

Abstract
The development and progression of cardiovascular disease (CVD) can mainly be attributed to the narrowing of blood vessels caused by atherosclerosis and thrombosis, which induces organ damage that will result in end-organ dysfunction characterized by events such as myocardial infarction or stroke. It is also essential to consider other contributory factors to CVD, including cardiac remodelling caused by cardiomyopathies and co-morbidities with other diseases such as chronic kidney disease. Besides, there is a growing amount of evidence linking the gut microbiota to CVD through several metabolic pathways. Hence, it is of utmost importance to decipher the underlying molecular mechanisms associated with these disease states to elucidate the development and progression of CVD. A wide array of systems biology approaches incorporating multi-omics data have emerged as an invaluable tool in establishing alterations in specific cell types and identifying modifications in signalling events that promote disease development. Here, we review recent studies that apply multi-omics approaches to further understand the underlying causes of CVD and provide possible treatment strategies by identifying novel drug targets and biomarkers. We also discuss very recent advances in gut microbiota research with an emphasis on how diet and microbial composition can impact the development of CVD. Finally, we present various biological network analyses and other independent studies that have been employed for providing mechanistic explanation and developing treatment strategies for end-stage CVD, namely myocardial infarction and stroke.
Keywords: systems biology, cardiovascular disease, omics integration, integrated networks, genome-scale metabolic model
Introduction
Cardiovascular disease (CVD), specifically coronary heart disease and stroke, remain the leading cause of death worldwide accounting for a combined annual total of over 17 million deaths [1]. A significant number of these deaths are linked to chronic kidney disease (CKD) with ~8.5% of global heart disease deaths and ~8% of stroke-related deaths attributable to decreased kidney function [2]. Most CVD-related complications are caused by ischemic CVD [3, 4], which is associated with the accumulation of fat inside the arteries (atherosclerosis) coupled with an increased risk of blood clots (thrombosis). Consequently, the rupture of atherosclerotic plaques and the formation of blood clots are the dominant cause of myocardial infarction, heart failure and stroke [5, 6]. Besides, heart failure can also occur through non-ischemic CVD, characterized by adverse cardiac remodelling and cardiomyopathies that mainly occur through left ventricular cavity remodelling [7], thus further emphasizing the complexity of CVD. The progression of these CVDs can primarily be attributed to disturbances in both global and cardiac-specific metabolism [8]. Furthermore, specific CVD components are influenced by genetic and epigenetic factors, whether it be mutations causing adverse cardiac remodelling in heart failure [9] or modifications that affect the susceptibility of the arterial wall to plaque formation [10].
Although the prevalence of CVD continues to significantly increase worldwide, a detailed understanding of the molecular mechanisms associated with the underlying causes of CVD is still lacking in some areas, particularly for non-ischemic CVD. To rectify this, extensive efforts and resources have been placed into systems biology approaches, which attempt to further understand the underlying conditions of CVD to improve early detection and treatment of CVD. The recent advances in high-throughput technology have enhanced researchers’ ability to analyse whole genomes, transcriptomes, proteomes, metabolomes and metagenomes, thus facilitating the integrative analysis of multi-omics data to identify causal genes and reveal underlying molecular mechanisms that are involved in the progression of cardiovascular events [11]. In particular, the development of genome-scale metabolic models (GEMs) allows for describing molecular variation and quantifying flux through metabolic pathways in different physiological systems [12, 13]. To date, several GEMs have been developed to further understand the heart-specific metabolic processes in health and disease [14–16]. Moreover, this wealth of data has highlighted the diverse pathological aspects of CVD, which will be vital in facilitating risk stratification of patients and optimizing intervention to prevent CVD progression.
There is a growing body of literature supporting the notion that specific metabolites secreted by the gut microbiome can increase cardiovascular risk based on recent studies associating elevated levels of lipopolysaccharides (LPS) [17], gammabutyrobetaine [18], trimethyllysine [19] and uremic toxins including indoxyl sulphate, indole-3-acetic acid and P-Cresyl sulphate [20], with increased risk of CVD events. Most notably, there is a substantial amount of evidence linking elevated levels of the gut microbe-derived trimethylamine-N-oxide (TMAO) to CVD [18, 21–23] and CKD [24] through a mechanism involving oxidation of trimethylamine (TMA) to TMAO in the liver. The subsequent circulation of TMAO in the bloodstream can induce mechanistic changes in several different cell types including (i) increased levels of scavenger receptors in macrophages which alter the cell phenotype and result in the production of foam cells, (ii) increases in NFκB activation, adhesion proteins and NLRP3 inflammasome activation coupled with a decrease in nitric oxide production in endothelial cells (ECs) and (iii) platelet hyper-reactivity through increased levels of Ca2+ release. Furthermore, these studies showed that the microbial production of TMA is strongly dependent on dietary precursors, and hence it is a combination of microbial metabolism and dietary interactions that are involved in the development of CVD. It has also recently been shown that the gut microbiota composition can alter various serum metabolite concentrations that are significantly associated with coronary artery disease (CAD) severity [25]. Hence, integrating metagenomics with other omics approaches (i.e. genomics, transcriptomics, proteomics, metabolomics) to study host-microbiome interactions will allow for advanced panomics enabling deep phenotyping that could prove invaluable in developing precision medicine for the treatment of CVD [26].
In this review, we first summarize how systems biology approaches have been applied towards mechanistically explaining the different underlying causes of CVD including atherosclerosis, thrombosis, adverse cardiac remodelling and CKD. We mainly focus on studies that identify biomarkers for stratification and promote the development of personalized treatment strategies based on a patient’s unique phenotype. Secondly, we assess the current state of gut microbiome research in explaining CVD pathophysiology accounting for the effect of diet. Moreover, we review the application of multi-omics analyses to the gut microbiota and discuss identification of gut microbe-derived metabolites that have been proposed as biomarkers for major adverse cardiac events. Finally, we comprehensively review the application of integrated multi-omics systems biology methods for end-stage CVD mainly characterized by CAD and ischaemic stroke.
Multi-omics approaches towards explaining the underlying causes of CVD
Atherosclerosis
Atherosclerosis is characterized by artery blockages that arise from plaque deposits consisting of cholesterol, lipid proteins, cellular waste products and fibrin [27]. The prerequisite for plaque formation is the presence of low-density lipoprotein (LDL)-cholesterol, particularly in oxidized form [28]. Over the past 50 years, the progression of atherosclerosis has been linked to a multitude of risk factors, including hyperlipidaemia, behavioural, metabolism-related, and genetic risk factors, together with systemic changes in the body (Figure 1A). Previous research centred around the observation of related clinical variables and autopsy studies showed that behavioural risk factors, including physical inactivity [29, 30], unhealthy diet [31, 32], stress, and alcohol and tobacco abuse [33, 34], play a significant role in the progression of atherosclerosis. Moreover, the aforementioned risk factors cause alterations in metabolism [35–37] related to the onset of hypertension, obesity, diabetes and hyperlipidaemia, which are also risk factors for atherosclerosis. In addition, it has been shown that genetic factors [38, 39], such as elevated levels of apolipoprotein B (apoB)-containing lipoproteins and to a lesser extent increased levels of LDL and very-LDLs, are associated with the disease, together with increased blood pressure, decreased high-density lipoproteins levels and family history. Considering several risk factors exist for atherosclerosis, there has been an increased focus on discovering pleiotropic effects of variants in lipoprotein metabolism genes with a recent phenome-wide association study (PheWAS) identifying two LDL receptor variants as having several significant lipid-related phenotype associations [40]. Furthermore, several studies have connected atherosclerosis with systemic inflammatory changes in the body, such as the increase of pro-inflammatory proteins in adipose [41] and liver [42]. Specifically, in adipose tissue, it has been shown that an increase in free fatty acid levels led to the increase of multiple lipoproteins that cause lipid deposits in the arteries.
Figure 1 .
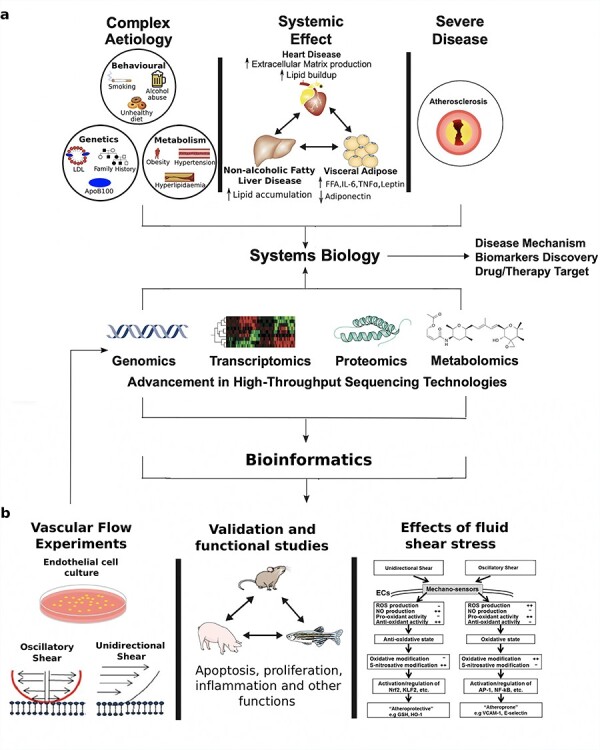
Schematic pathway for systems biology studies that utilize different omics data (genomics, transcriptomics, proteomics and metabolomics) to (A) further elucidate the complex aetiology and systemic effects arising from different tissues (heart, liver and visceral adipose) that contribute towards the development of atherosclerosis, and (B) investigate endothelial cell responses to flow and determine how different fluid shear stress states can promote either an ‘atheroprotective’ or ‘atheroprone’ state.
The complex aetiology and systemic effects of atherosclerosis, combined with its severity, has contributed towards its current status as one of the most studied diseases. Recent advancements in high-throughput sequencing and systems biology have allowed for a greater understanding of the underlying molecular mechanisms that contribute to atherosclerosis development and progression. As previously discussed, atherosclerosis is associated with genetic factors, and this has resulted in several genome-wide association studies (GWAS) with subsequent identification of multiple loci that are associated with atherosclerosis [43–45]. Moreover, studies with transcriptomics technologies have provided an overview of the global transcriptome map of the disease and revealed important candidate biomarker genes, biological processes, and pathways related to the disease in several species [46–49]. For example, an in vitro study detailing the profile and functions of long non-coding RNAs (lncRNA) and their association with atherosclerosis revealed the molecular mechanism surrounding ANRIL’s pro-atherogenic effect, which was validated in primary human cells from patients [50]. Furthermore, circular non-coding RNA of this same gene has been shown to induce processes that lead to apoptosis and inhibition of cell proliferation in human vascular cell and tissues, which are critical cellular functions closely related to atherosclerosis [51]. A separate study showed 180 differentially expressed lncRNA when comparing healthy samples with those from atherosclerosis-induced ischemic stroke patients, with SCARNA8 and SNRPN-2 being the most significant differentially expressed lncRNAs between the two sample types. In addition to tissue-level transcriptomics analysis, single-cell maps of atherosclerotic plaques have recently been built [52].
Other omics approaches, including proteomics [53, 54], lipidomics [55, 56] and metabolomics [57, 58], have been leveraged to provide overall disease profile and reveal possible early-stage biomarkers that can be used to further our understanding regarding the molecular mechanism of the disease (Figure 1A). This includes a very recent proteomics-based assessment which identified NPC2 and IGFBP7 as novel candidate biomarkers for atherosclerotic aortic aneurysm in humans [54]. In addition, a large-scale metabolomics study revealed 42 significantly altered metabolites in coronary atherosclerosis patients compared to controls, with nine of these metabolites combinatorically identifying the early stage of the disease with high accuracy (AUC = 0.890) [58] and belonging to a phospholipid, fatty acid, fructose and fucose, and tryptophan metabolism.
The continuous frictional force of blood flow across the vascular endothelium surface results in hemodynamic stress and pressure changes throughout each cardiac cycle. This interaction between the endothelium and fluid shear stress (FSS) is essential in maintaining vascular homeostasis and in protecting against or promoting atherosclerosis. Sustained unidirectional laminar shear (US) forces upregulate expression of genes and proteins in the endothelium that are protective against atherosclerosis. In contrast, oscillatory shear (OS) forces result in atherogenesis through the upregulation of EC genes and proteins that promote oxidative conditions and inflammatory states in the artery wall [59–61]. It is also now commonly accepted that FSS induces intimal hyperplasia and increased retention of subendothelial accumulation of apoB-containing lipoproteins [62, 63].
The study of EC mechanobiology mainly takes place through in vitro experiments using techniques such as microarray [64] and next-generation sequencing [65], thus enabling a global view of the multiple signalling events occurring in response to US or OS (Figure 1B). For example, experimental data gleaned from a systematic time-series RNA-sequencing dataset was used for pathway analysis and construction of tissue factor (TF)-to-gene networks with the results providing insights into the dynamics of functional response of ECs to shear stress over time [65]. This included upregulation of many genes in several pathways involving inflammation, oxidative stress, cell cycle and angiogenesis, with initiation and termination of pathway upregulation occurring at different time points. For example, several early response genes involved in inflammation (IL8, MCP-1, SELE and VCAM1) showed changes during 2–9 h following the activation of TF nuclear factor-κB (NF-κB) by upregulation of genes contributing to reactive oxygen species production by 2 h. Hence, this combined gene-TF-phenotypic network was able to identify transcriptional regulation mechanisms for cellular functions that lead to defined endothelial phenotypes. Consequently, this provided insights into the causality of mechanisms leading to stress responses.
Further high-throughput, omics-based approaches include studies of shear stress regulation of the endothelial proteome [66] and DNA methylome [67]. Interestingly, flow-regulated proteins included several transforming growth factor-β (TGFβ) superfamily members, angiopoietin 2 (ANG2), vascular endothelial growth factor (VEGF) [68] and Notch receptors [69]. A previous PheWAS also found that a single nucleotide polymorphism in the NOTCH2 gene was associated with increased risk of hypertension [70]. The regulation of genome-wide DNA methylation patterns by blood flow has been shown in pig ECs with differentially methylated regions being associated with oxidative stress, thus implicating this process in early-stage atherogenesis [67]. Flow has also been shown to alter the methylation of homeobox family genes in both mouse and pig models [67, 71]. To summarize, these different omics studies have revealed that FSS regulates various molecules that control diverse physiological processes, hence providing invaluable clues to the molecular mechanisms underlying atherosclerosis.
The large data sets produced by omics approaches can also be integrated to increase the power towards discovering novel molecules and pathways, such as the integration of microarray flow data with a bisulfite sequencing genome-wide methylation array, which identified genes downregulated by OS and containing hypermethylated promoter regions [71]. Of the identified genes, HOXA5 and KLF3 encode TFs containing cAMP response elements, supporting the notion that these loci’s methylation status could be acting as a mechanosensitive master switch in gene expression. These novel findings demonstrated that OS controls epigenomic DNA methylation patterns, which alters endothelial gene expression and promotes atherosclerosis. Future studies should explore the dynamics of proteins from distinctly expressed genes in shear, which would likely produce novel insights into mechanisms of atherosclerosis. Current limitations in the field:
Platelet signalling and thrombosis
In addition to ECs and circulating coagulation proteins, platelets are crucial mediators of thrombosis and vascular haemostasis with the latter process maintaining regulation of blood flow and vascular integrity. Pathological factors may overwhelm normal haemostasis, leading to uncontrolled clot formation and blood vessel occlusion [72]. Traditional epidemiological studies identified several arterial thrombotic risk factors, including hypertension, high LDL-cholesterol levels and smoking. Family cohort studies have also identified hereditary thrombophilia as an additional risk factor associated with an increased risk of arterial thromboembolism [73].
Furthermore, a meta-analysis of cohort studies revealed that mean platelet volume (MPV) might be a useful prognostic marker in patients with CVD with elevated MPV values found in patients with acute MI or coronary angioplasty [74]. The human platelet proteome contains ~4000 unique proteins with the majority of these defined in a protein interaction network (PlateletWeb) [75] consisting of 13 652 interactions and 1704 detected phosphorylation events. Besides, a platelet GEM [76] has been constructed to aid further understanding of pathophysiological conditions through data-driven, systems analysis of platelet metabolism. Numerous genetically modified mouse models have been previously developed to explore genotype/phenotype linkages with regards to clot formation with defects in various platelet receptors, and membrane proteins (e.g. GPVI [glycoprotein VI] and P2Y12 [P2Y purinergic receptor, type Y12]) found to slow and reduce clot growth whilst enhancing embolization [77]. There have been several applications of transcriptomics approaches towards understanding platelet responsiveness, including a previous study which examined individual heterogeneity in platelet response to ADP and collagen-related peptide with 63 different genes being implicated in influencing platelet responsiveness [78]. Furthermore, an association study of several of these genes showed a putative association with myocardial infarction for COMMD7 (COMM domain-containing protein 7) and a highly significant association for LRRFIP1 (leucine-rich repeat (in FLII) interacting protein 1). Both genes were positive regulators of thrombus formation with proteomics analysis indicating LRRFIP1 functions as a component of the platelet cytoskeleton and interacts with actin-remodelling proteins Flighless-1 and Drebrin [78]. A separate population study [79] assessing platelet hyperresponsiveness to arachidonic acid, ADP and PAR1/4-activating peptides through integrated plateletomics revealed many mRNAs and microRNAs (miRNA) which were differentially expressed by age and gender. Networks of miRNAs targeting mRNAs were identified for both categories, and the inverse relationship in these RNA pairs suggested that miRNAs regulate mRNA levels based on age and sex, hence implying that future platelet RNA association studies must account for these variables.
The multiscale and hierarchical nature of thrombosis requires a systems biology approach that can describe platelet and plasma function through events including platelet membrane receptor function, intracellular signalling and assembly of extracellular processes occurring in plasma or on platelet membranes [80]. There have been successful attempts in silico to model platelet signalling through kinetic models, including a full bottom-up model of platelet intracellular metabolism that incorporated phosphoinositide and calcium regulation [81]. This model accurately predicted steady-state resting concentrations and transient increases in intracellular calcium and inositol trisphosphate in response to ADP. Furthermore, several kinetic models of protease cascades have been developed to predict blood coagulation, the most widely used being the Kuharsky–Fogelson model [82] which considers platelet activation, deposition and mass transfer of reactive species. This model was also able to confirm the exceptional sensitivity of coagulation to initial conditions of tissue factor levels through experimentally validated [83] predictions of threshold concentrations of surface tissue factor that triggered clotting in human blood.
The aforementioned bottom-up signalling approaches were applied to individual receptor pathways and did not address signalling through combinatorial and time-dependent activators. Hence, another approach is required to capture the full process of platelets forming a core region through interaction with thrombus and collagen, with subsequent platelets in the growing thrombus shell being regulated by ADP and thromboxane signalling [84]. This integration of these numerous signals in human platelets (Figure 2A) was captured using a high-throughput assay that measured intracellular calcium levels in response to pairwise agonist scanning (PAS) and predicted cellular signalling responses to combinatorial stimuli through the training of a neural network (NN) model (Figure 2B) for prediction of entire platelet response space [85] (Figure 2C). This NN model could then successfully be applied to predict responses to sequential additions of agonists and ternary stimulation with experimental validation in a clinical setting confirming predictions such as a high-dimensional thrombotic risk at high thromboxane A2/prostaglandin I2 ratio consistent with the known cardiovascular risks of COX2 inhibitors [86]. The PAS approach in this study was limited to six agonists and further expansion of this PAS set would be required to map a major portion of the entire platelet response space by including agonists such as epinephrine and nitric oxide donors.
Figure 2 .
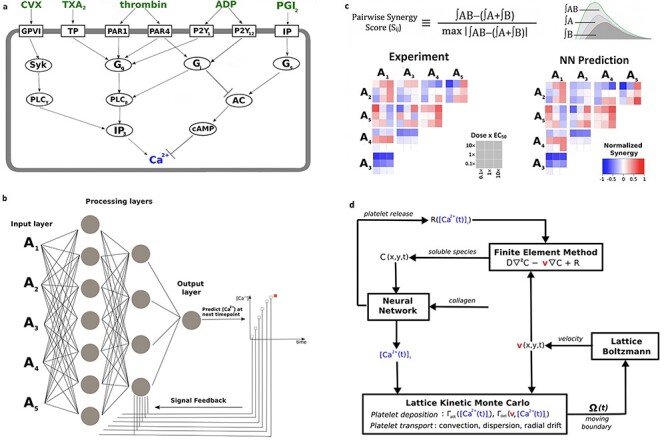
(A) Several signalling pathways in platelets converge on intracellular calcium release. (B) A dynamic neural network is used to train platelet response to combinatorial agonist activation. An input layer consisting of agonist concentrations is introduced to the 2-layer network at each time point and processing layers integrate input values with feedback signals for prediction of [Ca2+]i at the next timepoint. (C) Pairwise agonist synergy scores reflect gains or losses in calcium response due to agonist cross talk and are calculated for both experimental and predicted time-course traces to assess efficiency of the neural network. (D) Multiscale model of platelet deposition and thrombus formation under flow—this requires simultaneous solution of (1) intracellular platelet state ([Ca2+]i) and release reactions (R) for ADP and TXA2 calculated by Neural Network, (2) the instantaneous velocity field over a complex evolving platelet boundary □(t) calculated by Lattice Boltzmann, (3) concentration fields of ADP and TXA2 calculated by Finite Element Method, and (4) all platelet positions and attachment/detachment by Lattice Kinetic Monte Carlo.
A more complete systems biology analysis of blood clotting requires comprehensive models which must account for extreme sensitivity to initial conditions, strong autocatalytic feedback and non-linearity of kinetic rates in order to successfully predict system outcomes that lead to thrombosis [87]. Patient-specific NN models of platelet activation allowed for larger scale simulations of thrombosis under flow through embedment of the NN model into a multiscale model [88] (Figure 2D) which incorporated three further sub-models: lattice Boltzmann, finite element method and lattice kinetic Monte Carlo [89]. These sub-models mentioned above accounted for changes in blood flow with increasing platelet deposition inside the blood vessel, the release of soluble platelet agonists (ADP and TXA2) and the platelets’ motion within the blood flow, respectively. This enabled multiscale prediction of donor-specific clotting events under different flow and pharmacologic modulation with a predictive accuracy of the simulations validated through real-time platelet deposition measurement using microfluidic devices. These findings offered a crucial breakthrough in describing how a myocardial infarction would progress based on a patient’s unique platelet phenotype. It was the first multiscale model approach that made donor-specific predictions of platelet function under flow in the presence of agonists and pharmacologic modulators of clinical relevance. For example, one of the study donors was insensitive to the COX-1 inhibitors aspirin and indomethacin under flow conditions with this trait exactly predicted in the multiscale modelling. Future studies should continue on this focus of developing tools to define platelet variations between patients and stratify them according to risk as determined by the relationship of platelet phenotype to prothrombotic events.
Cardiac remodelling
Mechanical changes in myocardial cells and the myocardial dysfunction that arises from such changes can activate left ventricular cavity remodelling serving as a prelude towards heart failure and ultimately multi-organ damage. The application of systems biology in this area is essential in defining the early mechanical stresses and relevant functional modules to elucidate ventricular remodelling and the complex pathophysiology of heart failure [90]. The most common type of ventricular remodelling is dilation of the left ventricle with the associated disease termed dilated cardiomyopathy (DCM) and origins of cause not yet known. Hypertrophic cardiomyopathy (HCM) is a genetic disease that causes the left ventricle walls to become thicker and contract harder through mutations in contractile sarcomeric proteins. The established causal genes for HCM, including MYH7 (myosin heavy chain 7) and MYBPC3 (Myosin Binding Protein C3), account for ≅60% of HCM cases meaning the causal genes have not yet been identified in ≅40% of HCM patients [91]. A multitude of systems biology approaches have been employed to further understand the underlying molecular mechanisms of these two conditions.
There have been several studies towards explaining the aetiology of DCM, including a recent transcriptome analysis of human heart failure that revealed dysregulated cell adhesion in DCM [92]. Additionally, the use of animal models has provided new insights into mutations responsible for the onset of DCM, including a mutation in the Integrin-linked kinase gene in zebrafish [93] that was later described in patients with DCM [94]. Another successful application of animal models in this area includes a time-course mouse model of DCM which showed perturbations in apoptosis, integrin and MAPK (mitogen-activated protein kinase) signalling at the early- and mid-disease stage of DCM through global proteomic profiling and enrichment maps [95]. Further human-based studies have employed network-based comparative approaches [96, 97] that integrated DCM related gene expression profiles with protein–protein interaction data and biological function annotations to produce condition-specific co-expression networks (Figure 3A). A comparative analysis was then employed to extract DCM exclusive subnetworks that could be used to classify normal and disease samples (Figure 3B). This approach revealed dynamic functional modules corresponding to muscle contraction and organ morphogenesis that are closely related to heart failure and hence provided new insights into the cause of DCM.
Figure 3 .
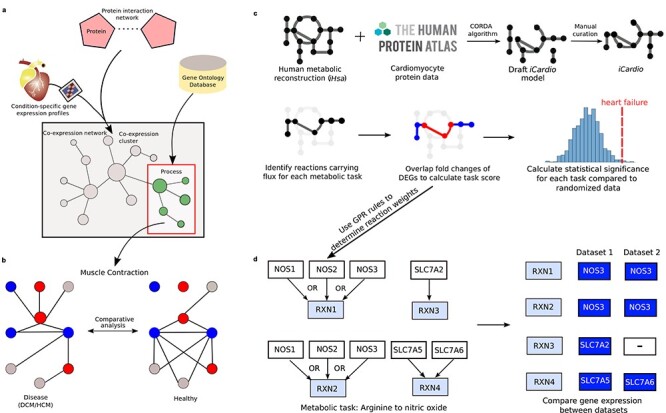
The integration of omics data and application of systems biology approaches towards providing mechanistic insights on heart failure. (A) Formation of a condition-specific co-expression network through integration of gene expression profiles with protein–protein interaction data and biological function annotations enable the identification of functional modules representative of different biological processes which are relevant towards the progression of heart failure. (B) A comparative analysis between identified functional modules can reveal dynamic variations in the modules between healthy and disease states which can be used to classify normal and disease samples, thus implying plausible molecular mechanisms that are involved in the progression of heart failure. (C) Construction of a cardiomyocyte metabolic model (iCardio) through integration with protein data and manual curation from metabolic tasks. Functional metabolic changes are identified from gene expression data using metabolic tasks. (D) The application of GPR rules in determining reaction weights based on gene expression data, thereby assigning the expression of one gene as governing the reaction. The selected genes can differ between different datasets while producing the same net result, as seen here with a statistically significant decrease in the conversion of arginine to nitric oxide.
The diverse clinical and pathologic phenotypes in HCM can be partially explained by the upregulation of various genes [98]. Population heterogeneity undermines efforts to uncover significant HCM-associated genes, and this has recently been addressed using a personalized multi-omics approach that identified Hes1 (hairy and enhancer of split-1) as a regulator of cardiac hypertrophy [99]. In an analogous manner to DCM, the complex phenotype associated with HCM is only partly explained by individual genetic variants, and more recent studies have focused on proteomic analysis of human myocardial tissue to explore the postgenomic phenotype [100]. The myocardial proteome of HCM revealed dysregulation of structural and metabolic proteins and raised lumican levels in HCM hearts, which provides further insight into the myocardial fibrosis characteristic of this disease. This finding was in agreement with an earlier study that utilized mouse models to show increased lumican production altering molecules essential for cardiac remodelling and fibrosis in cardiac fibroblasts [101].
Metabolic alterations that induce changes in substrate utilization have also been noted in diseased states associated with cardiac remodelling, such as the remodelling of glucose metabolism preceding pressure overload-induced HCM [102]. A recently developed kinetic model, CardioGlyo, was utilized to study the metabolic control of myocardial glycolysis and mathematical simulations revealed that reduction of phosphoglucose isomerase (PGI) activity directly affects myocyte growth through glucose-6-phosphate (G6P) accumulation, which is correlated with increased activation of the mTOR (mammalian target of rapamycin) signalling pathway that is involved in structural remodelling of the heart [103]. This association may be explained by G6P being redirected into the pentose phosphate pathway upon PGI inhibition, thus generating high amounts of NADPH (nicotinamide adenine dinucleotide phosphate, reduced), which has been linked to mTOR activation [104, 105]. A more comprehensive overview of metabolism in cardiomyocytes has been captured in models [14, 15] that utilized the global human metabolic network, termed Recon1, as a template when building cardiomyocyte-specific models. However, human models have greatly expanded since Recon1 to enable a more comprehensive description of human metabolism, and this has been captured in a recently published human GEM, termed iHsa [106], which was built in parallel as an expansion of the Human Metabolic Reaction 2.0 database [107]. This GEM offered the opportunity to create a more comprehensive cardiomyocyte-specific metabolic model (Figure 3C), termed iCardio [108], built using iHsa and data from the Human Protein Atlas (HPA) [16]. The model was integrated with multiple heart failure omics datasets to identify standard shifts in metabolic functions that are associated with heart failure, and this approach identified decreased NO (Figure 3D) and Neu5Ac synthesis as common metabolic markers of heart failure across transcriptomics datasets [108]. The GEM approach can also be utilized to identify causal factors for cardiac remodelling whether it is loss of function mutations in cardiovascular-related genes altering metabolic fluxes [109] or different diets leading to profound changes in the cardiac mitochondria, which may ultimately result in cell damage and heart failure [110].
Chronic kidney disease
The high comorbidity of CKD and CVD has been well established with a substantial number of CKD mortalities having registered cardiovascular problems; whilst CVD mortalities in stage 3 and stage 4 CKD patients are 2-fold and 3-fold higher than controls, i.e. patients with normal renal function, respectively [111, 112]. Both diseases also share numerous common risk factors, such as smoking, hypertension, abnormal mineral metabolism and vascular calcification, increased inflammation, sympathetic nervous system overactivity, hypertension, obesity and diabetes mellitus [113–115]. In particular, hypertension may occur in the early stages of CKD and lead to the onset of more severe conditions.
Several systems biology studies have shown a system-wide linkage between the kidney, heart and other internal organs. For example, an earlier study applied an interactome analysis approach (Figure 4A) and identified leptin, tumour necrosis factor, interleukin-6 (IL-6) and parathyroid hormone as candidate biomarkers for CVDs and bone metabolism disorders in CKD patients [116]. Besides, collagens (COL1A1, COL1A2), fibronectin, TGF-β or components of fibrinogen (FG-α, FG-β, FG-γ) were components of highly connected subnetworks of proteins associated with CKD and CVD. A separate systems biology approach investigated the role of fibroblastic growth factor (FGF23) in CKD and on the cardiovascular system, including possible crosstalk with the renin-angiotensin system (RAS) [117]. This study was motivated by previous findings that identified increasing circulating levels of fibroblastic growth factor 23 (FGF23), which regulates phosphate and bone metabolism, as a decisive independent risk factor for both CKD and CVD [118]; whilst the RAS is activated in CKD [119] and contributes to increased cardiovascular mortality and renal failure progression. Ingenuity Pathway Analysis performed on genes in the FGF23 and RAS endocrine networks showed that multiple interconnections exist between the respective networks [117].
Figure 4 .
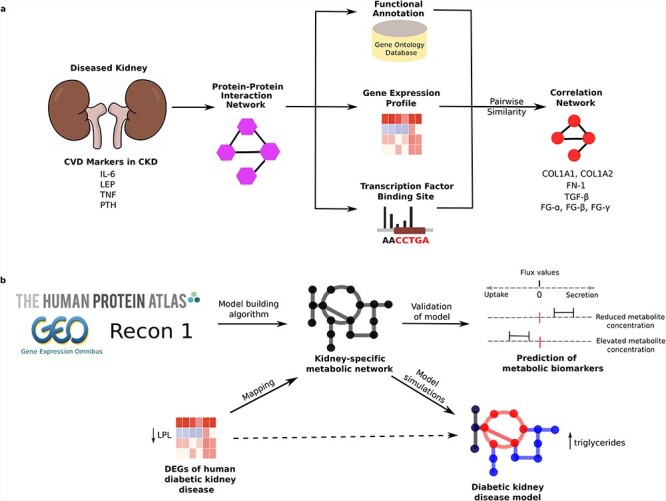
(A) Overview of a systems biology approach that links singular biomarker candidates towards deriving functional dependencies among chronic kidney disease (CKD) and cardiovascular disease (CVD). A set of genes associated with CVD in patients with CKD is used to construct a network of interacting proteins using reference data on known protein interactions. The functional interplay between interacting proteins was estimated by linking properties reflected by gene ontology terms, gene expression data characterizing CKD and TF binding sites with this methodology enabling detection of highly connected subnetworks associated with CKD and CVD. (B) The application of a kidney-specific metabolic model in determining metabolic process that are upregulated and downregulated in diabetic kidney disease resulting in the uptake and secretion of metabolites which may have an effect on the development and progression of CVD. Construction of the model was achieved through integration of proteome data from the Human Protein Atlas, transcriptome data from the Gene Expression Omnibus and application of a model-building algorithm that utilized Recon1 as a template to select only genes that were relevant to kidney.
The most common cause of CKD is diabetes meaning the linkages between kidney and heart involving energy metabolism should be considered [120]. For example, plasma mannose levels have been associated with type 2 diabetes (T2D), CVD, and the onset and progression of diabetic kidney disease [121, 122]. A comprehensive human kidney metabolic model was built [123] by integrating proteome data from the HPA [124] with transcriptome data from the Gene Expression Omnibus [125] and applying a model-building algorithm [126, 127] that used Recon1 as a template to select only those genes that are relevant to kidney (Figure 4B). This model showed upregulated metabolic processes in the kidney that are tightly linked to diabetes, including energy metabolism, extracellular transport and lipid metabolism. Furthermore, flux variability analysis of differentially expressed genes for diabetic kidney disease revealed several essential genes in the disease process. This included downregulation of lipoprotein lipase, an enzyme that breaks down triglycerides [128], with the strong possibility of this observation being partially responsible for elevated levels of circulating triglycerides conferring increased risk of CVD. Besides, there was upregulation of nicotinamide N-methyltransferase, an activity change associated with obesity and T2D [129]. More recently, an alternative molecular process model was developed to predict the progression of renal function decline in T2D patients using systems biology-derived biomarkers [130]. This approach identified a small set of serum protein biomarkers that enhanced the prediction of renal function loss in such patients.
Future work should continue to focus on gaining further understanding of the ‘kidney-heart’ axis through a systems biology approach that posits circular communication loops amid the heart and kidney with the notion that perturbation at any level can propagate dysregulation throughout the circuit. It may also be beneficial to include the gut microbiome in this axis owing to the importance of the gut as a potential contributor to CKD and CVD-related complications [131], the latter of which we discuss in much more detail in this review.
Role of the gut microbiota in CVD
The gut microbiota has emerged as a novel regulator of cardiovascular function and disease. Dysbiosis in the gut microbiome increases circulating microbial metabolites and bacterial structural components that may modify the function of metabolically relevant tissues and facilitate the development of CVD [132]. In particular, gut dysbiosis has been implicated in the pathology of numerous disturbances to the cardiovascular system, including atherosclerosis [133, 134], thrombosis [135], arterial hypertension and vascular dysfunction [136]. There are a variety of external factors that affect the composition and function of the gut microbiome, and these include most of the major risk factors for CVD, i.e. ageing, obesity, an inactive lifestyle and certain dietary patterns. From these external factors, the dietary pattern is widely accepted as one of the most critical factors in determining gut microbiota composition and function [137–140]. Hence, several studies have investigated the effects of dietary patterns in altering the gut microbiome and modifying signalling events that mediate CVD downstream of gut dysbiosis.
There is evidence that dietary interventions may improve cardiovascular health and this has been confirmed through the application of multi-omics approaches (Figure 5A). For example, a diet high in fibre was associated with changes in the gut microbiota that protected against CVD development, including a decreased ratio of Firmicutes to Bacteroidetes, and an increased prevalence of Bacteroides acidifaciens [141]. The favourable effect of the high-fibre diet was also explained by an increased generation and distribution of acetate, with this short-chain fatty acid affecting several molecular changes that are associated with improved cardiovascular function. Besides, the cardiac transcriptome revealed that a high-fibre diet or acetate supplementation resulted in the upregulation of genes (Tcap [titin-cap] [142] and Timp4 [tissue inhibitor of metalloprotease 4] [143]) that are strongly considered to have a preventive role for heart disease; whilst EGR1 (early growth response protein 1), a master cardiovascular regulator [144–146], was downregulated in both the heart and kidney. Collectively, this data suggests that acetate production is an essential mechanism for regulation of cardiovascular function by the gut microbiota, and provide mechanistic insight into how dietary fibre protects against the development of CVD. These findings are supported by increasing evidence that insufficient consumption of dietary fibre results in a loss of bacterial species in the gut microbiome [147].
Figure 5 .
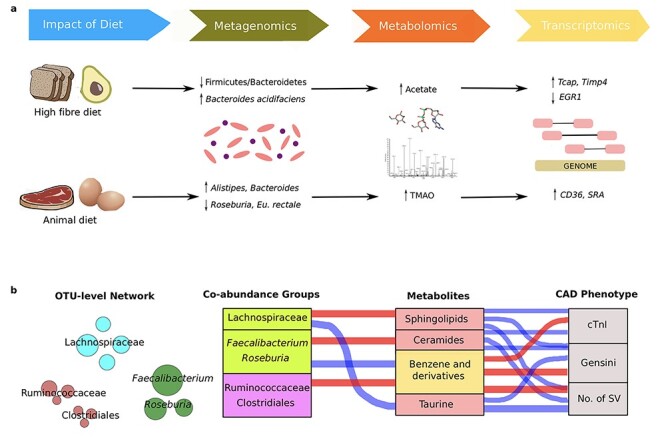
(A) The application of different fields of study (metagenomics, metabolomics and transcriptomics) in assessing how the impact of diet can alter the composition of the gut microbiota, plasma metabolite concentrations and expression of genes which protect against or promote biological processes related to cardiovascular disease. (B) Investigating the interrelationship between gut microbiota composition, host metabolic profile and main CAD phenotype. The OTU-level network indicates the abundance of each OTU in a CAG based on node size. Specific CAGs that are mainly composed of certain OTUs correlate with serum metabolite concentrations which in turn correlate with parameters representing CAD severity. Red connections indicate a positive correlation and blue connections represent negative correlations according to Spearman correlation test (FDR < 0.05). In the CAG column, the green boxes indicate CAGs that were highly enriched in the control group and the purple box represents a CAG that was increased in the severe CAD group. In the metabolomics column, the pink boxes represent CAD-negative metabotypes and the yellow box represents a CAD-positive metabotype.
Alternatively, the effects of an animal-based protein diet have also been examined through multi-omics analyses (Figure 5A). Here, multiple studies [148–150] revealed that animal-based protein consumption affected the gut microbiota by increasing the counts of bile-tolerant anaerobes such as Alistipes and Bacteroides. There have been links between Alistipes and CVD risk factors such as hypertension, with a positive correlation observed between systolic blood pressure and the abundance of several species belonging to the Alistipes genus [151]. However, a number of studies have also indicated a protective role for Alistipes in CVDs with a drastic decrease of this genus in the intestinal tract of patients with atrial fibrillation [152], atherosclerotic CVDs [153] and congestive heart failure [154]. Hence, it is still unclear if associations with Alistipes are protective, beneficial or pathogenic. This exposes a severe limitation of taxon-based analysis, which overlooks variations of bacterial strains belonging to the same taxon. A separate study showed that a high protein/low carbohydrate diet resulted in reduced levels of Roseburia and Eubacterium rectale in the gut microbiota of subjects, accompanied by a decreased proportion of butyrate in their faeces [155]. In addition, several microbial genera that are promoted by the intake of red meat were associated with increased levels of TMAO [156], thus increasing the risk of CVD through upregulation of scavenger receptors including CD36 (cluster of differentiation 36) [22, 157] and SRA (scavenger receptor A) [22]. These data support the established link between red meat consumption and CVD risk attributed to the high concentration of TMAO-producing nutrients, such as choline, phosphatidylcholine and L-carnitine, all of which are present in high concentrations in an animal diet including red meat, fish, milk and eggs.
Although several correlations have been established between CVD and the gut microbiota, it is much more challenging to identify potential causal relationships. An interdisciplinary approach is required to further understand gut bacterial-mediated mechanisms. Recent efforts have focussed on establishing causal relationships between the gut microbiota and CAD development via interactions with the host metabolic pathways. One such study revealed several alterations in gut microbial functional modules in CAD patients representative of amino acid transporters, LPS biosynthesis, the phosphotransferase system, vitamin metabolism, and the activities of short-chain fatty acids and TMA lyases [153]. These findings were in partial agreement with a separate study [154], which identified an elevation in microbial genes for LPS biosynthesis, TMAO and tryptophan generation in patients with chronic heart failure. This is of particular importance when considering the primary source of LPSs is the gut/gut microbiome, which provides an explanation for the greater plasma LPS levels in heart failure [158].
In addition to distinguishing between a CAD and healthy phenotype, the integration of multi-omics network analyses revealed that the composition of the gut microbiota and level of metabolites were significantly associated with CAD severity [25]. It was shown that several bacterial co-abundance groups (CAGs) might affect atherosclerosis by modulating several metabolic pathways in the host, which are positively or negatively associated with the CAD phenotype (Figure 5B). The basis for this approach is that key members of a CAG either thrive or decline together in response to a changing physiological environment and hence exist as functional groups [159] For example, a CAG that was enriched in the healthy control group, mainly composed of Faecalibacterium and Roseburia, was positively associated with ‘CAD-negative associated’ ceramides and negatively associated with ‘CAD-positive associated’ benzene derivatives. In contrast, a CAG mainly composed of operational taxonomic units from the Ruminoccocaceae family was higher in patients with CAD, which may partially be explained by the positive association between this CAG and the presence of benzene derivatives in the host metabolic profile. Hence, this CAG-based approach enabled the identification of functionally essential members of the gut microbiota in CVD and is recommended to be employed for further studies in place of the more conventional taxon-based analysis when investigating the links between the gut microbiota and the development of CVD. Future studies should focus on developing predictive systems-level approaches for each of these CAGs to elucidate causalities and quantify interactions between host, microbes and diet. In addition, this CAG approach could be used to investigate the influence of the microbiome on the plasma proteome with particular focus on signalling and inflammatory proteins pertinent to the cardiovascular system.
Biological network analyses of end-stage CVD cohorts
Coronary artery disease
CAD is the most common cause of CVD-related death and is caused by the accumulation of plaques in the coronary arteries, more widely known as atherosclerosis. Similar to atherosclerosis, CAD has a wide range of risk factors, including behavioural, metabolism-related and genetic risk factors. Patients diagnosed with CAD have also shown to be more prevalent to other chronic diseases, such as CKDs and end-stage renal diseases [160]. Statins are the most commonly used drugs used to treat CAD, especially in the early phase, due to their property of LDL reduction and their capability to decelerate the inflammation process [161] as demonstrated through various clinical trials (Table 1). They have also been shown to be beneficial in treating post-surgical CAD patients, both for cardiac stent and artery bypass graft patients [162–164].
Table 1.
Survey of completed studies investigating different causes and treatments for coronary artery disease
| Claim | Method | Result | Ref |
|---|---|---|---|
| Statins reduce LDL and are beneficial for post-surgical patients | Clinical trial | Rosuvastatin reduces LDL level by 50% and hCRP by 37% | [189] |
| Clinical trial | Group with pre- and post-operative statins has significantly lower in-hospital deaths compared to placebo group (1.9% versus 6.91%, P = 0.002) | [162] | |
| Clinical trial | Risk of death rate was lower for statin treated patients by 54% | [164] | |
| Genomic analysis reveals heritability and genetic variance of CAD | GWAS | Susceptibility to death from the disease was higher in male twins compared to female twins (0.57, 0.38, respectively) | [174] |
| GWAS | Identification of 304 genomic variants that explain 21.2% of CAD heritability with ~80% related to blood vessel morphogenenis, lipid metabolism, NOS and inflammation | [175] | |
| Urinary proteomic biomarkers for CAD | Proteomics | Identification of 15 peptides from urinary proteomics that showed CAD-specific characteristics | [178] |
| Network analysis explained the underlying mechanism of CAD | Network | Functional CAD network with genetic variants and transcriptomic data | [179] |
| Generation of CAD-specific regulatory genes and proteins network | Network | Integration of gene–gene networks with protein–protein interactions | [180] |
| Network analysis revealed shared regulatory pathways with T2D | Network | Genetic regulatory network | [181] |
CAS, coronary artery disease; GWAS, genome-wide association study; hCRP, human C-reactive protein; LDL, low-density lipoprotein; NOS, nitric oxide synthase; T2D, type 2 diabetes.
Early detection of CAD is required to perform adequate interventions due to the severity of the disease. Non-invasive diagnosis methods, such as exercise electrocardiogram (ECG), have been widely preferred in clinical settings for suspected CAD patients. These methods have been combined by assessing sociodemographic data, patient description of the pain [165, 166] such as angina and patient age to diagnose obstructive CAD, with this being the most common form of CAD. The less prevalent non-obstructive CAD is also generally diagnosed in patients with angina, and this type of CAD can be accompanied by coronary microvascular dysfunction [167]. In this context, diagnostic tests have been developed to assess this type of microvascular dysfunction to identify patients at risk of future abnormal cardiac events [168]. A number of these tests include the use of ECG for monitoring and interpretation purposes. Although the ECG’s sensitivity has been questioned [169], several computational methods employing artificial intelligence have been developed to significantly increase the sensitivity and accuracy of the ECG [170, 171]. The development of coronary computed tomographic angiography has been implemented to minimize invasive testing whilst improving accuracy of the testing. However, it was shown not to improve clinical outcomes over the early stage of CAD compared to functional testing (ECG, nuclear stress testing or stress echocardiography) [172].
Several studies have amplified the role of genetics in being one of the main risk factors of CAD with earlier studies showing that CAD has a high degree of heritability, especially in male subjects [173, 174]. Recent advances in high-throughput sequencing technologies have allowed for GWAS studies (Table 1) finding more than 300 genetic variants associated with CAD [175, 176]. A further study combined a two-stage GWAS and targeted metabolomics approach to show an association between carbamoyl-phosphate synthase 1 (CPS1) and CAD [177]. In addition, the variant localized to CPS1 was associated with decreased risk of CAD in female subjects. Furthermore, the application of urinary proteomics has been able to find linkages between CKD and CAD by identifying 15 signature peptides of CAD [178].
Several biological networks have been used to explain CAD (Table 1), including a recently generated functional network that integrated genetic variants and transcriptomic data from 600 CAD subjects in the STARTNET consortium [179]. This study successfully uncovered the mechanism of cis- and trans-acting loci in cardiometabolic disease, a precursor of CAD, and showed a gene-regulatory site for blood lipids. A separate study integrated gene-networks with known protein–protein interactions to generate regulatory-gene and protein networks across multiple tissues related to CAD [180]. The networks were combined with known GWAS loci associated with CAD to reveal the disease’s mechanism and propose novel drug targets. An additional study also generated genetic regulatory networks for CAD [181] and identified critical regulatory pathways shared between CAD and T2D. Moreover, this study discovered 15 essential driver genes that showed a high degree of connectivity with known GWAS hits in both CAD and T2D.
Ischaemic stroke
Ischaemic strokes account for 84% of all stroke events but only slightly fewer deaths than the other primary stroke type, haemorrhagic stroke [182]. Ischaemic strokes occur when a blood vessel leading to the brain is blocked. This can be attributed to several factors and causes include thrombosis, cardioembolism and stenosis. Carotid artery stenosis (CAS) narrows the carotid artery and is responsible for around 7% of ischaemic strokes [183]. A multitude of CAS risk factors, including age, hypertension, diabetes, dyslipidaemia, hypercholesterolaemia and smoking, account for the observed increase in the global burden of CAS, particularly in ageing populations. Middle cerebral artery occlusion (MCAO) refers to the blockage of the middle cerebral artery and is another cause of ischaemic stroke. Standard treatment includes IV thrombolysis, to be administered within hours of the stroke, and intra-arterial therapy, to be sustained over years [184]. In addition to these, aggressive mechanical clot disruption [185] and stenting [186] may be employed if drug treatments fail. This section summarizes the findings of completed studies that have identified different causes and treatments for CAS and MCAO (Table 2).
Table 2.
Survey of completed studies investigating different causes and treatments for carotid artery stenosis and middle cerebral artery occlusion
| Claim | Method | Result | Ref |
|---|---|---|---|
| Carotid artery stenosis (CAS) | |||
| Statins lower LDL and stroke risk | Clinical trial | Atorvastatin lowered LDL compared to placebo (1.9 mmol/L down from 3.3 mmol/L, P < 0.001) | [188] |
| Atorvastatin reduced stroke risk compared to placebo (11.2% incidence down from 13.1%, P = 0.03) | |||
| Clinical trial | Rosuvastatin lowered LDL by 50% (P < 0.001) | [189] | |
| Rosuvastatin reduced cardiovascular events by 43% (P < 0.00001) | |||
| Surgical procedures lower stroke risk | Clinical trial | In asymptomatics, CEA lowered cardiovascular event risk compared to deferred CEA (4.1% down from 10.0%) | [192] |
| Greatest net benefit from CEA was identified in those also on lipid-lowering therapy | |||
| Clinical trial | In asymptomatics, CEA + aspirin/medical management lowered cardiovascular event risk compared to aspirin/medical management alone (5.1% down from 11.0%) | [193] | |
| Antibody action lowers LDL | Clinical trial | Evolocumab lowers LDL by 59% (0.78 mmol/L down from 2.4 mmol/L, P < 0.001) | [190] |
| Clinical trial | Inclisiran lowers PCSK9 and LDL cholesterol levels (reductions of 35.5–52.6% after two doses, P < 0.001) for patients at high cardiovascular risk | [208] | |
| Genomics predicts carotid artery lumen diameter | GWAS | In the Arizona Strong Heart Family Study, genomic loci on chromosomes 7 and 12 were found to be significantly associated with left carotid artery diastolic and systolic lumen diameters | [194] |
| Genomics predicts moyamoya disease susceptibility | GWAS | Ten genomic loci were identified as significantly linked to moyamoya disease, including previously characterized loci on chromosome 17q25 | [195] |
| GWAS | In a Japanese cohort, the RNF213 gene on chromosome 17q25 was found to be highly associated with moyamoya disease | [196] | |
| miRNA transcriptomics predicts stroke risk | miRNA association study | In asymptomatics, five miRNAs were identified as significantly overexpressed in patients with disease progression compared to those without | [197] |
| Biological modelling of CAS is in its infancy | Network | fMRI-based functional network | [198] |
| Network | Doppler sonogram-based neural network | [199] | |
| Middle cerebral artery occlusion (MCAO) | |||
| Brain metabolic activity is altered post-MCAO | NMR | In rats, MCAO resulted in increased lactose, alanine, glutamine and GABA and decreased aspartate, glutamate, succinate and creatine due to increased glycolysis and decreased TCA flux | [203] |
| MS imaging | In mice, MCAO resulted in increased phosphocreatine, creatine and ceramide. Effects were most pronounced in the caudoputamen and cortex and least pronounced in the hippocampus | [204] | |
| Brain RNA content is altered post-MCAO | RNA-seq | lncRNA-mRNA co-expression network based on rat data, showing lncRNA regulatory changes post MCAO | [202] |
| Microarray | Circular RNAs, a lesser-studied class of ncRNA, are differentially expressed in mice post-MCAO | [201] | |
| RNA-seq | In mice, astrocytes respond to MCAO by upregulation of genes associated with complement and coagulation cascades, scar formation, inflammation, and apoptosis | [200] | |
| Jak/Stat signalling was upregulated; knocking out STAT3 resulted in increased neuron survival after MCAO | |||
| MCAO induces protein aggregation | MS | In mice, following MCAO, proteins associated with DNA/RNA processing and signal transduction increased in insolubility, indicating protein aggregation | [209] |
| Neuroprotective drugs mitigate MCAO effects | 2DGE/MS | In mice, quercetin mitigated the drop in ICDH, adenosylhomocysteinase, pyruvate kinase, and UCH-L1, and the increase in HSP60 and CRMP-2 protein expression as a result of MCAO | [205] |
| 2DGE/MS | In rats, resveratrol mitigated the drop in Prx-5, ICDH, ApoA-1 and UCH-L1, and the increase in CRMP-2 as a result of MCAO | [210] | |
ApoA-1, apolipoprotein A1; CAS, carotid artery stenosis; CEA, carotid endarterectomy; CRMP-2, collapsing response mediator protein 2; fMRI, functional magnetic resonance imaging; GABA, gamma-aminobutyric acid; GWAS, genome-wide association study; HSP60, heat shock protein 60; ICDH, isocitrate dehydrogenase; LDL, low-density lipoprotein; lncRNA, long non-coding RNA; MCAO, middle cerebral artery occlusion; miRNA, microRNA; MS, mass spectrometry; ncRNA, non-coding RNA; NMR, nuclear magnetic resonance, PRX-5, peroxisome assembly factor 5; RNF213, Ring Finger Protein 213; STAT3, signal transducer and activator of transcription 3; TCA, tricarboxylic acid cycle; UCH-L1, ubiquitin carboxy-terminal hydrolase L1; 2DGE, 2D Gel Electrophoresis.
Current diagnosis and risk stratification techniques for CAS include duplex ultrasound, computed tomography angiography, magnetic resonance angiography, digital subtraction angiography and transcranial Doppler (TCD) ultrasound [187]. Of these, TCD is the common technique of choice and has been used extensively to detect microembolic signals and stratification of high- and low-risk carotid lesions. Statins are commonly used to treat CAS by reducing blood LDL levels, thereby decreasing stroke risk, as demonstrated in clinical trials [188, 189]. More recently, antibody action against PCSK9 has also shown the ability to lower LDL levels [190] and have a positive impact on stroke prevention [191]. Finally, surgical procedures such as carotid endarterectomy and stenting are also commonly performed in high-grade stenosis cases, and these procedures confer increased benefits compared to therapeutic solutions alone [192, 193].
Genetic and genomic features in CAS have been markedly less studied than the contribution of diet, smoking and other environmental factors. However, a few studies are noteworthy. For instance, the Strong Heart Family Study, which investigated the heritability of CAS risk among American Indian tribes, found significant genetic linkage on chromosome 7q with left carotid artery lumen diameters in Arizona participants, suggesting candidate genes such as KCND and Kv4 as associated with CAS [194]. GWAS studies have also uncovered susceptibility loci for the related moyamoya disease, which is characterized by CAS [195, 196]; and microRNA biomarkers of CAS progression have been described [197]. Therefore, modelling CAS as an outcome of a biological network model should be considered. CAS has also been modelled as a functional network [198] as well as a NN [199]. In the functional network, fMRI data from 90 pre-specified regions of interest were processed, and correlations in signal intensities formed edges in the network. In the NN, parameters from Doppler sonogram spectroscopy were selected as the input layer. The two models described significant differences in fMRI and Doppler signatures between CAS patients and healthy controls. However, the accuracy of the NN diminished when used to predict CAS severity, and neither of these approaches considers genetic risk factors to CAS progression.
In contrast to CAS research, MCAO research has focused on biological effects post-stroke, and many studies have used mice and rats as a model. MCAO is modelled by anaesthetizing animals and then transient blocking off the middle cerebral artery by ligation or the use of a vascular clip, thereby inducing MCAO. The effects of MCAO on these animals can then be compared to the sham experiment in which animals undergo the same procedure of anaesthesia and exposure of the middle cerebral artery but without occlusion. Recent studies using this method have uncovered biochemical changes on the RNA, protein, and metabolic levels. Furthermore, these models have been useful in characterizing the effects of drugs on mitigating the modifications induced by MCAO. MCAO models have shown differential expression of coding and non-coding transcripts compared to animals undergoing the sham operation. In particular, genes associated with coagulation, migration, scar formation, inflammation and apoptosis were upregulated in mouse astrocytes, whereas cell adhesion, signalling and metabolism genes were downregulated [200]. This suggests a stress response in these cells, which agrees with studies on lncRNA expression in MCAO rats [195] and circular RNA expression in MCAO mice [201]. These studies used microarrays and RNA-seq to find differentially expressed non-coding RNAs known to regulate genes associated with reactive oxygen species, oxidative stress, and apoptosis. A co-expression network of upregulated and downregulated mRNAs and lncRNAs in the MCAO rat further implicates diverse pathways, such as chemokine signalling, Parkinson’s disease, the cell cycle and metabolism [202].
Mass spectrometry and nuclear magnetic resonance studies on MCAO mice and rats have shown metabolic changes post-MCAO. In rats, MCAO increased lactose, alanine, glutamine and GABA compared to the sham procedure rat, whereas aspartate, glutamate, succinate, and creatine decreased [203]. In mice, MCAO accumulates phosphocreatine and ceramide in an ischaemic core, whereas creatine levels decreased [204]. Taken together, this suggests that the restriction of blood flow due to MCAO significantly impacts energy metabolism, resulting in an increase in glycolytic flux and decrease in TCA flux, thus bringing about the observed metabolic changes and gene expression responses to oxidative stress and neuroinflammation. Quercetin and resveratrol are plant-based antioxidants drugs frequently used in Alzheimer’s disease and Parkinson’s disease treatment. Briefly, quercetin works as an inhibitor of acetylcholinesterase and an inhibitor of amyloid-beta filbril formation, whereas resveratrol is an activator of the anti-ageing factor SIRT-1. In experiments with MCAO mice, quercetin mitigated MCAO-related pyruvate kinase decreases as well as decreases in isocitrate dehydrogenase and adenosylhomocysteinase, restoring expression to sham-like levels [205]. Resveratrol also mitigated declines in the latter two proteins in experiments with MCAO rats.
Taken together, the insights from whole omics studies in CAS and MCAO research show that the field would greatly benefit from further work. As we have illustrated, entire omics research in CAS is relatively unexplored. Still, new avenues such as investigating multi-omic responses to drugs and treatments exist and have been more clearly explored for MCAO. There is also real potential for the integration of multiple data types as insightful genomic studies and studies based on imaging and/or physical data have been carried out separately. Current limitations in the field include little to no investigation in personalization of treatment despite the known heterogeneity of symptoms and risk factors for CAS and MCAO; whereas methods for studying blockage of these arteries have not significantly advanced in recent decades.
Conclusion
The complexity and heterogeneity of CVD strongly suggest that personalized treatments are required depending on the underlying cause that is contributing towards the development and progression of CVD. This includes atherosclerosis, thrombosis, adverse cardiac remodelling, CKD and the gut microbiota. We highlight recent systems biology approaches that have analysed high-throughput omics data to explain these underlying causes further and give examples of how these approaches have contributed significantly towards providing a prognosis for CVD subjects. Such an approach might predict how a myocardial infarction progresses based on a patient’s unique platelet phenotype or predict the development of atherosclerosis through several factors including genetics, behavioural and metabolism-related disorders. The latter of these aforementioned factors can be linked to the composition of the gut microbiome. Hence, we summarized how several bacterial CAGs may affect atherosclerosis by modulating several metabolic pathways in the host. We also assessed how GEMs have enabled us to gain a further understanding of heart-specific metabolic processes through the construction of the iCardio GEM and subsequent integration with transcriptomics data to reveal decreased NO and Neu5Ac synthesis as common metabolic markers of heart failure across multiple datasets.
In this review, we also focused on the application of biological networks and association studies in identifying causes and providing treatments for end-stage CVD, namely CAD and stroke, with our chosen examples identifying essential genes for stratifying and treating these two main types of CVD. Future studies should combine all of the fundamental biological mechanisms that underlie the complex pathophysiology of CVD for early prognosis preceding the occurrence of an atherothrombotic event. This could be achieved by integrating current systems biology approaches that have been applied towards explaining the underlying causes of CVD.
We expect future systems biology approaches in cardiovascular research to integrate disease-spanning data from the cardiovascular field with data from other chronic diseases, including the integration of data from CKD and CVD to identify shared commonalities of risk factors and further assess the high comorbidity of these diseases. There is also the need to further understand tissue-specific metabolic crosstalk through integration of models of the heart and other human tissues with the gut microbiota for the simulation of whole-body metabolic functions in health and CVD, with such roadmaps having already been designed for the study of liver-associated diseases [206]. This multi-tissue approach has been limited to date for CVD [207] and future efforts should focus on the generation of a final predictive model incorporating essential metabolic interactions or signalling pathways in each individual tissue/microbiota model that are known to either protect against or contribute towards the development and progression of CVD.
Authors’ contribution
SD, MA, SL and AB wrote the manuscript, and all authors were involved in the editing and reviewing of the manuscript.
Supplementary Material
Stephen Doran is a PhD student at King’s College London supervised by Prof. Adil Mardinoglu. He is interested in the multi-omics modelling of cardiovascular disease.
Muhammad Arif is a PhD student at KTH Royal Institute of Technology, Science for Life Laboratory supervised by Prof. Adil Mardinoglu. He is interested in the multi-omics modelling of cardiovascular disease.
Simon Lam is a PhD student at King’s College London supervised by Prof. Adil Mardinoglu. He is interested in the discovery of novel drug targets for effective treatment of Parkinson’s disease.
Abdulahad Bayraktar is a PhD student at King’s College London supervised by Prof. Adil Mardinoglu. He is interested in the discovery of novel drug targets for the effective treatment of Alzheimer’s disease.
Professor Hasan Türkez currently works at the Department of Medical Biology, Atatürk University. Hasan does research on Genetics, Drug R&D and Nano-Biotechnology.
Professor Mathias Uhlen currently works at KTH Royal Institute of Technology Stockholm. His research is focused on protein science, antibody engineering and precision medicine. It ranges from basic research in human and microbial biology to more applied research, including clinical applications in cancer, infectious diseases, cardiovascular diseases, autoimmune diseases and neurobiology.
Professor Jan Borén is head of the Institute of Medicine, Sahlgrenska Academy at the University of Gothenburg. His main research interest is lipid metabolism. He has focused on understanding the consequences of and underlying mechanisms that lead to lipid accumulation in the liver, arterial wall and heart, to translate this knowledge into effective treatment.
Professor Adil Mardinoglu leads a computational group in the Centre for Host-Microbiome Interactions, King’s College London and also works as a group leader in Science for Life Laboratory, KTH Royal Institute of Technology. His research activities include the generation of the context-specific genome-scale metabolic models for human cell-types including hepatocytes, adipocytes and myocytes as well as certain types of cancer, e.g. liver, prostate, glioblastoma and colon cancers.
Contributor Information
Stephen Doran, Centre for Host-Microbiome Interactions, Faculty of Dentistry, Oral & Craniofacial Sciences, King's College London, London, SE1 9RT, United Kingdom.
Muhammad Arif, Science for Life Laboratory, KTH - Royal Institute of Technology, Stockholm, Sweden.
Simon Lam, Centre for Host-Microbiome Interactions, Faculty of Dentistry, Oral & Craniofacial Sciences, King's College London, London, SE1 9RT, United Kingdom.
Abdulahad Bayraktar, Centre for Host-Microbiome Interactions, Faculty of Dentistry, Oral & Craniofacial Sciences, King's College London, London, SE1 9RT, United Kingdom.
Hasan Turkez, Department of Medical Biology, Faculty of Medicine, Atatürk University, Erzurum, Turkey.
Mathias Uhlen, Science for Life Laboratory, KTH - Royal Institute of Technology, Stockholm, Sweden.
Jan Boren, Institute of Medicine, Department of Molecular and Clinical Medicine, University of Gothenburg and Sahlgrenska University Hospital Gothenburg, Sweden.
Adil Mardinoglu, Centre for Host-Microbiome Interactions, Faculty of Dentistry, Oral & Craniofacial Sciences, King's College London, London, SE1 9RT, United Kingdom; Science for Life Laboratory, KTH - Royal Institute of Technology, Stockholm, Sweden.
Funding
Knut and Alice Wallenberg Foundation (grant number: 2017.0303).
References
- 1.Moodie DS. The global burden of cardiovascular disease: EDITORIAL. Congenit Heart Dis 2016;11:213–3. [DOI] [PubMed] [Google Scholar]
- 2.Jager KJ, Fraser SDS. The ascending rank of chronic kidney disease in the global burden of disease study. Nephrol Dial Transplant 2017;32:ii121–8. [DOI] [PubMed] [Google Scholar]
- 3.Nowbar AN, Gitto M, Howard JP, et al. . Mortality from ischemic heart disease: analysis of data from the World Health Organization and coronary artery disease risk factors from NCD risk factor collaboration. Circ Cardiovasc Qual Outcomes 2019;12:e005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ojaghihaghighi S. Comparison of neurological clinical manifestation in patients with hemorrhagic and ischemic stroke. World J Emerg Med 2017;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med 2013;11:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ISTH Steering Committee for World Thrombosis Day . Thrombosis: a major contributor to the global disease burden. J Thromb Haemost 2014;12:1580–90. [DOI] [PubMed] [Google Scholar]
- 7.Azevedo PS, Polegato BF, Minicucci MF, et al. . Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol 2016;106:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGarrah RW, Crown SB, Zhang G-F, et al. . Cardiovascular metabolomics. Circ Res 2018;122:1238–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet 2005;6:185–216. [DOI] [PubMed] [Google Scholar]
- 10.Borén J, Chapman MJ, Krauss RM, et al. . Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European atherosclerosis society consensus panel. Eur Heart J 2020;41:2313–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leon-Mimila P, Wang J, Huertas-Vazquez A. Relevance of multi-omics studies in cardiovascular diseases. Front Cardiovasc Med 2019;6:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cook DJ, Nielsen J. Genome-scale metabolic models applied to human health and disease: genome-scale metabolic models. Wiley Interdiscip Rev Syst Biol Med 2017;9:e1393. [DOI] [PubMed] [Google Scholar]
- 13.Robinson JL, Kocabaş P, Wang H, et al. . An atlas of human metabolism. Sci Signal 2020;13:eaaz1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Huang J. Reconstruction and analysis of human heart-specific metabolic network based on transcriptome and proteome data. Biochem Biophys Res Commun 2011;415:450–4. [DOI] [PubMed] [Google Scholar]
- 15.Karlstädt A, Fliegner D, Kararigas G, et al. . CardioNet: a human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst Biol 2012;6:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uhlen M, Fagerberg L, Hallstrom BM, et al. . Tissue-based map of the human proteome. Science 2015;347:1260419–9. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Q, Gao R, Zhang Y, et al. . Dysbiosis signatures of gut microbiota in coronary artery disease. Physiol Genomics 2018;50:893–903. [DOI] [PubMed] [Google Scholar]
- 18.Koeth RA, Wang Z, Levison BS, et al. . Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19:576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li XS, Obeid S, Wang Z, et al. . Trimethyllysine, a trimethylamine N-oxide precursor, provides near- and long-term prognostic value in patients presenting with acute coronary syndromes. Eur Heart J 2019;40:2700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velasquez MT, Centron P, Barrows I, et al. . Gut microbiota and cardiovascular uremic toxicities. Toxins 2018;10:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffin JL, Wang X, Stanley E. Does our gut microbiome predict cardiovascular risk?: a review of the evidence from metabolomics. Circ Cardiovasc Genet 2015;8:187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Klipfell E, Bennett BJ, et al. . Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang WHW, Wang Z, Levison BS, et al. . Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013;368:1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang WHW, Wang Z, Kennedy DJ, et al. . Gut microbiota-dependent trimethylamine N -oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 2015;116:448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H, Chen X, Hu X, et al. . Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 2019;7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leopold JA, Loscalzo J. Emerging role of precision medicine in cardiovascular disease. Circ Res 2018;122:1302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lusis AJ. Atherosclerosis. Nature 2000;407:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiolino G, Rossitto G, Caielli P, et al. . The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm 2013;2013:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Assmann G, Cullen P, Jossa F, et al. . Coronary heart disease: reducing the risk: the scientific background to primary and secondary prevention of coronary heart disease a worldwide view. Arterioscler Thromb Vasc Biol 1999;19:1819–24. [DOI] [PubMed] [Google Scholar]
- 30.Mayer-Davis EJ. Intensity and amount of physical activity in relation to insulin sensitivity: the insulin resistance atherosclerosis study. JAMA 1998;279:669. [DOI] [PubMed] [Google Scholar]
- 31.Keys A. Human atherosclerosis and the diet. Circulation 1952;5:115–8. [DOI] [PubMed] [Google Scholar]
- 32.Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part V: the discovery of the statins and the end of the controversy. J Lipid Res 2006;47:1339–51. [DOI] [PubMed] [Google Scholar]
- 33.Sackett DL, Gibson RW, Bross IDJ, et al. . Relation between aortic atherosclerosis and the use of cigarettes and alcohol: an autopsy study. N Engl J Med 1968;279:1413–20. [DOI] [PubMed] [Google Scholar]
- 34.Henkin L, Zaccaro D, Haffner S, et al. . Cigarette smoking, environmental tobacco smoke exposure and insulin sensitivity. Ann Epidemiol 1999;9:290–6. [DOI] [PubMed] [Google Scholar]
- 35.Kojda G. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res 1999;43:562–71. [DOI] [PubMed] [Google Scholar]
- 36.McGill HCJ, McMahan C, Herderick EE, et al. . Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 2002;105:2712–8. [DOI] [PubMed] [Google Scholar]
- 37.Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract 2013;7:e330–41. [DOI] [PubMed] [Google Scholar]
- 38.Lusis AJ, Fogelman AM, Fonarow GC. Genetic basis of atherosclerosis: part I: new genes and pathways. Circulation 2004;110:1868–73. [DOI] [PubMed] [Google Scholar]
- 39.Ference BA, Ginsberg HN, Graham I, et al. . Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017;38:2459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Safarova MS, Satterfield BA, Fan X, et al. . A phenome-wide association study to discover pleiotropic effects of PCSK9, APOB, and LDLR. NPJ Genom Med 2019;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol 2007;27:996–1003. [DOI] [PubMed] [Google Scholar]
- 42.Kleemann R, Verschuren L, van Erk MJ, et al. . Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: a combined transcriptomics and metabolomics analysis. Genome Biol 2007;8:R200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Li S, Yang Y, et al. . Genome-wide association study validation identifies novel loci for atherosclerotic cardiovascular disease: a two-stage genetic association study. J Thromb Haemost 2012;10:1508–14. [DOI] [PubMed] [Google Scholar]
- 44.Erbilgin A, Civelek M, Romanoski CE, et al. . Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. J Lipid Res 2013;54:1894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, Johnson AD, Hendricks AE, et al. . Genetic associations with expression for genes implicated in GWAS studies for atherosclerotic cardiovascular disease and blood phenotypes. Hum Mol Genet 2014;23:782–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Yao H, Hui JY, et al. . Global transcriptomic study of atherosclerosis development in rats. Gene 2016;592:43–8. [DOI] [PubMed] [Google Scholar]
- 47.Wuttge DM, Sirsjö A, Eriksson P, et al. . Gene expression in atherosclerotic lesion of ApoE deficient mice. Mol Med Camb Mass 2001;7:383–92. [PMC free article] [PubMed] [Google Scholar]
- 48.Lutgens E, Faber B, Schapira K, et al. . Gene profiling in atherosclerosis reveals a key role for small inducible cytokines: validation using a novel monocyte chemoattractant protein monoclonal antibody. Circulation 2005;111:3443–52. [DOI] [PubMed] [Google Scholar]
- 49.Auclair S, Milenkovic D, Besson C, et al. . Catechin reduces atherosclerotic lesion development in apo E-deficient mice: a transcriptomic study. Atherosclerosis 2009;204:e21–7. [DOI] [PubMed] [Google Scholar]
- 50.Holdt LM, Hoffmann S, Sass K, et al. . Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet 2013;9:e1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holdt LM, Stahringer A, Sass K, et al. . Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun 2016;7:12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez DM, Rahman AH, Fernandez NF, et al. . Single-cell immune landscape of human atherosclerotic plaques. Nat Med 2019;25:1576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herrington DM, Mao C, Parker SJ, et al. . Proteomic architecture of human coronary and aortic atherosclerosis. Circulation 2018;137:2741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yagi H, Nishigori M, Murakami Y, et al. . Discovery of novel biomarkers for atherosclerotic aortic aneurysm through proteomics-based assessment of disease progression. Sci Rep 2020;10:6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y, Wen S, Jiang M, et al. . Atherosclerotic dyslipidemia revealed by plasma lipidomics on ApoE−/− mice fed a high-fat diet. Atherosclerosis 2017;262:78–86. [DOI] [PubMed] [Google Scholar]
- 56.Mishra BH, Mishra PP, Mononen N, et al. . Lipidomic architecture shared by subclinical markers of osteoporosis and atherosclerosis: the cardiovascular risk in young Finns study. Bone 2020;131:115160. [DOI] [PubMed] [Google Scholar]
- 57.Dang VT, Huang A, Zhong LH, et al. . Comprehensive plasma metabolomic analyses of atherosclerotic progression reveal alterations in glycerophospholipid and sphingolipid metabolism in apolipoprotein E-deficient mice. Sci Rep 2016;6:35037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao X, Ke C, Liu H, et al. . Large-scale metabolomic analysis reveals potential biomarkers for early stage coronary atherosclerosis. Sci Rep 2017;7:11817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chiu J-J, Chen LJ, Lee PL, et al. . Shear stress inhibits adhesion molecule expression in vascular endothelial cells induced by coculture with smooth muscle cells. Blood 2003;101:2667–74. [DOI] [PubMed] [Google Scholar]
- 60.Chien S. Molecular and mechanical bases of focal lipid accumulation in arterial wall. Prog Biophys Mol Biol 2003;83:131–51. [DOI] [PubMed] [Google Scholar]
- 61.Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol-Heart Circ Physiol 2007;292:H1209–24. [DOI] [PubMed] [Google Scholar]
- 62.Bentzon JF, Otsuka F, Virmani R, et al. . Mechanisms of plaque formation and rupture. Circ Resume 2014;114:1852–66. [DOI] [PubMed] [Google Scholar]
- 63.Steffensen LB, Mortensen MB, Kjolby M, et al. . Disturbed laminar blood flow vastly augments lipoprotein retention in the artery wall: a key mechanism distinguishing susceptible from resistant sites. Arterioscler Thromb Vasc Biol 2015;35:1928–35. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J, Friedman MH. Adaptive response of vascular endothelial cells to an acute increase in shear stress magnitude. Am J Physiol-Heart Circ Physiol 2012;302:H983–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ajami NE, Gupta S, Maurya MR, et al. . Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc Natl Acad Sci 2017;114:10990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Firasat S, Hecker M, Binder L, et al. . Advances in endothelial shear stress proteomics. Expert Rev Proteomics 2014;11:611–9. [DOI] [PubMed] [Google Scholar]
- 67.Jiang Y-Z, Manduchi E, Jiménez JM, et al. . Endothelial epigenetics in biomechanical stress: disturbed flow–mediated epigenomic plasticity in vivo and in vitro. Arterioscler Thromb Vasc Biol 2015;35:1317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burghoff S, Schrader J. Secretome of human endothelial cells under shear stress. J Proteome Res 2011;10:1160–9. [DOI] [PubMed] [Google Scholar]
- 69.Wang X-L, Fu A, Raghavakaimal S, et al. . Proteomic analysis of vascular endothelial cells in response to laminar shear stress. Proteomics 2007;7:588–96. [DOI] [PubMed] [Google Scholar]
- 70.Pendergrass SA, Brown-Gentry K, Dudek S, et al. . Phenome-wide association study (PheWAS) for detection of pleiotropy within the population architecture using genomics and epidemiology (PAGE) network. PLoS Genet 2013;9:e1003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dunn J, Qiu H, Kim S, et al. . Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest 2014;124:3187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koupenova M, Kehrel BE, Corkrey HA, et al. . Thrombosis and platelets: an update. Eur Heart J 2016;ehw550. doi: 10.1093/eurheartj/ehw550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mahmoodi BK, Veeger NJGM, Middeldorp S, et al. . Interaction of hereditary thrombophilia and traditional cardiovascular risk factors on the risk of arterial thromboembolism: pooled analysis of four family cohort studies. Circ Cardiovasc Genet 2016;9:79–85. [DOI] [PubMed] [Google Scholar]
- 74.Chu SG, Becker RC, Berger PB, et al. . Mean platelet volume as a predictor of cardiovascular risk: a systematic review and meta-analysis. J Thromb Haemost 2010;8:148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boyanova D, Nilla S, Birschmann I, et al. . PlateletWeb: a systems biologic analysis of signaling networks in human platelets. Blood 2012;119:e22–34. [DOI] [PubMed] [Google Scholar]
- 76.Thomas A, Rahmanian S, Bordbar A, et al. . Network reconstruction of platelet metabolism identifies metabolic signature for aspirin resistance. Sci Rep 2014;4:3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cosemans JMEM, Angelillo-Scherrer A, Mattheij NJA, et al. . The effects of arterial flow on platelet activation, thrombus growth, and stabilization. Cardiovasc Res 2013;99:342–52. [DOI] [PubMed] [Google Scholar]
- 78.Goodall AH, Burns P, Salles I, et al. . Transcription profiling in human platelets reveals LRRFIP1 as a novel protein regulating platelet function. Blood 2010;116:4646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Simon LM, Edelstein LC, Nagalla S, et al. . Human platelet microRNA-mRNA networks associated with age and gender revealed by integrated plateletomics. Blood 2014;123:e37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diamond SL. Systems biology to predict blood function. J Thromb Haemost 2009;7:177–80. [DOI] [PubMed] [Google Scholar]
- 81.Purvis JE, Chatterjee MS, Brass LF, et al. . A molecular signaling model of platelet phosphoinositide and calcium regulation during homeostasis and P2Y1 activation. Blood 2008;112:4069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuharsky AL, Fogelson AL. Surface-mediated control of blood coagulation: the role of binding site densities and platelet deposition. Biophys J 2001;80:1050–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okorie UM, Denney WS, Chatterjee MS, et al. . Determination of surface tissue factor thresholds that trigger coagulation at venous and arterial shear rates: amplification of 100 fM circulating tissue factor requires flow. Blood 2008;111:3507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stalker TJ, Traxler EA, Wu J, et al. . Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013;121:1875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chatterjee MS, Purvis JE, Brass LF, et al. . Pairwise agonist scanning predicts cellular signaling responses to combinatorial stimuli. Nat Biotechnol 2010;28:727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. ACC Curr J Rev 2002;11:15. [DOI] [PubMed] [Google Scholar]
- 87.Chatterjee MS, Denney WS, Jing H, et al. . Systems biology of coagulation initiation: kinetics of thrombin generation in resting and activated human blood. PLoS Comput Biol 2010;6:e1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Flamm MH, Colace TV, Chatterjee MS, et al. . Multiscale prediction of patient-specific platelet function under flow. Blood 2012;120:190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Flamm MH, Diamond SL, Sinno T. Lattice kinetic Monte Carlo simulations of convective-diffusive systems. J Chem Phys 2009;130:094904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Louridas GE, Lourida KG. Systems biology and biomechanical model of heart failure. Curr Cardiol Rev 2012;8:220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017;121:749–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sweet ME, Cocciolo A, Slavov D, et al. . Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genomics 2018;19:812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bendig G, Grimmler M, Huttner IG, et al. . Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev 2006;20:2361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Knöll R, Postel R, Wang J, et al. . Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation 2007;116:515–25. [DOI] [PubMed] [Google Scholar]
- 95.Isserlin R, Merico D, Alikhani-Koupaei R, et al. . Pathway analysis of dilated cardiomyopathy using global proteomic profiling and enrichment maps. Proteomics 2010;10:1316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin C-C, Hsiang JT, Wu CY, et al. . Dynamic functional modules in co-expressed protein interaction networks of dilated cardiomyopathy. BMC Syst Biol 2010;4:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xiao J, Li F, Yang Q, et al. . Co-expression analysis provides important module and pathways of human dilated cardiomyopathy. J Cell Physiol 2020;235:494–503. [DOI] [PubMed] [Google Scholar]
- 98.Lim D-S, Roberts R, Marian AJ. Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: insight into the pathogenesis of phenotypes. J Am Coll Cardiol 2001;38:1175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Santolini M, Romay MC, Yukhtman CL, et al. . A personalized, multiomics approach identifies genes involved in cardiac hypertrophy and heart failure. Npj Syst Biol Appl 2018;4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Coats CJ, Heywood WE, Virasami A, et al. . Proteomic analysis of the myocardium in hypertrophic obstructive cardiomyopathy. Circ Genomic Precis Med 2018;11:e001974. [DOI] [PubMed] [Google Scholar]
- 101.Engebretsen KVT, Lunde IG, Strand ME, et al. . Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J 2013;280:2382–98. [DOI] [PubMed] [Google Scholar]
- 102.Kundu BK, Zhong M, Sen S, et al. . Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: review of a hypothesis. Cardiology 2015;130:211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karlstaedt A, Khanna R, Thangam M, et al. . Glucose 6-phosphate accumulates via Phosphoglucose isomerase inhibition in heart muscle. Circ Res 2020;126:60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nakano H, Minami I, Braas D, et al. . Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. Elife 2017;6:e29330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Düvel K, Yecies JL, Menon S, et al. . Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 2010;39:171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blais EM, Rawls KD, Dougherty BV, et al. . Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat Commun 2017;8:14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mardinoglu A, Agren R, Kampf C, et al. . Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:3083. [DOI] [PubMed] [Google Scholar]
- 108.Dougherty, B. V., Rawls, K.D., Kolling G.L.et al. . Identifying functional metabolic shifts in heart failure with the integration of omics data and a cardiomyocyte-specific, genome-scale model. bioRxiv. (2020) doi: 10.1101/2020.07.20.212274. [DOI] [PubMed] [Google Scholar]
- 109.Fouladiha H, Marashi S-A. Biomedical applications of cell- and tissue-specific metabolic network models. J Biomed Inform 2017;68:35–49. [DOI] [PubMed] [Google Scholar]
- 110.Thiele I, Price ND, Vo TD, et al. . Candidate metabolic network states in human mitochondria: impact of diabetes, ischemia, and diet. J Biol Chem 2005;280:11683–95. [DOI] [PubMed] [Google Scholar]
- 111.Chronic Kidney Disease Prognosis Consortium; Matsushita K, van der Velde MAstor BC, et al.. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. The Lancet 2010;375:2073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Velde M, Matsushita K, Coresh J, et al. . Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int 2011;79:1341–52. [DOI] [PubMed] [Google Scholar]
- 113.Roberts MA, Hare DL, Ratnaike S, et al. . Cardiovascular biomarkers in CKD: pathophysiology and implications for clinical management of cardiac disease. Am J Kidney Dis 2006;48:341–60. [DOI] [PubMed] [Google Scholar]
- 114.Said S. The link between chronic kidney disease and cardiovascular disease. Link Chronic Kidney Dis Cardiovasc Dis 2014. doi: 10.12860/jnp.2014.19 [DOI] [Google Scholar]
- 115.Rubin C, Nolin TD, Himmelfarb J. Are biomarkers useful for assessing cardiovascular risk in patients with chronic kidney disease? Curr Opin Nephrol Hypertens 2007;16:506–11. [DOI] [PubMed] [Google Scholar]
- 116.Perco P, Wilflingseder J, Bernthaler A, et al. . Biomarker candidates for cardiovascular disease and bone metabolism disorders in chronic kidney disease: a systems biology perspective. J Cell Mol Med 2008;12:1177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Quarles LD. A systems biology preview of the relationships between mineral and metabolic complications in chronic kidney disease. Semin Nephrol 2013;33:130–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Silver J, Rodriguez M, Slatopolsky E. FGF23 and PTH--double agents at the heart of CKD. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc 2012;27:1715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eräranta A, Riutta A, Fan M, et al. . Dietary phosphate binding and loading alter kidney angiotensin-converting enzyme mRNA and protein content in 5/6 nephrectomized rats. Am J Nephrol 2012;35:401–8. [DOI] [PubMed] [Google Scholar]
- 120.Pyram R, Kansara A, Banerji MA, et al. . Chronic kidney disease and diabetes. Maturitas 2012;71:94–103. [DOI] [PubMed] [Google Scholar]
- 121.Mardinoglu A, Stančáková A, Lotta LA, et al. . Plasma mannose levels are associated with incident type 2 diabetes and cardiovascular disease. Cell Metab 2017;26:281–3. [DOI] [PubMed] [Google Scholar]
- 122.Lee S, Zhang C, Kilicarslan M, et al. . Integrated network analysis reveals an association between plasma mannose levels and insulin resistance. Cell Metab 2016;24:172–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang A-D, Dai S-X, Huang J-F. Reconstruction and analysis of human kidney-specific metabolic network based on omics data. Biomed Res Int 2013;2013:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Uhlen M, Oksvold P, Fagerberg L, et al. . Towards a knowledge-based human protein atlas. Nat Biotechnol 2010;28:1248–50. [DOI] [PubMed] [Google Scholar]
- 125.Barrett T, Wilhite SE, Ledoux P, et al. . NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 2012;41:D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jerby L, Shlomi T, Ruppin E. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol Syst Biol 2010;6:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zur H, Ruppin E, Shlomi T. iMAT: an integrative metabolic analysis tool. Bioinformatics 2010;26:3140–2. [DOI] [PubMed] [Google Scholar]
- 128.Pirahanchi Y, Sharma S. Biochemistry, Lipase. In: StatPearls. StatPearls Publishing, Treasure Island (FL), 2020, ID: NBK537346. [PubMed] [Google Scholar]
- 129.Kraus D, Yang Q, Kong D, et al. . Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014;508:258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mayer G, Heerspink HJ, Aschauer C, et al. . Systems biology–derived biomarkers to predict progression of renal function decline in type 2 diabetes. Diabetes Care 2017;40:391–7. [DOI] [PubMed] [Google Scholar]
- 131.Sumida K, Kovesdy CP. The gut-kidney-heart axis in chronic kidney disease. Physiol Int 2019;106:195–206. [DOI] [PubMed] [Google Scholar]
- 132.Battson ML, Lee DM, Weir TL, et al. . The gut microbiota as a novel regulator of cardiovascular function and disease. J Nutr Biochem 2018;56:1–15. [DOI] [PubMed] [Google Scholar]
- 133.Gregory JC, Buffa JA, Org E, et al. . Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem 2015;290:5647–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hartiala J, Bennett BJ, Tang WH, et al. . Comparative genome-wide association studies in mice and humans for trimethylamine N -oxide, a Proatherogenic metabolite of choline and l -carnitine. Arterioscler Thromb Vasc Biol 2014;34:1307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhu W, Gregory JC, Org E, et al. . Gut microbial metabolite TMAO enhances platelet Hyperreactivity and thrombosis risk. Cell 2016;165:111–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Karbach SH, Schönfelder T, Brandão I, et al. . Gut microbiota promote angiotensin II–induced arterial hypertension and vascular dysfunction. J Am Heart Assoc 2016;5:e003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang C, Zhang M, Wang S, et al. . Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J 2010;4:232–41. [DOI] [PubMed] [Google Scholar]
- 138.Zhang C, Li S, Yang L, et al. . Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat Commun 2013;4:2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Voreades N, Kozil A, Weir TL. Diet and the development of the human intestinal microbiome. Front Microbiol 2014;5:494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Carmody RN, Gerber GK, LuevanoJM, Jr, et al. . Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015;17:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Marques FZ, Nelson E, Chu PY, et al. . High-Fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 2017;135:964–77. [DOI] [PubMed] [Google Scholar]
- 142.Knöll R, Linke WA, Zou P, et al. . Telethonin deficiency is associated with maladaptation to biomechanical stress in the mammalian heart. Circ Res 2011;109:758–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Koskivirta I, Kassiri Z, Rahkonen O, et al. . Mice with tissue inhibitor of metalloproteinases 4 (Timp4) deletion succumb to induced myocardial infarction but not to cardiac pressure overload. J Biol Chem 2010;285:24487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circ Res 2006;98:186–91. [DOI] [PubMed] [Google Scholar]
- 145.Ho L-C, Sung JM, Shen YT, et al. . Egr-1 deficiency protects from renal inflammation and fibrosis. J Mol Med 2016;94:933–42. [DOI] [PubMed] [Google Scholar]
- 146.Wang N-P, Pang XF, Zhang LH, et al. . Attenuation of inflammatory response and reduction in infarct size by postconditioning are associated with downregulation of early growth response 1 during reperfusion in rat heart. Shock 2014;41:346–54. [DOI] [PubMed] [Google Scholar]
- 147.Brunkwall L, Orho-Melander M. The gut microbiome as a target for prevention and treatment of hyperglycaemia in type 2 diabetes: from current human evidence to future possibilities. Diabetologia 2017;60:943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.David LA, Maurice CF, Carmody RN, et al. . Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014;505:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.ANR MicroObes consortium, Cotillard A, Kennedy SP, et al. . Dietary intervention impact on gut microbial gene richness. Nature 2013;500:585–8. [DOI] [PubMed] [Google Scholar]
- 150.de Filippo C, Cavalieri D, di Paola M, et al. . Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci 2010;107:14691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim S, Goel R, Kumar A, et al. . Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci 2018;132:701–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zuo K, Li J, Li K, et al. . Disordered gut microbiota and alterations in metabolic patterns are associated with atrial fibrillation. GigaScience 2019;8:giz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jie Z, Xia H, Zhong SL, et al. . The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun 2017;8:845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cui X, Ye L, Li J, et al. . Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci Rep 2018;8:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Russell WR, Gratz SW, Duncan SH, et al. . High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr 2011;93:1062–72. [DOI] [PubMed] [Google Scholar]
- 156.de Filippis F, Pellegrini N, Vannini L, et al. . High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016;65:1812–21. [DOI] [PubMed] [Google Scholar]
- 157.Geng J, Yang C, Wang B, et al. . Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed Pharmacother 2018;97:941–7. [DOI] [PubMed] [Google Scholar]
- 158.Sharma R, von Haehling S, Rauchhaus M, et al. . Whole blood endotoxin responsiveness in patients with chronic heart failure: the importance of serum lipoproteins. Eur J Heart Fail 2005;7:479–84. [DOI] [PubMed] [Google Scholar]
- 159.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol 2012;10:538–50. [DOI] [PubMed] [Google Scholar]
- 160.Virani SS, Alonso A, Benjamin EJ, et al. . Heart disease and stroke statistics—2020 update: a report from the American Heart Association. Circulation 2020;141:e139–596. [DOI] [PubMed] [Google Scholar]
- 161.Diamantis E, Kyriakos G, Quiles-Sanchez LV, et al. . The anti-inflammatory effects of statins on coronary artery disease: an updated review of the literature. Curr Cardiol Rev 2017;13:209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gan H-L, Zhang J-Q, Bo P, et al. . Statins decrease adverse outcomes in coronary artery bypass for extensive coronary artery disease as well as left main coronary stenosis. Cardiovasc Ther 2010;28:70–9. [DOI] [PubMed] [Google Scholar]
- 163.Conway AM, Musleh G. Which is the best statin for the postoperative coronary artery bypass graft patient? Eur J Cardiothorac Surg 2009;36:628–32. [DOI] [PubMed] [Google Scholar]
- 164.Schömig A, Mehilli J, Holle H, et al. . Statin treatment following coronary artery stenting and one-year survival. J Am Coll Cardiol 2002;40:854–61. [DOI] [PubMed] [Google Scholar]
- 165.Skelly AC, Hashimoto R, Buckley DI, et al. . Noninvasive Testing for Coronary Artery Disease. Agency for Healthcare Research and Quality, Comparative Effectiveness Reviews, No. 171. US, 2016. [PubMed] [Google Scholar]
- 166.Stangl V, Witzel V, Baumann G, et al. . Current diagnostic concepts to detect coronary artery disease in women. Eur Heart J 2008;29:707–17. [DOI] [PubMed] [Google Scholar]
- 167.Crea F. Angina pectoris and normal coronary arteries: cardiac syndrome X. Heart 2004;90:457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Radico F, Cicchitti V, Zimarino M, et al. . Angina pectoris and myocardial ischemia in the absence of obstructive coronary artery disease: practical considerations for diagnostic tests. JACC Cardiovasc Interv 2014;7:453–63. [DOI] [PubMed] [Google Scholar]
- 169.Mahmoodzadeh S, Moazenzadeh M, Rashidinejad H, et al. . Diagnostic performance of electrocardiography in the assessment of significant coronary artery disease and its anatomical size in comparison with coronary angiography. J Res Med Sci Off J Isfahan Univ Med Sci 2011;16:750–5. [PMC free article] [PubMed] [Google Scholar]
- 170.Goto S, Kimura M, Katsumata Y, et al. . Artificial intelligence to predict needs for urgent revascularization from 12-leads electrocardiography in emergency patients. Plos One 2019;14:e0210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Acharya UR, Fujita H, Lih OS, et al. . Automated detection of coronary artery disease using different durations of ECG segments with convolutional neural network. Knowl-Based Syst 2017;132:62–71. [Google Scholar]
- 172.Douglas PS, Hoffmann U, Patel MR, et al. . Outcomes of anatomical versus functional testing for coronary artery disease. N Engl J Med 2015;372:1291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Marenberg ME, Risch N, Berkman LF, et al. . Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 1994;330:1041–6. [DOI] [PubMed] [Google Scholar]
- 174.Zdravkovic S, Wienke A, Pedersen NL, et al. . Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med 2002;252:247–54. [DOI] [PubMed] [Google Scholar]
- 175.EPIC-CVD Consortium, Goel A, Butterworth AS, et al. . Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 2017;49:1385–91. [DOI] [PubMed] [Google Scholar]
- 176.The CARDIoGRAMplusC4D Consortium . A comprehensive 1000 genomes–based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hartiala JA, Wilson Tang WH, Wang Z, et al. . Genome-wide association study and targeted metabolomics identifies sex-specific association of CPS1 with coronary artery disease. Nat Commun 2016;7:10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zimmerli LU, Schiffer E, Zürbig P, et al. . Urinary proteomic biomarkers in coronary artery disease. Mol Cell Proteomics 2008;7:290–8. [DOI] [PubMed] [Google Scholar]
- 179.Franzen O, Ermel R, Cohain A, et al. . Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science 2016;353:827–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.CVgenes@target consortium, Lempiäinen H, Brænne I, et al. . Network analysis of coronary artery disease risk genes elucidates disease mechanisms and druggable targets. Sci Rep 2018;8:3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shu L, Chan KHK, Zhang G, et al. . Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet 2017;13:e1007040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Johnson CO, Nguyen M, Roth GA, et al. . Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol 2019;18:439–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dharmakidari S, Bhattacharya P, Chaturvedi S. Carotid artery stenosis: medical therapy, surgery, and stenting. Curr Neurol Neurosci Rep 2017;17:77. [DOI] [PubMed] [Google Scholar]
- 184.Barreto AD. Intravenous thrombolytics for ischemic stroke. Neurotherapeutics 2011;8:388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Noser EA, Shaltoni HM, Hall CE, et al. . Aggressive mechanical clot disruption: a safe adjunct to thrombolytic therapy in acute stroke? Stroke 2005;36:292–6. [DOI] [PubMed] [Google Scholar]
- 186.Nayak S, Ladurner G, Killer M. Treatment of acute middle cerebral artery occlusion with a solitaire AB stent: preliminary experience. Br J Radiol 2010;83:1017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Paraskevas KI, Spence JD, Veith FJ, et al. . Identifying which patients with asymptomatic carotid stenosis could benefit from intervention. Stroke 2014;45:3720–4. [DOI] [PubMed] [Google Scholar]
- 188.The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators . High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 2006;355:549–59.16899775 [Google Scholar]
- 189.Ridker PM, Danielson E, Fonseca FAH, et al. . Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008;359:2195–207. [DOI] [PubMed] [Google Scholar]
- 190.Sabatine MS, Giugliano RP, Keech AC, et al. . Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med 2017;376:1713–22. [DOI] [PubMed] [Google Scholar]
- 191.Giugliano RP, Pedersen TR, Saver JL, et al. . Stroke prevention with the PCSK9 (proprotein convertase subtilisin-kexin type 9) inhibitor evolocumab added to statin in high-risk patients with stable atherosclerosis. Stroke 2020;51:1546–54. [DOI] [PubMed] [Google Scholar]
- 192.Halliday A, Harrison M, Hayter E, et al. . 10-year stroke prevention after successful carotid endarterectomy for asymptomatic stenosis (ACST-1): a multicentre randomised trial. The Lancet 2010;376:1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Walker MD. Endarterectomy for asymptomatic carotid artery stenosis. JAMA J Am Med Assoc 1995;273:1421. [PubMed] [Google Scholar]
- 194.Bella JN, Cole SA, Laston S, et al. . Genome-wide linkage analysis of carotid artery lumen diameter: the strong heart family study. Int J Cardiol 2013;168:3902–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Duan L, Wei L, Tian Y, et al. . Novel susceptibility loci for Moyamoya disease revealed by a genome-wide association study. Stroke 2018;49:11–8. [DOI] [PubMed] [Google Scholar]
- 196.Kamada F, Aoki Y, Narisawa A, et al. . A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 2011;56:34–40. [DOI] [PubMed] [Google Scholar]
- 197.Dolz S, Górriz D, Tembl JI, et al. . Circulating MicroRNAs as novel biomarkers of stenosis progression in asymptomatic carotid stenosis. Stroke 2017;48:10–6. [DOI] [PubMed] [Google Scholar]
- 198.Chang T-Y, Huang KL, Ho MY, et al. . Graph theoretical analysis of functional networks and its relationship to cognitive decline in patients with carotid stenosis. J Cereb Blood Flow Metab 2016;36:808–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ergün U, Serhatlıoǧlu S, Hardalaç F, et al. . Classification of carotid artery stenosis of patients with diabetes by neural network and logistic regression. Comput Biol Med 2004;34:389–405. [DOI] [PubMed] [Google Scholar]
- 200.Rakers C, Schleif M, Blank N, et al. . Stroke target identification guided by astrocyte transcriptome analysis. Glia 2019;67:619–33. [DOI] [PubMed] [Google Scholar]
- 201.Mehta SL, Pandi G, Vemuganti R. Circular RNA expression profiles alter significantly in mouse brain after transient focal ischemia. Stroke 2017;48:2541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Duan X, Han L, Peng D, et al. . Bioinformatics analysis of a long non-coding RNA and mRNA regulation network in rats with middle cerebral artery occlusion based on RNA sequencing. Mol Med Rep 2019. doi: 10.3892/mmr.2019.10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hu Z, Xia H‑H, Yang Y‑J, et al. . Metabolic alterations in the rat cerebellum following acute middle cerebral artery occlusion, as determined by 1H NMR spectroscopy. Mol Med Rep 2017. doi: 10.3892/mmr.2017.7918. [DOI] [PubMed] [Google Scholar]
- 204.Abe T, Niizuma K, Kanoke A, et al. . Metabolomic analysis of mouse brain after a transient middle cerebral artery occlusion by mass spectrometry imaging. Neurol Med Chir (Tokyo) 2018;58:384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Shah F-A, Park D-J, Koh P-O. Identification of proteins differentially expressed by quercetin treatment in a middle cerebral artery occlusion model: a proteomics approach. Neurochem Res 2018;43:1608–23. [DOI] [PubMed] [Google Scholar]
- 206.Mardinoglu A, Boren J, Smith U, et al. . Systems biology in hepatology: approaches and applications. Nat Rev Gastroenterol Hepatol 2018;15:365–77. [DOI] [PubMed] [Google Scholar]
- 207.Arif, M., Klevstig Martina, Benfeitas Ruiet al. . Revealing tissue-specific metabolic crosstalk after a myocardial infarction. bioRxiv (2020) doi: 10.1101/2020.10.28.358556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ray KK, Landmesser U, Leiter LA, et al. . Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med 2017;376:1430–40. [DOI] [PubMed] [Google Scholar]
- 209.Kahl A, Blanco I, Jackman K, et al. . Cerebral ischemia induces the aggregation of proteins linked to neurodegenerative diseases. Sci Rep 2018;8:2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shah F-A, Gim S-A, Kim M-O, et al. . Proteomic identification of proteins differentially expressed in response to resveratrol treatment in middle cerebral artery occlusion stroke model. J Vet Med Sci 2014;76:1367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.