

Abstract
Sleep disorders are increasingly being characterized in modern society as contributing to a host of serious medical problems, including obesity and metabolic syndrome. Changes to the microbial community in the human gut have been reportedly associated with many of these cardiometabolic outcomes. In this study, we investigated the impact of sleep length on the gut microbiota in a large cohort of 655 participants of African descent, aged 25–45, from Ghana, South Africa (SA), Jamaica, and the United States (US). The sleep duration was self-reported via a questionnaire. Participants were classified into 3 sleep groups: short (<7hrs), normal (7-<9hrs), and long (≥9hrs). Forty-seven percent of US participants were classified as short sleepers and 88% of SA participants as long sleepers. Gut microbial composition analysis (16S rRNA gene sequencing) revealed that bacterial alpha diversity negatively correlated with sleep length (p<0.05). Furthermore, sleep length significantly contributed to the inter-individual beta diversity dissimilarity in gut microbial composition (p<0.01). Participants with both short and long-sleep durations exhibited significantly higher abundances of several taxonomic features, compared to normal sleep duration participants. The predicted relative proportion of two genes involved in the butyrate synthesis via lysine pathway were enriched in short sleep duration participants. Finally, co-occurrence relationships revealed by network analysis showed unique interactions among the short, normal and long duration sleepers. These results suggest that sleep length in humans may alter gut microbiota by driving population shifts of the whole microbiota and also specific changes in Exact Sequence Variants abundance, which may have implications for chronic inflammation associated diseases. The current findings suggest a possible relationship between disrupted sleep patterns and the composition of the gut microbiota. Prospective investigations in larger and more prolonged sleep researches and causally experimental studies are needed to confirm these findings, investigate the underlying mechanism and determine whether improving microbial homeostasis may buffer against sleep-related health decline in humans.
Background
Disrupted sleep has been associated with disturbances of hormone secretion and metabolism [1], as well as affecting physical, mental and emotional functions [2–7]. Recent data has shown that modern society as whole is severely sleep deprived, and that this chronic state of deprivation has consequences on the persons and society [8–11]. The underlying mechanisms of by which disrupted sleep patterns alter disease risk, especially cardiometabolic risks, have been demonstrated in several well-conducted sleep restriction experiments in healthy volunteers [12–15]. These include increased inflammatory markers [14], increased sympathetic nervous system activity [13], abnormal cortisol rhythmicity [13], alterations in appetite regulating hormones and food intake [16], and adipocyte dysfunction [15] which altogether contribute to insulin resistance, diabetes and obesity risks. Many of these findings have been confirmed in laboratory studies, demonstrating that disrupted sleep may be a risk factor for incident diabetes and obesity [17, 18]. According to the American Academy of Sleep Medicine and Sleep Research Society, the recommended sleep duration in adults is between 7–9 hours [19]. Despite strong data suggesting a causal association between disrupted sleep patterns and cardiometabolic risk, only a few sleep extensional studies have been conducted to date [20–23]. While preliminary evidence supports a possible benefit in glucose metabolism and dietary patterns, the ability to increase sleep time varied among participants [21–23]. Thus, development of preventive strategies based on new, modifiable risk factors is therefore imperative.
One such novel factor is the gut microbiota, important for regulating human physiology [24]. Structural and functional configurations of the gut bacterial community are associated with a series of metabolic and immune diseases, which are also adverse health consequences of disrupted sleep patterns [2, 25–28]. Emerging data from animal and human experimental studies have shown that sleep restriction or fragmentation led to significant changes in the structure of their intestinal microbial communities [29–31], although this was not consistently observed in all the studies related to sleep restriction and intestinal microbiota [32]. Structural changes in the gut microbiota may promote an increase in dietary fat intake and an increase in fat storage through a series of signal transductions [33]. Gut microbiota dysbiosis has recently been associated with systemic inflammation by producing butyrate with amino-acids (lysine, glutarate and 4-aminobytyrate/succinate) as substrates via the 4-aminobutyrate pathway, lipopolysaccharide (LPS) or hydrogen sulfide (H2S) [34–37], etc. Thus, alterations in gut microbiota may also contribute to the systemic inflammation which is a known consequence of disrupted sleep patterns [38].
Given the intersection between the gut microbiota, cardiometabolic disease and disrupted sleep patterns, it is possible that the gut microbiota is a potential mediator linking disrupted sleep and adverse health outcomes. Indeed, these associations between disrupted sleep patterns and changes in the gut microbiota community are not well understood. Notably, sleep-derived variations in the composition of gut microbiota has only been explored in small groups involving experimental sleep restriction [31]. Studies in large cohorts that explore this association across different geographic regions, with their own lifestyle and geographic idiosyncrasies could help elucidate the general characteristics that determine the differences in the microbiota across gradients of sleep duration. Therefore, to examine the potential impact of sleep disruption on the gut microbiota, this study leveraged African-origin participants enrolled in the Modeling the Epidemiologic Transition Study” (METS) cohort to investigate the structural and predicted metabolic dynamics of the gut (stool-derived) microbiota as a function of sleep quality in 655 adults of African descent aged 22 to 45 years.
Results
Participant characteristics
The first METS cohort consisted of 2,506 participants enrolled in December 2010 and January 2011, and 655 participants from this original cohort were additionally asked to provide stool samples for this current microbiome study. These 655 participants were recruited from Ghana (N = 196), South Africa (N = 176), Jamaica (N = 92) and US (N = 191) (Table 1). Prevalence of overweight and obesity was significantly higher in the US cohort (81.3%) when compared to the other sites (i.e., Ghana, 33.2%, South Africa, 55.7%, Jamaica 65.2%). South Africans and Americans had significantly higher blood pressure than Ghanaians and Jamaicans (Table 1). Americans also slept the least number of hours (6.7 ± 1.4 hours) compared to Ghanaians (7.9 ± 1.4 hours, p<0.001), Jamaicans (7.3 ± 2.1 hours, p<0.05), and South Africans (10.5 ± 1.7 hours, p<0.001). The normal range of sleep for adults defined by the American Academy of Sleep Medicine is 7–9 hours [19], therefore if a participant slept 7–9 hours, then he/she was identified as a “normal sleeper”. If a participant slept<7 hours of per 24-hour period, then he/she was considered to be a “short sleeper” and if >9 hours: a “long sleeper”. Overall, the US had the greatest proportion of short sleepers, while South Africa had highest proportion of long-sleepers. Table 2 presents a summary of the distribution across the 3 groups.
Table 1. Participant characteristics and CM risk factors by site (mean, std. dev).
| Ghana | South Africa | Jamaica | United States | Overall | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (N = 196) | (N = 175) | (N = 90) | (N = 191) | (N = 652) | |||||||||||
| Sleep Category | Short | Normal | Long | Short | Normal | Long | Short | Normal | Long | Short | Normal | Long | Short | Normal | Long |
| N | N = 28 | N = 113 | N = 55 | N = 3 | N = 18 | N = 154 | N = 33 | N = 33 | N = 24 | N = 90 | N = 86 | N = 15 | N = 154 | N = 250 | N = 248 |
| Age (y) | 37.4 ± 6.1 | 36.0 ± 6.4 | 34.6 ± 7.2 | 37.7 ± 3.1 | 36.6 ± 5.1 | 32.9 ± 6.0 ** | 32.3 ± 6.2 | 34.3 ± 6.4 | 35.8 ± 5.6 | 36.2 ± 5.9 | 35.8± 6.7 | 34.3 ± 6.5 | 35.6 ± 6.2 | 35.7 ± 6.4 | 33.6 ± 6.3 ** |
| Weight (kg) | 62.7 ± 7.7 | 63.7 ± 12.8 | 62.3 ± 12.3 | 84.4 ± 21.7 | 87.5 ± 23.6 | 74.8 ± 19.9 * | 81.3 ± 21.3 | 83.6 ± 23.8 | 74.5 ± 16.0 | 95.5 ± 27.4 | 94.1 ± 22.8 | 86.9 ± 22.2 | 86.3 ± 26.6 | 78.5 ± 23.5 | 72.7 ± 19.2 * |
| Height (cm) | 163.4 ± 8.7 | 161.4 ± 7.2 | 161.3 ± 7.9 | 168.4 ± 8.1 | 166.2 ± 8.3 | 163.3 ± 7.6 | 172.2 ± 10.0 | 166.1 ± 8.3 | 161.1 ± 12.2 | 170.0 ± 9.5 | 169.3 ± 7.2 | 169.2 ± 8.0 | 169.2 ± 9.8 | 165.1 ± 8.2 | 163.0 ± 8.4 |
| BMI (kg/m2) | 23.5 ± 2.9 | 24.6 ± 5.6 | 24.0 ± 4.6 | 30.3 ± 10.5 | 32.1 ± 9.6 | 28.3 ± 8.0 * | 27.5 ± 7.2 | 30.5 ± 9.3 | 29.6 ± 10.5 | 33.2 ± 9.6 | 32.9 ± 8.1 | 30.5 ± 8.1 | 30.2 ± 9.1 | 28.8 ± 8.3 | 27.6 ± 7.9 * |
| Sleep hours (hrs/night) | 5.6 ± 0.69 | 7.6 ± 0.48 | 9.5 ± 0.77 | 5.7 ± 0.58 | 7.7 ± 0.46 | 11.0 ± 1.3 | 5.1 ± 1.0 | 7.6 ± 0.50 | 9.9 ± 1.1 | 5.4 ± 0.72 | 7.5 ± 0.51 | 9.2 ± 0.41 | 5.4 ± 0.80 | 7.6 ± 0.50 | 10.4 ± 1.4 |
| SBP (mm Hg) | 114.0 ± 9.9 | 113.6 ± 14.9 | 111.0 ±12.0 | 129.2 ± 22.5 | 118.0 ± 13.8 | 125.1 ± 21.0 | 112.1 ± 10.5 | 113.6 ± 15.0 | 112.1 ± 12.1 | 123.1 ± 18.9 | 123.5 ± 15.8 | 122.9 ± 20.0 | 119.2 ±16.8 | 117.3 ± 15.8 | 120.6 ± 19.6 |
| DBP (mm Hg) | 65.3 ± 7.4 | 67.1 ± 12.1 | 66.2 ± 10.3 | 81.6 ± 20.0 | 75.0 ± 11.3 | 79.9 ± 12.9 | 66.3 ± 8.6 | 80.4 ± 55.2 | 68.8 ± 8.1 | 79.9 ± 14.3 | 81.2 ± 11.8 | 80.0 ± 13.9 | 74.4 ± 14.0 | 74.3 ± 23.6 | 75.8 ± 13.4 |
| HDL (mg/dL) | 42.3 ± 14.5 | 46.4 ± 11.4 | 47.5 ± 16.8 | 40.0 ± 11.8 | 49.8 ± 15.3 | 50.1 ± 15.0 | - | - | - | 52.5 ± 15.7 | 51.8 ± 14.9 | 52.3 ± 13.1 | 49.7 ± 16.0 | 48.8 ± 13.4 | 49.6 ± 15.3 |
| LDL (mg/dL) | 94.4 ± 26.7 | 102.2 ± 29.6 | 101.1 ± 29.4 | 83.7 ±37.7 | 93.5 ± 35.0 | 93.0 ± 32.4 | - | - | - | 107.6 ± 30.9 | 114.4 ± 40.0 | 116.7 ± 34.1 | 103.8 ± 30.5 | 105.1 ± 34.8 | 96.6 ± 32.3 * |
| Trigs (mg/dL) | 77.2 ± 33.7 | 82.3 ± 33.4 | 84.2 ± 43.5 | 135.8 ± 53.4 * | 76.3 ± 38.2 | 88.7 ± 56.5 | - | - | - | 91.2 ± 50.9 | 103.0 ± 73.6 | 115.1 ± 46.6 | 89.0 ± 48.0 | 90.0 ± 54.1 | 89.4 ± 53.3 |
If data were normal, student t- testing was used to analyze differences between short and long sleepers with normal sleepers (i.e. short sleepers were compared to normal sleepers and long sleepers were compared to normal sleepers) within each site and overall for each characteristic. Non-parametric testing was used for non-normal data.
*p<0.05
**p<0.01.
Table 2. Participant characteristics by site (N,%).
| Ghana | South Africa | Jamaica | United States | Overall | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (N = 196) | (N = 175) | (N = 90) | (N = 191) | (N = 652) | |||||||||||
| Sleep Category | Short | Normal | Long | Short | Normal | Long | Short | Normal | Long | Short | Normal | Long | Short | Normal | Long |
| N | N = 28 | N = 113 | N = 55 | N = 3 | N = 18 | N = 154 | N = 33 | N = 33 | N = 24 | N = 90 | N = 86 | N = 15 | N = 154 | N = 250 | N = 248 |
| Sex 2 | |||||||||||||||
| Women | 15, 53.6% | 74, 65.5% | 38, 69.1% | 1, 33.3% | 12, 66.6% | 98, 63.6% | 15, 45.5% | 27, 81.8% | 19, 79.2% | 44, 48.9% | 50, 58.1% | 8, 53.3% | 75, 48.7% | 163, 64.9 | 163, 65.7% |
| Men | 13, 46.4% | 39, 34.5% | 17, 30.9% | 2, 66.7% | 6, 33.3% | 56, 36.4% | 18, 54.6% | 6, 18.2% | 5, 20.8% | 46, 51.1% | 36, 41.9% | 7, 46.7% | 79, 51.3% | 88, 35.1 | 85, 34.3% |
| BMI class | |||||||||||||||
| 1Normal weight | 21, 75.0% | 74, 65.5% | 36, 65.5% | 1, 33.3% | 6, 33.3% | 71, 46.1% | 13, 39.4% | 9, 27.3% | 9, 37.5% | 20, 22.2% | 12, 14.0% | 4, 26.7% | 55, 35.7% | 101, 40.2% | 120, 48.4% |
| 1Overweight | 6, 21.4% | 24, 21.2% | 11, 20.0% | 1, 33.3% | 2, 11.1% | 24, 15.6% | 11, 33.3% | 10, 30.3% | 6, 25.0% | 23, 25.6% | 24, 27.9% | 5, 33.3% | 41, 26.6% | 61, 24.3% | 46, 18.6% |
| 1Obese | 1, 3.6% | 15, 13.3% | 8, 14.6% | 1, 33.3% | 10, 55.6% | 59, 38.3% | 9, 27.3% | 14, 42.4% | 9, 37.5% | 47, 52.2% | 50, 58.1% | 6, 40.0% | 58, 37.7% | 89, 35.5% | 82, 33.1% |
| Smokers | 0, 0% | 1, 0.9% | 3, 5.5% | 0, 0% | 5, 26.3% | 43, 27.9% | 5, 15.2% | 4, 12.1% | 1, 4.2% | 32, 35.6 | 31, 36.1 | 9, 60.0 | 31, 27.0% | 37, 17.4% | 55, 24.7% |
χ2 testing used to analyze differences between normal and short sleepers within each site and overall for each characteristic.
1Where “normal weight”, “overweight” and “obese” are defined as BMI<25 kg/m2, BMI≥25 kg/m2-<30 kg/m2 and BMI≥30 kg/m2, respectively.
2χ2 testing reached significance in Jamaica (p<0.05) and “Overall” (p<0.001).
Sleep length significantly impact gut-derived bacterial diversity and composition
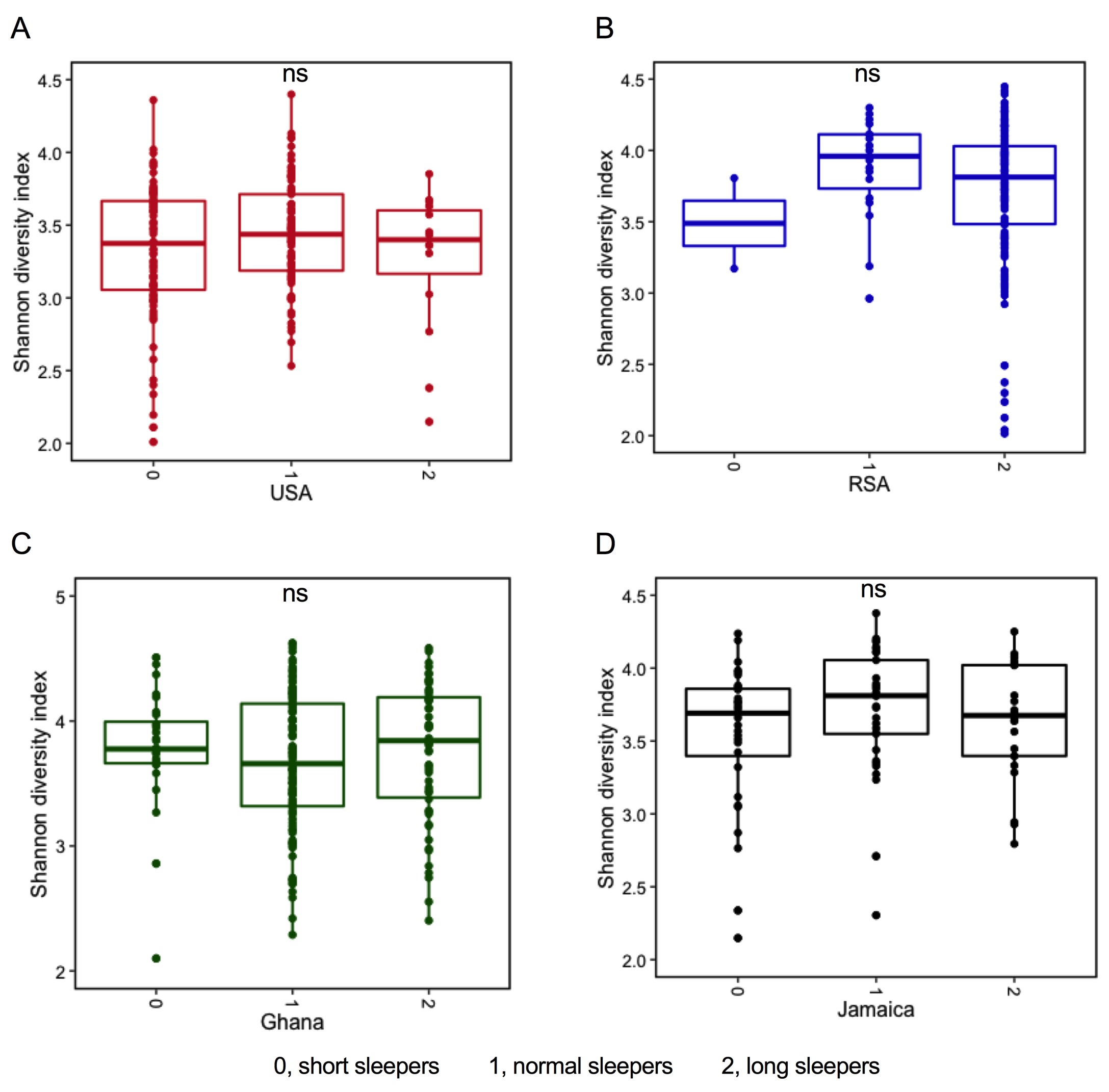
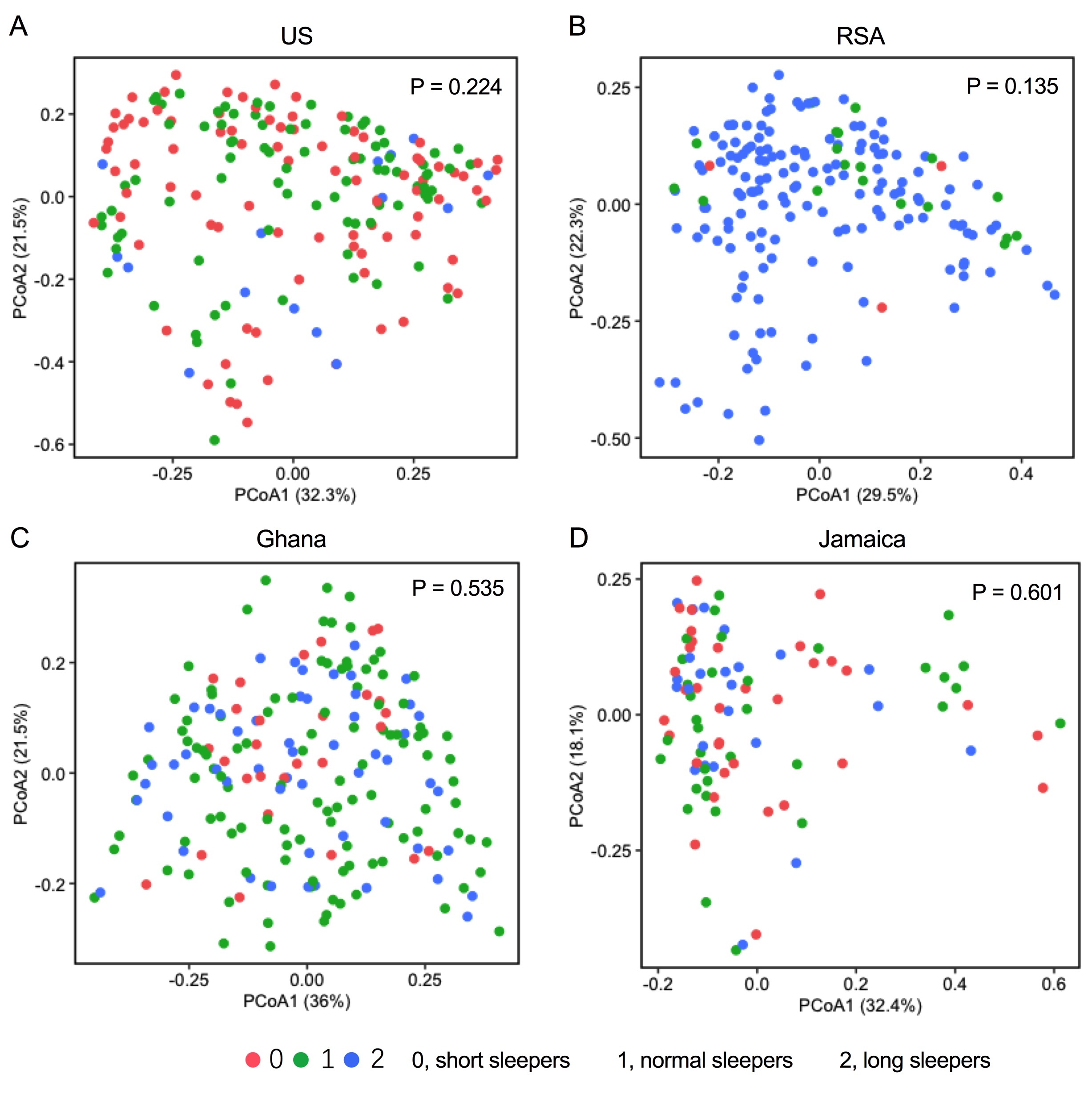
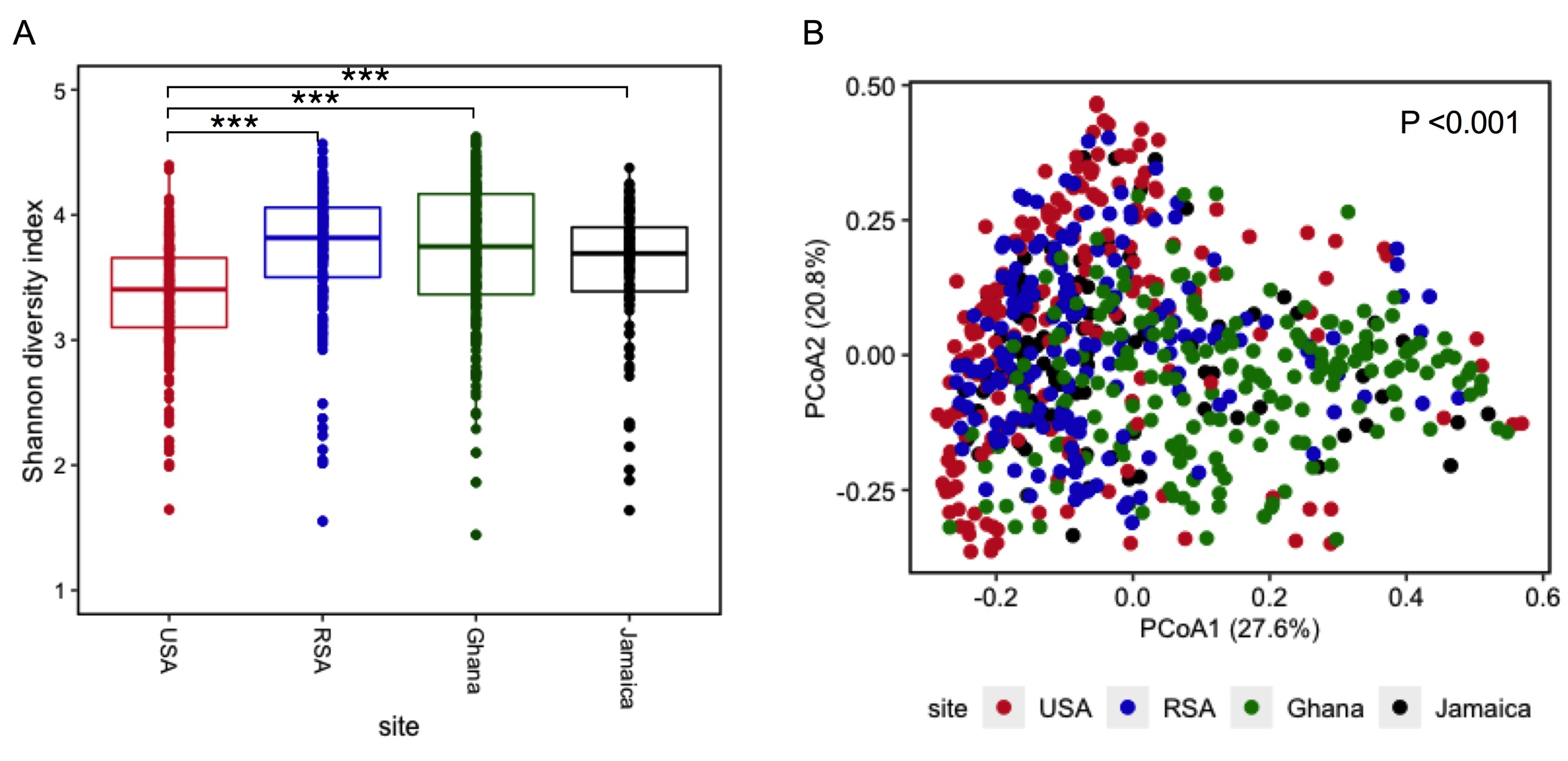
We used 16S rRNA amplicon sequencing to define shifts in microbial community structure and composition associated with sleep length. A total of 10,805,592 16S ribosomal RNA gene sequences with good quality were generated from the 655 fecal samples, which clustered into 6, 909 Exact Sequence Variants (ESVs). Alpha diversity was calculated by Shannon’s diversity index diversity [39], which is a quantitative measure of community richness, and observed otu diversity, which a qualitative measure of community richness [39]. Gut microbial alpha diversity was significantly different between normal sleepers, short sleepers, and long sleepers, whereby short sleepers had a significantly lower alpha diversity, and long sleepers had a significantly higher alpha diversity compared with normal sleepers (Fig 1A, Shannon index, p<0.05. S1 Fig. Observed otu diversity). Principal coordinate analysis (PCoA) was performed based on weighted and unweighted UniFrac distances, a method for computing differences between microbial communities based on phylogenetic information [40]. 1000 reads/samples were considered to calculate the inter-individual beta diversity dissimilarity. Weighted UniFranc considered both ESVs presence/absence and abundance distances, and unweighted UniFrac only considered ESVs presence/absence. Permutational multivariate analysis of variance (PERMANOVA, R function adonis (vegan, 999 permutations)) was used to analyze statistical differences in beta diversity [41]. Similarly, we found that sleep length significantly contributed to the inter-individual beta diversity dissimilarity in Exact Sequence Variants (ESV)-level gut microbial composition, both in the weighted and unweighted UniFrac dissimilarity index (permutational multivariate analysis of variance (PERMANOVA), p<0.001) (Fig 1B and 1C). Pairwise comparation based on permutation tests showed that both weighted and unweighted UniFrac distances between samples within short sleepers and long sleepers are significantly different from that between samples within normal sleepers (p<0.01). These data suggest that ESV features contribute markedly to the dissimilarity in microbial community structure in response to sleep length. However, neither alpha diversity (S2 Fig) nor beta diversity (weighted and unweighted UniFrac dissimilarity index, S3 and S4 Figs) within each country site (Ghana, South Africa, Jamaica or US) were significantly different among participants with different sleep length (p > 0.05), which is most likely due to small size of the cohorts. Since geographic location contributed significantly to the distribution of CM risk factors and sleep duration, e.g. Americans have the highest prevalence of overweight and obesity and slept the least number of hours, when compared to the other sites, we also investigate its effect in gut microbial community structure. Gut microbial alpha diversity was significantly lower in Americans compared to Ghanaians, Jamaicans, and South Africans (p<0.001) (S5A Fig). Geographic location also significantly contributed to the inter-individual beta diversity dissimilarity gut microbial composition in the weighted UniFrac dissimilarity index (PERMANOVA, p<0.001) (S5B Fig). These observations suggest that a country-related physiological or environmental influence change in the host is conducive to a less complex microbiota.
Fig 1. Sleep length can significantly impact intestinal microbiota community structure.

(A) Alpha diversity (Shannon index) between different sleep groups; * p < 0.05; *** p < 0.001. (B) Beta diversity analysis (weighted UniFrac distance) between different sleep groups; (C) Beta diversity analysis (unweighted UniFrac distance); and (D) Differential ESV abundance among short, normal and long sleepers, adjusted for BMI, age, gender and countries. ESVs with relative abundance ≥ 1% in at least one group shown. Data shown are means ± S.E.M.; * p(fdr-corrected) < 0.05. fdr, false discovery rate.
The taxonomic features that differentiated the gut microbiota among short, normal and long sleepers were determined using Analysis of Composition of Microbiota (ANCOM, adjusted for country, age, gender and BMI, p (false discovery rate (fdr)-corrected) < 0.05) (Fig 1D). An ESV assigned to genus Dialister was significantly enriched in short sleepers, while another ESV also assigned to genus Dialister was at a significant greater proportion in the long sleepers, compared to the normal sleepers. 2 ESVs belonging to genus Prevotella were both more abundant in the long sleepers, compared to short and normal sleepers. Furthermore, long sleepers were also significantly enriched with 4 ESVs belonging to family Erysipelotrichaceae, family Ruminococcaceae, genus Oscillospira, and genus Catenicacterium, compared to the other two groups. And short sleepers had significantly higher proportion of one ESV from genus Bacteroides, compared to normal and long sleepers (Fig 1D).
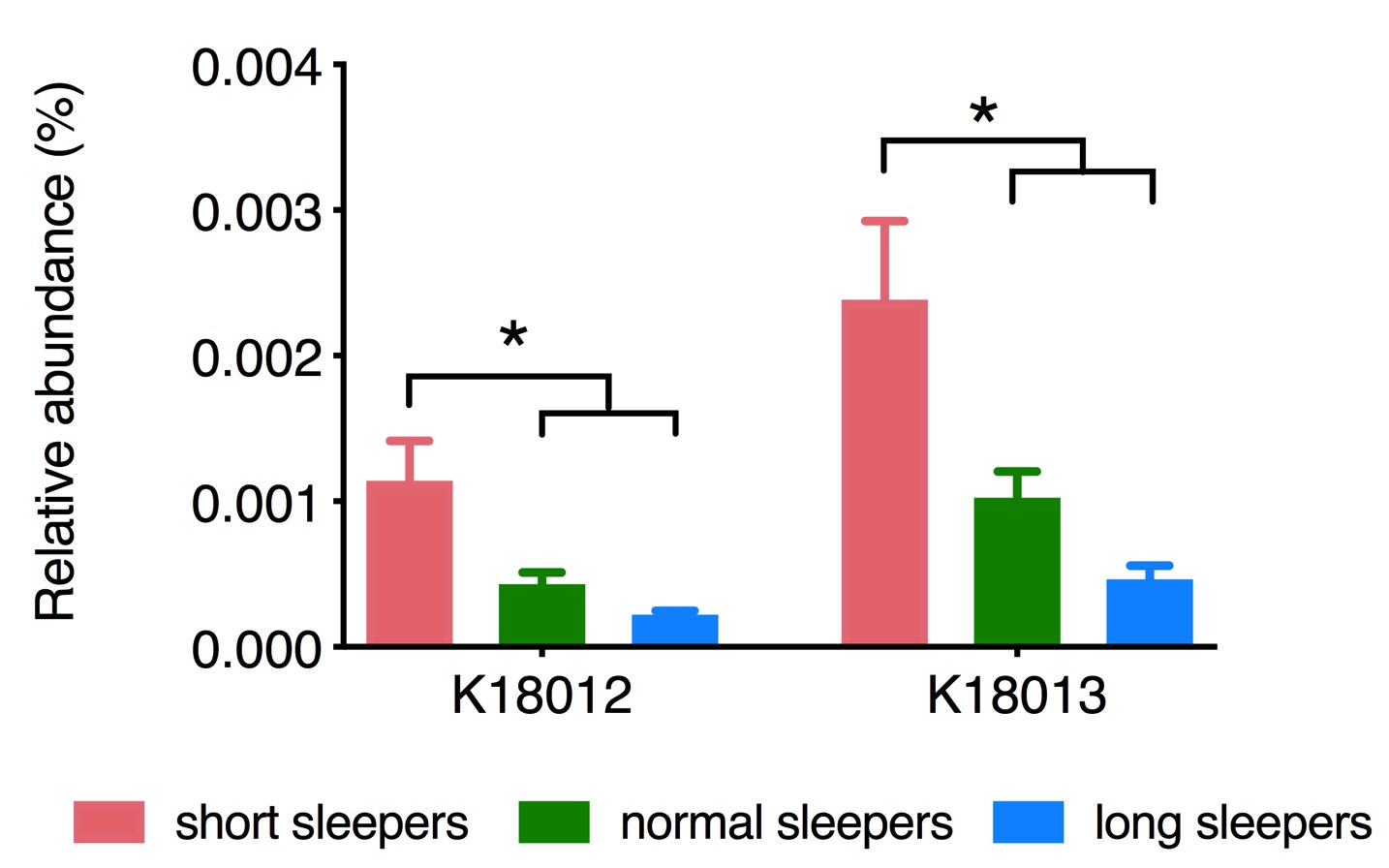
Piphillin [42] was used to predict the gene abundances for gut microbial metabolic pathways based on the 16S rRNA gene abundance. Specifically, we wanted to determine the differences in the predicted abundance of gut microbial genes associated with butyrate metabolism (via less-dominant pathway-the 4-aminobutyrate pathway, include amino-acids (lysine, glutarate and 4-aminobytyrate/succinate) as substrates [38], H2S and LPS synthesis between short, normal and long sleepers, which are involved in the low-grade systemic inflammation in metabolic diseases [34–37]. Predicted genes involved in butyrate biosynthesis pathways showed that only 3,5-diaminohexanoate dehydrogenase (K18012) and 3-keto-5-aminohexanoate cleavage enzyme (K18013) in the Lysine pathway was enriched in short sleepers, compared to normal sleepers and long sleepers (S6 Fig; pfdr<0.05, adjusted for age, gender, country and BMI). No statistically significant differences in the genes involved in the LPS or H2S synthesis pathway were observed between short sleepers and normal sleepers (pfdr>0.05, adjusted for age, gender, country and BMI).
A correlation network was constructed by calculating Spearman correlations between ESVs using the gut microbiota of short, normal and long sleepers’ datasets, and only robust correlations were considered for network construction (-0.60 ≥ ρ≥ 0.60, pfdr <0.05). The vertices in this network represent ESVs and the edges that connect these nodes represent correlations between ESVs. The network of the short and long sleepers’ microbiota has a higher number of vertices and edges (short sleepers, 312 vertices and 566 edges, long sleepers, 315 vertices and 376 edges) in comparison to that of normal sleepers (272 vertices and 265 edges) (Fig 2A–2C). Using the co-occurrence network outlined above, we also examined whether ESVs associated with a special sleep length exhibited unique node-level topological features. This feature set included betweenness centrality (a measure of centrality based on the number of times a node acts as a bridge along the shortest path between two other nodes.), closeness centrality (a measure of centrality based on the sum of the length of the shortest paths between this node and connected nodes), and degree (the number of connections a single node has). The betweenness centrality feature was used to measure the centrality of each node in the network. Significantly higher betweenness centrality scores were observed for ESVs associated with short sleepers’ microbiota than those associated with normal or long sleepers’ microbiota (pfdr <0.05, Fig 2D). This suggests that the gut microbes from the short sleepers were more often located in central positions within the network, compared to those from the normal sleepers. We next examined additional node-level topological measures, degree and closeness, for each ESV in the co-occurrence network. Degree and Closeness only take into account only the immediate neighborhood of ESVs, and hence capturing a different aspect of network topological features. Closeness and degree of ESVs associated with short sleepers’ microbiota were also significantly higher than those associated with normal or long sleepers’ microbiota (p <0.001, Fig 2E and 2F). Thus, co-occurrence relationships revealed by network analysis showed unique interactions among the short, normal and long duration sleepers, which the gut microbiota of short sleepers had a closer connected network, compared to those of normal and longer sleepers.
Fig 2. The network analysis revealing the co-occurrence patterns among short sleepers, normal sleepers and long sleepers.

A connection represents a strong (Spearman’s correlation coefficient ρ>0.6) and significant (P <0.05) correlation. A-C, co-occurrence network of short sleepers (A) normal sleepers (B) and long sleepers (C) The nodes represented unique ESV feature in the data sets. The size of each node is proportional to the relative abundance. Node color corresponds to phylum taxonomic classification. The edge thickness is equivalent to the correlation values. D-F, Topological features for each node in the network, (D) betweenness centralization, (E) closeness centrality and (F) node degree values. *** p< 0.0001.
Discussion
Recently, there has been some evidence that a relationship between disrupted sleep and the gut microbiota exists because both sleep alterations as well as a gut dysbiosis are associated with similar diseases [24, 43]. However, this relationship is not well understood, and studies have been limited by small sample sizes. Here, we performed the first investigation into how sleep length may alter the human gut microbiota in a large cohort of African-origin adults. Overall, we demonstrate that the structure of the gut microbiota is remarkably impacted by sleep length. Indeed, it may well be that many of the adverse effects that have been observed in sleep disorder may be due in part to gut microbial dysbiosis.
Most of the health decline in humans have been reported to be associated with reduced overall bacterial community diversity and richness [44–47], which is identified as one of the characters of gut microbial dysbiosis. In our current study, we saw that sleep length significantly associated with the alpha diversity in humans. Participants with short sleep duration exhibited lower alpha diversity; however, Participants with long sleep duration showed higher alpha diversity compared to those with normal sleep duration, which implied that not only too low, but too high gut bacterial community diversity may also induce the gut microbial dysbiosis. This suggest that chronic sleep disruption may also promote an intestinal microbial dysbiosis [29–31]. Certainly disrupted sleep may serve as another novel environmental factor that induce gut bacterial diversity, besides diet, urbanization, and other lifestyle changes in modern society [48].
In this study, we observed an increased relative abundance of one ESV assigned to genus Dialister in the gut community of participants with short or long sleep duration separately, compared to normal length sleepers. An increase of the species or strains in this genus, has been found to be associated with many kinds of diseases in humans, such as spondyloarthritis [49], periodontitis [50], inflammatory bowel disease and their unaffected relatives [51, 52], pulmonary disease [53] and so on. Such changes may promote increased chronic inflammatory state and could therefore constitute a possible mechanism through which chronic sleep loss can increase the risk of human diseases. We also observed decreased levels of two ESVs assigned to the genus Prevotella in the gut community of participants with short sleep duration. Reduced incidence of genus Prevotella have been associated previously with autistic children [54], and Parkinson’s disease [55]. However, one ESV assigned to genus Prevotella was significantly higher in the gut community of participants with long sleep duration, and it should also be noted that there are a handful of studies that suggest that Prevotella are significantly enriched in patients with metabolic diseases [56–58]. This was the first study to show that short sleep participants with harbored lower proportions of ESVs assigned to genus Prevotella, while long sleepers harbored higher proportions of ESVs assigned to genus Prevotella. Furthermore, this study also showed that participants with long sleep duration were also significantly enriched with 4 ESVs belonging to family Erysipelotrichaceae, family Ruminococcaceae, genus Oscillospira, and genus Catenicacterium, compared to the participants with normal or short sleep duration. The importance of Erysipelotrichaceae in inflammation-related disorders of the gastrointestinal tract is highlighted by the fact that they have been found to be enriched in individuals with colorectal cancer or IBD in both human and mouse studies [59–62]. In addition, higher levels of Erysipelotrichaceae have been observed in human disease related to metabolic disorders [63–65]. However, several recent studies have observed that members of the Oscillospira genus were highly enriched in lean subjects, and positively associated with the leanness in human and mouse studies [66, 67], and decreased in individuals with inflammatory diseases, nonalcoholic steatohepatitis [68], Crohn’s disease [69]. Members of the family Ruminococcaceae, which are involved in degrading dietary indigestible fibers and polysaccharides and in producing butyrate, were also important in improving weaning weight [70]. An increase of the relative abundance in the genus Catenicacterium has been found to be associated with obesity with metabolic syndrome [71], however, it was also shown that cholorectal cancer patients harbor less Catenicacterium in the gut [72]. We also observed that participants with short sleep duration had significantly higher proportion of one ESV from genus Bacteroides, compared to normal and long sleepers. Bacteroides, an abundant genus in the intestines of mammals, play essential roles in host immunity, glucose and lipid metabolism and the prevention or induction of diseases [73–80]. The specific role the members in genus Bacteroides found in this study requires more experimental evidence to interpret their role. Given that many members of these gut microbiota above have not yet been grown in culture and more experimental evidence need to be collected, it remains a conundrum in gut microbiota research and the association between gut microbiota and sleep disruption. However, in light of the strong evidence linking these gut microbiol members to human health and disease, these bacteria should be cultivated to characterize both genome and function to investigate its potential link to the sleep disruption in future studies.
Similarly, we observed that sleep length was associated with the gut microbiota functionality. The predicted functional potential of the bacterial communities showed significant elevated abundance of two predicted enzymes involved in the butyrate synthesis via lysine pathway in short sleep participants. It has shown the beneficial effects of butyrate on host homeostasis by enhancing gut barrier function and reducing inflammation [81], however, butyrate synthesis via less-dominant pathway-the 4-aminobutyrate pathway, include amino-acids (lysine, glutarate and 4-aminobytyrate/succinate) as substrates, can produce pro-inflammatory byproducts and increase the risk of human diseases [37].
The co-occurrence networks generated for the gut microbiota from participants with short, normal and long sleep duration showed significant differences in the node-level features, specifically between the participants with short sleep duration, compared to the other 2 cohorts. Participants with short sleep duration have more being center nodes, that were closely connected within the network, and nodes with increasing influence in connecting different parts of the network, compared to the less connected and centered co-occurrence networks, as found in participants with normal and long sleep duration. Co-occurrence relationships revealed by network analysis showed unique interactions among the participants with different duration.
At present, it is still not known that if there’s a causal relationship between disrupted sleep and disrupted gut microbiota in humans. It is not clear there is a common mechanism mediates the effects of disrupted gut microbiota and sleep disorder either. There are several potential mechanisms that are listed in previous publications and reviews in the field, such as alterations in interaction of gut microbiome with bile acid metabolism [82, 83], microbiota-mediated molecular signaling, like SCFA [84–86] or altered nutrient uptake through the intestinal wall [87, 88], etc. Chronic inflammation also appears to be an important mechanism for the deleterious health consequences of insufficient sleep and gut microbial dysbiosis, as exemplified by increased risk and severity of obesity and related other metabolic syndrome [89], cardiovascular disease [90], and inflammatory bowel disease [91]. Notably, when considered alongside previous studies, the current research suggests that disrupted sleep may be associated with dysbiosis of gut microbiota in humans (including overall decreased diversity, changed abundance of some taxa and unique co-occurrence network). However, this study only observes an association and doesn’t prove causality between the sleep disruption and gut microbial dysbiosis with the composition of gut microbiota. In future, prospective investigations in larger and more prolonged sleep researches and causally experimental studies are needed to confirm these findings, investigate the underlying mechanism and determine whether improving microbial homeostasis may buffer against sleep-related health decline in humans. Lastly, considering that the significant geographically difference of sleep duration and the microbial composition showed in this study, the geographic effect need to be considered to investigate the potential relationship between sleep disruption and gut microbiota dysbiosis in the future studies
Our study is not without several limitations, including that sleep lengths were self-reported and do not include the time of day when sleep occurred. In fact, a single question was asked about sleep, thus excluding an evaluation of sleep. However, these weaknesses were balanced by several study strengths, including the large sample size, the use of cohorts from diverse countries and measured clinical data. Future directions of this study will include increasing the number of questions asked about sleep routines and quality. Also sleep will be objectively measured using sleep monitors.
Conclusion
To our knowledge, our study is the first to provide information in a large human cohort that sleep length alters gut microbiota. The changes in the structural and functional dysbiosis of microbiota that we observed in the present study could be due, at least in part, to sleep length. These findings should be considered along with the limitations of the study. The sleep disorder-gut microbiota connection will require numerous larger studies, including longitudinal studies, to fully elucidate the role of the gut microbiota in relation to sleep length and associated deleterious health consequences. Similarly, further animal studies are needed to understand causal mechanisms by which sleep duration affects the gut microbial dysbiosis. Metagenomic sequencing might also need to be considered to determine whether disrupted sleep patterns functionally alter gut microbiota.
Materials and methods
Participant selection
The original METS cohort consisted of 2,506 male and female participants of African descent aged 25–45 years old. Participants were enrolled in METS between January 2010 and December 2011 and followed annually for three years. A detailed description of the METS protocol has previously been published [92]. For the current study of METS, the microbiome study, both fecal and saliva samples were collected in 2014 from 655 and 620 male and female participants from the first METS cohort, respectively, from Ghana (N = 196), South Africa (N = 176), Jamaica (N = 92) and the US (N = 191). These sites were chosen as they span the epidemiologic transition, and each site is at a different stage of development as defined by the United Nation’s Human Development Index. Exclusion criteria included self-reported infectious disease (e.g., HIV), pregnancy, current breast-feeding and inability to participate in normal physical activities. METS was approved by the Institutional Review Board of Loyola University Chicago, IL, US; the Committee on Human Research Publication and Ethics of Kwame Nkrumah University of Science and Technology, Kumasi, Ghana; the Research Ethics Committee of the University of Cape Town, South Africa; the Board for Ethics and Clinical Research of the University of Lausanne, Switzerland; and the Ethics Committee of the University of the West Indies, Kingston, Jamaica. All study procedures were explained to participants in their native languages, and participants provided written informed consent after being given the opportunity to ask any questions.
Anthropometric and biochemical measurements and stool collection
All measurements were performed by site staff in their local research clinics. Participants wore light clothing and no shoes when weight and height were measured. Participants were asked to provide an early morning fecal sample, using a standard collection kit (EasySampler stool collection kit, Alpco, NH) at their home. Fecal samples were immediately brought to the site clinics and stored at -80°C. Participants were asked to fast 10–12 hours prior to clinic visit, so that fasting capillary glucose concentrations could be determined using finger stick (Accu-check Aviva, Roche).
Sleep and demographic data
Sleep, age and smoking data were obtained via questionnaire. Participants were asked how many hours they slept each night. Neither the times at which they slept nor whether they slept during the day or evening was recorded.
Statistical analysis
Statistical analyses for Tables 1and 2 were performed in Stata (version 12, Manufacturer, College Station, TX, USA). Table 1 summarized continuous participant characteristics and risk factors using mean ± SD, and Table 2 summarized dichotomous and categorical variables using proportions which were presented as N, %. Comparisons of participant characteristics and risk factors within sites and overall were performed using t-tests for normally distributed continuous variables, Wilcoxon rank-sum scores for non-normally distributed continuous variables and Pearson’s chi-squared test for categorical variables at a significance level (α) of 0.05.
DNA isolation and 16S ribosomal RNA (rRNA) gene sequencing
The microbial genomic DNA from the human stool samples was extracted using the DNeasy PowerSoil DNA Isolation Kit (Qiagen) (Mo Bio Laboratories, Carlsbad, CA, USA) following the protocol of Flores et al [93]. The V4 region of 16S rRNA gene was amplified and sequenced using the Illumina MiSeq platform [94]. The primers used for amplification (515F-806R) contain adapters for MiSeq sequencing and single-end barcodes allowing pooling and direct sequencing of PCR products [95]. Each 25 μl PCR reaction contained the following mixture: 12 μl of MoBio PCR Water (Certified DNA-Free; Mo Bio Laboratories), 10 μl of 5-Prime HotMasterMix (1×), 1 μl of forward primer (5 μM concentration, 200 pM final), 1 μl of Golay Barcode Tagged Reverse Primer (5 μM concentration, 200 pM final), and 1 μl of template DNA [96]. The conditions for PCR were as follows: 94 °C for 3 min to denature the DNA, with 35 cycles at 94 °C for 45 s, 50 °C for 60 s, and 72 °C for 90 s, with a final extension of 10 min at 72 °C to ensure complete amplification. Amplicons were quantified using PicoGreen (Invitrogen, Grand Island, NY, USA) assays and a plate reader, followed by clean-up using UltraClean® PCR Clean-Up Kit (Mo Bio Laboratories) and then quantification using Qubit readings (Invitrogen). The 16S rRNA gene samples were sequenced on an Illumina MiSeq platform (2 × 150 paired-end sequencing, V3 chemistry) at Argonne National Laboratory core sequencing facility according to Earth Microbiome Project (EMP) standard protocols [97].
16S rRNA gene sequencing data preprocessing and analysis
Raw sequences were pre-processed, quality filtered and analyzed using the next-generation microbiome bioinformatics platform (QIIME2 version 2019.1 pipeline) according to the developer’s suggestion [98]. We used the DADA2 algorithm [99] a software package wrapped in QIIME2, to identify exact sequence variants (ESVs). Quality control, filtering low quality regions of the sequences, by truncating them to 120 base pair length, identification and removal of chimera sequences, merging paired end reads, which yielded the ESV feature table (ESV table). Chloroplast and mitochondrial DNA were removed. Alpha and beta-diversity analyses were performed in R using the phyloseq package [100]. Alpha diversity was calculated by Shannon’s diversity index diversity [39]. Results were adjusted for BMI, age, gender and country. Principal coordinate analysis (PCoA) was performed based on weighted and unweighted UniFrac distances, a method for computing differences between microbial communities based on phylogenetic information [40]. Weighted UniFranc considered both ESVs presence/absence and abundance distances, and unweighted UniFrac only considered ESVs presence/absence. Permutational multivariate analysis of variance (PERMANOVA, R function adonis (vegan, 999 permutations)) was used to analyze statistical differences in beta diversity [41]. Pairwise tests were performed to determine which specific pairs of groups (e.g., normal sleepers and short sleepers) differ from one another using the beta-group-significance command with the—p-pairwise parameter in QIIME2.
For taxonomic comparisons, relative abundances based on all obtained reads were used. We used the QIIME2 plugin “q2-feature-classifier” and the Naïve Bayes classifier that was trained on the metagenome annotation package Greengenes13.8 99% operational taxonomic units (OTUs) full-length sequences to obtain the taxonomy for each ESV [101]. Significantly differential ESVs were determined using the statistical framework called analysis of composition of microbiomes (ANCOM) [102] for two group comparisons. Benjamini–Hochberg false discovery rate (fdr) correction was used to correct for multiple hypothesis testing [103]. Results were adjusted for BMI, age, gender and country.
Metagenome functional predictions of the microbial pathways
We used Piphillin algorithm to predict the functional profiles of the microbiome [42]. Briefly, this algorithm uses direct nearest-neighbor matching between 16S rRNA gene sequencing datasets and microbial genomic databases to infer the metagenomic content of the samples. Gene prediction was performed on ESVs table using online Piphillin (http://secondgenome.com/Piphillin.).), with KEGG (2017) as reference database and 97% identity cut-off. Predicted gene content and gene copy numbers within each genome were retrieved and classified in terms of KEGG orthology (KOs) [104]. Results were adjusted for BMI, age, gender and country. Statistical analyses were performed in R. Student’s t-test (normally distributed) or Mann-Whitney U test (not normally distributed) was used for to detect differentially abundant KOs between two groups. FDR correction was used to correct for multiple hypothesis testing.
Network construction and topological feature analysis
To visualize the correlations in the network interface, we constructed a correlation matrix by calculating all possible pairwise Spearman’s rank correlations between the normal-, short- and long sleepers in the present study. To reduce rare ESVs in the data set, we removed ESVs with relative abundances less than 0.01% of the total number of sequences. A correlation between two ESVs was considered statistically robust if the Spearman’s correlation coefficient (ρ) was >0.6 and the P-value was <0.05. To reduce the chances of obtaining false-positive results, the P-values were adjusted with a multiple testing correction using the Benjamini–Hochberg method [103]. The robust pairwise correlations formed their co-occurrence networks. The nodes in this network represent ESVs and the edges that connect these nodes represent correlations between ESVs. Network analyses were performed in R environment using VEGAN [41], and igraph packages [105]. We calculated topological features for each node in the network with the igraph package [105]. This feature set included betweenness centrality (the number of shortest paths going through a node), closeness centrality (the number of steps required to access all other nodes from a given node), and degree (the number of adjacent edges). The betweenness centrality feature was used to measure the centrality of each node in the network.
Supporting information
(JPG)
{kind=link}
(A) USA, the United States of America, (B) RSA, South Africa (C) Ghanaian and (D) Jamaican populations.
(JPG)
{kind=link}
(A) USA, the United States of America, (B) RSA, South Africa (C) Ghanaian and (D) Jamaican populations.
(JPG)
{kind=link}
(A) USA, the United States of America, (B) RSA, South Africa (C) Ghanaian and (D) Jamaican populations.
(JPG)
{kind=link}
(A) Alpha diversity analysis (Shannon Index); (B) beta diversity analysis (weighted UniFrac distance metric) from *** p < 0.001.
(JPG)
{kind=link}
K18012, 3,5-diaminohexanoate dehydrogenase; K18013, 3-keto-5-aminohexanoate cleavage enzyme. * p < 0.05.
(JPG)
{kind=link}
Acknowledgments
The authors would like to acknowledge the original METS study participants and site-specific clinic staff members.
Data Availability
All 16S rRNA sequences and sample metadata are publicly available in EBI under accession no. ERP115612 (Study: PRJEB32880).
Funding Statement
This research was funded by the National Institute of Diabetes and Digestive and Kidney Diseases in the form of NIH R01 grants awarded to AL (R01-DK080763), RD and AL (R01-DK090360), and LD (R01-DK111848), and the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development in the form of a grant awarded to LD (1I01BX003382-01-A1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Reutrakul S, Van Cauter E. Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metabolism. 2018;84:56–66. doi: 10.1016/j.metabol.2018.02.010 [DOI] [PubMed] [Google Scholar]
- 2.Spiegel K, Knutson K, Leproult R, Tasali E, Van Cauter E. Sleep loss: a novel risk factor for insulin resistance and Type 2 diabetes. Journal of applied physiology (Bethesda, Md: 1985). 2005;99(5):2008–19. doi: 10.1152/japplphysiol.00660.2005 [DOI] [PubMed] [Google Scholar]
- 3.Morriss RK, Wearden AJ, Battersby L. The relation of sleep difficulties to fatigue, mood and disability in chronic fatigue syndrome. Journal of psychosomatic research. 1997;42(6):597–605. doi: 10.1016/s0022-3999(97)89895-9 [DOI] [PubMed] [Google Scholar]
- 4.Barcelo A, Barbe F, Llompart E, Mayoralas LR, Ladaria A, Bosch M, et al. Effects of obesity on C-reactive protein level and metabolic disturbances in male patients with obstructive sleep apnea. The American journal of medicine. 2004;117(2):118–21. doi: 10.1016/j.amjmed.2004.01.025 [DOI] [PubMed] [Google Scholar]
- 5.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. The New England journal of medicine. 2005;353(19):2034–41. doi: 10.1056/NEJMoa043104 [DOI] [PubMed] [Google Scholar]
- 6.Swanson GR, Burgess HJ, Keshavarzian A. Sleep disturbances and inflammatory bowel disease: a potential trigger for disease flare? Expert review of clinical immunology. 2011;7(1):29–36. doi: 10.1586/eci.10.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goes ACJ, Reis LAB, Silva MBG, Kahlow BS, Skare TL. Rheumatoid arthritis and sleep quality. Revista brasileira de reumatologia. 2017;57(4):294–8. doi: 10.1016/j.rbre.2016.07.011 [DOI] [PubMed] [Google Scholar]
- 8.Kronholm E, Partonen T, Laatikainen T, Peltonen M, Harma M, Hublin C, et al. Trends in self-reported sleep duration and insomnia-related symptoms in Finland from 1972 to 2005: a comparative review and re-analysis of Finnish population samples. J Sleep Res. 2008;17(1):54–62. doi: 10.1111/j.1365-2869.2008.00627.x [DOI] [PubMed] [Google Scholar]
- 9.Stamatakis KA, Kaplan GA, Roberts RE. Short sleep duration across income, education, and race/ethnic groups: population prevalence and growing disparities during 34 years of follow-up. Ann Epidemiol. 2007;17(12):948–55. doi: 10.1016/j.annepidem.2007.07.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luckhaupt SE, Tak S, Calvert GM. The prevalence of short sleep duration by industry and occupation in the National Health Interview Survey. Sleep. 2010;33(2):149–59. doi: 10.1093/sleep/33.2.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hale L, Do DP. Racial differences in self-reports of sleep duration in a population-based study. Sleep. 2007;30(9):1096–103. doi: 10.1093/sleep/30.9.1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buxton OM, Pavlova M, Reid EW, Wang W, Simonson DC, Adler GK. Sleep restriction for 1 week reduces insulin sensitivity in healthy men. Diabetes. 2010;59(9):2126–33. doi: 10.2337/db09-0699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354(9188):1435–9. doi: 10.1016/S0140-6736(99)01376-8 [DOI] [PubMed] [Google Scholar]
- 14.Leproult R, Holmback U, Van Cauter E. Circadian misalignment augments markers of insulin resistance and inflammation, independently of sleep loss. Diabetes. 2014;63(6):1860–9. doi: 10.2337/db13-1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broussard JL, Ehrmann DA, Van Cauter E, Tasali E, Brady MJ. Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Ann Intern Med. 2012;157(8):549–57. doi: 10.7326/0003-4819-157-8-201210160-00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spiegel K, Tasali E, Penev P, Van Cauter E. Brief communication: Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med. 2004;141(11):846–50. doi: 10.7326/0003-4819-141-11-200412070-00008 [DOI] [PubMed] [Google Scholar]
- 17.Cappuccio FP, Taggart FM, Kandala NB, Currie A, Peile E, Stranges S, et al. Meta-analysis of short sleep duration and obesity in children and adults. Sleep. 2008;31(5):619–26. doi: 10.1093/sleep/31.5.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anothaisintawee T, Reutrakul S, Van Cauter E, Thakkinstian A. Sleep disturbances compared to traditional risk factors for diabetes development: Systematic review and meta-analysis. Sleep Med Rev. 2015;30:11–24. doi: 10.1016/j.smrv.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 19.Watson NF, Badr MS, Belenky G, Bliwise DL, Buxton OM, Buysse D, et al. Recommended Amount of Sleep for a Healthy Adult: A Joint Consensus Statement of the American Academy of Sleep Medicine and Sleep Research Society. Sleep. 2015;38(6):843–4. doi: 10.5665/sleep.4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Killick R, Hoyos CM, Melehan KL, Dungan GC, Poh J, Liu PY. Metabolic and hormonal effects of ’catch-up’ sleep in men with chronic, repetitive, lifestyle-driven sleep restriction. Clin Endocrinol (Oxf). 2015. doi: 10.1111/cen.12747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leproult R, Deliens G, Gilson M, Peigneux P. Beneficial Impact of Sleep Extension on Fasting Insulin Sensitivity in Adults with Habitual Sleep Restriction. Sleep. 2015;35(5):707–15. doi: 10.5665/sleep.4660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.So-ngern A, Chirakalwasan N, Saetung S, Chanprasertyothin S, Thakkinstian A, Reutrakul S. Effects of sleep extension on glucose metabolism in chronically sleep-deprived individuals Journal of Clinical Sleep Medicine. 2019;in press. doi: 10.5664/jcsm.7758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al Khatib HK, Hall WL, Creedon A, Ooi E, Masri T, McGowan L, et al. Sleep extension is a feasible lifestyle intervention in free-living adults who are habitually short sleepers: a potential strategy for decreasing intake of free sugars? A randomized controlled pilot study. Am J Clin Nutr. 2018;107(1):43–53. doi: 10.1093/ajcn/nqx030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258–70. doi: 10.1016/j.cell.2012.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–9. doi: 10.1038/nature11552 [DOI] [PubMed] [Google Scholar]
- 26.Buxton OM, Cain SW, O’Connor SP, Porter JH, Duffy JF, Wang W, et al. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med. 2012;4(129):129ra43. doi: 10.1126/scitranslmed.3003200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buxton OM, Marcelli E. Short and long sleep are positively associated with obesity, diabetes, hypertension, and cardiovascular disease among adults in the United States. Social science & medicine (1982). 2010;71(5):1027–36. [DOI] [PubMed] [Google Scholar]
- 28.Anothaisintawee T, Reutrakul S, Van Cauter E, Thakkinstian A. Sleep disturbances compared to traditional risk factors for diabetes development: systematic review and meta-analysis. Sleep medicine reviews. 2016;30:11–24. doi: 10.1016/j.smrv.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 29.Poroyko VA, Carreras A, Khalyfa A, Khalyfa AA, Leone V, Peris E, et al. Chronic Sleep Disruption Alters Gut Microbiota, Induces Systemic and Adipose Tissue Inflammation and Insulin Resistance in Mice. Sci Rep. 2016;6:35405. doi: 10.1038/srep35405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds AC, Paterson JL, Ferguson SA, Stanley D, Wright KP Jr., Dawson D. The shift work and health research agenda: Considering changes in gut microbiota as a pathway linking shift work, sleep loss and circadian misalignment, and metabolic disease. Sleep Med Rev. 2017;34:3–9. doi: 10.1016/j.smrv.2016.06.009 [DOI] [PubMed] [Google Scholar]
- 31.Benedict C, Vogel H, Jonas W, Woting A, Blaut M, Schurmann A, et al. Gut microbiota and glucometabolic alterations in response to recurrent partial sleep deprivation in normal-weight young individuals. Molecular metabolism. 2016;5(12):1175–86. doi: 10.1016/j.molmet.2016.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang SL, Bai L, Goel N, Bailey A, Jang CJ, Bushman FD, et al. Human and rat gut microbiome composition is maintained following sleep restriction. Proc Natl Acad Sci U S A. 2017;114(8):E1564–e71. doi: 10.1073/pnas.1620673114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, Hooper LV. The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science. 2017;357(6354):912–6. doi: 10.1126/science.aan0677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Current pharmaceutical design. 2009;15(13):1546–58. doi: 10.2174/138161209788168164 [DOI] [PubMed] [Google Scholar]
- 35.Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nature immunology. 2011;12(1):5–9. doi: 10.1038/ni0111-5 [DOI] [PubMed] [Google Scholar]
- 36.Blaut M, Clavel T. Metabolic diversity of the intestinal microbiota: implications for health and disease. J Nutr. 2007;137(3 Suppl 2):751s–5s. doi: 10.1093/jn/137.3.751S [DOI] [PubMed] [Google Scholar]
- 37.Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio. 2014;5(2):e00889. doi: 10.1128/mBio.00889-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullington JM, Simpson NS, Meier-Ewert HK, Haack M. Sleep loss and inflammation. Best Pract Res Clin Endocrinol Metab. 2010;24(5):775–84. doi: 10.1016/j.beem.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lozupone CA, Knight R. Species divergence and the measurement of microbial diversity. FEMS microbiology reviews. 2008;32(4):557–78. doi: 10.1111/j.1574-6976.2008.00111.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology. 2005;71(12):8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson MJ. A new method for non‐parametric multivariate analysis of variance. Austral ecology. 2001;26(1):32–46. [Google Scholar]
- 42.Iwai S, Weinmaier T, Schmidt BL, Albertson DG, Poloso NJ, Dabbagh K, et al. Piphillin: Improved Prediction of Metagenomic Content by Direct Inference from Human Microbiomes. PloS one. 2016;11(11):e0166104. doi: 10.1371/journal.pone.0166104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamdar BB, Needham DM, Collop NA. Sleep deprivation in critical illness: its role in physical and psychological recovery. J Intensive Care Med. 2012;27(2):97–111. doi: 10.1177/0885066610394322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–6. doi: 10.1038/nature12506 [DOI] [PubMed] [Google Scholar]
- 45.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, et al. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. The Journal of infectious diseases. 2008;197(3):435–8. doi: 10.1086/525047 [DOI] [PubMed] [Google Scholar]
- 46.Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, et al. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. Journal of clinical microbiology. 2013;51(9):2884–92. doi: 10.1128/JCM.00845-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bisgaard H, Li N, Bonnelykke K, Chawes BL, Skov T, Paludan-Muller G, et al. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. The Journal of allergy and clinical immunology. 2011;128(3):646–52.e1-5. doi: 10.1016/j.jaci.2011.04.060 [DOI] [PubMed] [Google Scholar]
- 48.Bello MGD, Knight R, Gilbert JA, Blaser MJ. Preserving microbial diversity. Science. 2018;362(6410):33–4. doi: 10.1126/science.aau8816 [DOI] [PubMed] [Google Scholar]
- 49.Tito RY, Cypers H, Joossens M, Varkas G, Van Praet L, Glorieus E, et al. Brief Report: Dialister as a Microbial Marker of Disease Activity in Spondyloarthritis. Arthritis & rheumatology (Hoboken, NJ). 2017;69(1):114–21. doi: 10.1002/art.39802 [DOI] [PubMed] [Google Scholar]
- 50.Contreras A, Doan N, Chen C, Rusitanonta T, Flynn MJ, Slots J. Importance of Dialister pneumosintes in human periodontitis. Oral microbiology and immunology. 2000;15(4):269–72. doi: 10.1034/j.1399-302x.2000.150410.x [DOI] [PubMed] [Google Scholar]
- 51.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–7. doi: 10.1136/gut.2010.223263 [DOI] [PubMed] [Google Scholar]
- 52.Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53(5):685–93. doi: 10.1136/gut.2003.025403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castellanos Monedero JJ, Prieto Gomez E, Carranza Gonzalez R. [Pneumonia due to Dialister pneumosintes in a patient with chronic obstructive pulmonary disease]. Archivos de bronconeumologia. 2010;46(12):663–4. doi: 10.1016/j.arbres.2009.05.003 [DOI] [PubMed] [Google Scholar]
- 54.Kang DW, Park JG, Ilhan ZE, Wallstrom G, Labaer J, Adams JB, et al. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PloS one. 2013;8(7):e68322. doi: 10.1371/journal.pone.0068322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Movement disorders: official journal of the Movement Disorder Society. 2015;30(3):350–8. doi: 10.1002/mds.26069 [DOI] [PubMed] [Google Scholar]
- 56.Neyrinck AM, Possemiers S, Druart C, Van de Wiele T, De Backer F, Cani PD, et al. Prebiotic effects of wheat arabinoxylan related to the increase in bifidobacteria, Roseburia and Bacteroides/Prevotella in diet-induced obese mice. PLoS One. 2011;6(6):e20944. doi: 10.1371/journal.pone.0020944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. doi: 10.1371/journal.pone.0009085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009;106(7):2365–70. doi: 10.1073/pnas.0812600106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, et al. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn’s disease. PloS one. 2012;7(7):e41594. doi: 10.1371/journal.pone.0041594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PloS one. 2012;7(6):e39743. doi: 10.1371/journal.pone.0039743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dey N, Soergel DA, Repo S, Brenner SE. Association of gut microbiota with post-operative clinical course in Crohn’s disease. BMC gastroenterology. 2013;13(1):1–11. doi: 10.1186/1471-230X-13-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell host & microbe. 2014;15(3):382–92. doi: 10.1016/j.chom.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell host & microbe. 2008;3(4):213–23. doi: 10.1016/j.chom.2008.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. The ISME journal. 2010;4(2):232–41. doi: 10.1038/ismej.2009.112 [DOI] [PubMed] [Google Scholar]
- 65.Martínez I, Wallace G, Zhang C, Legge R, Benson AK, Carr TP, et al. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Applied and environmental microbiology. 2009;75(12):4175–84. doi: 10.1128/AEM.00380-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, et al. Microbiota conservation and BMI signatures in adult monozygotic twins. The ISME journal. 2013;7(4):707–17. doi: 10.1038/ismej.2012.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–99. doi: 10.1016/j.cell.2014.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57(2):601–9. doi: 10.1002/hep.26093 [DOI] [PubMed] [Google Scholar]
- 69.Walters WA, Xu Z, Knight R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS letters. 2014;588(22):4223–33. doi: 10.1016/j.febslet.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang S, Chen X, Zhou L, Wang C, Chen Q, Lin R, et al. Faecal microbiota and functional capacity associated with weaning weight in meat rabbits. Microbial biotechnology. 2019;12(6):1441–52. doi: 10.1111/1751-7915.13485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gallardo-Becerra L, Cornejo-Granados F, García-López R, Valdez-Lara A, Bikel S, Canizales-Quinteros S, et al. Metatranscriptomic analysis to define the Secrebiome, and 16S rRNA profiling of the gut microbiome in obesity and metabolic syndrome of Mexican children. Microbial cell factories. 2020;19(1):1–18. doi: 10.1186/s12934-019-1269-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang J, McDowell A, Kim EK, Seo H, Lee WH, Moon C-M, et al. Development of a colorectal cancer diagnostic model and dietary risk assessment through gut microbiome analysis. Experimental & molecular medicine. 2019;51(10):1–15. doi: 10.1038/s12276-019-0313-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Troy EB, Kasper DL. Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Frontiers in bioscience: a journal and virtual library. 2010;15:25. doi: 10.2741/3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hickey CA, Kuhn KA, Donermeyer DL, Porter NT, Jin C, Cameron EA, et al. Colitogenic Bacteroides thetaiotaomicron antigens access host immune cells in a sulfatase-dependent manner via outer membrane vesicles. Cell host & microbe. 2015;17(5):672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, Lee Y, Kim Y, Lee S, Ryu S, Fukuda S, et al. Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal immunology. 2017;10(1):104–16. doi: 10.1038/mi.2016.42 [DOI] [PubMed] [Google Scholar]
- 76.Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141(7):1241–52. doi: 10.1016/j.cell.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zakharzhevskaya NB, Vanyushkina AA, Altukhov IA, Shavarda AL, Butenko IO, Rakitina DV, et al. Outer membrane vesicles secreted by pathogenic and nonpathogenic Bacteroides fragilis represent different metabolic activities. Scientific reports. 2017;7(1):1–16. doi: 10.1038/s41598-016-0028-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Slots J, Listgarten MA. Bacteroides gingivalis, Bacteroides intermedius and Actinobacillus actinomycetemcomitans in human periodontal diseases. Journal of clinical periodontology. 1988;15(2):85–93. doi: 10.1111/j.1600-051x.1988.tb00999.x [DOI] [PubMed] [Google Scholar]
- 79.Prindiville TP, Sheikh RA, Cohen SH, Tang YJ, Cantrell MC, Silva J Jr. Bacteroides fragilis enterotoxin gene sequences in patients with inflammatory bowel disease. Emerging infectious diseases. 2000;6(2):171. doi: 10.3201/eid0602.000210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell host & microbe. 2011;9(5):390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bedford A, Gong J. Implications of butyrate and its derivatives for gut health and animal production. Animal Nutrition. 2018;4(2):151–9. doi: 10.1016/j.aninu.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Staley C, Weingarden AR, Khoruts A, Sadowsky MJ. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Applied microbiology and biotechnology. 2017;101(1):47–64. doi: 10.1007/s00253-016-8006-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramírez-Pérez O, Cruz-Ramón V, Chinchilla-López P, Méndez-Sánchez N. The role of the gut microbiota in bile acid metabolism. Annals of hepatology. 2018;16(1):21–6. [DOI] [PubMed] [Google Scholar]
- 84.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut microbes. 2016;7(3):189–200. doi: 10.1080/19490976.2015.1134082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Den Besten G, Van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. Journal of lipid research. 2013;54(9):2325–40. doi: 10.1194/jlr.R036012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nature communications. 2020;11(1):1–18. doi: 10.1038/s41467-019-13993-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, et al. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell host & microbe. 2018;23(4):458–69. e5. doi: 10.1016/j.chom.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yadav S, Jha R. Strategies to modulate the intestinal microbiota and their effects on nutrient utilization, performance, and health of poultry. Journal of animal science and biotechnology. 2019;10(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Heredia FP, Gomez-Martinez S, Marcos A. Obesity, inflammation and the immune system. The Proceedings of the Nutrition Society. 2012;71(2):332–8. doi: 10.1017/S0029665112000092 [DOI] [PubMed] [Google Scholar]
- 90.Myasoedova E, Crowson CS, Kremers HM, Roger VL, Fitz-Gibbon PD, Therneau TM, et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Annals of the rheumatic diseases. 2011;70(3):482–7. doi: 10.1136/ard.2010.135871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Frontiers in immunology. 2012;3:107. doi: 10.3389/fimmu.2012.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luke A, Bovet P, Forrester TE, Lambert EV, Plange-Rhule J, Schoeller DA, et al. Protocol for the modeling the epidemiologic transition study: a longitudinal observational study of energy balance and change in body weight, diabetes and cardiovascular disease risk. BMC public health. 2011;11:927. doi: 10.1186/1471-2458-11-927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Flores GE, Bates ST, Caporaso JG, Lauber CL, Leff JW, Knight R, et al. Diversity, distribution and sources of bacteria in residential kitchens. Environmental microbiology. 2013;15(2):588–96. doi: 10.1111/1462-2920.12036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walters W, Hyde ER, Berg-Lyons D, Ackermann G, Humphrey G, Parada A, et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems. 2016;1(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal. 2012;6(8):1621–4. doi: 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leung MH, Tong X, Wilkins D, Cheung HH, Lee PK. Individual and household attributes influence the dynamics of the personal skin microbiota and its association network. Microbiome. 2018;6(1):26. doi: 10.1186/s40168-018-0412-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551(7681):457. doi: 10.1038/nature24621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hall M, Beiko RG. 16S rRNA Gene Analysis with QIIME2. Methods in molecular biology (Clifton, NJ). 2018;1849:113–29. doi: 10.1007/978-1-4939-8728-3_8 [DOI] [PubMed] [Google Scholar]
- 99.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nature methods. 2016;13(7):581–3. doi: 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 2006;72(7):5069–72. doi: 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microbial ecology in health and disease. 2015;26:27663. doi: 10.3402/mehd.v26.27663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boca SM, Leek JT. A direct approach to estimating false discovery rates conditional on covariates. PeerJ. 2018;6:e6035. doi: 10.7717/peerj.6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic acids research. 2012;40(Database issue):D109–14. doi: 10.1093/nar/gkr988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal, Complex Systems. 2006;1695(5):1–9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(JPG)
(A) USA, the United States of America, (B) RSA, South Africa (C) Ghanaian and (D) Jamaican populations.
(JPG)
(A) USA, the United States of America, (B) RSA, South Africa (C) Ghanaian and (D) Jamaican populations.
(JPG)
(A) USA, the United States of America, (B) RSA, South Africa (C) Ghanaian and (D) Jamaican populations.
(JPG)
(A) Alpha diversity analysis (Shannon Index); (B) beta diversity analysis (weighted UniFrac distance metric) from *** p < 0.001.
(JPG)
K18012, 3,5-diaminohexanoate dehydrogenase; K18013, 3-keto-5-aminohexanoate cleavage enzyme. * p < 0.05.
(JPG)
Data Availability Statement
All 16S rRNA sequences and sample metadata are publicly available in EBI under accession no. ERP115612 (Study: PRJEB32880).