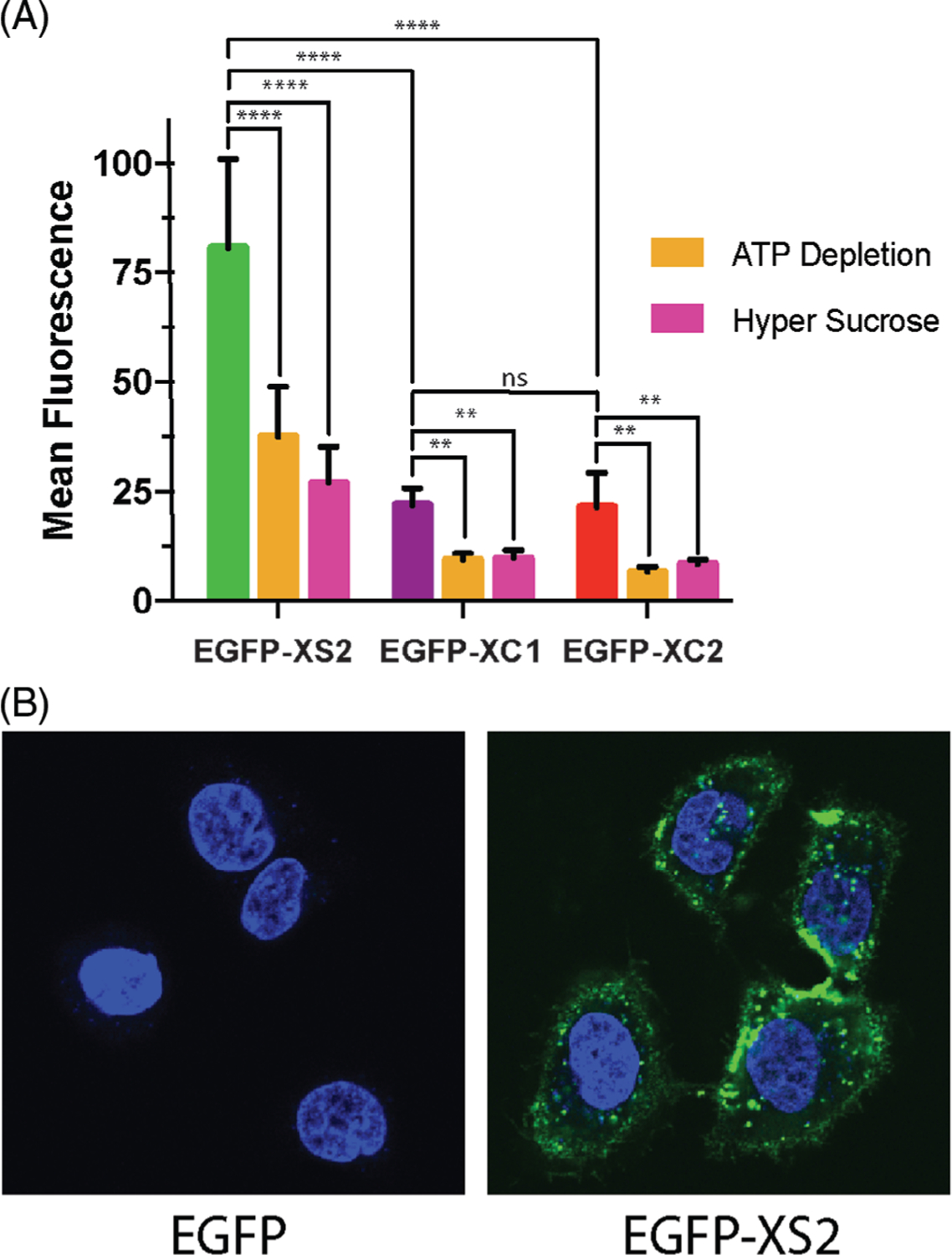
Assessment of protein delivery into A549 cells by (A) flow cytometry and (B) confocal microscopy. ****P < .0001; ***P < .001; **P < .01; ns = not significant
4 |. CONCLUSIONS
We have taken two previously developed CPPs and modified the composition of their central β-turn domain to replace the non-natural sequence (containing D-proline) with natural turns that are amenable to standard protein expression. These changes within the turn region not only affect the way the peptides fold and interact with the surface of negatively charged membranes, but ultimately impact a variety of important CPP-related properties such as cytotoxicity, total uptake, and mechanism of cell entry. We then designed and generated expression constructs that directly fused the natural turn-containing CPPs to EGFP and demonstrated intracellular protein delivery. As one might expect, the uptake mechanisms for these new CPPs differ depending on whether the attached cargo is a small molecule or a protein, with the later even more reliant on endocytosis. Intuitively, this seems reasonable since it should be difficult for a large protein to enter a cell via direct translocation without compromising cell membrane integrity. In general, endosomal entrapment limits the therapeutic potential of most CPP-based delivery approaches. One possible strategy forward may include the incorporation of domains that facilitate rapid escape from endosomes.[36,46,47] In addition to these possible design modifications, we are currently working toward sequence optimizations within our family of CPPs that reduce the toxicity of our well-folded β-hairpins (SVS1-like derivatives) and improve the overall uptake of the intrinsically disordered peptides (CLIP6-like derivatives), both of which could further improve the efficiency of cytosolic protein delivery.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health. We would like to thank Dr. Dominic Esposito for kindly providing the initial unmodified EGFP plasmid and the empty cloning vectors. S.E.M. would also like to thank Dr. Cem Somnez and Dr. Scott Medina for useful discussions and guidance at the outset of these studies.
Funding information
National Cancer Institute, Grant/Award Number: 1ZIABC011313–06
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- [1].Frankel AD, Pabo CO, Cell 1988, 55, 1189. [DOI] [PubMed] [Google Scholar]
- [2].Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A, Proc. Natl. Acad. Sci 1991, 88, 1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Johnson RM, Harrison SD, Maclean D, in Cell-Penetrating Peptides: Methods and Protocols (Ed: Langel Ü), Humana Press, Totowa, NJ: 2011, p. 535. [DOI] [PubMed] [Google Scholar]
- [4].Copolovici DM, Langel K, Eriste E, Langel Ü, ACS Nano 2014, 8, 1972. [DOI] [PubMed] [Google Scholar]
- [5].Rhodes CA, Pei D, Chem. Eur. J 2017, 23, 12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sánchez-Navarro M, Teixidó M, Giralt E, Acc. Chem. Res 2017, 50, 1847. [DOI] [PubMed] [Google Scholar]
- [7].Jones AT, Sayers EJ, Control Release J 2012, 161, 582. [DOI] [PubMed] [Google Scholar]
- [8].Futaki S, Nakase I, Acc. Chem. Res 2017, 50, 2449. [DOI] [PubMed] [Google Scholar]
- [9].Walrant A, Cardon S, Burlina F, Sagan S, Acc. Chem. Res 2017, 50, 2968. [DOI] [PubMed] [Google Scholar]
- [10].Henriques ST, Melo MN, Castanho MARB, Biochem. J 2006, 399, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Almeida PF, Pokorny A, Biochemistry 2009, 48, 8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bobone S, Piazzon A, Orioni B, Pedersen JZ, Nan YH, Hahm K-S, Shin SY, Stella L, J. Pept. Sci 2011, 17, 335. [DOI] [PubMed] [Google Scholar]
- [13].Bahnsen JS, Franzyk H, Sandberg-Schaal A, Nielsen HM, Biochim. Biophys. Acta 2013, 1828, 223. [DOI] [PubMed] [Google Scholar]
- [14].Gaspar D, Veiga AS, Castanho MARB, Front. Microbiol 2013, 4, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rodriguez Plaza JG, Morales-Nava R, Diener C, Schreiber G, Gonzalez ZD, Lara Ortiz MT, Ortega Blake I, Pantoja O, Volkmer R, Klipp E, Herrmann A, Del Rio G, J. Biol. Chem 2014, 289, 14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ponnappan N, Budagavi DP, Chugh A, Biochim. Biophys. Acta 2017, 1859, 167. [DOI] [PubMed] [Google Scholar]
- [17].Sinthuvanich C, Veiga AS, Gupta K, Gaspar D, Blumenthal R, Schneider JP, J. Am. Chem. Soc 2012, 134, 6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gaspar D, Veiga AS, Sinthuvanich C, Schneider JP, Castanho MARB, Biochemistry 2012, 51, 6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ishikawa K, Medina SH, Schneider JP, Klar AJS, Cell Chem. Biol 2017, 24, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Medina SH, Schneider JP, J. Control Release 2015, 209, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kapur A, Medina SH, Wang W, Palui G, Schneider JP, Mattoussi H, ACS Omega 2018, 3, 12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kapur A, Medina SH, Wang W, Palui G, Ji X, Schneider JP, Mattoussi H, ACS Omega 2018, 3, 17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Medina SH, Miller SE, Keim AI, Gorka AP, Schnermann MJ, Schneider JP, Angew. Chem. Int. Ed 2016, 55, 3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Akishiba M, Takeuchi T, Kawaguchi Y, Sakamoto K, Yu H-H, Nakase I, Takatani-Nakase T, Madani F, Gräslund A, Futaki S, Nat. Chem 2017, 9, 751. [DOI] [PubMed] [Google Scholar]
- [25].Safa N, Anderson JC, Vaithiyanathan M, Pettigrew JH, Pappas GA, Liu D, Gauthier TJ, Melvin AT, Pept. Sci 2019, 111, e24092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nischan N, Herce HD, Natale F, Bohlke N, Budisa N, Cardoso MC, Hackenberger CPR, Angew. Chem. Int. Ed 2015, 54, 1950. [DOI] [PubMed] [Google Scholar]
- [27].Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AEM, Ploegh HL, Nat. Protoc 2013, 8, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Theile CS, Witte MD, Blom AEM, Kundrat L, Ploegh HL, Guimaraes CP, Nat. Protoc 2013, 8, 1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yu H-H, Nakase I, Pujals S, Hirose H, Tanaka G, Katayama S, Imanishi M, Futaki S, Biochim. Biophys. Acta 2010, 1798, 2249. [DOI] [PubMed] [Google Scholar]
- [30].Herce HD, Schumacher D, Schneider AFL, Ludwig AK, Mann FA, Fillies M, Kasper M-A, Reinke S, Krause E, Leonhardt H, Cardoso MC, Hackenberger CPR, Nat. Chem 2017, 9, 762. [DOI] [PubMed] [Google Scholar]
- [31].Sonmez C, Nagy KJ, Schneider JP, Biomaterials 2015, 37, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Aravinda S, Raghavender US, Rai R, Harini VV, Shamala N, Balaram P, Org. Biomol. Chem 2013, 11, 4220. [DOI] [PubMed] [Google Scholar]
- [33].Griffiths-Jones SR, Maynard AJ, Sharman GJ, Chem. Commun 1998, 789. [Google Scholar]
- [34].Stanger HE, Gellman SH, J. Am. Chem. Soc 1998, 120, 4236. [Google Scholar]
- [35].Blandl T, Cochran AG, Skelton NJ, Protein Sci 2003, 12, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wadia JS, Stan RV, Dowdy SF, Nat. Med 2004, 10, 310. [DOI] [PubMed] [Google Scholar]
- [37].Massodi I, Bidwell GL 3rd, Raucher D, J. Control Release 2005, 108, 396. [DOI] [PubMed] [Google Scholar]
- [38].Caron NJ, Torrente Y, Camirand G, Bujold M, Chapdelaine P, Leriche K, Bresolin N, Tremblay JP, Mol. Ther 2001, 3, 310. [DOI] [PubMed] [Google Scholar]
- [39].McNaughton BR, Cronican JJ, Thompson DB, Liu DR, Proc. Natl. Acad. Sci 2009, 106, 6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Andersen KA, Smith TP, Lomax JE, Raines RT, ACS Chem. Biol 2016, 11, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Iwasaki T, Tokuda Y, Kotake A, Okada H, Takeda S, Kawano T, Nakayama Y, J. Control Release 2015, 210, 115. [DOI] [PubMed] [Google Scholar]
- [42].Takeshima K, Chikushi A, Lee K-K, Yonehara S, Matsuzaki K, J. Biol. Chem 2003, 278, 1310. [DOI] [PubMed] [Google Scholar]
- [43].Guimaraes CP, Carette JE, Varadarajan M, Antos J, Popp MW, Spooner E, Brummelkamp TR, Ploegh HL, Cell Biol J. 2011, 195, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cormack BP, Valdivia RH, Falkow S, Gene 1996, 173, 33. [DOI] [PubMed] [Google Scholar]
- [45].Hsu S-TD, Blaser G, Jackson SE, Chem. Soc. Rev 2009, 38, 2951. [DOI] [PubMed] [Google Scholar]
- [46].Zhang P, Monteiro da Silva G, Deatherage C, Burd C, DiMaio D, Cell 2018, 174, 1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pei D, Buyanova M, Bioconjug. Chem 2019, 30, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.