

Abstract
Background
Juvenile forms of parkinsonism are rare conditions with onset of bradykinesia, tremor and rigidity before the age of 21 years. These atypical presentations commonly have a genetic aetiology, highlighting important insights into underlying pathophysiology. Genetic defects may affect key proteins of the endocytic pathway and clathrin‐mediated endocytosis (CME), as in DNAJC6‐related juvenile parkinsonism.
Objective
To report on a new patient cohort with juvenile‐onset DNAJC6 parkinsonism‐dystonia and determine the functional consequences on auxilin and dopamine homeostasis.
Methods
Twenty‐five children with juvenile parkinsonism were identified from a research cohort of patients with undiagnosed pediatric movement disorders. Molecular genetic investigations included autozygosity mapping studies and whole‐exome sequencing. Patient fibroblasts and CSF were analyzed for auxilin, cyclin G–associated kinase and synaptic proteins.
Results
We identified 6 patients harboring previously unreported, homozygous nonsense DNAJC6 mutations. All presented with neurodevelopmental delay in infancy, progressive parkinsonism, and neurological regression in childhood. 123I‐FP‐CIT SPECT (DaTScan) was performed in 3 patients and demonstrated reduced or absent tracer uptake in the basal ganglia. CSF neurotransmitter analysis revealed an isolated reduction of homovanillic acid. Auxilin levels were significantly reduced in both patient fibroblasts and CSF. Cyclin G–associated kinase levels in CSF were significantly increased, whereas a number of presynaptic dopaminergic proteins were reduced.
Conclusions
DNAJC6 is an emerging cause of recessive juvenile parkinsonism‐dystonia. DNAJC6 encodes the cochaperone protein auxilin, involved in CME of synaptic vesicles. The observed dopamine dyshomeostasis in patients is likely to be multifactorial, secondary to auxilin deficiency and/or neurodegeneration. Increased patient CSF cyclin G–associated kinase, in tandem with reduced auxilin levels, suggests a possible compensatory role of cyclin G–associated kinase, as observed in the auxilin knockout mouse. DNAJC6 parkinsonism‐dystonia should be considered as a differential diagnosis for pediatric neurotransmitter disorders associated with low homovanillic acid levels. Future research in elucidating disease pathogenesis will aid the development of better treatments for this pharmacoresistant disorder. © 2020 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: auxilin, DNAJC6, dopamine, dystonia, parkinsonism
Classical Parkinson's disease (PD) is an age‐related neurodegenerative disorder, mainly affecting adults aged >50 years. Patients typically present with resting tremor, bradykinesia, rigidity, and postural instability. To date, a number of early‐onset genetic forms of PD (Parkin, PINK1, and DJ‐1)1, 2, 3 and complex parkinsonism syndromes (ATP13A2, PLA2G6, FBXO7, SLC6A3, SLC39A14, and PANK2) have been described.4, 5, 6, 7, 8, 9 Importantly, the study of such monogenic forms of disease have provided significant insight into the pathogenic mechanisms underlying sporadic PD.10 More recently, two genes, namely SYNJ1 11 and DNAJC6,12, 13, 14 encoding proteins involved in postendocytotic recycling of synaptic vesicles, have been identified in early‐onset parkinsonism.
In this study, we report on 6 children from three families, presenting with juvenile parkinsonism‐dystonia associated with novel, biallelic DNAJC6 mutations. We delineate their clinical phenotype, neuroimaging features (including 123I‐FP‐CIT single‐photon emission computed tomography [SPECT; DaTScan]) and pattern of cerebrospinal fluid (CSF) neurotransmitter metabolites. Furthermore, we utilized patient fibroblasts and CSF to investigate secondary effects on auxilin, cyclin G–associated kinase (GAK), and dopaminergic proteins.
Materials and Methods
Subject Recruitment:
A cohort of 232 children with undiagnosed movement disorders were recruited for research between 2012 and 2016 at UCL Great Ormond Street–Institute of Child Health (London, UK). A subgroup of 25 patients were identified with juvenile parkinsonism, defined as onset of bradykinesia before 21 years of age, and at least one of the following signs: resting tremor, rigidity, and postural instability. All patients had detailed clinical assessment, undertaken by a movement disorder specialist. Review of (1) the clinical history, (2) features on neuroimaging, and (3) video recordings of the movement disorder at different time points was undertaken. Written informed consent was obtained from participating families, and the study was approved by the local ethics committees (Reference 13/LO/0168).
Diagnostic CSF Neurotransmitter Analysis:
In order to rule out a primary neurotransmitter disorder, where possible, patients had a routine diagnostic lumbar puncture for CSF neurotransmitter analysis. Using standardized protocols,15 CSF samples were collected, snap frozen in liquid nitrogen, and stored at –80°C. Analysis was undertaken using high‐pressure liquid chromatography (HPLC) with electrochemical detection and reversed‐phase column.15 Seven anonymized control pediatric CSF samples (with normal CSF neurotransmitter profiles) were obtained from the Neurometabolic Laboratory (National Hospital for Neurology and Neurosurgery, London, UK). All samples were processed and stored in accordance with the UK Royal College of Pathologists guidelines.
Molecular Genetic Investigation:
From the subgroup of 25 patients with juvenile parkinsonism (16 singletons and 9 familial cases from 3 kindreds), we prioritized two consanguineous families (Family A, 3 affected children; Family B, 2 affected children) for initial analysis. These families were investigated using an autozygosity mapping approach, given that the affected children were phenotypically similar and both families originated from the same region in Pakistan. Single‐nucleotide polymorphism (SNP) genotyping was performed as previously described.16 In addition, whole‐exome sequencing (WES) was performed for 2 children (A:III‐1 and B:IV‐2) by UCL Genomics (average WES coverage as previously reported17), with an average DNAJC6 coverage of 30×, with minimum coverage 10× for 82% of the gene. WES data were probed for putative disease‐causing DNAJC6 mutations in the remaining 20 cases (16 sporadic patients, 4 familial cases from a single kindred). This was undertaken through UCL Genomics (8 patients) and Wellcome Trust Sanger Institute (12 patients) within the Wellcome Trust UK10K Rare Diseases project, as previously reported.18 For patients where DNAJC6 mutations were identified, whole‐exome data were also probed for other genes associated with early‐onset dystonia‐parkinsonism (Table 1).
Table 1.
Clinical characteristics of patients with DNAJC6 mutations from this cohort and literature
| Pat. 1 | Pat. 2 | Pat. 3 | Pat. 4 | Pat. 5 | Pat. 6 | Previously Described DNAJC6‐Related Childhood Parkinsonism Dystonia Phenotypes in the Literature | Previously Described DNAJC6‐Related Early Onset PD in the Literature | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A‐III:1 | A‐III:4 | A‐III:5 | B‐IV:2 | B‐IV:4 | C | II‐212 | II‐4 | 40213 | 502 | 504 | 505 | 4202921 | GPS31314 | GPS314 | PAL50 | PAL54 | BR‐2652 | |
| Presenting age (years)/sex | 20/F | 12/M | 10/M | 28/F | 19/F | 18/F | 18/M | 13/M | 44/F | 24/F | 31/F | 17/M | 10/F | 48/M | 44/F | 62/M | 46/F | 57/M |
| Consanguinity | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | N | N | N | N |
| Country of origin | PK | PK | PK | PK | PK | PR | PL | PL | TR | TR | TR | TR | YM | NL | NL | BR | BR | BR |
| Onset of parkinsonism (y) | 11 | 10 | 9 | 13 | 7 | 10 | 7 | 11 | 10 | 11 | 10 | 10 | 10 | 21 | 29 | 42 | 31 | 24 |
| Parkinsonism at presentation | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | Y | N | Y | Y | N |
| Early development | D | D | D | D | D | D | N | N | N | D | D | D | N | N | N | N | N | N |
| Bradykinesia | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Tremor | + | + | ‐ | + | ‐ | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Rigidity | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Hypomimia | + | + | + | + | ‐ | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Postural instability | + | + | +/‐ | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Dystonic posturing | + | + | ‐ | + | ‐ | + | ‐ | ‐ | + | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Dysarthria | Anarthric | + | +/‐ | Anarthric | NR | + | + | + | + | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Eye movements abnormal | + | ‐ | ‐ | NR | ‐ | ‐ | ‐ | + | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| Loss of ambulation (y) | 13 | Unsteady gait | ‐ | 13 | 10 | 15 | 18 | 13 | 39 | 21‐26 | 31 | 20‐25 | 12 | Wheelchair bound | ‐ | ‐ | ‐ | ‐ |
| Motor fluctuations | + | ‐ | ‐ | ‐ | ‐ | + | NR | NR | NR | NR | NR | NR | NR | ‐ | ‐ | + | + | + |
| Seizures | ‐ | ‐ | ‐ | + | ‐ | + | ‐ | ‐ | + | ‐ | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ |
| Cognition | CI | CI | CI | CI | CI | CI | N | N | CI | CI | CI | CI | CI | NR | NR | CI | NR | NR |
| Spasticity | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | + | + | + | + | + | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Psychiatric features |
Ax |
Ax, PsB |
‐ | Ax | ‐ | AD, AgB | NR | NR | ‐ | Psy after l‐dopa, AgB, Hall | ‐ | ‐ | Psy, visual and auditory Hall | NR | Psy | NR | NR | NR |
| Other clinical features | MC, hypothyroid | MC, SD | MC | SD | SD | Mild BP | NR | NR | Scol | Mcl | Negative Mcl, Scol | ‐ | ‐ | NR | NR | NR | NR | NR |
|
Brain imaging (MRI/CT) (Atrophy) |
Perisylvian, cerebellar | N | N | Mild, generalized | Mild generalized, cerebellar | Mild, generalized, thin corpus callosum | N | N | Generalized | N | N | N | N | N | N | N | N | N |
|
123I‐FP‐CIT SPECT (DaTScan™) (Uptake in BG) |
Absent |
Reduced | Not performed | Not performed |
Absent |
Not performed | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| Genetics | ||||||||||||||||||
| DNAJC6 mutation |
c.766 C>T |
c.766 C>T |
c.766 C>T |
c.766 C>T |
c.766 C>T |
c.2416 C>T |
c.801‐2A>G | c.801‐2A>G |
c.2200 C>T |
c.2200C>T |
c.2200 C>T |
c.2200 C>T |
c.2365 C>T |
c.2779 A>G |
c.2779 A>G |
c.2223 A>T |
c.2223 A>T |
c.2038+3 A>G and c.1468+83del/‐ |
| Protein change | R256* | R256* | R256* | R256* | R256* | R806* | Q734X | Q734X | Q734X | Q734X | Gln789* | R927G | R927G | Thr741* | Thr741* | |||
| Variants in other PD genesa | N | N | N | N | N | N | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR |
|
Response to l‐dopa/carbidopa (maximum dose) |
Unsustained (150 mg/d) | Not tried | Not tried | Unsustained (5.5 mg/kg/d) | No response (10 mg/kg/d) | Some response (200 mg/d) | NR | No response | Good | Good | Therapy refused | Good | Mild | Good, dosages limited by psychiatric side effects |
Good, dosages limited by psychiatric side effects |
Good | Good | Good |
ATP13A2, ATP1A3, ATP7B, ATXN2 (SCA2), ATXN3 (SCA3), C19orf12, C9orf72, CSF1R, DCTN1, DDC, DJ1 (PARK7), EIF4G1, FBXO7, FMR1, FTL, GCH1, GIGYF2, GRN, HTRA2, HTT, LRRK2, MAPT, ND4, NPC1, PANK2, PARKIN (PARK2), PINK1, PITX3, PLA2G6, POLG, PPP2R2B, PRKRA, PTS, QDPR, SLC6A3, SLC18A2, SLC30A10, SNCA, SPATACSIN (SPG11), SR (SNCG), SYNJ1, TAF1, TBP, TH, and UCHL1.
Y, years of age; F, female; M, male; N, no; Y, yes; NR, not reported; PK, Pakistan; PL, Palestine; TR, Turkey; YM, Yemen; NL, Netherlands; BR, Brasil; Cl, cognitive impairment; AD, attention deficit; AgB, aggressive behavior; Ax, anxiety; PsB, perseveration behavior; Psy, psychosis; Hall, hallucinations; MC, microcephaly; SD, sleep disturbance; BP, behavioral problems; Mcl, myoclonus; Scol, scoliosis; BG, basal ganglia.
Direct Sanger Sequencing
Sanger sequencing was used to confirm variants identified on WES and to establish familial segregation. A genomic DNAJC6 sequence (Ensembl transcript: ENST00000371069; NCBI reference sequence: NM_001256864) was utilized to design primers, using Primer3 software (http://bioinfo.ut.ee/primer3/). Primers and polymerase chain reaction (PCR) amplification conditions are available on request. PCR products were cleaned up with MicroCLEAN (Web Scientific) and directly sequenced using Big Dye Terminator Cycle Sequencing System (Applied Biosystems Inc., Foster City, CA). Sequencing reactions were run on an ABI PRISM 3730 DNA Analyzer (Applied Biosystems Inc.) and analyzed with Chromas (http://www.technelysium.com.au/chromas.html).
Fibroblast and CSF Immunoblotting:
Methods to assess protein expression in patient fibroblasts were as previously reported.17 In brief, primary fibroblast lines were cultured from skin biopsies taken from Patients A‐III:1 and B‐IV:4 (c.766C>T; p.R256*) and 2 age‐matched healthy donor controls. Antibodies for auxilin and GAK (gift from Professor Green, National Institutes of Health, Washington, DC) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) horseradish peroxidase (HRP) conjugate (Cell Signaling Technology, Inc., Danvers, MA) were used.
Patients A‐III:1, A‐III:4, and B‐IV:4 underwent lumbar puncture for diagnostic CSF neurotransmitter analysis, and three CSF aliquots were snap frozen and stored at −80°C. Seven age‐matched control CSF samples were identified, from subjects without movement disorders on no medication. Patient and control CSF samples were immunoblotted for auxilin, GAK, and dopaminergic proteins as described previously.19 CSF protein was probed with the following antibodies: auxilin, GAK, tyrosine hydroxylase (MilliporeSigma, Burlington, MA), dopamine receptor 2 (MilliporeSigma), dopamine transporter (MilliporeSigma), vesicular monoamine transporter 2 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and transferrin (Santa Cruz Biotechnology, Inc) as the loading control. Relative protein levels were quantified using ImageJ software (National Institutes of Health, Bethesda, MD) and normalized to the loading control and the mean percentage of optical densitometry of three replicates analyzed with standard error of the mean.
Statistical Analysis
Statistical analysis was performed using Prism software (version 8; GraphPad Software Inc., La Jolla, CA), with data tested for Gaussian distribution and compared by the Student t test.
Data Availability Statement:
All clinical and experimental data relevant to this study are contained within the article. For Families A, B, and C, there is no ethical approval in place for deposition of whole‐exome sequencing genomic data into a public repository. Genomic data from UK10K are available at the EGA European Genomen Phenome Archive (https://www.ebi.ac.uk/ega/home), EGAS00001000128(UK10K RARE FIND). Details of statistical analysis can be shared upon request.
Results
Patient Cohort
A total of 232 children were referred with undiagnosed movement disorders for genetic research (Fig. 1A). Of these, 25 children (10.7%) had juvenile parkinsonism, 16 females and 9 males with a current median age of 14 years (range, 4–28). Fourteen of 25 had additional neurological features, including dystonia (14 patients), developmental delay/learning difficulties (14 patients), and seizures (4 patients; Fig. 1B).
Figure 1.

Juvenile parkinsonism cohort: clinical features and molecular genetic investigation. (A) Flowchart demonstrating the pathway of molecular genetic investigations in a subcohort of 25 children with juvenile parkinsonism. (B) Clinical characteristics of 20 children from 17 families. Early infancy <3 months; infancy 3 to 12 months; toddler 12 to 24 months; childhood 2 to 13 years; adolescence 13 to 18 years. *Consanguineous family. M, male; F, female; N, normal; NP, not performed; NR, not reported. [Color figure can be viewed at wileyonlinelibrary.com]
Molecular Genetic Investigations
Families A and B (Fig. 1A) were prioritized for autozygosity mapping studies. SNP genotyping revealed a 4.33‐Mb region of common homozygosity in both families on chromosome 1, between rs640407 (64,267,606 base pairs [bp]) and rs2566784 (68,602,735 bp; Fig. 2A,B). This region showed a common haplotype in all affected children whereas unaffected siblings had a different haplotype. It was therefore considered to be the likely disease locus (Fig. 2B).
Figure 2.
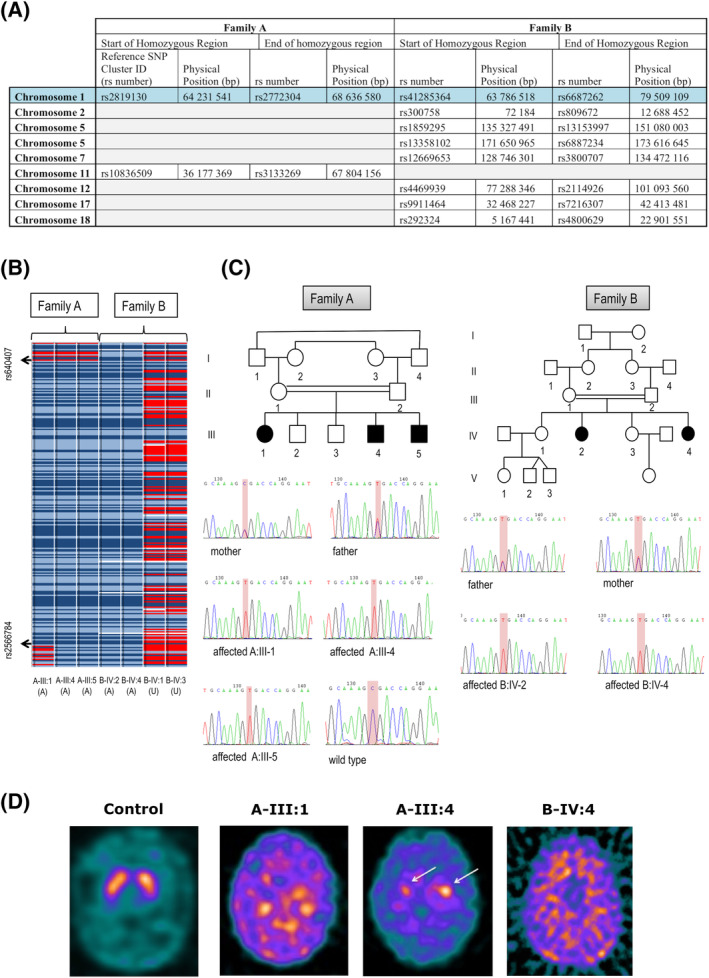
Molecular genetic investigations and DaTSCAN imaging. (A) Family A and B SNP array results showing homozygous regions detected. For each chromosome, the start and end point is specified using the Reference SNP Cluster ID (rs number) and physical position. (B) Homozygous SNPs are represented in light blue (AA) and dark blue (BB), heterozygous SNPs in red (AB), and “no calls” in white. (C) Sanger Sequencing confirms a homozygous DNAJC6 mutation, c.766C > T (p.R256*), in all affected children of Family A (A‐III:1, A‐III:4, and A‐III:5) and Family B (B‐IV:2, B‐IV:4). Parents are heterozygous carriers. (D) I‐123‐DaTSCAN™ with SPECT imaging in a control subject, Patient A‐III:1 (19 years 3 months), Patient A‐III:4 (11 years 4 months), and Patient B‐IV:4 (17 years). In Patients A‐III:1 and B‐IV:4, DaTSCAN findings indicate virtually complete absence of tracer uptake in the basal ganglia, with very high background activity, suggesting loss of presynaptic dopaminergic terminals, whereas Patient A‐III:4 showed significantly reduced, albeit still visible, uptake in the head of caudate (left better than right, white arrows). [Color figure can be viewed at wileyonlinelibrary.com]
WES performed in Patients A‐III:1 and B‐IV:2 revealed 23,365 and 23,549 variants, respectively. Given familial consanguinity and the autozygosity mapping results, targeted analysis for recessive pathogenic variants within the putative disease locus on chromosome 1 was undertaken. A single homozygous nonsense variant c.766C>T (p.R256*) in DNAJC6 (Chr1, 65,248,219–65,415,869) was identified both in A‐III:1 and B‐IV:2 on WES, located within the common region of homozygosity. No other pathogenic changes in previously reported genes causing juvenile parkinsonism‐dystonia phenotypes were identified from the WES data. Direct Sanger sequencing confirmed the homozygous c.766C>T mutation in all 5 affected patients, and familial segregation studies revealed that all parents were obligate carriers in both kindreds, with unaffected siblings either wild type or heterozygous for the identified variant (Fig. 2C). WES/whole‐genome sequencing data for the remainder of the parkinsonism‐dystonia cohort (n = 20) was interrogated for DNAJC6 mutations. This led to the identification of a homozygous nonsense variant (c.2416C>T) in a sixth unrelated patient (Patient C), which was confirmed on Sanger sequencing.
Delineation of the Clinical Phenotype of DNAJC6 Patients
Family A (3 Affected Patients)
Patients A‐III:1, A‐III:4, and A‐III:5 are 3 affected children born to first‐cousin parents, currently 20, 12, and 10 years old (Table 1). Two other brothers (A‐III:2 and A‐III:3), aged 17 and 15 years, have mild learning difficulties without evidence of a movement disorder. The paternal grandfather was diagnosed with PD in his fifties.
All 3 children were born at term after an uneventful antenatal period. Microcephaly was evident at birth (head circumference: <0.4th centile), but nonprogressive over time. All siblings had early neurodevelopmental delay and moderate learning difficulties.
A‐III:1 is the eldest daughter, aged 20 years. She presented at 10 years, with a 6‐week history of feeding difficulties, vomiting, and weight loss. Over time, she developed fever, unsteady gait, facial asymmetry, left‐sided tremor, and generalized seizures and was diagnosed with an encephalitis of uncertain etiology. She recovered from this acute illness, but subsequently had progressive bradykinesia, with tremor and rigidity, and loss of independent ambulation at 13 years, associated with cognitive decline. She is now wheelchair dependent, with generalised cogwheel‐rigidity, severe bradykinesia, multiple limb contractures and emotional lability (Video 1). She also has severe gut dysmotility, with recurrent vomiting, and required a gastrostomy for deteriorating bulbar dysfunction. CSF neurotransmitter analysis (age 11 years), while on levodopa therapy, revealed an isolated low 5‐hydroxyindoleacetic acid (5‐HIAA; Fig. 3A). At 12 years 11 months, when off l‐dopa, CSF HVA, and HVA:5‐HIAA ratio were low. Brain MRI showed evidence of right‐sided atrophy of the perisylvian region and right cerebellum by 19 years of age (Supporting Information Fig. S1). At 19 years, 123I‐FP‐CIT SPECT (DaTScan) showed absent uptake in the basal ganglia when compared to normal subjects (Fig. 2D). At this stage, while on l‐dopa treatment, her CSF HVA levels normalized (Fig. 3A). Her condition is refractory to medical treatment, with no clinical response to trihexyphenidyl, benzhexol, procyclidine, clobazam, rotigotine, and apomorphine. l‐dopa has proven difficult to titrate because of marked drug sensitivity. She experiences an improvement in motor function and speech 30 minutes postdose, after which she returns to the off state. l‐dopa dosages >150 mg/d have resulted in drug‐related dyskinesias.
Figure 3.
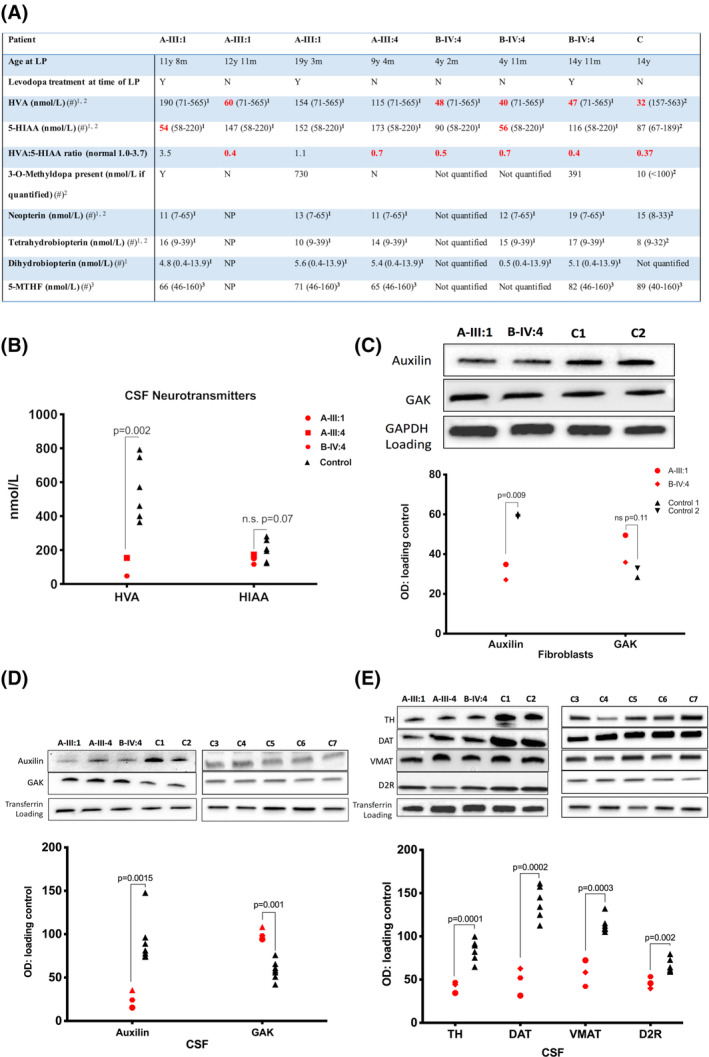
CSF neurotransmitter analysis and patient fibroblast and CSF immunoblotting. (A) CSF neurotransmitter analysis. Age‐related reference ranges indicated in brackets after each value. Red: abnormal result. Gray: borderline result. Symbol (“#”) indicates reference range: 1Keith Hyland, Robert A.H. Surtees, et al. Pediatr Res 1993;34:10–14; 2Keith Hyland, Future Neurol 2006;1:593–603; 3Surtees R, Hyland K. Biochem Med Metab Biol 1990;44:192–199. (B) Scatterplot of CSF HVA and 5‐HIAA levels (nmol/L) measured by high performance liquid chromatography (patient = red shapes, control = black triangles). Medication at time of CSF sampling: A‐III‐1: co‐careldopa, melatonin, glycopyronium; A‐III‐4: none; B‐IV‐4: l‐dopa, pyridoxine; Control 1: none; Control 2: none. Immunoblot of auxilin and GAK in patient fibroblasts (C) and CSF (D) compared to controls. (E) Immunoblot of patient CSF for TH, DAT, VMAT, and D2R protein levels measured compared to controls. Graphs show mean protein percent optical density (OD) normalized to loading control in patients (red) and controls (black). LP, lumbar puncture; y, years; m, months; 5‐MTHF, 5‐methyltetrahydrofolate; NP, not performed. [Color figure can be viewed at wileyonlinelibrary.com]
Her two brothers (A‐III:4 and A‐III:5) presented with fine motor difficulties at 8 years of age. They subsequently developed positional tremor, upper limb dystonic posturing, hypophonia, hypomimia, bradykinesia, cogwheel rigidity, and postural instability over 12 months (Videos 2 and 3). Like their sister, both have gastrointestinal complications with sialorrhea, recurrent vomiting, and feeding difficulties, necessitating gastrostomy insertion. A‐III:4 is currently 12 years old and suffers from anxiety and perseveration. His CSF‐HVA levels are at the lower limit of normal, with a low HVA:5‐HIAA ratio <1.0 (Fig. 3A). MRI brain scan was normal. 123I‐FP‐CIT SPECT (DaTScan) at 11 years showed profound reduction in tracer uptake in the basal ganglia (Fig. 2D). Both boys responded to treatment with transdermal rotigotine and oral trihexyphenidyl, but with increasing doses, both experienced dyskinesias, necessitating dose reduction.
Family B (2 Affected Patients)
Patients B‐IV:2 and B‐IV:4 are 2 affected girls, born to first‐cousin parents, and currently 28 and 19 years old. Both were born uneventfully following a normal pregnancy, presenting with early feeding difficulties, hypotonia, and delayed milestones by 6 months old. Both made slow developmental progress, achieving independent ambulation and spoken language by 3 years of age.
B‐IV:2 presented at 9 years with generalized seizures that stabilized with lamotrigine therapy. From 13 years of age, motor and cognitive deterioration ensued, with onset of parkinsonism and loss of speech and ambulation. She experienced anxiety and recurrence of seizures. She has severe antecollis, hypomimia, tremor, generalised cogwheel rigidity, bradykinesia, and positive glabellar tap (Video 4). MRI was normal until 18 years, after which there was radiological evidence of mild generalized atrophy. Several medications were tried without clinical benefit, including l‐dopa (maximum, 10 mg/kg/d), selegiline, rotigotine, and trihexyphenidyl. The younger sibling, B‐IV:4, presented at 7 years with gait deterioration, bradykinesia, and cogwheel rigidity. She lost independent ambulation and speech by 10 years (Video 5). She has developed dystonic posturing, bulbar dysfunction (necessitating gastrostomy), and a disrupted sleep pattern. The MRI brain scan was initially normal, but by 16 years showed subtle global cerebral atrophy (particularly in the posterior regions) as well as cerebellar atrophy (Supporting Information Fig. S2). 123I‐FP‐CIT SPECT (DaTScan) at 17 years showed profound reduction in tracer uptake in the basal ganglia (Fig. 2D). CSF HVA and HVA:5‐HIAA ratio were reduced at ages 4 and 14 years. CSF‐HIAA levels were reduced at age 4 years (Fig. 3A). She showed an initial response to l‐dopa, but developed emotional lability at 5.5 mg/kg/d, leading to drug withdrawal. There was no clinical improvement observed with trihexyphenidyl or chloral hydrate. She had a modest response to pramipexole, with improved facial expression, reduced tremor, and increase in voluntary movements.
Family C
This 18‐year‐old girl is the third child of distantly related Latin American parents, with 2 healthy siblings. She initially presented with neonatal feeding difficulties and hypotonia. In infancy, she showed delay in attaining milestones and developed seizures characterized by staring episodes with loss of tone. She walked independently from 2 years, but by 10 years of age her gait deteriorated, leading to frequent falls, postural instability, and losing the ability to run. Over the next 4 years, she continued to deteriorate with worsening antecollis and bradykinesia (Video 6). She developed severe bulbar dysfunction with sialorrhea, dysarthria, and, dysphagia, leading to considerable weight loss. At 12 years, she developed generalized tonic‐clonic seizures and atypical absences, responsive to lamotrigine and zonisamide therapy. MRI demonstrated subtle generalized cerebral atrophy and CSF HVA was low (Fig. 3A). Her movement disorder responded to l‐dopa, with improved tremor, gait, and a reduction in drooling. A maximum of 200 mg/d was tolerated, but further increases led to intolerable drug‐induced dyskinesias. After 4 months of treatment, she developed aggressive behavior and received treatment with quetiapine. By 16 years, she became increasingly sensitive to l‐dopa, with peak‐dose agitation, restlessness, and dyskinesia. Lowering the dose improved side effects, and continued to provide motor benefit, although the effects wore off 2 to 3 hours after administration. On‐off phenomena were commonly reported, and in the off state, she was often akinetic and rigid. Introduction of trihexyphenidyl improved rigidity, but not immobility.
Patient CSF and Fibroblast Analysis
CSF HPLC analysis of the DNAJC6 patient cohort showed reduction in CSF‐HVA levels (P = 0.002) compared to controls (but not 5‐HIAA levels) in 3 patients (Fig. 3A,B). Patient fibroblasts showed reduced auxilin (P = 0.009) and a trend for increased GAK protein (P = 0.11; Fig. 3C). Patient CSF auxilin levels were even more significantly reduced (P = 0.0015; Fig. 3D). Notably, CSF‐GAK levels were significantly increased in patients (P = 0.0014; Fig. 3D). CSF immunoblotting studies showed that several key components of the dopaminergic synapse were significantly reduced, including tyrosine hydroxylase (TH; P = 0.0001), vesicular monoamine transporter (VMAT; P = 0.0002), dopamine transporter (DAT; P = 0.0003), and D2 receptor (D2R; P = 0.002; Fig. 3E).
Discussion
Juvenile parkinsonism attributed to DNAJC6 mutations has only recently been reported. Here, we report on a further 6 patients from three families, with two previously unreported homozygous nonsense mutations in DNAJC6. Moreover, our findings on 123I‐FP‐CIT SPECT (DaTScan) imaging, CSF analysis, and immunoblotting suggest downstream dyshomeostasis of auxilin, GAK, and dopaminergic proteins in DNAJC6‐related disease.
Our data confirms that all reported cases of juvenile‐onset DNAJC6‐parkinsonism have core clinical characteristics (Table 1), including (1) clinical presentation of progressive parkinsonism toward of the first decade (median, 10 years; range, 7–13), (2) significant neurological regression thereafter, and (3) loss of ambulation in mid‐adolescence.12, 13, 14, 21 In contrast to adult‐onset PD, childhood parkinsonian disorders rarely present with a “pure” parkinsonian phenotype, as illustrated by the classical primary pediatric monoamine neurotransmitter disorders.20 Similarly, in early‐onset DNAJC6‐related disease, parkinsonism is commonly present in tandem with a multitude of other clinical features, including dystonia, moderate learning difficulties, epilepsy, and neuropsychiatric features12, 13, 14, 21 (Table 1). Furthermore, many of our patients had evidence of bulbar dysfunction, gut dysmotility, and sleep disturbance. The majority of our patients showed limited response to l‐dopa and other standard therapies for parkinsonism‐dystonia. They experienced severe, often intolerable, side effects with dopaminergic agents, including on‐off phenomenon and severe dyskinesia, particularly at higher drug dosages.
123I‐FP‐CIT SPECT (DaTScan) was performed in 3 patients, demonstrating reduced tracer uptake in the basal ganglia, suggestive of impaired presynaptic dopamine uptake and striatonigral neurodegeneration. Postmortem studies have confirmed striatal dopamine deficiency in patients with parkinsonism.22, 23 Together, these observations suggest a neurodegenerative process in DNAJC6 patients. MRI brain imaging further corroborates this hypothesis; the mild generalized cerebral and/or cerebellar atrophy in 4 patients suggests that DNAJC6‐related disorders may also be associated with neuronal loss in other regions of the central nervous system.
All 6 cases fit the juvenile phenotype associated with this gene, though more recently, DNAJC6 mutations have been reported in early adult‐onset PD.14 Although there are a number of overlapping features (progressive parkinsonism, psychiatric features), affected patients presented later (range, 21–42 years) and seizures and cognitive decline are not reported.
Homozygous and compound heterozygous mutations in DNAJC6 are predicted to result in loss of protein function. To date, splice‐site variants,12 large multiexonic deletions,24 truncating mutations,13 and missense mutations14 have been reported. All 6 patients in our cohort had nonsense mutations, predicted to cause nonsense‐mediated decay or premature protein truncation. Five of the 6 reported patients are from two consanguineous families originating from the same region in Pakistan, and all have the same nonsense mutation. SNP array confirmed a common haplotype at this disease locus for all affected children, suggesting a possible founder effect.
DNAJC6 encodes for auxilin, a neuronally expressed J‐chaperone protein involved in the uncoating of clathrin‐coated vesicles25, 26 (Fig. 4). Auxilin modifies the three‐dimensional conformation of heavy‐chain clathrin triskelions, leading to clathrin coat distortion, instability, and subsequent disassembly.25, 26 Neurotransmission involves rapid continuous recycling of synaptic vesicles through CME. Deficiency in auxilin ultimately results in impairment of synaptic vesicle recycling and impaired neurotransmission. Similarly, aberrant synaptic vesicular trafficking is also evident in other forms of early‐onset parkinsonism, including LRRK2,27, 28 VMAT2,29 and SNCA‐related disease.30 Clathrin‐mediated endocytosis is crucial for the regulation of developmental signaling pathways through internalization of receptors or ligands and is required for axon and dendrite outgrowth.31 Presence of developmental delay well before onset of parkinsonism in patients with DNAJC6 mutations further corroborates the notion that auxilin is likely to have a central role in neurodevelopment, given its role in CME. In Drosophila, auxilin is crucial for Notch signaling, a developmental pathway that regulates neural stem‐cell proliferation, survival, renewal, and differentiation, as well as neuronal specification of dopaminergic neurons.32, 33, 34
Figure 4.
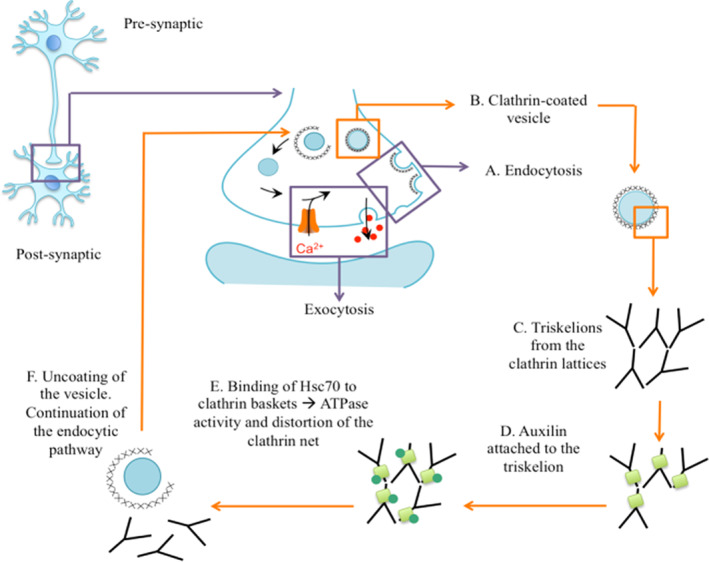
Schematic representation of DNAJC6‐encoded auxilin protein function. The role of auxilin in synaptic vesicle recycling and endocytic pathway. (A) A nascent clathrin‐coated pit is formed at the presynaptic membrane, followed by membrane invagination; (B) the pit develops into a clathrin‐coated vesicle, where the clathrin lattice consists of (C) clathrin triskelions formed by three crossed ankle regions. (D) Auxilin (pale green square) binds to an exposed domain in the heavy chain of the clathrin. (E) Auxilin facilitates a conformational change in clathrin that allows binding of Hsc70 and by ATPase‐mediated activity, and the clathrin lattice is disrupted and distorted, leading to (F) clathrin disassembly allowing subsequent delivery of cargo neurotransmitters to the membrane or other vesicle in the endocytic pathway. Hsc70, heat shock cognate protein 70. [Color figure can be viewed at wileyonlinelibrary.com]
To investigate the downstream effects of DNAJC6 mutations, we studied auxilin and GAK protein levels in 2 patients using patient fibroblasts and CSF. Auxilin is a neuron‐specific protein, enriched in presynaptic terminals, whereas GAK is an ubiquituously expressed protein.35, 36 Auxilin and GAK are highly homologous proteins that both have the ability to bind clathrin and clathrin adaptor protein 2 in order to initiate clathrin uncoating of endocytosed vesicles.37 In the auxilin knockout mouse model, it is reported that upregulation of GAK can partially compensate for the loss of auxilin and decrease mortality.35 We therefore wished to determine whether a similar compensatory mechanism was evident in our patients. In our study, we observed that patient fibroblast auxilin protein levels were significantly reduced when compared to controls, as previously reported.14 We found that patient fibroblast GAK levels were slightly, but not significantly, increased, whereas patient CSF GAK protein levels were significantly increased. Our findings support upregulation of brain GAK levels in DNAJC6 patients, partially compensating for auxilin reduction, as evident in the auxilin knockout mouse model.35
Diagnostic CSF neurotransmitter analysis revealed that levels of the stable dopamine metabolite (HVA) were either below the age‐related reference ranges or close to the lower limit of normal in our patients, indicating impaired dopamine turnover. Indeed, CSF‐HVA levels and HVA:5‐HIAA ratios were comparable to those observed in TH deficiency, an inherited dopamine synthesis defect associated with central dopamine deficiency.20 In order to determine how DNAJC6 mutations may impact the dopaminergic system, we used patient CSF to analyze proteins involved in dopamine signaling and homeostasis. We observed that patient CSF had significantly reduced levels of VMAT, DAT, TH, and D2R when compared to controls. 123I‐FP‐CIT SPECT (DaTScan) imaging additionally provides in vivo evidence of impaired DAT function in DNAJC6 patients. VMAT and DAT are both synaptic transporters recycled through clathrin‐mediated endocytosis.38, 39 The reduction in HVA associated with low VMAT/DAT protein levels may imply that the observed dopamine deficiency is associated with impaired clathrin‐mediated neurotransmitter recycling. D2R is also postulated to be internalized through clathrin‐mediated endocytosis.38, 39 Neurons internalize receptors to adjust excitability and degrade, resensitize, and recycle desensitized receptors.38, 39 DNAJC6 mutations thus may affect D2R protein levels and normal postsynaptic function. It is likely that presynaptic D2R autoreceptor function will also be affected, leading to aberrant TH regulation.40
Overall, our findings suggest that the mechanisms governing DNAJC6‐associated parkinsonism are likely to be multifactorial. Another plausible explanation for the reduction in synaptic protein levels may be as a result of neurodegeneration secondary to defective chaperone function. Auxilin and other J‐chaperone proteins play a crucial role in regulating the folding and conformational change of proteins to maintain integrity in the neuron.41, 42 Indeed, in the auxilin knockout mouse model, there is sequestration of clathrin cages in the cerebellum.35 With impaired auxilin function, a cumulative effect of sequestered misfolded proteins and accumulation of clathrin coat components in assembled coats and cages may lead to apoptotic cascades and neurodegeneration. There is growing interest in the role of such chaperone proteins in human disease and mutations in eight distinct J proteins (DNAJB2, DNAJB6, DNAJC5, DNAJC6, DNAJC12, DNAJC13, DNAJC19, and DNAJC29) have been described.43, 44 Future research into such “chaperonopathies” may provide further insights into neurodegenerative disorders.
In conclusion, we report on a cohort of patients with previously unreported DNAJC6 mutations associated with early neurodevelopmental delay, juvenile parkinsonism, and neurological regression in the second decade of life. We further demonstrate disturbance of dopamine homeostasis in patient‐derived CSF and report on a possible GAK‐mediated compensatory mechanism for auxilin deficiency. Mutations in DNAJC6 are rare, but a likely under‐recognized cause of parkinsonism‐dystonia in infants and children. Elucidating the genetic diagnosis has important implications for families given that early diagnosis negates the need for extensive invasive investigations, facilitates treatment strategies, and aids genetic counseling for future pregnancies. The early clinical features and CSF neurotransmitter signature observed in our patients can mimic primary neurotransmitter disorders, and DNAJC6 mutations should thus be considered as a differential diagnosis. We observed reduced auxilin and increased GAK protein levels, suggesting a possible compensatory role for GAK in this condition. Study of CSF synaptic proteins suggest downstream effects on dopamine synthesis, recycling, homeostasis, and signaling that may result from a combination of primary auxilin deficiency and neurodegeneration. Abnormal synaptic vesicle dynamics are increasingly recognized as a disease mechanism in neurodegenerative parkinsonian disorders, and future research into elucidating the pathogenesis of such conditions will no doubt assist the development of novel targeted treatments.
Author Roles
(1) Research Project: A. Conception and Design; B. Acquisition of Data; C. Analysis and Interpretation of Data; (2) Manuscript: A. Writing of the First Draft, B. Review and Critique; (3) Other: A. Statistical Analysis (for Figure 3); B. Drafting of Figures; C. CSF Neurotransmitter Analysis and Interpretation.
J.N.: 1A, 1B, 1C, 2A, 3A
E.C.‐S.: 1A, 1B, 1C, 2A, 3B
L.A.: 1B, 1C, 2A, 3B
P.T.: 1B, 1C
K.M.: 1B, 1C
S.S.: 1B, 1C
K.M.G.: 1B
S.J.R.H.: 3C
S.P.: 3C
L.B.: 1B, 1C
B.C.: 1B, 1C
J.C.: 1B, 1C
K.R.: 1B, 1C
H.C.: 1B, 1C
S.J.: 1B, 1C
R.J.: 1B, 1C
D.H.: 1B, 1C
À.G.‐C.: 1B, 1C
D.G.: 1B, 1C
F.L.R.: 1B, 1C
B.P.‐D.: 1B, 1C
C.D.G.: 1B, 1C
T.S.P.: 1B, 1C
E.M.: 1A, 1B, 1C, 2A
M.A.K.: 1A, 1B, 1C, 2A
Financial Disclosures
Nothing to report.
Supporting information
Video 1 Patient A‐III:1. She is now 20 years old. Examination in the off state, >3 hours after taking medication. She is hypomimic, has resting tremor more evident in her right foot, and has contractures and deformities in both hands and feet. She is markedly bradykinetic, and this is evident when she tries to smile.
Video 2 Patient A‐III:4. He is now almost 12 years old. Tremor is evident in resting state. Bradykinesia is evident when grasping. Mild hypomimia is present. There is no evidence of oculomotor abnormalities or upgaze palsy. He regularly touches his earlobe as a sensory trick attempting to control his tremor. Rigidity and striatal toe are evident when walking. Improvement of dystonia is seen upon running. Dystonic posturing (especially in the right hand) is also present when ambulant. He has difficulties in maintaining normal balance when he is standing.
Video 3 Patient A‐III:5. He has recently turned 10 years old. He has evidence of resting tremor and bradykinesia. No abnormal eye movements are present. The gait is mildly affected as he can run and walk, but has dystonic posturing of both hands. Improvement of dystonic posturing is seen upon running. He has trouble in trying to walk on a line, showing difficulties in maintaining balance.
Video 4 Patient B‐IV:2. She is now 27 years old. She has significant disability and is completely wheelchair bound. She is very hypomimic and has poor head control. She is able to understand simple commands, but she is extremely hypokinetic. She exhibits some sudden muscle contractions from both arms and legs. She has fixed distal deformities in her hands and feet, with dystonic postures. No signs of spasticity are present.
Video 5 Patient B‐IV:4. She is now 17 years old. She has poor head control. She is also very hypomimic. She can understand commands. She exhibits a clear tremor. She has episodic whole‐body shaking, almost always related to new situations when she is anxious or nervous. These episodes are short and rarely last for more than 1 minute. Bradykinesia is evident on finger‐finger testing. Cogwheel rigidity is more evident than in her older sister, B‐IV:2. Distal tremor from her left foot is shown.
Video 6 Patient C. Segment 1 demonstrates examination in the off state at age 14. There is moderate facial masking with prominent lower facial dystonia. She has moderate‐to‐severe dysarthria. Hand grasping, finger tapping, and foot tapping is moderately impaired bilaterally, left slightly greater than right. There is moderate generalized bradykinesia. Her gait is characterized by mildly reduced stride length, reduced bilateral arm swing, some festination, and a tendency to fall forward. Pull test is negative. Segment 2 illustrates examination in the off state at age 17, which shows progression from segment 1, taken 3 years earlier. She has marked anterocollis with more prominent lower facial dystonia. She is almost mute. She can follow some simple commands, but her movements are markedly bradykinetic with long latency to respond. She has prominent drooling. Finger‐to‐nose testing, hand grasping, and foot tapping reveal marked bradykinesia bilaterally. She requires assistance from one person to stand up from a chair and has a tendency to fall backward. She can take only a few steps very slowly, requiring assistance from other people.
FIG. S1 Brain MRI in patient A‐III:1. Patient A‐III:1: axial T2 and coronal FLAIR images at ages 12 and 19 years showing progressive right frontoparietal and perisylvian (black star) atrophy over time. There is also evidence of right cerebellar atrophy between the two studies (white arrow). FLAIR, fluid‐attenuated inversion recovery.
FIG. S2 Brain MRI in Patient B‐IV:4. Patient B‐IV:4: Sagittal T1 and axial T2 MR images at age 12 and 16 years show a subtle, but definite, global brain parenchymal volume loss over time, particularly in the posterior aspects of the cerebral hemispheres. The occipital horns of the lateral ventricles are, as a result, more dilated over time. There is also subtle cerebellar atrophy between the two studies as shown on the sagittal T1 images.
Acknowledgments
We thank the patients and families for their participation in this study. We acknowledge Kerra Pearce, Mark Kristiansen, and Alan Pittman (UCL Genomics) for assistance with the SNP arrays and whole‐exome sequencing. We thank Professor Lois Greene for kindly providing the anti‐auxilin and GAK antibodies.
Dr. Ng, Dr. Cortès‐Saladelafont, and Dr. Abela contributed equally to the manuscript.
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
Funding agencies: This work was funded by the UK Medical Research Council Doctoral Fellowship (to J.N.), a Wellcome Intermediate Fellowship and NIHR Professorship (to M.A.K.), Sir Jules Thorn Charitable Trust (to M.A.K.), Great Ormond Street Hospital Children's Charity (to J.N., E.M., and M.A.K.), Rosetrees Trust (to J.N., M.A.K.), Agustí Pedró i Pons Foundation, Universitat de Barcelona and Río Hortega 2015‐2017 Institute of Health Carlos III (to E.C.S.), Dystonia Medical Research Foundation (to P.T.), PS09/01132 ISCIII‐FEDER, Institute of Health Carlos III (to A.G.C.), European Union Marie Curie training network (to S.J.R.H.), NIHR Biomedical Research Centres, Swiss National Science Foundation Advanced Postdoc. Mobility Fellowship (to L.A.), Cambridge Biomedical Research Centre and Wellcome Trust for UK10K (to F.L.R., D.G.). This study made use of data generated by the UK10K Project award WT091310. A full list of consortium members can be found at the UK10K Project website (http://www.uk10k.org). This research was supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust, University College London.
References
- 1.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608. [DOI] [PubMed] [Google Scholar]
- 2.Valente EM, Abou‐Sleiman PM, Caputo V, et al. Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science 2004;304:1158–1160. [DOI] [PubMed] [Google Scholar]
- 3.Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset parkinsonism. Science 2003;299:256–259. [DOI] [PubMed] [Google Scholar]
- 4.Ramirez A, Heimbach A, Gründemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat Genet 2006;38:1184–1191. [DOI] [PubMed] [Google Scholar]
- 5.Paisan‐Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia‐parkinsonism. Ann Neurol 2009;65:19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fonzo AD, Dekker MCJ, Montagna P, et al. FBXO7 mutations cause autosomal recessive, early‐onset parkinsonian‐pyramidal syndrome. Neurology 2009;72:240–245. [DOI] [PubMed] [Google Scholar]
- 7.Ng J, Zhen J, Meyer E, et al. Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain 2014;137:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuschl K, Meyer E, Valdivia LE, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood‐onset parkinsonism‐dystonia. Nat Commun 2016;7:11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider SA, Bhatia KP, Hardy J. Complicated recessive dystonia parkinsonism syndromes. Mov Disord 2009;24:490–499. [DOI] [PubMed] [Google Scholar]
- 10.Bonifati V.Genetics of Parkinson's disease—state of the art, 2013. Parkinsonism Relat Disord 2014;20(Suppl. 1):S23–S28. [DOI] [PubMed] [Google Scholar]
- 11.Quadri M, Fang MY, Picillo M, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early‐onset parkinsonism. Hum Mutat 2013;34:1208–1215. [DOI] [PubMed] [Google Scholar]
- 12.Edvardson S, Cinnamon Y, Ta‐Shma A, et al. A deleterious mutation in DNAJC6 encoding the neuronal‐specific clathrin‐uncoating co‐chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 2012;7:e36458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koroglu C, Baysal L, Cetinkaya M, Karasoy H, Tolun A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Related Disord 2013;19:320–324. [DOI] [PubMed] [Google Scholar]
- 14.Olgiati S, Quadri M, Fang MY, et al. DNAJC6 mutations associated with early‐onset Parkinson's disease. Ann Neurol 2016;79:244–256. [DOI] [PubMed] [Google Scholar]
- 15.Hyland K, Surtees RA, Heales SJ, Bowron A, Howells DW, Smith I. Cerebrospinal fluid concentrations of pterins and metabolites of serotonin and dopamine in a pediatric reference population. Pediatr Res 1993;34:10–14. [DOI] [PubMed] [Google Scholar]
- 16.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer E, Carss KJ, Rankin J, et al. Mutations in the histone methyltransferase gene KMT2B cause complex early‐onset dystonia. Nat Genet 2017;49:223–237. [DOI] [PubMed] [Google Scholar]
- 18.Consortium UK, Walter K, Min JL, et al. The UK10K project identifies rare variants in health and disease. Nature 2015;526:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortez C, Duarte ST, Ormazábal A, et al. Cerebrospinal fluid synaptic proteins as useful biomarkers in tyrosine hydroxylase deficiency. Mol Genet Metab 2015;114:34–40. [DOI] [PubMed] [Google Scholar]
- 20.Willemsen MA, Verbeek MM, Kamsteeg EJ, et al. Tyrosine hydroxylase deficiency: a treatable disorder of brain catecholamine biosynthesis. Brain 2010;133:1810–1822. [DOI] [PubMed] [Google Scholar]
- 21.Elsayed LE, Drouet V, Usenko T, et al. A novel nonsense mutation in DNAJC6 expands the phenotype of autosomal‐recessive juvenile‐onset Parkinson's disease. Ann Neurol 2016;79:335–337. [DOI] [PubMed] [Google Scholar]
- 22.Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burke RE, O'Malley K. Axon degeneration in Parkinson's disease. Exp Neurol 2013;246:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vauthier V, Jaillard S, Journel H, Dubourg C, Jockers R, Dam J. Homozygous deletion of an 80 kb region comprising part of DNAJC6 and LEPR genes on chromosome 1P31.3 is associated with early onset obesity, mental retardation and epilepsy. Mol Genet Metab 2012;106:345–350. [DOI] [PubMed] [Google Scholar]
- 25.Ungewickell E, Ungewickell H, Holstein SEH, et al. Role of auxilin in uncoating clathrin‐coated vesicles. Nature 1995;378:632–635. [DOI] [PubMed] [Google Scholar]
- 26.Eisenberg E, Greene LE. Multiple roles of auxilin and Hsc70 in clathrin‐mediated endocytosis. Traffic 2007;8:640–646. [DOI] [PubMed] [Google Scholar]
- 27.Migheli R, Del Giudice MG, Spissu Y, et al. LRRK2 affects vesicle trafficking, neurotransmitter extracellular level and membrane receptor localization. PLoS One 2013;8:e77198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong Y, Dawson Valina L, Dawson Ted M. LRRK2 GTPase dysfunction in the pathogenesis of Parkinson's disease. Biochem Soc Trans 2012;40:1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rilstone JJ, Alkhater RA, Minassian BA. Brain dopamine‐serotonin vesicular transport disease and its treatment. N Engl J Med 2013;368:543–550. [DOI] [PubMed] [Google Scholar]
- 30.Alter SP, Lenzi GM, Bernstein AI, Miller GW. Vesicular integrity in Parkinson's disease. Curr Neurol Neurosci Rep 2013;13:362–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yap CC, Winckler B. Adapting for endocytosis: roles for endocytic sorting adaptors in directing neural development. Front Cell Neurosci 2015;9:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eun SH, Banks SML, Fischer JA. Auxilin is essential for delta signaling. Development 2008;135:1089–1095. [DOI] [PubMed] [Google Scholar]
- 33.Lathia JD, Mattson MP, Cheng A. Notch: from neural development to neurological disorders. J Neurochem 2008;107:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tio M, Toh J, Fang W, Blanco J, Udolph G. Asymmetric cell division and Notch signaling specify dopaminergic neurons in Drosophila. PLoS One 2011;6:e26879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yim YI, Sun T, Wu LG, et al. Endocytosis and clathrin‐uncoating defects at synapses of auxilin knockout mice. Proc Natl Acad Sci U S A 2010;107:4412–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahle S, Ungewickell E. Auxilin, a newly identified clathrin‐associated protein in coated vesicles from bovine brain. J Cell Biol 1990;111:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheele U, Kalthoff C, Ungewickell E. Multiple interactions of auxilin 1 with clathrin and the AP‐2 adaptor complex. J Biol Chem 2001;276:36131–36138. [DOI] [PubMed] [Google Scholar]
- 38.Ferguson SSG, Caron MG. G protein‐coupled receptor adaptation mechanisms. Semin Cell Dev Biol 1998;9:119–127. [DOI] [PubMed] [Google Scholar]
- 39.Sorkin A, Von Zastrow M. Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol 2002;3:600–614. [DOI] [PubMed] [Google Scholar]
- 40.Blackstone C.Infantile parkinsonism‐dystonia: a dopamine “transportopathy”. J Clin Invest 2009;119:1455–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kakkar V, Prins LCB, Kampinga HH. DNAJ proteins and protein aggregation diseases. Curr Top Med Chem 2012;12:2479–2490. [DOI] [PubMed] [Google Scholar]
- 42.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 2010;11:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koutras C, Braun JEA. J protein mutations and resulting proteostasis collapse. Front Cell Neurosci 2014;8:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anikster Y, Haack TB, Vilboux T, et al. Biallelic mutations in DNAJC12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am J Hum Genet 2017;100:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1 Patient A‐III:1. She is now 20 years old. Examination in the off state, >3 hours after taking medication. She is hypomimic, has resting tremor more evident in her right foot, and has contractures and deformities in both hands and feet. She is markedly bradykinetic, and this is evident when she tries to smile.
Video 2 Patient A‐III:4. He is now almost 12 years old. Tremor is evident in resting state. Bradykinesia is evident when grasping. Mild hypomimia is present. There is no evidence of oculomotor abnormalities or upgaze palsy. He regularly touches his earlobe as a sensory trick attempting to control his tremor. Rigidity and striatal toe are evident when walking. Improvement of dystonia is seen upon running. Dystonic posturing (especially in the right hand) is also present when ambulant. He has difficulties in maintaining normal balance when he is standing.
Video 3 Patient A‐III:5. He has recently turned 10 years old. He has evidence of resting tremor and bradykinesia. No abnormal eye movements are present. The gait is mildly affected as he can run and walk, but has dystonic posturing of both hands. Improvement of dystonic posturing is seen upon running. He has trouble in trying to walk on a line, showing difficulties in maintaining balance.
Video 4 Patient B‐IV:2. She is now 27 years old. She has significant disability and is completely wheelchair bound. She is very hypomimic and has poor head control. She is able to understand simple commands, but she is extremely hypokinetic. She exhibits some sudden muscle contractions from both arms and legs. She has fixed distal deformities in her hands and feet, with dystonic postures. No signs of spasticity are present.
Video 5 Patient B‐IV:4. She is now 17 years old. She has poor head control. She is also very hypomimic. She can understand commands. She exhibits a clear tremor. She has episodic whole‐body shaking, almost always related to new situations when she is anxious or nervous. These episodes are short and rarely last for more than 1 minute. Bradykinesia is evident on finger‐finger testing. Cogwheel rigidity is more evident than in her older sister, B‐IV:2. Distal tremor from her left foot is shown.
Video 6 Patient C. Segment 1 demonstrates examination in the off state at age 14. There is moderate facial masking with prominent lower facial dystonia. She has moderate‐to‐severe dysarthria. Hand grasping, finger tapping, and foot tapping is moderately impaired bilaterally, left slightly greater than right. There is moderate generalized bradykinesia. Her gait is characterized by mildly reduced stride length, reduced bilateral arm swing, some festination, and a tendency to fall forward. Pull test is negative. Segment 2 illustrates examination in the off state at age 17, which shows progression from segment 1, taken 3 years earlier. She has marked anterocollis with more prominent lower facial dystonia. She is almost mute. She can follow some simple commands, but her movements are markedly bradykinetic with long latency to respond. She has prominent drooling. Finger‐to‐nose testing, hand grasping, and foot tapping reveal marked bradykinesia bilaterally. She requires assistance from one person to stand up from a chair and has a tendency to fall backward. She can take only a few steps very slowly, requiring assistance from other people.
FIG. S1 Brain MRI in patient A‐III:1. Patient A‐III:1: axial T2 and coronal FLAIR images at ages 12 and 19 years showing progressive right frontoparietal and perisylvian (black star) atrophy over time. There is also evidence of right cerebellar atrophy between the two studies (white arrow). FLAIR, fluid‐attenuated inversion recovery.
FIG. S2 Brain MRI in Patient B‐IV:4. Patient B‐IV:4: Sagittal T1 and axial T2 MR images at age 12 and 16 years show a subtle, but definite, global brain parenchymal volume loss over time, particularly in the posterior aspects of the cerebral hemispheres. The occipital horns of the lateral ventricles are, as a result, more dilated over time. There is also subtle cerebellar atrophy between the two studies as shown on the sagittal T1 images.
Data Availability Statement
All clinical and experimental data relevant to this study are contained within the article. For Families A, B, and C, there is no ethical approval in place for deposition of whole‐exome sequencing genomic data into a public repository. Genomic data from UK10K are available at the EGA European Genomen Phenome Archive (https://www.ebi.ac.uk/ega/home), EGAS00001000128(UK10K RARE FIND). Details of statistical analysis can be shared upon request.