

ABSTRACT
The pH 6 antigen (PsaA) of Yersinia pestis is a virulence factor that is expressed in response to high temperature (37°C) and low pH (6.0). Previous studies have implicated the PsaE and PsaF regulators in the temperature- and pH-dependent regulation of psaA. Here, we show that PsaE levels are themselves controlled by pH and temperature, explaining the regulation of psaA. We identify hundreds of binding sites for PsaE across the Y. pestis genome, with the majority of binding sites located in intergenic regions bound by the nucleoid-associated protein H-NS. However, we detect direct regulation of only two transcripts by PsaE, likely due to displacement of H-NS from the corresponding promoter regions; our data suggest that most PsaE binding sites are nonregulatory or that they require additional environmental cues. We also identify the precise binding sites for PsaE that are required for temperature- and pH-dependent regulation of psaA and psaE. Thus, our data reveal the critical role that PsaE plays in the regulation of psaA and suggest that PsaE may have many additional regulatory targets.
IMPORTANCEY. pestis, the etiologic agent of plague, has been responsible for high mortality in several epidemics throughout human history. The plague bacillus has been used as a biological weapon during human history and is currently one of the most likely biological threats. PsaA and PsaE appear to play important roles during Y. pestis infection. Understanding their regulation by environmental cues would facilitate a solution to impede Y. pestis infection.
KEYWORDS: PsaA (pH 6 antigen), PsaE, Yersinia pestis
INTRODUCTION
Yersinia pestis is the etiologic agent of plague, a zoonotic disease that also occurs in human populations. Y. pestis is one of the most lethal bacterial pathogens and has caused several huge catastrophes in human history (1). Y. pestis uses a range of virulence factors that confer efficient adherence to host cells/tissues, subvert host functions, and enable it to combat host defenses. Several virulence determinants in Y. pestis for responsiveness to environmental cues have been identified (2–4). Among them, PsaA (“pH 6 antigen”) was first characterized in 1961 by Ben-Efraim and coworkers and exists in all strains of Y. pestis. PsaA has been reported to be synthesized only at temperatures above 34°C and at pH values below 6.7 (5). Y. pestis PsaA was reported to be a surface-exposed structure mediating agglutination of erythrocytes from several mammalian species (6). PsaA forms a fimbrial structure with a 3- to 5-nm diameter, and expression was reported to be induced by intracellular association with macrophages (4). Y. pestis PsaA does not enhance adhesion to mouse macrophages but promotes resistance to phagocytosis (7). Yersinia PsaA is responsible for thermo-inducible adhesion to cultured mammalian cells in addition to mediating hemagglutination (4, 6, 8–13).
Synthesis of PsaA is positively regulated by low pH combined with mammalian temperature and by the PsaE and PsaF proteins encoded in the upstream locus (psaEF) (14–16). A previous study reported that in-frame deletion of psaE and psaF greatly reduced synthesis of PsaA and caused defective hemagglutination, presumably due to loss of PsaA synthesis (16). During the course of this study, Quinn et al. (17, 18) also investigated the mechanism of psaA regulation. Consistent with our study, Quinn et al. showed that PsaE and PsaF are transcriptionally and translationally regulated by temperature and posttranslationally regulated by pH (17). In a follow-up study, Quinn et al. showed that PsaF senses pH and protects PsaE from proteolysis (18). Here, we further investigate the mechanism of psaA regulation by PsaE/F in response to pH and temperature, and we map the global PsaE regulon. Our data show that PsaE is a highly specific regulator despite binding hundreds of DNA sites and that psaABC expression responds to environmental signals due to transcriptional and posttranscriptional regulation of PsaE protein levels.
RESULTS
Transcription of Y. pestis psaA is controlled by temperature and pH.
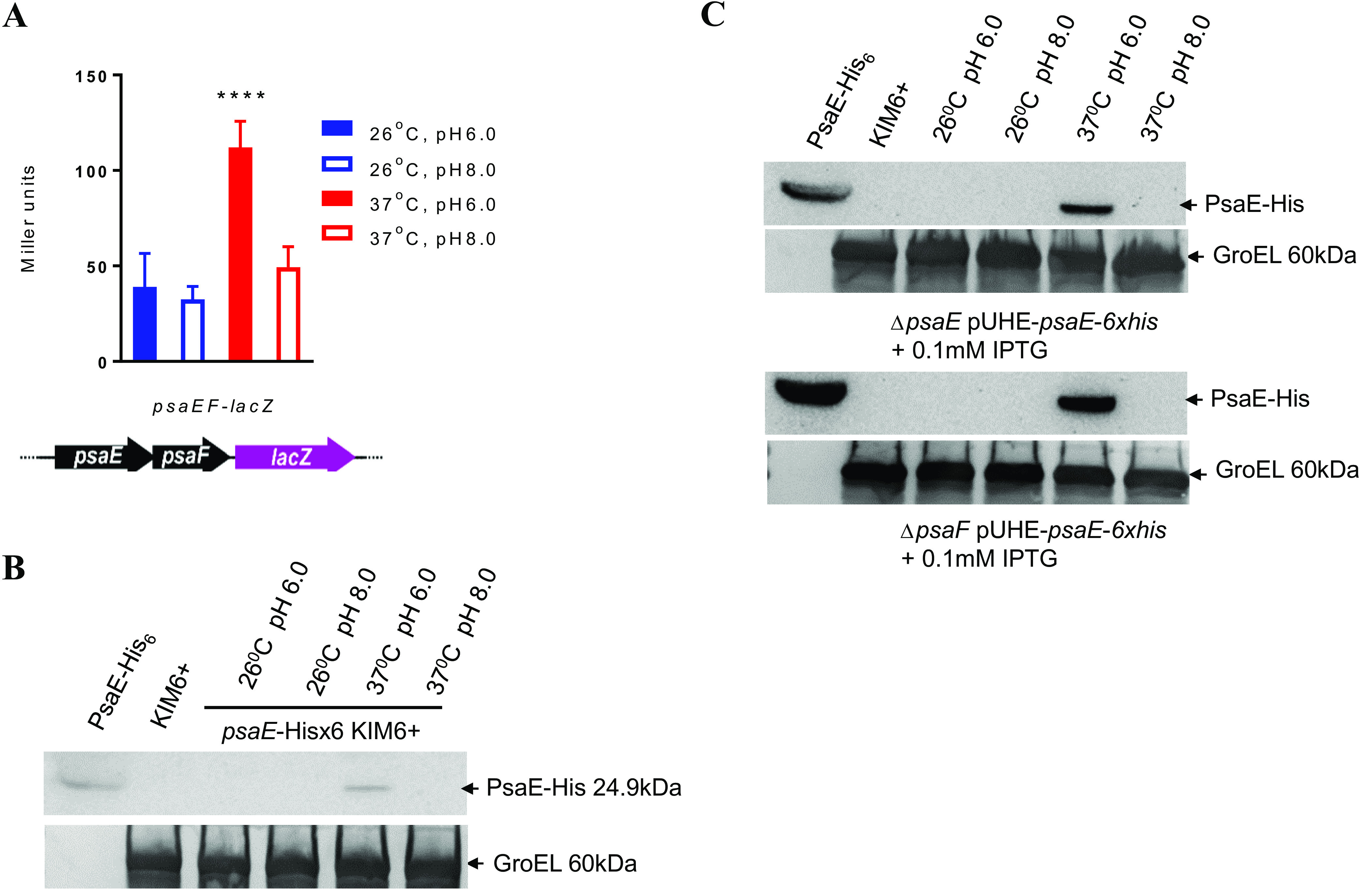
Previous studies showed that Y. pestis psaA expression is only detected in cells grown at temperatures above 36°C and at pH values below 6.7 (5, 14, 15). Using Western blotting with a PsaA-specific antibody, we confirmed this result; we were unable to detect psaA expression from Y. pestis KIM6+ cells grown at 28°C and pH 6.0 (28°C/pH 6.0), 28°C and pH 8.0 (28°C/pH 8.0), and 37°C and pH 8.0 (37°C/pH 8.0), but we detected robust expression at 37°C and pH 6.0 (37°C/pH 6.0) (Fig. 1A).
FIG 1.

Synthesis of PsaA is regulated at the level of transcription by temperature and pH. (A) Western blot showing PsaA protein for wild-type KIM6+ cells grown at the indicated temperature and pH. (B) β-Galactosidase assay of a psaA::lacZY fusion for wild-type KIM6+ cells grown at the indicated temperature and pH. (C) β-Galactosidase assay of a plasmid-borne psaA::lacZ transcriptional fusion (P1, shown in the schematic above the graph) for wild-type KIM6+ cells grown at the indicated temperature and pH. Panel A shows a representative example from three biological replicates. Data in panels B and C are from three independent assays conducted in duplicate. The values shown are means, and error bars show one standard deviation; ****, P < 0.0001.
To determine whether temperature-dependent, pH-dependent regulation of PsaA occurs at the transcriptional or posttranscriptional level, we inserted the lacZY reporter construct downstream of the psaA gene in the Y. pestis KIM6+ chromosome. In this strain, lacZ expression, and hence β-galactosidase activity, is completely dependent upon transcription of the upstream psaA. We detected robust β-galactosidase activity for cells grown at 37°C/pH 6.0 but not for cells grown at 28°C/pH 6.0, 28°C/pH 8.0, or 37°C/pH 8.0 (Fig. 1B). We conclude that temperature and pH modulate expression of psaA at the level of transcription, consistent with previous studies in Y. pestis (14) and Y. pseudotuberculosis (16).
To investigate the chromosomal region required for transcriptional regulation of psaA, we transcriptionally fused the full 529-bp psaA upstream region to a promoterless lacZY in a reporter plasmid (referred to as P1 in Table 1). We then transformed the P1 plasmid into Y. pestis KIM6+ and determined lacZY expression using a β-galactosidase assay. We detected robust β-galactosidase activity for cells grown at 37°C/pH 6.0 but not for cells grown at 28°C/pH 6.0, 28°C/pH 8.0, or 37°C/pH 8.0 (Fig. 1C). We conclude that the cis-acting elements required for temperature- and pH-dependent modulation of psaA expression are located in the upstream region.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E. coli Top10 | F– mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Lab collection |
| χ7213 | thi-1 thr-1 leuB6 fhuA21 lacY1 glnV44 ΔasdA4 recA1 RP4-2-Tc::Mu [λpir] Kmr | 40 |
| Y. pestis KIM6+ | Pgm+, pMT1, pPCP1, cured of pCD1 | 51 |
| YPS21 | psaA::lacZY | Y. pestis KIM6+ |
| YPS22 | ΔpsaE | Y. pestis KIM6+ |
| YPS23 | ΔpsaF | Y. pestis KIM6+ |
| YPS24 | psaEF::lacZY | Y. pestis KIM6+ |
| YPS25 | psaE-6×his | Y. pestis KIM6+ |
| YPS26 | psaF-ha | Y. pestis KIM6+ |
| YPS27 | psaE-3×flag | Y. pestis KIM6+ |
| Plasmids | ||
| pUC18 | For cloning and sequencing | Invitrogen |
| pKD46 | Apr, λ Red recombinase expression | 52 |
| pKD3 | Apr Cmr, cat cassette template | 52 |
| pPCP20 | Apr Cmr, FLP recombinase expression | 52 |
| pKG136 | repR6Kγ Kmr FRT lacZY this | 41 |
| pRE112 | Suicide vector, Cmr Mob− (RP4) R6K ori sacB | 53 |
| pEU730 | 15.2 kb, Spcr Smr, low-copy-number vector with a promoterless lacZ | 54 |
| pUHE21-2lacq | reppMB1 Apr lacIq | 55 |
| pYA4454 | Apr, pSC101 ori | 56 |
| pBAD-HisB | Expression vector | Lab collection |
| pSMV33 | The flanking regions for stop codon of psaA were cloned into pUC18 | This study |
| pSMV34 | FRT-Cm-FRT DNA fragment amplified from pKD3 was cloned into PstI and SacI sites of pSMV30 | This study |
| pSMV35 | The flanking regions for stop codon of psaEF were cloned into pUC18 | This study |
| pSMV36 | FRT-Cm-FRT DNA fragment amplified from pKD3 was cloned into PstI and SacI sites of pSMV32 | This study |
| pSMV37 | The flanking regions for psaE deletion were cloned into KpnI and XmaI sites of pRE112 | This study |
| pSMV38 | The flanking regions for psaF deletion were cloned into KpnI and XmaI sites of pRE112 | This study |
| pSMV39 | The flanking regions for psaE-his were cloned into KpnI and XmaI sites of pRE112 | This study |
| pSMV40 | The flanking regions for psaF-ha was cloned into KpnI and XmaI sites of pRE112 | This study |
| pSMV41 | The psaE-his DNA fragment was cloned into pUHE21-2lacq | This study |
| pSMV42 | The psaF-his DNA fragment was cloned into pUHE21-2lacq | This study |
| pSMV43 (referred to as P1) | The full promoter of psaA (544 bp in front of psaA start codon, PpsaA1) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV44 (referred to as P2) | The promoter of psaA (515 bp in front of psaA start codon, PpsaA2) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV45 (referred to as P3) | The promoter of psaA (475 bp in front of psaA start codon, PpsaA3) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV46 (referred to as P4) | The promoter of psaA (444 bp in front of psaA start codon, PpsaA4) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV47 (referred to as P5) | The promoter of psaA (377 bp in front of psaA start codon, PpsaA5) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV48 (referred to as P6) | The promoter of psaA (333 bp in front of psaA start codon, PpsaA6) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV49 (referred to as P7) |
The promoter of psaA (291 bp in front of psaA start codon, PpsaA7) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV50 (referred to as P8) | The promoter of psaA (240 bp in front of psaA start codon, PpsaA8) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV51 (referred to as P5m) | PpsaA5 promotor with four point mutations cloned into PmeI and KpnI sites of pEU730 | This study |
| pSMV52 (referred to as PEF1) |
The promoter of psaEF (620 bp in front of psaE start codon) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV53 (referred to as PEF2) |
The promoter of psaEF (491 bp in front of psaE start codon) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV54 (referred to as PEF3) |
The promoter of psaEF (390 bp in front of psaE start codon) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV55 (referred to as PEF4) |
The promoter of psaEF (234 bp in front of psaE start codon) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV56 (referred to as PEF5) |
The promoter of psaEF (146 bp in front of psaE start codon) cloned into AscI and KpnI sites of pEU730 | This study |
| pSMV57 (referred to as PEF5m) |
The promoter of psaEF (146 bp in front of psaE start codon with four point mutations) cloned into AscI and KpnI sites of pEU730 | This study |
PsaE and PsaF are required for transcription of psaA.
Previous studies have shown that expression of psaA is dependent upon PsaE in Y. pestis KIM5 and is dependent upon PsaE and PsaF in Y. pseudotuberculosis (14–16). To determine whether PsaE and PsaF regulate Y. pestis psaA expression via the cis-acting element(s) in the upstream region, we introduced the P1 psaA::lacZ reporter plasmid into wild-type Y. pestis KIM6+ and into ΔpsaE and ΔpsaF derivates. We detected robust β-galactosidase activity at 37°C/pH 6.0, and activity was abolished in the ΔpsaE and ΔpsaF mutants (Fig. 2A). Similarly, we detected robust synthesis of PsaA by Western blotting in wild-type Y. pestis KIM6+ but not in the ΔpsaE or ΔpsaF mutants (Fig. 2B). Expression of the psaA::lacZ reporter was restored in the ΔpsaE and ΔpsaF mutants, respectively, upon induced synthesis of respective PsaE or PsaF from a plasmid (Fig. 2C and D). In all cases, expression of the psaA::lacZ reporter was only detected at 37°C/pH 6.0. We conclude that both PsaE and PsaF are required for the transcriptional induction of Y. pestis psaA by temperature and pH and that PsaE and PsaF function through cis-acting elements in the psaA upstream region.
FIG 2.
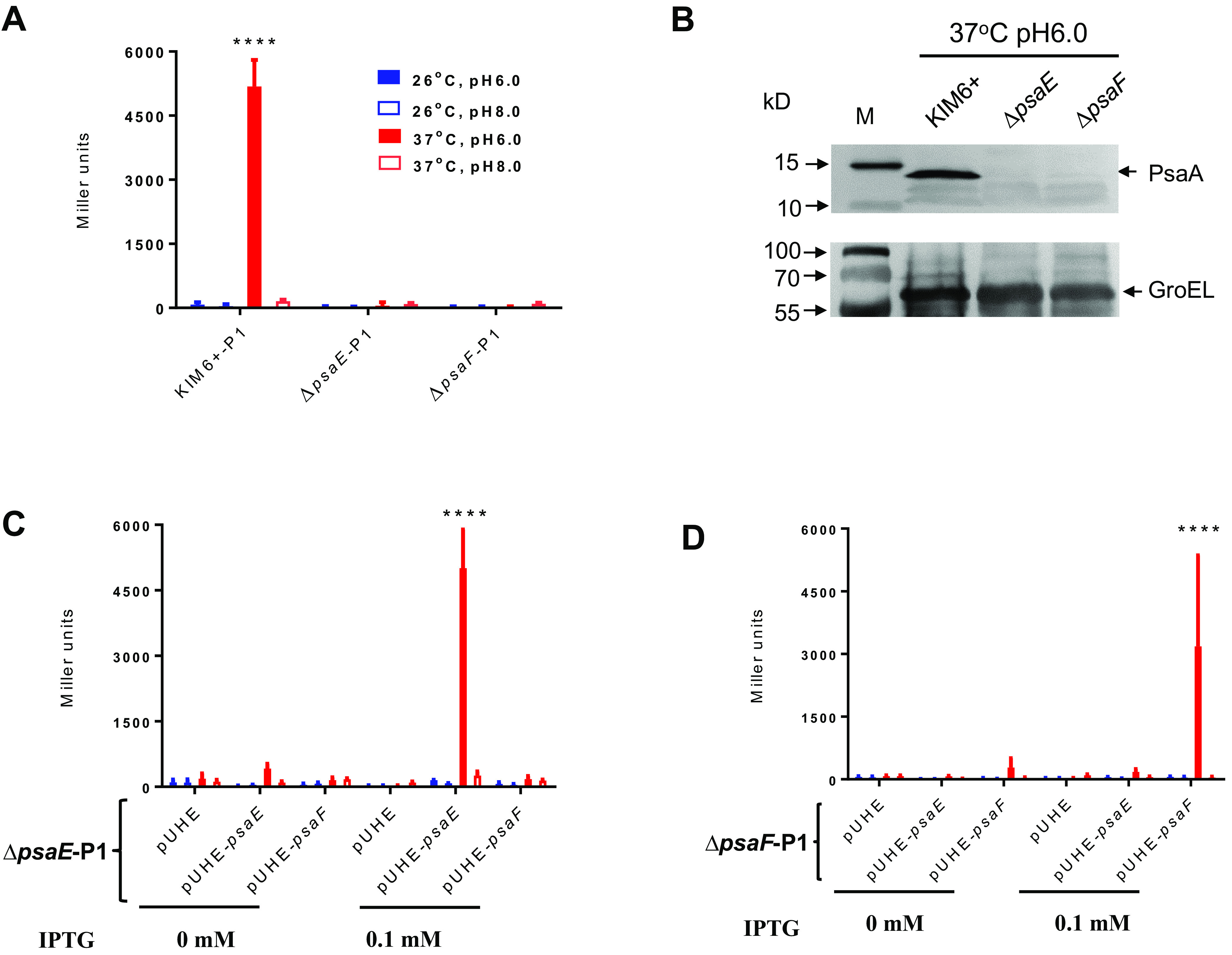
PsaE and PsaF are required for transcriptional activation of psaA. (A) β-Galactosidase activity of the P1 psaA::lacZ transcriptional fusion in wild-type Y. pestis KIM6+, ΔpsaE (YPS22), and ΔpsaF (YPS23) cells grown at the indicated temperature and pH. (B) Western blot showing PsaA protein for wild-type KIM6+ cells, KIM6+ ΔpsaE cells, and KIM6+ ΔpsaF cells grown at 37°C/pH 6.0. (C) β-Galactosidase activity of the P1 psaA::lacZ transcriptional fusion in Y. pestis KIM6+ ΔpsaE (YPS22) cells complemented with empty vector (pUHE), vector expressing psaE (pUHE-psaE), or vector expressing psaF (pUHE-psaF), with or without IPTG inducer and grown at the indicated temperature and pH. (D) β-Galactosidase activity of the P1 psaA::lacZ transcriptional fusion in Y. pestis KIM6+ ΔpsaF (YPS23) cells complemented with empty vector (pUHE), vector expressing psaE (pUHE-psaE), or vector expressing psaF (pUHE-psaF), with or without IPTG inducer and grown at the indicated temperature and pH. Y. pestis KIM6+ cells were used as a positive control. Data are from three independent assays conducted in duplicate; values shown are means, and error bars show one standard deviation; ****, P < 0.0001.
PsaE synthesis is controlled transcriptionally and posttranscriptionally by temperature and pH.
Since regulation of psaA transcription by temperature and pH is dependent on PsaE and PsaF (Fig. 2), we speculated that regulation might be due to changes in PsaE and/or PsaF levels. We inserted the lacZY reporter construct downstream of the psaEF operon in the Y. pestis KIM6+ chromosome. We then measured expression of the psaEF::lacZY fusion using a β-galactosidase assay. We detected β-galactosidase activity for cells grown at 28°C/pH 6.0, 28°C/pH 8.0, 37°C/pH 6.0, and 37°C/pH 8.0. However, activity was ∼3-fold higher for cells grown at 37°C/pH 6.0 than for cells grown under the other conditions (Fig. 3A). We observed a similar, but more pronounced, increase in PsaE protein levels by Western blotting for cells grown at 37°C/pH 6.0 (Fig. 3B).
FIG 3.
The transcription and expression of psaE under four different conditions. (A) β-Galactosidase activity of the psaEF::lacZ transcriptional fusion in wild-type Y. pestis KIM6+ cells grown at the indicated temperature and pH. (B) Western blot showing His-tagged protein synthesis in untagged KIM6+ cells or in KIM6+ cells expressing PsaE-His from its native locus (YPS25). Cells were grown at the indicated temperature and pH. PsaE-His overexpressed and purified from E. coli was run as a control. (C) Western blot showing His-tagged protein synthesis in KIM6+ ΔpsaE (YPS22) or KIM6+ ΔpsaF (YPS23) cells expressing PsaE-His from an inducible plasmid (pUHE-psaE-6xhis) in the presence of the IPTG inducer and grown at the indicated temperature and pH. PsaE-His overexpressed and purified from E. coli was run as a control. Data in panel A are from three independent assays conducted in duplicate. Values shown are means, and error bars show one standard deviation. Panels B and C show representative examples from three biological replicates; ****, P < 0.0001.
A previous study showed that psaE and psaF transcription in Y. pseudotuberculosis is not subject to regulation by variations of temperature or pH (16). Moreover, Y. pestis PsaE protein levels were undetectable under the noninducing conditions, whereas expression of the psaEF::lacZY fusion was only modestly induced by temperature and pH (compare Fig. 3A and B). Therefore, we speculated that PsaE synthesis might also be controlled posttranscriptionally. To test whether PsaE synthesis is regulated at the level of protein production and/or stability, we induced psaE overexpression in Y. pestis KIM6+ ΔpsaE harboring pUHE-psaE-6×his (pSMV41) by isopropyl-β-d-thiogalactopyranoside (IPTG) and measured PsaE protein levels by Western blotting. We only detected PsaE in cells grown at 37°C/pH 6.0 (Fig. 3C), indicating that PsaE is regulated posttranscriptionally by temperature and pH. Posttranscriptional regulation of PsaE was also observed in a Y. pestis KIM6+ ΔpsaF strain, indicating that regulation is not dependent on PsaF (Fig. 3C).
Genome-wide detection of PsaE binding and regulation.
The N-terminal region of PsaE (amino acids 1 to 94) is predicted to bind DNA (P68588), suggesting that PsaE regulates psaA transcription by functioning as a DNA-binding transcription factor. To determine the PsaE regulon, we used a combination of chromatin immunoprecipitation sequencing (ChIP-seq) and RNA sequencing (RNA-seq) to identify sites of PsaE binding and regulation on a genomic scale. ChIP-seq identifies binding sites for DNA-binding proteins but does not provide information on whether binding DNA is associated with transcriptional regulation. RNA-seq can identify changes in RNA levels that are dependent on a transcription factor but cannot be used to determine whether instances of regulation are direct or indirect. By combining ChIP-seq and RNA-seq, it is possible to identify sites of direct regulation.
We respectively constructed PsaE-FLAG and PsaE-His tagged Y. pestis KIM6+ mutant strains, with the tags introduced at the native psaE locus. Insertion of the tags did not affect psaA expression levels (see Fig. S1 in the supplemental material). We then performed ChIP-seq for each of the tagged strains. We detected 261 sites of PsaE enrichment in 218 regions across the chromosome (pairs of sites ≤100 bp apart were considered to be in the same region) and one site of PsaE enrichment on the pMT-1 plasmid, with the pattern of enrichment being very similar for each of the two tagged strains (Fig. 4A). No equivalent enrichment was detected in control ChIP-seq experiments using an untagged strain (Fig. 4A). The lack of enrichment in the control data sets and the similarity in ChIP-seq profiles for the differentially tagged strains indicates that the identified binding sites are genuine. One of the most highly enriched regions was identified upstream of psaE, with strong enrichment also observed nearby upstream of psaA (Fig. 4B). We identified a highly enriched sequence motif within the 219 PsaE-bound regions (Fig. 4C; Table S1), with an instance of the motif being found in each of the 219 regions. The sequence motif was significantly centrally enriched with respect to the peaks of ChIP-seq signal (P = 1.8E−10), strongly suggesting that the motif corresponds to the PsaE-binding sequence.
FIG 4.

ChIP-seq of PsaE identifies 219 binding sites that are strongly associated with a sequence motif. (A) Overview of the ChIP-seq data for PsaE-His6, Psa-FLAG, and untagged controls. For the untagged control data sets, the antibody used in the ChIP-seq is indicated in parentheses. Graphs show sequence read coverage on the plus strand from ChIP-enriched samples (y axis) across the entire Y. pestis chromosome (x axis). Peaks correspond to PsaE binding sites. Values on the y axis are capped at one-tenth the level of the most enriched region in the PsaE-His data set, and the maximum value shown in each plot is normalized between data sets based on the total number of sequence reads that align to the reference genome. (B) Zoomed-in view of the psa locus. ChIP-seq data are shown for the PsaE-FLAG data set, and RNA-seq data are shown for wild-type KIM6+ and KIM6+ ΔpsaE strains. Gene positions are indicated in the schematic above the graph. The black box indicates the location of the putative PsaE binding site mutated in the P5m construct (see Fig. 6). (C) A strongly enriched motif was associated with PsaE ChIP-seq peaks.
Most ChIP-seq studies of bacterial DNA-binding transcription factors identify less than 50 sites. However, in some cases, transcription factors have been observed to bind at hundreds of DNA sites, as is the case for PsaE (19). In cases where transcription factors bind more than 100 sites, most sites are usually located within genes, suggesting a random distribution of nonregulatory sites across the chromosome (19). Strikingly, 60% of chromosomal PsaE-bound sites fall in intergenic regions, a far higher fraction than the 14% of the chromosome that is intergenic. Thus, PsaE binding is strongly enriched for intergenic regions. Moreover, PsaE binding is strongly associated with A/T-rich sequence, with only four of the 219 PsaE-bound regions having an A/T content below 50%; the median A/T content of PsaE-bound regions is 67.3%. By contrast, the A/T content of the Y. pestis KIM6+ chromosome is 52.3%. Consistent with PsaE binding in regions of high A/T content, the PsaE-binding site motif is strongly A/T-rich (Fig. 4C).
We next determined which genes are regulated by PsaE by using RNA-seq to compare RNA levels genome wide in wild-type Y. pestis KIM6+ and a ΔpsaE mutant grown at 37°C/pH 6.0. Thus, we identified 125 differentially regulated genes between the two strains (false-discovery rate [q value] of ≤0.01, ≥2-fold difference in RNA levels between wild-type and ΔpsaE cells) (Fig. 5; Table S2). The most strongly differentially regulated genes were psaA, psaB, psaC, and psaF (Fig. 5). A previous study suggested that psaA is transcribed as a monocistronic RNA (14). Although we observed PsaE-dependent regulation of the two genes downstream of psaA (i.e., psaB and psaC), the absolute RNA level for psaA is ∼1,000 times higher than that for psaB and psaC, strongly suggesting that the observed regulation of psaB and psaC by PsaE is due to low-level readthrough of the psaA transcription terminator. Of the remaining 121 PsaE-regulated genes, only 8 are located within a window from −100 to +500 relative to PsaE-bound regions identified by ChIP-seq (Fig. 5; Table S2), and these genes are associated with relatively weak PsaE binding sites, as determined by the level of ChIP-seq enrichment. Given the large number of PsaE binding sites identified by ChIP-seq, we estimate that ∼4 differentially expressed genes would be found within a window from −100 to +500 relative to a PsaE-bound region by chance. We conclude that PsaE directly regulates very few genes under the growth conditions tested, despite the large number of intergenic binding sites.
FIG 5.

RNA-seq of wild-type and ΔpsaE cells reveals regulation of few transcripts. Scatter plot showing relative RNA levels for all genes as determined by RNA-seq for wild-type cells (x axis) and ΔpsaE mutant cells (y axis). The psaA, psaB, psaC, psaE, and psaF genes are shown in orange.
Overlap of PsaE binding sites with H-NS-bound regions.
The nucleoid-associated protein H-NS has been shown to bind long stretches of A/T-rich sequence in the Escherichia coli and Salmonella Typhimurium genomes (20–24). Given the high A/T content around most PsaE binding sites, we speculated that these sites might overlap with regions bound by H-NS. We used ChIP-seq to map the association of H-NS with the Y. pestis genome for cells grown at 37°C/pH 6.0. Consistent with studies of E. coli and S. Typhimurium, we observed H-NS association across long stretches of DNA, with high H-NS occupancy covering regions of >10 kb in some cases. We determined a set of 552 “high H-NS occupancy” regions, corresponding to 10% of the Y. pestis chromosome (Table S3). A previous study identified several genes in Yersinia enterocolitica that are strongly bound by H-NS (25). Six of these genes have homologs in Y. pestis: y0517, y0666, y3192, y3490, y3791, and psaF, and all except y0666 overlap with high H-NS occupancy regions. Moreover, we observed a strong correlation between H-NS occupancy and A/T content, consistent with the known propensity of H-NS to bind A/T-rich DNA; the A/T content of high H-NS occupancy regions is 64.2%, compared to 52.3% for the complete genome. Remarkably, most PsaE binding sites are associated with regions of high H-NS occupancy (Fig. 6), with 50% of all chromosomal PsaE sites located in high H-NS occupancy regions. Thus, there is a high degree of overlap between sites of PsaE binding and sites of H-NS binding.
FIG 6.

H-NS association with chromosomal regions ± psaE. Scatter plot showing normalized H-NS ChIP-seq sequence coverage around PsaE ChIP-seq peaks and 1,000 random chromosomal coordinates for wild-type (wt) and ΔpsaE cells. Data points are highlighted for PsaE ChIP-seq peaks upstream of psaE (“psaE us”) and psaA (“psaA us”) and within psaF (“psaF”).
Previous studies have shown that transcription factors can activate transcription by displacing H-NS bound to promoter regions (reviewed in references 26 and 27). Given that most PsaE sites are associated with high H-NS occupancy, we hypothesized that PsaE could function to displace H-NS in regions around PsaE binding sites. To test this hypothesis, we mapped the association of H-NS with the genome of a ΔpsaE strain grown at 37°C/pH 6.0. Comparison of H-NS occupancy between the psaE+ and ΔpsaE strains revealed an almost identical binding profile of H-NS across the chromosome, with H-NS occupancy unaffected by deletion of psaE regardless of whether the H-NS-bound region included a binding site for PsaE (Fig. 6). The one clear exception to this rule is the H-NS-bound region that covers the psaE, psaF, psaA, and psaB genes; H-NS occupancy in this region was substantially higher in ΔpsaE cells than in psaE+ cells (Fig. 6). It is formally possible that the absence of the DNA corresponding to the psaE gene is the cause of the increase in H-NS occupancy in this region; however, this absence of psaE DNA would be unlikely to affect H-NS occupancy across the entire >2.5-kb H-NS-bound region covering psaF, psaA, and psaB.
Identifying the site of PsaE binding in the psaA upstream region.
Our data suggest that PsaE regulates transcription of psaA using a cis-acting element in the psaA upstream region. Given that we identified binding of PsaE upstream of psaA, we speculated that one or more PsaE binding sites in the psaA upstream region is required for regulation by PsaE. There are several good matches to the PsaE binding motif in the psaA upstream region. Consistent with this, the profile of ChIP-seq enrichment suggests the presence of at least two PsaE binding sites (Fig. 4B). To begin to map the site of PsaE binding, we made derivatives of the P1 psaA::lacZ reporter plasmid with progressive truncations of the start of the psaA upstream region. We refer to these reporter plasmids as P2, P3, P4, P5, P6, P7, and P8 (Fig. 7A). We introduced these plasmids into Y. pestis KIM6+ and measured β-galactosidase activity in cells grown at 28°C/pH 6.0, 28°C/pH 8.0, 37°C/pH 6.0, or 37°C/pH 8.0. For all reporter fusions, no activity was observed under noninducing conditions (i.e., 28°C/pH 6.0, 28°C/pH 8.0, and 37°C/pH 8.0) (Fig. 7B). We detected robust β-galactosidase activity under inducing conditions (i.e., 37°C/pH 6.0) for P1 to P6 but not for P7 or P8 (Fig. 7B). These data strongly suggest that the key binding site(s) for PsaE is located in the region from −86 to −44 relative to the transcription start site of psaA (+1). Consistent with this idea, the region from −86 to −44 contains two strong matches to the PsaE-binding motif identified from ChIP-seq data (Fig. 7A), and the profile of ChIP-seq enrichment matches the position of these sequences (Fig. 4B).
FIG 7.

Mapping the PsaE binding region of the psaA promoter. (A) Schematic representation of constructs used for analyzing the sequence requirements for transcription activation of psaA by PsaE. Bent arrows indicate truncations of the P1 reporter construct (P2 to P8). Pink-highlighted bases indicate predicted PsaE binding sites that were mutated in the P5m construct. The yellow-highlighted base indicates the transcription start site. A line under nucleotides indicates potential binding site of PsaE. The black arrows indicate nucleotide replacements in P5m. (B) β-Galactosidase activity of the P1 to P8 constructs or empty vector (pEU730) in wild-type KIM6+ cells grown at the indicated temperature and pH. (C) β-Galactosidase activity of the P5 and P5m constructs in wild-type KIM6+ cells grown at the indicated temperature and pH. Data are from three independent assays conducted in duplicate; values shown are means, and error bars show one standard deviation; ****, P < 0.0001.
To test whether the predicted PsaE binding sites are required for transcription activation of psaA by PsaE, we modified the P5 plasmid by introducing two substitutions in each putative site that alter key positions that we predict are required for PsaE binding based on the binding site motif (compare Fig. 7A and Fig. 4C). We refer to this mutated plasmid as P5m. We then introduced the P5 and P5m plasmids into Y. pestis KIM6+ and measured β-galactosidase activity in cells grown at 28°C/pH 6.0, 28°C/pH 8.0, 37°C/pH 6.0, or 37°C/pH 8.0. Expression of the psaA::lacZ fusion under inducing conditions was abolished in the P5m mutant construct (Fig. 7C). These data strongly suggest that PsaE binds to one or both of the putative sites in the region from −333 to −292 and that binding to one or both of these sites is critical for transcription activation of psaA.
Characterization of psaE autoregulation.
PsaE protein levels are tightly controlled by temperature and pH (Fig. 3C), but differences in psaEF transcription for the same growth conditions are modest in comparison (Fig. 3A). Since we observed strong association of PsaE upstream of the psaE gene, and we detected lower expression of psaF in ΔpsaE cells than in wild-type cells by RNA-seq, we speculated that the transcriptional induction of psaEF by high temperature/low pH is due to autoregulation. To test this hypothesis, we cloned different lengths of the psaEF promoter region into a promoterless lacZ reporter plasmid (Fig. 8A). We introduced these plasmids into Y. pestis KIM6+ and measured β-galactosidase activity in cells grown at 28°C/pH 6.0, 28°C/pH 8.0, 37°C/pH 6.0, or 37°C/pH 8.0. We detected expression of all reporter fusions under all growth conditions, but expression was highest for all fusions at 37°C/pH 6.0. and lowest at 28°C regardless of the pH (Fig. 8B).
FIG 8.

Mapping the PsaE binding region of the psaEF promoter. (A) Schematic representation of constructs used for analyzing the sequence requirements for transcription activation of psaEF by PsaE. Bent arrows indicate truncations of the P1 reporter construct (PEF 2 to 5). Pink-highlighted bases indicate predicted PsaE binding sites that were mutated in the P5m1 construct. The cyan-highlighted base indicates the transcription start site. Purple-highlighted text indicates the predicted binding site for PsaE, with nucleotide replacements in PEF5m indicated by green-highlighted text. (B) β-Galactosidase activity of the PEF1 to 5 constructs or empty vector (pEU730) in wild-type KIM6+ cells grown at the indicated temperature and pH. (C) β-Galactosidase activity of the PEF5 and PEF5m constructs in wild-type KIM6+ cells grown at the indicated temperature and pH. Data are from three independent assays conducted in duplicate. Values shown are means, and error bars show one standard deviation; ****, P < 0.0001.
The shortest reporter fusion (PEF-5) extends 62 bp upstream of the expected transcription start site (28) and includes the predicted PsaE binding site (Fig. 8A). To test whether transcription of psaEF is activated by PsaE binding to this predicted site, we compared expression of the PEF-5 reporter fusion in wild-type and ΔpsaE cells under all four growth conditions. Deletion of psaE led to an ∼2-fold reduction in PEF-5 expression at 26°C regardless of pH. Moreover, deletion of psaE led to an ∼2-fold reduction of PEF-5 expression at 37°C/pH 6.0; expression of PEF-5 was similar between psaE+ and ΔpsaE cells at 37°C/pH 8.0. Thus, deletion of psaE abolishes the effect of pH on expression of psaEF. To test whether the effect of psaE on psaEF expression is due directly to PsaE binding upstream of psaE, we introduced three substitutions in the predicted PsaE binding site in the PEF-5 reporter fusion; these substitutions alter key positions that we predict are required for PsaE binding based on the binding site motif (compare Fig. 8A and Fig. 4C).The effect of mutating the putative PsaE binding site on expression of psaEF mirrored the effect of deleting psaE (Fig. 8C), consistent with PsaE directly regulating psaEF.
DISCUSSION
Our data support a model in which PsaE activates transcription of psaA by binding to one or more DNA sites in the psaA upstream region. Moreover, we have shown that PsaE levels are controlled posttranscriptionally by temperature and pH, explaining why psaA transcription is activated at 37°C/pH 6.0. During the course of this study, Quinn et al. (17) also investigated the mechanism of psaA regulation. Consistent with our study, Quinn et al. showed that PsaE and PsaF are transcriptionally and translationally regulated by temperature and posttranslationally regulated by pH. However, in contrast to our study, Quinn et al. did not observe transcriptional regulation of psaEF by pH (Fig. 3A). Additionally, a previous study of psaA regulation in Y. pseudotuberculosis showed that psaEF transcription is not regulated by temperature or pH (16). Our data support a model in which psaEF is autoregulated, such that changes in psaEF transcription caused by temperature and pH reflect changes in PsaE protein levels. Given the differences between studies, we suggest that the mechanisms by which temperature and pH control PsaE activity may differ between strains. Nonetheless, control of PsaE activity by temperature and pH appears to be a widespread phenomenon, since a psaA homolog in Yersinia enterocolitica, myfA, is also transcriptionally activated by high temperature and low pH and requires homologs of psaE and psaF for maximal expression (29).
PsaE has an unusually large number of binding sites across the Y. pestis genome (219 putative binding sites). For comparison, ChIP-seq data for cAMP receptor protein (CRP) in enterotoxigenic E. coli identified 111 sites, and CRP is considered to be a global regulator with one of the largest regulons of any E. coli transcription factor (30). Some bacterial transcription factors bind large numbers of sites that appear to lack regulatory activity (31, 32). However, these sites are typically distributed proportionally between intergenic and genic locations, which means that most fall within genes since bacterial genomes are composed mainly of genic sequence. By contrast, PsaE sites are highly enriched for intergenic regions (binomial test, P < 1E−15), strongly suggesting that PsaE regulates transcription of the downstream genes and may be a global regulator, directly controlling the transcription of hundreds of genes. However, we observed strong PsaE-dependent regulation of only two operons, psaABC and psaEF; the proximity of a few weakly PsaE-regulated genes to PsaE binding sites is likely coincidental.
A potentially simple explanation for the large proportion of intergenic PsaE binding sites is that the binding motif is very A/T rich, and intergenic regions tend to be more A/T rich than genes. To test this possibility, we scored every 11-nucleotide (nt) sequence in the Y. pestis genome for similarity to a scrambled version of the PsaE binding motif (Fig. 4C). We selected the sequences with the top 1,000 scores and discarded 4 sequences that mapped to plasmid sequence. We reasoned that the remaining 996 sequences have similar sequence content to PsaE-bound sequences. We refer to these sequences as “motif-like.” Consistent with intergenic regions being more A/T rich than genes, 42% of motif-like sequences are located in intergenic regions, a higher proportion than the 14% of the genome that is intergenic. However, 61% of PsaE sites are intergenic, a significantly higher proportion than the 42% of motif-like sequences (Fisher’s exact test P = 7E−7). Put another way, there are ∼41 more intergenic PsaE binding sites than expected by chance given the sequence content of the motif, suggesting that many intergenic PsaE binding sites are under purifying selection.
One possible explanation for why PsaE binding is enriched in intragenic regions but is associated with so little detectable regulation is that regulation may require additional factors that are inactive under the in vitro growth conditions we used. Consistent with this idea, several other transcription factors have been implicated in controlling expression of psaA, including RovA (25, 28, 33), CpxR (34), PhoP (28), and Fur (35). Intriguingly, several PsaE binding sites, including the site with the most enrichment in the ChIP-seq data, are upstream of genes associated with iron acquisition, feoABC, yiuA, and ftn, suggesting that PsaE may collaborate with the same transcription factor to regulate these genes.
An alternative possibility to explain the lack of detected regulation by PsaE is that the majority of PsaE binding sites are nonregulatory and that regulation by PsaA requires strict spacing requirements relative to promoter sequences. The psaA transcription start site has been mapped (28), and our data (Fig. 7) indicate that PsaE binds ∼50 bp upstream of this site. This is consistent with the location of activator proteins relative to the binding site of RNA polymerase. Many activator proteins need to be precisely positioned on the DNA relative to promoter sequences to be effective (36). Hence, it is possible that the majority of PsaE sites are not suitably positioned to activate transcription. However, this does not explain the enrichment of PsaE sites in intergenic regions.
PsaE binding sites show a remarkable degree of overlap with regions bound by H-NS (Fig. 6). This is likely due, at least in part, to the high A/T content of the PsaE binding motif since H-NS also binds A/T-rich sequence. Previous studies of species, including Y. pestis, have shown that DNA-binding transcription factors can displace H-NS bound to promoter regions, resulting in transcription activation of the associated genes (26). Our data indicate that at almost all of its binding sites, PsaE does not displace H-NS, consistent with the lack of transcriptional regulation associated with these sites. However, deletion of psaE does lead to a substantial increase in the association of H-NS across psaF and psaABC, suggesting that PsaE binding upstream of psaE and psaA displaces H-NS, leading to transcription activation. An alternative explanation for the effect of psaE deletion on H-NS occupancy at psaF and psaABC is that PsaE activates transcription independent of H-NS, but the act of transcription itself displaces H-NS as RNA polymerase transcribes through these regions, as has been described previously for H-NS-silenced genes in E. coli (37).
In conclusion, we have characterized the regulation of Y. pestis psaA by temperature and pH, revealing a key role for PsaE binding to one or more promoter-proximal sites. Several key unanswered questions remain. First, what is the role of PsaA during infection? Second, how does pH impact the stability of PsaE and PsaF? Third, are there additional PsaE-regulated genes that are activated in a condition-specific manner?
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains used in this study and their sources are listed in Table 1. Conditions and media used for growing strains of Y. pestis and E. coli were described previously (38). For determination of PsaA synthesis under various growth conditions, Y. pestis was grown in heart infusion broth (HIB) (Difco) adjusted to either pH 8.0 or pH 6.0 with NaOH or HCl, respectively. After autoclaving, 2.5 mM CaCl2 and 0.2% (wt/vol) xylose were added (15). Chloramphenicol (Cm), spectinomycin (Sp), and ampicillin (Ap) were added to the medium at final concentrations of 20, 50, and 100 μg/ml, respectively.
Plasmid construction.
All plasmids used in this study are described in Table 1. Primers used for PCR amplification are listed in Table S4 in the supplemental material. DNA manipulations were performed following standard methods (39), and all constructed plasmids were confirmed by DNA sequencing.
Strain construction.
To construct Y. pestis mutant strains, the corresponding suicide plasmid was conjugationally transferred from E. coli χ7213 (40) to the Y. pestis KIM6+ strain. Single-crossover insertion strains were isolated on tryptose blood agar (TBA) plates containing Cm. Loss of the suicide vector sequence after the second recombination between homologous regions (i.e., allelic exchange) was selected by using the SacB-based sucrose sensitivity counterselection system (38). A lacZY gene cassette was integrated in the deleted chromosomal psaA or psaEF location using plasmid pKG136, which was inserted into the flippase (FLP) recombination target sequence generated after the flippase recognition target (FRT)-Cmr-FRT cassette was removed using plasmid pCP20 (41, 42).
Western blotting.
Bacteria grown under different conditions were pelleted, boiled in gel loading dye, and separated by SDS-PAGE with 12.5% polyacrylamide. Proteins were transferred to nitrocellulose membranes for Western blotting analysis. Loading was normalized based on optical density at 600 nm (OD600) of bacterial culture. Membranes were blocked with 5% skim milk in phosphate-buffered saline (PBS) and incubated with primary antibody, specifically rabbit polyclonal anti-PsaA (1:10,000) and monoclonal antibody (Mab) anti-His (1:2,000) to probe for PsaA and PsaE-His6, respectively. Membranes were then incubated with goat anti-rabbit IgG alkaline phosphatase-conjugated (1:10,000) or goat anti-mouse IgG horseradish peroxidase (HRP)-conjugated (1:10,000) secondary antibody (Sigma, St. Louis, MO). Immunoreactive bands were detected by the addition of nitroblue tetrazolium−5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP) (Sigma, St. Louis, MO) and chemiluminescent substrates (Bio-Rad, CA).
β-Galactosidase assays.
β-Galactosidase assays (43) were performed in triplicate, and activity was determined using a SpectraMax 340PC plate reader (Molecular Devices). The data correspond to three independent assays conducted in duplicate, and values shown are the means, with error bars indicating one standard deviation.
ChIP-seq.
For ChIP-seq of PsaE, KIM6+ and YPS24 (psaE-his) were grown in HIB at 37°C, pH 6, to an OD600 of 0.6 to 0.8. For ChIP-seq of H-NS, KIM6+ and YPS22 (ΔpsaE) were grown in HIB at 37°C, pH 6, to an OD600 of 0.6 to 0.8. Sonicated lysates were prepared as previously described (44). ChIP-seq libraries were prepared as previously described (44) using 2 μg of anti-His tag monoclonal antibody and 25 μl of protein G-Sepharose slurry, 2 μl of M2 anti-FLAG monoclonal antibody (Sigma), and 25 μl of protein A-Sepharose slurry or 5 μl of anti-H-NS polyclonal antibody (Cusabio Technology, CSB-PA359764EA11ENV) and 25 μl of protein A-Sepharose slurry. Sequencing was performed using an Illumina Next-Seq instrument (Wadsworth Center Applied Genomic Technologies Core). Sequence reads were aligned to the Y. pestis KIM6+ chromosome sequence using the CLC Genomics Workbench (version 9). Statistically significantly (false-discovery rate of 0.01) enriched regions were identified as previously described (45), treating the PsaE-His and PsaE-FLAG data sets as independent replicates. Sequence reads for PsaE-FLAG, PsaE-His, and the corresponding untagged control ChIP-seq data sets were also aligned to the Y. pestis pMT-1 plasmid sequence using the CLC Genomics Workbench (version 9). Plasmid copy number was determined by comparing the read density on the pMT-1 plasmid to that on the chromosome for control ChIP-seq data from an untagged strain. Statistically significantly (false-discovery rate of 0.01) enriched regions were identified as previously described (45), except that coverage thresholds were normalized to account for plasmid copy number.
Analysis of H-NS occupancy.
Sequence reads were aligned to the Y. pestis KIM6+ chromosome using Rockhopper (version 2.03) to generate wiggle files with normalized coverage at all chromosome positions on each strand. Regions of high H-NS occupancy (Table S3) were identified by requiring at least 100 bp of contiguous sequence with normalized read counts of >499 at each position on each strand for each ChIP-seq replicate data set (wild-type KIM6+ data only).
To compare H-NS occupancy between wild-type Y. pestis KIM6+ and ΔpsaE, we first extracted 101-bp sequences centered on each chromosomal PsaE ChIP-seq peak. In cases where adjacent ChIP-seq peaks were less than 100 bp apart, the 101-bp sequences were merged, and a 101-bp sequence from the center of the region was extracted. We also extracted 1,000 101-bp sequences from randomly selected chromosomal coordinates. For the sets of sequences from PsaE ChIP-seq peaks and random coordinates, we determined the normalized sequence read coverage across each window in wild-type Y. pestis KIM6+ cells and in ΔpsaE cells for each replicate experiment. Values plotted in Fig. 6 are the average of two replicates.
ChIP-seq motif analysis.
We extracted 101 bp of DNA sequence centered around each ChIP-seq peak. We merged ChIP-seq peaks within 100 bp of each other. We used MEME (version 5.3.3; default settings run through the MEME-ChIP server) to identify an enriched sequence motif (46, 47). The positions of motifs identified by MEME were compared to the ChIP-seq peak center positions using Centrimo (48) (version 5.3.3; default settings).
The motif derived from MEME was scrambled by randomly repositioning each of the positions, thereby maintaining the overall nucleotide content. Genomic sequences were scored for their match to the scrambled motif using FIMO (49) (version 5.3.3; default parameters except 0.001 was used as the P value threshold). The top 1,000 sequences were selected, and 4 were discarded because they matched plasmid sequence.
RNA-seq.
RNA-seq was performed in strains KIM6+ and YPS21 (ΔpsaE). Bacteria cultured overnight in pH 8.0 HIB medium at 28°C were collected, reinoculated in pH 6 HIB medium, and cultured at 37°C for 4 h. Cells (1 ml) were pelleted in a microcentrifuge for 1 min at full speed and washed once with Tris-buffered saline. RNA was purified from bacterial cells using the Quick-RNA Miniprep Plus kit (Zymo Research). Duplicate samples were prepared from independent biological replicates for each condition/strain. RNA (6 μg) was treated with 3 U of Turbo DNase I (Invitrogen) for 45 min at 37°C. RNA was then acid phenol extracted and ethanol precipitated. rRNA was removed using the RiboZero kit (Illumina). RNA-seq libraries were prepared using the ScriptSeq 2.0 kit (Illumina). Sequencing was performed using an Illumina Next-Seq instrument (Wadsworth Center Applied Genomic Technologies Core). Differential RNA expression analysis was performed using Rockhopper (version 2.03) with default parameters (50). Differences in RNA levels were considered to indicate regulation for genes, with false-discovery rate (q) values of ≤0.01 and fold change values of ≥2.
Statistical analyses.
Statistical analyses of data among groups were evaluated by two-way analysis of variance (ANOVA) and Tukey’s multiple-comparison test. Data were analyzed using GraphPad Prism 8.0 software. Data are represented as mean values ± standard deviation.
Data availability.
Raw ChIP-seq and RNA-seq data are available from European Bioinformatics Institute (EBI) ArrayExpress with accession numbers E-MTAB-8369, E-MTAB-10694, and E-MTAB-8370.
ACKNOWLEDGMENTS
We thank Roy Curtiss III for providing E. coli and Y. pestis strains and different plasmids in this study. We thank the Wadsworth Center Applied Genomic Technologies Core Facility for DNA sequencing. We thank the Wadsworth Center Bioinformatics Core Facility, Tissue Culture and Media Core Facility, and Glassware Facility for technical support. We thank Pascal Lapierre for technical assistance.
This work was primarily supported by Albany Medical College start-up funds and was partially supported by National Institutes of Health grants R01AI125623 and R21AI139703 to W.S. This work was also supported by National Institutes of Health grant R01GM114812 to J.T.W.
W.S., Y.S., and J.T.W. conceived and designed the experiments. P.L., X.W., C.S., and W.S. performed the experiments. W.S., Y.S., C.S., and J.T.W. analyzed the data. W.S., Y.S., and J.T.W. contributed reagents/materials/analysis tools. W.S., Y.S., and J.T.W. wrote the paper.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Joseph T. Wade, Email: joseph.wade@health.ny.gov.
Wei Sun, Email: sunw@amc.edu.
Laurie E. Comstock, Brigham and Women’s Hospital/Harvard Medical School
REFERENCES
- 1.Perry RD, Fetherston JD. 1997. Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev 10:35–66. 10.1128/CMR.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cornelis GR. 2002. Yersinia type III secretion: send in the effectors. J Cell Biol 158:401–408. 10.1083/jcb.200205077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamashita S, Lukacik P, Barnard TJ, Noinaj N, Felek S, Tsang TM, Krukonis ES, Hinnebusch BJ, Buchanan SK. 2011. Structural insights into Ail-mediated adhesion in Yersinia pestis. Structure 19:1672–1682. 10.1016/j.str.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindler LE, Tall BD. 1993. Yersinia pestis pH 6 antigen forms fimbriae and is induced by intracellular association with macrophages. Mol Microbiol 8:311–324. 10.1111/j.1365-2958.1993.tb01575.x. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Efraim S, Aronson M, Bichowsky-Slomnicki L. 1961. New antigen component of Pasteurella pestis formed under specified conditions of pH and temperature. J Bacteriol 81:704–714. 10.1128/jb.81.5.704-714.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bichowsky-Slomnicki L, Ben-Efraim S. 1963. Biological activities in extracts of Pasteurella pestis and their relation to the “pH 6 antigen”. J Bacteriol 86:101–111. 10.1128/jb.86.1.101-111.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang XZ, Lindler LE. 2004. The pH 6 antigen is an antiphagocytic factor produced by Yersinia pestis independent of yersinia outer proteins and capsule antigen. Infect Immun 72:7212–7219. 10.1128/IAI.72.12.7212-7219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Felek S, Runco LM, Thanassi DG, Krukonis ES. 2007. Characterization of six novel chaperone/usher systems in Yersinia pestis. Adv Exp Med Biol 603:97–105. 10.1007/978-0-387-72124-8_8. [DOI] [PubMed] [Google Scholar]
- 9.Galvan EM, Chen H, Schifferli DM. 2007. The Psa fimbriae of Yersinia pestis interact with phosphatidylcholine on alveolar epithelial cells and pulmonary surfactant. Infect Immun 75:1272–1279. 10.1128/IAI.01153-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Merriam JJ, Mueller JP, Isberg RR. 1996. The psa locus is responsible for thermoinducible binding of Yersinia pseudotuberculosis to cultured cells. Infect Immun 64:2483–2489. 10.1128/iai.64.7.2483-2489.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Payne D, Tatham D, Williamson ED, Titball RW. 1998. The pH 6 antigen of Yersinia pestis binds to β1-linked galactosyl residues in glycosphingolipids. Infect Immun 66:4545–4548. 10.1128/IAI.66.9.4545-4548.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zav'yalov VP, Abramov VM, Cherepanov PG, Spirina GV, Chernovskaya TV, Vasiliev AM, Zav'yalova GA. 1996. pH 6 antigen (PsaA protein) of Yersinia pestis, a novel bacterial Fc-receptor. FEMS Immunol Med Microbiol 14:53–57. 10.1111/j.1574-695X.1996.tb00267.x. [DOI] [PubMed] [Google Scholar]
- 13.Makoveichuk E, Cherepanov P, Lundberg S, Forsberg A, Olivecrona G. 2003. pH 6 antigen of Yersinia pestis interacts with plasma lipoproteins and cell membranes. J Lipid Res 44:320–330. 10.1194/jlr.M200182-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Price SB, Freeman MD, Yeh KS. 1995. Transcriptional analysis of the Yersinia pestis pH 6 antigen gene. J Bacteriol 177:5997–6000. 10.1128/jb.177.20.5997-6000.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindler LE, Klempner MS, Straley SC. 1990. Yersinia pestis pH 6 antigen: genetic, biochemical, and virulence characterization of a protein involved in the pathogenesis of bubonic plague. Infect Immun 58:2569–2577. 10.1128/iai.58.8.2569-2577.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Isberg RR. 1997. Transcriptional regulation of the Yersinia pseudotuberculosis pH 6 antigen adhesin by two envelope-associated components. Mol Microbiol 24:499–510. 10.1046/j.1365-2958.1997.3511719.x. [DOI] [PubMed] [Google Scholar]
- 17.Quinn JD, Weening EH, Miner TA, Miller VL. 2019. Temperature control of psaA expression by PsaE and PsaF in Yersinia pestis. J Bacteriol 201:e00217-19. 10.1128/JB.00217-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quinn JD, Weening EH, Miller VL. 2021. PsaF is a membrane-localized pH sensor that regulates psaA expression in Y. pestis. J Bacteriol 10.1128/JB.00165-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, Gomes A, Rustad T, Dolganov G, Glotova I, Abeel T, Mahwinney C, Kennedy AD, Allard R, Brabant W, Krueger A, Jaini S, Honda B, Yu WH, Hickey MJ, Zucker J, Garay C, Weiner B, Sisk P, Stolte C, Winkler JK, Van de Peer Y, Iazzetti P, Camacho D, Dreyfuss J, Liu Y, Dorhoi A, Mollenkopf HJ, Drogaris P, Lamontagne J, Zhou Y, Piquenot J, Park ST, Raman S, Kaufmann SH, Mohney RP, Chelsky D, Moody DB, Sherman DR, Schoolnik GK. 2013. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499:178–183. 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grainger DC, Hurd D, Goldberg MD, Busby SJ. 2006. Association of nucleoid proteins with coding and non-coding segments of the Escherichia coli genome. Nucleic Acids Res 34:4642–4652. 10.1093/nar/gkl542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC. 2006. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog 2:e81. 10.1371/journal.ppat.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navarre WW, Porwollik S, Wang Y, McClelland M, Rosen H, Libby SJ, Fang FC. 2006. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313:236–238. 10.1126/science.1128794. [DOI] [PubMed] [Google Scholar]
- 23.Oshima T, Ishikawa S, Kurokawa K, Aiba H, Ogasawara N. 2006. Escherichia coli histone-like protein H-NS preferentially binds to horizontally acquired DNA in association with RNA polymerase. DNA Res 13:141–153. 10.1093/dnares/dsl009. [DOI] [PubMed] [Google Scholar]
- 24.Shen AB, Hustmyer MC, Roston D, Wolfe BM, Jessen DE, Landick R. 2021. Multimodal multiscale analysis reveals flexible DNA contacts of bacterial nucleoprotein filaments. bioRxiv 10.1101/2020.06.11.146589. [DOI]
- 25.Cathelyn JS, Ellison DW, Hinchliffe SJ, Wren BW, Miller VL. 2007. The RovA regulons of Yersinia enterocolitica and Yersinia pestis are distinct: evidence that many RovA-regulated genes were acquired more recently than the core genome. Mol Microbiol 66:189–205. 10.1111/j.1365-2958.2007.05907.x. [DOI] [PubMed] [Google Scholar]
- 26.Stoebel DM, Free A, Dorman CJ. 2008. Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology (Reading) 154:2533–2545. 10.1099/mic.0.2008/020693-0. [DOI] [PubMed] [Google Scholar]
- 27.Will WR, Navarre WW, Fang FC. 2015. Integrated circuits: how transcriptional silencing and counter-silencing facilitate bacterial evolution. Curr Opin Microbiol 23:8–13. 10.1016/j.mib.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Wang L, Fang N, Qu S, Tan Y, Guo Z, Qiu J, Zhou D, Yang R. 2013. Reciprocal regulation of pH 6 antigen gene loci by PhoP and RovA in Yersinia pestis biovar Microtus. Future Microbiol 8:271–280. 10.2217/fmb.12.146. [DOI] [PubMed] [Google Scholar]
- 29.Iriarte M, Cornelis GR. 1995. MyfF, an element of the network regulating the synthesis of fibrillae in Yersinia enterocolitica. J Bacteriol 177:738–744. 10.1128/jb.177.3.738-744.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haycocks JR, Sharma P, Stringer AM, Wade JT, Grainger DC. 2015. The molecular basis for control of ETEC enterotoxin expression in response to environment and host. PLoS Pathog 11:e1004605. 10.1371/journal.ppat.1004605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galagan J, Lyubetskaya A, Gomes A. 2013. ChIP-seq and the complexity of bacterial transcriptional regulation. Curr Top Microbiol Immunol 363:43–68. 10.1007/82_2012_257. [DOI] [PubMed] [Google Scholar]
- 32.Wade JT, Struhl K, Busby SJ, Grainger DC. 2007. Genomic analysis of protein-DNA interactions in bacteria: insights into transcription and chromosome organization. Mol Microbiol 65:21–26. 10.1111/j.1365-2958.2007.05781.x. [DOI] [PubMed] [Google Scholar]
- 33.Cathelyn JS, Crosby SD, Lathem WW, Goldman WE, Miller VL. 2006. RovA, a global regulator of Yersinia pestis, specifically required for bubonic plague. Proc Natl Acad Sci USA 103:13514–13519. 10.1073/pnas.0603456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JF, Obi IR, Thanikkal EJ, Kieselbach T, Francis MS. 2011. Phosphorylated CpxR restricts production of the RovA global regulator in Yersinia pseudotuberculosis. PLoS One 6:e23314. 10.1371/journal.pone.0023314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou D, Qin L, Han Y, Qui J, Chen Z, Li B, Song Y, Wang J, Guo Z, Zhai J, Du Z, Wang X, Yang R. 2006. Global analysis of iron assimilation and fur regulation in Yersinia pestis. FEMS Microbiol Lett 258:9–17. 10.1111/j.1574-6968.2006.00208.x. [DOI] [PubMed] [Google Scholar]
- 36.Lee DJ, Minchin SD, Busby SJ. 2012. Activating transcription in bacteria. Annu Rev Microbiol 66:125–152. 10.1146/annurev-micro-092611-150012. [DOI] [PubMed] [Google Scholar]
- 37.Rangarajan AA, Schnetz K. 2018. Interference of transcription across H-NS binding sites and repression by H-NS. Mol Microbiol 108:226–239. 10.1111/mmi.13926. [DOI] [PubMed] [Google Scholar]
- 38.Sun W, Wang S, Curtiss R. 2008. Highly efficient method for introducing successive multiple scarless gene deletions and markerless gene insertions into the Yersinia pestis chromosome. Appl Environ Microbiol 74:4241–4245. 10.1128/AEM.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambrook JE, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed.Cold Spring Harbor Laboratory Press, New York, NY. [Google Scholar]
- 40.Wang S, Li Y, Scarpellini G, Kong W, Shi H, Baek CH, Gunn B, Wanda SY, Roland KL, Zhang X, Senechal-Willis P, Curtiss R III, 2010. Salmonella vaccine vectors displaying delayed antigen synthesis in vivo to enhance immunogenicity. Infect Immun 78:3969–3980. 10.1128/IAI.00444-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. 10.1016/s0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- 42.Song HW, Kong W, Weatherspoon N, Qin GZ, Tyler W, Turk J, Curtiss R, Shi YX. 2008. Modulation of the regulatory activity of bacterial two-component systems by SlyA. J Biol Chem 283:28158–28168. 10.1074/jbc.M801058200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller JH (ed). 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 44.Bonocora RP, Wade JT. 2015. ChIP-seq for genome-scale analysis of bacterial DNA-binding proteins. Methods Mol Biol 1276:327–340. 10.1007/978-1-4939-2392-2_20. [DOI] [PubMed] [Google Scholar]
- 45.Fitzgerald DM, Bonocora RP, Wade JT. 2014. Comprehensive mapping of the Escherichia coli flagellar regulatory network. PLoS Genet 10:e1004649. 10.1371/journal.pgen.1004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2:28–36. [PubMed] [Google Scholar]
- 47.Machanick P, Bailey TL. 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27:1696–1697. 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey TL, Machanick P. 2012. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40:e128. 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-seq data. Nucleic Acids Res 41:e140. 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gong S, Bearden SW, Geoffroy VA, Fetherston JD, Perry RD. 2001. Characterization of the Yersinia pestis Yfu ABC inorganic iron transport system. Infect Immun 69:2829–2837. 10.1128/IAI.67.5.2829-2837.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwards RA, Keller LH, Schifferli DM. 1998. Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207:149–157. 10.1016/s0378-1119(97)00619-7. [DOI] [PubMed] [Google Scholar]
- 54.Froehlich B, Husmann L, Caron J, Scott JR. 1994. Regulation of rns, a positive regulatory factor for pili of enterotoxigenic Escherichia coli. J Bacteriol 176:5385–5392. 10.1128/jb.176.17.5385-5392.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soncini FC, Vescovi EG, Groisman EA. 1995. Transcriptional autoregulation of the Salmonella typhimurium phoPQ operon. J Bacteriol 177:4364–4371. 10.1128/jb.177.15.4364-4371.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun W, Six DA, Reynolds CM, Chung HS, Raetz CR, Curtiss R III.. 2013. Pathogenicity of Yersinia pestis synthesis of 1-dephosphorylated lipid A. Infect Immun 81:1172–1185. 10.1128/IAI.01403-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental text, Table S4, and Fig. S1. Download JB.00237-21-s0001.pdf, PDF file, 0.1 MB (152.5KB, pdf)
Tables S1 to S3. Download JB.00237-21-s0002.xlsx, XLSX file, 0.3 MB (349.5KB, xlsx)
Data Availability Statement
Raw ChIP-seq and RNA-seq data are available from European Bioinformatics Institute (EBI) ArrayExpress with accession numbers E-MTAB-8369, E-MTAB-10694, and E-MTAB-8370.